 Michal Cohen1
Michal Cohen1 Sharon Guger2 and Jill Hamilton1*
Sharon Guger2 and Jill Hamilton1*- 1 Division of Endocrinology, Department of Pediatrics, The Hospital for Sick Children, University of Toronto, Toronto, ON, Canada
- 2 Department of Psychology, The Hospital for Sick Children, University of Toronto, Toronto, ON, Canada
Craniopharyngioma are rare histologically benign brain tumors that develop in the pituitary–hypothalamic area. They may invade nearby anatomical structures causing significant rates of neurological, neurocognitive, and endocrinological complications including remarkable hypothalamic damage. Information regarding long term implications of the tumors and treatment in the pediatric population is accumulating, and treatment goals appear to be changing accordingly. In this review we aim to present data regarding long term complications of craniopharyngioma in children and adolescents and our experience from a large tertiary center. Hypothalamic dysfunction was noted to be the most significant complication, adversely affecting quality of life in survivors. Obesity, fatigue, and sleep disorders are the most notable manifestations of this dysfunction, and treatment is extremely difficult. Changes in management in recent years show a potential for improved long term outcomes; we found a trend toward less aggressive surgical management and increasing use of adjuvant treatment, accompanied by a decrease in complication rates.
Introduction
Craniopharyngioma (Cp) tumors are rare epithelial brain tumors that develop from remnants of Rathke’s pouch. They can be located along an axis extending from the sella turcica, through the pituitary stalk, to the hypothalamus. Tumors are partly cystic and have benign histological appearance (Bunin et al., 1998; Muller, 2010). As craniopharyngiomas enlarge they invade nearby anatomical structures and despite their histology can have an aggressive clinical course. Both the tumor and its treatment can lead to significant neurological and endocrinological complications; the most devastating being related to hypothalamic dysfunction (Sughrue et al., 2011). Long term survival is high (Poretti et al., 2004); however complications have serious implications on quality of life. These tumors present a great challenge to the neurosurgeon due to their location and proximity to vital structures, and controversy as to the preferable method for treatment exists. In the past decade treatment goals appear to be changing, with many centers adopting a less aggressive approach. Advancements in radiation technology as well as in chemotherapy methods and medications have been remarkable and these provide a variety of potential adjuvant treatments. In this report we aim to review the long term complications of Cp tumors and their treatment in the pediatric population; including data from the literature as well as our own experience.
Craniopharyngioma Treatment
The preferable approach for treatment of Cp tumors in children remains controversial despite many years of experience. In the past, gross total resection (GTR) of the tumor was the method of choice (Hoffman et al., 1992; De Vile et al., 1996; Van Effenterre and Boch, 2002; Poretti et al., 2004; Caldarelli et al., 2005) and it remains so in some centers (Caldarelli et al., 2005; Elliott and Wisoff, 2010). Based on the benign histology of the tumor this approach is aimed at achieving cure. However, these patients were found to have a considerable rate of recurrences, occurring in up to one-third of patients (Hoffman et al., 1992; Jung et al., 2010; Steno et al., 2011) as well as a high rate of complications and morbidity (Crom et al., 2010; Sughrue et al., 2011). Hypopituitarism, hypothalamic dysfunction, and neurocognitive impairments in areas of memory, learning, and school performance, are prevalent in series using primarily GTR (Hoffman et al., 1992; De Vile et al., 1996; Poretti et al., 2004). Taking into consideration the great significance of these morbidities, in recent years a surgical approach that is more patient and tumor specific is gaining acceptance; reserving extensive resections for favorably located tumors, not involving the hypothalamus (De Vile et al., 1996; Poretti et al., 2004; Steno et al., 2011). Endoscopic trans-sphenoidal surgery is used for smaller, primary intrasellar tumors (Elliott et al., 2011). Characteristically Cp tumors have single or multiple cysts within the tumor (Steinbok and Hukin, 2010) and limited resection or cyst decompression is an alternative surgical approach that addresses Cp more as a chronic disease. In cases of emergent decompression, this procedure may allow time for better planning of future treatment, and a catheter implanted in the cyst can be used later for administering intra-cystic treatment. In our experience, even with decompression as a sole treatment, patients can have a stable course not requiring additional treatment for prolonged periods.
Adjuvant treatment: In patients with predominantly cystic tumors, installation of medications, or radioisotopes to shrink the tumor may be of benefit (Hukin et al., 2007; Cavalheiro et al., 2010; Steinbok and Hukin, 2010). Intra-cystic bleomycin (ICB) for pediatric Cp has been in use in Canada since 1995 (Hukin et al., 2007), although side effects such as local leakage into adjacent brain tissue have raised some concerns and warrants close monitoring (Lafay-Cousin et al., 2007). Intra-cystic interferon-α for treatment of patients with Cp has shown good results, with a reduced complication rate and minimal side effects (Cavalheiro et al., 2010; Steinbok and Hukin, 2010). Radiation therapy for pediatric Cp has been in use for more than half a century (Kiehna and Merchant, 2010); it can be used both as an adjuvant in primary treatment and for disease recurrence. Limited surgery followed by radiation might induce favorable outcomes when compared to more aggressive solely surgical treatments (De Vile et al., 1996; Merchant et al., 2002; Kiehna and Merchant, 2010). Radiotherapy might have significant short and long term adverse effects and these pose a major limitation to the use of this treatment modality in children (Merchant et al., 2002; Caldarelli et al., 2005; Kiehna and Merchant, 2010).
Determining the exact prevalence of outcomes for the different treatment modalities is complicated by multiple factors and no randomized control trials exist in the pediatric population. Decisions regarding surgery and adjuvant therapy are done based on tumor and child characteristics as well as the treating team’s approach and experience. Late outcomes are also influenced by treatments given for disease recurrence. In addition, outcomes are not uniformly assessed or presented. Despite these limitations, there are data from the literature suggesting improved outcomes for less aggressive surgical approaches combined with radiation in terms of disease recurrence, neurocognitive functioning, and prevalence of diabetes insipidus (DI) and severe obesity. These are presented in more detail in the following sections.
Over the past decade, the surgical approach to Cp in our institution has changed to become less aggressive (Figure 1). In the years 1975–1989 90% of children had a total resection (Hoffman et al., 1992), between 1990 and 2001 86% (Ahmet et al., 2006) had a total resection and from 2001 to 2011 only 10% had a total resection. In recent years radiation and intra-cystic chemotherapy are given to a larger percent of patients.
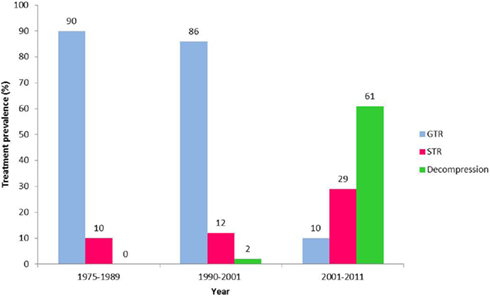
Figure 1. Trends in the surgical approach to craniopharyngioma 1975–2011. Prevalence of gross total resection (GTR – blue), subtotal resection (STR – red), and cyst decompression (Decompression – green) surgery as the primary treatment for pediatric craniopharyngioma in our institution over more than three decades; 1975–1989 (Hoffman et al., 1992), 1990–2001 (Ahmet et al., 2006), 2001–2011.
Tumor Recurrence and Regrowth
Recurrence and regrowth in Cp are not uncommon and can be expected even with extensive resection. Tumor recurrences are often asymptomatic and diagnosed on routine follow-up imaging (Hoffman et al., 1992; Elliott and Wisoff, 2010). Rates of recurrence following GTR range between 17 and 36% (Hoffman et al., 1992; Merchant et al., 2002; Poretti et al., 2004; Ahmet et al., 2006; Elliott and Wisoff, 2010; Jung et al., 2010; Elliott et al., 2011; Steno et al., 2011); however some report much lower rates of 7.7–10% following neuroradiologically confirmed total resection by trans-cranial approach (De Vile et al., 1996; Caldarelli et al., 2005), or with trans-sphenoidal GTR (Elliott et al., 2011). Regrowth rates after incomplete removal are higher, ranging between 43 and 67% (Jung et al., 2010; Steno et al., 2011). The 10-year recurrence rate after combined surgical and radiation treatment is reported to be lower, ranging between 0 and 30%, with some reports demonstrating improved disease control for a combination of biopsy or cyst resection and radiation compared with subtotal resection and radiation (Clayton et al., 1988; Stripp et al., 2004; Lin et al., 2008; Kiehna and Merchant, 2010). Recurrences occur at variable time intervals from the initial surgery, including many years after surgery (Hoffman et al., 1992; De Vile et al., 1996; Merchant et al., 2002; Caldarelli et al., 2005; Elliott and Wisoff, 2010) and occasionally appear in a location away from the original Cp site (Caldarelli et al., 2005). Mean interval from treatment to recurrence is most commonly reported to be approximately 3 years (Hoffman et al., 1992; Poretti et al., 2004; Caldarelli et al., 2005; Elliott and Wisoff, 2010). Risk factors for recurrence include a large tumor size, young age, and severe hydrocephalus at presentation; incomplete tumor resection and previous recurrence are additional risk factors (Hoffman et al., 1992; De Vile et al., 1996; Van Effenterre and Boch, 2002; Poretti et al., 2004; Caldarelli et al., 2005; Elliott and Wisoff, 2010; Jane et al., 2010; Jung et al., 2010; Steno et al., 2011).
Pituitary Deficiencies
Pituitary hormone deficiencies are common in children treated for Cp. At diagnosis 40–87% of patients (Hoffman et al., 1992; Caldarelli et al., 2005; Muller, 2008) have been identified to have at least one hormone deficiency and 17–27% (Hoffman et al., 1992; Muller, 2008; Elliott and Wisoff, 2010) have been reported to have DI. Post surgery, the rate of pituitary hormone deficiencies increases and has been reported to be 80–100% (Hoffman et al., 1992; De Vile et al., 1996; Merchant et al., 2002; Muller et al., 2004; Poretti et al., 2004; Caldarelli et al., 2005; Elliott and Wisoff, 2010; Jung et al., 2010; Steno et al., 2011). Prevalence is comparable even with pituitary stalk preservation (Jung et al., 2010). We have found rates similar to those reported in the literature for individual deficiencies and an encouraging decrease in rates of panhypopituitarism with less aggressive treatment (Figure 2). A majority of patients need both anterior and posterior pituitary hormonal replacement. The evaluation of anterior pituitary hormone deficiencies is not uniform in reports from different studies. Those based on rates of hormone replacement might underestimate the prevalence of growth hormone (GH) and gonadotropin deficiencies or overestimate ACTH deficiency if reporting hormone replacement at any time point. Overall, ACTH deficiency is described to range between 70 and 95%, with no clear trend related to treatment modality. TSH deficiency ranges between 80 and 96% in different series with a slightly higher rate for the combination of surgery and radiation compared to more aggressive surgery. Gonadotropin deficiency is most commonly described to be around 30–40%. Clinical or biochemical GH deficiency has been described at diagnosis in 26–75% of children with Cp (Ahmet et al., 2006; Muller, 2008), and decreases in height SD score (SDS) may occur years before diagnosis (Muller et al., 2004). GH deficiency following treatment for Cp is common, found in about 70–92% of patients, the literature does not clearly delineate a significant difference between the treatment modalities (Muller et al., 2004, 2011; Halac and Zimmerman, 2005; Crom et al., 2010) and a good response to GH treatment is described (Geffner et al., 2004).
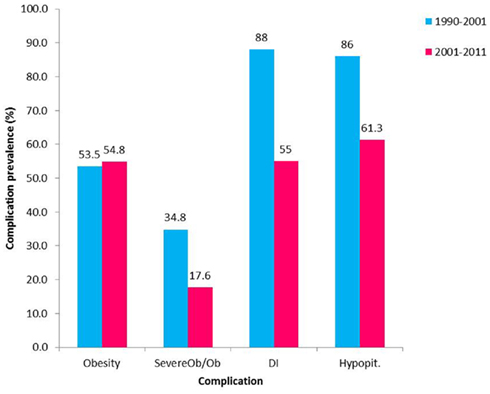
Figure 2. Prevalence of complications following Cp treatment in the past two decades. Prevalence of hypopituitarism (Hypopit), diabetes insipidus (DI), and obesity between the years 1990–2001 (Ahmet et al., 2006; blue) and 2001–2011 (red). Obesity (Ob) defined as BMI > 95th percentile for age and gender. Severe obesity defined as BMI > 99th percentile for age and gender or BMI > 40 kg/m2 if more than 18 years old); Severe Ob/Ob – the percent of obese patients with severe obesity.
Transient post-surgical DI is a prevalent finding that has been described to occur in almost all patients in some series (Poretti et al., 2004; Caldarelli et al., 2005). In our experience, the rapid shifts in serum sodium that commonly complicated the course post GTR and significantly prolonged the post-surgical admission have decreased remarkably in magnitude when using less aggressive surgical procedures. We also noted a decrease in prevalence of permanent DI (Figure 2). Permanent DI after treatment was found in different series to range between 60–90% after aggressive surgery and 50–55% after less aggressive surgery combined with radiation therapy (Hoffman et al., 1992; De Vile et al., 1996; Merchant et al., 2002; Poretti et al., 2004; Caldarelli et al., 2005; Ahmet et al., 2006; Elliott and Wisoff, 2010; Elliott et al., 2011; Muller et al., 2011). Replacement treatment for pituitary hormones is in most instances readily accessible; however hypothalamic injury in this population might interfere with thirst regulation complicating the DI management and putting the children at risk for life threatening electrolyte imbalances. Pituitary hormone deficiencies might also occur late, particularly when adjuvant treatment is used, therefore monitoring pituitary function is a life-long requirement.
Linear Growth
Despite the common occurrence of GH deficiency after treatment for Cp not all require GH replacement for growth induction. In a study of Cp-related hypothalamic obesity (CRHO) conducted by our group, one-third of the patients were growing despite GH deficiency and no supplementation (O’gorman et al., 2010b; Simoneau-Roy et al., 2010); another study found an even higher rate of normal growth without GH (Srinivasan et al., 2004). Patients with hypothalamic involvement were found to achieve normal height at last follow-up more often than patients without hypothalamic involvement (Muller et al., 2004). This phenomenon of “growth without GH” has been described in Cp patients almost five decades ago (Matson, 1964). The physiology of growth in these cases is not fully understood. Insulin and leptin are thought to induce growth in the fetus and in obese children and are hypothesized to induce growth in Cp (Costin et al., 1976; Geffner, 1996; Phillip et al., 2002). Leptin was shown to have characteristics of a bone growth factor acting directly at the level of bone growth centers, independently of GH (Phillip et al., 2002). Mechanisms by which insulin stimulates growth include its known anabolic effects; at high levels it may bind to the insulin-like growth factor-1 (IGF-1) receptor and induce growth, and through its actions to decrease IGF-binding protein 1 levels it may increase levels of free IGF-1 (Phillip et al., 2002). In support of this theory, obese Cp patients were found to have a higher height SDS at diagnosis and at last follow-up with no difference in the usage of GH, thyroxine, or hydrocortisone (Muller et al., 2001); and increased insulin levels were found in children with Cp. In contrast, another study found that children who were growing despite GH deficiency were not different from those requiring GH treatment in terms of anthropometrical measures, body composition, and metabolic indexes; including insulin levels (Srinivasan et al., 2004). Sex hormones may also induce growth in these patients (Phillip et al., 2002). Finally, the effects of other local growth factors acting on bone have been hypothesized to contribute to this phenomenon (Geffner, 1996; Phillip et al., 2002).
Hypothalamic Dysfunction
Hypothalamic dysfunction in children with Cp is common and has been found at diagnosis in 35% of patients (Elliott and Wisoff, 2010). It dramatically increases following treatment; in some series occurring in up to 65–80% of patients (Poretti et al., 2004; Elliott and Wisoff, 2010). This complication can significantly compromise health-related quality of life (HRQoL) and be extremely resilient to treatment. Some of the manifestations such as obesity can be quantified by relatively simple anthropometric measures; however, others may be more elusive in nature. Fatigue, behavioral changes, circadian rhythm, and sleep irregularities and imbalances in regulation thirst, body temperature, heart rate, or blood pressure might be more difficult for both patients and physicians to appreciate. These dysfunctions are inter-related in a way that can worsen outcome. The pre-surgical evaluation of hypothalamic damage might be difficult both clinically and radiologically (Steno et al., 2011); tumor involvement of the third ventricle, or obstructive hydrocephalus are suggestive findings (Caldarelli et al., 2005).
Sleep Dysregulation
Hypothalamic injury may result in disturbances in sleep–wake patterns. Our and others’ clinical experience is that patients with CRHO commonly suffer from an altered pattern of circadian rhythm with early morning awakening, followed by an extra period of sleep during the afternoon. Thus, sleep fragmentation and increased daytime sleepiness are common. Increased daytime sleepiness has been reported by one-third of children with Cp that were surveyed with a prevalence of 40% in the severely obese children (Muller et al., 2002, 2006b; Snow et al., 2002; Poretti et al., 2004). Actigraph recording in three patients with Cp and daytime hypersomnolence demonstrated irregular bedtimes, frequent night-time activity, and inappropriate daytime episodes of rest, consistent with our clinical experience (Lipton et al., 2009). Melatonin secretion occurs during hours of darkness; it affects sleep patterns and potentially has a role in circadian rhythm regulation. An altered pattern of diurnal melatonin secretion with decreased night-time melatonin levels was detected in severely obese Cp patients (Muller et al., 2002; Lipton et al., 2009). Nocturnal levels had a negative relation to both the degree of obesity and the sleepiness scoring. Melatonin treatment in Cp patients with severe daytime sleepiness resulted in an increment in morning and night melatonin levels, improvement in daytime sleepiness and physical activity (Muller et al., 2006b).
Modafinil is a wake-promoting agent that has shown beneficial effects in adults with Cp-related hypersomnolence (Crowley et al., 2011). We have offered this medication to two patients with hypothalamic obesity (HO) and significant daytime fatigue related to treatment for tumors in the hypothalamic area. Both patients reported a remarkable improvement in alertness and activity tolerance, and the medication was well tolerated; no significant effect on weight has been noticed. Future research on the effects of this medication in children with Cp is warranted.
In addition to the effect of hypothalamic damage, the obesity and metabolic dysfunction in patients with Cp might be associated with sleep-disordered breathing (Redline et al., 2007). Our group found sleep-disordered breathing to be increased in adolescents with Cp-related obesity compared with BMI-matched controls (O’gorman et al., 2010b). Specifically, increased sleep fragmentation and reduced sleep efficiency were found in children with Cp; the obstructive apnea–hypopnea index was higher and correlated negatively with adiponectin levels, an adipokine with insulin sensitizing properties. We propose considering routine polysomnography in obese patients with craniopharyngioma.
Activity and Energy Expenditure
Weight gain in patients with Cp was traditionally thought to be induced by hyperphagia; however a number of studies suggest that although subjective hunger may be higher, energy intake is not increased when compared to control subjects (Harz et al., 2003; Holmer et al., 2010). A decreased metabolic rate (both resting and total energy expenditure) has been suggested to contribute to weight gain in this population. Adults with childhood onset Cp as well as children with Cp were found to have a lower resting energy expenditure (REE) compared to controls (Shaikh et al., 2008b; Holmer et al., 2010; Kim et al., 2010). This difference was not explained by differences in body composition. The energy intake/REE ratio was significantly lower in those with tumors involving the third ventricle (Holmer et al., 2010). Patients with childhood Cp were also found to have decreased physical activity compared to healthy obese and normal weight controls contributing to overall lowering of total energy expenditure (Harz et al., 2003; Shaikh et al., 2008b; Holmer et al., 2009, 2010). Factors that could potentially contribute to decreased activity are neurological and visual deficits, increased daytime sleepiness, low levels of testosterone and GH and psychosocial difficulties such as teasing and bullying leading to self-consciousness about engaging in physical activity.
Weight Gain and Obesity
Of the manifestations of hypothalamic damage in children treated for Cp, rapid weight gain is the most concerning complication for patients, their families, and practitioners. This occurs despite adequate replacement of pituitary hormone deficiencies. The hypothalamic disturbance in energy management contributes to obesity and is further augmented by factors limiting activity. There is evidence demonstrating increased weight gain in children with Cp occurring years before diagnosis (Muller et al., 2004). At diagnosis 12–19% of patients were reported to be obese (Hoffman et al., 1992; Poretti et al., 2004; Ahmet et al., 2006; Muller, 2008). Obesity develops early after treatment and weight gain was found to rapidly increase in the first 6–12 months after treatment (Muller et al., 2001; Ahmet et al., 2006); later on BMI stabilizes but obesity continues to remain a problem (Ahmet et al., 2006; Holmer et al., 2010). Following treatment, the prevalence of obesity is higher, reaching up to 55% (Hoffman et al., 1992; Muller et al., 2001, 2003b; Poretti et al., 2004; Srinivasan et al., 2004; Ahmet et al., 2006; Lek et al., 2010; O’gorman et al., 2010b; Elliott et al., 2011). Review of our patients indicates a lower prevalence of severe obesity associated with less aggressive surgery (Figure 2). This is supported by more recent reports of lower rates of severe obesity after combined surgical and radiation approaches (Lin et al., 2008; Crom et al., 2010). However the literature does not demonstrate a significant change in the prevalence of overall obesity with the change in treatment approach. This might be related to the small number of patients in studies as well as to the approach to assessment of obesity; more detailed stratification of the degree of obesity appears to provide additional information. When evaluating body composition and fat distribution in children with Cp, results are conflicting; some studies have demonstrated patients to have increased fat free mass and muscle mass when compared to controls (Holmer et al., 2010; Kim et al., 2010) while others found comparable fat mass in Cp patients and controls (Srinivasan et al., 2004; Shaikh et al., 2008a). Excess weight has a significantly adverse effect on HRQoL and self-esteem (Hoffman et al., 1992; Muller et al., 2001) and is associated with difficulties at school as well as at home. Adults with childhood onset Cp and obesity also reported a higher tendency to restrict food intake for controlling body weight compared with controls (Holmer et al., 2010). Risk factors for HO include hypothalamic involvement of the tumor or treatment, tumor growth inside the cavity of the third ventricle, pre-operative hydrocephalus, large tumor size, a lower rate of successful GTR, higher radiation doses to the hypothalamus (>51 Gy), higher BMI in early childhood, higher BMI, and height SDS at diagnosis and familial predisposition for obesity (Muller et al., 2001, 2003b, 2004, 2011; Lustig, 2002; Lustig et al., 2003b; Caldarelli et al., 2005; Ahmet et al., 2006; Holmer et al., 2009, 2010; Lek et al., 2010; Roth et al., 2011; Steno et al., 2011). The dose and duration of post-operative dexamethasone treatment was found to have only a short term effect on weight gain and treatment correlated with weight gain during the first post-operative year but not with long term development of obesity (Muller et al., 2003b).
Metabolic Syndrome
In a study conducted by our group, children and adolescents with CRHO were found to have an increased rate of the metabolic syndrome (MS) occurring in 10 out of 15 subjects studied (66%) compared to 3 out of 15 BMI-matched controls (20%; Simoneau-Roy et al., 2010). Impaired glucose tolerance was more prevalent in the Cp subjects; they also demonstrated higher levels of free fatty acids and TNF-alpha, molecules associated with MS (Figure 3). In agreement with our results, children and adolescents following Cp removal were found to have features of the MS, including higher abdominal fat and less favorable lipid profiles when compared with matched controls (Srinivasan et al., 2004). In contrast, adults with childhood onset Cp, were demonstrated increased risk for cardiovascular disease compared to a control group; however, after adjustment for BMI, these differences disappeared (Holmer et al., 2009). Further work is needed to assess longer term outcomes related to risk for type 2 diabetes and cardiovascular disease in this population.
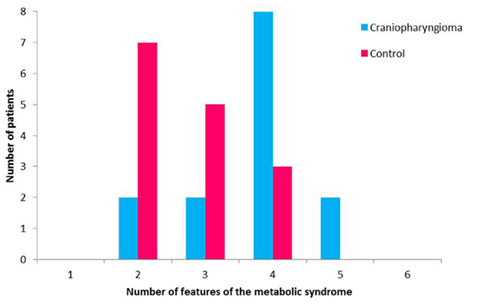
Figure 3. Features of the metabolic syndrome in patients with craniopharyngioma and control subjects.
Autonomous Nervous System Balance in Craniopharyngioma
The hypothalamic regulation of appetite and energy homeostasis is a complex system involving hormonal and neuronal pathways connecting the hypothalamus with the alimentary system and periphery, including adipose tissue which also produces hormones and cytokines. The hypothalamus receives input regarding adipose accumulation, satiety, and metabolism from peripheral hormones including leptin, ghrelin, and insulin respectively (Lustig, 2002); it exerts its efferent signal via the autonomic nervous system (ANS). Damage that lowers the sympathetic nervous system (SNS) activity and/or increases the parasympathetic nervous system (PNS) activity could contribute to a positive energy balance and weight gain and both conditions have been theorized to contribute to the development of HO (Lustig, 2002; Hochberg and Hochberg, 2010). The common symptom of increased fatigue in these patients could also be related to a decreased sympathetic tone.
In support of decreased activity of the SNS, urine catecholamine levels were found to be lower in children with Cp compared to controls (Coutant et al., 2003) and levels negatively associate with the degree of obesity and physical activity (Roth et al., 2007). Further support comes from the improvement seen in terms of weight, activity, and attention when treating Cp patients with medications that increase sympathetic tone (Mason et al., 2002; Ismail et al., 2006; Danielsson et al., 2007; Schultes et al., 2009). SNS activity is thought to decrease insulin secretion through inhibition of pancreatic beta-cells and to inhibit insulin stimulated leptin release from adipocytes (Cammisotto and Bukowiecki, 2002; Lustig, 2003). Elevated levels of both hormones have been reported in Cp-related obesity, even after adjusting for fat mass, and may in part be due to an decreased sympathetic activity (Goldstone et al., 2005; Shaikh et al., 2008a; Guran et al., 2009; Holmer et al., 2009, 2010; O’gorman et al., 2010a; Simoneau-Roy et al., 2010; Roth et al., 2011).
Increased vagal activity as a result of hypothalamic damage may lead to increased pancreatic beta-cell stimulation and hypersecretion of insulin (Lustig, 2002). This is supported by the aforementioned increased insulin levels in patients with Cp-related obesity and by improvement in terms of weight gain with treatments targeted at suppressing beta-cell activity. Octreotide has been found to attenuate glucose induced insulin excursions; and to result in reduced weight gain, or in some cases, weight loss (Lustig et al., 1999, 2003a; Inge et al., 2007). Our group has investigated the combined effect of two medications on children with Cp and HO; diazoxide in order to decrease insulin secretion and metformin to enhance insulin action. A decrease in weight gain and BMI was also noted and the best response was seen in subjects with the highest glucose stimulated insulin secretion prior to receiving the medication (Hamilton et al., 2011).
Neurological and Visual Outcomes
Due to the tumor’s location, visual deficits in children with Cp are relatively common including impairments in both visual acuity and visual fields. Visual impairment was found to exist as an initial manifestation of pediatric Cp in more than half of the patients (Muller, 2008) with some improvement in vision in 41–48% of patients post-surgically (Caldarelli et al., 2005; Elliott et al., 2011). Risk factors for post-surgical visual impairment include severe pre-surgical visual deficits and tumors located in the prechiasmatic area (Caldarelli et al., 2005; Steno et al., 2011). Despite potential visual improvement after treatment overall post treatment rates of visual impairment are reported in about 45–70% of patients; different treatment modalities result in comparable rates (De Vile et al., 1996; Stripp et al., 2004; Merchant et al., 2006; Lin et al., 2008; Crom et al., 2010). Improved results were found in patients that underwent a trans-sphenoidal procedure (Elliott et al., 2011).
Neurological abnormalities include hemiparesis, epilepsy, cranial nerve deficits, learning problems, hearing loss, cerebrovascular disease manifestations, and headaches (Merchant et al., 2002; Crom et al., 2010; Elliott and Wisoff, 2010). A significant part of these are transient and the total prevalence of long term neurological complications is reported to be 8% (Caldarelli et al., 2005), however rises to 36% for giant tumors (Elliott and Wisoff, 2010) and 30% when including visual and neurological complications together (Poretti et al., 2004).
Health-Related Quality of Life, Psychosocial Functioning and Neurocognitive Outcome
Health-related quality of life in children can be affected by both the Cp tumor itself and the treatment received. Reports assessing psychosocial and physical functioning, show variable results ranging from excellent in a majority of subjects to impaired function in almost half of the patients (Hoffman et al., 1992; Van Effenterre and Boch, 2002; Poretti et al., 2004). The most common areas of difficulty reported include social and emotional functioning, with patients rating their psychosocial health to be lower than their physical health (Poretti et al., 2004). Other challenges included somatic complaints such as pain, mobility, and self-care (Merchant et al., 2002; Poretti et al., 2004). Children with Cp were found to have lower HRQoL in all domains; and parental reports of children’s social and total functioning were significantly lower compared to scores from parents of survivors of other cancers (Rakhshani et al., 2010). Behavioral questionnaires indicate more frequent presence of psychopathological symptoms, including depression, anxiety, and withdrawal. The most frequent problems in children’s everyday functioning included inability to control emotions, difficulties in learning, unsatisfactory peer relationships, and concerns regarding physical appearance (Crom et al., 2010; Ondruch et al., 2011).
Factors associated with worse outcomes in terms of HRQoL as well as psychosocial and neurocognitive functioning include younger age at diagnosis and pre-operative functional impairment; tumor characteristics including larger size, hypothalamic, and third ventricle involvement at presentation. Treatment type has also been implicated, with worse outcomes for surgery alone compared to limited surgery and radiotherapy and for multiple operations for tumor recurrence (Merchant et al., 2002; Poretti et al., 2004). Neurological, ophthalmological, and endocrine sequelae adversely affect outcome (Hoffman et al., 1992; Merchant et al., 2002; Van Effenterre and Boch, 2002; Muller et al., 2003a; Poretti et al., 2004; Elliott and Wisoff, 2010; Steno et al., 2011) and hypothalamic dysfunction was found to have an important negative effect on physical ability, social functioning, and body image (Muller et al., 2001, 2003a; Poretti et al., 2004).
In addition to psychosocial function, long term neurocognitive complications following treatment for Cp may include cognitive problems, particularly affecting attention, executive function, working memory, and episodic memory (Cavazzuti et al., 1983; Riva et al., 1998; Carpentieri et al., 2001; Poretti et al., 2004; Sands et al., 2005; Kiehna et al., 2006; Crom et al., 2010; Ondruch et al., 2011). Various neurocognitive outcomes were assessed in different studies making comparison difficult. Series including children treated primarily with GTR report 57% of patients exhibit post treatment memory deficits (Hoffman et al., 1992; Poretti et al., 2004). A significant number of long term survivors of pediatric Cp treated primarily with radiation and subtotal surgical resection, were found to have psychological and educational deficits (Crom et al., 2010). Neurocognitive deficits have ranged to include memory disturbances, slower cognitive speed, attention problems, and behavioral lability (Cavazzuti et al., 1983; Colangelo et al., 1990; Carpentieri et al., 2001; Kiehna et al., 2006; Crom et al., 2010; Ondruch et al., 2011). While intact intellectual functioning has been reported in up to 82% of patients, visual memory is reduced, despite normal visual–spatial abilities (Crom et al., 2010; Ondruch et al., 2011). The acquired deficits in higher cognitive processing such as attention problems are considered precursors to poor academic achievement and vocational failure. Following radiation therapy IQ was found to remain stable overall, with improved results for older children (Kiehna and Merchant, 2010). The impact of conformal radiation therapy and clinical variables on measures of attention among children and young adults with brain tumors was prospectively examined (Kiehna et al., 2006). Over the first 5 years after treatment, patients with Cp demonstrated increased inattentiveness and profound inattentiveness was accompanied by markedly slower reaction time. Overall, newer focal beam radiation techniques demonstrated improved outcomes in terms of functional performance in up to 85% of patients (Merchant et al., 2002; Kiehna and Merchant, 2010).
Despite over a quarter of century of literature documenting the neurocognitive challenges encountered by individuals treated for Cp, intervention efforts have lagged. Recent case studies have examined the efficacy of cognitive rehabilitation for dysexecutive problems and behavioral lability (Metzler-Baddeley and Jones, 2010; Hammond and Hall, 2011). In a case report of a 2-month intervention using a combination of goal management therapy and naturally occurring distractions within the patients’ work environment, significant improvements in cognitive tests requiring organized behavior were reported. Social, emotional, and/or behavior problems, most notably aggression, have been reported (Anderson et al., 1997; Riva et al., 1998; Poretti et al., 2004). Despite their occurrence, the assumption of the biological underpinning of the behavioral disturbance appears to have limited attempts to effectively manage it with intervention. Behavioral treatment was used for severe aggressive behaviors demonstrated by a 6-year-old post-Cp resection (Hammond and Hall, 2011); the intervention included functional behavioral analysis followed by differential reinforcement of alternative behaviors and extinction with the goal of decreasing the frequency of aggressive behavior. Aggressive behavior subsequently decreased to below 88% of baseline levels and adaptive behaviors were found to increase significantly. These results suggest that the patient’s aggression was maintained by inadvertent social reinforcement. Taken together, these case studies suggest that cognitive rehabilitation approaches such as goal management therapy and functional behavioral analysis may be useful in supporting survivors of Cp adept to and compensate for cognitive and psychosocial challenges.
In light of the cognitive, behavioral, and emotional sequelae associated with treatment for Cp, monitoring and support is warranted. Ongoing counseling, educational, and advocacy for this unique group of brain tumor survivors will be key to optimizing their potential.
General Follow-Up Management of Children with Craniopharyngioma
Follow-up surveillance of the residual tumor or assessment for recurrence is addressed by the neurosurgery and oncology teams (see other reviews in this issue). Endocrine follow-up and surveillance should be conducted at least annually in those with no pituitary abnormalities as these may evolve over time. During periods of active treatment, or for those with residual pituitary deficits follow-up should occur more frequently, usually every 3–6 months, to reassess hormone replacement status. When required, replacement with GH appears to be safe and has not been associated with an increased risk of tumor recurrence in children and adults with Cp (Clayton et al., 1988; Price et al., 1998; Karavitaki et al., 2006). However since there are potential concerns regarding GH mitogenic activity (Taguchi et al., 2010), ongoing surveillance is advised. For those patients with hypothalamic symptoms related to Cp management is more of a challenge. Obesity develops rapidly and early after treatment and appears to be resilient to traditional weight management methods. Visual deficits and increased fatigue add further obstacles for those attempting to increase physical activity. Cognitive and psychological challenges can further impact daily functioning at home and school. A multi-disciplinary approach is essential to address these issues for the patient and their family.
In 2005, we established an interdisciplinary, comprehensive care clinic at the Hospital for Sick Children in Toronto (Rakhshani et al., 2010). The clinic provided family-centered treatment with medical, behavioral, dietary, and exercise support. Patients attended as frequently as once per month to routine visits every 6 months and at other times telephone or e-mail communication was available. Treatment was patient specific and included group sessions as well as individual therapy. In certain situations, pharmacologic agents or specific diets have been prescribed.
The rate of increase in percent BMI slowed in patients attending the clinic compared to their prior BMI change while in standard care. The majority of patients entering the clinic shortly after diagnosis exhibited stable weight as opposed to the early rapid weight gain seen in historical controls. There were also significant increases in the children’s reported quality of life, physical functioning, and school functioning after 1 year in the program with a trend toward less time spent in sedentary activity. Parents reported improvement in coordination of health care and understanding of their child’s disease. These results demonstrate the benefit of coordinated health care in improving the physical and psychological well-being of patients and their families.
As discussed earlier, additional therapies to target HO have been studied. Their mechanisms of action include increasing sympathetic tone (Mason et al., 2002; Ismail et al., 2006; Danielsson et al., 2007; Schultes et al., 2009), reducing insulin secretion, and improving sensitivity (Lustig et al., 1999, 2003a; Inge et al., 2007; Hamilton et al., 2011). Patients have experienced a decrease in weight gain or weight stabilization but without dramatic weight reduction. The effect of GH on weight gain has also been sought; however it was noted to have only a slight beneficial effect on BMI gain (Geffner et al., 2004). Similar to other populations with severe obesity, there is growing interest in bariatric surgery as a mode of therapy in HO. There is only sparse information about this treatment in Cp individuals with three case reports published on four patients (three adolescent, one adult) undergoing intestinal bypass surgery and one case series of four individuals (two adolescent, two young adult) receiving a laparoscopic adjustable gastric band (LAGB; Inge et al., 2007; Muller et al., 2007; Rottembourg et al., 2009; Schultes et al., 2009). Gastric bypass surgery induced significant and sustained weight loss, reduction in feelings of hunger and remarkable improvement in comorbidities (Inge et al., 2007; Rottembourg et al., 2009; Schultes et al., 2009). LAGB was reported to induce a continuous decrease in BMI in four patients with major improvements in eating behavior noted and improved ability to focus on non-food matters (Muller et al., 2007). In recent years three additional adolescents followed in our comprehensive care clinic underwent LAGB procedures with good results in terms of weight stabilization in one and weight reduction in two (unpublished data). One patient lost 51% of her excess body weight and was able to sustain a reduced weight with 2 years follow-up. A major caution, however, is that bariatric surgery of any kind is a relatively new treatment for adolescents and extensive pre-surgical assessment and close medical and psychological short and long term follow-up is needed to ensure that no untoward negative consequences arise. As such, these procedures should only be performed in experienced centers and with full support of an interdisciplinary team and careful assessment and follow-up of biomedical and psychosocial outcomes.
Survival and Late Mortality
Despite a relatively high rate of tumor regrowth, survival in children treated for Cp is generally good. However, disease related mortality can still occur many years after treatment. Data regarding survival includes primarily surgically treated patients. The reported post-surgical 5-year overall survival is 88–94% (De Vile et al., 1996; Muller et al., 2001, 2006a; Van Effenterre and Boch, 2002), and the reported 10-year overall survival is 70–92% (Hoffman et al., 1992; Van Effenterre and Boch, 2002; Poretti et al., 2004; Elliott and Wisoff, 2010; Visser et al., 2010) with a 20-year survival of 76%. Survival rates of patients treated with combined surgery and radiation are comparable (Stripp et al., 2004; Merchant et al., 2006; Lin et al., 2008; Crom et al., 2010). Causes of late mortality include causes directly related to the tumor or treatment such as progressive disease with multiple recurrences, hormonal deficiencies, chronic hypothalamic insufficiency, cerebrovascular disease, and seizure related (De Vile et al., 1996; Poretti et al., 2004; Elliott and Wisoff, 2010; Visser et al., 2010; Steno et al., 2011). Other causes have been described including head trauma, drug abuse, or liver failure (Poretti et al., 2004; Caldarelli et al., 2005; Visser et al., 2010).
In a study that compared the primary treatment modality used in a group of survivors to that used in patients who died, no particular pattern emerged. This might be related the small numbers of patients in each survival–treatment group (Visser et al., 2010), however, this finding is further supported by similar 5 year survival rates in patients who did or did not receive radiotherapy (Muller et al., 2006a). Based on other reports, risk factors for decreased survival were recurrent resections of tumor regrowth (Elliott and Wisoff, 2010), and tumor location, with improved survival for tumors that grow beneath the sellar diaphragm (Steno et al., 2011).
Conclusion
Although treatment for pediatric Cp tumors remains challenging, changes in management show potential for improved long term outcomes. Common long term complications include endocrine, neurologic, psychosocial, and neurocognitive and metabolic morbidities. However, the most significant adverse effect on quality of life are those related to hypothalamic dysfunction and in particular, post-surgical weight gain and increased fatigue with daytime somnolence. In recent years there is a trend toward individualizing treatment based on patient and tumor characteristics as well as managing Cp more as a chronic disease with less aggressive surgical treatment and/or adjuvant radiation or chemotherapy. A coordinated interdisciplinary approach to management, beginning at the time of diagnosis, is very important to identify risk factors for adverse outcomes, and provide support and education for these patients and their families.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ahmet, A., Blaser, S., Stephens, D., Guger, S., Rutkas, J. T., and Hamilton, J. (2006). Weight gain in craniopharyngioma – a model for hypothalamic obesity. J. Pediatr. Endocrinol. Metab. 19, 121–127.
Anderson, C., Wilkening, G., Filley, C., Reardon, M., and Kleinschmidt-Demasters, B. (1997). Neurobehavioral outcome in pediatric craniopharyngioma. Pediatr. Neurosurg. 26, 255–260.
Bunin, G. R., Surawicz, T. S., Witman, P. A., Preston-Martin, S., Davis, F., and Bruner, J. M. (1998). The descriptive epidemiology of craniopharyngioma. J. Neurosurg. 89, 547–551.
Caldarelli, M., Massimi, L., Tamburrini, G., Cappa, M., and Di Rocco, C. (2005). Long-term results of the surgical treatment of craniopharyngioma: the experience at the Policlinico Gemelli, Catholic University, Rome. Childs Nerv. Syst. 21, 747–757.
Cammisotto, P. G., and Bukowiecki, L. J. (2002). Mechanisms of leptin secretion from white adipocytes. Am. J. Physiol. Cell Physiol. 283, C244–C250.
Carpentieri, S., Waber, D., Scott, R., Goumnerova, L., Kieran, M., Cohen, L., Kim, F., Billett, A., Tarbell, N., and Pomeroy, S. (2001). Memory deficits among children with craniopharyngioma. Neurosurgery 49, 1053–1058.
Cavalheiro, S., Di Rocco, C., Valenzuela, S., Dastoli, P. A., Tamburrini, G., Massimi, L., Nicacio, J. M., Faquini, I. V., Ierardi, D. F., Silva, N. S., Pettorini, B. L., and Toledo, S. R. (2010). Craniopharyngiomas: intratumoral chemotherapy with interferon-alpha: a multicenter preliminary study with 60 cases. Neurosurg. Focus 28, E12.
Cavazzuti, V., Fischer, E., Welch, K., Belli, J., and Winston, K. (1983). Neurological and psychophysiological sequelae following different treatments of craniopharyngioma in children. J. Neurosurg. 59, 409–417.
Clayton, P. E., Price, D. A., Shalet, S. M., and Gattemaneni, H. R. (1988). Craniopharyngioma recurrence and growth hormone therapy. Lancet 1, 642.
Colangelo, M., Ambrosio, A., and Ambrosio, C. (1990). Neurological and behavioral sequelae following different approaches to craniopharyngioma. Long-term follow-up review and therapeutic guidelines. Childs Nerv. Syst. 6, 379–382.
Costin, G., Kogut, M. D., Phillips, L. S., and Daughaday, W. H. (1976). Craniopharyngioma: the role of insulin in promoting postoperative growth. J. Clin. Endocrinol. Metab. 42, 370–379.
Coutant, R., Maurey, H., Rouleau, S., Mathieu, E., Mercier, P., Limal, J. M., and Le Bouil, A. (2003). Defect in epinephrine production in children with craniopharyngioma: functional or organic origin? J. Clin. Endocrinol. Metab. 88, 5969–5975.
Crom, D., Smith, D., Xiong, Z., Onar, A., Hudson, M., Merchant, T., and Morris, E. (2010). Health status in long-term survivors of pediatric craniopharyngiomas. Am. Assoc. Neurosci. Nurses 42, 323–328.
Crowley, R. K., Woods, C., Fleming, M., Rogers, B., Behan, L. A., O’sullivan, E. P., Kane, T., Agha, A., Smith, D., Costello, R. W., and Thompson, C. J. (2011). Somnolence in adult craniopharyngioma patients is a common, heterogeneous condition that is potentially treatable. Clin. Endocrinol. (Oxf.) 74, 750–755.
Danielsson, P., Janson, A., Norgren, S., and Marcus, C. (2007). Impact sibutramine therapy in children with hypothalamic obesity or obesity with aggravating syndromes. J. Clin. Endocrinol. Metab. 92, 4101–4106.
De Vile, C. J., Grant, D. B., Kendall, B. E., Neville, B. G., Stanhope, R., Watkins, K. E., and Hayward, R. D. (1996). Management of childhood craniopharyngioma: can the morbidity of radical surgery be predicted? J. Neurosurg. 85, 73–81.
Elliott, R. E., Jane, J. A. Jr., and Wisoff, J. H. (2011). Surgical Management of craniopharyngiomas in children: meta-analysis and comparison of transcranial and transsphenoidal approaches. Neurosurgery 69, 630–643.
Elliott, R. E., and Wisoff, J. H. (2010). Surgical management of giant pediatric craniopharyngiomas. J. Neurosurg. Pediatr. 6, 403–416.
Geffner, M., Lundberg, M., Koltowska-Haggstrom, M., Abs, R., Verhelst, J., Erfurth, E. M., Kendall-Taylor, P., Price, D. A., Jonsson, P., and Bakker, B. (2004). Changes in height, weight, and body mass index in children with craniopharyngioma after three years of growth hormone therapy: analysis of KIGS (Pfizer International Growth Database). J. Clin. Endocrinol. Metab. 89, 5435–5440.
Geffner, M. E. (1996). The growth without growth hormone syndrome. Endocrinol. Metab. Clin. North Am. 25, 649–663.
Goldstone, A. P., Patterson, M., Kalingag, N., Ghatei, M. A., Brynes, A. E., Bloom, S. R., Grossman, A. B., and Korbonits, M. (2005). Fasting and postprandial hyperghrelinemia in Prader-Willi syndrome is partially explained by hypoinsulinemia, and is not due to peptide YY3-36 deficiency or seen in hypothalamic obesity due to craniopharyngioma. J. Clin. Endocrinol. Metab. 90, 2681–2690.
Guran, T., Turan, S., Bereket, A., Akcay, T., Unluguzel, G., Bas, F., Gunoz, H., Saka, N., Bundak, R., Darendeliler, F., Isguven, P., Yildiz, M., Adal, E., Sarikaya, S., Baygin, L. A., Memioglu, N., Onal, H., Ercan, O., and Haklar, G. (2009). The role of leptin, soluble leptin receptor, resistin, and insulin secretory dynamics in the pathogenesis of hypothalamic obesity in children. Eur. J. Pediatr. 168, 1043–1048.
Halac, I., and Zimmerman, D. (2005). Endocrine manifestations of craniopharyngioma. Childs Nerv. Syst. 21, 640–648.
Hamilton, J. K., Conwell, L. S., Syme, C., Ahmet, A., Jeffery, A., and Daneman, D. (2011). Hypothalamic obesity following craniopharyngioma surgery: results of a pilot trial of combined diazoxide and metformin therapy. Int. J. Pediatr. Endocrinol. 2011, 417949.
Hammond, J., and Hall, S. (2011). Functional analysis and treatment of aggressive behavior following resection of a craniopharyngioma. Dev. Med. Child Neurol. 53, 369–374.
Harz, K. J., Muller, H. L., Waldeck, E., Pudel, V., and Roth, C. (2003). Obesity in patients with craniopharyngioma: assessment of food intake and movement counts indicating physical activity. J. Clin. Endocrinol. Metab. 88, 5227–5231.
Hochberg, I., and Hochberg, Z. (2010). Expanding the definition of hypothalamic obesity. Obes. Rev. 11, 709–721.
Hoffman, H. J., De Silva, M., Humphreys, R. P., Drake, J. M., Smith, M. L., and Blaser, S. I. (1992). Aggressive surgical management of craniopharyngiomas in children. J. Neurosurg. 76, 47–52.
Holmer, H., Ekman, B., Bjork, J., Nordstom, C. H., Popovic, V., Siversson, A., and Erfurth, E. M. (2009). Hypothalamic involvement predicts cardiovascular risk in adults with childhood onset craniopharyngioma on long-term GH therapy. Eur. J. Endocrinol. 161, 671–679.
Holmer, H., Pozarek, G., Wirfalt, E., Popovic, V., Ekman, B., Bjork, J., and Erfurth, E. M. (2010). Reduced energy expenditure and impaired feeding-related signals but not high energy intake reinforces hypothalamic obesity in adults with childhood onset craniopharyngioma. J. Clin. Endocrinol. Metab. 95, 5395–5402.
Hukin, J., Steinbok, P., Lafay-Cousin, L., Hendson, G., Strother, D., Mercier, C., Samson, Y., Howes, W., and Bouffet, E. (2007). Intracystic bleomycin therapy for craniopharyngioma in children: the Canadian experience. Cancer 109, 2124–2131.
Inge, T. H., Pfluger, P., Zeller, M., Rose, S. R., Burget, L., Sundararajan, S., Daniels, S. R., and Tschop, M. H. (2007). Gastric bypass surgery for treatment of hypothalamic obesity after craniopharyngioma therapy. Nat. Clin. Pract. Endocrinol. Metab. 3, 606–609.
Ismail, D., O’connell, M. A., and Zacharin, M. R. (2006). Dexamphetamine use for management of obesity and hypersomnolence following hypothalamic injury. J. Pediatr. Endocrinol. Metab. 19, 129–134.
Jane, J. A. Jr., Prevedello, D. M., Alden, T. D., and Laws, E. R. Jr. (2010). The transsphenoidal resection of pediatric craniopharyngiomas: a case series. J. Neurosurg. Pediatr. 5, 49–60.
Jung, T. Y., Jung, S., Moon, K. S., Kim, I. Y., Kang, S. S., and Kim, J. H. (2010). Endocrinological outcomes of pediatric craniopharyngiomas with anatomical pituitary stalk preservation: preliminary study. Pediatr. Neurosurg. 46, 205–212.
Karavitaki, N., Warner, J. T., Marland, A., Shine, B., Ryan, F., Arnold, J., Turner, H. E., and Wass, J. A. (2006). GH replacement does not increase the risk of recurrence in patients with craniopharyngioma. Clin. Endocrinol. (Oxf.) 64, 556–560.
Kiehna, E., Mulhern, R., Li, C., Xiong, X., and Merchant, T. (2006). Changes in attentional performance of children and young adults with localized primary brain tumors after conformal radiation therapy. J. Clin. Oncol. 24, 5283–5290.
Kiehna, E. N., and Merchant, T. E. (2010). Radiation therapy for pediatric craniopharyngioma. Neurosurg. Focus 28, E10.
Kim, R. J., Shah, R., Tershakovec, A. M., Zemel, B. S., Sutton, L. N., Grimberg, A., and Moshang, T. (2010). Energy expenditure in obesity associated with craniopharyngioma. Childs Nerv. Syst. 26, 913–917.
Lafay-Cousin, L., Bartels, U., Raybaud, C., Kulkarni, A. V., Guger, S., Huang, A., and Bouffet, E. (2007). Neuroradiological findings of bleomycin leakage in cystic craniopharyngioma. Report of three cases. J. Neurosurg. 107, 318–323.
Lek, N., Prentice, P., Williams, R. M., Ong, K. K., Burke, G. A., and Acerini, C. L. (2010). Risk factors for obesity in childhood survivors of suprasellar brain tumours: a retrospective study. Acta Paediatr. 99, 1522–1526.
Lin, L. L., El Naqa, I., Leonard, J. R., Park, T. S., Hollander, A. S., Michalski, J. M., and Mansur, D. B. (2008). Long-term outcome in children treated for craniopharyngioma with and without radiotherapy. J. Neurosurg. Pediatr. 1, 126–130.
Lipton, J., Megerian, J. T., Kothare, S. V., Cho, Y. J., Shanahan, T., Chart, H., Ferber, R., Adler-Golden, L., Cohen, L. E., Czeisler, C. A., and Pomeroy, S. L. (2009). Melatonin deficiency and disrupted circadian rhythms in pediatric survivors of craniopharyngioma. Neurology 73, 323–325.
Lustig, R. H. (2002). Hypothalamic obesity: the sixth cranial endocrinopathy. Endocrinologist 12, 8.
Lustig, R. H. (2003). Autonomic dysfunction of the beta-cell and the pathogenesis of obesity. Rev. Endocr. Metab. Disord. 4, 23–32.
Lustig, R. H., Hinds, P. S., Ringwald-Smith, K., Christensen, R. K., Kaste, S. C., Schreiber, R. E., Rai, S. N., Lensing, S. Y., Wu, S., and Xiong, X. (2003a). Octreotide therapy of pediatric hypothalamic obesity: a double-blind, placebo-controlled trial. J. Clin. Endocrinol. Metab. 88, 2586–2592.
Lustig, R. H., Post, S. R., Srivannaboon, K., Rose, S. R., Danish, R. K., Burghen, G. A., Xiong, X., Wu, S., and Merchant, T. E. (2003b). Risk factors for the development of obesity in children surviving brain tumors. J. Clin. Endocrinol. Metab. 88, 611–616.
Lustig, R. H., Rose, S. R., Burghen, G. A., Velasquez-Mieyer, P., Broome, D. C., Smith, K., Li, H., Hudson, M. M., Heideman, R. L., and Kun, L. E. (1999). Hypothalamic obesity caused by cranial insult in children: altered glucose and insulin dynamics and reversal by a somatostatin agonist. J. Pediatr. 135, 162–168.
Mason, P. W., Krawiecki, N., and Meacham, L. R. (2002). The use of dextroamphetamine to treat obesity and hyperphagia in children treated for craniopharyngioma. Arch. Pediatr. Adolesc. Med. 156, 887–892.
Merchant, T. E., Kiehna, E. N., Kun, L. E., Mulhern, R. K., Li, C., Xiong, X., Boop, F. A., and Sanford, R. A. (2006). Phase II trial of conformal radiation therapy for pediatric patients with craniopharyngioma and correlation of surgical factors and radiation dosimetry with change in cognitive function. J. Neurosurg. 104, 94–102.
Merchant, T. E., Kiehna, E. N., Sanford, R. A., Mulhern, R. K., Thompson, S. J., Wilson, M. W., Lustig, R. H., and Kun, L. E. (2002). Craniopharyngioma: the St. Jude Children’s Research Hospital experience 1984-2001. Int. J. Radiat. Oncol. Biol. Phys. 53, 533–542.
Metzler-Baddeley, C., and Jones, R. (2010). Brief communication: cognitive rehabilitation of executive functioning in a case of craniopharyngioma. Appl. Neuropsychol. 17, 299–304.
Muller, H. L. (2008). Childhood craniopharyngioma. Recent advances in diagnosis, treatment and follow-up. Horm. Res. 69, 193–202.
Muller, H. L. (2010). Childhood craniopharyngioma – current concepts in diagnosis, therapy and follow-up. Nat. Rev. Endocrinol. 61, 609–618.
Muller, H. L., Bueb, K., Bartels, U., Roth, C., Harz, K., Graf, N., Korinthenberg, R., Bettendorf, M., Kuhl, J., Gutjahr, P., Sorensen, N., and Calaminus, G. (2001). Obesity after childhood craniopharyngioma – German multicenter study on pre-operative risk factors and quality of life. Klin. Padiatr. 213, 244–249.
Muller, H. L., Emser, A., Faldum, A., Bruhnken, G., Etavard-Gorris, N., Gebhardt, U., Oeverink, R., Kolb, R., and Sorensen, N. (2004). Longitudinal study on growth and body mass index before and after diagnosis of childhood craniopharyngioma. J. Clin. Endocrinol. Metab. 89, 3298–3305.
Muller, H. L., Faldum, A., Etavard-Gorris, N., Gebhardt, U., Oeverink, R., Kolb, R., and Sorensen, N. (2003a). Functional capacity, obesity and hypothalamic involvement: cross-sectional study on 212 patients with childhood craniopharyngioma. Klin. Padiatr. 215, 310–314.
Muller, H. L., Heinrich, M., Bueb, K., Etavard-Gorris, N., Gebhardt, U., Kolb, R., and Sorensen, N. (2003b). Perioperative dexamethasone treatment in childhood craniopharyngioma – influence on short-term and long-term weight gain. Exp. Clin. Endocrinol. Diabetes 111, 330–334.
Muller, H. L., Gebhardt, U., Pohl, F., Flentje, M., Emser, A., Warmuth-Metz, M., Kolb, R., Calaminus, G., and Sorensen, N. (2006a). Relapse pattern after complete resection and early progression after incomplete resection of childhood craniopharyngioma. Klin. Padiatr. 218, 315–320.
Muller, H. L., Handwerker, G., Gebhardt, U., Faldum, A., Emser, A., Kolb, R., and Sorensen, N. (2006b). Melatonin treatment in obese patients with childhood craniopharyngioma and increased daytime sleepiness. Cancer Causes Control 17, 583–589.
Muller, H. L., Gebhardt, U., Teske, C., Faldum, A., Zwiener, I., Warmuth-Metz, M., Pietsch, T., Pohl, F., Sorensen, N., and Calaminus, G. (2011). Post-operative hypothalamic lesions and obesity in childhood craniopharyngioma: results of the multinational prospective trial KRANIOPHARYNGEOM 2000 after 3-year follow-up. Eur. J. Endocrinol. 165, 17–24.
Muller, H. L., Gebhardt, U., Wessel, V., Schroder, S., Kolb, R., Sorensen, N., Maroske, J., and Hanisch, E. (2007). First experiences with laparoscopic adjustable gastric banding (LAGB) in the treatment of patients with childhood craniopharyngioma and morbid obesity. Klin. Padiatr. 219, 323–325.
Muller, H. L., Handwerker, G., Wollny, B., Faldum, A., and Sorensen, N. (2002). Melatonin secretion and increased daytime sleepiness in childhood craniopharyngioma patients. J. Clin. Endocrinol. Metab. 87, 3993–3996.
O’gorman, C. S., Simoneau-Roy, J., Pencharz Mb, P., Adeli, K., and Hamilton, J. (2010a). Delayed ghrelin suppression following oral glucose tolerance test in children and adolescents with hypothalamic injury secondary to craniopharyngioma compared with obese controls. Int. J. Pediatr. Obes. 6, 285–288.
O’gorman, C. S., Simoneau-Roy, J., Pencharz, P., Macfarlane, J., Maclusky, I., Narang, I., Adeli, K., Daneman, D., and Hamilton, J. (2010b). Sleep-disordered breathing is increased in obese adolescents with craniopharyngioma compared with obese controls. J. Clin. Endocrinol. Metab. 95, 2211–2218.
Ondruch, A., Maryniak, A., Kropiwnicki, T., Roszkowski, M., and Daszkiewicz, P. (2011). Cognitive and social functioning in children and adolescents after the removal of craniopharyngioma. Childs Nerv. Syst. 27, 391–397.
Phillip, M., Moran, O., and Lazar, L. (2002). Growth without growth hormone. J. Pediatr. Endocrinol. Metab. 15(Suppl. 5), 1267–1272.
Poretti, A., Grotzer, M. A., Ribi, K., Schonle, E., and Boltshauser, E. (2004). Outcome of craniopharyngioma in children: long-term complications and quality of life. Dev. Med. Child Neurol. 46, 220–229.
Price, D. A., Wilton, P., Jonsson, P., Albertsson-Wikland, K., Chatelain, P., Cutfield, W., and Ranke, M. B. (1998). Efficacy and safety of growth hormone treatment in children with prior craniopharyngioma: an analysis of the Pharmacia and Upjohn International Growth Database (KIGS) from 1988 to 1996. Horm. Res. 49, 91–97.
Rakhshani, N., Jeffery, A. S., Schulte, F., Barrera, M., Atenafu, E. G., and Hamilton, J. K. (2010). Evaluation of a comprehensive care clinic model for children with brain tumor and risk for hypothalamic obesity. Obesity (Silver Spring) 18, 1768–1774.
Redline, S., Storfer-Isser, A., Rosen, C. L., Johnson, N. L., Kirchner, H. L., Emancipator, J., and Kibler, A. M. (2007). Association between metabolic syndrome and sleep-disordered breathing in adolescents. Am. J. Respir. Crit. Care Med. 176, 401–408.
Riva, D., Pantaleoni, C., Devoti, M., Saletti, V., Nichelli, F., and Giorgi, C. (1998). Late neuropsychological and behavioral outcome of children surgically treated for craniopharyngioma. Childs Nerv. Syst. 14, 179–184.
Roth, C. L., Gebhardt, U., and Muller, H. L. (2011). Appetite-regulating hormone changes in patients with craniopharyngioma. Obesity (Silver Spring) 19, 36–42.
Roth, C. L., Hunneman, D. H., Gebhardt, U., Stoffel-Wagner, B., Reinehr, T., and Muller, H. L. (2007). Reduced sympathetic metabolites in urine of obese patients with craniopharyngioma. Pediatr. Res. 61, 496–501.
Rottembourg, D., O’gorman, C. S., Urbach, S., Garneau, P. Y., Langer, J. C., Van Vliet, G., Hamilton, J., and Huot, C. (2009). Outcome after bariatric surgery in two adolescents with hypothalamic obesity following treatment of craniopharyngioma. J. Pediatr. Endocrinol. Metab. 22, 867–872.
Sands, S., Milner, J., Goldberg, J., Mukhi, V., Moliterno, J., Maxfield, C., and Wisoff, J. (2005). Quality of life and behavioral follow-up study of pediatric survivors of craniopharyngioma. J. Neurosurg. 103, 302–311.
Schultes, B., Ernst, B., Schmid, F., and Thurnheer, M. (2009). Distal gastric bypass surgery for the treatment of hypothalamic obesity after childhood craniopharyngioma. Eur. J. Endocrinol. 161, 201–206.
Shaikh, M. G., Grundy, R. G., and Kirk, J. M. (2008a). Hyperleptinaemia rather than fasting hyperinsulinaemia is associated with obesity following hypothalamic damage in children. Eur. J. Endocrinol. 159, 791–797.
Shaikh, M. G., Grundy, R. G., and Kirk, J. M. (2008b). Reductions in basal metabolic rate and physical activity contribute to hypothalamic obesity. J. Clin. Endocrinol. Metab. 93, 2588–2593.
Simoneau-Roy, J., O’gorman, C., Pencharz, P., Adeli, K., Daneman, D., and Hamilton, J. (2010). Insulin sensitivity and secretion in children and adolescents with hypothalamic obesity following treatment for craniopharyngioma. Clin. Endocrinol. (Oxf.) 72, 364–370.
Snow, A., Gozal, E., Malhotra, A., Tiosano, D., Perlman, R., Vega, C., Shahar, E., Gozal, D., Hochberg, Z., and Pillar, G. (2002). Severe hypersomnolence after pituitary/hypothalamic surgery in adolescents: clinical characteristics and potential mechanisms. Pediatrics 110, e74.
Srinivasan, S., Ogle, G. D., Garnett, S. P., Briody, J. N., Lee, J. W., and Cowell, C. T. (2004). Features of the metabolic syndrome after childhood craniopharyngioma. J. Clin. Endocrinol. Metab. 89, 81–86.
Steinbok, P., and Hukin, J. (2010). Intracystic treatments for craniopharyngioma. Neurosurg. Focus 28, E13.
Steno, J., Bizik, I., Steno, A., and Matejcik, V. (2011). Craniopharyngiomas in children: how radical should the surgeon be? Childs Nerv. Syst. 27, 41–54.
Stripp, D. C., Maity, A., Janss, A. J., Belasco, J. B., Tochner, Z. A., Goldwein, J. W., Moshang, T., Rorke, L. B., Phillips, P. C., Sutton, L. N., and Shu, H. K. (2004). Surgery with or without radiation therapy in the management of craniopharyngiomas in children and young adults. Int. J. Radiat. Oncol. Biol. Phys. 58, 714–720.
Sughrue, M. E., Yang, I., Kane, A. J., Fang, S., Clark, A. J., Aranda, D., Barani, I. J., and Parsa, A. T. (2011). Endocrinologic, neurologic, and visual morbidity after treatment for craniopharyngioma. J. Neurooncol. 101, 463–476.
Taguchi, T., Takao, T., Iwasaki, Y., Pooh, K., Okazaki, M., Hashimoto, K., and Terada, Y. (2010). Rapid recurrence of craniopharyngioma following recombinant human growth hormone replacement. J. Neurooncol. 100, 321–322.
Van Effenterre, R., and Boch, A. L. (2002). Craniopharyngioma in adults and children: a study of 122 surgical cases. J. Neurosurg. 97, 3–11.
Keywords: child, craniopharyngioma, complications, hypothalamic obesity, sleep, pituitary, neurocognitive, recurrence
Citation: Cohen M, Guger S and Hamilton J (2011) Long term sequelae of pediatric craniopharyngioma – literature review and 20 years of experience. Front. Endocrin. 2:81. doi: 10.3389/fendo.2011.00081
Received: 05 August 2011; Paper pending published: 29 August 2011;
Accepted: 09 November 2011; Published online: 28 November 2011.
Edited by:
Hermann Lothar Mueller, Klinikum Oldenburg gGmbH, GermanyReviewed by:
Laurence Katznelson, Stanford University, USASara DiVall, Johns Hopkins University School of Medicine, USA
Copyright: © 2011 Cohen, Guger and Hamilton. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Jill Hamilton, Division of Endocrinology, The Hospital for Sick Children, 555 University Avenue, Toronto, ON, Canada M5G 1×8. e-mail:amlsbC5oYW1pbHRvbkBzaWNra2lkcy5jYQ==