 Deepak P. Srivastava1,2*
Deepak P. Srivastava1,2* Peter Penzes1,3
Peter Penzes1,3
- 1 Department of Physiology, Feinberg School of Medicine, Northwestern University, Chicago, IL, USA
- 2 Department of Neuroscience and Centre for the Cellular Basis of Behaviour, The James Black Centre, Institute of Psychiatry, King’s College London, London, UK
- 3 Department of Psychiatry and Behavioral Sciences, Feinberg School of Medicine, Northwestern University, Chicago, IL, USA
Estrogens have multiple actions in the brain including modulating synaptic plasticity, connectivity, and cognitive behaviors. While the classical view of estrogens are as endocrine signals, whose effects manifest via the regulation of gene transcription, mounting evidence has been presented demonstrating that estrogens have rapid effects within specific areas of the brain. The emergence that 17 β-estradiol can be produced locally in the brain which can elicit rapid (within minutes) cellular responses has led to its classification as a neurosteroid. Moreover, recent studies have also begun to detail the molecular and cellular underpinnings of how 17 β-estradiol can rapidly modulate spiny synapses (dendritic spines). Remodeling of dendritic spines is a key step in the rewiring of neuronal circuitry thought to underlie the processing and storage of information in the forebrain. Conversely, abnormal remodeling of dendritic spines is thought to contribute to a number of psychiatric and neurodevelopmental disorders. Here we review recent molecular and cellular work that offers a potential mechanism of how 17 β-estradiol may modulate synapse structure and function of cortical neurons. This mechanism allows cortical neurons to respond to activity-dependent stimuli with greater efficacy. In turn this form of plasticity may provide an insight into how 17 β-estradiol can modulate the rewiring of neuronal circuits, underlying its ability to influencing cortically based behaviors. We will then go on to discuss the potential role of 17 β-estradiol modulation of neural circuits and its potential relevance for the treatment of psychiatric and neurodevelopmental disorders.
Introduction
Neuronal connections in the adult brain are plastic and change in response to a number of environmental and extracellular stimuli, ranging from activity-dependent stimuli to neuromodulator and neurosteroids (Alvarez and Sabatini, 2007; Bhatt et al., 2009; Brinton, 2009; Holtmaat and Svoboda, 2009). This structural plasticity is essential for normal cognitive function (Chklovskii et al., 2004; DeBello, 2008; Bhatt et al., 2009; Holtmaat and Svoboda, 2009) and may ameliorate progression of psychiatric and neurodegenerative disorders such as schizophrenia, depression, and Alzheimer’s disease (Penzes et al., 2011a,b). However, excessive or aberrant reorganization of neural circuits has been proposed to contribute to psychiatric and neurodegenerative disorders, by providing a pathological cellular mechanism by which cognitive deficits seen in such disorders may emerge (van Spronsen and Hoogenraad, 2010; Penzes et al., 2011a). Therefore, harnessing structural plasticity for active therapeutic intervention requires an understanding of the cellular mechanisms that underpin cortical plasticity.
It is well established that estrogens have a wide range of effects on physiological functions ranging from reproduction, development, and cardiovascular function. Estrogens refer to a class of steroid compounds, of which 17 β-estradiol (herein referred to as estradiol) is considered to be the most biologically active form. Over recent years there has been a growing appreciation of the complex actions of estrogens within the brain. In addition to their actions in the hypothalamus (Kelly et al., 2005), it has become clear that estradiol can exert effects in multiple regions of the brains including the cerebral cortex and hippocampus (McEwen and Alves, 1999; Brinton, 2009). The emergence of estradiol’s actions within these areas has also been accompanied by clinical and basic studies implicating estrogens in regulating cognitive and memory processing in both animal models and humans (Luine, 2008; Brinton, 2009; Henderson, 2009). The effects of estrogens on cognitive function is of significant interest due to the growing evidence that estrogens may delay the onset or ameliorate the severity of a number of psychiatric and neurodegenerative disorders, such as schizophrenia, anxiety, depression, and Alzheimer’s disease (Hughes et al., 2009). Therefore, elucidating the molecular and cellular mechanisms that underlie estradiol’s effects within the brain, and how they may be relevant for neuropathologies is essential for understanding the potential role of estrogens in these disorders.
Despite the fact that rapid estrogenic effects within the brain were first reported over 40 years ago (Bueno and Pfaff, 1976), the majority of studies to date have focused on the long-term actions of estrogens as a circulating hormone. This classical mode of action for estrogens, also referred to as genomic actions, occurs via the regulation of gene transcription, whose actions take hours to days to manifest. However, recent studies have demonstrated that estradiol can modulate cognitive function within minutes to hours (Balthazart and Ball, 2006; Sinopoli et al., 2006; Phan et al., 2011). This is paralleled with findings that estradiol can increase synaptic plasticity in several areas of the brain, including the hippocampus, cortex, and prefrontal cortex (PFC), also within minutes to hours (Woolley, 2007; Srivastava et al., 2008). However, the precise molecular mechanisms that are required for estradiol’s ability to rapidly influence synaptic plasticity, and thus lead to long-term changes in the cellular responses to subsequent stimuli, are only just becoming evident. Here we will focus on recent insights into the actions of estradiol in the cortex. Specifically, we will first consider the evidence that estradiol is a neurosteroid and may even function as a neuromodulator before going on to examine the cellular and molecular mechanisms underlying rapid estrogenic-dependent remodeling of cortical circuits. Finally we will discuss how this mechanism in part, may underlie estradiol’s ability to influence cognitive function, and its potential relevance for psychiatric and neurodegenerative disorders.
Estradiol as a Neuromodulator
The classic mode of action for estradiol in the brain is as an endocrine signal, which travels over long distances before entering the target tissue where it exerts its effects via gene regulation (McEwen and Alves, 1999). However, there is increasing evidence that significant concentrations of estradiol are produced locally in discrete regions of the brain which can rapidly induce electrophysiological, cellular, molecular, and behavioral effects (Woolley, 2007; Srivastava et al., 2008, 2010; Saldanha et al., 2011). Brain-synthesized estradiol has been identified in several brain regions of a number of avian and mammalian species (Yague et al., 2006; Boon et al., 2010; Saldanha et al., 2011). Moreover, this de novo production of estradiol demonstrates that it is a neurosteroid and has led to suggestions that it may be considered a neuromodulator (Balthazart and Ball, 2006; Saldanha et al., 2011). In order for estradiol to be considered as a neuromodulator, a number of criteria must be met.
(1) Rapid biosynthesis and breakdown of estradiol within discrete regions of the brain.
(2) Production of estradiol must be spatially and temporally restricted, and regulated by electrochemical events, to allow for specific actions of the neurosteroid.
(3) Specific receptors for estradiol must be ideally located to allow for the coupling of estrogenic signal to elicit physiological and cellular effects within seconds to minutes via ion channels and second messenger pathways.
Indeed, numerous studies have demonstrated that estradiol meets these criteria.
De novo Synthesis of Estradiol
Studies where the sex organs of rodents have been removed or direct measurements of estradiol production in the zebra finch song birds have shown that estradiol is produced rapidly within the brain (Remage-Healey et al., 2008; Konkle and McCarthy, 2011). Furthermore, the presence of aromatase, the major enzyme required for the synthesis of estradiol from androgen has been identified in a number of brain regions including the hypothalamus, hippocampus, visual cortex, and temporal cortex in avian, mammalian, and human brains (Rune and Frotscher, 2005; Yague et al., 2006; Boon et al., 2010; Saldanha et al., 2011). Based on experimental studies in the avian and mammalian brains, brain-aromatase activity has been shown to be required for the rapid synthesis of estradiol (Garcia-Segura, 2008; Yague et al., 2008; Saldanha et al., 2011). The enzymes StAR (steroidogenic acute regulatory protein) 3β-HSD (3β-hydroxysteroid dehydrogenase) and SCC (side-chain-cleavage enzyme), enzymes required for the conversation of cholesterol into estrogenic precursors, have also been reported to be expressed in neurons (Wehrenberg et al., 2001). The presence of these enzymes in neurons provides a potential direct pathway for the biosynthesis of estrogens from cholesterol. On the other hand, enzymes including 2- and 4-hydroxylase, and catechol-O-methyltransferase, which are involved in the metabolism of estrogens into inactive (or less active) water-soluble metabolites, have also been detected within the brain (Zhu and Conney, 1998). Together these studies demonstrate that specific mechanisms required for the rapid de novo synthesis and metabolism of estrogens in the brain exist, supporting the hypothesis that estradiol is a neurosteroid.
Pre-Synaptic Localization and Synthesis of Aromatase
A number of studies have also localized aromatase within pre-synaptic terminals. In the hypothalamus EM studies have demonstrated a synaptic localization for aromatase in avian, mammalian, and human tissue (Naftolin et al., 1996). Furthermore, biochemical studies have detected high levels of aromatase activity in isolated pre-synaptic tissue preparations (Mak et al., 1985). Using mature cultured cortical neurons derived from embryonic rats, we find that aromatase is present at synapses (Srivastava et al., 2010). Aromatase co-localizes with the post-synaptic marker, PDS-95 (post-synaptic density protein 95), and the pre-synaptic marker bassoon in cortical neurons (Srivastava et al., 2010). In addition, aromatase was detected in tau5-positive axonal processes, indicating that a portion of aromatase is present at pre-synaptic terminals (Srivastava et al., 2010). More recently, evidence has been presented that acute fluctuations in brain-synthesized estradiol levels mediated by aromatase activity in the cortex of zebra finch, is controlled by specific depolarization-sensitive calcium-dependent events (Remage-Healey et al., 2011). This study presents strong evidence that locally produced estradiol within the brain is regulated by electrochemical signals, strongly supporting a hypothesis that estradiol may be considered a neuromodulator. Collectively, the presence and regulation of aromatase at pre-synaptic terminals places the machinery required for the de novo production of estradiol at an ideal location for this neurosteroid to act on post-synaptic structures.
Localization of Estrogen Receptors in the Brain
The presence of receptors specific for estrogens in the brain has been well documented (Brinton, 2009; Hughes et al., 2009). Estrogen receptor (ER) α, ERβ, and the GPCR, GPR30 (also known as G protein-coupled ER, GPER) have been shown to be expressed in several regions of the brain. ERα is highly expressed in the hypothalamus and in the mammalian forebrain, whereas ERβ has lower expression in the hypothalamus, but is thought to be expressed highly in the cortex and hippocampus (Milner et al., 2001, 2005; Kritzer, 2002; Mitra et al., 2003). Furthermore, recent studies have identified both ERα and β to reside in both pre- and post-synaptic compartments (McEwen et al., 2001; Milner et al., 2001, 2005). Some controversy surrounds whether estrogens are the true ligand for GPR30 (Langer et al., 2010), nevertheless there are numerous reports that have indicated that this receptor is highly responsive to acute estradiol treatment (Prossnitz and Maggiolini, 2009; Maggiolini and Picard, 2010; Nadal et al., 2011). Investigations into the localization of this receptor within the brain have shown expression in several areas including the cortex, hippocampus as well as the hypothalamus (Brailoiu et al., 2007; Hazell et al., 2009). All three receptors have been shown to be able to rapidly initiate second messenger pathways in response to acute estradiol treatment within neurons (Brinton, 2009). This provides a mechanism to allow the coupling of rapid estrogenic signaling with intracellular signaling cascades, resulting in acute cellular effects. Collectively, these data suggest that brain-synthesized estradiol does fulfill the criteria required for the consideration of estradiol as a neuromodulator as outlined above. Moreover, these studies support a hypothesis that brain-synthesized estradiol can initiate rapid cellular effects, altering post-synaptic structures, and may be a mechanism whereby cognition and behavior can be regulated.
Role of Estradiol in the Cortex
Neuromodulation of synaptic plasticity is thought to be a key mechanism influencing information storage (Bavelier et al., 2010). Recent evidence has demonstrated that estrogens can influence behaviors, cellular mechanisms, and signaling pathways within minutes (Balthazart and Ball, 2006; Srivastava et al., 2008, 2010; Brinton, 2009). Multiple studies have shown that estrogens influence hippocampally based behaviors. However, estrogens can also exert powerful actions on cognitive processes that require information processing in the cortex and PFC. For example, studies in non-human primates have investigated the role of estradiol in cognitive ability mediated by the PFC (see Bailey et al., 2011 for a recent review). In a delay response task, which is mediated by the dorsolateral PFC, long-term treatment with estradiol in hormone depletion, ovariectomized (OVX) aged female rhesus monkeys, perform significantly better than OVX vehicle-treated animals (Hao et al., 2007; Bailey et al., 2011). Interestingly, younger OVX monkeys perform equally well when treated chronically with either estradiol or vehicle, suggesting an age-dependent effect in this task (Hao et al., 2007; Bailey et al., 2011).
Multiple studies have also investigated the role of acute estradiol treatment in a number of tasks involving cortical processing. In zebra finch songbirds, estradiol is rapidly (within minutes) produced in the cortex in response to social interactions (Remage-Healey et al., 2008). Furthermore, acute blockade of estradiol production within the cortex disrupts songbird social interactions (Remage-Healey et al., 2010). Studies using female rats have shown that OVX impaired the acquisition of the working memory component of a delay match-to-sample task, as assessed by a T maze paradigm (Gibbs and Johnson, 2008; Sherwin and Henry, 2008). Interestingly, treatment with 17β-estradiol was sufficient to prevent the loss of the working memory component in this task (Gibbs and Johnson, 2008; Sherwin and Henry, 2008). Similar results were also observed in a study examining the effect of OVX on the performance of female rats in a two-way active avoidance paradigm, which is dependent on the frontal cortex as well as the hippocampus. OXV animals displayed a reduced performance compared to control, intact animals, whereas OVX animals treated with 17β-estradiol performed better than both groups in the active avoidance task (Singh et al., 1994). In another study using a win-shift version of the radial arm maze to test spatial working memory, the local administration of estradiol benzoate into the frontal cortex of OVX female mice 40 min prior to testing significantly improved performance compared to OVX mice (Sinopoli et al., 2006). Acute estradiol treatment has also been shown to increase aggression (Trainor et al., 2007), which is also associated with PFC processing.
Several studies have used object recognition, which relies on both cortical and hippocampal processing (Ennaceur et al., 1997; Barker and Warburton, 2011), to test the ability of estradiol to rapidly influence memory processing. In female OVX rats, administration of 17α-estradiol or 17β-estradiol, either 30 min prior or 30 min post-training, was able to enhance memory recognition/retention (Luine et al., 2003). Interestingly, this enhancement was not observed when either estradiol isomer was administered 2 h post-training (Luine et al., 2003). Similar results were also observed with intact female rats; female rats in behavioral estrus and estrus, when circulating estradiol levels are increased compared to disestrus, displayed enhanced memory recognition compared to disestrus female rats (Walf et al., 2006). Moreover, the same study demonstrated that treatment with propyl pyrazole triol (PPT) or diarylpropionitrile (DPN), synthetic ERs modulators (SERMs) with higher affinities for ERα or ERβ respectively, were also able to enhance memory performance in object recognition when administered 30 min post-training (Walf et al., 2006). In mice, treatment with 17β-estradiol immediately post-training, injected directly into the dorsal third ventricle or dorsal hippocampus, was sufficient to enhance object recognition in middle-aged female OVX mice (Fan et al., 2010). Importantly, this enhancement was lost following administration of inhibitors for the kinases ERK (extracellular signal-regulated kinase) 1/2 and PI3K (phosphatidylinositol 3-kinase; Fan et al., 2010). Conversely to data shown in OVX female rats, another study has shown that PPT, but not DPN, is able to enhance object recognition in OVX female mice (Walf et al., 2006; Phan et al., 2011). It is of note that animals were treated 15 min prior to training, and that PPT and DPN were shown to enhance object recognition in female rats when administered 30 min post-training. Thus it is not clear whether the differences between these studies are due to the timing of estradiol administration. Several studies have also investigated the most effective dose of estradiol in the enhancement of object recognition (Luine et al., 2003; Inagaki et al., 2010; Phan et al., 2011). These studies have demonstrated that there is an inverted U-shaped dose response curve as opposed to a more traditional S-shaped dose response curve. In OVX female rats, post-training treatment with 5 μg/kg of 17β-estradiol and 1–2 μg/kg of 17α-estradiol, but not lower or high concentrations were effective in enhancing object recognition (Inagaki et al., 2010). In female OVX mice, pre-training treatment with 50 or 75 μg (per 30 g mouse) PPT was sufficient to enhance object recognition, whereas lower or higher concentrations were not (Phan et al., 2011). Interestingly an inverse U-shaped dose response curve have also been reported in estradiol-mediated enhancement of spatial memory (Gresack and Frick, 2006). This suggests that an inverse U-shaped pattern of estradiol effects on memory may be applicable to a number of memory tasks, and not limited to those involving cortical processing. Together, these studies demonstrate that acute estradiol treatment can rapidly influence behaviors that require cortical processing, and more so, that this enhancement occurs in a time-dependent or time-critical manner, and demonstrate an inverse U-shaped pattern of responsiveness according to the dose of estradiol used.
Dendritic Spines, a Link between Structural and Functional Plasticity
Synaptic plasticity is the process by which the strength of communication between neurons is regulated in response to stimuli. This process has emerged as the strongest candidate cellular mechanism that underlies information storage and processing in the brain, and is thought to be achieved by altering the strength and number of synaptic connections (Chklovskii et al., 2004; DeBello, 2008; Holtmaat and Svoboda, 2009). In the mammalian forebrain, the majority of glutamatergic excitatory synapses found on pyramidal neurons occur on highly specialized dendritic profusions called dendritic spines (Figures 1A,B). Dendritic spines are highly dynamic structures and undergo many forms of structural modification (Figure 1C). Spine remodeling occurs in concert with changes in post-synaptic function, driven by the trafficking of synaptic glutamate receptors. During development, dendritic spines are required for the formation of synapses, and thus formation of neural circuits. In the mature brain, dendritic spines change shape and number in response to plasticity or in response to a number of extracellular stimuli (Figure 1D). Regulation of dendritic spine structure modulates synaptic properties of neurons, and the ability of synapses to undergo plasticity (Bourne and Harris, 2008). It is this dynamic remodeling of dendritic spine shape and number that is thought to be essential for the ability of synapses to undergo long-lasting functional and structural changes, underlying the rewiring of neuronal circuits (Malenka and Nicoll, 1999; Luscher et al., 2000; Cline, 2003; Figure 1D) thus contributing to memory and cognition (Holtmaat and Svoboda, 2009; Kasai et al., 2010). Indeed, recent in vivo imaging studies have provided strong evidence that concurrent with the acquisition of learned behaviors there are coordinated changes in synaptic structure and protein content, leading to the rewiring of neuronal circuits required for information processing and storage (Holtmaat and Svoboda, 2009; Yang et al., 2009). More recently, extrinsic signals such as neuromodulators have been shown to induce changes in dendritic spine morphology (Jones et al., 2009; Woolfrey et al., 2009; Bavelier et al., 2010), but how such signals regulate dendritic spine morphology, synapse function, and ultimately modulate the rewiring of neuronal circuits is only now becoming evident.
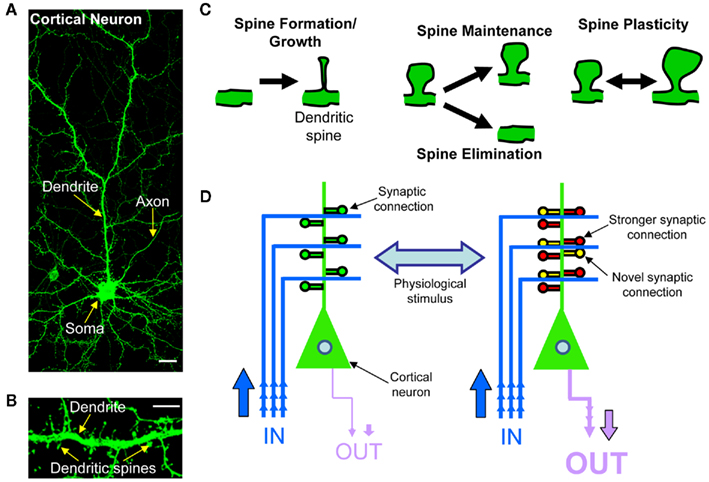
Figure 1. Rewiring of cortical circuitry. (A) Confocal image of a cortical neuron expressing green fluorescent protein (GFP), exemplifying cortical neuron morphology of a cortical neuron. The main dendrite is branched and has dendritic spines along its‘length. The neuron’s axon is much thinner than the dendrite, and has no spines. Scale bar, 5 μm. (B) High magnification confocal image of dendritic spine and dendrite of a cortical neurons. (C) Schematic of dendritic spine plasticity: spines form and grow, undergo either maintenance or elimination, or change shape and size. Dendritic spines change morphology in response to numerous extracellular stimuli. (D) Schematic of cortical circuitry rewiring. The strengthening or weakening of existing synaptic connections and the addition or elimination of synaptic connections induced by physiological stimuli, allows for the bi-directional rewiring of cortical circuits.
Functional Correlates of Dendritic Spine Structure
Dendritic spines are found in all shapes and forms. All types of dendritic spines, large or thin, have the potential to make connections with pre-synaptic partners (Bourne and Harris, 2008). Larger dendritic spines on average feature larger post-synaptic densities (PSDs; Bourne and Harris, 2008), persist for longer periods of time (Trachtenberg et al., 2002; Holtmaat and Svoboda, 2009; Kasai et al., 2010) and are resistant to plasticity-inducing stimuli. These large, stable spines have thus been labeled “memory spines” (Kasai et al., 2003). Conversely, smaller spines are often short-lived but can be readily potentiated to become stable spines, thus acting as highly dynamic “learning spines” (Kasai et al., 2003). Therefore, according to this model, spine morphology modulates synaptic properties and the ability to undergo plasticity (Segal, 2005; Bourne and Harris, 2008). The shape/size of a dendritic spine is also thought to describe the functional properties of synapses. Larger dendritic spines are also associated with larger α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) content, and have larger AMPA responses, whereas thinner/smaller spines have fewer AMPA receptors and small AMPA responses (Matsuzaki et al., 2001; Noguchi et al., 2011). Thus larger spines are more stable and form stronger synapses, while thinner spines are more dynamic and form weaker synapses. However, it is of note that this is not always the case; recent studies have demonstrated that in a few circumstances changes in spine size do not always correlate with synaptic strength (Segal, 2010). Importantly, structural morphologies are not static, but are highly dynamic, and can change shape and number in response to a number of extracellular stimuli. As extrinsic signals, such as neuromodulator are now known to be plasticity-inducing stimuli (Srivastava et al., 2008; Jones et al., 2009; Woolfrey et al., 2009; Bavelier et al., 2010), a major focus of current cellular and molecular neuroscience is to uncover the mechanisms underlying the control of dendritic spine shape, size and number, synapse function, via the trafficking of glutamate receptors, and how these changes integrate to modulate the rewiring of neuronal circuits.
Dendritic Spines Play Key Roles in Normal Brain Function
Neural circuits need to exhibit functional plasticity to encode information about the environment. A variety of stimuli, both physiological and pathological, can influence spine dynamics (Alvarez and Sabatini, 2007; Tau and Peterson, 2010; Figures 1C,D). Sensory experience, as modeled by mouse whisker stimulation, resulted in a transient increase in dendritic spine density in the corresponding primary cortical sensory region (Wilbrecht et al., 2010). On the other hand, selective sensory deprivation elicited through whisker trimming increased the fraction of transient spines (Trachtenberg et al., 2002). Rearing animals in enriched environments also results in elevated spine density in forebrain (Bose et al., 2009). Very recent evidence has shown rapid spine formation and stabilization in association with motor learning, followed by homeostatic pruning of synapses in layer V motor cortex (Yang et al., 2009). Thus structural reorganization of forebrain spiny synapses appears to be a powerful mechanism for information storage.
Wiring Plasticity is a Mechanism for the Rewiring of Cortical Circuits
Based on evidence from in vivo studies, as well as in vitro and theoretical work (Chklovskii et al., 2004; Bourne and Harris, 2008; Kasai et al., 2010; Segal, 2010), two methods of circuit rewiring have been proposed. In the first instance, changes in the shape and size of existing synapses, and therefore, the amount of information that passes across the synapse, is believed to be a critical mechanism in underlying information processing and storage in the cortex. However, in addition to this mechanism, it has been proposed that information storage can be accomplished through altering neuronal connectivity, which can be achieved by increasing or decreasing the number of functional synapses between two cells (Chklovskii et al., 2004; Le Be and Markram, 2006; DeBello, 2008). Together, these two mechanisms have been referred to as “wiring plasticity,” whereby altering the amount of information flow between existing synapses and altering the connectivity between cells are the underlying mechanisms of neuronal circuit rewiring, and thus involved in the acquisition of behaviors (cognitive function; DeBello, 2008; Figure 1D). Indeed, a recent in vivo imaging study of cortical motor neurons has demonstrated that there is a concurrent increase in dendritic spine density with the acquisition of a learned motor behavior (Yang et al., 2009). Thus the rewiring of cortical neurons appears to be a powerful mechanism for information storage in the cortex.
Dendritic Spine Dysfunction in Disease
Deficits in cognitive function, notably in working, spatial and reference memory, as well as social interactions, are core features of a great number of neurological disorders (DSM-IV, 2000). As dendritic spine morphology has been intimately linked to cognitive function (Ramakers, 2002; Holtmaat and Svoboda, 2009; Kasai et al., 2010), it is not surprising that multiple neuropathologies are strongly associated with disruptions of neural circuits (van Spronsen and Hoogenraad, 2010; Penzes et al., 2011a). Indeed, numerous neuropathological postmortem studies have strongly linked abnormal spine morphology with the pathogenesis of a number of neuropsychiatric disorders and neurodevelopmental disorders (Fiala et al., 2002; Penzes et al., 2011a); such as mental retardation (MR; Dierssen and Ramakers, 2006), fragile-X (Irwin et al., 2000), Down’s syndrome (Takashima et al., 1989), autism spectrum disorders (ASDs; Zoghbi, 2003; Pickett and London, 2005; Hutsler and Zhang, 2010), schizophrenia (Glantz and Lewis, 2000; Lewis and Sweet, 2009), depression (Gorman and Docherty, 2010), and Alzheimer’s disease (DeKosky and Scheff, 1990; Selkoe, 2002). It is currently posited that dendritic spine dysmorphogenesis can lead to defective or excessive synapse function and connectivity, resulting in disruptions in neural circuitry. This topic has recently been reviewed in depth (Tau and Peterson, 2010; van Spronsen and Hoogenraad, 2010; Penzes et al., 2011a). Dysregulation of the complex mechanisms that control dendritic spine structure and function may contribute to these synaptic irregularities. Understanding the mechanisms by which dendritic spine morphogenesis occurs will therefore, not only expand our knowledge of normal brain function, but that of abnormal brain function as well.
Molecular Control of Dendritic Spine Morphology
Recently major advances have been made into our understanding of the mechanisms that underlie changes in synapse structure. The post-synaptic density contains hundreds of distinct proteins, and the organizational complexity of this structure is becoming increasingly apparent (Figure 2A). A significant challenge is to identify the proteins that are responsible for determining post-synaptic ultrastructure. Dendritic spines are actin rich structures (Figure 1B); actin is the primary cytoskeletal component in spines, and actin modulation is essential for changes in spine morphology (Frost et al., 2010; Hotulainen and Hoogenraad, 2010). Multiple signaling pathways are know to converge on the actin cytoskeleton, and stimuli that induce filamentous actin rearrangements are associated with the formation, elimination, and changes in morphology of dendritic spines (Lise and El-Husseini, 2006; Tada and Sheng, 2006; Xie et al., 2007, 2008; Penzes et al., 2008; Srivastava et al., 2008; Jones et al., 2009; Woolfrey et al., 2009; Yoshihara et al., 2009). One family of proteins that have been shown to have potent effects on the actin cytoskeleton are the small GTPases.
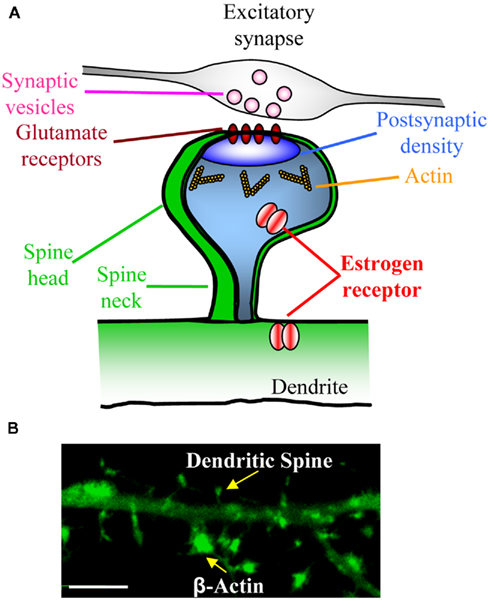
Figure 2. Dendritic spines are small protrusions along dendrites that contain post-synaptic densities. (A) Schematic of a mature dendritic spine making contact with an axon. Dendritic spines display a typical morphology of a spine neck and spine head. Cross section of spine reveals that a number of proteins ranging from neurotransmitter receptors (e.g., glutamate receptors) and scaffold proteins (e.g., PSD-95) reside within the post-synaptic density. Furthermore, actin, signaling proteins and other signaling proteins (e.g., small GTPases) are also found in dendritic spines, demonstrating the organizational complexity of dendritic spines. (B) High magnification confocal image of a cortical neuron expressing GFP-fused β-actin. Note the enrichment of β-actin in dendritic spines.
Small GTPases are G proteins that comprise a superfamily of more than 100 proteins with diverse cellular functions. As their name implies, these proteins are of low molecular weight (20–40 kD) and are related to heterotrimeric G-proteins (e.g., Gs and Gi). These proteins are highly evolutionarily conserved and expressed in many tissues including brain (Takai et al., 2001). There are five families (Ras, Rho, Rab, Sar1/Arf, and Ran) within the small GTPase superfamily which are categorized by structure and function. All small GTPases have a similar structure which permits binding to GDP and GTP (Takai et al., 2001). Diversity in small GTPases arises from unique effector-interacting domains and C-terminal regions which can be modified by post-translational modification. The ability of Rho- and Ras-family GTPases to regulate cytoskeleton dynamics and gene transcription places them as idea candidates to control fundamentally important neuronal functions.
Estradiol Rapidly Remodels Dendritic Spines of Cortical Neurons
As described above, there is ample evidence that estrogens can rapidly modulate cognitive processes that are mediated by distinct cortical regions. Previous studies have investigated the effect of estradiol on dendritic spines of pyramidal neurons in the dorsolateral cortex of young and aged rhesus monkeys (Hao et al., 2007). Long-term replacement with estradiol in OVX rhesus monkeys was sufficient to increase spine number compared to vehicle-treated animals (Hao et al., 2007; Dumitriu et al., 2010). While these study demonstrates a role of long-term estrogenic treatment in controlling dendritic spine number, it is not clear what effects acute estradiol has on synapses. To understand whether estradiol can acutely modulate dendritic spine morphology of cortical neurons, we investigated the ability of 17 β-estradiol (estradiol), to alter the shape/size or number of dendritic spines in mature (adolescent) cultured cortical neurons; at this time point, dendritic spines display a mature morphology, exhibiting a distinct head structure, and containing PSD-95 (Xie et al., 2007; Srivastava et al., 2008, 2010; Jones et al., 2009; Woolfrey et al., 2009; Figures 1B and 2A).
Treatment with a physiologically relevant concentration of estradiol (10 nM) resulted in an increase in the number of dendritic protrusions 30 min following treatment. Interestingly, this increase in spine number disappeared after 60 min, and the number of dendritic protrusions return to control levels. Time-lapse imaging further demonstrated that it was the estradiol-induced dendritic protrusions that were selectively eliminated, indicating that estradiol’s effects on spines were transient (Figure 3A). Importantly, these estradiol-induced protrusions were shown to be functional dendritic spines as they were juxtaposed to pre-synaptic terminals; non-functional dendritic protrusions, known as filopodial protrusions, do not make synaptic contacts. Close examination of dendritic spine morphology following estradiol treatment, revealed that estradiol-induced spines had a “thin” like morphology. This spine morphology is associated with highly dynamic synapses that can be either readily potentiated and stabilized, or eliminated (Xie et al., 2005; Penzes et al., 2011b). Owing to the suggestion that estradiol may act as a neuromodulator, we speculated that the time that estradiol is present at synapses may be tightly regulated. Indeed, several enzymes that are involved in metabolism of estradiol have been detected with the brain (Balthazart and Ball, 2006). Exposure of cortical neurons with estradiol for 5 min to mimic the rapid removal of the neurosteroid from synapses, elicited the same transient increase in spine density as the continued presence of estradiol in the experimental medium.
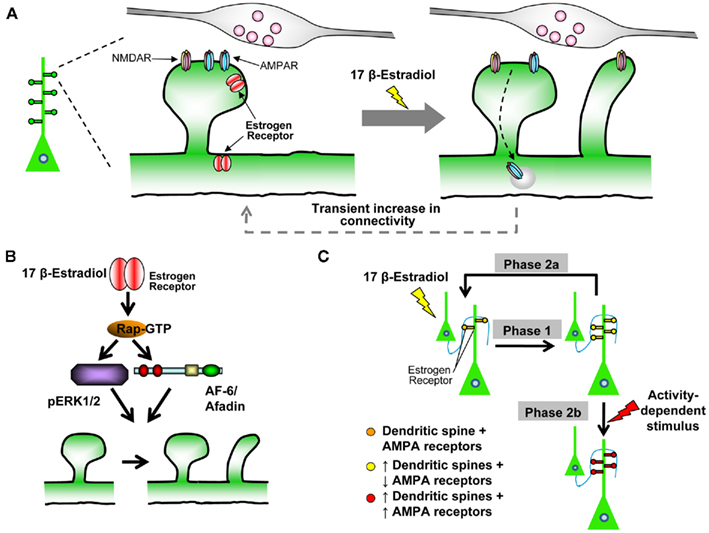
Figure 3. Estradiol-induced “two-step wiring plasticity.”(A) Schematic of estradiol-induced spinogenesis and glutamate receptor trafficking. Treatment with 17 β-estradiol induces the formation of novel spines, which form connections with pre-synaptic partners within 30 min. Concurrently GluA1-containing AMPARs are removed from existing spines, and GluN1-containing NMDARs are trafficking into nascent spines. The effect is transient, as by 60 min, estradiol-induced spines are preferentially eliminated, GluA1-containing AMPARs and GluN1-containing NMDARs return to control levels. (B) Schematic representation of Rap/ERK1/2/AF-6 pathway required for estradiol-induced spinogenesis. Note, both ERK1/2 and AF-6 are downstream of Rap signaling, but may act in a parallel manner to increase spine density. (C) Model of Two-Step Wiring Plasticity (TSWP). TSWP can be divided into three distinct phases Phase 1: estradiol increases the number of connections between cells, synaptic communication is reduced. Phase 2a: number of connections return to control levels, synaptic communication returns to normal. OR Phase 2b: synaptic activity-like stimulus leads to long-lasting (24 h) increase in connectivity and increase synaptic communication.
Together, these data suggest that acute estradiol treatment is able to induce a transient increase in the number of dendritic spines, without affecting existing dendritic spines (Figure 3A). These results parallel a recent study of estradiol remodeling of dendritic spines in CA1 cells of hippocampal slices cultures, which demonstrated that estradiol induces spine formation without affecting pre-established networks (Mendez et al., 2010). Interestingly, this rapid and transient effect of estradiol on mature cortical neurons has also been described in young, developing cortical neurons. Treatment of young neurons with the same concentration of estradiol resulted in a rapid, yet transient, increase in the number of dendritic protrusions, in a similar time frame (Sanchez et al., 2009). It is also of note that rapid estradiol-induced spinogenesis in mature cortical neurons were not dependent on N-methyl-D-aspartic acid receptor (NMDAR) activity, as estradiol-induced spinogenesis occurred in the presence or absence of NMDAR blockade (Srivastava et al., 2008). In hippocampal neurons NMDAR activity is required for rapid estradiol-induced spinogenesis (Mukai et al., 2007), indicating that the actions of estradiol in cortical neurons employ distinct molecular mechanism(s) that those initiated in hippocampal neurons to induce changes in spine morphology.
Molecular Mechanisms of Estradiol Modulation of Dendritic Spine Structure
Dendritic spines are highly specialized signaling compartments containing numerous signaling proteins, and a major component of these structures is the actin cytoskeleton (Figures 2A,B). Rearrangements of the cytoskeleton, driven by actin polymerization and depolymerization, are thought to be a key mechanism underlying the dynamic regulation of spine structure (Tada and Sheng, 2006; Yoshihara et al., 2009). Multiple signaling pathways, particularly those involving Rho- and Ras-family small GTPases, have been shown to converge on the cytoskeleton to regulate actin rearrangement, thus underlying the formation, elimination, and stabilization of dendritic spines (Tada and Sheng, 2006; Penzes et al., 2008, 2011b; Yoshihara et al., 2009). Estradiol has been shown to regulate both small GTPase signaling and actin dynamics in non-neuronal cells (Sanchez et al., 2010a), but it is not clear if these pathways are required for the remodeling of dendritic spines of cortical neurons.
The morphology of estradiol-induced nascent spines provided us with the first insight into the potential underlying molecular pathways required for the increase in dendritic spine density. Previous studies have shown that activation of Rap, a member of the Ras-family of small GTPases, results in the formation of highly dynamic and “thin” dendritic spines (Xie et al., 2005; Woolfrey et al., 2009; Penzes et al., 2011b). Following estradiol treatment active Rap levels were increased in a time-dependent manner mimicking the effect of estradiol on spine density. In situ inhibition of Rap signaling by overexpression of RapGAP, a protein that inhibits Rap activity, or of a dominant-negative Rap construct, blocked estradiol-induced spinogenesis. In contrast, pharmacological inhibition of the closely related small GTPase Ras did not block estradiol-induced nascent spines. When the activity of Rac, a Rho-family small GTPase, was examined, a modest decrease in Rac levels after 60 min of estradiol treatment was observed. It is possible that this small inhibition in Rac activity is sufficient to drive estradiol-induced spine numbers back to a level similar to control, as previous studies have shown that inhibition of Rac reduces spine numbers (Tashiro and Yuste, 2004).
Further investigation of the downstream targets of Rap signaling revealed that the MAP kinases, ERK1/2 (extracellular signal-regulated kinase) was required for estradiol-induced spinogenesis. Importantly, ERK1/2 activation has been shown to be required for the remodeling of dendritic spines, acquisition of learning and memory, and is a common downstream target of estradiol in a number of cell types (Thomas and Huganir, 2004; Raz et al., 2008). The protein AF-6 (also known as afadin), which is a PDZ domain-containing and F-actin-binding protein, previously shown to be regulated by Rap and is required for Rap-dependent spine plasticity (Xie et al., 2005), was clustered to synapses in response to estradiol treatment, paralleling estradiol’s effects on spine density. Furthermore, estradiol-dependent spine formation was prevented by the expression of a mutant AF-6 with an inactive PDZ domain. Taken together, these data suggested that estradiol signaling via a Rap/ERK/AF-6-dependent pathway is required for increased dendritic spine number in mature cortical neurons (Figure 3B).
In young, developing cortical neurons, estradiol-mediated increases in dendritic protrusions required the activation of a c-SRC/Rac1/Cdk5/WAVE1/Arp2/3 pathway, in parallel with activation of a RhoA/ROCK-2/moesin cascade (Sanchez et al., 2009). Elucidation of these pathways directly link estradiol signaling with the rearrangement of the actin cytoskeleton via WAVE1/Arp2/3 and moesin. It is of note that these pathways are different from those activated in mature cortical neurons by estradiol. One potential explanation of this difference could be attributed to the differential expressing of signaling proteins and thus the coupling of estradiol to different intracellular pathways dependent on the developmental time point. Upstream regulators of small GTPase pathways have been shown to be developmentally regulated (Penzes et al., 2008), and the local environment of signaling pathways that link ERs with such small GTPase pathways may also be developmentally regulated. Put together, it is possible that during a young developmental time point, parallel activation of a c-SRC/Rac1/Cdk5/WAVE1/Arp2/3 pathway and a RhoA/ROCK-2/moesin cascade is required for the formation of potential synaptic connections, whereas mature cortical neurons, with pre-existing synaptic connections, require an interplay between a Rap/ERK1/2/AF-6 and Rac-dependent pathways in order to induce a transient increase in synaptic connectivity. Interestingly, a recent study of rapid estradiol actions in CA1 hippocampal cells demonstrated that estradiol increased the polymerization of actin in a RhoA/ROCK/cofilin-dependent pathway (Kramar et al., 2009). Moreover, estradiol has been shown to regulate the expression and activity of LIM kinase (LIMK) in CA1 neurons (Yildirim et al., 2008; Yuen et al., 2011). As activation (phosphorylation) of LIMK can control the polymerization of actin through the actin binding protein, cofilin, estradiol regulation of LIMK could provide another mechanism by which estradiol can control actin dynamics in pyramidal neurons. The disparities in the elucidated mechanisms required for estradiol-mediated spinogenesis in cortical neurons and hippocampal could be due to several reasons. It is possible that differences in the cell preparations (i.e., ex vivo versus in vitro) or age of neurons underlie these differences. However, it is more likely that these differences arise due to cell type specificity, thus suggesting that neurons from separate regions of the brain employ distinct molecular mechanism to transduce rapid estrogenic signaling into morphological changes. Ultimately, these discrete mechanisms may be important for how estradiol modulates neuronal circuitry in each area.
The Role of ERβ in Spine Formation in Cortical Neurons
Investigations into the expression profile of ERs have indicated that ERβ is highly expressed in the mammalian cortex, but less so in the hippocampus; the expression profile of ERα is thought to be higher in the hippocampus compared to the cortex (Kritzer, 2002). Previous studies investigating which ER is required for rapidly induced morphological changes in hippocampal neurons have suggested that ERα but not ERβ is required for rapid increases in dendritic spine density (Mukai et al., 2007). However, owing to the increased expression of ERβ compared to ERα in cortical neurons, we hypothesized that activation of ERβ may be sufficient to induced rapid morphological changes in mature cultured cortical neurons. Treatment of cortical neurons with WAY-200070, an ERβ-specific agonist (Liu et al., 2008), resulted in a rapid (within 30 min) increase in dendritic spine density (Srivastava et al., 2010). The majority of these spines were positive for overlap with the pre-synaptic marker bassoon, suggesting that ERβ-induced spines were making connections with pre-synaptic termini (Srivastava et al., 2010). Moreover, these spines also contained the post-synaptic protein, PSD-95, further indicating that ERβ-induced spines were forming functional connections. Interestingly, there was no increase in the overall expression of PSD-95 following acute treatment with WAY-200070. Rather, a redistribution of PSD-95 was observed following treatment with WAY-200070; a concurrent increase in membrane PSD-95 was seen with a reduction in cytosolic PSD-95, suggesting that the recruitment of PSD-95 from cytosolic clusters within the dendritic shaft to dendritic spines resulted in the enrichment of ERβ-induced nascent spines with PSD-95 (Srivastava et al., 2010). Examination of the signaling pathways activated following ERβ stimulation revealed an increase in the phosphorylation (activation) of PAK and ERK1/2, providing a potential signaling cascade linking ERβ with cytoskeleton rearrangements underlying changes in dendritic spine density. Together these data suggest that activation of ERβ in cortical neurons is capable of increasing functional dendritic spine numbers, via a PAK/ERK1/2 pathway, in a rapid time frame.
Estradiol Rapidly Modulates Glutamate Receptor Trafficking
A widely accepted view is that the rapid trafficking of the glutamate receptors N-methyl-D-aspartic acid (NMDARs) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) is a key mechanism in controlling synaptic communication. Multiple studies have indicated that structural and functional changes in synapses often go hand in hand (Matsuzaki et al., 2001). However, this is not always the case (Segal, 2010). Recent studies have demonstrated that synapses can contain NMDARs, but lack AMPARs. These synapses, termed “silent synapses” are thought to be rapidly potentiated during plasticity, and are a major mechanism for the remodeling of neuronal circuits (Ashby and Isaac, 2011). Estradiol has been shown to increase the responses to long-term potentiation (LTP) and long-term depression (LTD) at hippocampal synapses (Mukai et al., 2007; Woolley, 2007; Kramar et al., 2009). But whether estradiol regulates the trafficking of NMDARs or AMPARs in cortical neurons is not clear.
To examine whether acute estradiol treatment affected glutamate receptor trafficking, we investigated with localization of NMDAR and AMPARs subunits following treatment with estradiol. Acute treatment with estradiol in cortical neurons induced the removal of the GluA1-containing AMPARs from synapses at 30 min, while inducing insertion of the GluN1-containing NMDARs. Remarkably, by 60 min of treatment, GluA1 and GluN1 content had returned to control levels. Time-lapse imaging of GFP-tagged GluA1 demonstrated that GluA1 was being internalized from pre-exiting dendritic spines, and returning into the same spine, without entering nascent spines (Figure 3A). To understand the functional outcome of this trafficking we measured AMPAR-mediated miniature excitatory post-synaptic currents (mEPSCs). Consistent with GluA1 trafficking, electrophysiological analysis demonstrated that estradiol-induced a transient reduction in AMPAR-mediated mEPSC frequency, but not amplitude, indicating a change in the number of active synapses (Srivastava et al., 2008). Together, these data indicated that estradiol may transiently increase in the number of synapses containing NMDARs but lacking AMPARS and thus the number of silent synapses (Figure 3A).
Previous studies have shown that activation of the small GTPase Rap can lead to the internalization of AMPARs (Xie et al., 2005; Woolfrey et al., 2009). As estradiol treatment results in the activation of Rap, it is likely that a Rap-dependent mechanism underlies the rapid internalization of GluA1 in cortical neurons. Interestingly estradiol treatment of hippocampal neurons resulted in the rapid increase in levels of membrane GluA1, via a MAP kinase/calpain pathway (Zadran et al., 2009). It is likely that the differences seen in GluA1 trafficking in hippocampal and cortical neurons is due to the different signaling mechanisms activated by estradiol in these different cell types.
Estradiol-Induced “Two-Step Wiring Plasticity”
The ability of estradiol to rapidly induce spine formation, and silence a subset of synapses, led us to question: what is the physiological role of such a transient increase in silent synaptic connections; what relevance it has for the rewiring of cortical circuits; how do these effects relate to estradiol’s ability to rapidly modulate information encoding?
In vivo imaging studies have very elegantly demonstrated that cortical circuits are remodeled in a physiologically relevant manner; however, the underlying molecular and cellular mechanisms are still somewhat of a mystery. Theoretical studies have posited that there is sufficient overlap between dendrites and axons (potential synapses) to allow additional synapses to occur (Chklovskii et al., 2004). In vivo work has also shown that neurons can spontaneously make transient synapses. These nascent connections are thought to “sample” pre-synaptic contacts, but are not considered to be functional. These novel connections are not permanent and retract unless a subsequent Hebbian-like, or activity-dependent stimulus, is provided which stabilizes or “holds” the nascent connections (Holtmaat et al., 2005). Furthermore, this activity-like stimulus is also thought to potentiate these connections, making them functional (Chklovskii et al., 2004; Holtmaat et al., 2005). Therefore, it has been posited that information can be stored through a “sample and hold” or “Two-Step” model (Holtmaat et al., 2005). Under this theoretical framework, novel dendritic spines “sample” pre-synaptic contacts, but will retract without subsequent Hebbian-like mechanisms that reinforce or “hold” nascent connections. Thus, this model represents a mechanism that features enormous capacity for cortical information storage (Chklovskii et al., 2004; Le Be and Markram, 2006). Such alterations in synapse number may result in both increased and stronger connectivity between neurons, and thus constitutes a novel form of “wiring plasticity” (Chklovskii et al., 2004; Holtmaat et al., 2005; Le Be and Markram, 2006; DeBello, 2008; Holtmaat and Svoboda, 2009; Figure 1D). Although this phenomenon has been observed in vivo (Holtmaat et al., 2005; Le Be and Markram, 2006), the physiological signals and molecular mechanisms that govern this form of plasticity are not known.
Based on these observations, we hypothesized that estradiol’s ability to transiently induce nascent silent synaptic connections may serve to “prime” neurons to respond to activity-dependent stimuli with greater efficacy thus participating in “Two-Step” model of wiring plasticity. Estradiol-induced thin spines would sample the pre-synaptic environment forming nascent synapses that contain NMDARs but lacked AMPARs. If a second stimulus is not applied, these novel connections retract, allowing the cell to return to its “resting state” (Figure 3A). However, if a second, activity-dependent stimulus is applied, these newly formed synapses may become stabilized and potentiated, leading to an increase in connectivity between cortical neurons.
Co-treatment of estradiol followed by an activity-dependent stimulus, achieved by activation of NMDARs by a chemical LTP paradigm (Xie et al., 2007; Srivastava et al., 2008), resulted in a stabilization of estradiol-induced spines lasting up to an hour. Moreover, this maintained increased spine density persisted for up to 24 h. Importantly, these spines overlapped with the pre-synaptic maker bassoon, demonstrating that the combined treatment of estradiol and an activity-dependent stimulus had induced a long-term increase in connectivity in neurons. Furthermore, investigations of the effect of these two treatments on synapse function revealed that there was an increase in the surface expression of GluA1 at synapses which was paralleled by an increase in AMPAR-mediated transmission (Figure 3C). Consistent with this finding, AMPAR-mediated mEPSCs were also increased compared to control levels, suggestive of an overall increase in synaptic transmission. Previous reports have demonstrated that CaMKII activity is required for NMDAR-dependent changes in spine morphology and GluA1 insertion into synapses (Xie et al., 2007). Indeed, we found that the combined treatment of estradiol and activity-dependent stimuli resulted in an increase in active levels of CaMKII (Srivastava et al., 2008), suggesting that this kinase plays a role in mediating the increase in connectivity and transmission by these two extracellular signals. In summary, these data demonstrate that the combined treatment of estradiol and activity-dependent stimulus was able to both induce a long-lasting increase in synaptic connectivity and enhance synaptic communication between neurons (Figure 3C).
Estradiol and Disease
Multiple studies from basic science have demonstrated neuroprotective effects by estradiol. Coupled with evidence that estradiol can induce synaptic enhancements and influence cognition in a range of animal models, the use of estradiol as a treatment in a number of disorders has been investigated. Moreover, estradiol has been implicated in the onset, clinical course, and symptomatology of a number of psychiatric and neurodegenerative disorders. While a number of clinical studies have indicated a beneficial role of estradiol in a number of disorders (Cyr et al., 2000; Kulkarni et al., 2008; Hughes et al., 2009; Kulkarni, 2009; Sanchez et al., 2010b), studies by the Women’s Health Initiative (WHI) and ancillary studies by WHI Memory Study (WHIMS) have reported conflicting results (Rossouw et al., 2002; Espeland et al., 2004; Shumaker et al., 2004). In these studies, a hormone treatment of conjugated equine estradiol and medroxyprogesterone were administered to women of 65 years of age or old. These randomized control trials did not find any effect in protection against cognitive decline. Conversely, they suggested a potential increase in cognitive decline and increased risk for a number of risk factors for cardiovascular problems, stroke, and cancer (Rossouw et al., 2002; Espeland et al., 2004; Shumaker et al., 2004). While there is controversy regarding the results of these findings (Craig et al., 2005), it is clear that caution must be taken when examining the potential beneficial effects of estradiol in a number of disorders. Moreover, it may also be suggested that the use of alternative approaches to mimic estradiol’s positive effects, by modulating specific ERs (Hughes et al., 2009) and/or regulating estradiol intracellular molecular targets, may offer an avenue of treatment that may allow for the beneficial effects of estradiol without the harmful side effects. Here we will highlight some of the evidence and potential molecular basis for estradiol’s role in a number of neurodegenerative and psychiatric disorders.
Estradiol and Schizophrenia
Schizophrenia is a devastating disorder that affects 0.5–1% of the population. Deficits in cognitive function, notably in working memory, are a core feature of this disorder, and dendritic spine morphology has been intimately linked to cognitive function (Lewis and Sweet, 2009). While current anti-psychotic treatments have had some success in the management of the positive symptoms of schizophrenia, cognitive functional deficits have been more difficult to treat (Lewis and Sweet, 2009). Numerous clinical, epidemiological, and postmortem studies have strongly linked abnormal spine morphology, and disruptions in endocrine signaling with the pathogenesis of schizophrenia (Hill et al., 2006; Lewis and Sweet, 2009). Multiple studies have suggested that estradiol may have neuroprotective properties against this disorder, and several clinical trials have found that estradiol, in conjunction with ongoing treatments with anti-psychotics, has partial success at ameliorating schizophrenic symptoms in females (Mendrek, 2007; Mortimer, 2007; Kulkarni et al., 2008). Interestingly, a recent clinical trial has also demonstrated a positive role for estradiol, administered in addition to anti-psychotics, in the treatment of schizophrenic symptoms in males, leading to the authors proposing that estradiol may be an effective treatment in males as well (Kulkarni et al., 2011). However, it is important to note that these trials were short-term, lasting up to 28 days. As such, the potential benefits of long-term effects of adjunct treatment of estradiol with ongoing anti-psychotic drugs have yet to be investigated. The etiology of schizophrenia appears to involve a number of genetic and environmental components, resulting in a pathogenic mechanism leading to molecular and cellular disturbances and altered brain function. While clinical studies have reported that estradiol can ameliorate schizophrenic symptoms, the mechanisms underlying the beneficial effects of estradiol are not well understood. Recent studies have shown sequence variation and lower mRNA of wild type ERα associated in patients with schizophrenia suggesting that deficient signaling via this receptor may contribute to the symptomatology of schizophrenia (Perlman et al., 2005; Weickert et al., 2008). This in turn may affect the ability of estradiol to regulate dendritic spine density in subjects with either reduced or variant forms of ERα. Recent studies have also shown that ERα can interact with ErbB4 in a neuronally derived cell line (Wong and Weickert, 2009), and furthermore, estradiol treatment has been shown to regulate ErbB4 expression (Ma et al., 1999). Importantly, genome-wide association studies have identified neuregulin-ErbB4 signaling as a major susceptibility pathway in schizophrenic patients (Jaaro-Peled et al., 2009). Furthermore, as neuregulin-ErbB4 signaling has been shown to indirectly regulate spine density (Li et al., 2007), estradiol-dependent regulation of ErbB4 expression may offer an indirect pathway in the regulation of dendritic spines in subjects with schizophrenia. Taken together, these preclinical and clinical studies suggest that targeting estrogenic signaling may be a viable option for the treatment of schizophrenia in male and female patients.
Estradiol and Depression
Major depression is a disease that is strongly associated with sex differences in the predominance of this disease being higher in females and the onset of this disease also being younger in female subjects compared to males. This has lead to the suggestion that estradiol signaling may be involved in this disease (Weissman et al., 1993; Hughes et al., 2009). Multiple lines of evidence have been provided suggesting that a loss of synaptic connections in subjects suffering from major depression contributes to the etiology of this disorder (Nestler et al., 2002; Gorman and Docherty, 2010; Soetanto et al., 2010). As with most psychiatric disorders, the underlying pathogenesis of this complex disease involves an integration of multiple factors ranging from environment to molecular and genetic components. Clinical studies have suggested that estradiol or hormone replacement in combination with antidepressants may be beneficial in some case (de novaes Soares et al., 2001; Morgan et al., 2005; Rasgon et al., 2007; Zanardi et al., 2007). However, these findings are inconsistent and more anecdotal observations. Preclinical studies of animal studies of depression have also shown an anti-depressive effect of estradiol. In a learned helplessness model of depression in rats, estradiol treatment was able to improve escape performance and rescue stress-induced loss of dendritic spines of CA1 neurons in OVX females (Hajszan et al., 2010). In aged intact female mice, administration of estradiol 1 h prior to testing demonstrated anti-anxiety and anti-depressive effects in a number of anxiety-like and depressive-like behaviors (Walf and Frye, 2010), suggesting that estradiol via a rapid mechanism may have anti-depressive effects. In addition, investigations in ER knockout mice have shown that loss of ERβ results in an increase in depressive and anxious behaviors (Krȩżel et al., 2001; Rocha et al., 2005). Moreover, treatments with estradiol, or ERβ agonists specifically, were enough to reduce these abnormal behaviors (Hughes et al., 2008; Walf et al., 2008). However, the mechanisms that underlie estradiol’s potential beneficial effects in depression are not clear. Several studies have shown that mRNA levels of ERα are reduced in subjects with major depression (Perlman et al., 2004, 2005) and that SNPs of the ER alpha gene are associated with patients with major depression (Mill et al., 2008). The reduction in ERα expression may offer a mechanism that is disrupted in major depression leading to a reduction in dendritic spine number. Interestingly, reduced levels of brain-derived neurotrophic factor (BDNF) have also been associated with major depression (Shalev et al., 2009; Gorman and Docherty, 2010). Estradiol has been shown to increase the production of BDNF (Scharfman and MacLusky, 2006), and a functional interaction between the TrkB receptor and ERα has also been demonstrated (Wong et al., 2011). This finding, in combination with evidence that BDNF can regulate dendritic spine morphology (Verpelli et al., 2010), suggests that an integration of these two signaling pathways may contribute to the beneficial role of estradiol in major depression.
Estradiol and Alzheimer’s Disease
Alzheimer’s disease is the most common form of dementia. Although amyloid plaques, neurofibrillary tangles and cell death are defining characteristics of Alzheimer’s disease, finding from neuropathological studies have consistently reported prominent synapse loss (DeKosky and Scheff, 1990; Selkoe, 2002). Familial Alzheimer’s disease has been associated with mutations in APP, PSEN1, and PSEN2, genes essential for the production of beta amyloid (Aβ). Mutations in these genes result in an increase in Aβ production, and several cellular studies have provided compelling evidence that soluble Aβ oligomers reduce spine density (Selkoe, 2008). On the other hand, the emergence of Alzheimer’s disease after age 65, referred to as late-onset Alzheimer’s disease (LOAD) accounts for the vast majority of Alzheimer’s disease cases. Although numerous genes have been proposed as LOAD risk factors, the gene encoding apolipoprotein E (APOE) is considered to be the most important risk factor for LOAD (Sleegers et al., 2010). The evidence of the involvement of estrogens in Alzheimer’s disease comes from epidemiological studies demonstrating that the risk for females developing Alzheimer’s disease significantly increases following menopause (Henderson, 2009). Furthermore, studies have also detected lower levels of estradiol and reduced aromatase expression within the brains of females with Alzheimer’s disease (Yue et al., 2005). In addition, genetic studies have also shown an association between variants of the gene CYP19a (aromatase; Iivonen et al., 2004), altered levels of ERα mRNA and protein (Ishunina and Swaab, 2008), and variants in the ERβ gene (Pirskanen et al., 2005) with risk for Alzheimer’s disease. Several clinical studies have suggested a beneficial role of estradiol plus progestin in the treatment of Alzheimer’s disease (Resnick et al., 2006); however, this beneficial role has also been challenged (Rossouw et al., 2002; Rapp et al., 2003; Espeland et al., 2004; Shumaker et al., 2004). Preclinical studies have also supported a role of estradiol in a neuroprotective role. Disruption of estradiol signaling in mouse models of Alzheimer’s disease resulted in an accumulation of Aβ (Carroll et al., 2007), whereas replacement of estradiol following ovariectomy, reduced the accumulation of Aβ, and improved cognitive performance (Carroll and Pike, 2008). Other studies have also shown that estradiol treatment can regulate APOE expression. Activation of ERα up-regulates APOE expression, whereas ERβ decreases APOE expression (Wang et al., 2006). Importantly, mice expressing APOE ε4 (the APOE allele associated with the greatest risk of developing Alzheimer’s disease), displayed reduced dendritic spine number compared with wild type mice (Dumanis et al., 2009). Conversely, expression of APOE ε2 in a mouse model of Alzheimer’s disease was reported to restore spine density to control levels (Lanz et al., 2003). Collectively, these studies suggest that estradiol may have a beneficial role in Alzheimer’s disease, and that these effects may be due to its ability to modulate dendritic spine density, either directly via activation of specific ERs, or in directly, by reducing levels of Aβ, or regulating the expression levels of specific APOE alleles. As described above, studies from the WHI and WHIMS have failed to support a positive role of estradiol in cognitive decline, and in contrast reported increased risk of cognitive decline and dementia. However, it has also been suggested that there is a critical period, or “window of opportunity,” in which estradiol can be effective in protecting again cognitive decline (Gibbs, 2000). Indeed evidence from both basic and clinical studies are beginning to support such a critical period hypothesis (Rocca et al., 2011). Furthermore, a recent study has suggested that sustaining a sufficient level of ERα expression is required for the critical period, thus offering an insight into the potential mechanisms underlying this window of opportunity (Zhang et al., 2011). Whether this reduction in ERα levels is a critical mechanism in the pathophysiology of Alzheimer’s disease, or whether this pertains solely to this hypothesis is also unclear at this point.
Potential Cellular Mechanisms Underlying Beneficial Roles of Estradiol in Disease
The pleiotropic effects of estrogens in the CNS demonstrate that there are potentially multiple different mechanisms by which it’s beneficial and neuroprotective effects may be exerted. Studies from primary neuronal cultures have revealed that estradiol is neuroprotective in response to glutamate toxicity. Moreover, both ERα and ERβ have been implicated in this role. Neuronal viability has been shown to be increased following acute treatment with estradiol, and is dependent on a SRC/ERK/CREB/BCL-2 pathway (Singer et al., 1999; Wu et al., 2005). In addition, the coupling of ERα to Gβ/γ G protein subunits has been shown to be required for hippocampal cell survival following NMDA-induced excitotoxicity (Dominguez et al., 2009). However, in addition to these mechanisms of neuroprotection, the potential beneficial role of estradiol in neurodegenerative and psychiatric disorders may also be mediated by estradiol’s effects on neuronal circuitry. As described above, several of these disorders display aberrant dendritic spine morphology, and thus being able to modulate these synaptic structures may offer a potential therapeutic avenue. Specifically, as our data suggest that estradiol is able to “prime” cortical neurons to respond to subsequent activity-dependent stimuli, it is interesting to speculate that such a two-step form of plasticity would allow neurons to make novel connections that have been lost due to a pathogenic insult. In agreement with this hypothesis, it is interesting to consider that the anti-depressive effect of estradiol in a learned helplessness model of depression is coupled with an increase in spinogenesis in CA1 neurons (Hajszan et al., 2010). Furthermore, activation of ERβ has anti-depressive-like effects (Walf et al., 2008), and activation of ERβ can increase spine density in cortical neurons (Srivastava et al., 2010). Moreover, a recent study has demonstrated that rapid estradiol signaling, acting via a mechanism almost identical to “two-step wiring plasticity,” has shown that estradiol treatment is sufficient to ameliorate spine loss induced by soluble Aβ (Logan et al., 2011). Despite the potential beneficial effects of estradiol in neurodegenerative and psychiatric disorders, it is important to take into account the potential harmful side effects. As such, a greater understanding of the molecular and cellular mechanisms underlying both estradiol’s effects on neuroprotection and on neuronal circuitry will hopefully identify new targets in the development of certain CNS disorders.
Conclusion and Future Directions
Our cellular and molecular studies into rapid action of estradiol on cortical neurons have led us to propose the following model: estradiol can rapidly “prime” neurons to respond to subsequent synaptic activity-like stimuli with greater efficacy (Srivastava et al., 2008). This is achieved by estradiol modulation of spine structure and functional in neural circuits. To understand the complex mechanisms underlying this form of wiring plasticity termed “Two-Step Wiring Plasticity,” we have divided it into three conceptual phases (see Figure 3C):
Phase 1: Estradiol transiently increases the number of dendritic spines, and generates silent synapses by removing GluA1-containing AMPARs from existing and inserting GluN1-containing NMDARs into nascent synapses. Increased connectivity and generation of silent synapses places the neurons in a “primed” state, ready to respond to subsequent stimuli with greater efficacy.
Phase 2a: Without a second stimulus, estradiol-induced novel spines are preferentially eliminated, and GluA1-containing AMPARs and GluN1-containing NMDARs return to pre-existing spines. This mechanism allows the cell to return a “resting state.”
Phase 2b: Addition of a subsequent activity-dependent stimulus leads to persistence of estradiol-induced spines and the trafficking of GluA1-containing AMPARs into both pre-existing and novel spines. This results in long-term (24 h) increased synaptic connectivity and transmission.
Elucidation of the molecular pathways activated following acute estradiol treatment may offer an insight into the critical signals required for this form of wiring plasticity to occur. Activation of a Rap/AF-6/ERK1/2 pathway is critical for the estradiol-mediated increase in spine density seen in phase 1 (Figure 3B). During phase 2a, it is possible that a decrease in Rac activity may be required to induce the retraction of estradiol-induced spines. Conversely, during phase 2b activation of Rac via an NMDAR/CaMKII pathway as previously shown (Xie et al., 2007) is required for the stabilization of nascent spines and the potentiation of silent synapses (Figure 3C). Together this model of two-step wiring plasticity demonstrates how estradiol may act to prepare a cell to respond to activity-dependent stimuli with greater efficacy, resulting in an increase in connectivity and communication between neurons, leading to a rewiring of cortical circuits. This in turn may contribute to the mechanism(s) by which estradiol influences cognitive function.
It is of note that these studies were performed using an in vitro system, which may also be the basis for the differences seen in estradiol’s actions in different brain areas. In the future it will be important to confirm the effects of two-step wiring plasticity within more intact systems, and eventually in vivo. However, the use of an in vitro system, such as cultured neurons offers an excellent platform for dissecting the potential molecular mechanisms underlying this form of plasticity. Based on our current data, it is appealing to speculate that the “priming” of dendritic spines in cortical neurons may serve to augment the acquisition or consolidation of certain behaviors. The evidence that acute estradiol administration in a time-specific manner pre- or post-training, is required for the enhancement of object recognition, may point to a role for this plasticity in the acquisition of learned behaviors. However, the relationship between estradiol-induced two-step wiring plasticity and the acquisition, consolidation, or retrieval of memory needs to be investigated.
Aberrant dendritic spine morphology and number in disease have implications for both synapse function and circuit-level connectivity. Although the deficits in dendritic spines may be the original insults contributing to the pathophysiology of a disease, it is possible that alterations in dendritic spine morphology or number may be a secondary effect or compensatory effect. Nevertheless, dendritic spines may serve as a common substrate in a number of psychiatric, neurodevelopmental, and neurodegenerative disorders, particularly those involving cognitive deficits. As modifications in spine morphology and number are associated with cognitive function, understanding how extracellular signals impact spine morphology, and moreover the underlying molecular mechanisms that are essential for the development of more effective and specific therapies for these disorders. The evidence that estradiol is able to influence a range of cognitive behaviors, coupled with the ability of this neurosteroid to exert a powerful effect on dendritic spine morphology and number, identifies estradiol as a good candidate as a therapeutic target.
The evidence provided by clinical and preclinical studies suggest that estradiol-based therapies are a potential therapeutic avenue for the treatment of certain complex disorders. However, as with most estradiol-based treatments, it is important to remember that the use of estradiol as a long-term treatment is associated with an increase in risk for the emergence of cardiovascular problems, estradiol-sensitive cancers, and stroke. Because of the potential detrimental effects of long-term estradiol treatment, the importance of understanding the molecular mechanism underlying estradiol’s effects on dendritic spines, cognition, and their potential relevance for both normal and abnormal cognitive function is necessary. Many of the proteins that are involved in pathways that regulate dendritic spine morphology are enzymes that could be targeted with “designer” small molecules and drugs that target trophic and morphogenic signaling pathways, leading to alterations in neuronal connectivity, inducing fewer side effects.
Moreover, as the complexity of estradiol signaling within the brain is now readily evident, with both rapid and long-term actions of the steroid, it is critical to understand how both modes of actions of this steroid integrate to influence synapse structure and function, leading to its influences on cognitive function. Furthermore, the discrete production of estradiol via the enzyme aromatase offers an alternative to directly targeting estradiol-dependent signaling pathways or ERs. By targeting the specific activity of brain-aromatase to regulate brain estradiol levels, it may be possible to produce the beneficial effects of estradiol within the brain, without the adverse side effects in other tissues. Finally, owing to the multiple lines of evidence that estradiol cannot only affect the onset, but the progression and the symptomatology of several diseases, new therapies based on estrogenic signaling could be used to delay the onset, slow the progression of disease in an early stage, or mitigate symptoms, or even promote functional recovery after a disease is fully manifested.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Kelly A. Jones for proofreading of this manuscript. This work was supported by grants from the American Heart Association HA and the National Alliance for Research on Schizophrenia and Depression (NARSAD) awarded to Deepak P. Srivastava, and grants from the National Alliance for Autism Research (NAAR), NARSAD, Alzheimer’s Association, and NIH grant MH 071316 to Peter Penzes.
References
Alvarez, V. A., and Sabatini, B. L. (2007). Anatomical and physiological plasticity of dendritic spines. Annu. Rev. Neurosci. 30, 79–97.
Ashby, M. C., and Isaac, J. T. (2011). Maturation of a recurrent excitatory neocortical circuit by experience-dependent unsilencing of newly formed dendritic spines. Neuron 70, 510–521.
Bailey, M. E., Wang, A. C., Hao, J., Janssen, W. G., Hara, Y., Dumitriu, D., Hof, P. R., and Morrison, J. H. (2011). Interactive effects of age and estrogen on cortical neurons: implications for cognitive aging. Neuroscience 191, 148–158.
Balthazart, J., and Ball, G. F. (2006). Is brain estradiol a hormone or a neurotransmitter? Trends Neurosci. 29, 241–249.
Barker, G. R., and Warburton, E. C. (2011). When is the hippocampus involved in recognition memory? J. Neurosci. 31, 10721–10731.
Bavelier, D., Levi, D. M., Li, R. W., Dan, Y., and Hensch, T. K. (2010). Removing brakes on adult brain plasticity: from molecular to behavioral interventions. J. Neurosci. 30, 14964–14971.
Bhatt, D. H., Zhang, S., and Gan, W. B. (2009). Dendritic spine dynamics. Annu. Rev. Physiol. 71, 261–282.
Boon, W. C., Chow, J. D., and Simpson, E. R. (2010). The multiple roles of estrogens and the enzyme aromatase. Prog. Brain Res. 181, 209–232.
Bose, M., Munoz-Llancao, P., Roychowdhury, S., Nichols, J. A., Jakkamsetti, V., Porter, B., Byrapureddy, R., Salgado, H., Kilgard, M. P., Aboitiz, F., Dagnino-Subiabre, A., and Atzori, M. (2009). Effect of the environment on the dendritic morphology of the rat auditory cortex. Synapse 64, 97–110.
Bourne, J. N., and Harris, K. M. (2008). Balancing structure and function at hippocampal dendritic spines. Annu. Rev. Neurosci. 31, 47–67.
Brailoiu, E., Dun, S. L., Brailoiu, G. C., Mizuo, K., Sklar, L. A., Oprea, T. I., Prossnitz, E. R., and Dun, N. J. (2007). Distribution and characterization of estrogen receptor G protein-coupled receptor 30 in the rat central nervous system. J. Endocrinol. 193, 311–321.
Brinton, R. D. (2009). Estrogen-induced plasticity from cells to circuits: predictions for cognitive function. Trends Pharmacol. Sci. 30, 212–222.
Bueno, J., and Pfaff, D. W. (1976). Single unit recording in hypothalamus and preoptic area of estrogen-treated and untreated ovariectomized female rats. Brain Res. 101, 67–78.
Carroll, J. C., and Pike, C. J. (2008). Selective estrogen receptor modulators differentially regulate Alzheimer-like changes in female 3xTg-AD mice. Endocrinology 149, 2607–2611.
Carroll, J. C., Rosario, E. R., Chang, L., Stanczyk, F. Z., Oddo, S., Laferla, F. M., and Pike, C. J. (2007). Progesterone and estrogen regulate Alzheimer-like neuropathology in female 3xTg-AD mice. J. Neurosci. 27, 13357–13365.
Chklovskii, D. B., Mel, B. W., and Svoboda, K. (2004). Cortical rewiring and information storage. Nature 431, 782–788.
Craig, M. C., Maki, P. M., and Murphy, D. G. (2005). The Women’s Health Initiative Memory Study: findings and implications for treatment. Lancet Neurol. 4, 190–194.
Cyr, M., Calon, F., Morissette, M., Grandbois, M., Di Paolo, T., and Callier, S. (2000). Drugs with estrogen-like potency and brain activity: potential therapeutic application for the CNS. Curr. Pharm. Des. 6, 1287–1312.
de novaes Soares, C., Almeida, O. P., Joffe, H., and Cohen, L. S. (2001). Efficacy of estradiol for the treatment of depressive disorders in perimenopausal women: a double-blind, randomized, placebo-controlled trial. Arch. Gen. Psychiatry 58, 529–534.
DeKosky, S. T., and Scheff, S. W. (1990). Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann. Neurol. 27, 457–464.
Dierssen, M., and Ramakers, G. J. (2006). Dendritic pathology in mental retardation: from molecular genetics to neurobiology. Genes Brain Behav. 5(Suppl. 2), 48–60.
Dominguez, R., Hu, E., Zhou, M., and Baudry, M. (2009). 17Beta-estradiol -mediated neuroprotection and ERK activation require a pertussis toxin-sensitive mechanism involving GRK2 and beta-arrestin-1. J. Neurosci. 29, 4228–4238.
DSM-IV. (2000). Diagnostic and Statistical Manual of Mental Disorders, 4th edn. Washington, DC: American Psychiatric Association.
Dumanis, S. B., Tesoriero, J. A., Babus, L. W., Nguyen, M. T., Trotter, J. H., Ladu, M. J., Weeber, E. J., Turner, R. S., Xu, B., Rebeck, G. W., and Hoe, H. S. (2009). ApoE4 decreases spine density and dendritic complexity in cortical neurons in vivo. J. Neurosci. 29, 15317–15322.
Dumitriu, D., Rapp, P. R., Mcewen, B. S., and Morrison, J. H. (2010). Estrogen and the aging brain: an elixir for the weary cortical network. Ann. N. Y. Acad. Sci. 1204, 104–112.
Ennaceur, A., Neave, N., and Aggleton, J. P. (1997). Spontaneous object recognition and object location memory in rats: the effects of lesions in the cingulate cortices, the medial prefrontal cortex, the cingulum bundle and the fornix. Exp. Brain Res. 113, 509–519.
Espeland, M. A., Rapp, S. R., Shumaker, S. A., Brunner, R., Manson, J. E., Sherwin, B. B., Hsia, J., Margolis, K. L., Hogan, P. E., Wallace, R., Dailey, M., Freeman, R., and Hays, J. (2004). Conjugated equine estrogens and global cognitive function in postmenopausal women: Women’s Health Initiative Memory Study. JAMA 291, 2959–2968.
Fan, L., Zhao, Z., Orr, P. T., Chambers, C. H., Lewis, M. C., and Frick, K. M. (2010). Estradiol-induced object memory consolidation in middle-aged female mice requires dorsal hippocampal extracellular signal-regulated kinase and phosphatidylinositol 3-kinase activation. J. Neurosci. 30, 4390–4400.
Fiala, J. C., Spacek, J., and Harris, K. M. (2002). Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res. Brain Res. Rev. 39, 29–54.
Frost, N. A., Kerr, J. M., Lu, H. E., and Blanpied, T. A. (2010). A network of networks: cytoskeletal control of compartmentalized function within dendritic spines. Curr. Opin. Neurobiol. 20, 578–587.
Garcia-Segura, L. M. (2008). Aromatase in the brain: not just for reproduction anymore. J. Neuroendocrinol. 20, 705–712.
Gibbs, R. B. (2000). Long-term treatment with estrogen and progesterone enhances acquisition of a spatial memory task by ovariectomized aged rats. Neurobiol. Aging 21, 107–116.
Gibbs, R. B., and Johnson, D. A. (2008). Sex-specific effects of gonadectomy and hormone treatment on acquisition of a 12-arm radial maze task by Sprague Dawley rats. Endocrinology 149, 3176–3183.
Glantz, L. A., and Lewis, D. A. (2000). Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch. Gen. Psychiatry 57, 65–73.
Gorman, J. M., and Docherty, J. P. (2010). A hypothesized role for dendritic remodeling in the etiology of mood and anxiety disorders. J. Neuropsychiatry Clin. Neurosci. 22, 256–264.
Gresack, J. E., and Frick, K. M. (2006). Post-training estrogen enhances spatial and object memory consolidation in female mice. Pharmacol. Biochem. Behav. 84, 112–119.
Hajszan, T., Szigeti-Buck, K., Sallam, N. L., Bober, J., Parducz, A., Maclusky, N. J., Leranth, C., and Duman, R. S. (2010). Effects of estradiol on learned helplessness and associated remodeling of hippocampal spine synapses in female rats. Biol. Psychiatry 67, 168–174.
Hao, J., Rapp, P. R., Janssen, W. G., Lou, W., Lasley, B. L., Hof, P. R., and Morrison, J. H. (2007). Interactive effects of age and estrogen on cognition and pyramidal neurons in monkey prefrontal cortex. Proc. Natl. Acad. Sci. U.S.A. 104, 11465–11470.
Hazell, G. G., Yao, S. T., Roper, J. A., Prossnitz, E. R., O’carroll, A. M., and Lolait, S. J. (2009). Localisation of GPR30, a novel G protein-coupled oestrogen receptor, suggests multiple functions in rodent brain and peripheral tissues. J. Endocrinol. 202, 223–236.
Henderson, V. W. (2009). Estrogens, episodic memory, and Alzheimer’s disease: a critical update. Semin. Reprod. Med. 27, 283–293.
Hill, J. J., Hashimoto, T., and Lewis, D. A. (2006). Molecular mechanisms contributing to dendritic spine alterations in the prefrontal cortex of subjects with schizophrenia. Mol. Psychiatry 11, 557–566.
Holtmaat, A., and Svoboda, K. (2009). Experience-dependent structural synaptic plasticity in the mammalian brain. Nat. Rev. Neurosci. 10, 647–658.
Holtmaat, A. J., Trachtenberg, J. T., Wilbrecht, L., Shepherd, G. M., Zhang, X., Knott, G. W., and Svoboda, K. (2005). Transient and persistent dendritic spines in the neocortex in vivo. Neuron 45, 279–291.
Hotulainen, P., and Hoogenraad, C. C. (2010). Actin in dendritic spines: connecting dynamics to function. J. Cell Biol. 189, 619–629.
Hughes, Z. A., Liu, F., Marquis, K., Muniz, L., Pangalos, M. N., Ring, R. H., Whiteside, G. T., and Brandon, N. J. (2009). Estrogen receptor neurobiology and its potential for translation into broad spectrum therapeutics for CNS disorders. Curr. Mol. Pharmacol. 2, 215–236.
Hughes, Z. A., Liu, F., Platt, B. J., Dwyer, J. M., Pulicicchio, C. M., Zhang, G., Schechter, L. E., Rosenzweig-Lipson, S., and Day, M. (2008). WAY-200070, a selective agonist of estrogen receptor beta as a potential novel anxiolytic/antidepressant agent. Neuropharmacology 54, 1136–1142.
Hutsler, J. J., and Zhang, H. (2010). Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 1309, 83–94.
Iivonen, S., Corder, E., Lehtovirta, M., Helisalmi, S., Mannermaa, A., Vepsalainen, S., Hanninen, T., Soininen, H., and Hiltunen, M. (2004). Polymorphisms in the CYP19 gene confer increased risk for Alzheimer disease. Neurology 62, 1170–1176.
Inagaki, T., Gautreaux, C., and Luine, V. (2010). Acute estrogen treatment facilitates recognition memory consolidation and alters monoamine levels in memory-related brain areas. Horm. Behav. 58, 415–426.
Irwin, S. A., Galvez, R., and Greenough, W. T. (2000). Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb. Cortex 10, 1038–1044.
Ishunina, T. A., and Swaab, D. F. (2008). Estrogen receptor-alpha splice variants in the human brain. Gynecol. Endocrinol. 24, 93–98.
Jaaro-Peled, H., Hayashi-Takagi, A., Seshadri, S., Kamiya, A., Brandon, N. J., and Sawa, A. (2009). Neurodevelopmental mechanisms of schizophrenia: understanding disturbed postnatal brain maturation through neuregulin-1-ErbB4 and DISC1. Trends Neurosci. 32, 485–495.
Jones, K. A., Srivastava, D. P., Allen, J. A., Strachan, R. T., Roth, B. L., and Penzes, P. (2009). Rapid modulation of spine morphology by the 5-HT2A serotonin receptor through kalirin-7 signaling. Proc. Natl. Acad. Sci. U.S.A. 106, 19575–19580.
Kasai, H., Fukuda, M., Watanabe, S., Hayashi-Takagi, A., and Noguchi, J. (2010). Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci. 33, 121–129.
Kasai, H., Matsuzaki, M., Noguchi, J., Yasumatsu, N., and Nakahara, H. (2003). Structure-stability-function relationships of dendritic spines. Trends Neurosci. 26, 360–368.
Kelly, M. J., Qiu, J., and Ronnekleiv, O. K. (2005). Estrogen signaling in the hypothalamus. Vitam. Horm. 71, 123–145.
Konkle, A. T., and McCarthy, M. M. (2011). Developmental time course of estradiol, testosterone, and dihydrotestosterone levels in discrete regions of male and female rat brain. Endocrinology 152, 223–235.
Kramar, E. A., Chen, L. Y., Brandon, N. J., Rex, C. S., Liu, F., Gall, C. M., and Lynch, G. (2009). Cytoskeletal changes underlie estrogen’s acute effects on synaptic transmission and plasticity. J. Neurosci. 29, 12982–12993.
Kritzer, M. F. (2002). Regional, laminar, and cellular distribution of immunoreactivity for ER alpha and ER beta in the cerebral cortex of hormonally intact, adult male and female rats. Cereb. Cortex 12, 116–128.
Krȩżel, W., Dupont, S., Krust, A., Chambon, P., and Chapman, P. F. (2001). Increased anxiety and synaptic plasticity in estrogen receptor β-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 98, 12278–12282.
Kulkarni, J. (2009). Oestrogen – a new treatment approach for schizophrenia? Med. J. Aust. 190, S37–S38.
Kulkarni, J., De Castella, A., Fitzgerald, P. B., Gurvich, C. T., Bailey, M., Bartholomeusz, C., and Burger, H. (2008). Estrogen in severe mental illness: a potential new treatment approach. Arch. Gen. Psychiatry 65, 955–960.
Kulkarni, J., De Castella, A., Headey, B., Marston, N., Sinclair, K., Lee, S., Gurvich, C., Fitzgerald, P. B., and Burger, H. (2011). Estrogens and men with schizophrenia: is there a case for adjunctive therapy? Schizophr. Res. 125, 278–283.
Langer, G., Bader, B., Meoli, L., Isensee, J., Delbeck, M., Noppinger, P. R., and Otto, C. (2010). A critical review of fundamental controversies in the field of GPR30 research. Steroids 75, 603–610.
Lanz, T. A., Carter, D. B., and Merchant, K. M. (2003). Dendritic spine loss in the hippocampus of young PDAPP and Tg2576 mice and its prevention by the ApoE2 genotype. Neurobiol. Dis. 13, 246–253.
Le Be, J. V., and Markram, H. (2006). Spontaneous and evoked synaptic rewiring in the neonatal neocortex. Proc. Natl. Acad. Sci. U.S.A. 103, 13214–13219.
Lewis, D. A., and Sweet, R. A. (2009). Schizophrenia from a neural circuitry perspective: advancing toward rational pharmacological therapies. J. Clin. Invest. 119, 706–716.
Li, B., Woo, R. S., Mei, L., and Malinow, R. (2007). The neuregulin-1 receptor erbB4 controls glutamatergic synapse maturation and plasticity. Neuron 54, 583–597.
Lise, M. F., and El-Husseini, A. (2006). The neuroligin and neurexin families: from structure to function at the synapse. Cell. Mol. Life Sci. 63, 1833–1849.
Liu, F., Day, M., Muniz, L. C., Bitran, D., Arias, R., Revilla-Sanchez, R., Grauer, S., Zhang, G., Kelley, C., Pulito, V., Sung, A., Mervis, R. F., Navarra, R., Hirst, W. D., Reinhart, P. H., Marquis, K. L., Moss, S. J., Pangalos, M. N., and Brandon, N. J. (2008). Activation of estrogen receptor-beta regulates hippocampal synaptic plasticity and improves memory. Nat. Neurosci. 11, 334–343.
Logan, S. M., Sarkar, S. N., Zhang, Z., and Simpkins, J. W. (2011). Estrogen-induced signaling attenuates soluble A[beta] peptide-mediated dysfunction of pathways in synaptic plasticity. Brain Res. 1383, 1–12.
Luine, V. N., Jacome, L. F., and Maclusky, N. J. (2003). Rapid enhancement of visual and place memory by estrogens in rats. Endocrinology 144, 2836–2844.
Luscher, C., Nicoll, R. A., Malenka, R. C., and Muller, D. (2000). Synaptic plasticity and dynamic modulation of the postsynaptic membrane. Nat. Neurosci. 3, 545–550.
Ma, Y. J., Hill, D. F., Creswick, K. E., Costa, M. E., Cornea, A., Lioubin, M. N., Plowman, G. D., and Ojeda, S. R. (1999). Neuregulins signaling via a glial erbB-2-erbB-4 receptor complex contribute to the neuroendocrine control of mammalian sexual development. J. Neurosci. 19, 9913–9927.
Maggiolini, M., and Picard, D. (2010). The unfolding stories of GPR30, a new membrane-bound estrogen receptor. J. Endocrinol. 204, 105–114.
Mak, P., Zeen, R., and Callard, G. V. (1985). Subcellular distribution and reaction kinetics of aromatase in goldfish brain. Program of the 67th Annual Meeting of the Endocrine Society, Baltimore, 265.
Malenka, R. C., and Nicoll, R. A. (1999). Long-term potentiation – a decade of progress? Science 285, 1870–1874.
Matsuzaki, M., Ellis-Davies, G. C., Nemoto, T., Miyashita, Y., Iino, M., and Kasai, H. (2001). Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat. Neurosci. 4, 1086–1092.
McEwen, B., Akama, K., Alves, S., Brake, W. G., Bulloch, K., Lee, S., Li, C., Yuen, G., and Milner, T. A. (2001). Tracking the estrogen receptor in neurons: implications for estrogen-induced synapse formation. Proc. Natl. Acad. Sci. U.S.A. 98, 7093–7100.
McEwen, B. S., and Alves, S. E. (1999). Estrogen actions in the central nervous system. Endocr. Rev. 20, 279–307.
Mendez, P., Garcia-Segura, L. M., and Muller, D. (2010). Estradiol promotes spine growth and synapse formation without affecting pre-established networks. Hippocampus.
Mendrek, A. (2007). Reversal of normal cerebral sexual dimorphism in schizophrenia: evidence and speculations. Med. Hypotheses 69, 896–902.
Mill, J., Kiss, E., Baji, I., Kapornai, K., Daroczy, G., Vetro, A., Kennedy, J., Kovacs, M., and Barr, C. (2008). Association study of the estrogen receptor alpha gene (ESR1) and childhood-onset mood disorders. Am. J. Med. Genet. B Neuropsychiatr. Genet. 147B, 1323–1326.
Milner, T. A., Ayoola, K., Drake, C. T., Herrick, S. P., Tabori, N. E., Mcewen, B. S., Warrier, S., and Alves, S. E. (2005). Ultrastructural localization of estrogen receptor beta immunoreactivity in the rat hippocampal formation. J. Comp. Neurol. 491, 81–95.
Milner, T. A., Mcewen, B. S., Hayashi, S., Li, C. J., Reagan, L. P., and Alves, S. E. (2001). Ultrastructural evidence that hippocampal alpha estrogen receptors are located at extranuclear sites. J. Comp. Neurol. 429, 355–371.
Mitra, S. W., Hoskin, E., Yudkovitz, J., Pear, L., Wilkinson, H. A., Hayashi, S., Pfaff, D. W., Ogawa, S., Rohrer, S. P., Schaeffer, J. M., Mcewen, B. S., and Alves, S. E. (2003). Immunolocalization of estrogen receptor beta in the mouse brain: comparison with estrogen receptor alpha. Endocrinology 144, 2055–2067.
Morgan, M. L., Cook, I. A., Rapkin, A. J., and Leuchter, A. F. (2005). Estrogen augmentation of antidepressants in perimenopausal depression: a pilot study. J. Clin. Psychiatry 66, 774–780.
Mortimer, A. M. (2007). Relationship between estrogen and schizophrenia. Expert Rev. Neurother. 7, 45–55.
Mukai, H., Tsurugizawa, T., Murakami, G., Kominami, S., Ishii, H., Ogiue-Ikeda, M., Takata, N., Tanabe, N., Furukawa, A., Hojo, Y., Ooishi, Y., Morrison, J. H., Janssen, W. G., Rose, J. A., Chambon, P., Kato, S., Izumi, S., Yamazaki, T., Kimoto, T., and Kawato, S. (2007). Rapid modulation of long-term depression and spinogenesis via synaptic estrogen receptors in hippocampal principal neurons. J. Neurochem. 100, 950–967.
Nadal, A., Alonso-Magdalena, P., Soriano, S., Ripoll, C., Fuentes, E., Quesada, I., and Ropero, A. B. (2011). Role of estrogen receptors alpha, beta and GPER1/GPR30 in pancreatic beta-cells. Front. Biosci. 16, 251–260.
Naftolin, F., Horvath, T. L., Jakab, R. L., Leranth, C., Harada, N., and Balthazart, J. (1996). Aromatase immunoreactivity in axon terminals of the vertebrate brain. An immunocytochemical study on quail, rat, monkey and human tissues. Neuroendocrinology 63, 149–155.
Nestler, E. J., Barrot, M., Dileone, R. J., Eisch, A. J., Gold, S. J., and Monteggia, L. M. (2002). Neurobiology of depression. Neuron 34, 13–25.
Noguchi, J., Nagaoka, A., Watanabe, S., Ellis-Davies, G. C., Kitamura, K., Kano, M., Matsuzaki, M., and Kasai, H. (2011). In vivo two-photon uncaging of glutamate revealing the structure-function relationships of dendritic spines in the neocortex of adult mice. J. Physiol. (Lond.) 589, 2447–2457.
Penzes, P., Cahill, M. E., Jones, K. A., and Srivastava, D. P. (2008). Convergent CaMK and RacGEF signals control dendritic structure and function. Trends Cell Biol. 18, 405–413.
Penzes, P., Cahill, M. E., Jones, K. A., Vanleeuwen, J. E., and Woolfrey, K. M. (2011a). Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 14, 285–293.
Penzes, P., Woolfrey, K. M., and Srivastava, D. P. (2011b). Epac2-mediated dendritic spine remodeling: implications for disease. Mol. Cell. Neurosci. 46, 368–380.
Perlman, W. R., Tomaskovic-Crook, E., Montague, D. M., Webster, M. J., Rubinow, D. R., Kleinman, J. E., and Weickert, C. S. (2005). Alteration in estrogen receptor alpha mRNA levels in frontal cortex and hippocampus of patients with major mental illness. Biol. Psychiatry 58, 812–824.
Perlman, W. R., Webster, M. J., Kleinman, J. E., and Weickert, C. S. (2004). Reduced glucocorticoid and estrogen receptor alpha messenger ribonucleic acid levels in the amygdala of patients with major mental illness. Biol. Psychiatry 56, 844–852.
Phan, A., Lancaster, K. E., Armstrong, J. N., Maclusky, N. J., and Choleris, E. (2011). Rapid effects of estrogen receptor alpha and beta selective agonists on learning and dendritic spines in female mice. Endocrinology 152, 1492–1502.
Pickett, J., and London, E. (2005). The neuropathology of autism: a review. J. Neuropathol. Exp. Neurol. 64, 925–935.
Pirskanen, M., Hiltunen, M., Mannermaa, A., Helisalmi, S., Lehtovirta, M., Hanninen, T., and Soininen, H. (2005). Estrogen receptor beta gene variants are associated with increased risk of Alzheimer’s disease in women. Eur. J. Hum. Genet. 13, 1000–1006.
Prossnitz, E. R., and Maggiolini, M. (2009). Mechanisms of estrogen signaling and gene expression via GPR30. Mol. Cell. Endocrinol. 308, 32–38.
Ramakers, G. J. (2002). Rho proteins, mental retardation and the cellular basis of cognition. Trends Neurosci. 25, 191–199.
Rapp, S. R., Espeland, M. A., Shumaker, S. A., Henderson, V. W., Brunner, R. L., Manson, J. E., Gass, M. L. S., Stefanick, M. L., Lane, D. S., Hays, J., Johnson, K. C., Coker, L. H., Dailey, M., Bowen, D., and Investigators, F. T. W. (2003). Effect of estrogen plus progestin on global cognitive function in postmenopausal women. JAMA 289, 2663–2672.
Rasgon, N. L., Dunkin, J., Fairbanks, L., Altshuler, L. L., Troung, C., Elman, S., Wroolie, T. E., Brunhuber, M. V., and Rapkin, A. (2007). Estrogen and response to sertraline in postmenopausal women with major depressive disorder: a pilot study. J. Psychiatr. Res. 41, 338–343.
Raz, L., Khan, M. M., Mahesh, V. B., Vadlamudi, R. K., and Brann, D. W. (2008). Rapid estrogen signaling in the brain. Neurosignals 16, 140–153.
Remage-Healey, L., Coleman, M. J., Oyama, R. K., and Schlinger, B. A. (2010). Brain estrogens rapidly strengthen auditory encoding and guide song preference in a songbird. Proc. Natl. Acad. Sci. U.S.A. 107, 3852–3857.
Remage-Healey, L., Dong, S., Maidment, N. T., and Schlinger, B. A. (2011). Presynaptic control of rapid estrogen fluctuations in the songbird auditory forebrain. J. Neurosci. 31, 10034–10038.
Remage-Healey, L., Maidment, N. T., and Schlinger, B. A. (2008). Forebrain steroid levels fluctuate rapidly during social interactions. Nat. Neurosci. 11, 1327–1334.
Resnick, S. M., Maki, P. M., Rapp, S. R., Espeland, M. A., Brunner, R., Coker, L. H., Granek, I. A., Hogan, P., Ockene, J. K., Shumaker, S. A., and Women’s Health Initiative Study of Cognitive Aging Investigators. (2006). Effects of combination estrogen plus progestin hormone treatment on cognition and affect. J. Clin. Endocrinol. Metab. 91, 1802–1810.
Rocca, W. A., Grossardt, B. R., and Shuster, L. T. (2011). Oophorectomy, menopause, estrogen treatment, and cognitive aging: clinical evidence for a window of opportunity. Brain Res. 1379, 188–198.
Rocha, B. A., Fleischer, R., Schaeffer, J. M., Rohrer, S. P., and Hickey, G. J. (2005). 17β-estradiol-induced antidepressant-like effect in the forced swim test is absent in estrogen receptor-β knockout (BERKO) mice. Psychopharmacology (Berl.) 179, 637–643.
Rossouw, J. E., Anderson, G. L., Prentice, R. L., Lacroix, A. Z., Kooperberg, C., Stefanick, M. L., Jackson, R. D., Beresford, S. A., Howard, B. V., Johnson, K. C., Kotchen, J. M., and Ockene, J. (2002). Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA 288, 321–333.
Rune, G. M., and Frotscher, M. (2005). Neurosteroid synthesis in the hippocampus: role in synaptic plasticity. Neuroscience 136, 833–842.
Saldanha, C. J., Remage-Healey, L., and Schlinger, B. A. (2011). Synaptocrine signaling: steroid synthesis and action at the synapse. Endocr. Rev. 32, 532–549.
Sanchez, A. M., Flamini, M. I., Baldacci, C., Goglia, L., Genazzani, A. R., and Simoncini, T. (2010a). Estrogen receptor-alpha promotes breast cancer cell motility and invasion via focal adhesion kinase and N-WASP. Mol. Endocrinol. 24, 2114–2125.
Sanchez, M. G., Bourque, M., Morissette, M., and Di Paolo, T. (2010b). Steroids-dopamine interactions in the pathophysiology and treatment of CNS disorders. CNS Neurosci. Ther. 16, e43–e71.
Sanchez, A. M., Flamini, M. I., Fu, X. D., Mannella, P., Giretti, M. S., Goglia, L., Genazzani, A. R., and Simoncini, T. (2009). Rapid signaling of estrogen to WAVE1 and moesin controls neuronal spine formation via the actin cytoskeleton. Mol. Endocrinol. 23, 1193–1202.
Scharfman, H. E., and MacLusky, N. J. (2006). Estrogen and brain-derived neurotrophic factor (BDNF) in hippocampus: complexity of steroid hormone-growth factor interactions in the adult CNS. Front. Neuroendocrinol. 27, 415–435.
Segal, M. (2010). Dendritic spines, synaptic plasticity and neuronal survival: activity shapes dendritic spines to enhance neuronal viability. Eur. J. Neurosci. 31, 2178–2184.
Selkoe, D. J. (2008). Soluble oligomers of the amyloid [beta]-protein impair synaptic plasticity and behavior. Behav. Brain Res. 192, 106–113.
Shalev, I., Lerer, E., Israel, S., Uzefovsky, F., Gritsenko, I., Mankuta, D., Ebstein, R. P., and Kaitz, M. (2009). BDNF Val66Met polymorphism is associated with HPA axis reactivity to psychological stress characterized by genotype and gender interactions. Psychoneuroendocrinology 34, 382–388.
Sherwin, B. B., and Henry, J. F. (2008). Brain aging modulates the neuroprotective effects of estrogen on selective aspects of cognition in women: a critical review. Front. Neuroendocrinol. 29, 88–113.
Shumaker, S. A., Legault, C., Kuller, L., Rapp, S. R., Thal, L., Lane, D. S., Fillit, H., Stefanick, M. L., Hendrix, S. L., Lewis, C. E., Masaki, K., and Coker, L. H. (2004). Conjugated equine estrogens and incidence of probable dementia and mild cognitive impairment in postmenopausal women: Women’s Health Initiative Memory Study. JAMA 291, 2947–2958.
Singer, C. A., Figueroa-Masot, X. A., Batchelor, R. H., and Dorsa, D. M. (1999). The mitogen-activated protein kinase pathway mediates estrogen neuroprotection after glutamate toxicity in primary cortical neurons. J. Neurosci. 19, 2455–2463.
Singh, M., Meyer, E. M., Millard, W. J., and Simpkins, J. W. (1994). Ovarian steroid deprivation results in a reversible learning impairment and compromised cholinergic function in female Sprague-Dawley rats. Brain Res. 644, 305–312.
Sinopoli, K. J., Floresco, S. B., and Galea, L. A. (2006). Systemic and local administration of estradiol into the prefrontal cortex or hippocampus differentially alters working memory. Neurobiol. Learn. Mem. 86, 293–304.
Sleegers, K., Lambert, J.-C., Bertram, L., Cruts, M., Amouyel, P., and Van Broeckhoven, C. (2010). The pursuit of susceptibility genes for Alzheimer’s disease: progress and prospects. Trends Genet. 26, 84–93.
Soetanto, A., Wilson, R. S., Talbot, K., Un, A., Schneider, J. A., Sobiesk, M., Kelly, J., Leurgans, S., Bennett, D. A., and Arnold, S. E. (2010). Association of anxiety and depression with microtubule-associated protein 2- and synaptopodin-immunolabeled dendrite and spine densities in hippocampal CA3 of older humans. Arch. Gen. Psychiatry 67, 448–457.
Srivastava, D. P., Woolfrey, K. M., Jones, K. A., Shum, C. Y., Lash, L. L., Swanson, G. T., and Penzes, P. (2008). Rapid enhancement of two-step wiring plasticity by estrogen and NMDA receptor activity. Proc. Natl. Acad. Sci. U.S.A. 105, 14650–14655.
Srivastava, D. P., Woolfrey, K. M., Liu, F., Brandon, N. J., and Penzes, P. (2010). Estrogen receptor ss activity modulates synaptic signaling and structure. J. Neurosci. 30, 13454–13460.
Tada, T., and Sheng, M. (2006). Molecular mechanisms of dendritic spine morphogenesis. Curr. Opin. Neurobiol. 16, 95–101.
Takai, Y., Sasaki, T., and Matozaki, T. (2001). Small GTP-binding proteins. Physiol. Rev. 81, 153–208.
Takashima, S., Ieshima, A., Nakamura, H., and Becker, L. E. (1989). Dendrites, dementia and the Down syndrome. Brain Dev. 11, 131–133.
Tashiro, A., and Yuste, R. (2004). Regulation of dendritic spine motility and stability by Rac1 and Rho kinase: evidence for two forms of spine motility. Mol. Cell. Neurosci. 26, 429–440.
Tau, G. Z., and Peterson, B. S. (2010). Normal development of brain circuits. Neuropsychopharmacology 35, 147–168.
Thomas, G. M., and Huganir, R. L. (2004). MAPK cascade signalling and synaptic plasticity. Nat. Rev. Neurosci. 5, 173–183.
Trachtenberg, J. T., Chen, B. E., Knott, G. W., Feng, G., Sanes, J. R., Welker, E., and Svoboda, K. (2002). Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature 420, 788–794.
Trainor, B. C., Lin, S., Finy, M. S., Rowland, M. R., and Nelson, R. J. (2007). Photoperiod reverses the effects of estrogens on male aggression via genomic and nongenomic pathways. Proc. Natl. Acad. Sci. U.S.A. 104, 9840–9845.
van Spronsen, M., and Hoogenraad, C. C. (2010). Synapse pathology in psychiatric and neurologic disease. Curr. Neurol. Neurosci. Rep. 10, 207–214.
Verpelli, C., Piccoli, G., Zibetti, C., Zanchi, A., Gardoni, F., Huang, K., Brambilla, D., Di Luca, M., Battaglioli, E., and Sala, C. (2010). Synaptic activity controls dendritic spine morphology by modulating eEF2-dependent BDNF synthesis. J. Neurosci. 30, 5830–5842.
Walf, A. A., and Frye, C. A. (2010). Estradiol reduces anxiety- and depression-like behavior of aged female mice. Physiol. Behav. 99, 169–174.
Walf, A. A., Koonce, C. J., and Frye, C. A. (2008). Estradiol or diarylpropionitrile decrease anxiety-like behavior of wildtype, but not estrogen receptor beta knockout, mice. Behav. Neurosci. 122, 974–981.
Walf, A. A., Rhodes, M. E., and Frye, C. A. (2006). Ovarian steroids enhance object recognition in naturally cycling and ovariectomized, hormone-primed rats. Neurobiol. Learn. Mem. 86, 35–46.
Wang, J. M., Irwin, R. W., and Brinton, R. D. (2006). Activation of estrogen receptor alpha increases and estrogen receptor beta decreases apolipoprotein E expression in hippocampus in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 103, 16983–16988.
Wehrenberg, U., Prange-Kiel, J., and Rune, G. M. (2001). Steroidogenic factor-1 expression in marmoset and rat hippocampus: co-localization with StAR and aromatase. J. Neurochem. 76, 1879–1886.
Weickert, C. S., Miranda-Angulo, A. L., Wong, J., Perlman, W. R., Ward, S. E., Radhakrishna, V., Straub, R. E., Weinberger, D. R., and Kleinman, J. E. (2008). Variants in the estrogen receptor alpha gene and its mRNA contribute to risk for schizophrenia. Hum. Mol. Genet. 17, 2293–2309.
Weissman, M. M., Bland, R., Joyce, P. R., Newman, S., Wells, J. E., and Wittchen, H. U. (1993). Sex differences in rates of depression: cross-national perspectives. J. Affect. Disord. 29, 77–84.
Wilbrecht, L., Holtmaat, A., Wright, N., Fox, K., and Svoboda, K. (2010). Structural plasticity underlies experience-dependent functional plasticity of cortical circuits. J. Neurosci. 30, 4927–4932.
Wong, J., and Weickert, C. S. (2009). Transcriptional interaction of an estrogen receptor splice variant and ErbB4 suggests convergence in gene susceptibility pathways in schizophrenia. J. Biol. Chem. 284, 18824–18832.
Wong, J., Woon, H. G., and Weickert, C. S. (2011). Full length TrkB potentiates estrogen receptor alpha mediated transcription suggesting convergence of susceptibility pathways in schizophrenia. Mol. Cell. Neurosci. 46, 67–78.
Woolfrey, K. M., Srivastava, D. P., Photowala, H., Yamashita, M., Barbolina, M. V., Cahill, M. E., Xie, Z., Jones, K. A., Quilliam, L. A., Prakriya, M., and Penzes, P. (2009). Epac2 induces synapse remodeling and depression and its disease-associated forms alter spines. Nat. Neurosci. 12, 1275–1284.
Woolley, C. S. (2007). Acute effects of estrogen on neuronal physiology. Annu. Rev. Pharmacol. Toxicol. 47, 657–680.
Wu, T. W., Wang, J. M., Chen, S., and Brinton, R. D. (2005). 17Beta-estradiol induced Ca2+ influx via L-type calcium channels activates the Src/ERK/cyclic-AMP response element binding protein signal pathway and BCL-2 expression in rat hippocampal neurons: a potential initiation mechanism for estrogen-induced neuroprotection. Neuroscience 135, 59–72.
Xie, Z., Huganir, R. L., and Penzes, P. (2005). Activity-dependent dendritic spine structural plasticity is regulated by small GTPase Rap1 and its target AF-6. Neuron 48, 605–618.
Xie, Z., Photowala, H., Cahill, M. E., Srivastava, D. P., Woolfrey, K. M., Shum, C. Y., Huganir, R. L., and Penzes, P. (2008). Coordination of synaptic adhesion with dendritic spine remodeling by AF-6 and kalirin-7. J. Neurosci. 28, 6079–6091.
Xie, Z., Srivastava, D. P., Photowala, H., Kai, L., Cahill, M. E., Woolfrey, K. M., Shum, C. Y., Surmeier, D. J., and Penzes, P. (2007). Kalirin-7 controls activity-dependent structural and functional plasticity of dendritic spines. Neuron 56, 640–656.
Yague, J. G., Munoz, A., De Monasterio-Schrader, P., Defelipe, J., Garcia-Segura, L. M., and Azcoitia, I. (2006). Aromatase expression in the human temporal cortex. Neuroscience 138, 389–401.
Yague, J. G., Wang, A. C., Janssen, W. G., Hof, P. R., Garcia-Segura, L. M., Azcoitia, I., and Morrison, J. H. (2008). Aromatase distribution in the monkey temporal neocortex and hippocampus. Brain Res. 1209, 115–127.
Yang, G., Pan, F., and Gan, W. B. (2009). Stably maintained dendritic spines are associated with lifelong memories. Nature 462, 920–924.
Yildirim, M., Janssen, W. G., Tabori, N. E., Adams, M. M., Yuen, G. S., Akama, K. T., Mcewen, B. S., Milner, T. A., and Morrison, J. H. (2008). Estrogen and aging affect synaptic distribution of phosphorylated LIM kinase (pLIMK) in CA1 region of female rat hippocampus. Neuroscience 152, 360–370. Psychopharmacology (Berl.) 179, 637–643.
Yoshihara, Y., De Roo, M., and Muller, D. (2009). Dendritic spine formation and stabilization. Curr. Opin. Neurobiol. 19, 146–153.
Yue, X., Lu, M., Lancaster, T., Cao, P., Honda, S., Staufenbiel, M., Harada, N., Zhong, Z., Shen, Y., and Li, R. (2005). Brain estrogen deficiency accelerates Abeta plaque formation in an Alzheimer’s disease animal model. Proc. Natl. Acad. Sci. U.S.A. 102, 19198–19203.
Yuen, G. S., Mcewen, B. S., and Akama, K. T. (2011). LIM kinase mediates estrogen action on the actin depolymerization factor cofilin. Brain Res. 1379, 44–52.
Zadran, S., Qin, Q., Bi, X., Zadran, H., Kim, Y., Foy, M. R., Thompson, R., and Baudry, M. (2009). 17-Beta-estradiol increases neuronal excitability through MAP kinase-induced calpain activation. Proc. Natl. Acad. Sci. U.S.A. 106, 21936–21941.
Zanardi, R., Rossini, D., Magri, L., Malaguti, A., Colombo, C., and Smeraldi, E. (2007). Response to SSRIs and role of the hormonal therapy in post-menopausal depression. Eur. Neuropsychopharmacol. 17, 400–405.
Zhang, Q. G., Han, D., Wang, R. M., Dong, Y., Yang, F., Vadlamudi, R. K., and Brann, D. W. (2011). C terminus of Hsc70-interacting protein (CHIP)-mediated degradation of hippocampal estrogen receptor-{alpha} and the critical period hypothesis of estrogen neuroprotection. Proc. Natl. Acad. Sci. U.S.A. 108, E617–E624.
Zhu, B. T., and Conney, A. H. (1998). Functional role of estrogen metabolism in target cells: review and perspectives. Carcinogenesis 19, 1–27.
Keywords: cortex, 17 β-estradiol, small GTPases, signal transduction, rapid, schizophrenia, depression, Alzheimer’s disease
Citation: Srivastava DP and Penzes P (2011) Rapid estradiol modulation of neuronal connectivity and its implications for disease. Front. Endocrin. 2:77. doi: 10.3389/fendo.2011.00077
Received: 15 July 2011;
Paper pending published: 08 August 2011;
Accepted: 04 November 2011;
Published online: 22 November 2011.
Edited by:
Hubert Vaudry, University of Rouen, FranceReviewed by:
Christian J. Pike, University of Southern California, USAVictoria Luine, Hunter College of Cuny, USA
Copyright: © 2011 Srivastava and Penzes. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Deepak P. Srivastava, Department of Physiology, Feinberg School of Medicine, Northwestern University, 303 E. Chicago Avenue, Chicago, IL 60611, USA. e-mail:ZC1zcml2YXN0YXZhQG5vcnRod2VzdGVybi5lZHU=