


- 1 Department of Basic Medical Sciences, University of Arizona College of Medicine – Phoenix, Phoenix, AZ, USA
- 2 Department of Pharmacology and Neuroscience and Institute for Aging and Alzheimers Disease Research, University of North Texas Health Sciences Center, Fort Worth, TX, USA
Activation of the hypothalamo-pituitary–adrenal (HPA) axis is a basic reaction of animals to environmental perturbations that threaten homeostasis. These responses are ultimately regulated by neurons residing within the paraventricular nucleus (PVN) of the hypothalamus. Within the PVN, corticotrophin-releasing hormone (CRH), vasopressin (AVP), and oxytocin (OT) expressing neurons are critical as they can regulate both neuroendocrine and autonomic responses. Estradiol (E2) and testosterone (T) are well known reproductive hormones; however, they have also been shown to modulate stress reactivity. In rodent models, evidence shows that under some conditions E2 enhances stress activated adrenocorticotropic hormone (ACTH) and corticosterone secretion. In contrast, T decreases the gain of the HPA axis. The modulatory role of testosterone was originally thought to be via 5 alpha reduction to the potent androgen dihydrotestosterone (DHT) and its subsequent binding to the androgen receptor, whereas E2 effects were thought to be mediated by estrogen receptors alpha (ERalpha) and beta (ERbeta). However, DHT has been shown to be metabolized to the ERbeta agonist, 5α- androstane 3β, 17β Diol (3β-Diol). The actions of 3β-Diol on the HPA axis are mediated by ERbeta which inhibits the PVN response to stressors. In gonadectomized rats, ERbeta agonists reduce CORT and ACTH responses to restraint stress, an effect that is also present in wild-type but not ERbeta-knockout mice. The neurobiological mechanisms underlying the ability of ERbeta to alter HPA reactivity are not currently known. CRH, AVP, and OT have all been shown to be regulated by estradiol and recent studies indicate an important role of ERbeta in these regulatory processes. Moreover, activation of the CRH and AVP promoters has been shown to occur by 3β-Diol binding to ERbeta and this is thought to occur through alternate pathways of gene regulation. Based on available data, a novel and important role of 3β-Diol in the regulation of the HPA axis is suggested.
Introduction
The hypothalamo-pituitary–adrenal (HPA) axis represents a complex series of neural signals that ultimately controls the hormonal response to stressors. Stress, as originally defined by Hans Selye (1936) is “…the non-specific response of the body to any demand for change….” Thus, the HPA axis represents one of several “non-specific” response systems that are used by the brain to adjust multiple physiological functions in an attempt to maintain homeostasis. Other “non-specific” responses activated by stressors may include the immune system and autonomic nervous system. Although stress may have positive (eustress) or negative (distress) connotations, the consequences of unresolved persistent stress responses can lead to severe neuropsychiatric disorders in susceptible individuals (Selye, 1975).
Regulation of the HPA axis by gonadal steroid hormones is an important consideration in anxiety and affective disorders. For example, major depressive disorder (MDD), and anxiety disorders, are current major public health concerns and are responsible for substantial social and economic burden in developed countries (Murray and Lopez, 1997; Ustun et al., 2004). Of interest, women are up to 2.5 times more likely than men to be diagnosed with MDD in their lifetime (Fava and Kendler, 2000; Kessler, 2003), and women have a significantly higher heritability of MDD than men (Kendler, 1998; Hettema et al., 2001). Clinical studies of depressed patients reveal that the sex differences in incidence arise at adolescence and more closely coincide with androgen and estrogen levels rather than physical changes associated with puberty (Warren and Brooks-Gunn, 1989; Angold and Worthman, 1993; Angold et al., 1999). As a result, changing patterns of hormones and hormone sensitivity may be implicated as potential etiological factors. In this review, we explore the steroid hormone influences on the regulation of HPA axis function that could influence susceptibility to disease states such as affective disorders, with particular emphasis on androgens and estrogenic metabolites of androgen.
The Hypothalamo-Pituitary–Adrenal Axis
Glucocorticoid hormones, the end-product of the HPA axis, are secreted by the adrenal cortex into the general circulation, under tight hypothalamic control. The HPA axis represents a cascade of neural and humoral signals organized by a central circadian pacemaker and activated in response to environmental triggers. Under non-stressed conditions, the release of corticosterone from the rodent adrenal cortex is linked to a circadian regulatory system (Chung et al., 2011). Circulating adrenal glucocorticoid levels are lowest during the inactive phase (light period in rodents, dark period in humans) and begin to rise several hours preceding the onset of activity or awakening (L:D transition in rodents) to peak shortly after activity onset. Thus, the rise in adrenal glucocorticoid secretion predicts and readies physiological systems for the onset of activity and feeding (Nader et al., 2010). The diurnal rise in glucocorticoids is allowed to occur by decreases in vasopressin (AVP) from the SCN that normally inhibit neurons in the paraventricular nucleus (PVN; Szafarczyk et al., 1983; Kalsbeek et al., 1996; Caldwell et al., 2008). Moreover, adrenal sensitivity to adrenocorticotropic hormone (ACTH) changes over the day thereby altering the corticosterone response to ACTH in a diurnal fashion (Oster et al., 2006).
In addition to diurnal signals regulating adrenal glucocorticoid secretion, changing environmental conditions or perceived threats to homeostasis activate the HPA through a common final pathway involving neurons located in the medial parvocellular division of the PVN of the hypothalamus. These neurons integrate excitatory and inhibitory inputs received from multiple brain areas (Cullinan et al., 1996; Herman et al., 1996; Choi et al., 2007). Thus, the motor neurons driving the HPA response to stress are neuroendocrine neurons that contain corticotrophin-releasing hormone (CRH) and that project to the median eminence. The release of CRH into the hypothalamo-hypophyseal portal system stimulates the release of ACTH from the anterior pituitary gland by acting upon the CRH-R1 receptor on corticotrophs (Aguilera et al., 2004).
Several additional neuropeptide phenotypes make up the PVN neurons that control HPA axis activity. Vasopressin and oxytocin are found at high levels in magnocellular and parvocellular neurons of the PVN. Although typically thought to regulate osmotic balance and parturition, vasopressin, and oxytocin have been shown to co-localize with CRH in discrete PVN populations and to be co-released with CRH (Whitnall, 1988; Bondy et al., 1989; Raadsheer et al., 1993). Vasopressin reportedly acts synergistically with CRH to potentiate CRH’s secretogogue activity at the level of the corticotroph (Rivier and Vale, 1983; Schlosser et al., 1994; Papadimitriou and Priftis, 2009), whereas a role for the oxytocin in CRH neurons has not been determined (Palkovits, 2000). Nonetheless, both vasopressin and oxytocin can stimulate ACTH secretion even in the absence of CRH (Gillies et al., 1982). In contrast, when applied to the PVN, or injected into the third ventricle, oxytocin inhibits HPA responses (Windle et al., 1997; Neumann et al., 2000) suggesting the possibility that oxytocin can be released locally from PVN neurons, perhaps through dendritic release (Landgraf and Neumann, 2004; Neumann, 2007).
Following the release from corticotrophs of the anterior pituitary, ACTH acts on the adrenal cortex to stimulate the synthesis and secretion of CORT. Circulating CORT has numerous effects throughout the body, one of which is to signal to the anterior pituitary, hypothalamus, and higher brain areas to limit further hormone secretion (Gomez et al., 1998) thus closing this neuroendocrine negative feedback loop.
Roles for Gonadal Steroids in Regulating the HPA Axis
Androgens and Androgen Receptors
Although testosterone and estradiol are classic reproductive hormones, both have been reported to regulate the HPA axis (Gaskin and Kitay, 1970, 1971; Coyne and Kitay, 1971). Gonadectomy of male rats increases corticosterone and ACTH responses to stress and correspondingly, c-fos mRNA expression in the PVN is elevated (Handa et al., 1994a; Viau et al., 2003; Lund et al., 2004a,b). The effects of testosterone (T) are not through aromatization to estradiol given that hormone replacement of castrated rats with the non-aromatizable androgen, dihydrotestosterone (DHT) returns stress-responsive plasma CORT, and ACTH levels back to that of the intact male. Hormone replacement also inhibits the stress-induction of c-fos mRNA in the PVN (Viau, 2002; Viau et al., 2003; Lund et al., 2004b, 2006). Additional evidence for an androgenic regulation of HPA axis reactivity comes from studies examining the hormonal stress response of male rats before and after puberty. Prior to puberty, when T levels are low, the CORT response to acute and chronic stress is high relative to the response seen after puberty (Viau et al., 2005; Romeo and McEwen, 2006; Follib et al., 2011). This correlates with the increases in T that occur during the pubertal transition of males. However, given that Romeo et al. (2004) have shown that T administration cannot shift the pattern of HPA regulation in pre-pubertal males to that of post-pubertal males, the involvement of T is not sufficient for the pre- and post-pubertal changes observed in HPA axis activity.
Androgens have been reported to inhibit HPA axis function (Handa et al., 1994a) and alter CRH-immunoreactivity (ir; Bingaman et al., 1994a) and vasopressin mRNA within the PVN (Viau et al., 2001). Nonetheless, androgen receptors (AR) are not localized in hypophysiotrophic CRH or AVP neurons within the PVN (Bingaman et al., 1994b) and have only been reported in the dorsal and the ventral medial parvocellular parts of the PVN, which are non-neuroendocrine neurons that project to spinal cord and brainstem pre-autonomic nuclei (Bingham et al., 2006). Consequently, it has been hypothesized that androgens regulate PVN neuropeptide expression and secretion trans-synaptically. Data supporting this hypothesis come from studies showing that AR are in neurons projecting to the PVN (Williamson and Viau, 2007) and that the implantation of testosterone into the medial preoptic nucleus (MPN) and bed nucleus of the stria terminalis (BnST), brain regions that provide afferent input to the PVN, can reduce the CORT response to acute stress (Viau and Meaney, 1996). Testosterone micro-implants to the MPN of gonadectomized rats can also decrease stress-induced expression of c-Fos in the PVN and lateral septum, an effect that is blocked by lesions of the MPN (Williamson et al., 2010). Further, retrograde tracing studies show that AR-ir can be found in neurons of the BnST, but not the septum, that project to the PVN (Suzuki et al., 2001). However, the MPN/BnST are likely not the only brain site(s) mediating androgen’s inhibitory effect on HPA reactivity given that stereotaxic application of DHT to a region just above the PVN (to prevent mechanical disruption of the PVN) is also effective in reducing CORT and ACTH responses to stress (Lund et al., 2006). We interpret such data as indicating that DHT can also have direct actions on PVN hypophysiotrophic neuron functions. However, based on AR distribution in brain, the local inhibitory action of DHT on PVN neurons likely occurs through a multisynaptic pathway that involves activation/inhibition of several neural pathways and the resulting feedback loops to inhibit activity of neurosecretory PVN neurons. An alternate, but not mutually exclusive possibility is that DHT may act through another receptor type found in the PVN or in neurons projecting to the PVN. Our recent results implicate ER beta (Lund et al., 2006), as an important receptor for DHT’s actions.
Estrogens and Estrogen Receptors
The role of estrogens in regulating stress reactivity remains controversial. Initial studies showing sex differences in corticosterone responses to a stressor (Gaskin and Kitay, 1970) demonstrated that sex steroid hormones could interact with the regulatory elements of the HPA axis. Gonadectomy of both males and females reduces the sex difference and hormone replacement to gonadectomized animals can reinstate the sex difference (Handa et al., 1994b). Initial studies indicated that estradiol treatment enhanced, and testosterone treatment inhibited HPA reactivity (Kitay, 1963; Viau and Meaney, 1991, 1996; Burgess and Handa, 1992; Handa et al., 1994a). For estradiol, the direction of effect has not always been consistent, as enhancement (Isgor et al., 2003) and inhibition (Orchedalski et al., 2007) of HPA activity following estradiol have also been reported. Moreover, evidence is present in the literature showing that estradiol and testosterone can act at the adrenal gland (Kitay, 1965), anterior pituitary (Coyne and Kitay, 1971; Viau and Meaney, 2004), and hypothalamus (Handa et al., 1994a; Viau and Meaney, 1996; Viau et al., 2003). Thus, contributions to each level of the axis may mediate the gonadal steroid effects on HPA function. Furthermore, amplitude and duration of hormone exposure could also influence the actions of gonadal steroid hormone effects on HPA axis function (Orchedalski et al., 2007).
Following the initial discovery of a second form of estrogen receptor, termed ERbeta (Kuiper et al., 1996), its mRNA and immunoreactivity were shown to be highly expressed by PVN neurons (Shughrue et al., 1997; Laflamme et al., 1998; Shughrue and Merchenthaler, 2001; Suzuki and Handa, 2004). A large number of ERbeta-ir cells in PVN are OT positive, and ERbeta is also found in AVP and prolactin expressing neurons (Alves et al., 1998; Hrabovsky et al., 1998; Somponpun and Sladek, 2003; Suzuki and Handa, 2005). Many fewer CRH neurons of the PVN express ERbeta (Laflamme et al., 1998; Suzuki and Handa, 2005; Miller et al., 2004). These data suggest that by binding to ERbeta, E2 might directly alter the function of PVN neuropeptide neurons. Indeed, the administration of ERbeta agonists to rats cause an inhibition of stress-induced corticosterone secretion (Lund et al., 2006; Weiser et al., 2009), coupled with increased anxiolytic-like behaviors (Walf et al., 2004; Lund et al., 2005; Weiser et al., 2009). A PVN site of action in the regulation of HPA reactivity has been demonstrated by the studies of Lund et al. (2006) who placed ERbeta agonists in an area adjacent to the PVN and demonstrated a reduction of stress-responsive corticosterone and ACTH secretion in ovariectomized female rats. In contrast, ERalpha agonists had an enhancing effect on corticosterone and ACTH. Little ERalpha is found in the PVN and it does not associate with CRH, AVP, or OXY neurons (Simerly et al., 1990; Estacio et al., 1996; Suzuki and Handa, 2005). Thus, a direct action of E2 that is mediated by ERalpha acting through neuropeptide neurons of the PVN to regulate stress reactivity appears unlikely.
Synthesis and Metabolism of Gonadal Steroid Hormones
In all steroid-synthesizing tissues, the conversion of cholesterol to pregnenolone by the side chain cleavage enzyme, P450scc or CYP11A1, is considered to be the rate-limiting step in steroidogenesis and occurs in the mitochondria of steroid-synthesizing cells. Pregnenolone is transported outside the mitochondria for subsequent synthetic steps. The weak androgen, androstenedione is produced via a delta4 pathway that first involves the conversion of pregnenolone to progesterone and then 17alpha hydroxyl progesterone by the enzymes 3β hydroxysteroid dehydrogenase (3β HSD) and 17alpha hydroxylase, respectively. Androstenedione is formed from 17alpha hydroxy-progesterone by 17,20 lyase activity. In humans, the pathway for androgen synthesis largely involves the delta5 pathway that produces dehydroepiandrosterone (DHEA) as the precursor for androstenedione. Depending on the tissue examined, androstenedione can be converted to testosterone by Type 3 17β hydroxysteroid dehydrogenase (17β HSD) or to estrone by the aromatase enzyme. Type 1 17β HSD is responsible for the conversion of estrone to estradiol whereas aromatase can also synthesize estradiol, using testosterone as the precursor (see Figure 1A; Rosen and Cedars, 2007).
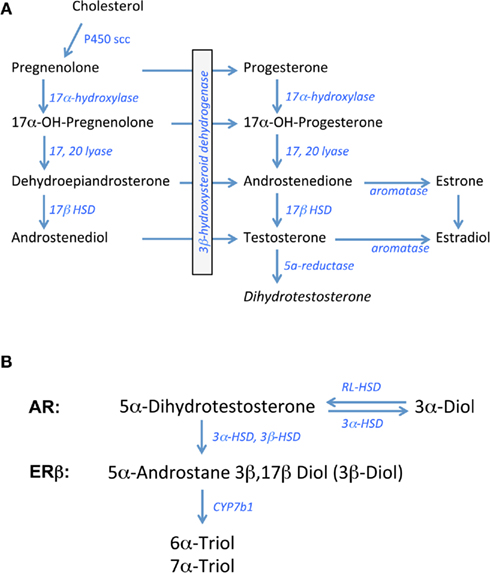
Figure 1. Biosynthetic pathway for gonadal steroid hormones. (A) Shows known enzymes and intermediates leading to the synthesis of estrogens and androgens from cholesterol. (B) Shows the metabolic steps of dihydrotestosterone processing putatively involved in pre-receptor regulation of ERbeta activation. 3α-Diol = 5α androstane 3α, 17β Diol; 3β Diol = 5α androstane 3β, 17β Diol, RL-HSD = 11-cis-retinol dehydrogenase like 3α-HSD.
In brain and other steroid sensitive tissues, there are overlapping functions that have been attributed to androgens and estrogens based on studies showing that the brain is a rich source of aromatase activity and testosterone is a precursor to estradiol in brain (Abdelgadire et al., 1994; Naftolin, 1994). Testosterone can also be converted to the more potent androgen, DHT, by the 5α reductase enzyme (5αR; Krieger et al., 1983; Melcangi et al., 1985). For many years, DHT was considered a prototypic androgen receptor (AR) agonist, with greater binding activity at the AR compared to testosterone (Zhou et al., 1995). Moreover, unlike testosterone, DHT cannot be aromatized to estradiol-like metabolites. In contrast, recent data indicate that DHT may be converted to products with estrogen-like activity, but by enzymes other than aromatase (Weihua et al., 2001, 2002).
The principal metabolites of DHT are 5α-androstane 3β, 17β Diol (3β-Diol), and 5α- androstane 3α, 17β Diol (3α-Diol). Both steroids have little activity at the AR, however 3β-Diol has a moderate ability to bind and activate ERbeta (Kuiper et al., 1997; Weihua et al., 2001). 3β-Diol is synthesized from DHT by the actions of multiple enzymes including 3αHSD, 17αHSD, and 3βHSD, whereas 3β Diol is metabolized to 6α- and 7α-triol by the actions of CYP7b1 (Figure 1B; Jin and Penning, 2001; Gangloff et al., 2003; Steckelbroeck et al., 2004; Penning et al., 2007). In contrast, 3α-Diol is synthesized from DHT through the actions of 3αHSD. This reaction is a reversible reaction through the actions of “RoDH Like” 3αHSD (RL–HSD; Bauman et al., 2006). Thus, any of these enzymes may be important in pre-receptor regulation of androgen action. Based on the metabolism of DHT to 3β-Diol, Weihua et al. (2001, 2002) suggested a role for ERbeta in mediating its actions in prostate gland. Given the important role that estrogens play in regulating the HPA axis, particularly those that bind ERbeta, these DHT metabolites could play an important role in the regulation of non-reproductive functions such as stress reactivity in the rodent.
3β-Diol and the Regulation of the HPA Axis
The ability of 3β-Diol to regulate the reactivity of the HPA axis was first described by Lund et al. (2004a) who tested the ability of peripherally administered 3β-Diol-diproprionate to alter stress-responsive CORT and ACTH secretion in castrated adult male mice. These studies revealed that 3β-Diol treatment was as effective as DHT in reducing the gain of stress-induced CORT and ACTH secretion. Moreover, the effects of 3β-Diol could be blocked by co-administration of the non-selective ER antagonist, tamoxifen, but not by the AR antagonist, flutamide, thus implicating estrogen receptors in the actions of both DHT and 3β-Diol. Furthermore, ERbeta agonists inhibited HPA reactivity in a fashion similar to DHT and 3β-Diol, whereas ERalpha agonists did not.
Evidence for the PVN being the primary neural target of 3β-Diol’s HPA inhibiting activity has also been demonstrated (Lund et al., 2006). Using small pellets of beeswax as a carrier for hormone, it was found that the stereotaxic application of 3β-Diol to the PVN of castrated male rats mimicked the actions of both central and peripherally administered DHT. Such local application of the ERbeta agonist, diarylpropionitrile (DPN), could also mimic the actions of DHT. The inhibitory actions of 3β-Diol and DPN were blocked by co-administration of tamoxifen, whereas the AR antagonist, flutamide had little effect. Although it is currently unknown whether 3β-Diol is produced by cells in or near the PVN, the fact that mRNAs for the steroid metabolizing enzymes such as 5αR, 3αHSD, 17αHSD, and CYP7b1 are found in the PVN suggests that local synthesis of the hormone may be responsible (Lund et al., 2006). Thus, the paracrine effects of 3β-Diol on nearby cells of the PVN likely impact the function of HPA reactivity to stressors through binding to ERbeta.
3β-Diol has also been shown to impact other neurobiological systems through ERbeta. For example, Osborne et al. (2009) demonstrated that 3β-Diol improved performance in the Morris water maze and Pendergast et al. (2008) demonstrated that 3β-Diol can regulate tyrosine hydroxylase expression in the locus coeruleus.
In contrast to the actions of 3β-Diol-bound ERbeta, estradiol appears to act primarily through ERalpha to augment HPA reactivity. Estradiol and the ERalpha selective agonist, propylpyrazole-triol (PPT), oppose the action of ERbeta agonists and consistently increase HPA reactivity to restraint stress (Lund et al., 2006). Given the co-localization of ERalpha in GABAergic neurons in the peri-PVN area, it has been hypothesized that ERalpha increases the gain of the HPA axis through modulation of local inhibitory circuits (Weiser and Handa, 2009).
Unknown at present are the mechanisms by which the HPA axis distinguishes enhancing from inhibiting actions of estradiol, a hormone that binds with similar high affinity to both ERalpha and ERbeta (Kuiper et al., 1997; Lund et al., 2005). It is possible that the ratio of ERalpha to ERbeta or the phenotype of neuron that expresses ERalpha and ERbeta may be altered under different physiological conditions. A greater alpha/beta ratio could cause a shift toward enhanced gain and the opposite would be true under conditions where ERbeta was elevated compared to ERalpha. Indeed, it has been demonstrated that levels of ERbeta change in response both to circulating glucocorticoid and estradiol levels (Isgor et al., 2003; Suzuki and Handa, 2004). This would allow the gain of the system to shift based on a given physiological state. An alternate hypothesis is that estradiol works predominantly by activating ERalpha, whereas other endogenous ligands, such as 3β-Diol are used to activate ERbeta.
Molecular Mechanism of Regulating CRH, AVP, and OT Expression
The receptors for estradiol (ERalpha and ERbeta) and for DHT (androgen receptor) belong to the nuclear receptor (NR) superfamily of proteins. The NR superfamily is a large group of transcription factors, most of which are ligand-activated. Characteristics of the steroid receptor branch of the family include their ability to interact with three types of response elements: (1) an inverted palindrome, (2) a composite element (Diamond et al., 1990), and (3) a “tethering” element (Lefstin and Yamamoto, 1998). Inverted palindromes are “classic” elements; they were the first described and for years were thought to be the sole means by which a receptor could interact with DNA to regulate transcription. Composite elements for steroid receptors consist of a hormone response element half-site, and a half-site for a monomer of another transcription factor. In fact, an example of this is the negative glucocorticoid response element in the proximal region of the CRH promoter (Malkoski et al., 1997; Malkoski and Dorin, 1999). The third type is a tethering or DNA-binding independent element, one in which a transcription factor regulates the activity of another transcription factor bound to its own DNA-binding site. This mechanism is also called an alternate pathway. A well-studied example is ER action mediated through an AP-1 or Sp-1 transcription factor bound to its DNA element (Kushner et al., 2000; Safe and Kim, 2008). Awareness of these three modes of regulation is critical for understanding ER regulation of CRH, VP, and OT.
3β-Diol-Regulated CRH Expression
E2 regulated CRH expression was first described by Vamvakopoulos and Chrousos (1993) who analyzed ER binding at estrogen response element (ERE) half-sites. At the time alternate pathways of gene regulation by ERs were only beginning to be explored and ERbeta had yet to be discovered. Thus, given that the CRH promoter is devoid of palindromic EREs, the ERE half-sites were a logical target for analysis.
By 2004, the ability of ER regulation of gene expression via alternate pathways was well established, particularly for SP-1 and AP-1 (Kushner et al., 2000; Safe and Kim, 2008). The cAMP regulatory element binding protein (CREB) binding protein (CBP) had been found to be a co-activator for AP-1, as well as CREB (Bannister et al., 1995) and steroid receptor co-activators (SRCs) were known to interact with CBP (reviewed in McKenna et al., 1999). Based on these and numerous other data, Kushner et al. (2000) proposed that ER regulation through AP-1 involved formation of an ER:SRC:CBP complex. The fact that CRH activation is critically dependent on the proximal cAMP regulatory element (CRE; Seasholtz et al., 1988) suggested that its activation could also be mediated via an alternate pathway, one that was regulated via CREB.
The discovery of ERbeta (Kuiper et al., 1996) greatly broadened the approach to CRH regulation. Using a widely used CRH promoter:reporter construct (−663 to +124; Seasholtz et al., 1988; Guardiola-Diaz et al., 1996), Miller et al. (2004) demonstrated that various splice variants of ERbeta activated the CRH promoter activity to different degrees. The most potent isoform was ERbeta 1δ3, which mediated a 12.5 fold increase in CRH promoter activity in the presence of E2. In the presence of Tmx, ERbeta 1, and ERbeta 2δ3 activated the promoter, as well. That an anti-estrogen could activate the promoter corroborated the hypothesis that CRH regulation involves an alternate pathway (Miller et al., 2004). This hypothesis was strengthened by examining the occupancy of the endogenous CRH promoter by ERalpha, ERbeta and associated coregulators in a CRH expressing amygdalar cell line (Kasckow et al., 1999; Lalmansingh and Uht, 2008). Results of chromatin immuno-precipitation analysis followed by quantitative PCR showed that ERalpha and beta occupancy in the region of CRE at the proximal promoter increased by approximately 22- and 12-fold respectively. The peak occupancy was different for the two receptors. ERalpha and beta peaked at 1 and 3 min, respectively. Furthermore, the pattern of ERalpha and beta occupancy correlated differentially with SRC-1 and CBP occupancy, suggesting that ERalpha and beta exist in distinct complexes (Lalmansingh and Uht, 2008). The increases in occupancy were tightly correlated with the expression profile of CRH mRNA, suggesting that the ER interactions with the CRH promoter are functional.
More recently, another neuronal cell line that constitutively expresses CRH was used to assess the effect of different estrogenic ligands on CRH promoter activity (Ogura et al., 2008). E2 and the ERbeta selective agonist diarylpropionitrile (DPN) increased reporter activity in hypothalamic IVB cells whereas the ERalpha agonist, propylpyrazole-triol (PPT) did not. These data suggest that in certain cellular contexts ERbeta, but not ERalpha, activates the CRH promoter. These data are germane to HPA axis regulation by 3β-Diol, which has been shown to act via ERbeta at the VP promoter (Pak et al., 2007).
Direct evidence for the regulation of CRH by 3β-Diol has been reported by Huang et al. (2008). Using CHO-K1 cells, they showed that E2 and 3β-Diol increased CRH promoter activity to the same extent, and did so through both ERalpha and ERbeta. In distinction Tmx reduced the E2 and 3β-Diol induced promoter activity to the level of vehicle in all cases, a finding consistent with an ER-mediated response. More recently, we have found that 3β-Diol treatment stimulates CRH expression rapidly; it increases mRNA levels between 1 and 5 mins of exposure (Stacey and Uht, unpublished data). Thus, emerging evidence demonstrates that 3β-Diol increases CRH expression and likely does so through an alternate pathway of gene regulation.
3β-Diol-Regulated AVP Expression
Prior to investigating the effects of 3β-Diol in AVP regulation, investigators had evaluated the role of ERalpha and ERbeta in regulating AVP expression. Using reporter assays, the differential regulation of AVP promoter activity by ERalpha and beta was studied (Shapiro et al., 2000). These investigators traced ERalpha and beta activity, in the presence of E2, to an upstream ERE, between −5.5 and −4.0 kb and ERbeta repression to a proximal region of the promoter that contains several AP-1 sites.
The effects of 3β-Diol at the AVP promoter, mediated through ERbeta1 and its splice variant, ERbeta2, has also been examined (Pak et al., 2007). In a fashion similar to that shown by Shapiro et al. (2000), ERbeta1 displayed constitutive activity in the proximal promoter. Maximal constitutive activity required the region between −740 b and 1.3 kb of the AVP promoter. ERbeta2 also displayed constitutive activity but approximately half of that exhibited by ERbeta1 (Pak et al., 2007). In distinction to the studies by Shapiro and colleagues, Pak et al. (2007) did not report that E2 down-regulated the constitutive activity of ERbeta1. Rather, in the presence of ERbeta1, E2 had no effect whereas DHT and 3β-Diol increased activity. In the presence of ERbeta2, however, E2 and 3β-Diol increased activity whereas DHT had no effect. That 3β-Diol increases AVP reporter activity through both ER-beta1 and -beta2 is striking, given that the difference between the two receptors has been traced to the ligand-binding domain. Lastly, Pak et al. (2007) showed that expression of a GRIP/SRC-2-NR box abrogated the 3β-Diol effect through ERbeta1, suggesting that 3β-Diol elicits an AF-2 conformation similar to E2. As the authors suggest, it will be interesting to determine whether the spectrum of co-activators required by 3β-Diol-bound ERbeta1 and ERbeta2 differ. Regardless of which co-activators are used, 3β-Diol clearly regulates AVP expression. Moreover, sufficient information is present in the literature to suggest that 3β-Diol could target AVP expressing neurons in the PVN.
3β-Diol-Regulated OT Expression
For years, the OT promoter has been known to be estrogen responsive (Richard and Zingg, 1990). However, dissection of the molecular mechanisms of OT regulation has been hampered by species differences in the upstream promoter and a complex composite hormone response element within the proximal promoter. A point in common across species is the location of the primary site of estrogen regulation at about −160 bp (Richard and Zingg, 1990; Adan et al., 1993). The composite response element contains sequences in common with a consensus ERE; however, the degree to which it binds ERalpha and/or beta is species dependent.
Common to all species is the ability of two “orphan” receptors to bind the response element. These are steroidogenic factor-1 (SF-1) and a chicken ovalbumin upstream promoter transcription factor COUP–TF (Wehrenberg et al., 1994). The site is also bound by other NRs such as the retinoic acid and thyroid hormone receptors and the orphan receptor ERRα; however, the extent to which they do so is again species dependent (Richard and Zingg, 1991; Adan and Burbach, 1992; Lipkin et al., 1992; Burbach et al., 1994; Lopes da Silva and Burbach, 1995; Chu and Zingg, 1999; Dellovade et al., 1999; Stedronsky et al., 2002; Koohi et al., 2005; Wang et al., 2006).
Although the OT ERE has been studied for its ability to interact with a number of different receptors, differences in regulation elicited by various ligands has not been well-studied. Koohi et al. (2005) compared E2 to Tamoxifen, raloxifene, and ICI 180,780 effects at the bovine OT promoter in the presence of ERalpha. They found that the profile of all of the “antagonists” was in keeping with their activity through alternate pathways. That is, when used alone they elicited activity at the OT response element but not at an individual vitellogenin ERE, a site which is very close to being a consensus sequence. With respect to 3β-Diol effects on OT expression, we have recently found that ERalpha and beta are differentially recruited to the OT promoter. Furthermore, this recruitment is a function of receptor ligand (Figure 2). A hypothalamic cell line derived from mice (mHypoE-38; Mayer et al., 2009) was used for ChIP analysis. By 30 min, 3β-Diol stimulated ERbeta occupancy to fourfold that of vehicle. At 60 min, both ligands reduced promoter occupancy by both receptors (Figure 2; unpublished data). Thus, 3β-Diol and E2 differentially regulate OT promoter occupancy by ERalpha and ERbeta in a time dependent manner.
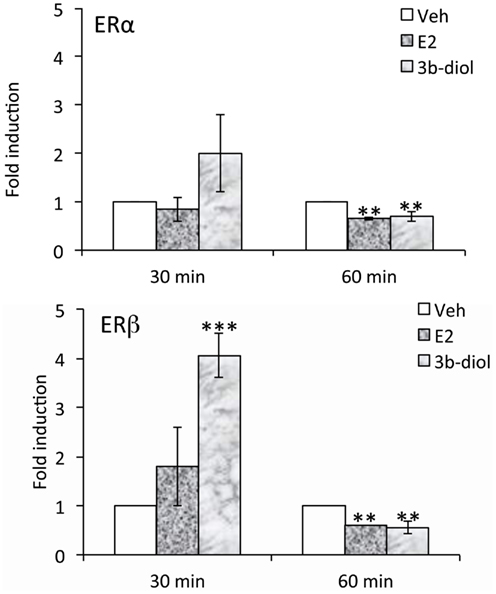
Figure 2. E2 and 3β-Diol lead to different patterns of promoter occupancy by ERα and ERβ. mHypoE-38 cells were treated with E2 (10−7 M) or 3β-Diol (10−7 M) for 30 or 60 min. Anti-ERα or anti-ERβ antiserum was used for chromatin immuno-precipitation followed by quantitative PCR of the OT promoter. n = 4; Bars represent the mean ± SEM and are presented as the fold difference of vehicle. **, p < 0.01, ***, p < 0.001.
Taken together, the available molecular data underscore the complexity of OT regulation. An important point to be taken is that the species differences are so striking that generalizations with respect to regulation of this stress regulatory neuropeptide can only be made with caution.
Significance
Clinical and preclinical studies of MDD provide evidence linking the reported dysregulation of the HPA axis and depressive neuropathology. This dysregulation is characterized by increased cortisol secretory responses, enlarged adrenal gland volume (Rubin et al., 1987, 1996) and elevated CRH in cerebrospinal fluid (Nemeroff et al., 1984) in depressed patients. Furthermore, 20–40% of depressed patients are dexamethasone (DEX) non-suppressors (i.e., DEX suppresses ACTH and cortisol to a much smaller extent than healthy controls) and the inability of DEX to suppress cortisol release following a CRH stimulation can distinguish greater than 90% of depressed patients from non-depressed controls (Heuser et al., 1994). Neurobiological evidence for HPA involvement in MDD comes from postmortem studies showing that the PVN of depressed patients contains four times the number of CRH expressing cells (Raadsheer et al., 1993). Increases in the number of oxytocin and AVP-ir neurons in the PVN have been reported as well (Raadsheer et al., 1994; Purba et al., 1996). Of interest, plasma vasopressin levels have been shown to be elevated in MDD patients (Van Londen et al., 1997). Similarly, nocturnal plasma OT levels have recently been reported to be elevated in individuals with major depression (Parker et al., 2010), although earlier studies show a negative correlation between oxytocin levels and depressive symptoms (Scantamburlo et al., 2007). Pulsatile patterns of oxytocin release have also been reported to be more variable in depressed women (Cyranowski et al., 2008). Consequently, estrogen and androgen signaling in brain may influence the regulation of HPA axis function and may impact the dysregulation of the HPA axis seen in patients with affective disorders thus underlying susceptibility to affective disorders. Further work is required to identify estrogen receptor regulated genes that may be targets for future pharmacological intervention.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Funding for the authors’ research program has been provided by grants by the National Institutes of Health R01 NS039951 (Robert J. Handa) and 1R01MH082900 (Rosalie Uht).
References
Abdelgadire, S. E., Resko, J. A., Ojeda, S. R., Lephart, E. D., McPhaul, M. J., and Roselli, C. E. (1994). Androgens regulate aromatase cytochrome P450 messenger ribonucleic acid in rat brain. Endocrinology 135, 395–401.
Adan, R. A., and Burbach, J. P. (1992). Regulation of vasopressin and oxytocin gene expression by estrogen and thyroid hormone. Prog. Brain Res. 92, 127–136.
Adan, R. A., Cox, J. J., Beischlag, T. V., and Burbach, J. P. (1993). A composite hormone response element mediates the transactivation of the rat oxytocin gene by different classes of nuclear hormone receptors. Mol. Endocrinol. 7, 47–57.
Aguilera, G., Nikodemova, M., Wynn, P. C., and Catt, K. J. (2004). Corticotropin releasing hormone receptors: two decades later. Peptides 25, 319–329.
Alves, S. E., Lopez, V., McEwen, B. S., and Weiland, N. G. (1998). Differential colocalization of estrogen receptor beta (ERbeta) with oxytocin and vasopressin in the paraventricular and supraoptic nuclei of the female rat brain: an immunocytochemical study. Proc. Natl. Acad. Sci. U.S.A. 95, 3281–3286.
Angold, A., Costello, E. J., Erkanll, A., and Worthman, C. M. (1999). Pubertal changes in hormone levels and depression in girls. Pyschol. Med. 29, 1043–1053
Angold, A., and Worthman, C. W. (1993). Puberty onset of gender differences in rates of depression: a developmental, epidemiologic and neuroendocrine perspective. J. Affect. Disord. 29, 145–158.
Bannister, A. J., Oehler, T., Wilhelm, D., Angel, P., and Kouzarides, T. (1995). Stimulation of c-Jun activity by CBP: c-Jun residues Ser63/73 are required for CBP induced stimulation in vivo and CBP binding in vitro. Oncogene 11, 2509–2514.
Bauman, D. R., Steckelbroeck, S., Williams, M. V., Peehl, D. M., and Penning, T. M. (2006). Identificatin of the major oxidative 3alpha-hydroxysteroid dehydrogenase in human prostate that converts 5alph-androstane-3alpha, 17beta-diiol to 5alpha-dihydrotestosterone: a potential therapeutic target for androgen-dependent disease. Mol. Endocrinol. 20, 444–458.
Bingaman, E. W., Magnuson, D. J., Gray, T. S., and Handa, R. J. (1994a). Androgen inhibits the increases in hypothalamic corticotropin-releasing hormone (CRH) and CRH-immunoreactivity following gonadectomy. Neuroendocrinology 59, 228–234.
Bingaman, E. W., Baeckman, L. M., Yracheta, J. M., Handa, R. J., and Gray, T. S. (1994b). Localization of androgen receptor within peptidergic neurons of the rat forebrain. Brain Res. Bull. 35, 379–382.
Bingham, B., Williamson, M., and Viau, V. (2006). Androgen and estrogen receptor-beta distribution within spinal-projecting and neurosecretory neurons in the paraventricular nucleus of the male rat. J. Comp. Neurol. 499, 911–923.
Bondy, C. A., Whitnall, M. H., Brady, L. S., and Gainer, H. (1989). Coexisting peptides in hypothalamic neuroendocrine systems: some functional implications. Cell. Mol. Neurobiol. 9, 427–446.
Burbach, J. P., Lopes da Silva, S., Cox, J. J., Adan, R. A., Cooney, A. J., Tsai, M. J., and Tsai, S. Y. (1994). Repression of estrogen-dependent stimulation of the oxytocin gene by chicken ovalbumin upstream promoter transcription factorI. J. Biol. Chem. 269, 15046–15053.
Burgess, L. H., and Handa, R. J. (1992). Chronic estrogen-induced alterations in adrenocorticotropin and corticosterone secretion and glucocorticoid receptor-mediated functions in female rats. Endocrinology 131, 1261–1269.
Caldwell, H. K., Lee, H. J., Macbeth, A. H., and Young, W. E. III. (2008). Vasopressin: behavioral roles of an “original” neuropeptide. Prog. Neurobiol. 84, 1–24.
Choi, D. C., Furay, A. R., Evanson, N. K., Ostrander, M. M., Ulrich-Lai, Y. M., and Herman, J. P. (2007). Bed nucleus of the stria terminalis subregions differentially regulate hypothalamic-pituitary-adrenal axis activity: implications for the integration of limbic inputs. J. Neurosci. 27, 2025–2034.
Chu, K., and Zingg, H. H. (1999). Activation of the mouse oxytocin promoter by the orphan receptor RORalpha. J. Mol. Endocrinol. 23, 337–346.
Chung, S., Son, G. H., and Kim, K. (2011). Circadian rhythm of adrenal glucocorticoid: its regulation and clinical implications. Biochem. Biophys. Acta 1812, 581–591.
Coyne, M. D., and Kitay, J. I. (1971). Effect of orchiectomy on pituitary secretion of ACTH. Endocrinology 89, 1024–1028.
Cullinan, W. E., Helmreich, D. L., and Watson, S. J. (1996). Fos expression in forebrain afferents to the hypothalamic paraventricular nucleus following swim stress. J. Comp. Neurol. 368, 88–99.
Cyranowski, J. M., Hofkens, T. L., Frank, E., Seltman, H., Cai, H. M., and Amicao, J. A. (2008). Evidence of dysregulated peripheral oxytocin release among depressed women. Psyhosom. Med. 70, 967–975.
Dellovade, T. L., Zhu, Y. S., and Pfaff, D. W. (1999). Thyroid hormones and estrogen affect oxytocin gene expression in hypothalamic neurons. J. Neuroendocrinol. 11, 1–10.
Diamond, M. I., Miner, J. N., Yoshinaga, S. K., and Yamamoto, K. R. (1990). Transcription factor interactions: selectors of positive or negative regulation from a single DNA element. Science 249, 1266–1272.
Estacio, M. A., Yamada, S., Tsukamura, H., Hirunagi, K., and Maeda, K. (1996). Effect of fasting and immobilization stress on estrogen receptor immunoreactivity in the brain in ovariectomized female rats. Brain Res. 717, 55–61.
Follib, A., Lui, P., and Romeo, R. (2011). The transformation of hormonal stress responses throughout puberty and adolescence. J. Endocrinol. 210, 391–398.
Gangloff, A., Shi, R., Nahoum, V., and Lin, S. X. (2003). Pseudo-symmetry of C19 steroids, alternative binding orientations, and multispecificity in human estrogenic 17beta-hydroxysteroid dehydrogenase. FASEB J. 17, 274–276.
Gaskin, J. H., and Kitay, J. I. (1970). Adrenocortical function in the hamster. Sex differences and effects of gonadal hormones. Endocrinology 87, 779–786.
Gaskin, J. H., and Kitay, J. I. (1971). Hypothalamic and pituitary regulation of adrenocortical function in the hamster: effects of gonadectomy and gonadal hormone replacement. Endocrinology 89, 1047–1053.
Gillies, G. E., Linton, E. A., and Lowry, P. J. (1982). Corticotropin releasing activity of the new CRF is potentiated several times by vasopressin. Nature 299, 355–357.
Gomez, F., De Kloet, E. R., and Armario, A. (1998). Glucocorticoid negative feedback on the HPA axis in five inbred rat strains. Am. J. Physiol. 274, R420–R427.
Guardiola-Diaz, H. M., Kolinske, J. S., Gates, L. H., and Seasholtz, A. F. (1996). Negative glucorticoid regulation of cyclic adenosine 3′, 5′-monophosphate-stimulated corticotropin-releasing hormone-reporter expression in AtT-20 cells. Mol. Endocrinol. 10, 317–329.
Handa, R. J., Nunley, K. M., Lorens, S. A., Louie, J. P., McGivern, R. F., and Bollnow, M. R. (1994a). Androgen regulation of adrenocorticotropin and corticosterone secretion in the male rat following novelty and foot shock stressors. Physiol. Behav. 55, 117–124.
Handa, R. J., Burgess, L. H., Kerr, J. E., and O’Keefe, J. A. (1994b). Gonadal steroid hormone receptors and sex differences in the hypothalamo-pituitary-adrenal axis. Horm. Behav. 28, 464–476.
Herman, J. P., Prewitt, C. M., and Cullinan, W. E. (1996). Neuronal circuit regulation of the hypothalamo-pituitary-adrenocortical stress axis. Crit. Rev. Neurobiol. 10, 371–394.
Hettema, J. M., Neale, M. C., and Kendler, K. S. (2001). A review and meta-analysis of the genetic epidemiology of anxiety disorders. Am. J. Psychiatry 158, 1568–1578.
Heuser, I., Yassouridis, A., and Holsboer, F. (1994). The combined dexamethasone/CRH test: a refined laboratory test for psychiatric disorders. J. Psychiatr. Res. 28, 341–356.
Hrabovsky, E., Kallo, I., Hajszan, T., Shughrue, P. J., Merchenthaler, I., and Liposits, Z. (1998). Expression of estrogen receptor beta messenger ribonucleic acid in oxytocin and vasopressin neurons of the rat supraoptic and paraventricular nuclei. Endocrinology 139, 2600–2604.
Huang, Q., Zhu, H., Fischer, D. F., and Zhou, J. N. (2008). An estrogenic effect of 5alpha-androstane-3beta, 17beta-diol on the behavioral response to stress and on CRH regulation. Neuropharmacology 54, 1233–1238.
Isgor, C., Cecchi, M., Kabbaj, M., Akil, H., and Watson, S. J. (2003). Estrogen receptor beta in the paraventricular nucleus of hypothalamus regulates the neuroendocrine response to stress and is regulated by corticosterone. Neuroscience 121, 837–845.
Jin, Y., and Penning, T. M. (2001). Steroid 5alpha-reductases and 3alpha-hydroxysteroid dehydrogenases: key enzymes in androgen metabolism. Best Pract. Res. Clin. Endocrinol. Metab. 15, 79–94.
Kalsbeek, A., van der Vliet, J., and Buijs, R. M. (1996). Release of endogenous vasopressin release necessary for the expression of the circadian rise in plasma corticosterone: a reverse microdialysis study. J. Neuroendocrinol. 8, 299–307.
Kasckow, J. W., Regmi, A., Seasholtz, A. F., and Mulchahey, J. J. (1999). Regulation of corticotropin-releasing factor-binding protein expression in amygdalar neuronal cultures. J. Neuroendocrinol. 11, 959–966.
Kendler, K. S. (1998). Gender differences in the genetic epidemiology of major depression. J. Gend. Specif. Med. 1, 28–31.
Kitay, J. I. (1963). Effects of estradiol on pituitary-adrenal function in male and female rats. Endocrinology 72, 947–954.
Kitay, J. I. (1965). Effects of oophorectomy and various doses of estroadiol-17beta on cortiosterone production by rat adrenal slices. Proc. Soc. Exp. Biol. Med. 120, 193–196.
Koohi, M. K., Ivell, R., and Walther, N. (2005). Transcriptional activation of the oxytocin promoter by oestrogens uses a novel non-classical mechanism of oestrogen receptor action. J. Neuroendocrinol. 17, 197–207.
Krieger, N. R., Scott, R. G., and Jurman, M. E. (1983). Testosterone 5 alpha-reductase in rat brain. J. Neurochem. 40, 1460–1464.
Kuiper, G. G., Carlsson, B., Grandien, K., Enmark, E., Haggblad, J., Nilsson, S., and Gustafsson, J. A. (1997). Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology 138, 863–870.
Kuiper, G. G., Enmark, E., Pelto-Huikko, M., Nilsson, S., and Gustafsson, J. A. (1996). Cloning of a novel receptor expressed in rat prostate and ovary. 93, 5925–5930.
Kushner, P. J., Agard, D. A., Greene, G. L., Scanlan, T. S., Shiau, A. K., Uht, R. M., and Webb, P. (2000). Estrogen receptor pathways to AP-1. J. Steroid Biochem. Mol. Biol. 74, 311–317.
Laflamme, N., Nappi, R. E., Drolet, G., Labrie, C., and Rivest, S. (1998). Expression and neuropeptidergi characterization of estrogen receptors (ERalpha and ERbeta) throughout the rat brain: anatomical evidence of distinct roles of each subtype. J. Neurobiol. 36, 357–378.
Lalmansingh, A. S., and Uht, R. M. (2008). Estradiol regulates corticotropin-releasing hormone gene (crh) expression in a rapid and phasic manner that parallels estrogen receptor-alpha and -beta recruitment to a 3′,5′-cyclic adenosine 5′-monophosphate regulatory region of the proximal crh promoter. Endocrinology 149, 346–357.
Landgraf, R., and Neumann, I. D. (2004). Vasopressin and oxytocin release within the brain: a dynamic concept of multiple and variable modes of neuropeptide communication. Front. Neuroendocrinol. 25, 150–176.
Lefstin, J. A., and Yamamoto, K. R. (1998). Allosteric effects of DNA on transcriptional regulators. Nature 392, 885–888.
Lipkin, S. M., Nelson, C. A., Glass, C. K., and Rosenfeld, M. G. (1992). A negative retinoic acid response element in the rat oxytocin promoter restricts transcriptional stimulation by heterologous transactivation domains. Proc. Natl. Acad. Sci. U.S.A. 89, 1209–1213.
Lopes da Silva, S., and Burbach, J. P. (1995). The nuclear hormone-receptor family in the brain: classics and orphans. Trends Neurosci. 18, 542–548.
Lund, T. D., Hinds, L. R., and Handa, R. J. (2006). The androgen 5alpha-dihydrotestosterone and its metabolite 5alpha androstan-3beta, 17beta-diol inhibit the hypothalamo-pituitary-adrenal response to stress by acting through estrogen receptor beta-expressing neurons in the hypothalamus. J. Neurosci. 26, 1448–1456.
Lund, T. D., Munson, D. J., Haldy, M. E., and Handa, R. J. (2004a). Dihydrotestosterone may inhibit hypothalamo-pituitary-adrenal activity by acting through estrogen receptor in the male mouse. Neurosci. Lett. 365, 43–47.
Lund, T. D., Munson, D. J., Haldy, M. E., and Handa, R. J. (2004b). Androgen inhibits, while oestrogen enhances, restraint-induced activation of neuropeptide neurons in the paraventricular nucleus of the hypothalamus. J. Neuroendocrinol. 16, 272–278.
Lund, T. D., Rovis, T., Chung, W. C., and Handa, R. J. (2005). Novel actions of estrogen receptor-beta on anxiety-related behaviors. Endocrinology 146, 797–807.
Malkoski, S. P., and Dorin, R. I. (1999). Composite glucocorticoid regulation at a functionally defined negative glucocorticoid response element of the human corticotropin-releasing hormone gene. Mol. Endocrinol. 13, 1629–1644.
Malkoski, S. P., Handanos, C. M., and Dorin, R. I. (1997). Localization of a negative glucocorticoid response element of the human corticotropin releasing hormone gene. Mol. Cell. Endocrinol. 127, 189–199.
Mayer, C. M., Fick, L. J., Gingerich, S., and Belsham, D. D. (2009). Hypothalamic cell lines to investigate neuroendocrine control mechanisms. Front. Neuroendocrinol. 30, 405–423.
McKenna, N. J., Xu, J., Nawaz, Z., Tsai, S. Y., Tsai, M.-J., and O’Malley, B. W. (1999). Nuclear receptor coactivators: multiple enzymes, multiple complexes, multiple functions. J. Steroid Biochem. Mol. Biol. 69, 3–12.
Melcangi, R. C., Celotti, F., Negri-Cesi, P., and Martini, L. (1985). Testosterone 5 alpha-reductase in discrete hypothalamic nuclear areas in the rat: effect of castration. Steroids 45, 347–356.
Miller, W. J., Suzuki, S., Miller, L. K., Handa, R., and Uht, R. M. (2004). Estrogen receptor (ER) beta isoforms rather than Eralpha regulate corticotropin-releasing hormone promoter activity through an alternate pathway. J. Neurosci. 24, 10628–10635.
Murray, C. J., and Lopez, A. D. (1997). Global mortality, disability and the contribution of risk factors: global burden of disease study. Lancet 349, 1436–1442.
Nader, N., Chrousos, G. P., and Kino, T. (2010). Interactions of the circadian CLOCK system and the HPA axis. Trends Endocrinol. Metab. 21, 277–286.
Nemeroff, C. B., Widerlov, E., Bissette, G., Walleus, H., Karlsson, I., Eklund, K., Kilts, C. D., Loosen, P. T., and Vale, W. (1984). Elevated concentrations of CSF corticotropin-releasing factor-like immunoreactivity in depressed patients. Science 226, 1342–1344.
Neumann, I. D. (2007). Stimuli and cnosequences of dendritic release of oxytocin within the brain. Biochem. Soc. Trans. 35, 1252–1257.
Neumann, I. D., Kromer, S. A., Toschi, N., and Ebner, K. (2000). Brain oxytocin inhibits the (re)activity of the hypothalamo-pituitary-adrenal axis in male rats: involvement of hypothalamic and limbic brain regions. Regul. Pept. 96, 31–38.
Ogura, E., Kageyama, K., Hanada, K., Kasckow, J., and Suda, T. (2008). Effects of estradiol on regulation of corticotropin-releasing factor gene and interleukin-6 production via estrogen receptor type beta in hypothalamic 4B cells. Peptides 29, 456–464.
Orchedalski, T., Subbaraju, S., Wynn, P. C., and Aguilera, G. (2007). Interaction between oestrogen and oxytocin on hypothalamic-pituitary-adrenal axis activity. J. Neuroendocrinol. 19, 189–197.
Osborne, D. M., Edinger, K., and Frye, C. A. (2009). Chronic administration of androgens with actions at estrogen receptor beta have anti-anxiety and cognitive enhancing effects in male rats. Age (Dordr.) 31, 191–198.
Oster, H., Damerow, S., Kiessling, S., Jakubcakova, V., Abraham, D., Tian, J., Hoffmann, M. W., and Eichele, G. (2006). The circadian rhythm of glucocorticoids is regulated by a gating mechanism residing in the adrenal cortical clock. Cell Metab. 4, 163–173.
Palkovits, M. (2000). Stress-induced expression of co-localized neuropeptides in hypothalamic and amygdaloid neurons. Eur. J. Pharmacol. 405, 161–166.
Pak, T. R., Chung, W. C., Hinds, L. R., and Handa, R. J. (2007). Estrogen receptor-beta mediates dihydrotestosterone-induced stimulation of the arginine vasopressin promoter in neuronal cells. Endocrinology 148, 3371–3382.
Papadimitriou, A., and Priftis, K. N. (2009). Regulation of the hypothalamic-pituitary-adrenal axis. Neuroimmunomodulation 16, 265–271.
Parker, K. J., Kenna, H. A., Zeitzer, J. M., Keller, J., Blasey, C. M., Amico, J. A., and Schatzberg, A. F. (2010). Preliminary evidence that plasma oxytocin levels are elevated in major depression. Pyschiatry Res. 178, 359–362.
Pendergast, J. S., Tuesta, L. M., and Bethea, J. R. (2008). Oestrogen receptor beta contributes to the transient sex difference in tyrosine hydroxylase expression in the mouse locus coeruleus. J. Neuroendocrinol. 20, 1155–1164.
Penning, T. M., Bauman, D. R., Jin, Y., and Rizner, T. L. (2007). Identification of the molecular switch that regulates access of 5alpha-DHT to the androgen receptor. Mol. Cell. Endocrinol. 265–266, 77–82.
Purba, J. S., Hoogendijk, W. J., Hofman, M. A., and Swaab, D. F. (1996). Increased number of vasopressin- and oxytocin-expressing neurons in the paraventricular nucleus of the hypothalamus in depression. Arch. Gen. Psychiatry 53, 137–143.
Raadsheer, F. C., Sluiter, A. A., Ravid, R., Tilders, F. J., and Swaab, D. F. (1993). Localization of corticotropin-releasing hormone (CRH) neurons in the paraventricular nucleus of the human hypothalamus; age-dependent colocalization with vasopressin. Brain Res. 615, 50–62.
Raadsheer, F. C., Tilders, F. J., and Swaab, D. F. (1994). Increased numbers of corticotropin-releasing hormone expressing neurons in the hypothalamic paraventricular nucleus of depressed patients. Neuroendocrinology 60, 436–444.
Richard, S., and Zingg, H. H. (1990). The human oxytocin gene promoter is regulated by estrogens. J. Biol. Chem. 265, 6098–6103.
Richard, S., and Zingg, H. (1991). Identification of a retinoic acid response element in the human oxytocin promoter. J. Biol. Chem. 266, 21428–21433.
Rivier, C., and Vale, W. (1983). Interaction of corticotropin-releasing factor and arginine vasopressin on adrenocorticotropin secretion in vivo. Endocrinology 113, 939–942.
Romeo, R. D., and McEwen, B. S. (2006). Stress and the adolescent brain. Ann. N. Y. Acad. Sci. 1094, 202–214.
Romeo, R. D., Lee, S. J., Chua, N., McPherson, C. R., and McEwen, B.S. (2004). Testosterone cannot activate an adult-like stress response in prepubertal male rats. Neuroendocrinology 79, 125–132.
Rosen, M. P., and Cedars, M. I. (2007). “Female reproductive endocrinology and infertility,” in Greenspans’s Basic and Clinical Endocrinology, 8th Edn, eds D. G. Gardner and D. Shoback (New York, NY: McGraw Hill Medical) 502–562.
Rubin, R. T., Phillips, J. J., McCracken, J. T., and Sadow, T. F. (1996). Adrenal gland volume in major depression: relationship to basal and stimulated pituitary-adrenal cortical axis function. Biol. Pyschiatry. 40, 89–97.
Rubin, R. T., Poland, R. E., Lesser, I. M., Winston, R. A., and Blodgett, A. L. (1987). Neuroendocrine aspects of primary endogenous depression. I. Cortisol secretory dynamics in patients and matched controls. Arch. Gen Psychiatry 44, 328–365.
Safe, S., and Kim, K. (2008). Non-classical genomic estrogen receptor (ER)/specificity protein and ER/activating protein-1 signaling pathways. J. Mol. Endocrinol. 41, 263–275.
Scantamburlo, G., Hansenne, M., Fuchs, S., Pitchot, W., Marechal, P., Pequeux, C., Ansseau, M., and Legros, J. J. (2007). Plasma oxytocin levels and anxiety in patients with major depression. Psychoneurendocrinology 32, 407–410.
Schlosser, S. F., Almeida, O. F., Patchev, V. K., Uassouridis, A., and Elands, J. (1994). Oxytocin-stimuated release of adrenocorticotropin from the rat pituitary is mediated by arginine vasopressin receptors of the V1b type. Endocrinology 135, 2058–2063.
Seasholtz, A. F., Thompson, R. C., and Douglass, J. O. (1988). Identification of a cyclic adenosine monophosphate-responsive element in the rat corticotropin-releasing hormone gene. Mol. Endocrinol. 2, 1311–1319.
Shapiro, R. A., Xu, C., and Dorsa, D. M. (2000). Differential transcriptional regulation of rat vasopressin gene expression by estrogen receptor alpha and beta. Endocrinology 141, 4056–4064.
Shughrue, P. J., Lane, M. V., and Merchenthaler, I. (1997). Comparative distribution of estrogen receptor alpha and beta mRNA in the rat central nervous system. J. Comp. Neurol. 388, 507–525.
Shughrue, P. J., and Merchenthaler, I. (2001). Distribution of estrogen receptor beta immunoreactivity in the rat central nervous system. J. Comp. Neurol. 436, 64–81.
Simerly, R. B., Chang, C., Muramatsu, M., and Swanson, L. W. (1990). Distribution of androgen and estrogen receptor mRNA-containing cells in the rat brain: an in situ hybridization study. J. Comp. Neurol. 294, 76–95.
Somponpun, S. J., and Sladek, C. D. (2003). Osmotic regulation of estrogen receptor-beta in rat vasopressin and oxytocin neurons. J. Neurosci. 23, 4261–4269.
Steckelbroeck, S., Jin, Y., Gopishetty, S., Oyesanmi, B., and Penning, T. M. (2004). Human cytosolic 3alpha-hydroxysteroid dehydrogenases of the aldo-keto reductase superfamily display significant 3beta-hydroxysteroid dehydrogenase activity: implications for steroid hormone metabolism and action. J. Biol. Chem. 279, 10784–10795.
Stedronsky, K., Telgmann, R., Tillmann, G., Walther, N., and Ivell, R. (2002). The affinity and activity of the multiple hormone response element in the proximal promoter of the human oxytocin gene. J. Neuroendocrinol. 14, 472–485.
Suzuki, S., and Handa, R. J. (2004). Regulation of estrogen receptor-beta expression in the female rat hypothalamus: differential effects of dexamethasone and estradiol. Endocrinology 145, 3658–3670.
Suzuki, S., and Handa, R. J. (2005). Estrogen receptor-beta, but not estrogen receptor-alpha is expressed in prolactin neurons of the female rat paraventricular and supraoptic nuclei: comparison with other neuropeptides. J. Comp. Neurol. 484, 28–42.
Suzuki, S., Lund, T. D., Price, R. H. Jr., and Handa, R. J. (2001). “Sex differences in the hypothalamo-pituitary-adrenal axis: novel roles for androgen and estrogen receptors,” in Recent Research Developments in Endocrinology, Vol. 2, ed. S. G. Pandalai (Trivandrum: Transworld Research Network), 69–86.
Szafarczyk, A., Ixart, G., Alonso, G., Malaval, F., Nouguier-Soule, J., and Assenmacher, I. (1983). CNS control of the circadian adrenocortical rhythm. J. Steroid Biochem. 19, 1009–1015.
Ustun, T. B., Ayuso-Mateos, J. L., Chatterji, S., Mathers, C., and Murray, C. F. (2004). Global burden of depressive disorders in the year (2000). Br. J. Psychiatry 184, 386–392.
Vamvakopoulos, N. C., and Chrousos, G. P. (1993). Evidence of direct estrogenic regulation of human corticotropin-releasing hormone gene expression. Potential implications for the sexual dimophism of the stress response and immune/inflammatory reaction . J. Clin. Invest. 92, 1896–1902.
Van Londen, L., Goekoop, J. G., van Kempen, G. M., Frankhuijzen-Sierevogel, A. C., Wiegant, V. M., van der Velde, E. A., and De Wied, D. (1997). Plasma levels of arginine vasopressin elevated in patients with major depression. Neuropsychopharmacology 17, 284–292.
Viau, V. (2002). Functional cross-talk between the hypothalamic-pituitary-gonadal and adrenal axes. J. Neuroendocrinol. 14, 506–513.
Viau, V., Bingham, B., Davis, J., Lee, P., and Wong, M. (2005). Gender and puberty interact on the stress-induced activation of parvocellular neurosecretory neurons and corticotropin-releasing hormone messenger ribonucleic acid expression in the rat. Endocrinology 146, 137–146.
Viau, V., Lee, P., Sampson, J., and Wu, J. (2003). A testicular influence on restraint-induced activation of medial parvocellular neurons in the paraventricular nucleus in the male rat. Endocrinology 144, 3067–3075.
Viau, V., and Meaney, M. J. (1991). Variations in the hypothalamic-pituitary-adrenal response to stress during the estrous cycle in the rat. Endocrinology 129, 2503–2511.
Viau, V., and Meaney, M. J. (1996). The inhibitory effect of testosterone on hypothalamic-pituitary-adrenal responses to stress is mediated by the medial preoptic area. J. Neurosci. 16, 1866–1876.
Viau, V., and Meaney, M. J. (2004). Testosterone-dependent variations in plasma and intrapituitary corticosteroid binding globulin and stress hypothalamic-pituitary-adrenal activity in the male rat. J. Endocrinol. 181, 223–231.
Viau, V., Soriano, L., and Dallman, M. F. (2001). Androgens alter corticotropin releasing hormone and arginine vasopressin mRNA within forebrain sites known to regulate activity in the hypothalamic-pituitary-adrenal axis. J. Neuroendocrinol. 13, 442–445.
Walf, A., Rhodes, M. E., and Frye, C. A. (2004). Antidepressant effects of ERbeta-selective estrogen receptor modulators in the forced swim test. Pharmacol. Biochem. Behav. 78, 523–529.
Wang, C. P., Lee, Y. F., Chang, C., and Lee, H. J. (2006). Transactivation of the proximal promoter of human oxytocin gene by TR4 orphan receptor. Biochem. Biophys. Res. Commun. 351, 204–208.
Warren, M. P., and Brooks-Gunn, J. (1989). Mood and behavior at adolescence: evidence for hormonal factors. J. Clin. Endocrinol. Metab. 69, 77–83.
Wehrenberg, U., Ivell, R., Jansen, M., Von Goedecke, S., and Walther, N. (1994). Two orphan receptors binding to a common site are involved in the regulation of the oxytocin gene in the bovine ovary. Proc. Natl. Acad. Sci. U.S.A. 91, 1440–1444.
Weihua, Z., Makela, S., Andersson, L. C., Salmi, S., Saji, S., Webster, J. I., Jensen, E. V., Nilsson, S., Warner, M., and Gustafsson, J. A. (2001). A role for estrogen receptor beta in the regulation of growth of the ventral prostate. Proc. Natl. Acad. Sci. U.S.A. 98, 6330–6335.
Weihua, Z., Warner, M., and Gustafsson, J. A. (2002). Estrogen receptor beta in the prostate. Mol. Cell. Endocrinol. 193, 1–5.
Weiser, M. J., and Handa, R. J. (2009). Estrogen impairs glucocorticoid dependent negative feedback on the hypothalamic-pituitary-adrenal axis via estrogen receptor alpha within the hypothalamus. Neuroscience 159, 883–895.
Weiser, M. J., Wu, T. J., and Handa, R. J. (2009). Estrogen receptor-beta agonist diarylpropionitrile: biological activities of R- and S-enantiomers on behavior and hormonal response to stress. Endocrinology 150, 1817–1825.
Whitnall, M. H. (1988). Distributions of pro-vasopressin expressing and pro-vasopresin deficient CRH neurons in the paraventricular hypothalamic nucleus of colchicine-treated normal and adrenalectomized rats. J. Comp. Neurol. 275, 13–28.
Williamson, M., Bingham, B., Gray, M., Innala, L., and Viau, V. (2010). The medial preoptic nucleus integrates the central influences of testosterone on the paraventricular nucleus of the hypothalamus and its extended circuitries. J. Neurosci. 30, 11762–11770.
Williamson, M., and Viau, V. (2007). Androgen receptor expressing neurons that project to the paraventricular nucleus of the hypothalamus in the male rat. J. Comp. Neurol. 503, 717–740.
Windle, R. J., Shanks, N., Lightman, S. L., and Ingram, C. D. (1997). Central oxytocin administration reduces stress-induced corticosterone release and anxiety behavior in rats. Endocrinology 138, 2829–2834.
Keywords: corticotropin releasing hormone, vasopressin, oxytocin, estrogen receptor beta, stress, androgen, paraventricular nucleus, 3β-Diol
Citation: Handa RJ, Sharma D and Uht R (2011) A role for the androgen metabolite, 5alpha androstane 3beta, 17beta Diol (3β-Diol) in the regulation of the hypothalamo-pituitary–adrenal axis. Front. Endocrin. 2:65. doi: 10.3389/fendo.2011.00065
Received: 01 August 2011;
Paper pending published: 07 September 2011;
Accepted: 13 October 2011;
Published online: 10 November 2011.
Edited by:
Kazuyoshi Tsutsui, Waseda University, JapanReviewed by:
Gustavo M. Somoza, Instituto de Investigaciones Biotecnologicas-Instituto Tecnologico de Chascomus, ArgentinaShaila Mani, Baylor College of Medicine, USA
Copyright: © 2011 Handa, Sharma and Uht. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Robert J. Handa, Department of Basic Medical Sciences, University of Arizona College of Medicine – Phoenix, 425 N. 5th Street, Phoenix, AZ 85004, USA. e-mail:cmhhbmRhQGVtYWlsLmFyaXpvbmEuZWR1