
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 09 September 2011
Sec. Cancer Endocrinology
volume 2 - 2011 | https://doi.org/10.3389/fendo.2011.00027
 Matthias Kroiss1 Miriam Reuss1 Dorothee Kühner1 Sarah Johanssen1 Melanie Beyer1 Martina Zink1 Michaela F. Hartmann2 Vivek Dhir3 Stefan A. Wudy2 Wiebke Arlt3 Silviu Sbiera1 Bruno Allolio1 Martin Fassnacht1*
Matthias Kroiss1 Miriam Reuss1 Dorothee Kühner1 Sarah Johanssen1 Melanie Beyer1 Martina Zink1 Michaela F. Hartmann2 Vivek Dhir3 Stefan A. Wudy2 Wiebke Arlt3 Silviu Sbiera1 Bruno Allolio1 Martin Fassnacht1*The multi-tyrosine kinase inhibitor sunitinib is used in the treatment of several solid tumors. Animal experiments pointed to an adrenotoxic effect of sunitinib. Therefore, we evaluated the expression of key targets of sunitinib in human adrenocortical carcinoma (ACC) tumor samples and investigated its in vitro effects in ACC cell lines. We carried out immunohistochemistry for vascular endothelial growth factor (VEGF) and its receptor (VEGF-R2) in 157 ACC samples and nine normal adrenal glands. VEGF and VEGF-R2 protein were expressed in 72 and 99% of ACC samples, respectively. Using NCI-H295 and SW13 ACC cell lines, we investigated the effects of sunitinib on cell proliferation. Sunitinib reduced dose-dependently cell viability of both NCI-H295 and SW13 cells (SW13: 0.1 μM 96 ± 7%, 1 μM 90 ± 9%*, 5 μM 62 ± 6%*, controls 100 ± 9%; *p < 0.05). To determine sunitinib effects on steroidogenesis, we measured steroid hormones in cell culture supernatant by gas chromatography–mass spectrometry. We observed a pronounced decrease of cortisol secretion (1 μM 90.1 ± 1.5%*, 5 μM 57.2 ± 0.3%*, controls 100 ± 2.4%) and a concomitant increase in the DHEA/4-androstenedione and 17-hydroxypregnenolone/17-hydroxyprogesterone ratios, indicating specific inhibition of 3β-hydroxysteroid dehydrogenase (HSD3B2). In yeast microsomes transformed with HSD3B2, no direct inhibition of HSD3B2 by sunitinib was detected. Sunitinib induced down-regulation of HSD3B2 mRNA and protein in ACC cell lines (mRNA: 1 μM 44 ± 16%*; 5 μM 22 ± 2%*; 10 μM 19 ± 4%*; protein: 1 μM 82 ± 8%; 5 μM 63 ± 8%*; 10 μM 55 ± 9%*). CYP11B1 was down-regulated at mRNA but not at protein level and CYP11A1 remained unchanged. In conclusion, target molecules of sunitinib are expressed in the vast majority of ACC samples. Sunitinib exhibits anti-proliferative effects in vitro, and appears to specifically block adrenal steroidogenesis by down-regulation of HSD3B2, rendering it a promising option for treatment of ACC.
Sunitinib, a multi-tyrosine kinase inhibitor, has yielded promising results in a number of tumors that respond poorly to conventional chemotherapy. It is currently used, e.g., as a first line therapy in metastasized renal cancer (Motzer et al., 2007) and as a second line treatment in advanced gastrointestinal stroma tumors (Younus et al., 2010). Sunitinib targets several tyrosine kinase receptors, foremost the vascular endothelial growth factor receptors 1 and 2 (VEGF-R1 and 2), but also c-KIT, Fms-like tyrosine kinase 3 (FLT-3), and platelet derived growth factor receptor (PDGF-R; Chow and Eckhardt, 2007; Fassnacht et al., 2009). Thus, the drug combines direct anti-tumor effects with anti-angiogenic activity.
Adrenocortical carcinoma (ACC) is a rare malignancy with an annual incidence of about one per million (Golden et al., 2009). Treatment options are still limited and new therapeutic approaches are urgently needed (Libe et al., 2007; Fassnacht et al., 2011). In one case report, a long lasting response after sunitinib treatment of ACC has been reported (Lee et al., 2009). We were intrigued by the fact that sunitinib has been shown to induce adrenal hemorrhage in animal experiments (Patyna et al., 2008). Furthermore, some patients with other solid tumors who received sunitinib during phase I–III clinical trials developed impaired responsiveness to ACTH indicative of adrenal insufficiency (Goodman et al., 2007; Lodish and Stratakis, 2010), which has resulted in a safety note in the summary of product characteristics (SPC) of sunitinib. These observations suggest that sunitinib may have specific activity in ACC.
To further investigate the putative adrenotoxic effects of sunitinib we analyzed the expression of the key sunitinib targets VEGF and its receptor VEGF-R2 in normal adrenal tissue and ACC tumor samples and tested the effect of sunitinib on cell proliferation in ACC cell lines. As many ACC patients suffer from tumor related steroid hormone excess, and with respect to impaired cortisol secretion in patients treated with sunitinib, we investigated the effects of sunitinib on steroid hormone secretion in the steroid-producing ACC cell line NCI-H295.
Sunitinib was provided by Pfizer (Karlsruhe, Germany) and dissolved in distilled water at pH 7. Forskolin was purchased from Sigma, Karlsruhe, Germany.
We collected a total of 195 adrenocortical tissue samples (163 ACC, 23 adenomas, and nine normal adrenal samples) with informed, written consent by the patients. The study was approved by the Ethics Committee of the University of Würzburg (# 93/02).
The adrenal tumor samples were assembled into three tissue arrays as described previously (Fenske et al., 2009; Ronchi et al., 2009). Immunohistochemical detection was performed using an indirect immunoperoxidase technique after high temperature antigen retrieval in 0.01 M citrate buffer (pH 6.0) for 25 min. Primary antibodies were VEGF-R2 monoclonal mouse antibody (Flk-1), used in a dilution of 1:250 and VEGF antibody A-20 in a dilution of 1:100 (both antibodies from Santa Cruz Biotechnology, Heidelberg, Germany). The slides were incubated with the secondary antibody (BioGenex, Neufahrn, Germany) for 20 min and washed in PBS followed by the application of the chromogen diaminobenzidine. After washing in PBS and water, the slides were counterstained with hematoxylin. As negative controls we employed an IgG isotype antibody and the omission of the primary antibody during immunohistochemical staining. All tissue array slides were analyzed independently by two investigators (Dorothee Kühner and Silviu Sbiera) by assigning staining intensity to the following categories: negative, low, intermediate, or strong. Where discrepancies were observed, results were double checked by both investigators together with a third observer (Martin Fassnacht). Tissue samples with parts of tumor vessels displayed on the slide were evaluated separately for endothelial VEGF-R2 expression in the same manner.
NCI-H295 ACC cells (Gazdar et al., 1990) were grown as monolayers in F12 HAM-medium supplemented with insulin, transferrin, and selenium, 1.2% BSA (Sigma-Aldrich, Deisenhofen, Germany) and 2.5% fetal calf serum (FCS, PAN Biotech, Aidenbach, Germany). The suspension cell variant of these cells was grown in RPMI 1640 medium supplemented with transferrin, insulin, selenium, and 5% FCS at 37°C under an atmosphere of 5% CO2/95% air as described before (Fassnacht et al., 2002, 2003).
The ACC cell line SW13 (Leibovitz et al., 1973) was cultured in F15 medium Leibowitz (Sigma-Aldrich) with 10% FCS and 2 mM L-glutamine (Invitrogen, Darmstadt, Germany) under low CO2 conditions (Hahner et al., 2010).
NCI-H295 ACC cells were plated in six-well-plates at a density of 5 × 105/ml and grown for 2, 4, and 6 days respectively at concentrations of 0 μM (control), 0.1 μM, 1 μM, and 5 μM sunitinib (each in triplicate). Aliquots were mixed 1:2 with trypan blue and cells excluding the dye counted in a Neubauer hemocytometer.
The MTT assay makes use of the conversion of color-less MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] into a blue formazan dye in vital cells only. Dye formation as measured by absorbance at 570 nm hence is a function of viable cell number (Mosmann, 1983).
Adherent NCI-H295 and SW13 cells were plated into 96-well-plates, grown over night and exposed to 0 μM (control), 0.1, 1, 2, 5, and 10 μM sunitinib for 5 days. Culture supernatants were removed and cells washed with PBS. MTT was dissolved in serum free DMEM F10 (0.5 mg/ml) and cells incubated for 4 h at 37°C. Formazan dye was dissolved using 0.04 N HCl/2-propanol and extinction was measured in an ELISA Reader at 570 nm (Fassnacht et al., 2002).
Cortisol and DHEA-S were measured in cell culture supernatant using an Immulite 2000 analyzer (Siemens Medical Solutions, Bad Nauheim, Germany). 17-hydroxyprogesterone (17OHP) and 4-androstenedione were measured with commercially available radioimmunoassay (Beckman-Coulter, Krefeld, Germany and Siemens Medical Solutions).
In addition, 4-androstenedione, cortisol, DHEA, 11-deoxycortisol, 17-hydroxypregnenolone (17OHPreg), and 17-hydroxyprogesterone (17OHP) were determined by isotope dilution gas chromatography–mass spectrometry (GC–MS) as described elsewhere (Wudy et al., 2001; Wudy and Hartmann, 2004). Briefly, cell supernatant was equilibrated with internal standard (deuterium-labeled analogs of analytes). After solvent extraction and purification, heptafluorobutyrate derivatives were prepared. Gas chromatography was carried out on an Agilent 6890 series gas chromatograph housing an OV-1 fused silica capillary (25 m × 0.2 mm, film 0.1 μm; Macherey-Nagel, Düren, Germany) coupled with an Agilent 5973 mass selective detector operated in the selected ion monitoring mode. For computerized quantification, peak areas of specific ions of analytes and their corresponding internal standards were recorded.
Enzyme inhibition experiments were conducted as described previously (Auchus et al., 1998; Arlt et al., 2004). Briefly, yeast microsomes transformed with human wild-type HSD3B2 cDNA were incubated in 50 mM potassium phosphate buffer pH 7.4 containing pregnenolone (10 μM), sodium pyruvate (5 mM), and lactate dehydrogenase (2 IU/ml). [3H]-pregnenolone (55.4 Ci/mol; 10,000 cpm) was added in 4 μl ethanol to a total reaction volume of 200 μl. For inhibition studies, sunitinib (0–5 μM) was added and catalysis initiated by the addition of NAD + (1 mM), with subsequent incubation at 37°C for 30 min which had been previously determined as within the linear range of the enzymatic reaction. Steroids were extracted with 2 ml dichloromethane, concentrated by evaporation at 55°C, and separated by thin layer chromatography on PE SIL G/UV silica gel plates (Whatman, Maidstone, UK), using a 1:4 dichloromethane/ethyl acetate solvent system. The HSD3B2 substrate pregnenolone and the conversion product progesterone were quantified using a Bioscan 2000 image analyzer (LabLogic, Sheffield, UK).
RNA was extracted from cultured cells using the respective RNeasy Kit (Qiagen, Hilden, Germany) according to manufacturer’s manual. Reverse transcription was performed using 500 ng RNA with QuantiTect reverse transcription kit (Qiagen) employing a mixture of random and oligo-dT primers. qRT-PCR was carried out using TaqMan Gene Expression Master Mix (Applied Biosystems, Darmstadt, Germany) and expression normalized to β-actin using the ΔΔCt-method. mRNA of β-actin was demonstrated to be expressed at similar levels in NCI-H295 and SW13 cells (Ct-values 22.35 ± 0.19 for NCI-H295 vs. 22.14 ± 0.023 in SW13, n = 3, p = 0.12 for difference in unpaired Student’s t-test) and hence chosen as reference gene. β-actin primer sequences were: 5′-tct aca atg agc tgc gtg tg-3′ (forward), 5′-cag agg cgt aca ggg ata gc-3′ (reverse). For the target transcripts, commercial probes from Applied Biosystems were used (HSD3B2: HS00605123_M1, CYP11B1: HS01596404_M1, CYP11A1: HS00167984_M1).
A number of 106 cells were lysed directly in 100 μl SDS-sample buffer by heating to 96°C. Samples were then separated on a discontinuous SDS-PAGE with 5% stacking gel and 10% separation gel in a Mini Protean II chamber (Bio-Rad, Munich, Germany). Blotting was done onto Hybond ECL nitrocellulose membrane (GE Healthcare, Munich, Germany) which was then blocked with 5% skim milk powder and 0.1% Tween 20 in Tris buffered saline pH 8.0. Primary polyclonal rabbit antibody against HSD3B2 (R1484) was a generous gift from Ian Mason, Edinburgh and used in a dilution of 1:2500 (Thomas et al., 2002). CYP11B1 antibody was obtained from Hiroshi Takemori, National Institute of Biomedical Innovation, Osaka, Japan (Nonaka et al., 1995) and employed diluted 1:1000. Secondary antibody was anti-rabbit IgG conjugated to horse radish peroxidase (HRP) from GE Healthcare diluted 1:1000. Chemoluminescence reaction was initiated by adding ECL Western Blotting Detection System reagent (GE Healthcare) reagent. The membrane was exposed for 1–5 s to Fuji Medical X-ray film and developed automatically. Quantification of the optical density was performed after digital capturing with BioDoc Analyze (Biometra, Göttingen, Germany) using the shareware program JImage (NIH, Bethesda, MD, USA) and normalized to β-actin staining as a loading control (anti-β-actin: Sigma A5441, anti-mouse IgG HRP-conjugate, GE Healthcare).
All assays were performed in at least three independent triplicate experiments. Data are presented as means ± SD. One-way ANOVA with Tukey’s post hoc test was done using GraphPad Prism version 3.00 for Windows (GraphPad Software, San Diego, CA, USA). Figures show one representative experiment.
Utilizing a tissue array we examined the protein expression of VEGF and VEGF-R2 by immunohistochemistry (Figure 1). VEGF expression was strong, intermediate, and low in 11, 45, and 57, respectively of 157 evaluable ACC samples while no expression was detected in 44 of 157 (28%) samples. Seven of nine normal adrenal glands showed low VEGF protein expression while in the two remaining samples no specific staining was visible.
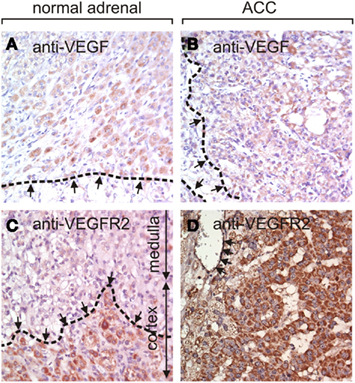
Figure 1. Immunohistochemistry of VEGF and VEGF-R2 in normal adrenal glands and ACC. (A) An example of a normal adrenal gland (zona fasciculata, dashed line) with low VEGF expression. Approximately 30% of ACC exhibit intermediate [(B), borders of VEGF-immunoreactivity dashed], or strong VEGF staining. VEGF-R2 is expressed in the cortex of normal adrenal gland but not in the medulla [(C), border of cortex with dashed line]. ACC frequently express high levels of VEGF-R2 in the tumor cells as well as in the endothelial cells (arrows) as shown in (D). The magnification in all images is 400×.
VEGF-R2 protein expression was strong in 40, intermediate in 74, low in 35, and absent in 2 of 151 evaluable samples. 113 tumor samples were evaluable for endothelial staining. Strong, intermediate, and low VEGF-R2 staining was found in the endothelium of 9, 68, and 21, respectively, whereas 15 samples (13%) did not stain for VEGF-R2. In the nine normal adrenal gland samples we found intermediate (n = 1), low (n = 7), or absent (n = 1) VEGF-R2 expression. Taken together, expression levels of VEGF and VEGF-R2 proteins are generally higher in ACC than in normal adrenal glands.
In order to determine the effect of sunitinib on in vitro proliferation of ACC cells, we first performed dye exclusion assays of NCI-H295 cells. A moderate, dose-dependent inhibition of cell proliferation was observed at sunitinib treatment (Figure 2A). Independently, dose-dependent inhibition of proliferation by sunitinib was confirmed by employing MTT testing in NCI-H295 and SW13 cells (Figure 2B).
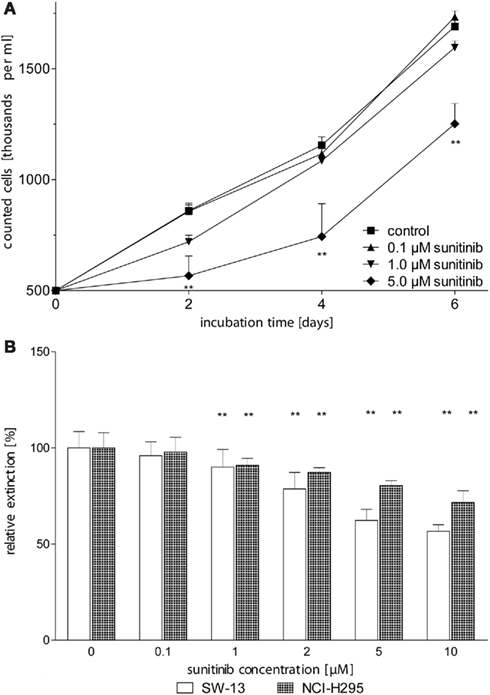
Figure 2. Sunitinib inhibits proliferation of ACC cell lines in a dose-dependent manner. (A) NCI-H295 cells were counted after 2, 4, and 6 days of incubation with up to 5 μM sunitinib. After 2 days of incubation, we observed a dose-dependent reduction of viable cells to 99.6 ± 7.5% (0.1 μM sunitinib, n.s.), 84 ± 6% (1 μM, n.s.), 66 ± 10%* (5 μM) as compared to untreated control incubations (100 ± 5.2%). (B) After 5 days of incubation, viable cells were determined using the MTT assay. Viable NCI-H295 cells were reduced to 98 ± 8% (0.1 μM, n.s.), 91 ± 4%** (1 μM), and 80 ± 3%** (5 μM) compared to controls (100 ± 8%). Similarly, SW13 vital cells declined to 96 ± 7% (0.1 μM), 90.0 ± 9.2%** (1 μM), and 62 ± 6%** (5 μM) compared to control (100 ± 9%). *p < 0.05, **p < 0.01 compared to control.
To test whether sunitinib impacts on steroidogenesis in ACC cell lines, we analyzed steroid concentrations in cell culture supernatants. To account for the cytotoxic effect described, results were normalized for cell numbers. Of note, cortisol changed as a function of sunitinib concentration to 103 ± 10% (0.1 μM, n.s.), 96 ± 7% (1 μM, n.s.), 73 ± 15% (5 μM, p < 0.05) compared to untreated controls (100 ± 6%) after 4 days of incubation. Conversely, DHEA-S increased to 110 ± 10% (0.1 μM), 156 ± 14% (1 μM), 189 ± 426% (5 μM, p < 0.01) as compared to untreated cells (100 ± 12%).
Next we asked whether these changes could be overcome by stimulation of steroidogenesis with the activator of adenylate cyclase forskolin (FSK, Figure 3). Three days of simultaneous incubation with 10 μM FSK led to a nearly 2.5-fold increase of cortisol secretion compared to control, whereas stimulation of androgen/precursor secretion was statistically not significant. The addition of 5 μM sunitinib decreased cortisol close to baseline concentrations of unstimulated cells. In contrast, production of precursor steroids and androgens was strongly increased. Hence we concluded that sunitinib leads to a specific inhibition of cortisol synthesis and accumulation of androgens and their precursors also after stimulation of steroidogenesis by FSK.
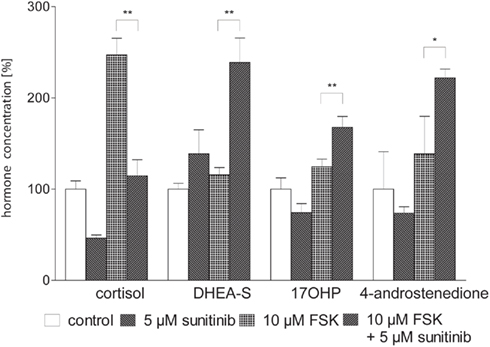
Figure 3. Sunitinib specifically alters basal and forskolin (FSK) stimulated steroidogenesis in NCI-H295 cells. Cortisol and androgen precursor secretion was assessed in cell culture supernatants of NCI-H295 cells after 3 days of incubation with sunitinib and/or FSK employing immunoassays. Representative data from one triplicate experiment are shown. 10 μM FSK led to an increase of cortisol secretion to 247.1 ± 18.3% compared to control (100 ± 9%, p < 0.01). Stimulation of androgen and precursor steroid secretion was statistically not significant (DHEA-S 116 ± 8%, control 100 ± 6%; 17OHP 125 ± 8%, control 100 ± 12%; 4-androstenedione 139 ± 41%, control 100 ± 18%). Co-incubation with 10 μM FSK and 5 μM sunitinib nearly completely reversed FSK-induced stimulation of cortisol secretion to 115 ± 18%** of unstimulated cells whereas production of precursor steroids and androgens was strongly increased (DHEA-S 238.8 ± 26.8%**, 17OHP 168 ± 12%**, 4-androstenedione 222 ± 10%*). *p < 0.05, **p < 0.01 compared to FSK alone.
To assess steroid hormone production more comprehensively we next conducted isotope dilution GC–MS profiling of cell culture supernatants with and without sunitinib (Figure 4A). In line with our previous results obtained with immunoassays, cortisol concentration exhibited a strong dose-dependent decrease. DHEA was found to be strongly enriched and we likewise noted a moderate increase in 4-androstenedione and 17OHP. In contrast, deoxycortisol, 17OHP, and testosterone remained unchanged.
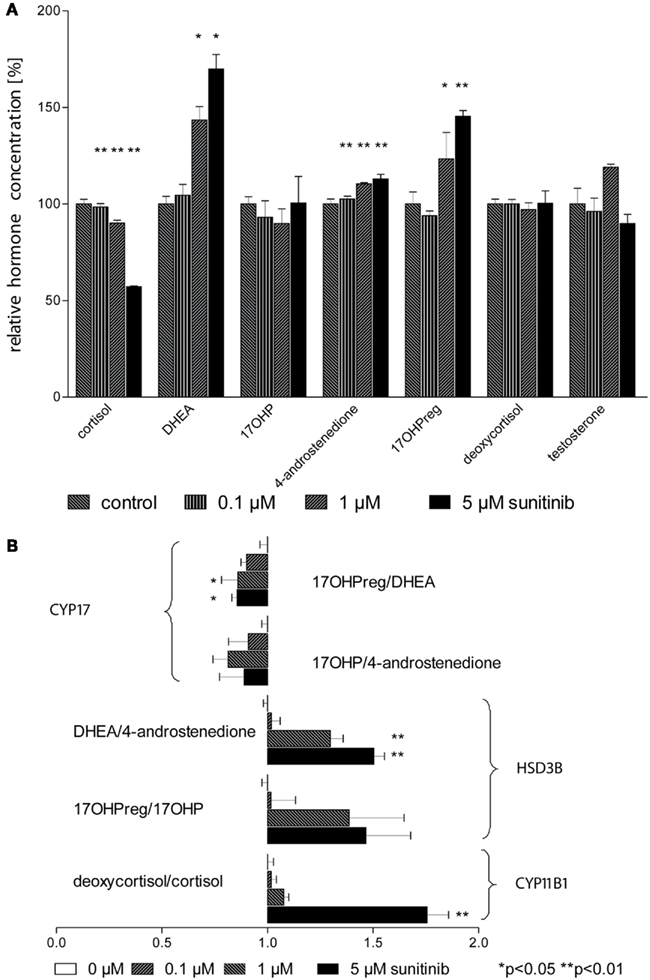
Figure 4. Sunitinib leads to characteristic changes in steroid profiles. (A) Isotope dilution GC–MS was used to study changes in steroid profiles of NCI-H295 cell culture supernatants after incubation with sunitinib for 4 days. Individual values were normalized to control cells (100%). Cortisol concentration was diminished to 98 ± 2%** (0.1 μM), 90 ± 2%** (1 μM), and 57 ± 0.3%** (5 μM) of control (100 ± 2%) but DHEA increased to 105 ± 6%, 143 ± 7%*, and 170 ± 8%* respectively compared to control incubations (100.0 ± 3.9%). However, a less pronounced increase was found for 4-androstenedione to 103 ± 2%**, 111 ± 1%**, and 113 ± 2%** (4-androstenedione, control 100 ± 3%) and for 17OHPreg to 94 ± 2% (n.s.), 123.3 ± 13.7%* (1 μM), and 145.4 ± 3.0%** (5 μM). (B) Steroid ratios were calculated for four characteristic substrates and products of HSD3B (3β-hydroxylase), CYP17 (17,20-lyase), and CYP11B1 (11β-hydroxylase). *p < 0.05, **p < 0.01 compared to control.
From these data, we calculated substrate/product ratios in order to define enzyme activities affected by sunitinib (Figure 4B). We found that the ratios DHEA/4-androstenedione, 17OHPreg/17OHP, and deoxycortisol/cortisol were significantly increased, pointing to specific inhibition of the steroidogenic enzymes 3β-hydroxysteroid dehydrogenase (HSD3B2) and 11β-hydroxylase (CYP11B1). Importantly, the ratios 17OHPreg/DHEA and 17OHP/4-androstenedione indicative of CYP17 activity were not altered.
In order to investigate whether sunitinib directly inhibits HSD3B2, we used isolated yeast microsomes transformed with human HSD3B2. Sunitinib did not exhibit any significant inhibition of HSD3B2 activity across the concentration range of 1–5 μM sunitinib in these enzymatic assays: 100 ± 6% (0 μM), 99 ± 2% (1 μM), 94 ± 2% (5 μM).
We therefore hypothesized that sunitinib may alter enzyme activities at the transcriptional level. Using quantitative PCR, we investigated the expression of HSD3B2, CYP11B1 (P450c11), and CYP11A1 (P450scc) mRNA in NCI-H295 cells treated with sunitinib. Whereas no change in expression was observed for CYP11A1 (data not shown), we found a moderate dose-dependent reduction in CYP11B1 after 24 h of incubation compared to control cells (Figure 5A). Interestingly, HSD3B2 mRNA (Figure 5B) exhibited an even more pronounced down-regulation on the mRNA level.
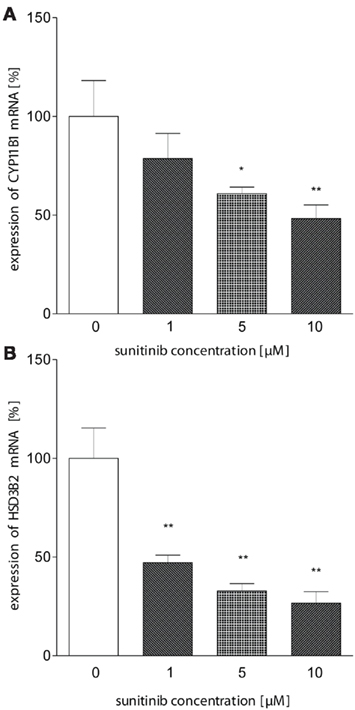
Figure 5. Sunitinib leads to reduced mRNA expression of HSD3B and CYP11B1. (A) CYP11B1 mRNA expression in NCI-H295 cells was investigated using quantitative PCR using β-actin as a standard. We observed a dose-dependent reduction to 78.6 ± 12.7% (1 μM), 61 ± 3%* (5 μM), and 48 ± 7%** (10 μM) compared to control (100 ± 18%). (B) HSD3B2 mRNA was demonstrated to be down-regulated to 47 ± 7%** (1 μM), 33 ± 7%** (5 μM), 27 ± 6%** (10 μM, control: 100 ± 27; p < 0.01). *p < 0.05, **p < 0.01 compared to control.
To further investigate this finding, we conducted immunoblot analysis in sunitinib-treated NCI-H295 cells. Here, we found a similar sunitinib-induced decrease in HSD3B2 protein expression compared to control (Figure 6). However, we did not detect significant changes of CYP11B1 protein expression (data not shown).
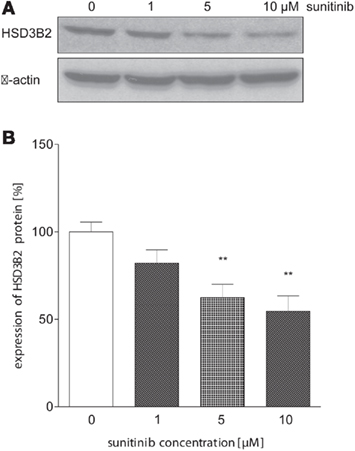
Figure 6. Sunitinib down-regulates HSD3B2 expression at protein level. (A) HSD3B2 protein expression in NCI-H295 cells after 24 h of incubation with sunitinib was assessed by immunoblot analysis using a specific antibody. (B) Densitometric quantification of immunoblot signals from three experiments confirmed a significant sunitinib-induced decrease of HSD3B2 to 82 ± 8% (1 μM, n.s.), 63 ± 8%** (5 μM), and 54.6 ± 8.8%** (10 μM) compared to control cells (100 ± 6%). **p < 0.01 compared to control.
Here we provide evidence for expression of target molecules of sunitinib in ACC samples and demonstrate a direct anti-proliferative effect of sunitinib on ACC cell lines NCI-H295 and SW13. In addition, we show specific inhibition of cortisol synthesis and concomitant accumulation of steroid precursors and androgens caused by down-regulation of HSD3B2 in hormonally active NCI-H295 cells.
We found low expression of VEGF and its receptor in normal adrenal glands whereas 72 and 99% of ACC samples showed expression of VEGF and VEGF-R2, respectively, with 36 and 75% of ACC specimens exhibiting strong or intermediate staining intensity. Of note, both endothelial cells and ACC tumor cells expressed VEGF-R2 in a large proportion of specimens. VEGF expression in both tumor cells and tumor vessels has been reported in one case before (Lee et al., 2009). Our findings indicate that VEGF and VEGF-R2 are up-regulated at protein level in a significant proportion of ACC compared to normal adrenal glands. In addition, expression of VEGF-R2 has been demonstrated in SW13 and NCI-H295 cells by immunoblotting (Mariniello et al., 2011). The expression of one of the key targets of sunitinib, VEGF-R2 and its ligand VEGF in ACC makes sunitinib a candidate molecule for further clinical investigation.
In both dye exclusion assays and MTT tests, sunitinib incubation resulted in a dose dependent, moderate but significant decrease of cell proliferation. Both assays cannot distinguish whether this is due to increased cell death or reduced cell proliferation. However, these data demonstrate a direct cytostatic effect of the drug on ACC cells, which is similar to those observed in other sunitinib-treated tumor cell lines in vitro (de Boüard et al., 2007) and which have been reported recently in ACC-cells with sorafenib (Mariniello et al., 2011). However, in vivo, sunitinib might affect ACC tumor growth more effectively by also targeting tumor-associated angiogenesis.
Using immunoassays and GC–MS, we found a specific effect of sunitinib on steroidogenesis, with a significant decrease of cortisol whereas 17OHPreg, 4-androstenedione, and DHEA-S concentrations increased. Furthermore, maximum stimulation of steroid biosynthesis by protein kinase A (PKA) activation with forskolin was effectively blocked by sunitinib. Calculation of substrate/product ratios clearly indicated that sunitinib specifically reduces HSD3B2 activity, resulting in the observed increase in androgens and decrease in cortisol. After exclusion of a direct inhibition of HSD3B2 in a yeast microsome assay, significant down-regulation of HSD3B2 was observed using quantitative PCR. We confirmed that HSD3B2 was reduced at the protein level. Altered mRNA stability (Balagopal and Parker, 2009) may account for HSD3B2 mRNA down-regulation but these processes have not been investigated in any detail for transcripts involved in steroidogenesis.
By contrast, transcriptional regulation of HSD3B2 in the adrenal gland has been studied using a variety of experimental settings. Key transcription factors include members of the NR5A (steroidogenic factor 1, SF1 and Liver Receptor Homolog 1, LRH-1) and the NR4A family (Nur77, Nur related protein 1, Nurr1, and neuron-derived orphan receptor 1, Nor1; Hammer et al., 2005). It appears that SF1 accounts for the basal transcriptional activity of HSD3B2 in NCI-H295 cells and can be stimulated with phorbol esters indicating a protein kinase C (PKC) dependent pathway (Leers-Sucheta et al., 1997). Further, it plays a pivotal role in adrenocortical cell proliferation and cancer (Beuschlein et al., 2002; Doghman et al., 2007; Lalli, 2010; Sbiera et al., 2010). On the other hand, Nur77 has been proposed to orchestrate HSD3B2 expression in response to hormonal stimulation via PKA (Bassett et al., 2004; Martin and Tremblay, 2005).
With respect to upstream pathways, insulin-like growth factors IGFI and II have been found to stimulate expression of CYP17 and also HSD3B2 in human adult adrenal fasciculata–reticularis cells (l’Allemand et al., 1996). Similarly to our observations, a pharmacologic study demonstrated reduced secretion of cortisol and increased DHEA and DHEA-S production in NCI-H295 cells upon inhibition of Src tyrosine kinase with the specific inhibitor PP2 (Sirianni et al., 2003). Induction of steroidogenic acute regulatory protein (StAR), CYP11A1, and 17a-hydroxylase/17,20-lyase (CYP17) were found to mediate these effects.
Although sunitinib and the Src-inhibitor PP2 finally lead to similar changes in steroid production, we did not find increased CYP11A1 expression indicating that the majority of sunitinib effects is transduced through a different pathway. In fact it is likely that the observed changes in hormone secretion result from alterations in more than one signaling pathway taking into account that VEGF-R2 is only one among several tyrosine kinases targeted by sunitinib. In addition, cross-talk to PKA and/or PKC dependent pathways is suggested by the results of forskolin stimulation.
Our study has obvious limitations: Since only one steroid hormone producing ACC cell line is currently available, we cannot exclude that the described effect on the steroidogenesis is cell line specific and not a general effect of sunitinib. However, NCI-H295 cell line has been firmly established as a model for hormonally active ACC (Rainey et al., 1994; Rainey and Nakamura, 2008). In our in vitro setting, the most pronounced effect was seen using a sunitinib concentration of 5 μM and more. In humans, administration of 50 mg sunitinib per day leads to plasma concentration of 50–100 ng/ml, corresponding to 0.1–0.2 μM (Faivre et al., 2006). Thus we administered sunitinib in a presumably “supra-therapeutical” concentration. In humans, sunitinib is metabolized in the intestine and liver by CYP3A4 monooxygenase to first form the active metabolite SU12662 which is then inactivated in a second CYP3A4-mediated step (Faivre et al., 2006). CYP3A4-mediated inactivation of sunitinib in SW13 and NCI-H295 cells in vitro is unlikely, since we found no expression of this enzyme using qRT-PCR (data not shown). However, it is well known that in vitro much higher concentrations are needed to yield a given effect compared to the situation in vivo. Accordingly, published cell culture data for sunitinib support that 0.1–10 μM are appropriate concentrations to mimic putative in vivo effects (de Boüard et al., 2007; Salem et al., 2008). Moreover, anti-angiogenic effects of sunitinib are supposed to enhance the anti-tumoral effects of the drug in vivo.
From a clinical perspective, our in vitro results support the possibility of sunitinib-induced adrenal insufficiency, primarily by inhibition of HSD3B2 transcription. Symptoms of adrenal insufficiency such as prostration, stiffness of joints or even gastrointestinal disturbances like vomiting and nausea may be easily attributed to non-specific side effects of sunitinib or the malignant disease itself. Although clinically evident adrenal insufficiency appears to be a rare event in patients treated with sunitinib (Lodish and Stratakis, 2010), physicians should be aware that sunitinib may unmask clinically inapparent adrenal insufficiency due to concomitant treatment with other drugs influencing steroidogenesis (e.g., ketoconazole, fluconazole) or heterozygous congenital adrenal hyperplasia. On the other hand, glucocorticoid excess is a hallmark of ACC leading to a high morbidity and in this situation inhibition of steroidogenesis will be a desired effect. Taken together, our results indicate that sunitinib may have significant therapeutic potential in the treatment of ACC, which should be explored further in clinical studies, e.g., in the ongoing Sunitinib in refractory adrenocortical carcinoma (SIRAC) trial.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are very grateful to Hiroshi Takemori and Ian Mason for generous gifts of antibodies and to Pfizer Oncology for providing sunitinib. We thank Patrick Adam (Institute of Pathology, Würzburg) for helping with the immunohistochemistry analysis. This study was supported by grants of the Deutsche Krebshilfe (grant # 107111 to Martin Fassnacht), the German Research Foundation DFG (grant # FA 466/3-1 to Martin Fassnacht), and the German Ministry of Research (grant 01KG0501 to Bruno Allolio and Martin Fassnacht). Wiebke Arlt is supported by the Medical Research Council UK (G0801473).
Arlt, W., Neogi, P., Gross, C., and Miller, W. L. (2004). Cinnamic acid based thiazolidinediones inhibit human P450c17 and 3beta-hydroxysteroid dehydrogenase and improve insulin sensitivity independent of PPARgamma agonist activity. J. Mol. Endocrinol. 32, 425–436.
Auchus, R. J., Lee, T. C., and Miller, W. L. (1998). Cytochrome b5 augments the 17,20-lyase activity of human P450c17 without direct electron transfer. J. Biol. Chem. 273, 3158–3165.
Balagopal, V., and Parker, R. (2009). Polysomes, P bodies and stress granules: states and fates of eukaryotic mRNAs. Curr. Opin. Cell Biol. 21, 403–408.
Bassett, M. H., Suzuki, T., Sasano, H., De Vries, C. J., Jimenez, P. T., Carr, B. R., and Rainey, W. E. (2004). The orphan nuclear receptor NGFIB regulates transcription of 3beta-hydroxysteroid dehydrogenase. implications for the control of adrenal functional zonation. J. Biol. Chem. 279, 37622–37630.
Beuschlein, F., Mutch, C., Bavers, D. L., Ulrich-Lai, Y. M., Engeland, W. C., Keegan, C., and Hammer, G. D. (2002). Steroidogenic factor-1 is essential for compensatory adrenal growth following unilateral adrenalectomy. Endocrinology 143, 3122–3135.
Chow, L. Q., and Eckhardt, S. G. (2007). Sunitinib: from rational design to clinical efficacy. J. Clin. Oncol. 25, 884–896.
de Boüard, S., Herlin, P., Christensen, J., Lemoisson, E., Gauduchon, P., Raymond, E., and Guillamo, J. (2007). Antiangiogenic and anti-invasive effects of sunitinib on experimental human glioblastoma. Neuro-oncology 9, 412–423.
Doghman, M., Karpova, T., Rodrigues, G. A., Arhatte, M., De Moura, J., Cavalli, L. R., Virolle, V., Barbry, P., Zambetti, G. P., Figueiredo, B. C., Heckert, L. L., and Lalli, E. (2007). Increased steroidogenic factor-1 dosage triggers adrenocortical cell proliferation and cancer. Mol. Endocrinol. 21, 2968–2987.
Faivre, S., Delbaldo, C., Vera, K., Robert, C., Lozahic, S., Lassau, N., Bello, C., Deprimo, S., Brega, N., Massimini, G., Armand, J., Scigalla, P., and Raymond, E. (2006). Safety, pharmacokinetic, and antitumor activity of SU11248, a novel oral multitarget tyrosine kinase inhibitor, in patients with cancer. J. Clin. Oncol. 24, 25–35.
Fassnacht, M., Franke, A., Dettling, A., Hahner, S., Zink, M., Wudy, S., and Allolio, B. (2002). Clodronate inhibits adrenocortical cell proliferation and P450c21 activity. J. Endocrinol. 174, 509–516.
Fassnacht, M., Hahner, S., Hansen, I. A., Kreutzberger, T., Zink, M., Adermann, K., Jakob, F., Troppmair, J., and Allolio, B. (2003). N-terminal proopiomelanocortin acts as a mitogen in adrenocortical tumor cells and decreases adrenal steroidogenesis. J. Clin. Endocrinol. Metab. 88, 2171–2179.
Fassnacht, M., Kreissl, M. C., Weismann, D., and Allolio, B. (2009). New targets and therapeutic approaches for endocrine malignancies. Pharmacol. Ther. 123, 117–141.
Fassnacht, M., Libé, R., Kroiss, M., Allolio, B., and Medscape. (2011). Adrenocortical carcinoma: a clinician’s update. Nat. Rev. Endocrinol. 7, 323–335.
Fenske, W., Volker, H. U., Adam, P., Hahner, S., Johanssen, S., Wortmann, S., Schmidt, M., Morcos, M., Muller-Hermelink, H. K., Allolio, B., and Fassnacht, M. (2009). Glucose transporter GLUT1 expression is an stage-independent predictor of clinical outcome in adrenocortical carcinoma. Endocr. Relat. Cancer 16, 919–928.
Gazdar, A. F., Oie, H. K., Shackleton, C. H., Chen, T. R., Triche, T. J., Myers, C. E., Chrousos, G. P., Brennan, M. F., Stein, C. A., and La Rocca, R. V. (1990). Establishment and characterization of a human adrenocortical carcinoma cell line that expresses multiple pathways of steroid biosynthesis. Cancer Res. 50, 5488.
Golden, S. H., Robinson, K. A., Saldanha, I., Anton, B., and Ladenson, P. W. (2009). Clinical review: prevalence and incidence of endocrine and metabolic disorders in the United States: a comprehensive review. J. Clin. Endocrinol. Metab. 94, 1853–1878.
Goodman, V., Rock, E., Dagher, R., Ramchandani, R., Abraham, S., Gobburu, J., Booth, B., Verbois, S., Morse, D., Liang, C., Chidambaram, N., Jiang, J., Tang, S., Mahjoob, K., Justice, R., and Pazdur, R. (2007). Approval summary: sunitinib for the treatment of imatinib refractory or intolerant gastrointestinal stromal tumors and advanced renal cell carcinoma. Clin. Cancer Res. 13, 1367–1373.
Hahner, S., Sturmer, A., Fassnacht, M., Hartmann, R. W., Schewe, K., Cochran, S., Zink, M., Schirbel, A., and Allolio, B. (2010). Etomidate unmasks intraadrenal regulation of steroidogenesis and proliferation in adrenal cortical cell lines. Horm. Metab. Res. 42, 528–534.
Hammer, G. D., Parker, K. L., and Schimmer, B. P. (2005). Minireview: transcriptional regulation of adrenocortical development. Endocrinology 146, 1018–1024.
l’Allemand, D., Penhoat, A., Lebrethon, M. C., Ardevol, R., Baehr, V., Oelkers, W., and Saez, J. M. (1996). Insulin-like growth factors enhance steroidogenic enzyme and corticotropin receptor messenger ribonucleic acid levels and corticotropin steroidogenic responsiveness in cultured human adrenocortical cells. J. Clin. Endocrinol. Metab. 81, 3892–3897.
Lalli, E. (2010). Adrenocortical development and cancer: focus on SF-1. J. Mol. Endocrinol. 44, 301–307.
Lee, J. O., Lee, K. W., Kim, C. J., Kim, Y. J., Lee, H. E., Kim, H., Kim, J. H., Bang, S. M., Kim, J. S., and Lee, J. S. (2009). Metastatic adrenocortical carcinoma treated with sunitinib: a case report. Jpn. J. Clin. Oncol. 39, 183–185.
Leers-Sucheta, S., Morohashi, K., Mason, J. I., and Melner, M. H. (1997). Synergistic activation of the human type II 3beta-hydroxysteroid dehydrogenase/delta5-delta4 isomerase promoter by the transcription factor steroidogenic factor-1/adrenal 4-binding protein and phorbol ester. J. Biol. Chem. 272, 7960–7967.
Leibovitz, A., McCombs, W. M. 3rd, Johnston, D., McCoy, C. E., and Stinson, J. C. (1973). New human cancer cell culture lines. I. SW-13, small-cell carcinoma of the adrenal cortex. J. Natl. Cancer Inst. 51, 691–697.
Libe, R., Fratticci, A., and Bertherat, J. (2007). Adrenocortical cancer: pathophysiology and clinical management. Endocr. Relat. Cancer 14, 13–28.
Lodish, M. B., and Stratakis, C. A. (2010). Endocrine side effects of broad-acting kinase inhibitors. Endocr. Relat. Cancer 17, R233–R244.
Mariniello, B., Rosato, A., Zuccolotto, G., Rubin, B., Pezzani, R., Cicala, M. V., Iacobone, M., Fassina, A., and Mantero, F. (2011). Combination of sorafenib and everolimus impacts therapeutically on adrenal tumor models. Endocr. Rev. 32, OR13.
Martin, L. J., and Tremblay, J. J. (2005). The human 3beta-hydroxysteroid dehydrogenase/Delta5-Delta4 isomerase type 2 promoter is a novel target for the immediate early orphan nuclear receptor Nur77 in steroidogenic cells. Endocrinology 146, 861–869.
Mosmann, T. (1983). Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 65, 55–63.
Motzer, R. J., Hutson, T. E., Tomczak, P., Michaelson, M. D., Bukowski, R. M., Rixe, O., Oudard, S., Negrier, S., Szczylik, C., Kim, S. T., Chen, I., Bycott, P. W., Baum, C. M., and Figlin, R. A. (2007). Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N. Engl. J. Med. 356, 115–124.
Nonaka, Y., Takemori, H., Halder, S. K., Sun, T., Ohta, M., Hatano, O., Takakusu, A., and Okamoto, M. (1995). Frog cytochrome P-450 (11 beta,aldo), a single enzyme involved in the final steps of glucocorticoid and mineralocorticoid biosynthesis. Eur. J. Biochem. 229, 249–256.
Patyna, S., Arrigoni, C., Terron, A., Kim, T. W., Heward, J. K., Vonderfecht, S. L., Denlinger, R., Turnquist, S. E., and Evering, W. (2008). Nonclinical safety evaluation of sunitinib: a potent inhibitor of VEGF, PDGF, KIT, FLT3, and RET receptors. Toxicol. Pathol. 36, 905–916.
Rainey, W., Bird, I., and Mason, J. (1994). The NCI-H295 cell line: a pluripotent model for human adrenocortical studies. Mol. Cell. Endocrinol. 100, 45–50.
Rainey, W., and Nakamura, Y. (2008). Regulation of the adrenal androgen biosynthesis. J. Steroid Biochem. Mol. Biol. 108, 281–286.
Ronchi, C. L., Sbiera, S., Kraus, L., Wortmann, S., Johanssen, S., Adam, P., Willenberg, H. S., Hahner, S., Allolio, B., and Fassnacht, M. (2009). Expression of excision repair cross complementing group 1 and prognosis in adrenocortical carcinoma patients treated with platinum-based chemotherapy. Endocr. Relat. Cancer 16, 907–918.
Salem, A. K., Fenton, M. S., Marion, K. M., and Hershman, J. M. (2008). Effect of sunitinib on growth and function of FRTL-5 thyroid cells. Thyroid 18, 631.
Sbiera, S., Schmull, S., Assie, G., Voelker, H. U., Kraus, L., Beyer, M., Ragazzon, B., Beuschlein, F., Willenberg, H. S., Hahner, S., Saeger, W., Bertherat, J., Allolio, B., and Fassnacht, M. (2010). High diagnostic and prognostic value of steroidogenic factor-1 expression in adrenal tumors. J. Clin. Endocrinol. Metab. 95, E161–E171.
Sirianni, R., Carr, B. R., Ando, S., and Rainey, W. E. (2003). Inhibition of Src tyrosine kinase stimulates adrenal androgen production. J. Mol. Endocrinol. 30, 287–299.
Thomas, J. L., Mason, J. I., Brandt, S., and Norris, W. (2002). Differences in substrate and inhibitor kinetics of human type 1 and type 2 3beta-hydroxysteroid dehydrogenase are explained by the type 1 mutant, H156Y. Endocr. Res. 28, 471–475.
Wudy, S. A., Hartmann, M., Solleder, C., and Homoki, J. (2001). Determination of 17alpha-hydroxypregnenolone in human plasma by routine isotope dilution mass spectrometry using benchtop gas chromatography-mass selective detection. Steroids 66, 759–762.
Wudy, S. A., and Hartmann, M. F. (2004). Gas chromatography-mass spectrometry profiling of steroids in times of molecular biology. Horm. Metab. Res. 36, 415–422.
Keywords: adrenal cancer, tyrosine kinase inhibitor, 3β-hydroxysteroid dehydrogenase
Citation: Kroiss M, Reuss M, Kühner D, Johanssen S, Beyer M, Zink M, Hartmann MF, Dhir V, Wudy SA, Arlt W, Sbiera S, Allolio B and Fassnacht M (2011) Sunitinib inhibits cell proliferation and alters steroidogenesis by down-regulation of HSD3B2 in adrenocortical carcinoma cells. Front. Endocrin. 2:27. doi: 10.3389/fendo.2011.00027
Received: 30 March 2011; Accepted: 18 August 2011;
Published online: 09 September 2011.
Edited by:
Stephen Plymate, University of Washington, USAReviewed by:
Ettore C. Degli Uberti, University of Ferrara, ItalyCopyright: © 2011 Kroiss, Reuss, Kühner, Johanssen, Beyer, Zink, Hartmann, Dhir, Wudy, Arlt, Sbiera, Allolio and Fassnacht. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Martin Fassnacht, Department of Internal Medicine I, University Hospital Würzburg, Oberdürrbacher Street 6, 97080 Würzburg, Germany. e-mail:ZmFzc25hY2h0X21Aa2xpbmlrLnVuaQ==- wuerzburg.de
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.