
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 16 May 2011
Sec. Cancer Endocrinology
volume 2 - 2011 | https://doi.org/10.3389/fendo.2011.00013
 Claire M. Perks* Carla Burrows
Claire M. Perks* Carla Burrows Jeff M. P. Holly
Jeff M. P. Holly
We established previously that IGFBP-3 could exert positive or negative effects on cell function depending upon the extracellular matrix composition and by interacting with integrin signaling. To elicit its pro-apoptotic effects IGFBP-3 bound to caveolin-1 and the beta 1 integrin receptor and increased their association culminating in MAPK activation. Disruption of these complexes or blocking the beta 1 integrin receptor reversed these intrinsic actions of IGFBP-3. In this study we have examined the signaling pathway between integrin receptor binding and MAPK activation that mediates the intrinsic, pro-apoptotic actions of IGFBP-3. We found on inhibiting protein kinase A (PKA), Rho associated kinase (ROCK), and ceramide, the accentuating effects of IGFBP-3 on apoptotic triggers were reversed, such that IGFBP-3 then conferred cell survival. We established that IGFBP-3 activated Rho, the upstream regulator of ROCK and that beta1 integrin and PKA were upstream of Rho activation, whereas the involvement of ceramide was downstream. The beta 1 integrin, PKA, Rho, and ceramide were all upstream of MAPK activation. These data highlight key components involved in the pro-apoptotic effects of IGFBP-3 and that inhibiting them leads to a reversal in the action of IGFBP-3.
The IGFs are present in the body almost entirely in association with specific binding proteins of which there are a family of six closely related but distinct proteins (IGFBPs 1–6): IGFBP-3 being the major IGF binding protein in human serum. IGFBPs-3 and -5 are the most structurally similar IGFBPs and share a number of common features (Jones and Clemmons, 1995; Perks and Holly, 2003). In addition to either enhancing or inhibiting the actions of IGFs, it is now clear that IGFBPs can also have IGF-independent effects on cell function that can also be both positive and negative. IGFBP-3 is known to mediate the growth inhibitory effects of a number of different agents including vitamin D3 and TGF-beta (Perks and Holly, 2003) and inhibits cell growth in human breast cancer cells (Oh et al., 1995). Conversely it has also been reported that IGFBP-3 can promote growth in non-malignant mammary epithelial cells (McCaig et al., 2002a), in colon cancer cells (Kansra et al., 2000), LnCaP prostate cancer cells (Martin and Pattison, 2000), and in human airway smooth muscle cells (Cohen et al., 2000). The intrinsic ability of IGFBP-3 to exert differential effects on cell growth is also reflected in its ability to modulate apoptosis in an IGF-independent manner. There have been many reports that IGFBP-3 can accentuate apoptosis of different cells including prostate (Rajah et al., 1997), breast (Gill et al., 1997), colorectal (Williams et al., 2000), and esophageal (Hollowood et al., 2000) epithelial cells and induce cell death alone in prostate cancer cells (Rajah et al., 1997) but IGFBP-3 can also inhibit apoptosis of non-malignant mammary epithelial cells (McCaig et al., 2002a) and of HUVEC cells (Granata et al., 2004).
In an attempt to determine how IGFBP-3 exerts its intrinsic actions, there was a report of an IGFBP-3 cell surface binding site (Oh et al., 1993) and this has recently been suggested to be a novel receptor for IGFBP-3 that mediates only pro-apoptotic effects (Ingermann et al., 2010). It was also reported that IGFBP-3 associated with the TGF-beta V receptor (Leal et al., 1997). Signal transduction initiated by IGFBP-3 in T47D breast cancer cells, was shown to require TGF-beta receptors and its signaling intermediates, Smad-2 and -3. IGFBP-3 possesses a nuclear localization sequence (Schedlich et al., 1998) which may allow it to enter the nucleus and in vitro studies have shown that it binds to the nuclear retinoid X receptor (RXR; Schedlich et al., 1998; Liu et al., 2000). Translocation of IGFBP-3 to the nucleus and subsequent effects on gene transcription may be a possible mechanism by which IGFBP-3 could exert some IGF-independent actions, but could not however explain the very rapid cellular responses such as IGFBP-3 modulating the phosphorylation status of signaling molecules such as MAPK (Martin et al., 2003; Burrows et al., 2006) and SMAD-2 and -3 (Fanayan et al., 2000) or the very rapid effects on cell adhesion (McCaig et al., 2002b; Martin and Jambazov, 2006).
Cellular responses such as growth, survival, and migration are regulated by soluble growth factors as well as interaction with the extracellular matrix. Integrins are a family of heterodimeric transmembrane receptors, which serve as a link between the extracellular matrix and the intracellular signaling network (Berman et al., 2003). There is evidence implicating the extracellular matrix and integrin signaling in the intrinsic actions of the IGFBPs. Both IGFBP-1 and -2 contain the classical integrin recognition sequence RGD and their cell surface binding has been shown to involve the α5β1 integrin, which results in both FAK and MAPK deactivation in Hs578T breast cancer and A67 sarcoma cells culminating in apoptosis (Jones et al., 1993; Perks et al., 1999b; Schutt et al., 2004).
Although IGFBP-3 does not contain an RGD sequence there is strong evidence to support the involvement of the extracellular matrix and integrins in its intrinsic actions (Perks and Holly, 2003). We have shown previously that the extracellular matrix modulates the IGF-independent actions of IGFBP-3. In breast tumors, disruption of the well-defined, normal ductal tissue architecture results in reduced exposure of epithelial cells to laminin and collagen but increased exposure to fibronectin in the stroma. Increased expression of fibronectin in breast cancers is associated with poor prognosis and metastasis (Oda et al., 1992). We found that on laminin and collagen, IGFBP-3 enhances triggers of apoptosis and reduces attachment of Hs578T cells, but when plated onto fibronectin these effects of IGFBP-3 were reversed and IGFBP-3 then promoted adhesion and acted as a survival factor (McCaig et al., 2002b). We have also recently reported that IGFBP-3 can inhibit EGF-induced growth of breast cancer cells when grown on plastic or laminin but promotes EGF-induced proliferation when cultured on fibronectin (McIntosh et al., 2010). Integrin receptors congregate within cholesterol-stabilized microdomains within the plasma membrane that form platforms for the clustering of cell signaling components (Brown, 2000) and caveolin 1 is a recognized marker of such microdomains. More recently we have shown that IGFBP-3 can bind caveolin-1 and the β1 integrin sub-unit, increase their association and the recruitment of a key integrin signaling molecule, focal adhesion kinase (Burrows et al., 2006). Furthermore, we determined that the critical determinant of whether IGFBP-3 had positive or negative effects on cell function was dependent on the status of such cholesterol-stabilized integrin receptor complexes (Burrows et al., 2006). These data showed that the intrinsic actions of IGFBP-3 depend not only on cell type but also importantly on cell context and this underlies whether IGFBP-3 will exert either a positive or negative effect on cell function. In this study we have tried to delineate further the pro-apoptotic signaling pathway of IGFBP-3, which may provide a better understanding of therapeutic responses to drugs that act in concert with IGFBP-3 and whose actions change with tumor progression.
Recombinant, human, non-glycosylated IGFBP-3 (ngIGFBP-3) was a kind gift from Dr. C. Maack, Celtrix, CA, USA. Recombinant, human, non-glycosylated IGFBP-5 (ngIGFBP-5) was bought from GroPep, Adelaide, SA, Australia. Serine phosphorylation domain peptide (SPD) is a 15 amino acid peptide sequence spanning the two mid-region serines of IGFBP-3 and mimics all the intrinsic actions of full-length IGFBP-3 (Hollowood et al., 2002). It was synthesized at the micro-chemical facility of the Babraham Institute (Cambridge, UK). The ceramide antibody was purchased from Alexis Corporation, UK. A negative control mouse IgM was bought from Dako, Cambridgeshire, UK. The PKA inhibitor, KT5720 was purchased from Tocris, Bristol, UK. The Rho kinase inhibitor ROCK, Y-27632, and C2-ceramide were purchased from Merck, Nottingham, UK. The anti-ERK-2 antibody was bought from Santa Cruz, CA, USA. The anti-phospho-MAP kinase antibody was purchased from Promega, Southampton, UK. The anti-Rho antibody and Rhotekin Rho binding domain bound to glutathione–agarose were purchased from Upstate Biotechnology, NY, USA. The anti-beta 1 integrin blocking antibody was bought from Millipore, Watford, Hertfordshire. All other chemicals including antimycin A, an inhibitor of ceramide synthase, fumonisin B1, an inhibitor of sphingosine kinase, N,N-dimethylsphingosine (DMS), anti-alpha-tubulin primary antibody, and TRITC phalloidin stain were bought from Sigma, Dorset, UK. Tissue culture plastics were obtained from Greiner Labortechnik Ltd., Tyne and Wear, UK. The BCA protein assay reagent kit was purchased from Pierce, Illinois, USA.
Human breast cancer cells, Hs578T were purchased from ECACC (Porton Down, Wiltshire, UK) and grown in a humidified 5% carbon dioxide atmosphere at 37°C. The cells were cultured in Dulbecco’s modified Eagles Medium (DMEM, BioWhittaker, Verviers, Belgium) supplemented with 10% fetal bovine serum (FBS, Gibco, Paisley, UK), penicillin (50 IU/ml; Britannia Pharmaceuticals, Redhill, UK), streptomycin (50 μg/ml; Celltech Pharmaceuticals, Slough, UK), and L-glutamine (2 mM; Sigma, Dorset, UK) – growth media (GM). For all cell lines experiments were performed in phenol red-and serum-free DMEM and Hams Nutrient Mix F12 with sodium bicarbonate (0.12%; Sigma, Dorset, UK), bovine serum albumin (BSA) (0.2 mg/ml; Sigma, Dorset, UK), transferrin (0.01 mg/ml; Sigma, Dorset, UK) and supplemented with penicillin (50 IU/ml), streptomycin (50 μg/ml), and L-glutamine (2 mM) – SFM.
Aliquots of cell suspension were loaded onto a haemocytometer (1:1) with Trypan blue dye. Viable cells exclude the dye. Both living and dead cells were counted from which the percentage of dead cells was calculated.
Hs578T cells were seeded at 0.1 × 106 cells/well in six-well plates in GM and then switched to SFM for 24 h prior to pre-dosing with SPD (10 ng/ml), IGFBP-3 or IGFBP-5 (each at 100 ng/ml) with or without either fumonisin B1 (1 μM), DMS (0.5 μM), a ceramide antibody (1 μg/ml), ROCK (5 μM), or a pre-dose for 30 min with KT5720 (0.5 μM). The cells were then re-dosed with IGFBP-3 or IGFBP-5 (each at 100 ng/ml) for a further 24 h in the presence or absence of an apoptotic dose of either antimycin A or C2-ceramide (doses chosen to give approximately 40–60% cell death) with either of the inhibitors listed above. We have shown previously that the amount of apoptosis as quantified by flow cytometry is directly comparable to the amount of cell death measured by Trypan blue cell counts in these models of cell death (Gill et al., 1997; Perks et al., 1999a).
To assess changes in the levels of apoptosis in the presence of apoptotic triggers and morphological alteration in the presence of Y-27632 (1, 2, and 5 μM), Hs578T cells were viewed under phase contrast and photomicrographs were captured (magnification ×100) using JVC TK-1281 color video camera on an Axiovert 100 microscope (Zeiss) coupled to a time-lapse video recorder using Adobe Premiere 4.1.
Hs578T cells were seeded at 0.1 × 106 cells per coverslip in GM for 24 h before being placed into SFM for a further 24 h. As the effects of Y-27632 were most marked between 2 and 6 h, Hs578T cells were treated with or without Y-27632 (5 μM) for 4 h. Following treatment cells were washed with PBS (×2), then fixed in a 4% paraformaldehyde solution for 10 min and then permeabilized with 0.5% triton X-100 for 10 min. The coverslips were blocked in 3% goat serum for 30 min and incubated with 250 μl (1:500) anti-alpha-tubulin primary antibody in 3% goat serum for 40 min. The coverslips were then rinsed in PBS (×3). Slides were then stained with 250 μl (1:500) TRITC phalloidin in goat serum in conjunction with incubation of the secondary antibody for tubulin [alexa flour 488; 250 μl (1:250)] in 3% goat serum for 40 min. The coverslips were washed in PBS (×3) before being removed from the plate and air dried for 10 min prior to mounting on slides using Vectasheild and DAPI (Vector Labs Inc, Peterborough, UK). Photomicrographs were taken using an Olympus BX40 fluorescent microscope under oil immersion at a magnification of ×40.
Hs578T cells (0.3 × 106) were grown to 80% confluency in T25 flasks and then washed twice with phosphate buffered saline (PBS). The GM was replaced with SFM for 24 h. The cells were then treated with ngIGFBP-3 for 30 min after a 30 min pre-incubation with fumonisin B1 (1 μM), and then lysed on ice for 10 min (1 ml; 10 mM Tris HCl, 5 mM EDTA, 50 mM NaCl, 30 mM Na pyrophosphate, 50 mM sodium fluoride, 100 μM sodium orthovanadate, 1% Triton and protease and inhibitor cocktails: 1:100). Lysates were then centrifuged at 14,000 g for 15 min at 4°C. The protein content of each sample was determined using a BCA Protein Assay Reagent Kit and equivalent volumes of protein used. Non-specific binding sites on the nitro-cellulose membranes were blocked overnight with 3% BSA in tris-buffered saline; 2% TWEEN (TBST) for probing with anti-p-MAPK (1:5000) and 5% milk in tris-buffered saline; 2% TWEEN (TBST) for probing with anti-ERK-2 (1:1000). Following the removal of excess unbound antibody, an anti-rabbit antibody (1:10,000 for p-MAPK), an anti-goat (1:5000 for ERK-2) conjugated to peroxidase was added for 1 h. Chemiluminescence was detected using the ChemiDoc-IT Imaging System (UVP) (Biorad, Hertfordshire, UK) and analyzed using Vision WorksTH ls Analysis Software (UVP Inc., Upland, CA, USA).
Activation assays were performed as described previously (Ren and Schwartz, 2000; Pellegrin and Mellor, 2008). In brief, Hs578T were seeded at 0.3 × 106 per T25 culture flasks for 24 h after which the GM was replaced with SFM for 24 h. Cells were treated with either IGFBP-3 (100 ng/ml) with or without a 30-min pre-dose with fumonisin B1 (1 μM), an anti-beta 1 integrin antibody (200 ng/ml) or KT5720 (0.5 μM), or with sphingosine-1-phosphate (S1P, 1 μM), ranging from 1 to 5 min used as a positive control. Following treatment, the supernatant was removed and the cells were then washed twice with Tris-buffered saline (TBS, 50 mM Tris base, 140 mM NaCl; pH7.6) prior to cell lysis (50 mM Tris HCl pH 7.2, 1% triton X-100, 0.1% SDS, 500 mM NaCl, 10 mM MgCl2, protease, and phosphatase inhibitor cocktails, 1:100). Samples were then centrifuged at 14,000 g at 4°C for 10 min. Of the cell lysates, equal volumes were incubated with 20–30 μg GST-TRBD beads and rotated at 4°C for 45 min and equal volumes were also removed for assessment of total Rho as a loading control. The samples were washed four times with 600 μl of tris buffer (TB, 50 mM Tris HCl pH 7.2, 1% triton X-100, 150 mM NaCl, 10 mM MgCl2 protease and phosphatase inhibitor cocktails, 1:100) centrifuged at 5000 rpm for 20 s at 4°C and the supernatant removed. The samples were eluted with 50 μl of Laemmli sample buffer containing 40 mM DTT and boiled for 10 min. Samples were spun at 5000 rpm for 20 s and the supernatant was then loaded onto the gel and SDS-PAGE and Western blotting performed. Non-specific binding sites on the nylon membranes were blocked with 5% milk in tris-buffered saline; 2% TWEEN (TBST) and incubated with anti-Rho antibody (1:1000) overnight with rotation. Following the removal of excess unbound primary antibody, blots were incubated with an anti-rabbit antibody (1:2000) conjugated to peroxidase for 1 h and processed as described above.
Hs578T cells were pre-dosed with IGFBP-3 as described above, and then dosed with the apoptotic trigger. Attached cells were then washed in 0.2% BSA-containing PBS, trypsinized, and then re-suspended in 0.2% BSA-containing PBS in the presence of 10% FCS. Cells were spun down and re-suspended in a 0.2% BSA-containing PBS solution of a ceramide antibody or a control IgM (10 μg/ml). The cells were incubated on ice for 30 min, spun down and washed in 0.2% BSA-containing PBS. Cells were then re-suspended in a 0.2% BSA-containing PBS solution of a FITC-labeled anti-mouse IgM antibody (7.8 μg/ml), incubated for 30 min on ice, spun down, washed, and then re-suspended in 0.2% BSA-containing PBS. Cell surface ceramide was assessed using a FACS Calibur flow cytometer (Becton Dickinson). Analysis was by Cell Quest software package (Becton Dickinson).
The data were analyzed with using SPSS 12.0.1 for windows software using ANOVA followed by least significant difference (LSD) post hoc test. A statistically significant difference was considered to be present at p < 0.05.
IGFBP-3 binds to caveolin-1 (Burrows et al., 2006) binding specifically to its scaffolding domain (CSD) (Lee et al., 2004; Singh et al., 2004). One of the main signaling molecules which binds to the CSD and is inactivated through binding is protein kinase A (Levin et al., 2007). We investigated if PKA was involved in the intrinsic effects of IGFBP-3. Figure 1A indicates that in Hs578T breast cancer cells, the accentuating effects of SPD on C2-induced apoptosis (p < 0.05) was reversed in the presence of a PKA inhibitor, KT5720, such that SPD then conferred cell survival (p < 0.05). In the presence of KT5720 the survival effect of IGFBP-5 against C2 (p < 0.05) was unaffected (Figure 1B). Preliminary data also showed that the apoptotic response to C2 was enhanced in the presence of an activator of PKA, 8-Bromo-cAMP (data not shown) consistent with activation of PKA being involved in the accentuation of C2-induced apoptosis by IGFBP-3.
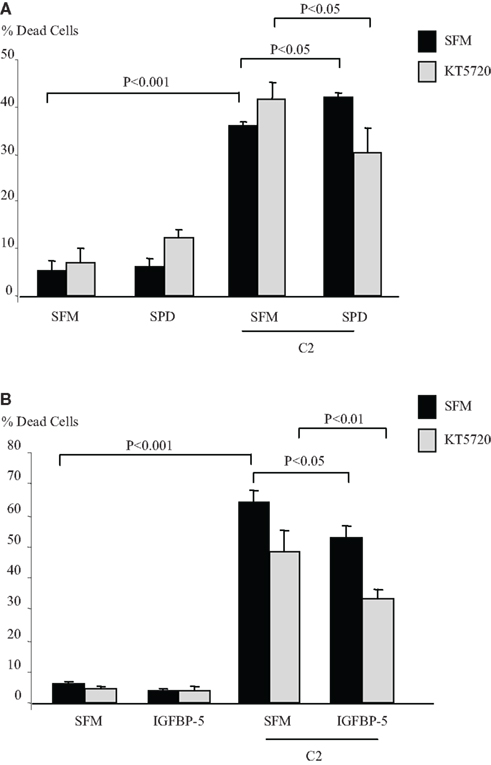
Figure 1. Hs578T cells were seeded in six-well plates at 0.1 × 106 cells in GM followed by 24 h in SFM prior to pre-dosing (A) with SPD (10 ng/ml) or (B) IGFBP-5 ± KT5720 (1 μM). The cells were then re-dosed with IGFBP-3 or IGFBP-5 (each at 100 ng/ml) for a further 24 h in the presence or absence of an apoptotic dose of C2-ceramide. The graphs represent changes in cell death and show the mean ± SEM of three experiments each repeated in triplicate.
One of the major downstream targets of PKA is Rho (Leve et al., 2008) and the assembly of integrin adhesion complexes requires both Rho GTPases and the extracellular matrix (Hotchin and Hall, 1995). ROCK is a serine/threonine kinase and a key target for Rho signaling, which induces actin bundling and contraction through increasing myosin light chain (MLC) phosphorylation (Bishop and Hall, 2000).
Figure 2A shows the morphology of the Hs578T cells in the presence of the Rho kinase inhibitor (1, 2, and 5 μM) at 4 h. Despite the more rounded phenotype and increased formation of long cell processes (lamellipodia/filopodia), there was no induction of cell death above basal levels over 24 or 48 h. With the use of the actin stain phalloidin, we show in Figure 2B that inhibition of Rho kinase (5 μM) resulted in a loss of organized actin containing stress fibers. Using antibodies against α-tubulin we also observed the loss of the microtubule mesh network as well as longitudinally orientated tubulin within the protruding cellular processes.
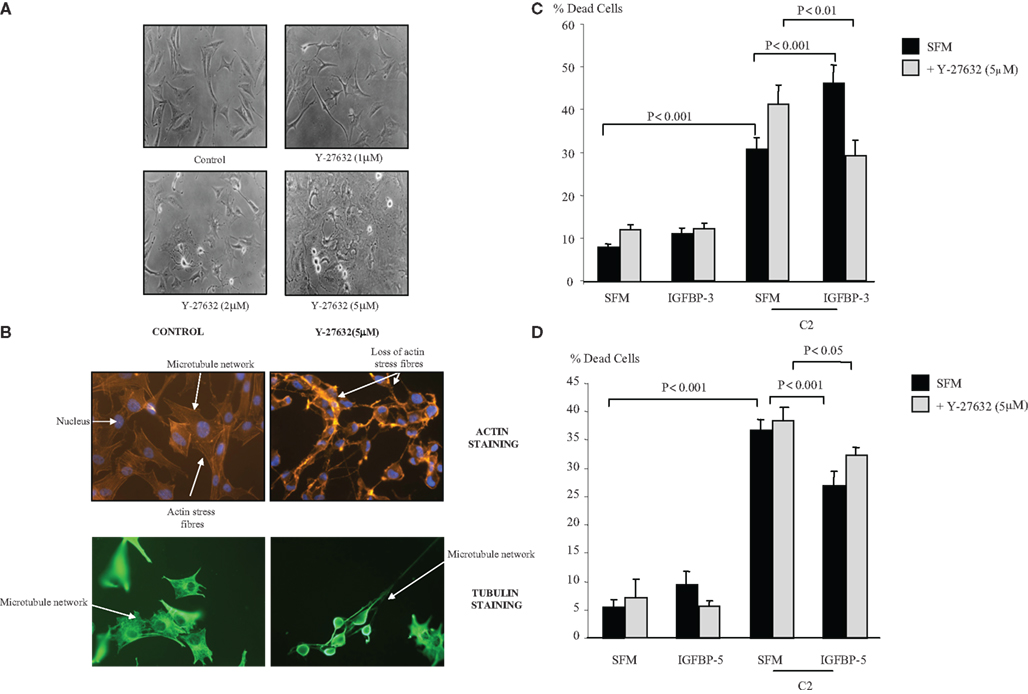
Figure 2. Photomicrographs of Hs578T cells showing (A) morphological alteration in the presence of Y-27632 (1, 2, and 5 μM). Cells were captured (magnification ×100) as described in Section “Materials and Methods” (B) changes in actin stress fibers and microtubule organization. Cells were seeded at 0.1 × 106 cells per coverslip in GM for 24 h before being placed into SFM for a further 24 h ± Y-27632 (5 μM) for 4 h. Cells were stained and captured (magnification of ×40) as described in Section “Materials and Methods.” Hs578T cells were seeded in six-well plates at 0.1 × 106 cells in GM followed by 24 h in SFM prior to pre-dosing with (C) IGFBP-3 (100 ng/ml) or (D) IGFBP-5 ± Y-27632 (5 μM). The cells were then re-dosed with IGFBP-3 or IGFBP-5 (each at 100 ng/ml) for a further 24 h in the presence or absence of an apoptotic dose of C2-ceramide. The graphs represent changes in cell death and show the mean ± SEM of three experiments each repeated in triplicate.
Figure 2C shows that the accentuating effect of IGFBP-3 on C2-induced apoptosis (p < 0.001) was reversed in the presence of the Rho kinase inhibitor, such that IGFBP-3 then conferred cell survival (p < 0.01). In the presence of Y-27632 IGFBP-5 was still able to significantly decrease C2-ceramide-induced cell death (p < 0.05) (Figure 2D).
With Hs578T cells we found that IGFBP-3 rapidly increased Rho activation after 1 min but activity had waned and was non-significant by 5 min. In contrast, the activation of Rho by a positive control, sphingosine-1-phosphate (S1P) at 1 min was still evident at 5 min (data not shown). Figure 3A confirms that IGFBP-3 significantly activated Rho after treatment for 1 min (p < 0.01). We showed previously that IGFBP-3 binds to the beta 1 integrin and that the accentuating effects of IGFBP-3 were reversed when the beta 1 integrin was blocked (Burrows et al., 2006). In this study we found that the ability of IGFBP-3 to activate Rho was blocked in the presence of a beta 1 blocking antibody and a PKA inhibitor with a tendency for these effects to be reversed (Figures 3B,C). These data indicated that the involvement of the beta 1 integrin and PKA occurred upstream of Rho. We have shown previously that IGFBP-3 dose-dependently increased the activation of MAPK and inhibiting MAPK with PD98059 blocked the accentuating effects of IGFBP-3 on apoptotic triggers (Burrows et al., 2006). Figure 3D shows that the activation of MAPK by IGFBP-3 (p < 0.01) was reversed in the presence of Y-27632, such that IGFBP-3 then inhibited MAPK phosphorylation (p < 0.05). These data suggest that MAPK is downstream of Rho kinase and hence of the beta 1 integrin and PKA.
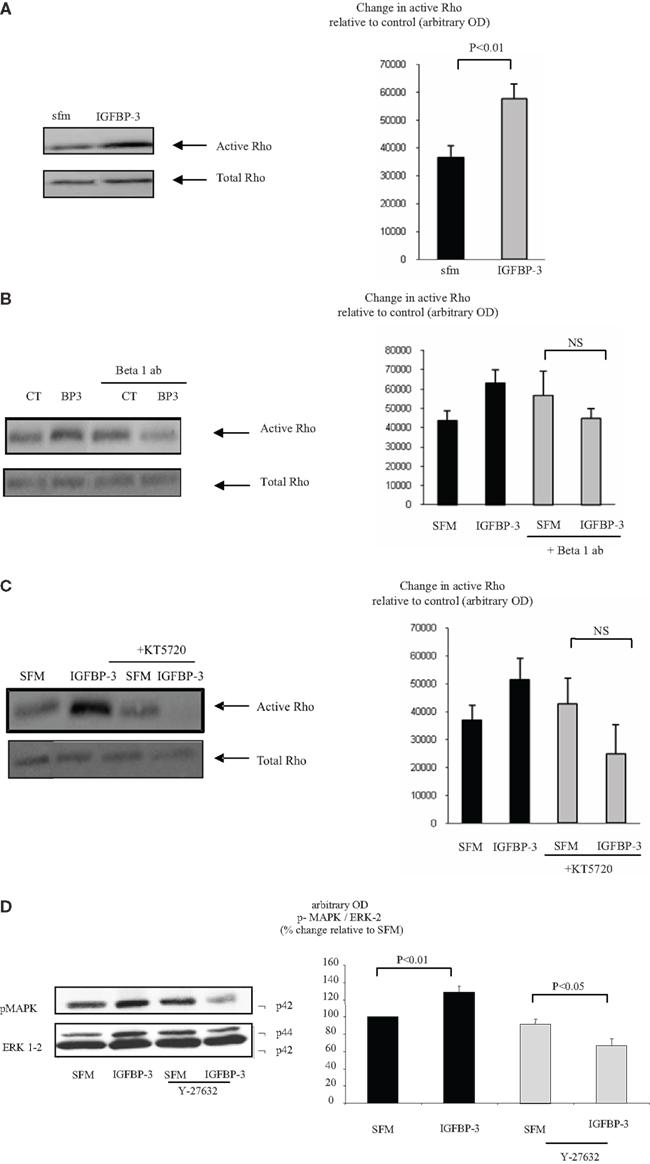
Figure 3. Hs578T were seeded at 0.3 × 106 per T25 culture flasks for 24 h after which the GM was replaced with SFM for 24 h. Cells were treated (A) with IGFBP-3 (100 ng/ml) for 30 min or (B) with IGFBP-3 (100 ng/ml) following a 30-min pre-dose with an anti-beta 1 integrin blocking antibody (200 ng/ml) or (C) a PKA inhibitor KT5720 (1 μM). Following treatment activation of Rho was assessed as described in Section “Materials and Methods.” (A–C) show Western immunoblots (of active and total Rho and are representative of experiments repeated three times) and graphs of the mean arbitrary optical density measurements from three experiments demonstrating the change in active Rho compared to the control. (D) Cells were treated with IGFBP-3 (100 ng/ml) for 30 min following a 30-min pre-dose with Y-27632 (5 μM) and shows Western immunoblots of p-MAPK and total ERK-2 and are representative of experiments repeated three times. The graph shows the mean arbitrary optical density measurements from three experiments demonstrating changes in p-MAPK corrected for ERK-2.
The sphingolipid rheostat describes the dynamic balance that exists between intracellular S1P and ceramide which have opposing actions. To investigate whether IGFBP-3 affected this balance we initially inferred the involvement of ceramide by using an inhibitor of ceramide synthase, fumonisin B1, which blocks endogenous production of ceramide. Figures 4A,B show that the enhancing effects of IGFBP-3 and SPD respectively on C2-ceramide-induced cell death (p < 0.01 for each) were reversed by fumonisin B1, such that IGFBP-3 and SPD each acted as a survival factor (p < 0.05 for each), consistent with an unveiling of the effect of increased S1P. This reversal was similarly observed when we used an alternative trigger of apoptosis, antimycin A (Figure 4C). We found no evidence to suggest that the activation of Rho by IGFBP-3 was modulated in the presence of fumonisin B1 (data not shown) but Figures 5A,B indicate that IGFBP-3 can no longer activate p-MAPK in the presence of fumonisin B1, suggesting that ceramide is acting upstream of MAPK in the enhancement of cell death by IGFBP-3, but downstream of Rho. As anticipated, we show in Figure 5C, that the survival effect of IGFBP-5 against C2-induced cell death was unaffected by blocking ceramide production with fumonisin B1.
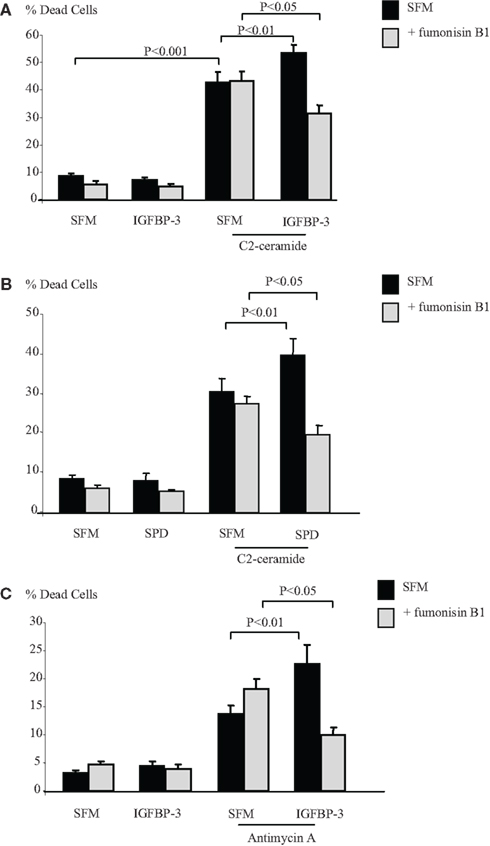
Figure 4. Hs578T cells were seeded in six-well plates at 0.1 × 106 cells in GM followed by 24 h in SFM prior to pre-dosing for 24 h (A) with IGFBP-3 (100 ng/ml), (B) SPD (10 ng/ml) or (C) IGFBP-3 ± fumonisin B1 (1 μM). Cells were then re-dosed for 24 h ± an apoptotic dose of C2-ceramide (A,B) or antimycin A (C). The graphs represent changes in cell death and show the mean ± SEM of three experiments each repeated in triplicate.
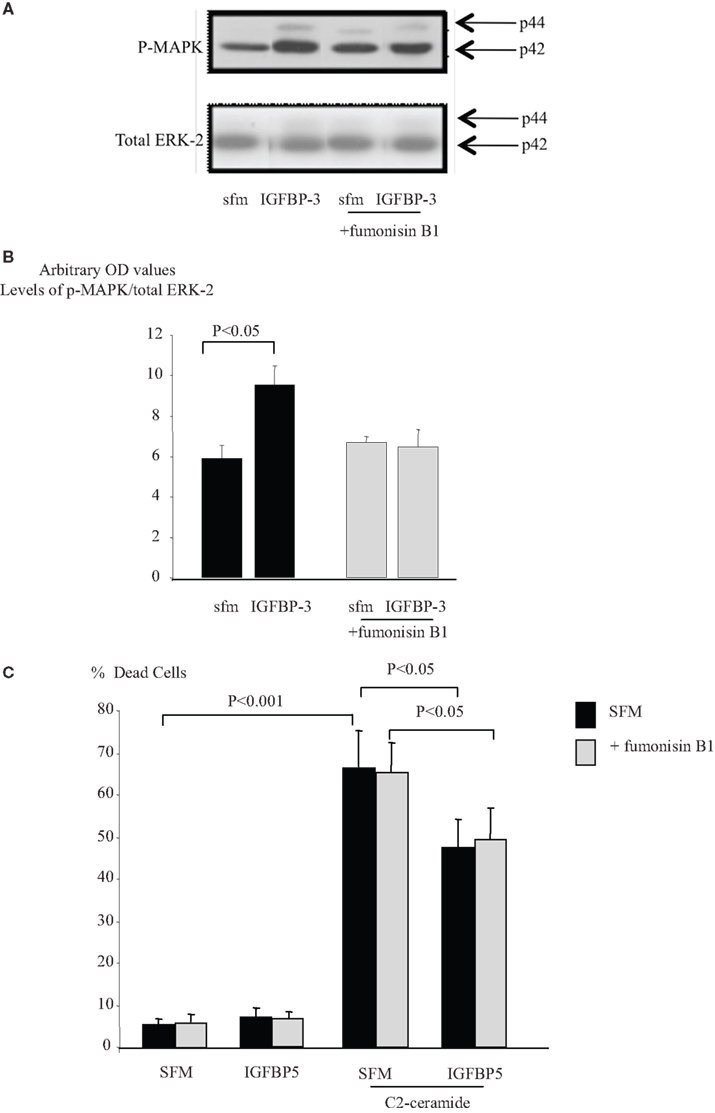
Figure 5. Hs578T were seeded at 0.3 × 106 per T25 culture flasks for 24 h after which the GM was replaced with SFM for 24 h. Cells were treated with IGFBP-3 (100 ng/ml) for 30 min following a 30-min pre-dose with fumonisin B1 (1 μM). (A) Shows Western immunoblots of p-MAPK and ERK-2 and are representative of experiments repeated three times. (B) Shows a graph of the mean arbitrary optical density measurements from three experiments demonstrating changes in p-MAPK corrected for total ERK-2. (C) Hs578T cells were seeded in six-well plates at 0.1 × 106 cells in GM followed by 24 h in SFM prior to pre-dosing for 24 h with IGFBP-5 (100 ng/ml) ± fumonisin B1 (1 μM). Cells were then re-dosed for 24 h as above ± an apoptotic dose of C2-ceramide. The graph represents changes in cell death and shows the mean ± SEM of three experiments each repeated in triplicate.
We also then measured levels of ceramide on the cell surface as described previously (Grassme et al., 2001). Figure 6A indicates that neither IGFBP-3 nor the apoptotic trigger antimycin A alone had any effect, but in combination IGFBP-3 caused a significant increase in the levels of ceramide detected on the cell surface (p < 0.05) We next neutralized surface ceramide using the ceramide antibody as a blocking antibody in the apoptosis assay as described previously (Grassme et al., 2001) and show in Figure 6B that this reversed the accentuating actions of IGFBP-3 on antimycin A (p < 0.001), such that IGFBP-3 conferred survival (p < 0.05). This would be consistent with a shift in the rheostat and a contemporaneous increase in S1P, the effect of which was unveiled when the ceramide was blocked. The control IgM antibody was without effect (data not shown). Figure 6C supports these results visually with photomicrographs of the cells. The ceramide antibody and IGFBP-3 alone had no effect on the appearance of the cells relative to the control panel. As anticipated with the induction of apoptosis Antimycin A caused cell detachment with an increase in floating cells. A number of cells remaining on the plate exhibited a more rounded appearance. The cells were unchanged when either IGFBP-3 or antimycin A were co-incubated with the ceramide antibody. The effects of antimycin A were exacerbated by IGFBP-3, coincident with the ability of IGFBP-3 to enhance antimycin A-induced cell death. Very few cells were left attached to the plate and of these a high number were rounded in appearance. In the presence of the ceramide antibody, it is clear that the effects of IGFBP-3 on antimycin A-induced apoptosis were reversed and that fewer of the cells were rounded and floating in comparison to antimycin A alone. We showed previously that the survival effect of IGFBP-5 was blocked when we inhibited the production of sphingosine-1-phosphate with a sphingosine kinase inhibitor, DMS (McCaig et al., 2002c). We then used the same approach to establish whether the other side of the sphingolipid rheostat was involved in the effects of IGFBP-3. Figure 6D indicates that the level of accentuation of cell death by IGFBP-3 on plastic was actually doubled when sphinogosine-1-phosphate production was blocked with the sphingosine kinase inhibitor (p < 0.001). In Figure 1A we showed that PKA was involved in the accentuating effects of SPD/IGFBP-3 on apoptotic triggers. Our data imply that the pro-apoptotic effect of IGFBP-3 involved the production of ceramide that was partly counterbalanced by increased production of S1P such that when the latter was blocked then the effect on cell death was enhanced.

Figure 6. Hs578T cells were seeded in six-well plates at 0.1 × 106 cells in GM followed by 24 h in SFM. Cells were pre-dosed for 24 h with IGFBP-3 (100 ng/ml) prior to redosing ± an apoptotic dose of antimycin A (A). Graph represents cell surface ceramide as assessed by flow cytometry and is the mean ± SEM of three experiments. Cells were pre-dosed with IGFBP-3 for 24 h ± a ceramide blocking antibody (1 μg/ml) prior to redosing ± an apoptotic dose of antimycin A (B). Graph represents changes in cell death and shows the mean ± SEM of three experiments each repeated in triplicate. (C) Shows phase contrast photomicrographs (magnification ×100) of Hs578T cells following treatment described in (B), which are representative of experiments repeated three times. Cells were pre-dosed for 24 h with IGFBP-3 (100 ng/ml) ± DMS (0.5 μM) and then re-dosed ± an apoptotic dose of C2-ceramide (D). The graph represents changes in cell death and shows the mean ± SEM of three experiments each repeated in triplicate.
Integrin receptors congregate in cholesterol-stabilized microdomains within the plasma membrane that form platforms for the clustering of cell signaling components (Brown, 2000). Caveolin 1 is a recognized marker of such domains and we have previously shown that IGFBP-3 can bind to both caveolin-1 and the beta 1 integrin and increase their association (Burrows et al., 2006). It has been reported that IGFBP-3 specifically binds to the scaffolding domain of caveolin 1 (CSD; Lee et al., 2004; Singh et al., 2004). This would require entry of IGFBP-3 into the cell, which has been shown to occur via both caveolae and clathrin-mediated pathways (Lee et al., 2004; Singh et al., 2004). One of the main signaling molecules that is bound and inhibited by the CSD is PKA (Levin et al., 2007). We have shown that the pro-apoptotic actions of IGFBP-3 were reversed when the activity of PKA was inhibited. These data suggest that IGFBP-3 may activate PKA by displacing it from the CSD and the active PKA is involved in the accentuating effects of IGFBP-3 on apoptotic triggers. We also found that the survival effect of IGFBP-5 was unaffected in the presence of the PKA inhibitor, in keeping with our previous work showing the survival effect of IGFBP-5 was PKC-dependent (McCaig et al., 2002c). Collectively we suggest that the accentuating effects of IGFBP-3 on apoptotic triggers are PKA-dependent and reversible, whereas the ability of IGFBP-5 to confer survival is PKC-dependent and not reversible.
The initiation of integrin signaling requires the clustering of integrins and the formation of focal contacts. PKA is a key activator of Rho (Howe, 2004) and the formation of integrin clusters requires re-modeling of the actin cytoskeleton, which is achieved primarily through controlled activation of the Rho GTPases (Ridley and Hall, 1992; Schwartz et al., 1995; Yamada and Miyamoto, 1995). However Rho GTPase activity is not limited to cytoskeletal re-arrangement, they are also involved in cell cycle progression, apoptosis, cytokinesis, and cell transformation (Ridley, 2001). Rho kinase (ROCK) is a serine/threonine kinase and a key target for Rho signaling (Bishop and Hall, 2000). By blocking ROCK we were again able to reverse the accentuating actions of IGFBP-3, whereas the survival effect of IGFBP-5 was unaffected. This suggested that the pro-apoptotic effects of IGFBP-3 required activation of ROCK and again this was reversible. Since ROCK contributed to the pro-apoptotic actions of IGFBP-3, we assessed and confirmed that IGFBP-3 activated its upstream regulator Rho. As the activation of Rho was inhibited by the PKA inhibitor, we concluded that PKA was upstream of Rho. We showed previously that blocking the beta 1 integrin receptor reversed the actions of IGFBP-3 such that it went from promoting to inhibiting triggers of apoptosis. We now confirm that this interaction is also upstream of Rho, since the beta 1 blocking antibody also blocked the effects of IGFBP-3 on Rho activation. Cholesterol-stabilized microdomains are sphingolipid rich and changes in the relative levels of ceramide and S-1P, which constitute the “sphingolipid rheostat,” can determine whether a cell survives or dies. A shift toward higher intracellular levels of S-1P is associated with growth and survival, and increased concentrations of ceramide leads to growth inhibition and/or apoptosis (Cuvillier et al., 1996). We found that IGFBP-3 was even more effective at accentuating apoptosis when sphingosine kinase was inhibited, suggesting that S1P production was opposing the effect of IGFBP-3 to enhance apoptosis and then when S1P production was inhibited this then tilted the balance in favor of ceramide generation.
Indeed inhibiting ceramide synthase, which blocks ceramide production and tilts the balance back in favor of the survival factor S1P, reversed the accentuating effects of both IGFBP-3 and SPD, such that they both then acted as survival factors. Since we originally suggested the involvement of the sphingolipid rheostat in the intrinsic effects of IGFBP-3 (Perks et al., 2004) there have been reports confirming that at least one side of this balance is involved in specific effects of IGFBP-3 on apoptosis (Granata et al., 2004) and the effects of IGFBP-3 on EGF-induced growth (Martin et al., 2009). We now provide evidence that IGFBP-3 can affect the sphingolipid rheostat in both directions and that this provides an explanation for the different reports of apparently contradicting actions. It is well established that ceramide can be generated at numerous sites within a cell (van Blitterswijk et al., 2003) and that ceramide in the plasma membrane plays an important role as a membrane structural component being involved in receptor clustering, vesicle formation and fusion (van Blitterswijk et al., 2003). Ligation of a number of cell surface receptors, including integrin receptors also results in rapid clustering, which is required for efficient receptor-mediated signaling (Schatzmann et al., 2003; Bollinger et al., 2005). Extracellularly orientated ceramide has been shown to play a key part in CD95 clustering that culminated in apoptosis, which was inhibited when the cell surface ceramide was neutralized (Grassme et al., 2001). We found that IGFBP-3 alone had no effect on cell surface ceramide, but it did enhance the levels in the presence of an apoptotic trigger, consistent with it having no effect on apoptosis alone but being able to enhance triggers of cell death. Neutralizing the cell surface ceramide caused IGFBP-3 action to be reversed from enhancing to inhibiting apoptosis, as we had observed when we inhibited ceramide synthase with fumonisin B1. These data indicate that cell surface ceramide contributes to the way in which IGFBP-3 modulates apoptosis, but does not preclude that intracellular ceramide might also play a part. Together our findings suggest that alterations in the specific spatial organization of integrin receptors, sphingolipids, and signaling molecules within cholesterol-stabilized complexes may modify the response of the cells to IGFBP-3. We found no evidence to suggest that fumonisin B1 affected the ability of IGFBP-3 to activate Rho, suggesting that the involvement of ceramide is downstream of Rho.
We have shown previously that the accentuating effects of IGFBP-3 on apoptotic triggers involving cholesterol-stabilized integrin receptor complexes culminated in the activation of MAPK (Burrows et al., 2006). We now show that the activation of MAPK is blocked in the presence of fumonisin B1, which suggests that MAPK is downstream of the beta 1 integrin, PKA, Rho, and ceramide in the pathway which IGFBP-3 uses to accentuate triggers of apoptosis.
We established previously that IGFBP-3 could exert positive or negative effects on cell function depending upon the extracellular matrix composition (McCaig et al., 2002b; Burrows et al., 2006). We also showed that IGFBP-3 was able to modulate downstream signaling events initiated by integrins through enhanced phosphorylation of MAPK (Burrows et al., 2006). However, although the actions of IGFBP-3 could be reversed by disrupting integrin receptor complexes (Burrows et al., 2006), they were not reversible when MAPK was inhibited; the actions of IGFBP-3 were just blocked. In this study we have identified key components of cholesterol-stabilized integrin receptor signaling, PKA, Rho, and ceramide, which are involved in the intrinsic, pro-apoptotic actions of IGFBP-3. Furthermore, we determined that they are upstream of MAPK and when each are blocked, the actions of IGFBP-3 are reversed (see Figure 7). It has been proposed that IGFBP-3 could be used therapeutically to treat cancers due to its ability to inhibit growth and promote apoptosis. These data highlight the importance of understanding the mechanism by which IGFBP-3 actions can be reversed from a negative to a positive regulator, since attempting to inhibit or promote IGFBP-3 function could actually lead to it having the opposite effect.
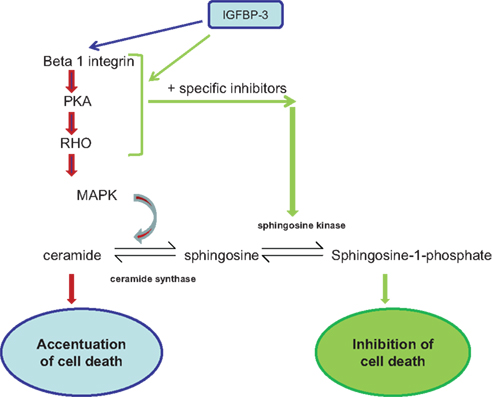
Figure 7. Signaling molecules involved in the pathway by which IGFBP-3 enhances apoptotic triggers: Inhibition of which can lead to IGFBP-3 having the opposite effect. IGFBP-3 is able to have either enhancing or inhibiting effects on apoptotic triggers depending upon the context.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We acknowledge Professor Harry Mellor and Dr. Catherine McCaig and we thank the University of Bristol Cancer Research Fund for supporting this work.
Berman, A. E., Kozlova, N. I., and Morozevich, G. E. (2003). Integrins: structure and signaling. Biochemistry Mosc. 68, 1284–1299.
Bishop, A. L., and Hall, A. (2000). Rho GTPases and their effector proteins. Biochem. J. 348(Pt 2), 241–255.
Bollinger, C. R., Teichgraber, V., and Gulbins, E. (2005). Ceramide-enriched membrane domains. Biochim. Biophys. Acta 1746, 284–294.
Brown, N. H. (2000). An integrin chicken and egg problem: which comes first, the extracellular matrix or the cytoskeleton? Curr. Opin. Cell Biol. 12, 629–633.
Burrows, C., Holly, J. M., Laurence, N. J., Vernon, E. G., Carter, J. V., Clark, M. A., McIntosh, J., McCaig, C., Winters, Z. E., and Perks, C. M. (2006). Insulin-like growth factor binding protein 3 has opposing actions on malignant and nonmalignant breast epithelial cells that are each reversible and dependent upon cholesterol-stabilized integrin receptor complexes. Endocrinology 147, 3484–3500.
Cohen, P., Rajah, R., Rosenbloom, J., and Herrick, D. J. (2000). IGFBP-3 mediates TGF-beta1-induced cell growth in human airway smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 278, L545–L551.
Cuvillier, O., Pirianov, G., Kleuser, B., Vanek, P. G., Coso, O. A., Gutkind, S., and Spiegel, S. (1996). Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature 381, 800–803.
Fanayan, S., Firth, S. M., Butt, A. J., and Baxter, R. C. (2000). Growth inhibition by insulin-like growth factor-binding protein-3 in T47D breast cancer cells requires transforming growth factor-beta (TGF-beta) and the type II TGF-beta receptor. J. Biol. Chem. 275, 39146–39151.
Gill, Z. P., Perks, C. M., Newcomb, P. V., and Holly, J. M. (1997). Insulin-like growth factor-binding protein (IGFBP-3) predisposes breast cancer cells to programmed cell death in a non-IGF-dependent manner. J. Biol. Chem. 272, 25602–25607.
Granata, R., Trovato, L., Garbarino, G., Taliano, M., Ponti, R., Sala, G., Ghidoni, R., and Ghigo, E. (2004). Dual effects of IGFBP-3 on endothelial cell apoptosis and survival: involvement of the sphingolipid signaling pathways. FASEB J. 18, 1456–1458.
Grassme, H., Jekle, A., Riehle, A., Schwarz, H., Berger, J., Sandhoff, K., Kolesnick, R., and Gulbins, E. (2001). CD95 signaling via ceramide-rich membrane rafts. J. Biol. Chem. 276, 20589–20596.
Hollowood, A. D., Lai, T., Perks, C. M., Newcomb, P. V., Alderson, D., and Holly, J. M. (2000). IGFBP-3 prolongs the p53 response and enhances apoptosis following UV irradiation. Int. J. Cancer 88, 336–341.
Hollowood, A. D., Stewart, C. E., Perks, C. M., Pell, J. M., Lai, T., Alderson, D., and Holly, J. M. (2002). Evidence implicating a mid-region sequence of IGFBP-3 in its specific IGF-independent actions. J. Cell. Biochem. 86, 583–589.
Hotchin, N. A., and Hall, A. (1995). The assembly of integrin adhesion complexes requires both extracellular matrix and intracellular rho/rac GTPases. J. Cell Biol. 131, 1857–1865.
Howe, A. K. (2004). Regulation of actin-based cell migration by cAMP/PKA. Biochim. Biophys. Acta 1692, 159–174.
Ingermann, A. R., Yang, Y. F., Han, J., Mikami, A., Garza, A. E., Mohanraj, L., Fan, L., Idowu, M., Ware, J. L., Kim, H. S., Lee, D.-Y., and Oh, Y. (2010). Identification of a novel cell death receptor mediating IGFBP-3-induced anti-tumor effects in breast and prostate cancer. J. Biol. Chem. 285, 30233–30246.
Jones, J. I., and Clemmons, D. R. (1995). Insulin-like growth factors and their binding proteins: biological actions. Endocr. Rev. 16, 3–34.
Jones, J. I., Gockerman, A., Busby, W. H. Jr., Wright, G., and Clemmons, D. R. (1993). Insulin-like growth factor binding protein 1 stimulates cell migration and binds to the alpha 5 beta 1 integrin by means of its Arg-Gly-Asp sequence. Proc. Natl. Acad. Sci. U.S.A. 90, 10553–10557.
Kansra, S., Ewton, D. Z., Wang, J., and Friedman, E. (2000). IGFBP-3 mediates TGF beta 1 proliferative response in colon cancer cells. Int. J. Cancer 87, 373–378.
Leal, S. M., Liu, Q., Huang, S. S., and Huang, J. S. (1997). The type V transforming growth factor beta receptor is the putative insulin-like growth factor-binding protein 3 receptor. J. Biol. Chem. 272, 20572–20576.
Lee, K. W., Liu, B., Ma, L., Li, H., Bang, P., Koeffler, H. P., and Cohen, P. (2004). Cellular internalization of insulin-like growth factor binding protein-3: distinct endocytic pathways facilitate re-uptake and nuclear localization. J. Biol. Chem. 279, 469–476.
Leve, F., de Souza, W., and Morgado-Diaz, J. A. (2008). A cross-link between protein kinase A and Rho-family GTPases signaling mediates cell-cell adhesion and actin cytoskeleton organization in epithelial cancer cells. J. Pharmacol. Exp. Ther. 327, 777–788.
Levin, A. M., Murase, K., Jackson, P. J., Flinspach, M. L., Poulos, T. L., and Weiss, G. A. (2007). Double barrel shotgun scanning of the caveolin-1 scaffolding domain. ACS Chem. Biol. 2, 493–500.
Liu, B., Lee, H. Y., Weinzimer, S. A., Powell, D. R., Clifford, J. L., Kurie, J. M., and Cohen, P. (2000). Direct functional interactions between insulin-like growth factor-binding protein-3 and retinoid X receptor-alpha regulate transcriptional signaling and apoptosis. J. Biol. Chem. 275, 33607–33613.
Martin, J. L., and Jambazov, S. (2006). Insulin-like growth factor binding protein-3 in extracellular matrix stimulates adhesion of breast epithelial cells and activation of p44/42 mitogen-activated protein kinase. Endocrinology 147, 4400–4409.
Martin, J. L., Lin, M. Z., McGowan, E. M., and Baxter, R. C. (2009). Potentiation of growth factor signaling by insulin-like growth factor-binding protein-3 in breast epithelial cells requires sphingosine kinase activity. J. Biol. Chem. 284, 25542–25552.
Martin, J. L., and Pattison, S. L. (2000). Insulin-like growth factor binding protein-3 is regulated by dihydrotestosterone and stimulates deoxyribonucleic acid synthesis and cell proliferation in LNCaP prostate carcinoma cells. Endocrinology 141, 2401–2409.
Martin, J. L., Weenink, S. M., and Baxter, R. C. (2003). Insulin-like growth factor-binding protein-3 potentiates epidermal growth factor action in MCF-10A mammary epithelial cells. Involvement of p44/42 and p38 mitogen-activated protein kinases. J. Biol. Chem. 278, 2969–2976.
McCaig, C., Fowler, C. A., Laurence, N. J., Lai, T., Savage, P. B., Holly, J. M., and Perks, C. M. (2002a). Differential interactions between IGFBP-3 and transforming growth factor-beta (TGF-beta) in normal vs cancerous breast epithelial cells. Br. J. Cancer 86, 1963–1969.
McCaig, C., Perks, C. M., and Holly, J. M. (2002b). Intrinsic actions of IGFBP-3 and IGFBP-5 on Hs578T breast cancer epithelial cells: inhibition or accentuation of attachment and survival is dependent upon the presence of fibronectin. J. Cell. Sci. 115, 4293–4303.
McCaig, C., Perks, C. M., and Holly, J. M. (2002c). Signalling pathways involved in the direct effects of IGFBP-5 on breast epithelial cell attachment and survival. J. Cell. Biochem. 84, 784–794.
McIntosh, J., Dennison, G., Holly, J. M., Jarrett, C., Frankow, A., Foulstone, E. J., Winters, Z. E., and Perks, C. M. (2010). IGFBP-3 can either inhibit or enhance EGF-mediated growth of breast epithelial cells dependent upon the presence of fibronectin. J. Biol. Chem. 28, 38788–38800.
Oda, K., Hori, S., Itoh, H., Osamura, R. Y., Tokuda, Y., Kubota, M., and Tajima, T. (1992). Immunohistochemical study of transforming growth factor beta, fibronectin, and fibronectin receptor in invasive mammary carcinomas. Acta Pathol. Jpn. 42, 645–650.
Oh, Y., Gucev, Z., Ng, L., Muller, H. L., and Rosenfeld, R. G. (1995). Antiproliferative actions of insulin-like growth factor binding protein (IGFBP)-3 in human breast cancer cells. Prog. Growth Factor Res. 6, 503–512.
Oh, Y., Muller, H. L., Pham, H., and Rosenfeld, R. G. (1993). Demonstration of receptors for insulin-like growth factor binding protein-3 on Hs578T human breast cancer cells. J. Biol. Chem. 268, 26045–26048.
Pellegrin, S., and Mellor, H. (2008). Rho GTPase activation assays. Curr. Protoc. Cell Biol. Chapter 14, Unit 14.8.
Perks, C. M., Vernon, E. G., Clark, M. A., Burrows, C., and Holly, J. M. P. (2004). “IGF-independent actions of IGFBP-3 involve cholesterol stabilised complexes and modulation of sphingolipid metabolites,” in Proceedings of the 86th Annual Meeting of the American Endocrine Society, New Orleans, P3–P94.
Perks, C. M., Bowen, S., Gill, Z. P., Newcomb, P. V., and Holly, J. M. (1999a). Differential IGF-independent effects of insulin-like growth factor binding proteins (1-6) on apoptosis of breast epithelial cells. J. Cell. Biochem. 75, 652–664.
Perks, C. M., Newcomb, P. V., Norman, M. R., and Holly, J. M. (1999b). Effect of insulin-like growth factor binding protein-1 on integrin signalling and the induction of apoptosis in human breast cancer cells. J. Mol. Endocrinol. 22, 141–150.
Perks, C. M., and Holly, J. M. (2003). The insulin-like growth factor (IGF) family and breast cancer. Breast Dis. 18, 45–60.
Rajah, R., Valentinis, B., and Cohen, P. (1997). Insulin-like growth factor (IGF)-binding protein-3 induces apoptosis and mediates the effects of transforming growth factor-beta1 on programmed cell death through a p53- and IGF-independent mechanism. J. Biol. Chem. 272, 12181–12188.
Ren, X. D., and Schwartz, M. A. (2000). Determination of GTP loading on Rho. Meth. Enzymol. 325, 264–272.
Ridley, A. J. (2001). Rho family proteins: coordinating cell responses. Trends Cell Biol. 11, 471–477.
Ridley, A. J., and Hall, A. (1992). The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 70, 389–399.
Schatzmann, F., Marlow, R., and Streuli, C. H. (2003). Integrin signaling and mammary cell function. J. Mammary Gland Biol. Neoplasia 8, 395–408.
Schedlich, L. J., Young, T. F., Firth, S. M., and Baxter, R. C. (1998). Insulin-like growth factor-binding protein (IGFBP)-3 and IGFBP-5 share a common nuclear transport pathway in T47D human breast carcinoma cells. J. Biol. Chem. 273, 18347–18352.
Schutt, B. S., Langkamp, M., Rauschnabel, U., Ranke, M. B., and Elmlinger, M. W. (2004). Integrin-mediated action of insulin-like growth factor binding protein-2 in tumor cells. J. Mol. Endocrinol. 32, 859–868.
Schwartz, M. A., Schaller, M. D., and Ginsberg, M. H. (1995). Integrins: emerging paradigms of signal transduction. Annu. Rev. Cell Dev. Biol. 11, 549–599.
Singh, B., Charkowicz, D., and Mascarenhas, D. (2004). Insulin-like growth factor-independent effects mediated by a C-terminal metal-binding domain of insulin-like growth factor binding protein-3. J. Biol. Chem. 279, 477–487.
van Blitterswijk, W. J., van der Luit, A. H., Veldman, R. J., Verheij, M., and Borst, J. (2003). Ceramide: second messenger or modulator of membrane structure and dynamics? Biochem. J. 369, 199–211.
Williams, A. C., Collard, T. J., Perks, C. M., Newcomb, P., Moorghen, M., Holly, J. M., and Paraskeva, C. (2000). Increased p53-dependent apoptosis by the insulin-like growth factor binding protein IGFBP-3 in human colonic adenoma-derived cells. Cancer Res. 60, 22–27.
Keywords: IGFBP-3, PKA, rho, ceramide, IGF-independent
Citation: Perks CM, Burrows C and Holly JMP (2011) Intrinsic, pro-apoptotic effects of IGFBP-3 on breast cancer cells are reversible: involvement of PKA, Rho, and ceramide. Front. Endocrin. 2:13. doi: 10.3389/fendo.2011.00013
Received: 06 January 2011;
Paper pending published: 24 January 2011;
Accepted: 03 May 2011;
Published online: 16 May 2011.
Edited by:
Antonino Belfiore, University Magna Graecia of Catanzaro, ItalyReviewed by:
Andrea Morrione, Thomas Jefferson University, USACopyright: © 2011 Perks, Burrows and Holly. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Claire M. Perks, IGF and Metabolic Endocrinology Group, Department of Clinical Sciences at North Bristol, The Medical School, Southmead Hospital, University of Bristol, Bristol, BS10 5NB, UK. e-mail:Y2xhaXJlLm0ucGVya3NAYnJpc3RvbC5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.