 Heather R. Willis1,2*†Medhini S. Sosale3*†Cody W. Edwards4,5Henrique V. Figueiró6Anna Zhuk7,8Klaus-Peter Koepfli4
Heather R. Willis1,2*†Medhini S. Sosale3*†Cody W. Edwards4,5Henrique V. Figueiró6Anna Zhuk7,8Klaus-Peter Koepfli4- 1School of Systems Biology, George Mason University, Fairfax, VA, United States
- 2Department of Reproductive Science, Smithsonian’s National Zoo and Conservation Biology Institute, Front Royal, VA, United States
- 3Department of Bioengineering, Volgenau School of Engineering, George Mason University, Fairfax, VA, United States
- 4Smithsonian-Mason School of Conservation, George Mason University, Front Royal, VA, United States
- 5Department of Biology, George Mason University, Fairfax, VA, United States
- 6Environmental Genomics Group, Vale Institute of Technology, Belém, Brazil
- 7Institute of Applied Computer Science, ITMO University, St. Petersburg, Russia
- 8Laboratory of Amyloid Biology, St. Petersburg State University, St. Petersburg, Russia
Introduction
Fourteen morphologically diverse small American mesocarnivores form the family Procyonidae, one of the sixteen comprising the mammalian order Carnivora (Helgen et al., 2013). Procyonids fall within the suborder Caniformia, infraorder Arctoidea, superclass Musteloidea; the latter also includes families Mephitidae (skunks and stink badgers), Ailuridae (red pandas), and Mustelidae (badgers, martens, weasels, and otters) (Hassanin et al., 2021; Wozencraft, 2005). The best-known procyonids are the charismatic food-washing masked bandits, the raccoons (genus Procyon). Other procyonids, however, include the solitary, monochromatic, arboreal olingos (genus Bassaricyon); the ringtail and cacomistle, nocturnal, opportunistic omnivores with striking black-and-white ringed tails (genus Bassariscus); the social, ground-dwelling coatis (genus Nasua) and mountain coatis (genus Nasuella); and the arboreal kinkajou (genus Potos) (Hunter and Barrett, 2018). According to the IUCN Red List of Threatened Species, Procyon pygmaeus and Nasuella meridensis are Critically Endangered and Endangered, respectively (Cuarón et al., 2016; Gonzalez-Maya and Arias-Alzate, 2016), while Bassaricyon neblina and Nasuella olivacea are categorized as Near Threatened (Gonzalez-Maya et al., 2016; Helgen et al., 2020). The other 10 species of procyonids are categorized as Least Concern, but except for the northern raccoon, these species’ population sizes are trending downward, raising concern for their long-term conservation.
During the last two decades, studies based on DNA sequences have clarified the systematics of the Procyonidae, revealing a pattern of evolutionary relationships at odds with expectations based on morphological characters (Koepfli et al., 2007). DNA-based studies consistently support a topology that groups Bassaricyon, Nasua, and Nasuella in one clade, Bassariscus and Procyon in another clade, and Potos as a distinct basal lineage, although morphology would suggest groupings of Bassaricyon + Potos and Nasua + Procyon (Eizirik et al., 2010; Hassanin et al., 2021; Koepfli et al., 2007; Law et al., 2018). The procyonids continue to undergo taxonomic revision, including the discovery of the olinguito (Bassaricyon neblina) in 2013, uncertainty about the species status of the coati found on Cozumel Island, Mexico (Jaramillo and Ruiz-García, 2022; Valenzuela-Galván et al., 2023), and repeated calls to synonymize the genera Nasua and Nasuella (Helgen et al., 2013; Jaramillo and Ruiz-García, 2022; Nigenda-Morales et al., 2019; Ruiz-García et al., 2021). Thus, more attention to evaluating phylogenetic relationships within this complex clade is appropriate.
Phylogenetic relationships can be elucidated relatively quickly using mitochondrial DNA, or mtDNA. mtDNA is haploid, considerably smaller than nuclear genomes, and exhibits a high rate of DNA substitution, making it a good benchmark for inferring phylogenetic relationships among and within species (Moritz et al., 1987). Though partial or complete sequences from individual mitochondrial genes are sometimes used for phylogenetic inference, whole mitochondrial genomes, or mitogenomes, are considered more informative (Boore et al., 2005). Due to their high copy number in the mitochondria of cells, mitogenomes can be assembled relatively easily and inexpensively from low coverage (≤5x) high-throughput sequencing data, an approach known as genome skimming (Hoban et al., 2022; Trevisan et al., 2019). This approach is growing in popularity for resolving systematic relationships (e.g., Quattrini et al., 2024; Taite et al., 2023). To date, complete mitogenomes of only four procyonid species have been deposited into NCBI’s GenBank database: northern raccoon, Procyon lotor (AB297804, AB291073, CM027276, MT410951); northern olingo, Bassaricyon gabbii (ON704723); South American coati, Nasua nasua (HM106331); and kinkajou, Potos flavus (MW257234). Complete mitogenomes of species such as white-nosed coati (Nasua narica) and western mountain coati (Nasuella olivacea) have, however, been reported in several studies but not deposited into public sequence databases (Hassanin et al., 2021; Jaramillo and Ruiz-García, 2022; Tsuchiya, 2017).
To address this paucity of publicly available procyonid mitogenomes, we report new annotated mitochondrial genome assemblies generated with genome skimming for the following species: eastern lowland olingo (Bassaricyon alleni), ringtail (Bassariscus astutus), white-nosed coati (Nasua narica), and crab-eating raccoon (Procyon cancrivorus). We also assembled and annotated complete mitogenomes of the olinguito (Bassaricyon neblina) and the little-known cacomistle (Bassariscus sumichrasti) using short-read sequencing data generated by Hawkins et al. (2016). These mitogenomes were used to develop an updated phylogeny, which was supplemented by a broader phylogenetic analysis of just the cytochrome b gene. Using the newly annotated complete mitogenomes, we were also able to infer sequence distances and codon usage bias in relation to previously available mitogenomes of procyonid species.
Materials and methods
Frozen tissue samples used to generate the new mitogenomes reported in this study were obtained from vouchered specimens deposited at the Museum of Southwestern Biology, University of New Mexico, New Mexico, USA (Nasua narica, Procyon cancrivorus) and the Museum of Vertebrate Zoology, University of California, Berkeley, California, USA (Bassaricyon alleni, Bassariscus astutus). Details about the sampled specimens, including their geographic origin, are found in Supplementary Table S1.
DNA extraction, library preparation, and sequencing were conducted at Psomagen, Inc. (Rockville, MD). The Mag-Bind Blood and Tissue Kit (Omega Bio-Tek Inc., Norcross, GA) was used for genomic DNA extraction, after which concentration was estimated with the Picogreen and Victor X2 fluorometry assay (Life Technologies, Carlsbad, CA) and quality was checked with an Agilent 4200 Tapestation (Agilent Technologies, Santa Clara, CA) and 1% agarose gel electrophoresis. A Covaris S220 ultrasonicator (Woburn, MA) was used to shear genomic DNA; fragments of ~350bp were enriched, and the TruSeq DNA PCR-free library kit (Illumina, San Diego, CA) was used to prepare a genomic library for each sample. Libraries were quality checked using an Agilent 4200 Tapestation and a Lightcycler quantitative PCR assay (Roche Life Science, St. Louis, MO). An Illumina NovaSeq 6000 instrument was used to paired-end sequence (2 x 150 bp) each library to a depth of 5x.
The FASTQ files from Bassaricyon alleni, Nasua narica, Procyon cancrivorus, Bassaricyon neblina (SRR2103277), and Bassariscus sumichrasti (SRR2104917) were trimmed with AdapterRemoval (Lindgreen, 2012) using default settings. The mitogenomes were assembled with GetOrganelle v1.7.7.1 (Jin et al., 2020) using the following parameter settings: 10 extending rounds, the animalmt reference database, SPAdes assembler (Prjibelski et al., 2020), k-mer values 21, 45, 65, 85, and 105, and automatic word size estimation. Bassaricyon neblina and Bassariscus sumichrasti were assembled with seed sequence Bassaricyon gabbii (ON704723) and Procyon lotor (AB291073), respectively. Reads from Bassariscus astutus were cleaned with fastp v0.22.0 detectadapterforpe-l30-q20–overrepresentationanalysis (Chen, 2023) and assembled with GetOrganelle v1.7.7.1, using k-mer values 21, 55, 85, and 115 and seed sequences Procyon lotor (NC_009126.1), Potos flavus (NC_053977.1, CM027365.1), Bassaricyon gabbii (NC_066722.1), and Nasua nasua (HM106331.1). All mitogenomes were annotated with MITOS2 (Donath et al., 2019) using the RefSeq 89 Metazoa database and genetic code 2.
The six mitogenomes assembled were aligned with four previously published procyonid mitogenomes downloaded from the NCBI GenBank database: ON704723 (Bassaricyon gabbii), AB291073 (Procyon lotor), HM106331 (Nasua nasua), and MW257234 (Potos flavus). For outgroups, we included one species from Mephitidae, Mephitis mephitis (HM106332), one species from Ailuridae, Ailurus fulgens (MK886830), and two species from Mustelidae, Mellivora capensis (MW257239) and Lutra lutra (MW573979), for a total alignment of 14 mitogenomes from distinct species. We aligned these sequences using the MAFFT v7.450 plugin in Geneious Prime 2023.1.1 with default settings: algorithm “Auto”, scoring matrix PAM200 with k = 2, gap open penalty = 1.53, and offset value = 0.123 (Katoh and Standley, 2013). The control region was trimmed due to poor alignment within the tandem repeat regions, resulting in a final alignment length of 15,541bp, which included sequences of 13 protein-coding genes (PCGs), 2 rRNAs and 22 tRNAs. We constructed a maximum-likelihood phylogeny with the RAxML version 8.2.11 (Stamatakis, 2014) plugin in Geneious Prime 2023.1.1 using the rapid hill-climbing algorithm, applying 1000 bootstrap replicates to evaluate node support. For both analyses, we used the GTR+GAMMA nucleotide substitution model, as this is one of the four applicable models in RAxML, and because our alignment contains a relatively small number of taxa, such that the alpha parameter of the gamma distribution sufficiently accounts for among-site rate heterogeneity (Yang, 2006).
We also constructed a Bayesian phylogeny using the MrBayes 3.2.6 (Huelsenbeck and Ronquist, 2001) plugin in Geneious Prime 2024.0.7 with default settings (chain length = 107, subsampling frequency = 103, heated chains = 4, heated chain temperature = 0.2, burn-in length = 103), the GTR+GAMMA substitution model, and designating Ailurus fulgens as the outgroup in the full mitogenome alignment.
To incorporate a more complete taxon set, we generated an additional phylogeny using only sequences of the cytochrome b (CYTB) gene. We extracted these sequences from all the complete mitogenomes used in our initial phylogeny except for A. fulgens and M. mephitis, which were removed to prevent error from long branch attraction. Additionally, we downloaded 25 CYTB sequences from GenBank, representing all procyonid species in our complete mitogenome phylogeny as well as Nasuella olivacea, Bassaricyon medius, and Bassaricyon beddardi, which has now been synonymized with Bassaricyon alleni (Helgen et al., 2013). Ultimately, our sample set for this analysis included one sample each for two outgroup species (M. capensis and L. lutra), B. neblina, B. neblina neblina, B. beddardi, two samples each for B. astutus, B. sumichrasti, N. olivacea, B. gabbii, B. alleni, and B. medius medius, three samples each for P. cancrivorus, P. lotor, N. narica, and N. nasua, and four samples each for P. flavus and B. medius orinomus, for a total of 37 sequences. We aligned these sequences using the same methods as the complete mitogenome and trimmed the resulting alignment to 1,140 bp. We then used the same RAxML and MrBayes settings as described above for the mitogenome alignment, including the GTR+GAMMA substitution model, to generate maximum-likelihood and Bayesian inference phylogenies from this alignment and evaluated node support with 1000 bootstrap replicates (RAxML) and posterior probabilities (MrBayes).
We estimated the absolute number of pairwise differences (p-distances) between procyonid taxa in the trimmed mitogenome alignment with MEGA 11.0.10 (Tamura et al., 2021) using the following settings: Variance Estimation Method: None, Substitutions Type: Nucleotide, Model/Method: No. of differences, Substitutions to Include: d: Transitions + Transversions, Rates among Sites: Uniform Rates, Pattern among Lineages: Same (Homogeneous), Gaps/Missing Data Treatment: Pairwise deletion. We repeated the analysis with a Kimura two-parameter model (Kimura, 1980), which accounts for different substitution rates between transitions and transversions. Settings were identical to those described above, except the Model/Method setting was changed to “Kimura 2-parameter model.”
To test for differences in codon usage in the 13 PCGs, protein-coding sequences from each procyonid mitogenome (except Bassariscus sumichrasti, which was done by hand due to technical difficulties) were concatenated with a homemade Python script, which is available upon request. Each concatenated sequence was analyzed with the Codon Usage calculator in the Sequence Manipulation Suite (Stothard, 2000) under the vertebrate mitochondrial genetic code.
Data description
We assembled and annotated full mitogenomes for six procyonid species: Eastern lowland olingo (Bassaricyon alleni), olinguito (Bassaricyon neblina), ringtail (Bassariscus astutus), cacomistle (Bassariscus sumichrasti), white-nosed coati (Nasua narica), and crab-eating raccoon (Procyon cancrivorus) (Figure 1). The coverage and lengths of the mitogenomes varied between 627 – 6758.1x and 16,486 – 16,653 bp, respectively (Supplementary Table S1). These lengths were consistent with those of previously published procyonid mitogenomes, which range from 16,388 – 16,600 bp. Each mitogenome contained the standard set of vertebrate mitochondrial genes, with 13 protein-coding genes, 22 tRNAs, 2 rRNAs, two origins of replication, and a control region, arranged in the order typical for therian mammals (Kocher et al., 1989).

Figure 1. The six newly generated mitogenomes: (A) Bassaricyon alleni; (B) Bassaricyon neblina; (C) Bassariscus sumichrasti; (D) Bassariscus astutus; (E) Nasua narica; (F) Procyon cancrivorus. Black outer rings show the relative nucleotide position of each gene in the mitogenomes. Inner rings are colored according to the annotated features: protein-coding genes (green), ribosomal RNAs (red), transfer RNAs (pink), control region (orange), light strand and heavy strand origins of replication (blue). Arrowed bars indicate transcription on either the plus strand (clockwise arrows) or minus strand (counterclockwise arrows). Source: (A) Photo 37986455 copyright MaoMorning Yip (https://www.inaturalist.org/photos/37986455), licensed under CC BY-NC; (B) Olinguito (Bassaricyon neblina) copyright blackdogto (https://www.inaturalist.org/observations/92111781), licensed under CC BY-NC; (C) Southern California Ringtail (Bassariscus astutus ssp. Octavus) copyright a-geoman (https://www.inaturalist.org/observations/149483825), licensed under CC BY-NC; (D) Cacomistle (Bassariscus sumichrasti) copyright jensanford (https://www.inaturalist.org/observations/154347289), licensed under CC BY-NC; (E) Photo 346763147 by Zygy (https://www.inaturalist.org/photos/346763147), public domain; (F) Crab-eating Raccoon (Procyon cancrivorus) copyright atdahl (https://www.inaturalist.org/observations/196664383), licensed under CC BY-NC.
Maximum-likelihood and Bayesian inference phylogenetic analysis of the 15,541 bp alignment yielded a tree in which Bassaricyon and Nasua, the olingos and coatis, formed one clade, while Bassariscus and Procyon, the ringtail, cacomistle, and raccoons, formed a second clade (Figure 2A and Supplementary Figure S1). The kinkajou (Potos flavus) was basal to these groupings. All labeled nodes had a bootstrap and posterior probability support of 100% and 1.0, respectively. This phylogeny is generally concordant with the relationships inferred in previous studies using partitioned supermatrices of mitochondrial and nuclear DNA sequences (Koepfli et al., 2007; Eizirik et al., 2010; Helgen et al., 2013; Law et al., 2018). The same groupings appear in the maximum likelihood and Bayesian inference CYTB trees (Figure 2B and Supplementary Figure S2), with two major exceptions. First, the kinkajou clade is positioned as the sister lineage to the clade containing the coatis and olingos (Nasua, Nasuella, and Bassaricyon) rather than basal to the raccoon+ringtail+cacomistle and coati+olingo clades. However, this deep node only had a 59% bootstrap and 0.89 posterior probability support, respectively. Second, the two species of Bassariscus are paraphyletic, with the cacomistle sister to the clade containing the two species of Procyon. The node uniting B. sumichrasti with P. cancrivorus+P. lotor had a bootstrap support of 42% and a posterior probability support of 0.51, suggesting this grouping is unstable.
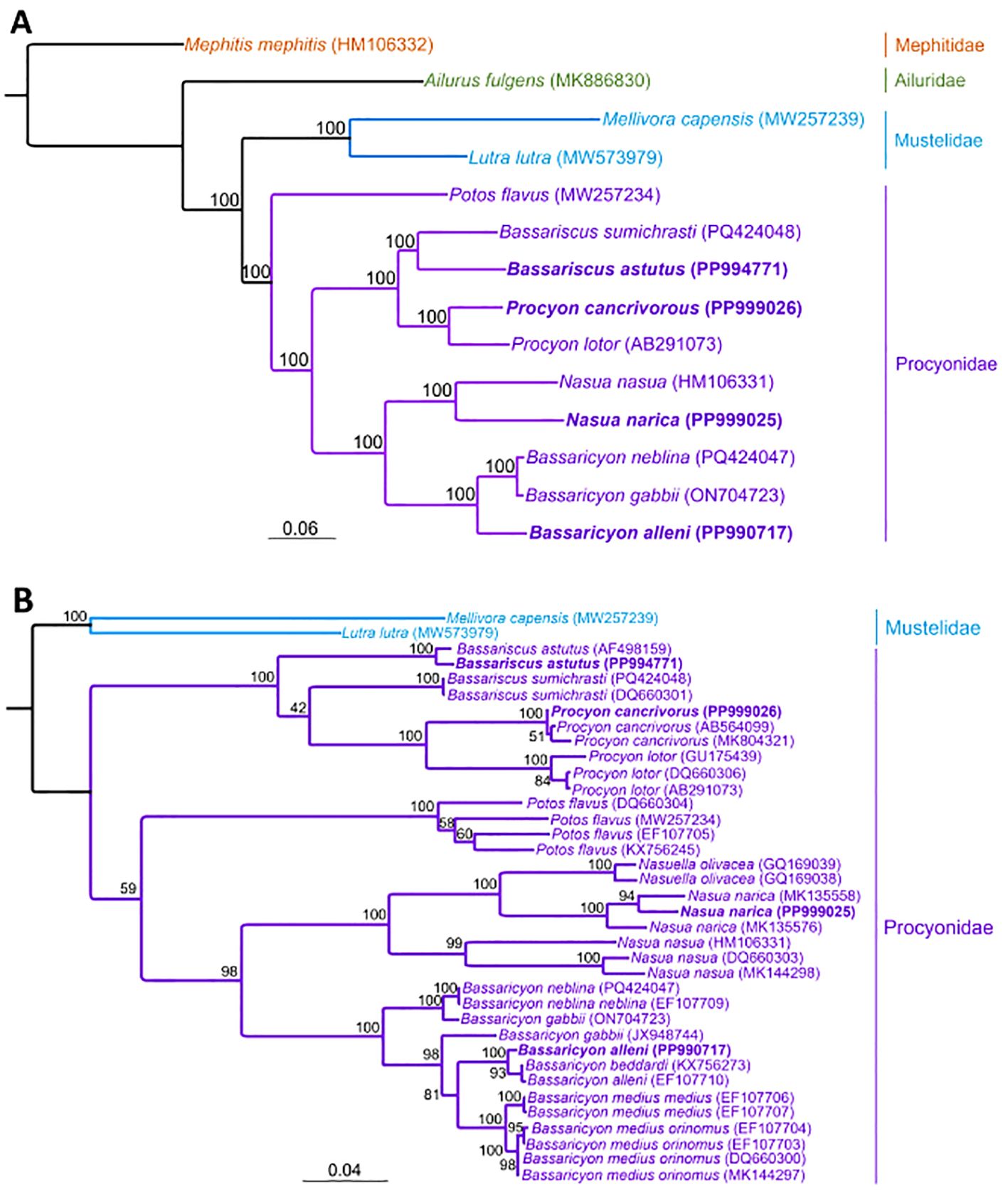
Figure 2. Phylogenetic trees generated with RAxML. (A) tree based on whole mitogenome alignment (15,541 bp), with 10 procyonid species and 4 outgroups; (B) tree based on cytochrome b gene (1140 bp), with 12 procyonid species and 2 outgroups. Note the placement of Bassaricyon gabbii (ON704723) in a clade containing two sequences of Bassaricyon neblina obtained from vouchered museum specimens in the cytochrome b tree. Newly generated mitogenomes (or CYTB genes from new mitogenomes) are indicated in bold.
The sole deviation from previous phylogenies in the complete mitogenome tree is the placement of Bassaricyon neblina. In Helgen et al. (2013), based on a combination of the complete cytochrome b (CYTB) gene and a nuclear intron of the CHRNA1 gene, B. neblina was placed basal to all other olingo species, while our phylogeny places B. neblina sister to B. gabbii, with B. alleni basal to both. Moreover, based on Kimura 2-Parameter distances among the CYTB sequences, B. neblina showed a 9-11% sequence divergence from other olingo taxa in Helgen et al., 2013. The sequence divergence, however, between the B. neblina mitogenome assembled in this study and the B. gabbii assembled by Winter et al. (2023) is only 0.966%, while B. alleni’s p-distances from B. neblina and B. gabbii are 7.44% and 7.37%, respectively.
Thus, our results strongly suggest that the mitogenome of B. gabbii reported by Winter et al. (2023) instead likely represents B. neblina. Our CYTB tree supports this assertion: this B. gabbii (ON704723) groups with our assembled B. neblina sequence (PQ424047) and with EF107709, another B. neblina CYTB sequence (Figure 2B and Supplementary Figure SX). These three are basal to all other olingos, including JX948744, a taxonomically verified B. gabbii sequence that occupies its own branch, separate from ON704723. The B. gabbii mitogenome from Winter et al. (2023) was derived from a cell culture with no information about the origin or source of the culture. This misidentification emphasizes the importance of reference genome resources being tied to vouchered specimens that have been taxonomically verified (Buckner et al., 2021).
In addition, the CYTB gene tree includes a sequence downloaded from GenBank (KX756273) and identified as Bassaricyon beddardi in Nascimento et al. (2017), which is nested among two B. alleni sequences, including the one from which we assembled the complete mitogenome (PP990717). However, Helgen et al. (2013) concluded that B. beddardi should be regarded as a synonym of B. alleni, based on the limited molecular and morphological differences between these taxa. Therefore, according to our data, B. alleni is a monophyletic species.
Intergeneric Kimura 2-Parameter distances between whole mitogenomes averaged 18.5%, ranging from 12.9% (B. sumichrasti versus P. cancrivorus) to 20.6% (N. narica versus P. flavus) – 1785 to 2708 base pairs different (Supplementary Table S3). Intrageneric distances range from 7.37-13.6%, averaging 9.7%. The highest intrageneric distance was observed between the two Nasua species at 13.6% (1864 base pairs different), while the lowest was found between Bassaricyon alleni and B. gabbii, at 7.37% (1064 base pairs different).
The codon usage results show a significant bias in favor of codons ending with pyrimidines (Supplementary Table S4), which is common among mammalian mitogenomes (Jia and Higgs, 2008). Although the preferred codon of a synonymous set is usually the same across the family, the magnitude of this preference varies considerably even within genera. For example, both Nasua narica and Nasua nasua prefer TGC for cysteine, but N. nasua uses this codon 75% of the time, while N. narica uses it only 58% of the time. N. narica consistently shows the least extreme bias, while B. neblina and B. gabbii frequently show the most extreme bias. These latter two mitogenomes have a highly similar codon usage profile, with an average difference of 0.6% between them. Bassaricyon spp. codon bias is 7-8% different from any other genus, while the other genera, regardless of phylogenetic distance, differ from each other by 4-6%. B. sumichrasti codon bias seems more closely correlated with that of P. cancrivorus than that of B. astutus: P. cancrivorus and B. sumichrasti have on average 2.8% difference in codon usage, while B. astutus and B. sumichrasti have on average 3.9% difference. These results show that similarities or differences in codon use bias do not correlate overall with species relatedness.
Our findings add context to prior knowledge of the evolutionary history of Procyonidae in the Americas and reveal the contribution of mitochondrial genomes in elucidating the relationships among species in this family. For instance, prior work that relied on morphological (teeth) and ecological (habitat type) data to separate these species suggested phylogenies that grouped Bassaricyon with Potos (both arboreal species that largely consume fruit) and grouped Bassariscus with a subclade containing both Nasua and Procyon (both omnivorous and terrestrial species) (Baskin, 2004; Decker and Wozencraft, 1991). These groupings are also reflective of coat patterns, as the latter group often demonstrates distinctive facial and tail markings not observed in the former (Koepfli et al., 2007). However, more recent research that used both partial mitogenomes and segments of nuclear genomes suggests that the phylogeny of the Procyonidae more closely resembles the topology we present in Figure 2A (Eizirik et al., 2010; Koepfli et al., 2007; Law et al., 2018) and does not agree with expected relationships based on morphology. In fact, the topologies based on DNA sequence data consistently suggest that Potos was the first species to diverge whereas the morphologically dissimilar taxa Bassaricyon + Nasua and Bassariscus + Procyon each comprise separate clades. This result indicates that classification via ecology and morphology should be supplemented by genetic studies. This information not only confirms evolutionary relationships but also helps detect phylogenetic errors that may arise from poor selection of morphological traits on which to base classifications.
Our dataset more than doubles the mitogenome resources available for the Procyonidae. Although two of the six mitogenomes we assembled have been previously reported, this study is the first to make this data publicly available. Our mitogenome analysis includes all procyonid genera except Nasuella, the mountain coatis. Recent papers, however, show that the genus Nasuella phylogenetically nests within Nasua (Helgen et al., 2009; Jaramillo and Ruiz-García, 2022; Nigenda-Morales et al., 2019; Ruiz-García et al., 2021), suggesting that Nasuella should be synonymized with Nasua. Since ON704723 appears to come from an olinguito, five species without full mitogenomes remain: the northern olingo (Bassaricyon gabbii), the western lowland olingo (Bassaricyon medius), the eastern mountain coati (Nasuella meridiensis), the western mountain coati (Nasuella olivacea), and the critically endangered pygmy raccoon (Procyon pygmaeus) (Cuarón et al., 2016). Although this dataset deepens our understanding of this understudied family and provides additional tools for taxonomy and conservation, creating a family-wide mitogenome dataset would improve our understanding of evolutionary innovations across procyonids.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
Ethical approval was not required for the study involving animals in accordance with the local legislation and institutional requirements because all DNA was extracted from tissues taken from specimens in museum collections. Appropriate loan request protocols were followed.
Author contributions
HW: Data curation, Formal analysis, Investigation, Visualization, Writing – original draft, Writing – review & editing. MS: Data curation, Formal analysis, Investigation, Visualization, Writing – original draft, Writing – review & editing. CE: Conceptualization, Funding acquisition, Methodology, Writing – review & editing. HF: Conceptualization, Methodology, Writing – review & editing. AZ: Data curation, Formal analysis, Writing – review & editing. K-PK: Conceptualization, Methodology, Project administration, Resources, Visualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. AZ was funded by Saint-Petersburg State University project #129660189.
Acknowledgments
We thank the staffs of the Division of Mammals at the Museum of Southwestern Biology, University of New Mexico, and Mammal Collections at the Museum of Vertebrate Zoology, University of California, Berkeley, for providing the tissue samples listed in Supplementary Table S1. We also thank the staff at Psomagen, Inc. (Rockville, Maryland, USA) for providing outstanding services in the generation of the genome skimming data. MS thanks A. DeLuycker and J. Davis at the Smithsonian-Mason School of Conservation for their early support of this project.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2025.1518714/full#supplementary-material
References
Baskin J. (2004). Bassariscus and probassariscus (Mammalia, carnivora, procyonidae) from the early barstovian (Middle miocene). J. Vertebrate Paleontology 24, 709–720. doi: 10.1671/0272-4634(2004)024[0709:BAPMCP]2.0.CO;2
Boore J. L., Macey J. R., Medina M. (2005). Sequencing and comparing whole mitochondrial genomes of animals. Methods Enzymology 395, 311–348). doi: 10.1016/S0076-6879(05)95019-2
Buckner J. C., Sanders R. C., Faircloth B. C., Chakrabarty P. (2021). The critical importance of vouchers in genomics. eLife 10, e68264. doi: 10.7554/eLife.68264
Chen S. (2023). Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using fastp. iMeta 2, e107. doi: 10.1002/imt2.107
Cuarón A. D., de Grammont P., McFadden K. (2016). IUCN Red List of Threatened Species: Procyon pygmaeus (IUCN Red List of Threatened Species). Available at: https://www.iucnredlist.org/en (Accessed June 29, 2024).
Decker D. M., Wozencraft W. C. (1991). Phylogenetic analysis of recent procyonid genera. J. Mammalogy 72, 42–55. doi: 10.2307/1381979
Donath A., Jühling F., Al-Arab M., Bernhart S. H., Reinhardt F., Stadler P. F., et al. (2019). Improved annotation of protein-coding genes boundaries in metazoan mitochondrial genomes. Nucleic Acids Res. 47, 10543–10552. doi: 10.1093/nar/gkz833
Eizirik E., Murphy W. J., Koepfli K.-P., Johnson W. E., Dragoo J. W., Wayne R. K., et al. (2010). Pattern and timing of diversification of the mammalian order Carnivora inferred from multiple nuclear gene sequences. Mol. Phylogenet. Evol. 56, 49–63. doi: 10.1016/j.ympev.2010.01.033
Gonzalez-Maya J., Arias-Alzate A. (2016). IUCN Red List of Threatened Species: Nasuella meridensis (IUCN Red List of Threatened Species). Available at: https://www.iucnredlist.org/en (Accessed July 19, 2024).
Gonzalez-Maya J., Reid F., Helgen K. M. (2016). IUCN Red List of Threatened Species: Nasuella olivacea (IUCN Red List of Threatened Species). Available at: https://www.iucnredlist.org/en (Accessed July 19, 2024).
Hassanin A., Veron G., Ropiquet A., Jansen van Vuuren B., Lécu A., Goodman S. M., et al. (2021). Evolutionary history of Carnivora (Mammalia, Laurasiatheria) inferred from mitochondrial genomes. PloS One 16, e0240770. doi: 10.1371/journal.pone.0240770
Hawkins M. T. R., Hofman C. A., Callicrate T., McDonough M. M., Tsuchiya M. T. N., Gutiérrez E. E., et al. (2016). In-solution hybridization for mammalian mitogenome enrichment: Pros, cons and challenges associated with multiplexing degraded DNA. Mol. Ecol. Resour. 16, 1173–1188. doi: 10.1111/1755-0998.12448
Helgen K. M., Kays R., Helgen L., Tsuchiya-Jerep M. T. N., Pinto M., Koepfli K.-P., et al. (2009). Taxonomic boundaries and geographic distributions revealed by an integrative systematic overview of the mountain coatis, Nasuella (Carnivora: Procyonidae). Small Carnivore Conserv. 41, 65–74.
Helgen K. M., Kays R., Pinto C., Schipper J., Gonzalez-Maya J. (2020). IUCN Red List of Threatened Species: Bassaricyon neblina (amended version of 2016 assessment) (IUCN Red List of Threatened Species). Available at: https://www.iucnredlist.org/en (Accessed October 9, 2023).
Helgen K. M., Pinto M., Kays R., Helgen L., Tsuchiya M., Quinn A., et al. (2013). Taxonomic revision of the olingos (Bassaricyon), with description of a new species, the Olinguito. ZooKeys 324, 1–83. doi: 10.3897/zookeys.324.5827
Hoban M. L., Whitney J., Collins A. G., Meyer C., Murphy K. R., Reft A. J., et al. (2022). Skimming for barcodes: Rapid production of mitochondrial genome and nuclear ribosomal repeat reference markers through shallow shotgun sequencing. PeerJ 10, e13790. doi: 10.7717/peerj.13790
Huelsenbeck J. P., Ronquist F. (2001). MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755. doi: 10.1093/bioinformatics/17.8.754
Hunter L., Barrett P. (2018). A field guide to the carnivores of the world, second edition (London, UK: Bloomsbury).
Jaramillo M. F., Ruiz-García M. (2022). Evidence of the genetic and spatial structure of Nasua narica in Central America and northern South America from mitogenomic analysis. Therya 13, 205–224. doi: 10.12933/therya-22-1173
Jia W., Higgs P. G. (2008). Codon usage in mitochondrial genomes: distinguishing context-dependent mutation from translational selection. Mol. Biol. Evol. 25, 339–351. doi: 10.1093/molbev/msm259
Jin J.-J., Yu W.-B., Yang J.-B., Song Y., dePamphilis C. W., Yi T.-S., et al. (2020). GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21, 241. doi: 10.1186/s13059-020-02154-5
Katoh K., Standley D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kimura M. (1980). A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120. doi: 10.1007/BF01731581
Kocher T. D., Thomas W. K., Meyer A., Edwards S. V., Pääbo S., Villablanca F. X., et al. (1989). Dynamics of mitochondrial DNA evolution in animals: Amplification and sequencing with conserved primers. Proc. Natl. Acad. Sci. 86, 6196–6200. doi: 10.1073/pnas.86.16.6196
Koepfli K.-P., Gompper M. E., Eizirik E., Ho C.-C., Linden L., Maldonado J. E., et al. (2007). Phylogeny of the Procyonidae (Mammalia: Carnivora): molecules, morphology and the Great American Interchange. Mol. Phylogenet. Evol. 43, 1076–1095. doi: 10.1016/j.ympev.2006.10.003
Law C. J., Slater G. J., Mehta R. S. (2018). Lineage diversity and size disparity in musteloidea: testing patterns of adaptive radiation using molecular and fossil-based methods. Systematic Biol. 67, 127–144. doi: 10.1093/sysbio/syx047
Lindgreen S. (2012). AdapterRemoval: Easy cleaning of next-generation sequencing reads. BMC Res. Notes 5, 337. doi: 10.1186/1756-0500-5-337
Moritz C., Dowling T. E., Brown W. M. (1987). Evolution of animal mitochondrial DNA: relevance for population biology and systematics. Annu. Rev. Ecology Evolution Systematics 18, 269–292. doi: 10.1146/annurev.es.18.110187.001413
Nascimento F. F., Oliveira-Silva M., Veron G., Salazar-Bravo J., Gonçalves P. R., Langguth A., et al. (2017). The evolutionary history and genetic diversity of kinkajous, Potos flavus (Carnivora, procyonidae). J. Mamm. Evol. 24, 439–451. doi: 10.1007/s10914-016-9354-9
Nigenda-Morales S. F., Gompper M. E., Valenzuela-Galván D., Lay A. R., Kapheim K. M., Hass C., et al. (2019). Phylogeographic and diversification patterns of the white-nosed coati (Nasua narica): Evidence for south-to-north colonization of North America. Mol. Phylogenet. Evol. 131, 149–163. doi: 10.1016/j.ympev.2018.11.011
Prjibelski A., Antipov D., Meleshko D., Lapidus A., Korobeynikov A. (2020). Using SPAdes de novo assembler. Curr. Protoc. Bioinf. 70, e102. doi: 10.1002/cpbi.102
Quattrini A. M., McCartin L. J., Easton E. E., Horowitz J., Wirshing H. H., Bowers H., et al. (2024). Skimming genomes for systematics and DNA barcodes of corals. Ecol. Evol. 14, e11254. doi: 10.1002/ece3.11254
Ruiz-García M., Jaramillo M. F., López J. B., Rivillas Y., Bello A., Leguizamon N., et al. (2021). Mitochondrial and karyotypic evidence reveals a lack of support for the genus Nasuella (Procyonidae, Carnivora). J. Vertebrate Biol. 71, 21040.1. doi: 10.25225/JVB.21040
Stamatakis A. (2014). RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stothard P. (2000). The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. BioTechniques 28, 1102. doi: 10.2144/00286ir01
Taite M., Fernández-Álvarez F.Á., Braid H. E., Bush S. L., Bolstad K., Drewery J., et al. (2023). Genome skimming elucidates the evolutionary history of Octopoda. Mol. Phylogenet. Evol. 182, 107729. doi: 10.1016/j.ympev.2023.107729
Tamura K., Stecher G., Kumar S. (2021). MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027. doi: 10.1093/molbev/msab120
Trevisan B., Alcantara D. M. C., MaChado D. J., Marques F. P. L., Lahr D. J. G. (2019). Genome skimming is a low-cost and robust strategy to assemble complete mitochondrial genomes from ethanol preserved specimens in biodiversity studies. PeerJ 7, e7543. doi: 10.7717/peerj.7543
Tsuchiya N. (2017). Evolutionary history of procyonidae (Mammalia, carnivora): integrating genomics, morphology and biogeographic modeling (George Mason University). Available at: https://www.proquest.com/docview/1999244770/abstract/584119F101E04E10PQ/1 (Accessed July 24, 2024).
Valenzuela-Galván D., Vázquez-Domínguez E., Cuarón A. D., Vázquez L.-B., Flores-Manzanero A., Nigenda-Morales S. (2023). A single sample is not enough to claim systematic conclusions, much less for taxa of conservation concern: Comments on Jaramillo and Ruiz-García, (2022). Therya 14, 197–199. doi: 10.12933/therya-23-2201
Winter S., Fennessy J., Janke A., Nilsson M. A. (2023). Northern olingo (Bassaricyon gabbi), zorilla (Ictonyx striatus), and honey badger (Mellivora capensis) mitochondrial genomes and a phylogeny of Musteloidea. Front. Ecol. Evol. 10. doi: 10.3389/fevo.2022.1089641
Wozencraft W. C. (2005). “Order carnivora,” in Mammal species of the world: A taxonomic and geographic reference. Eds. Wilson D. E., Reeder D. M. (Baltimore, MD: Johns Hopkins University Press), 279–348.
Keywords: procyonidae, mitogenomes, genome skimming, phylogeny, systematics, olinguito, cacomistle, vouchers
Citation: Willis HR, Sosale MS, Edwards CW, Figueiró HV, Zhuk A and Koepfli K-P (2025) Complete mitochondrial genomes of the Eastern lowland olingo (Bassaricyon alleni), ringtail (Bassariscus astutus), white-nosed coati (Nasua narica), and crab-eating raccoon (Procyon cancrivorus) elucidate the phylogeny of the Procyonidae. Front. Ecol. Evol. 13:1518714. doi: 10.3389/fevo.2025.1518714
Received: 28 October 2024; Accepted: 27 January 2025;
Published: 18 February 2025.
Edited by:
Fangluan Gao, Fujian Agriculture and Forestry University, ChinaReviewed by:
Sergio F. Nigenda-Morales, National Polytechnic Institute of Mexico (CINVESTAV), MexicoJun Deng, Fujian Agriculture and Forestry University, China
Copyright © 2025 Willis, Sosale, Edwards, Figueiró, Zhuk and Koepfli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Heather R. Willis, hwillis2@gmu.edu; Medhini S. Sosale, medhini.sosale@gmail.com
†These authors have contributed equally to this work and share first authorship