 Guillaume Schwob1*
Guillaume Schwob1* Léa Cabrol1,2,3
Léa Cabrol1,2,3 Paula M. Vidal1
Paula M. Vidal1 Yasna C. Tapia1
Yasna C. Tapia1 Fernando Moya1
Fernando Moya1 Tamara Contador1,4,5
Tamara Contador1,4,5 Julieta Orlando1,6*
Julieta Orlando1,6* Claudia S. Maturana1,4
Claudia S. Maturana1,4- 1Millennium Institute Biodiversity of Antarctic and Subantarctic Ecosystems (BASE), Santiago, Chile
- 2Aix-Marseille University, Mediterranean Institute of Oceanography (M.I.O, UMR 110), CNRS, IRD, Marseille, France
- 3Institute of Ecology and Biodiversity, Santiago, Chile
- 4Cape Horn International Center (CHIC), Puerto Williams, Chile
- 5Wankara Laboratory, Cape Horn International Center, University of Magallanes, Puerto Williams, Chile
- 6Department of Ecological Sciences, Faculty of Sciences, University of Chile, Santiago, Chile
Metazoans comprise multiple physical niches (“microenvironments”), each colonized by unique microbiomes that contribute to their hosts’ evolutionary dynamics, influencing their health, physiology, and adaptation to changing environments. Most wildlife microbiome studies focus on higher metazoans and multiple host microenvironments, while studies of lower species often concentrate on a single microenvironment, sometimes pooling whole bodies or specimens. This is particularly evident in small-sized animals, such as freshwater meiofaunal invertebrates, thus impeding a holistic understanding of microbiome assembly across host microenvironments and its relation with host population genetics. Leveraging the anostracan fairy shrimp Branchinecta, which has easily discernible organs and expected high levels of intraspecific genetic divergence, we aimed to investigate the microbiome assembly processes and test the phylosymbiosis signal in two microenvironments (gill and intestine) across four host populations of Branchinecta gaini within Maritime Antarctica, using 16S rRNA metabarcoding. Our results showed that the gill and intestine harbor strikingly different microbiomes resulting from the B. gaini ecological filtering of the surrounding environment microbial community. Both microenvironments exhibit their respective core microbiomes, yet the gill’s core microbiome is narrower and constitutes a smaller proportion of the overall bacterial community compared to that of the intestine. Within each host population (i.e. each sampling site), the microbiome assembles through distinct eco-evolutionary processes in both microenvironments, mostly stochastically (ecological drift) in the gill and deterministically (variable selection) in the intestine. Across different B. gaini populations, variable selection dominates in driving compositional divergence of both microenvironment microbiomes, although to a lesser extent in the gill. Lastly, our study reveals robust correlation between host intraspecific genetic structure and intestine microbiome composition, providing evidence of phylosymbiosis in anostracans. Contrastingly, phylosymbiosis was less pronounced in the gill microbiome. We discuss the potential differences in ecological filtering between each host microenvironment that may underlie the difference in the strength of phylosymbiosis. Our study highlights the relevance of considering host microenvironment and intraspecific levels in testing the phylosymbiosis hypothesis to better understand the intricate eco-evolutionary relationships between hosts and their microbiomes.
1 Introduction
Most macroorganisms exist in close association with diverse microbial partners, colonizing various physical niches provided by their hosts, thereby conforming what is termed metaorganisms (McFall-Ngai et al., 2013; Ludington, 2024). The variability in microbiome composition and function across different body parts has been linked to various facets of the host ecology (Zilber-Rosenberg and Rosenberg, 2008; Alberdi et al., 2022; Ludington, 2024). An iconic example is the human body, where each organ constitutes a distinct ecological niche that hosts a unique microbiome carrying out specific functions essential for host health (Costello et al., 2009; Dekaboruah et al., 2020). However, most of the animal-associated microbiome studies remain confined to compositional description of single-body sites (Rosenberg and Zilber-Rosenberg, 2018; Woodhams et al., 2020; Wang et al., 2023). Considering various body sites within the same host is necessary to gain a holistic understanding of the eco-evolutionary processes driving microbiome assembly (Stegen et al., 2013; Zhou and Ning, 2017). One of these eco-evolutionary processes is the contribution of the host evolution to microbiome assembly. Indeed, the intimate and prolonged ecological interdependency of hosts and their microbiomes can result in continuous mutual adaptation, also known as co-evolution (Wilson and Duncan, 2015; Rosenberg and Zilber-Rosenberg, 2018; Hayward et al., 2021; Mazel et al., 2023). Co-evolution can leave a phylogenetic footprint of the host in the microbiome composition, meaning that closely related host species exhibit more similar microbiomes, which is known as phylosymbiosis (Bordenstein and Theis, 2015; Brooks et al., 2016; Groussin et al., 2020). Although the detection of phylosymbiosis signature does not explicitly underpin a common evolutionary history between hosts and their microbiomes (Moran and Sloan, 2015; Mazel et al., 2018; Mallott and Amato, 2021), it constitutes a relevant hypothesis to delve into the eco-evolutionary mechanisms that shape metaorganisms-associated microbiome (Lim and Bordenstein, 2020; Mazel et al., 2023; Mallott, 2024).
Phylosymbiosis has been extensively tested across a biased fraction of metazoan species (Woodhams et al., 2020; Mazel et al., 2023), primarily vertebrates, including mammals (Groussin et al., 2017; Moeller et al., 2017; Youngblut et al., 2019) and fishes (Doane et al., 2020; Pan et al., 2023; Sadeghi et al., 2023; Schwob et al., 2024a), and to a lesser extent in invertebrates, like insects (Qin et al., 2023), sponges and corals (Pollock et al., 2018; O’Brien et al., 2020; Pankey et al., 2022). Despite a growing body of research, the universality of phylosymbiosis still remains to be fully tackled (Mazel et al., 2018; Lim and Bordenstein, 2020). The frequency and magnitude of phylosymbiosis appear to be inconsistent among taxonomic groups (Lim and Bordenstein, 2020), being relatively common and strong across mammals (Mallott and Amato, 2021), while looser in other vertebrates, such as birds (Trevelline et al., 2020; Bodawatta et al., 2022), and even absent in most (though not all) of the terrestrial and aquatic invertebrates studied (Hammer et al., 2019; Huot et al., 2019; Eckert et al., 2021; Boscaro et al., 2022; Eckert et al., 2022). Consequently, general conclusions about the underlying mechanisms of phylosymbiosis may be skewed by the restricted range of investigated models (Hammer et al., 2019), emphasizing the need to consider understudied species (Leasi et al., 2023). Some authors suggested that the phylosymbiosis signal might be obscured by extended divergence time among studied species (Groussin et al., 2017; Kohl et al., 2018; Ross et al., 2018), as well as by taxonomically rich microbiomes (Mallott and Amato, 2021), which contain a relatively high proportion of transient microorganisms (Ross et al., 2018). Conversely, the phylosymbiosis signal seems to be more frequent in internal rather than external host body sites (Moran et al., 2019). Moreover, considering the intraspecific variability of hosts offers an essential, yet often overlooked, resolution level for generalizing conclusions about the consistency of phylosymbiosis across taxonomic resolution and understanding how intraspecific host genetic variation associates with microbiome variation (Mazel et al., 2018; Couch and Epps, 2022). To the best of our knowledge, no study combines intraspecific resolution and multiple body microenvironments to test the eco-evolutionary processes in general, and phylosymbiosis in particular.
Characterizing the microbiome assembly across multiple body sites can be challenging, particularly in small-sized animals. To date, the microbiome of meiofaunal freshwater invertebrates like copepod and cladoceran has only been investigated at the whole individual-scale. These invertebrates host a specific microbiome discernable from the surrounding water microbiome (Samad et al., 2020), confirming the host-driven ecological filtering (Mazel et al., 2018). In Daphnia (Anamopoda), specific bacteria were consistently associated with geographically and genetically separated host populations, suggesting a stable “core microbiome” shaped by host-microbiome interactions (Qi et al., 2009; Guivier et al., 2018). Additionally, the gut microbiome of Daphnia magna is shaped by host genotype and enhances host fitness by providing essential nutrients (Cooper and Cressler, 2020; Macke et al., 2020; Motiei et al., 2020). In the branchiopod genus Artemia (Anostraca), host species has been identified as a key determinant of microbiome composition. The Artemia microbiome influences host tolerance to low salinity, thereby potentially contributing to its evolutionary trajectory (Nougué et al., 2015; Lee et al., 2024). Despite the ecological significance of branchiopoda-associated microbiomes, their variability across body sites and their eco-evolutionary drivers are unclear (Eckert et al., 2021; Boscaro et al., 2022). In previous phylosymbiosis studies focusing on invertebrates, the relatively small size imposed to use the whole individuals (Eckert et al., 2021; Leasi et al., 2023), or even to pool several individuals for microbiome assembly (Callens et al., 2018; Eckert et al., 2022). Besides ignoring the microbiome variability across host body site and among individuals, these approaches may have masked the impact of host phylogeny on microbiome assembly (Mazel et al., 2018).
The anostracan fairy shrimp Branchinecta gaini Daday (1910) is the largest (adult body size c. 16 mm) and most abundant crustacean species of Antarctic lacustrine habitats. B. gaini is widely distributed across Maritime Antarctica (i.e., South Shetland Islands, the Antarctic Peninsula, and the islands of the Scotia Arc) and southern South America (Hawes, 2009). It plays a key role in the trophic chain of these freshwater ecosystems (Peck, 2004; Hawes, 2009). B. gaini is considered as a primarily non-selective detritivore and herbivorous filter feeder (Lukic et al., 2023), although occasional predation on copepods like Boeckella poppei, larval stages of Parochlus steineni and even conspecific organisms has been reported (Paggi, 1996). B. gaini is characterized by two main microenvironments: a prominent branchial lamina (referred to as gill) and a digestive tract (referred to as intestine) easily discernable through body transparency (Lynch, 1964), facilitating the dissection of these tissues despite the host’s small size. The microbiome of B. gaini remains unexplored to date. Similarly, the intraspecific genetic variation of B. gaini in the Maritime Antarctica is uncharacterized, although other Branchinectidae species exhibit high intraspecific genetic divergences (Aguilar et al., 2017). Given the relative isolation of freshwater lentic ecosystems in Maritime Antarctica (i.e., patchiness of suitable habitat), B. gaini is likely to exhibit geographic and genetic structuring at an intra-species level, as previously reported in other freshwater invertebrates [e.g., B. poppei, Maturana et al. (2020)]. Thus, the fairy shrimp B. gaini represents a suitable model for comprehensively investigating the microbiome assembly processes and testing the phylosymbiosis signal across different host microenvironments in relation with the intraspecific genetic structure of host populations.
This study aims to (i) evaluate the distinguishability of B. gaini microbiomes between gill and intestine, (ii) identify the core microbiomes of gill and intestine, and their keystone Operational Taxonomic Units (OTUs), across geographically-distant sites in Maritime Antarctica, (iii) compare the eco-evolutionary processes governing gill and intestine microbiome assembly at local and regional scales, and (iv) elucidate the relative effects of host phylogenetic diversity, geography and climate in shaping both B. gaini microbiomes.
2 Methods
2.1 Host and environmental samples recollection and preparation
Specimens of B. gaini were sampled from Maritime Antarctica lake water between 2018 and 2023, during the austral summer (January), using a hand-crafted net, with mesh size < 1mm (specimen body size of 15–20 mm) (Table 1). The sampling sites encompass four shallow (0.5–1 m-deep) freshwater lakes, located in Signy Island (PUM) in the South Orkney Islands, King George Island (KIT, WUK) in the South Shetland Islands, and Avian Island (ANT) in the West Antarctic Peninsula (Figure 1). Immediately following sampling, entire individuals of B. gaini were preserved in absolute ethanol with one 50-ml Falcon tube dedicated per sampling site, for subsequent morphological and molecular analyses. The ethanol-preserved B. gaini individuals (8 specimens per site) were dissected under binocular magnifier to separate the head, the gill and the entire intestine with its content. Due to logistical constraints, superficial sediment was collected at a single site (ANT) in sterile Ziploc® bags. Samples from all the microenvironments (i.e., sediment, head, gill and intestine) were conserved into 2-ml sterile cryotubes at -20°C until further molecular analyses (Table 1).
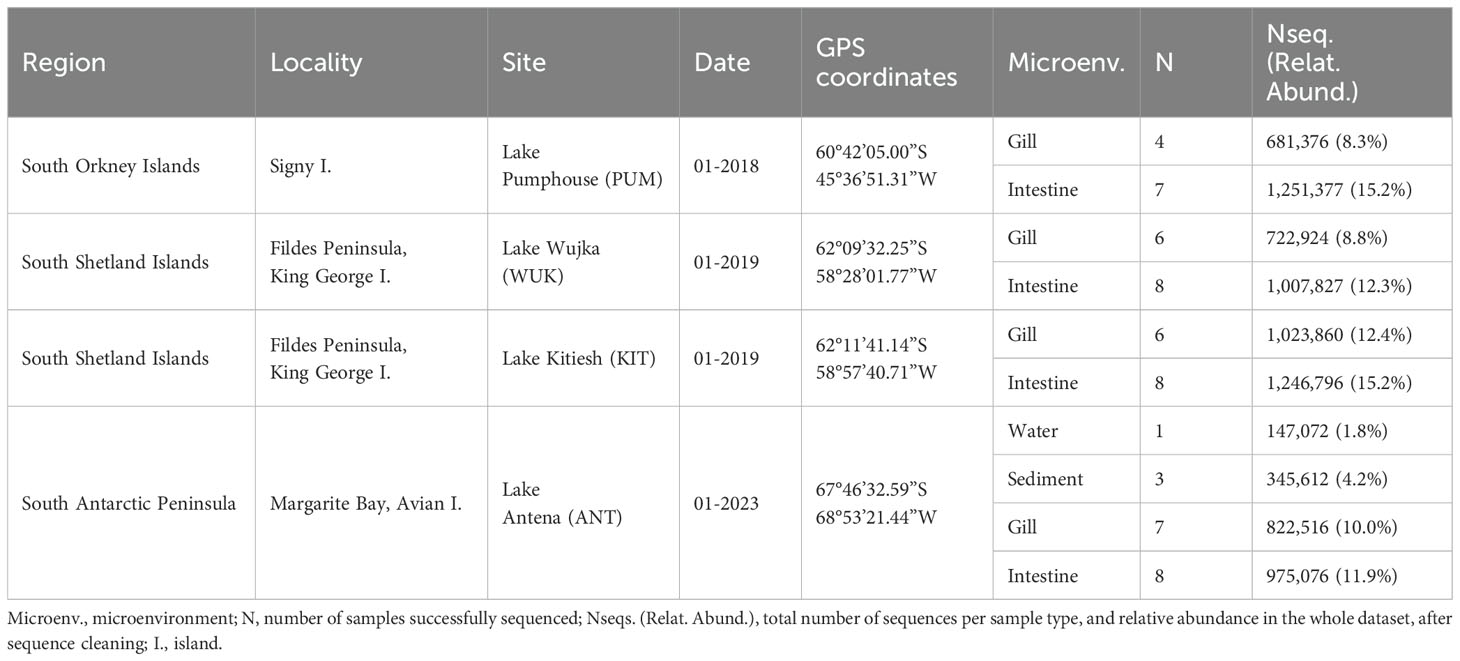
Table 1. Sampling locations of Branchinecta gaini and overview of metabarcoding sequencing data.
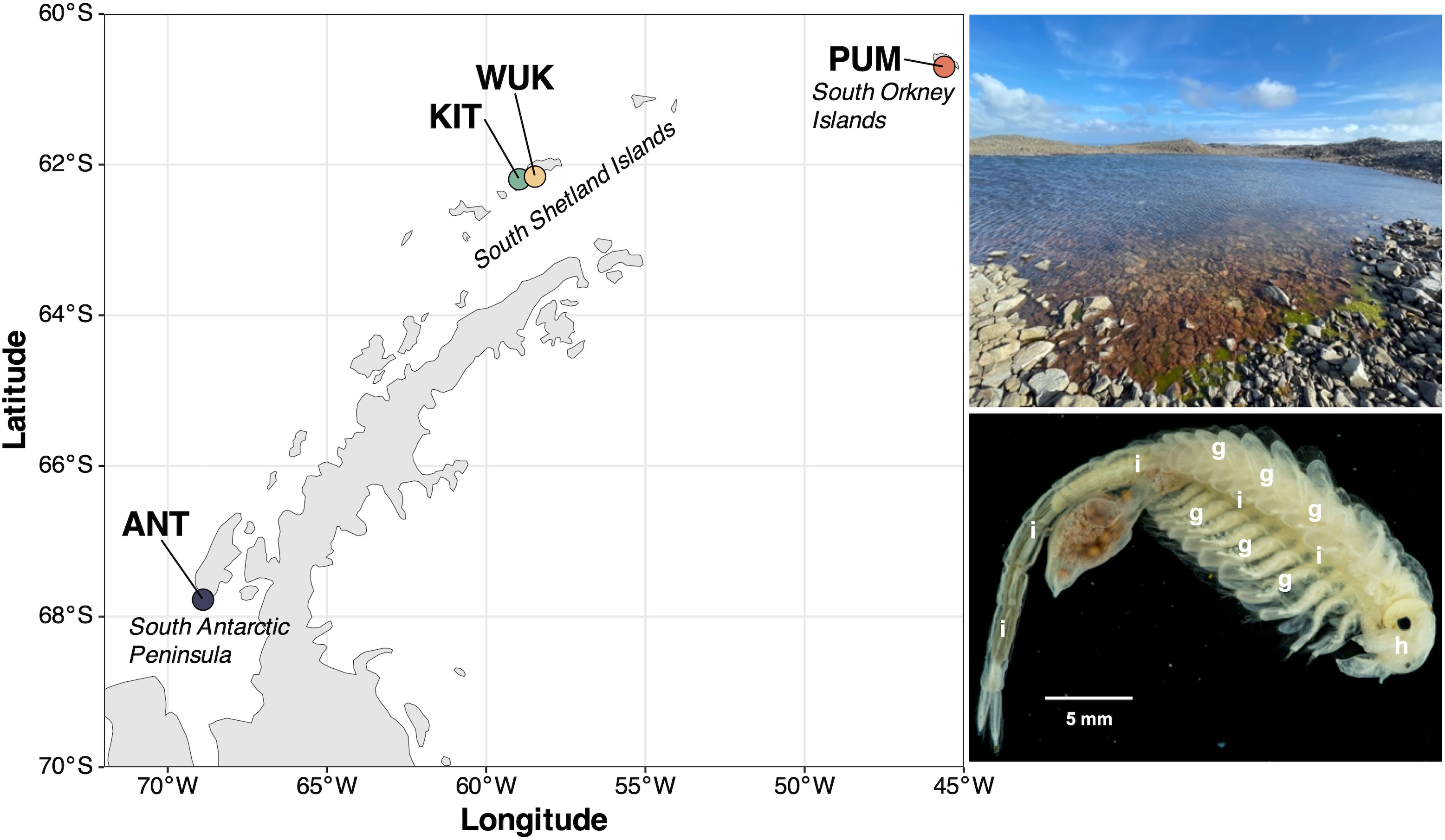
Figure 1. Localization of the sampling sites of Branchinecta gaini across Maritime Antarctica. From North to South, PUM: Lake ‘Pumphouse’ from South Orkney Islands, WUK and KIT: Lakes ‘Wukja’ and ‘Kitiesh’ from South Shetland Islands, and ANT: Lake ‘Antena’, from South Antarctic Peninsula (see Table 1 for details). The photos illustrate the characteristic habitat of B. gaini (above) and an individual of B. gaini (below). White letters “i”, “g” and “h” indicate the intestine, the gill and the head, respectively. More detailed photos are provided in Supplementary Figure S1.
2.2 Molecular procedures
Prior to molecular processing, host individuals were carefully rinsed with sterile water to remove the ethanol and minimize the risk of cross-contamination. For host analysis, the genomic DNA from B. gaini head samples was extracted using the DNAeasy Blood & Tissue Kit (Qiagen, CA, USA) protocol for small tissue samples with previous optimizations, including a preliminary incubation in proteinase K+ATL buffer solution overnight at 37°C (Maturana et al., 2020). One segment of the mitochondrial cytochrome c oxidase subunit I (cox1) was amplified using the universal primer LCO1490 and HCO2198 (Folmer et al., 1994). Detailed PCR protocol is given in Supplementary Material (Supplementary Material S1). The obtained PCR products obtained were sequenced in both directions at Macrogen Chile, using Sanger technology.
For microbiome analysis, the genomic DNA from gill and intestine samples was extracted using the DNAeasy PowerSoil Pro Kit (Qiagen, CA, USA) with a previous FastPrep-24® homogenization step (20 s at 4 m s-1) (MP Biomedicals, USA) according to manufacturer’s recommendations. Amplification, library preparation, and amplicon sequencing was carried out by Novogene (Beijing, China). Briefly, the hypervariable V3-V4 region of the 16S rRNA gene was amplified using the 341F-806R primers (Klindworth et al., 2013), and the Phusion® High-Fidelity PCR Master Mix (New England Biolabs, USA), following the PCR protocol detailed in Supplementary Material (Supplementary Material S2).
2.3 Bioinformatic treatment of sequencing data
Host cox1 partial sequences were manually quality controlled, assembled and edited using GENEIOUS 10.2.2 (Kearse et al., 2012). Alignments were obtained using MUSCLE (Edgar, 2004) with standard setting. The host phylogenetic tree was reconstructed from cox1 sequences with NGPhylogeny (Lemoine et al., 2019), using the HKY85+I model recommended by the implemented Smart Model Selection tool (SMS), and complemented by non-parametric SH-like branch supports. Subsequently, the pairwise phylogenetic distances between the pairs of host cox1 sequences within the host were calculated with the cophenetic.phylo function implemented in the ape R package (v5.7-1) (Paradis and Schliep, 2019).
Bacterial paired-end raw sequences were analyzed through the Mothur pipeline (v1.48.0) (Schloss et al., 2009), following the previously described procedure (Schwob et al., 2020). Chimeras were removed using Uchime implemented in Mothur (Edgar et al., 2011). Cleaned sequences were clustered into OTUs at 97%-identity, and low-abundance (< 0.0001%) OTUs were discarded according to (Bokulich et al., 2013). The final OTU table, representing a total of 688 OTUs, was rarefied at 80,000 sequences per sample and converted into a Bray-Curtis dissimilarity distance matrix using the vegdist function of the Vegan R package (v2.6-2) (Oksanen et al., 2011). The representative sequences of the OTUs were used to calculate the phylogenetic tree with NGPhylogeny (Lemoine et al., 2019), using the GTR+G+I model recommended by the implemented Smart Model Selection tool (SMS), and complemented by non-parametric SH-like branch supports. The bacterial phylogenetic tree was rooted on the branch separating Cyanobacteria from the rest of the sequences (Coleman et al., 2021).
2.4 Microbiome diversity analysis
The observed richness, the Chao1, and the Shannon indices were estimated using the plot_richness function from the Phyloseq R package (version 1.46) (McMurdie and Holmes, 2013), and Faith’s phylogenetic diversity was estimated using the estimate_pd function from the btools R package (v0.0.1) (Battaglia, 2021). Mean values of alpha-diversity indices among microenvironments were compared using a non-parametric pairwise Wilcoxon test through the geom_signif function of the ggpubr R package (Kassambara, 2020).
The beta-diversity pattern of B. gaini microbiomes across sites and microenvironments was visualized through a hierarchical clustering and a non-metric multidimensional scaling (NMDS) ordination, using the hclust function from the dendextend R package and the plot_ordination function in the phyloseq R package, respectively. Differences in community structure among sites and microenvironments were tested with PERMANOVA and pairwise PERMANOVA using the adonis2 and pairwise.adonis functions (vegan and funfuns R packages, respectively). The contribution of individual OTUs to the distinguishability of the microbiome among microenvironments was assessed using Linear Discriminant Analysis (LDA) Effect Size (LEfSe) analysis, performed via the run_lefse function in the microbiomeMarker R package (Cao et al., 2022).
The ecological niche breadth (BN) was computed for each OTU within the gill and intestine microbiome using the Levins’ method implemented in levins.Bn function of the microniche R package (Finn et al., 2020). To classify OTUs as generalists, specialists, or neutralists within gill and intestine microbiomes, their Levins’ BN values were compared to a null distribution, following the methodology described in Gao et al. (2023). The proportions of neutralists, generalists, and specialists were statistically compared between gill and intestine microenvironments using the dunn.test R package (Dinno, 2017). The mean community-level niche breadth was calculated for each sample and the significance of the difference in means between the two microenvironments was assessed using the Wilcoxon test.
To characterize the core microbiome of B. gaini gill and intestine, we applied the ‘common core’ definition (Risely, 2020), which encompasses the OTUs present in a large proportion of hosts (i.e., in > 75% of all replicates from each site), including rare OTUs (i.e. no abundance criteria) (Lahti et al., 2017). OTUs with an occurrence frequency of less than 75% were classified as part of the flexible microbiome. The OTU tables of gill and intestine core microbiomes were transformed into relative abundances (normalized by each total core abundance). Co-occurrence networks were computed for each core community using the SPIEC-EASI algorithm with default parameters in R (Kurtz et al., 2015). Nodes’ features, including closeness centrality (i.e., the proximity of a node to all other nodes in the network), betweenness centrality (i.e., the extent to which a node lies on the shortest paths among other nodes in the network), and degree count, were used to assess their potential hub taxa status (Berry and Widder, 2014). The microbiome assembly processes within the gill and intestine of B. gaini were analyzed both at each individual site and across all sites, and then averaged by microenvironments, using an in-house R function of the framework from Stegen et al. (2015), and previously published in Delleuze et al. (2024).
For phylosymbiosis analysis, Mantel tests with 9,999 permutations were conducted for each microenvironment (gill, intestine) to independently test the correlation between the microbiome Bray-Curtis dissimilarity distance and the host phylogenetic distance matrix using the cophenetic function implemented in the ape R package. The Mantel correlations were visualized under scatterplots generated with the ggplot R package (Wickham, 2016). Tanglegrams were generated using the tanglegram function in the dendextend R package to illustrate the correspondence of samples’ order between the dendrograms of gill and intestine microbiomes and the host tree. Partial Mantel tests were employed to determine the contribution of host phylogeny to beta-diversity in gill and intestine microbiomes, while controlling for geographic and climatic distances (Martiny et al., 2006). Geographic distances among sites were computed from the longitude and latitude coordinates using the earth.dist function of the fossil R package (Vavrek, 2011). Climatic variables, including precipitation amount (pr), mean daily maximum air temperature (tasmax), mean daily minimum air temperature (tasmin), mean daily air temperature (tas), and surface downwelling shortwave flux (rsds) were obtained from the CHELSA database v2.1 (Karger et al., 2017), selecting January 2018 data as the most coincident with our sampling time. Standardization of climatic variables was performed using the decostand function (Vegan). Next, automatic stepwise model selection for redundancy analysis was performed through the ordiR2step function (Vegan) to select the climatic variables that explained most of the variance in the OTU composition of gill and intestine samples. To address collinearity among explanatory factors, principal component analysis (PCA) was conducted on the selected climatic variables (i.e., pr, tasmax, and rsds), and the PCA axis scores were transformed into Euclidean distances using the vegdist function (Vegan) that will be further used as the climatic distance matrix.
3 Results
3.1 Significant alpha-diversity variations across B. gaini microenvironments
Microbiome alpha diversity varied significantly across B. gaini microenvironments. Except for the WUK site, the observed and Chao1-estimated OTU richness was approximately twice higher in intestinal samples compared to gill samples (Figure 2; Supplementary Figure S2). Likewise, greater phylogenetic diversity was observed in intestine than in gill (Figure 2). Contrarily, the Shannon diversity values were more homogenous between gill and intestinal samples across sites (Supplementary Figure S2). At the ANT site, alpha diversity indices of the lake sediment (excluding the Shannon index) were comparable to those of the intestine and significantly higher than those of the gill samples (Supplementary Figure S3).
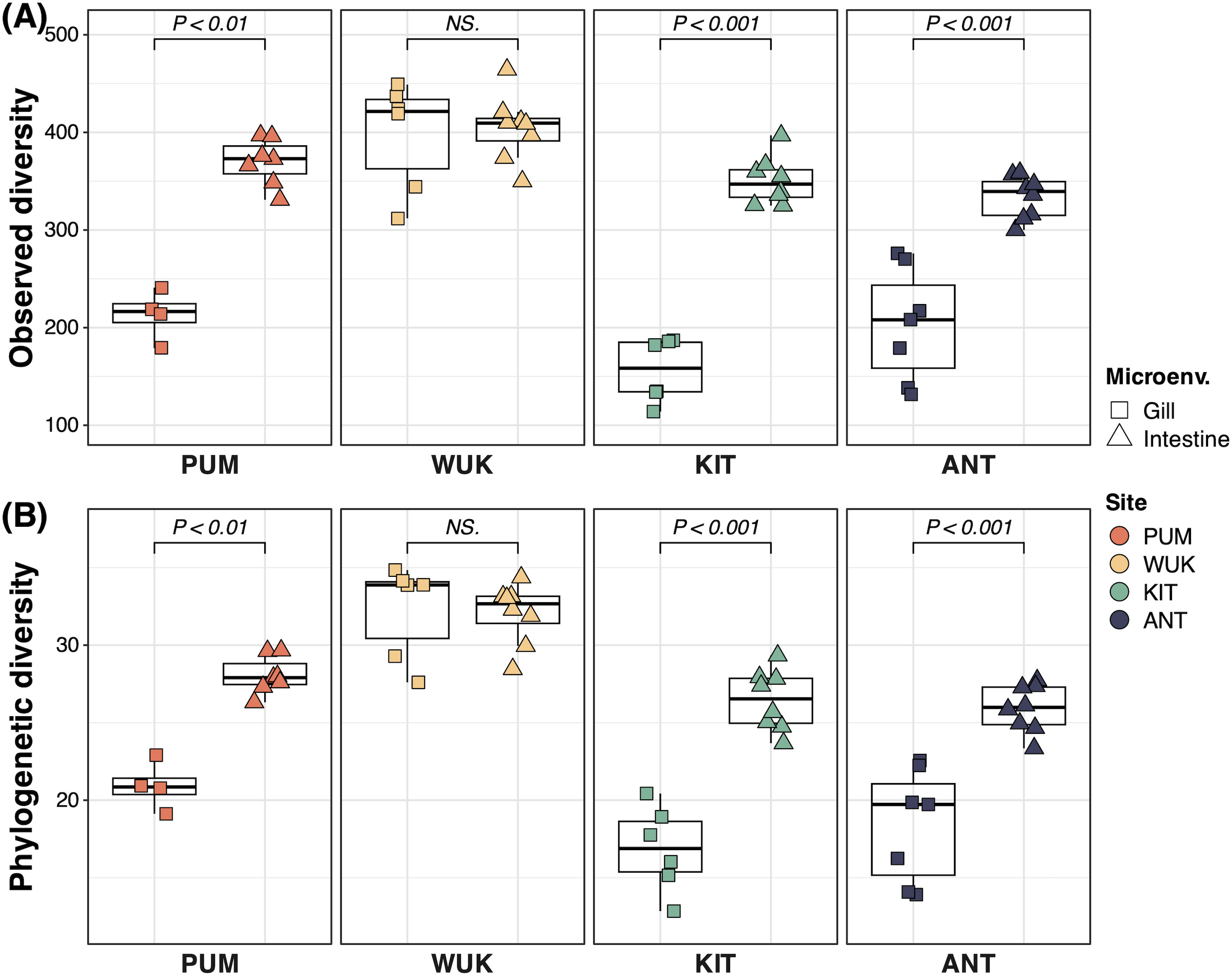
Figure 2. Bacterial alpha diversity in gill and intestine microbiomes of Branchinecta gaini. The shape and color correspond to the microenvironment and to the site, respectively. The significance of the differences was inferred through Wilcoxon tests, and p-values (P) are displayed. ‘NS.’ indicates non-significant differences. (A) Observed diversity; (B) Chao Index.
3.2 Unique microbiome features in gill vs. intestine of B. gaini
Within each site, clear dissimilarities in microbiome compositions were observed between gill and intestinal microenvironments (pairwise PERMANOVA, p-values < 0.005, Table 2; Figures 3A, B). For ANT and KIT, the microbial communities clustered primarily by microenvironments and secondly by site, indicating that microbiome compositions among samples from the same microenvironment (despite coming from different sites) were more similar than among samples from the same site (Figures 3A, B). Intestinal communities were on average more homogeneous than gill communities (mean pairwise dissimilarity in gill: 0.71 ± 0.006, vs. in intestine: 0.56 ± 0.006, Wilcoxon test, p < 0.001, Supplementary Figure S4). The effect of the sampling site in shaping B. gaini microbiome composition was twice as strong as that of the microenvironment (R2 = 0.39, p = 0.001 and R2 = 0.19, p = 0.001), and was about 25% stronger in intestine than in gill (R2 = 0.78, p = 0.001 and R2 = 0.51, p = 0.001, respectively, Table 2; Figure 3B). The effect of the host microenvironment on community composition differed depending on the levels of the site factor, as evidenced by the significant interaction in the PERMANOVA (R2 = 0.13, p = 0.001, Table 2), and by the hierarchical clustering in which communities clustered either according to the microenvironment (i.e., KIT and ANT) or by site (i.e., PUM and WUK, Figure 3A). At the ANT site, sediment microbiome composition was more similar to intestine than gill samples (Figures 3A, B).
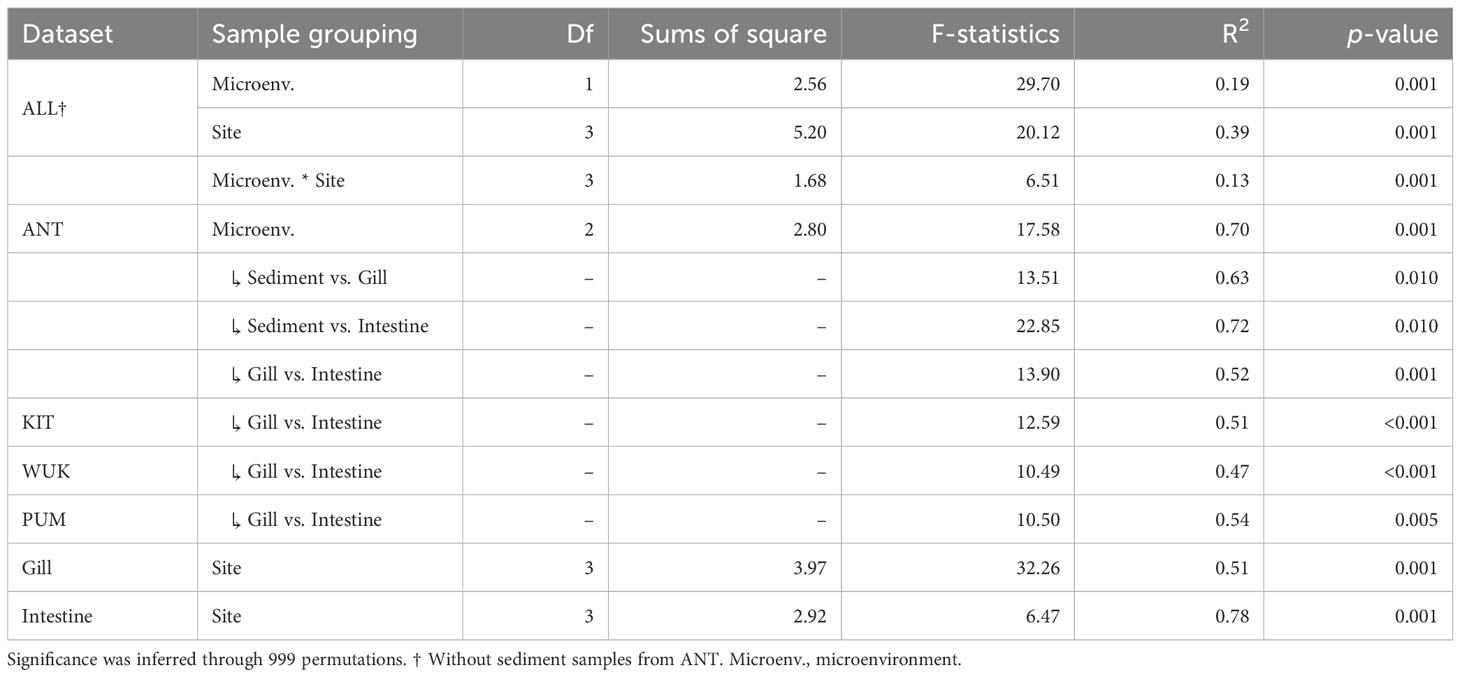
Table 2. Individual and pairwise PERMANOVA on Bray-Curtis dissimilarities assessing the effect of microenvironment and site on microbiome composition.
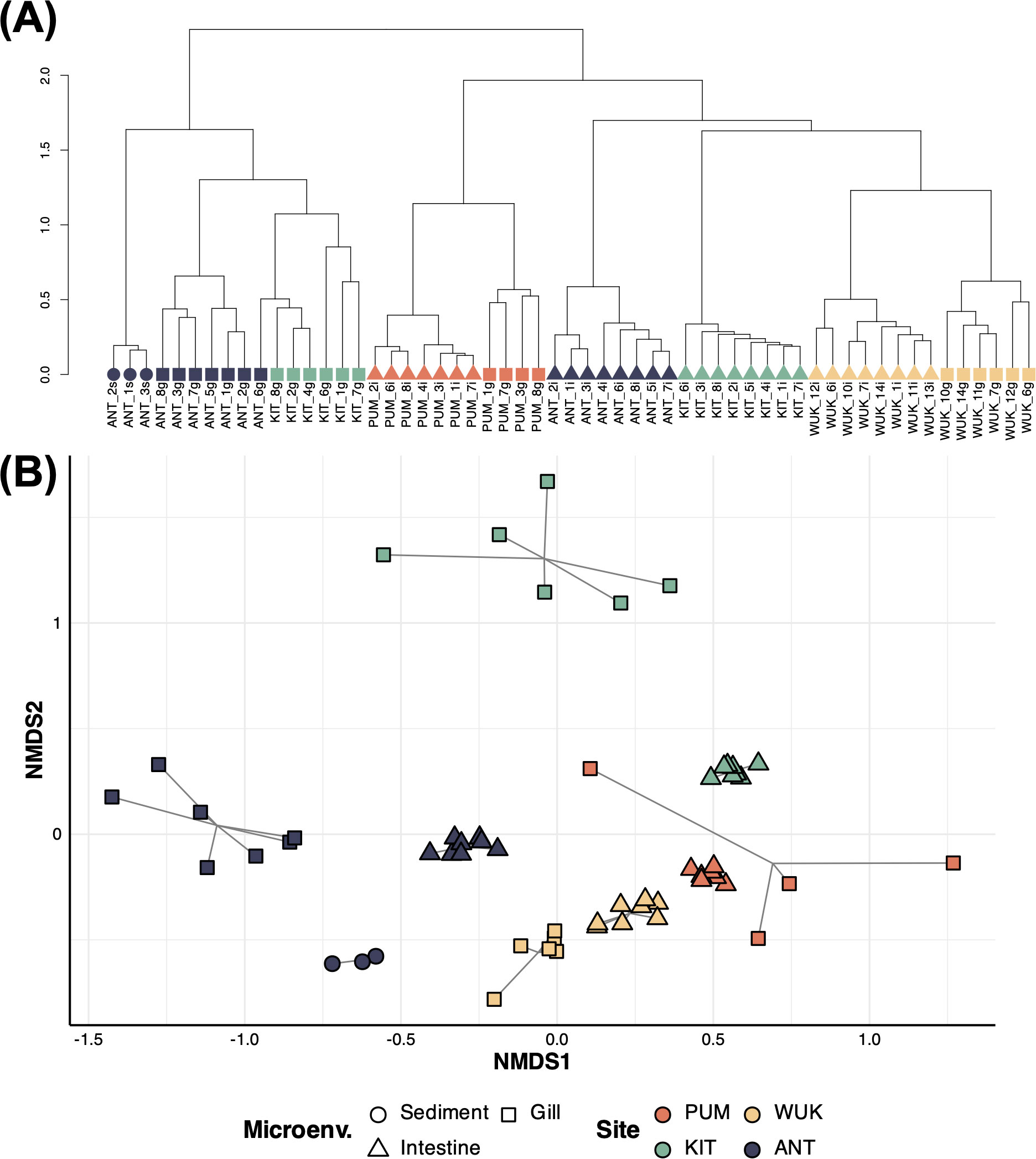
Figure 3. Bacterial beta diversity in Branchinecta gaini-associated and lake sediment microbiomes. Hierarchical clustering (A) and non-metric multidimensional scaling ordination (B) are based on Bray-Curtis dissimilarities among B. gaini microenvironments. Clustering is performed using the ward. D2 agglomeration method. The shapes and colors correspond to the microenvironment and to the site, respectively. In sample nomenclature, the first three letters, the number, and the last lowercase letter correspond to the site, the sample number, and the microenvironment, respectively.
The partitioning of gill, intestinal, and sediment microbiomes was explained by the significant enrichment of discriminant bacterial taxa within each microenvironment, with a total of 375 discriminant OTUs identified through LEfSe analysis. The taxonomic affiliations of the top 20 OTUs with highest LDA scores within each microenvironment encompassed 12 bacterial classes (Figure 4). In the gill microbiome, discriminant OTUs were mainly from the Bacteroidia class, predominantly represented by Flavobacterium. In the intestine microbiome, most of discriminant OTUs were assigned to the Clostridia and Alphaproteobacteria classes, represented by Clostridium and Tabrizicola, respectively (Figure 4). The discriminant OTUs of sediment microbiomes predominantly belonged to the Gammaproteobacteria and Bacteroidia classes, represented by various taxa such as Rhodoferax and Lentimicrobiaceae, among others.
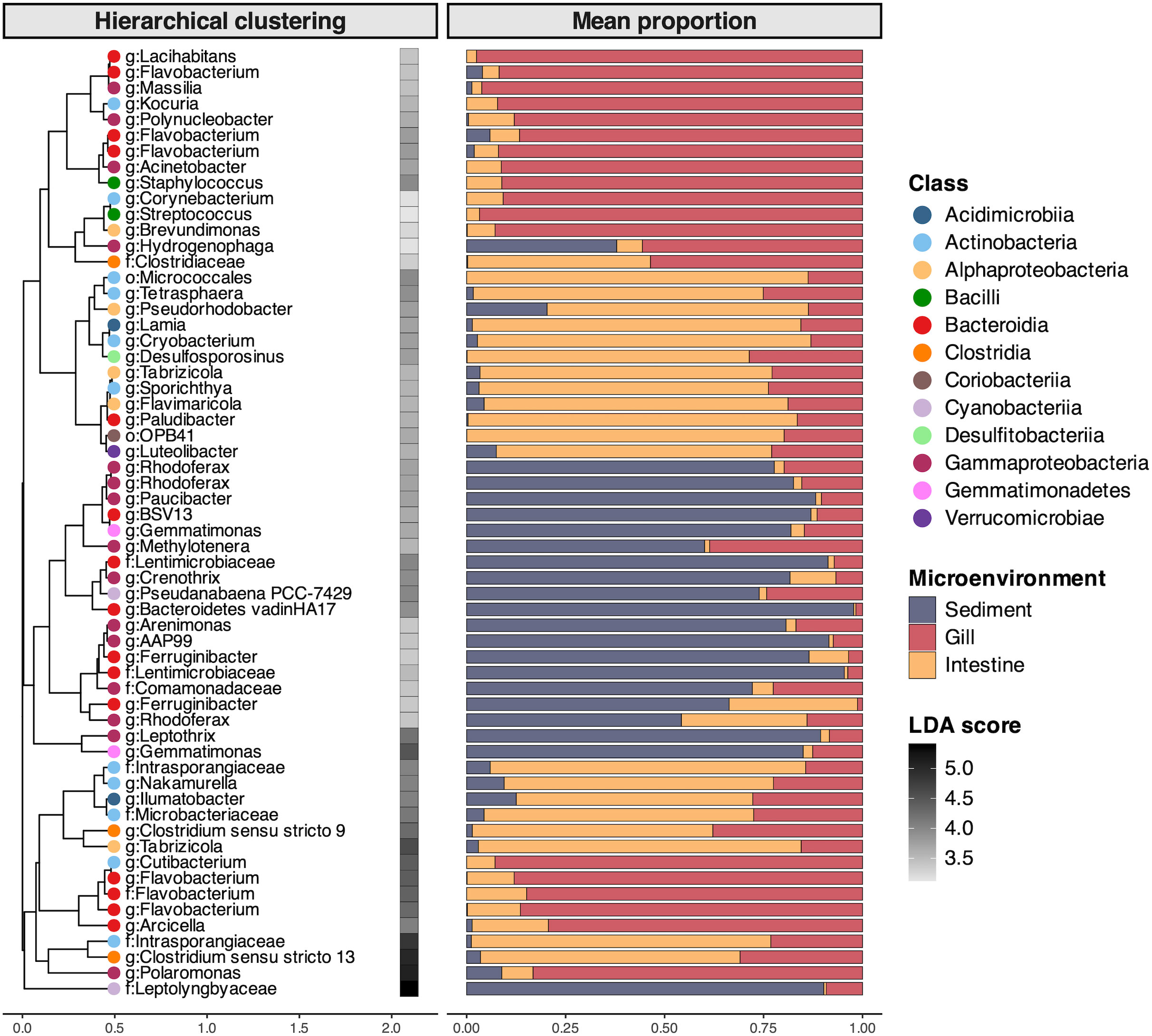
Figure 4. The twenty most enriched bacterial OTUs within each B. gaini and lake sediment microenvironments identified through Linear discriminant analysis (LDA) effect size (LEfSe) analysis. Taxonomic affiliations at the genus level (g): or at the family level (f): when genus was undetermined, and LDA score are presented for each OTU. The LDA score is represented by the color gradient in the heatmap. The barplot indicates the mean relative abundance of the indicator bacterial OTUs to the microbial community within each microenvironment (Microenv.). The hierarchical clustering is based on complete method from OTU relative abundance across microenvironments.
The Levins’ niche breadth index (Bj) was roughly 30% higher in the intestine compared to the gill microenvironment (Wilcoxon test, p < 0.001, Figure 5, left panel). The proportion of specialist OTUs was strongly higher in the gill community (96%) than in the intestine community (52%). Generalist OTUs were nearly absent in both microenvironments (< 0.01%), and around half of the intestinal OTUs were neutralists (Figure 5, right panel).
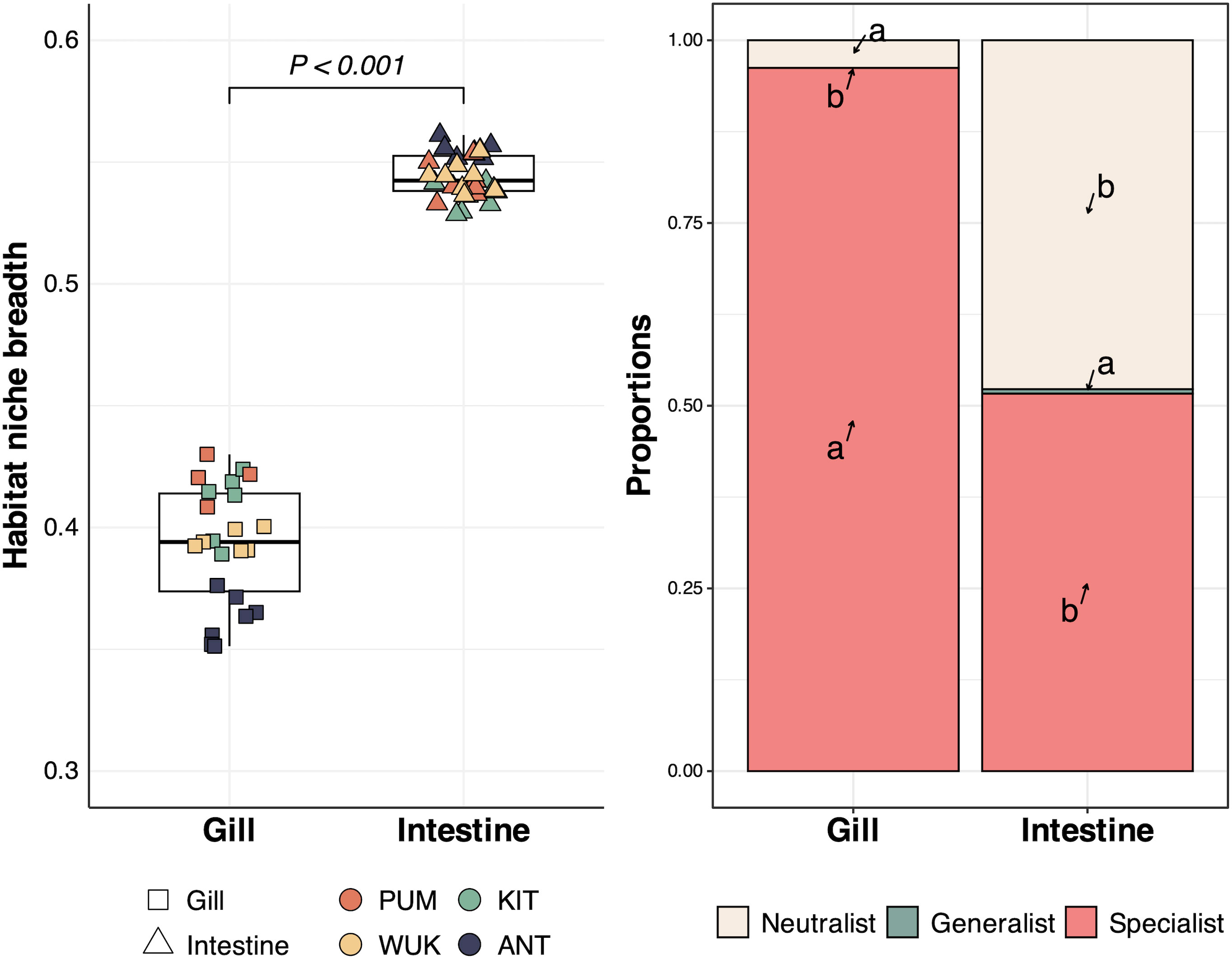
Figure 5. Habitat niche breadth (left) and proportions of habitat neutralists, generalists, and specialists (right) of the microbiome of Branchinecta gaini microenvironments. In the left panel, means of the Levins niche breadth index (Bj) were calculated for each OTU within a community (i.e., sample). Differences between microenvironments were assessed for significance using the Wilcoxon test. In the right panel, arrows are used to indicate the different category within each microenvironment. Different lowercase letters between gill and intestine for the same category (e.g., neutralists) indicate significant difference, as determined by the Dunn test. All comparisons were statistically different (p-values < 0.001).
3.3 Core microbiome detected but no hub taxa in gill and intestine of B. gaini
All core OTUs detected had a minimum prevalence of 87.5% across all samples and variable abundances, particularly within the intestine, as indicated by the abundance-occupancy curves (Figure 6A). Gill samples revealed a more flexible microbiome with only 12 core OTUs detected, whereas intestinal samples displayed 107 core OTUs (Figure 6B). In both microenvironments, the core OTUs accounted for substantial fractions of the entire bacterial communities, with cumulative relative abundances of 45.5% (gill) and 73.3% (intestine) of the community (Figure 5). The core composition was mostly distinct between both microenvironments. In line with the microenvironment-specific OTUs detected in the preceding section, the gill core microbiome was mostly dominated in abundance by OTUs belonging to genera like Polaromonas and Flavobacterium. Conversely, the intestine core microbiome was primarily dominated by OTUs belonging to Intrasporangiaceae and Tabrizicola (Figure 4; Figure 6C). The only bacterial genera (with relative abundance > 0.015) represented in both gill and intestine were Clostridium sensu stricto 9 and 13 (Figure 6C), previously detected as significantly enriched solely within intestinal samples (Figure 4).
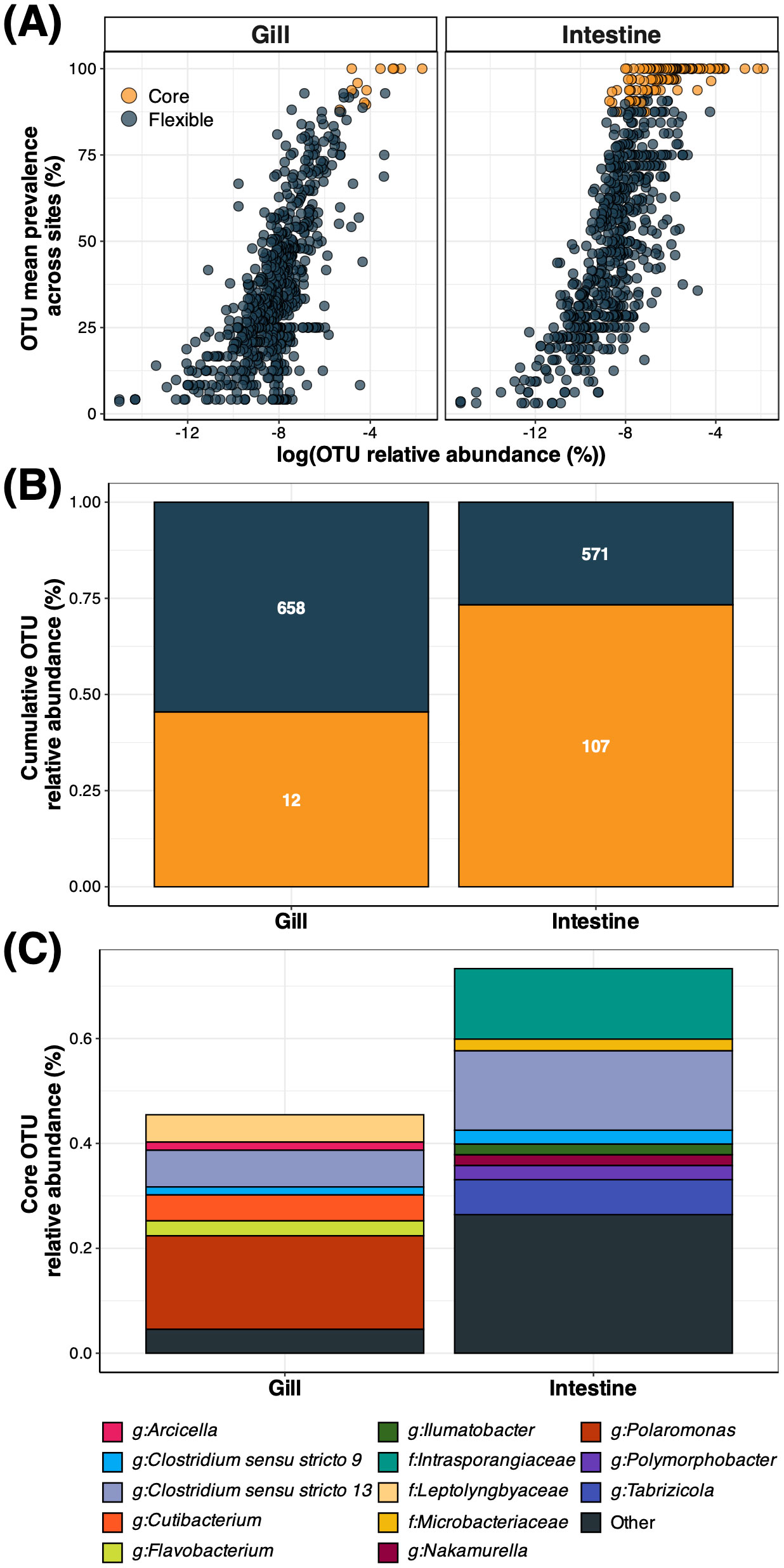
Figure 6. Bacterial core and flexible microbiomes of B. gaini gill and intestine. (A) represents the abundance-occupancy curves of all OTUs in B gaini gill and intestine. Colors indicate either the core (carrot orange) or flexible OTUs (charcoal blue). (B) represents the cumulative relative abundance of core (carrot orange) and flexible OTUs (charcoal blue) in the gill and intestine. The number inside each bar section indicates the number of OTUs. (C) displays the taxonomic composition at the genus level (or family level when genus information was not available) of the gill and intestine core microbiome OTUs. Core OTUs with relative abundance < 0.015 are represented under the label “Other”.
Co-occurrence networks computed for both gill and intestine core microbiomes did not reveal any hub taxa in either microenvironment. In the gill core microbiome, a single significant interaction was identified between Acinetobacter and Microbacteriaceae taxa, with betweenness centrality values below the threshold to be considered as hub taxa. Contrastingly, in the intestine microbiome, co-occurrence interactions among core OTUs were more abundant, with average betweenness and closeness centrality of 209.2 ± 20.0 and 0.002 ± 2.5e-5, respectively, and a degree count of 3.3 ± 0.2; however, none of the nodes in the core co-occurrence network reached the 97th percentile of closeness and betweenness centrality values, precluding them from being classified as hub OTUs (Supplementary Figure S5).
3.4 Divergence-driving processes dominate in B. gaini microbiome assembly
Ecological processes leading to microbiome divergence (i.e., ecological drift, dispersal limitation, and variable selection) accounted for most (> 95%) of the community composition turnover in both microenvironments, regardless of the geographical scale (i.e., intra- and inter-site) (Figures 7A, B). However, the respective contributions of these ecological processes were markedly different between the gill and intestine. Notably, at the intra-site scale, variable selection was the dominant process within the intestinal microbiome across B. gaini individuals, accounting for 60% of the community composition turnover, while it only explains about 6% of the turnover observed in the gill microbiome. In contrast, ecological drift was the most important process in the gill microbiome, driving more than 70% of the community composition turnover but only 36% in the intestine microenvironment. Additionally, the dispersal limitation contribution, absent in the intestine microbiome assembly processes, drove 18% of the intra-site turnover of the gill bacterial community (Figure 7A).
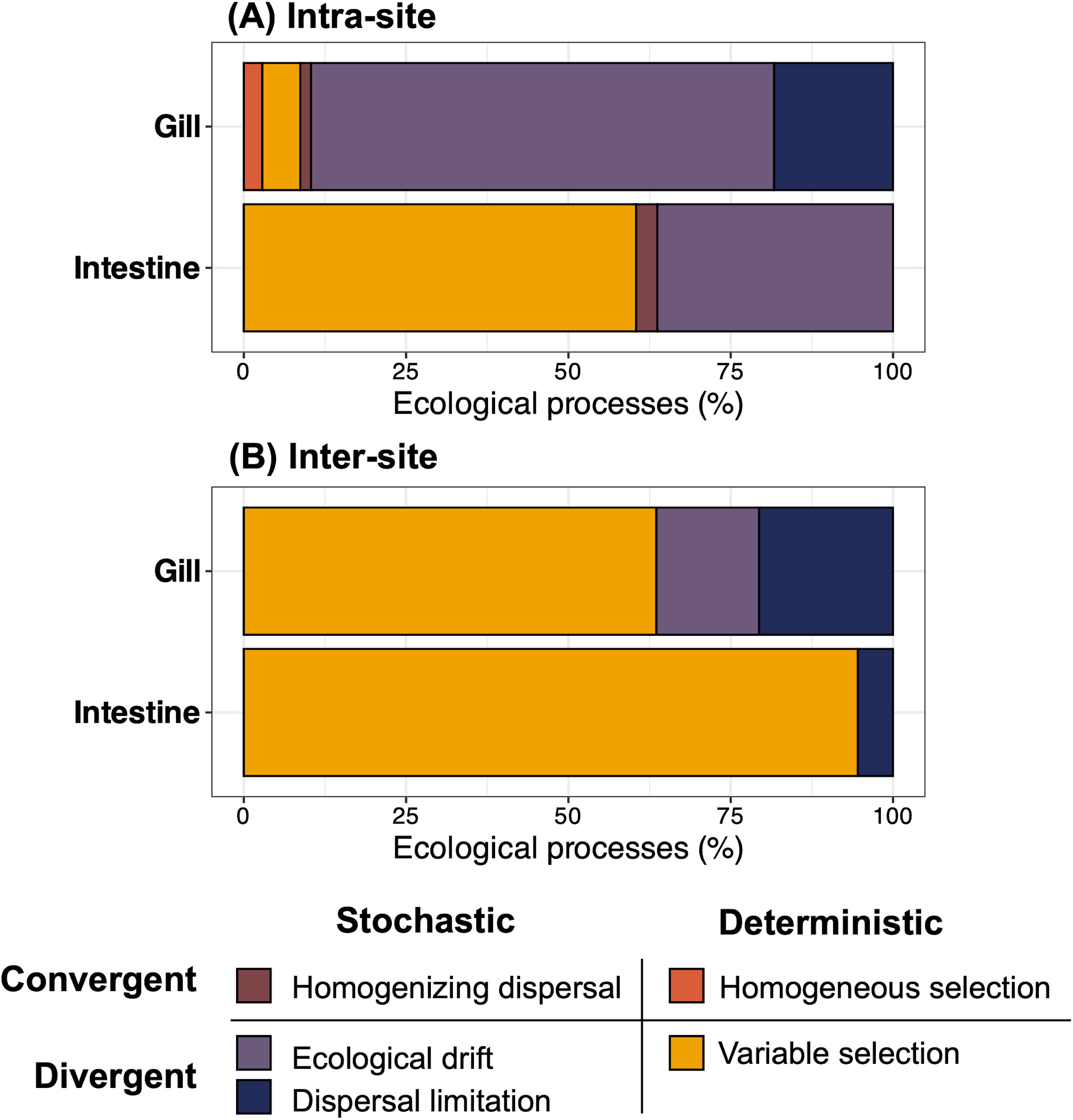
Figure 7. Estimation of the ecological processes governing the microbiome of Branchinecta gaini microenvironments within (A) and among (B) sites. The significance of the contribution changes in ecological processes was determined using a permutation test. The ‘ecological processes’ axis refers to the mean contribution of the community assembly processes.
When examining the inter-site turnover, the contribution of variable selection increased to 95% and 58% in the gill and intestine microbiomes, respectively. The contribution of dispersal limitation slightly increased, accounting for 21% and 5% in the gill and intestine microbiomes, respectively. Meanwhile, the contribution of ecological drift decreased to 16% and 0% in the gill and intestine microbiomes, respectively (Figure 7B).
3.5 Stronger host phylogenetic footprint in intestinal vs. gill microbiome of B. gaini
A substantial congruence was detected between microbiome composition dissimilarity and B. gaini phylogenetic distances, with a stronger correlation in the intestine (R2 = 0.64, p = 0.0001) compared to the gill (R2 = 0.49, p < 0.0001) (Figure 8A). Upon controlling for either climatic or geographic variability, the correlation strength slightly weakened for both microenvironments, but remained higher in the intestine than in the gill microbiome (Supplementary Table S3). In addition, while the p-values of partial Mantel tests remained largely significant in case of the intestine, they became less significant for the gill, and even marginally significant in the case of climatic distance-controlled correlation (Supplementary Table S3). Finally, the degree of alignment between the microbiome dendrogram and the host phylogenetic tree labels (i.e., entanglement) was twice higher in the intestine compared to the gill (Figure 8B).
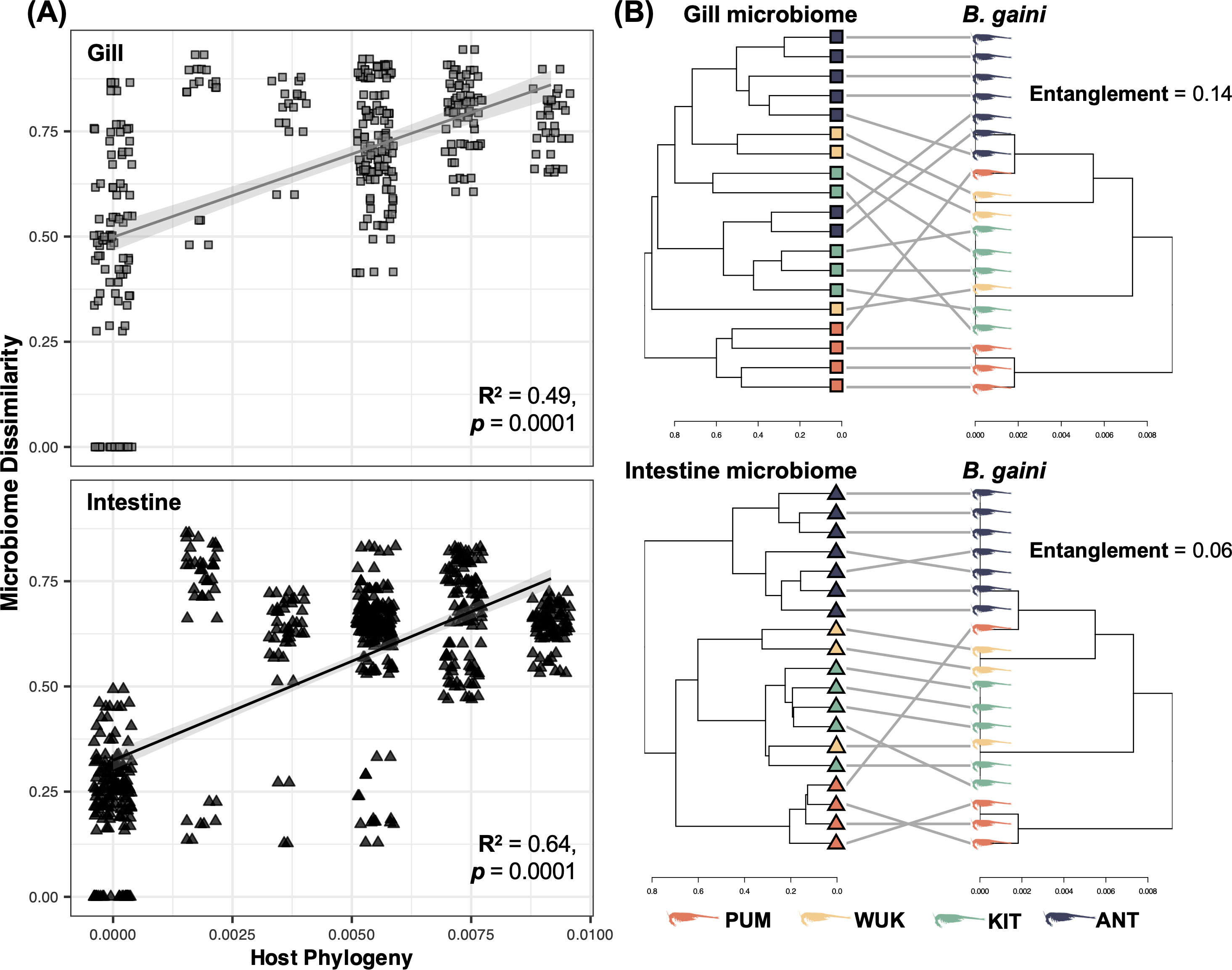
Figure 8. Congruence between Branchinecta gaini phylogeny and its microbiome composition. (A) Scatter plots of partial Mantel test results based on microbiome dissimilarity of B. gaini gill (gray square) and intestine (black triangle), using Bray-Curtis distances and host phylogeny. R2 and p-values based on 10,000 permutations are provided. (B) Tanglegrams showing the association between the estimated B. gaini phylogeny and its gill (square) or intestine (triangle) microbiome composition. The scale and direction of the divergence are indicated by the scale bars, representing Bray-Curtis dissimilarity and pairwise phylogenetic distances for microbiome and host, respectively. The degree of alignment between the dendrograms is presented under a value of entanglement (0: fully aligned labels, 1: fully mismatched labels). Colors represent the sampling site as follows: Terra cotta; PUM, goldenrod yellow: WUK, asparagus green; KIT, dark slate blue; ANT.
4 Discussion
We have limited understanding of how microbiome assembly vary across host microenvironments and among intraspecific populations of the same species. In this study, we surveyed the microbiome associated with two microenvironments of the fairy shrimp B. gaini across four populations distributed throughout Maritime Antarctica, and explored the eco-evolutionary mechanisms influencing the assembly of these microbiomes. Our results refine previous findings on meiofaunal host-microbiome relationships and verify several general hypotheses regarding the detection of phylosymbiosis (Woodhams et al., 2020).
4.1 B. gaini microbiome is shaped by host microenvironments
Assessing the distinguishability of bacterial community across host microenvironments and between the host and the surrounding environment is an essential first step to further unravel the microbiome significance in host ecology and evolution (Hammer et al., 2017; Schwob et al., 2024b). The composition of gill and intestine microbiomes of B. gaini significantly differed from each other. This finding demonstrates that fairy shrimps host body-site specific microbiomes, as previously observed in bigger crustaceans from Malacostraca class such as shrimps and crabs (Zhang et al., 2016; Cornejo-Granados et al., 2018). Our results underscore the importance of examining distinct organs individually, even in small and seemingly simple organisms like branchiopods. Moreover, the gill and intestine microbiomes were distinct from the lacustrine sediment environment. This first evidence of host ecological filtering in B. gaini suggests a selective enrichment of specific bacterial taxa able to colonize the gill and intestine tissues within B. gaini (Moran and Sloan, 2015; Mazel et al., 2018). A similar pattern has been previously reported through whole individual analysis in other small and microscopic freshwater organisms (Samad et al., 2020; Eckert et al., 2021; Boscaro et al., 2022).
4.2 Greater specificity and stochasticity in gill versus intestine microbiome assembly of B. gaini
Based on alpha-diversity results, host filtering seems to be stronger in the gill than in the intestine, limiting its colonization to a fewer number of, and more phylogenetically homogeneous, bacterial taxa compared to the intestine samples. This suggests that the gill may be a more selective habitat with a lower microbial carrying capacity compared to the intestine. Consistently, almost all the gill OTUs were identified as specialists according to the Levin’s niche breadth framework. For instance, the most enriched and prevalent bacterial taxa in the gills is Polaromonas, a psychrophilic genus previously isolated from saline pond water, and various glacial environments, including lake microbial mat and glacier surfaces in Antarctica (Kapardar et al., 2010). Some authors propose that this genus can form dormant cells, facilitating its survival and dispersion over time and space (Darcy et al., 2011). Moreover, the Polaromonas genus is described as metabolically versatile due to high levels of horizontal gene transfer, allowing it to thrive during transient periods of higher temperatures and substrate availability (Yagi et al., 2009). This “opportunitroph” lifestyle (Polz et al., 2006) echoes the ecological strategy observed in B. gaini, which can survive extreme environmental fluctuations (Peck, 2004). B. gaini gills were also enriched in Flavobacterium, a genus associated with gill rot symptoms in various cold freshwater fish species (Farkas, 1985). The enrichment of these genera in the gills of freshwater invertebrates from the surrounding habitat is unprecedented and warrants further dedicated study to uncover their potential ecological roles.
The apparently stronger specificity observed in the gill versus the intestine aligns with the greater contribution of dispersal limitation processes in the gill, as previously shown in other organ-associated microbiomes of invertebrates and mammals (Mazel et al., 2024). This suggests that gill-associated OTUs possess phenotypic bacterial traits associated with reduced dispersal abilities (Mallott, 2024). Additionally, we identified a narrow core microbiome in the gill consisting of only 12 OTUs, yet accounting for almost 50% of all reads in the gill. This finding suggests that at least a fraction of the gill microbiome may be less transitory, reflecting some degree of host-symbiont fidelity over time and space (Risely, 2020).
However, the relatively higher variability in gill microbiome composition compared to the intestine, coupled with the greater proportion of flexible microbiome, challenges the notion of a highly specific gill microenvironment and prompts distinct scenarios to explain this discrepancy. First, in line with observations from other crustacean species, we propose that the microbial community colonizing B. gaini gill is periodically eliminated and gradually re-establishes after each host molting event (Corbari et al., 2008; Middlemiss et al., 2015; Zhang et al., 2021). Although the molting rate in B. gaini remains uncertain, estimates for the closely related species B. giga suggest approximately 18 molts per life cycle (Daborn, 1975). It is thus plausible that asynchronicity in molting and differences in the gill recolonization outcomes across individuals introduce variability in the microbiome composition.
Alternatively, the unique anatomical structure of the anastrocan gills, which lack a protective gill chamber unlike larger invertebrates, results in a more direct and continuous exposure to the environment (Paggi, 1996). Consequently, the gill microbiome may be more influenced by the lake water column than the intestine, leading to greater variability across sampling sites, potentially reflecting environmental differences among lakes. This aligns with the stronger increase of variable selection from intra-site to inter-sites in the gill compared to the more stable intestine microbiome. The high flexibility of microbiome in response to the external environment has been previously described in freshwater zooplankton, including rotifers, copepods, and cladocerans (Eckert et al., 2021). Although the lack of direct water sampling in this study limits our understanding of the water microbiome’s influence on the gill microbiome, Branchinecta species primarily inhabit the surface of the nutrient-rich substrates at the bottom of Antarctic lakes, rather than the water column. These sediments can be considered as their primary habitat, as Branchinecta actively plow through superficial sediments using their thoracopods (Cáceres and Rogers, 2015). Additionally, the interstitial water present in the sediment samples provides an indirect representation of the water environment, compensating to some extent for the absence of explicit water samples in this study.
Finally, stochastic processes, primarily ecological drift, dominates in B. gaini gill microbiome assembly at a local scale (intra-site), suggesting that the microbiome is mostly acquired horizontally through pervasiveness uptake of environmental bacteria (Hammer et al., 2017, 2019; Rosenberg and Zilber-Rosenberg, 2021). Ecological assembly processes have not been specifically estimated in the past in crustacean models, but this finding intuitively contrasts with the typical pattern described in gill of other crustaceans, such as crabs, where the microbiome coating the gills lamella is dominated by stable bacterial taxa across host populations (Bacci et al., 2023; Fusi et al., 2023). The importance of ecological drift during establishment of symbioses has been demonstrated to weaken/counteract deterministic selection through mechanisms such as priority or founder effect (Hagen and Hamrick, 1996; Kohl, 2020). Thus, the colonization outcome (i.e., symbiont community composition) of a selective microenvironment may vary within and between host populations after the coarse ecological filter against taxa unable to colonize the tissue, even in the case of highly specific and vertically-transmitted symbionts (Vega and Gore, 2017; Lange et al., 2023; Chen et al., 2024). Such mechanisms may explain the detection of relatively variable community composition, the detection of stochastic assembly processes in the gill, in spite of the specificity of this microenvironment, which initially seemed contradictory.
4.3 Greater consistency and determinism in intestine versus gill microbiome assembly of B. gaini
We detected a predominant core microbiome across the four populations of B. gaini accounting for almost two thirds of the total community abundance, suggesting the existence of stable, permanent, and abundant microbial partners in the intestine of B. gaini (Astudillo-Garcia et al., 2017). Among the core microbial taxa, we reported the Intrasporangiaceae family, which has been characterized as highly abundant in the intestine of terrestrial earthworms and correlated with the digestive capacity of the host (Liu et al., 2023). This family has also been repeatedly detected in Antarctic ecosystems, mainly in soils and seawater (Giudice et al., 2007), but also associated with metazoan hosts such as tardigrades living on the surface of glaciers (Zawierucha et al., 2022). The Clostridium sensu stricto 13 and 9 genera were also particularly prevalent and enriched in B. gaini intestines. Representatives of the Clostridia class, including Clostridium, are typically detected in the gut of freshwater invertebrates and fish (Zhao et al., 2018; Weingarten et al., 2019; Savard et al., 2023). Clostridium is a fermentative anaerobe known for its chitinolytic activity (Olsen et al., 1999). We suggest that the enrichment of these genera in the intestine of B. gaini might be attributed to their chitinolytic activity, enabling them to leverage the chitin-rich peritrophic matrix.
As in the gill microenvironment, the microbiome assembly processes within the intestine of B. gaini revealed a predominance of processes leading to a divergent microbiome. Yet, in contrast with the gill, these processes were mostly deterministic, dominated by variable selection and ecological drift at intra-site scale, and almost exclusively by variable selection at inter-sites scale. Our findings contrast with several studies of the macrofauna gut microbiomes, which have identified either deterministic processes leading to phylogenetically convergent gut microbiome (i.e., homogenizing selection) (Xiong et al., 2017; Li et al., 2019), or mostly stochastic processes leading to divergent microbiome (i.e., mainly ecological drift) (Yan et al., 2016; Ge et al., 2021; Schwob et al., 2021). Our results also diverge from the typical pattern observed in the micro-eukaryotic community of freshwater lakes on the King George Island (South Shetland Islands, Maritime Antarctica) (including the same sampling site as ours), which are dominated by homogenizing dispersal process (Zhang et al., 2022). In our case, the high contribution of variable selection, which causes the variability of intestinal microbiome, might be somewhat linked to B. gaini host-related factors (e.g., ecology, diet, genetics, physiology) that selected, enriched or maintained bacteria adapted to the intestine microenvironment with site-specific differences (Stegen et al., 2013, 2015). We speculate that the trophic plasticity of B. gaini, coupled with potentially significant inter-sites variations in nutrient resources, may drive the gut microbial diversity toward various directions associated with different dietary intakes. This could lead to distinct enrichments within the intestine and consequently high intraspecific variations in microbiome compositions, similar to patterns observed in Tibetan herdsmen and birds (Li et al., 2018; Bodawatta et al., 2021). In line with this, the niche breadth in the intestine was wider than in the gill, with half of the OTUs being either neutralists or generalists, suggesting that the diet flexibility of B. gaini promotes a metabolically more flexible microbial community in the intestine (Jiao et al., 2020).
4.4 Intestinal microbiomes exhibit stronger phylosymbiosis than the gill microbiomes
Our study addressed three significant gaps in the current phylosymbiosis literature by (i) comparing the strength of phylosymbiosis in two host microenvironments, (ii) through host intraspecific resolution, and (iii) within an unexplored taxonomic group (Brachiopoda). To date, only a single phylosymbiosis study has been reported in this region (Schwob et al., 2024a). We detected robust correlations between within-species phylogenetic distances of B. gaini and its microbiomes’ compositions. This result formally confirms the feasibility of detecting phylosymbiosis among genetically-structured intraspecies populations of freshwater invertebrates, thereby highlighting the importance of preserving the individual identity when considering different populations (Mazel et al., 2018; Lim and Bordenstein, 2020). Our results contrast with previous findings that mostly report the absence of phylosymbiosis in meiofaunal invertebrates, including rotifers, crustaceans, and flatworms, among others (Turgay et al., 2020; Eckert et al., 2021; Boscaro et al., 2022; Eckert et al., 2022; Rosa and Loreto, 2023). Several major methodological variances may explain the difference in phylosymbiosis detection with these previous works. First, ancient divergence time among different hosts tend to obscure the phylogenetic signal within their microbiomes, thus limiting the detection of phylosymbiosis (Brooks et al., 2016; Groussin et al., 2017). Consistently, Leasi et al. (2023) did not detect phylosymbiosis signal among the microbiomes of seven marine interstitial nemertean genera, but the correlation was positive when tested within the single genus Ototyphlonemertes. Secondly, we distinguished two different host microenvironments within B. gaini, while all the aforementioned studies used complete individuals for inspecting phylosymbiosis association. Mixing different microbiomes from whole individuals may lead to spurious conclusions about phylosymbiosis. Here, the strength of the phylosymbiosis pattern was markedly different between gill and intestine microbiomes. Specifically, the phylosymbiosis signal was weaker (and even marginally significant) in the B. gaini gill compared to intestine microenvironment when controlling for climatic and geographic distances. These findings suggest that, independently of climatic and geographic variables, host genetics shape more strongly the intestine microbiome than the gill microbiome, reflecting deterministic assemblages of bacteria. This is in accordance with the predominance of the variable selection process observed for the intestine microbiome assembly across sampling sites. This pattern is consistent with the general trend in phylosymbiosis studies showing that internal microbiomes (e.g., gut) tend to harbor stronger and more frequent phylosymbiosis signals than external microbiomes (e.g., skin), as reported in fishes (Minich et al., 2022), due to likely more direct influence of host physiology, immune system, and trophic diet in shaping the microbial communities (Moran and Sloan, 2015; Mazel et al., 2018; Woodhams et al., 2020).
We cannot discard a non-adaptive origin of the phylosymbiosis through host deterministic ecological filtering. Indeed, more phylogenetically closely related hosts might share more similar habitat, ecology and physiology, thus indirectly selecting similar microbial taxa from the surrounding environment (Moran and Sloan, 2015; Mazel et al., 2018). This hypothesis likely applies in the case of the intestine microenvironment, as the phylosymbiosis may result from diverging dietary intakes, related to diet preferences and/or the available resources (Hammer et al., 2020), which tend to be more homogenous within and between geographically closer sites, as observed in lake plankton (Soininen et al., 2011). This is in line with previous microscopic observations in another branchiopod genus (Artemia), suggesting that the intestinal microbial community is mostly transient and associated with the ingested material (Martin et al., 2020). The localization and density of the microbes in the intestine, as well as the functional host-symbiont complementarity remain to be fully resolved in B. gaini, and constitute promising avenues of research to further understand the gut microbial roles in freshwater crustaceans inhabiting lacustrine ecosystems in the rapidly changing region of Maritime Antarctica.
5 Conclusions
This study examines the microbiome assembly processes and phylosymbiosis signal in the Antarctic fairy shrimp Branchinecta gaini, relating to the host’s intraspecific genetic structure. We found that the gill and intestine harbor distinct microbiomes, each with specific core OTUs that form site-dependent interaction networks, assembled through eco-evolutionary processes that result in divergent microbial communities. The host genetic structure correlates with the composition of both intestine and gill microbiomes of fairy shrimp, validating the phylosymbiosis hypothesis. The stronger phylosymbiosis signal in the intestine is linked to deterministic microbiome assembly processes associated with site-specific dietary variations that reflect the host’s genetic structure. In contrast, the greater variability observed in the gill microbiome–due to stochastic assembly processes likely influenced by environmental exposure and/or random colonization–obscures this signal. These findings emphasize the importance of distinguishing between host microenvironments, even in small organisms, when studying the microbiome assembly.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA1090016 https://www.ncbi.nlm.nih.gov/, PP506203-PP506230.
Ethics statement
The animal study was approved by Comité de Bioética, Instituto de Ecología y Biodiversidad (IEB), Santiago, Chile. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
GS: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing, Funding acquisition, Project administration, Resources. LC: Writing – review & editing, Validation. PV: Methodology, Writing – review & editing. YT: Methodology, Writing – review & editing. FM: Visualization, Writing – review & editing, Methodology. TC: Writing – review & editing, Funding acquisition, Project administration, Resources. JO: Funding acquisition, Project administration, Resources, Writing – review & editing, Validation. CM: Project administration, Resources, Writing – review & editing, Validation, Conceptualization, Data curation, Investigation, Methodology, Supervision, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the ANID-Millennium Science Initiative Program (ICN2021_002), the ANID FONDECYT Postdoctoral Grant No. 3210063, and the Cape Horn International Center (CHIC) funded by ANID/BASAL FB210018.
Acknowledgments
The authors express their gratitude to the BAS station staff on Signy Island and acknowledge the NERC core funding provided to the BAS “Biodiversity, Evolution, and Adaptation” program. Powered@NLHPC: This research was partially supported by the supercomputing infrastructure of the NLHPC (CCSS210001).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2024.1438057/full#supplementary-material
References
Aguilar A., Maeda-Martínez A. M., Murugan G., Obregón-Barboza H., Christopher Rogers D., McClintock K., et al. (2017). High intraspecific genetic divergence in the versatile fairy shrimp Branchinecta lindahli with a comment on cryptic species in the genus Branchinecta (Crustacea: Anostraca). Hydrobiologia 801, 59–69. doi: 10.1007/s10750-017-3283-3
Alberdi A., Andersen S. B., Limborg M. T., Dunn R. R., Gilbert M. T. P. (2022). Disentangling host-microbiota complexity through hologenomics. Nat. Rev. Genet. 23, 281–297. doi: 10.1038/s41576-021-00421-0
Astudillo-Garcia C., Bell J. J., Webster N. S., Glasl B., Jompa J., Montoya J. M., et al. (2017). Evaluating the core microbiota in complex communities: A systematic investigation. Environ. Microbiol. 19, 1450–1462. doi: 10.1111/1462-2920.13647
Bacci G., Fratini S., Meriggi N., Cheng C. L., Ng K. H., Pindo M., et al. (2023). Conserved organ-specific microbial assemblages in different populations of a terrestrial crab. Front. Microbiol. 14, 1113617. doi: 10.3389/fmicb.2023.1113617
Battaglia T. (2021). btools: A suite of R function for all types of microbial diversity analyses. R package version 0.0.1. Available online at: https://scholar.google.com/scholar?hl=fr&as_sdt=0%2C5&q=btools%3A+A+suite+of+R+function+for+all+types+of+microbial+diversity+analyses.&btnG=#d=gs_cit&t=1729863033392&u=%2Fscholar%3Fq%3Dinfo%3AQyGUFtacEcAJ%3Ascholar.google.com%2F%26output%3Dcite%26scirp%3D0%26hl%3Dfr
Berry D., Widder S. (2014). Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front. Microbiol. 5. doi: 10.3389/fmicb.2014.00219
Bodawatta K. H., Hird S. M., Grond K., Poulsen M., Jønsson K. A. (2022). Avian gut microbiomes taking flight. Trends Microbiol. 30, 268–280. doi: 10.1016/j.tim.2021.07.003
Bodawatta K. H., Koane B., Maiah G., Sam K., Poulsen M., Jønsson K. A. (2021). Species-specific but not phylosymbiotic gut microbiomes of New Guinean passerine birds are shaped by diet and flight-associated gut modifications. Proc. R. Soc. B 288, 20210446. doi: 10.1098/rspb.2021.0446
Bokulich N. A., Subramanian S., Faith J. J., Gevers D., Gordon J. I., Knight R., et al. (2013). Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 10, 57–59. doi: 10.1038/nmeth.2276
Bordenstein S. R., Theis K. R. (2015). Host biology in light of the microbiome: ten principles of holobionts and hologenomes. PloS Biol. 13, e1002226. doi: 10.1371/journal.pbio.1002226
Boscaro V., Holt C. C., Van Steenkiste N. W. L., Herranz M., Irwin N. A. T., Alvarez-Campos P., et al. (2022). Microbiomes of microscopic marine invertebrates do not reveal signatures of phylosymbiosis. Nat. Microbiol. 7, 810–819. doi: 10.1038/s41564-022-01125-9
Brooks A. W., Kohl K. D., Brucker R. M., van Opstal E. J., Bordenstein S. R. (2016). Phylosymbiosis: relationships and functional effects of microbial communities across host evolutionary history. PloS Biol. 14, e2000225. doi: 10.1371/journal.pbio.2000225
Cáceres C. E., Rogers D. C. (2015). Class branchiopoda. In Thorp and Covich’s Freshwater Invertebrates. Academic Press, 687–708. Available online at: https://scholar.google.com/scholar?hl=fr&as_sdt=0%2C5&q=Cáceres%2C+C.E.%2C+and+Rogers%2C+D.C.+%282015%29.+%22Class+branchiopoda%2C%22+in+Thorp+and+Covich%27s+Freshwater+Invertebrates.+Elsevier%29%2C+687-708.&btnG=.
Callens M., Watanabe H., Kato Y., Miura J., Decaestecker E. (2018). Microbiota inoculum composition affects holobiont assembly and host growth in Daphnia. Microbiome 6, 1–12. doi: 10.1186/s40168-018-0444-1
Cao Y., Dong Q., Wang D., Zhang P., Liu Y., Niu C. (2022). microbiomeMarker: an R/Bioconductor package for microbiome marker identification and visualization. Bioinformatics 2022, btac438. doi: 10.1093/bioinformatics/btac438
Chen J. Z., Kwong Z., Gerardo N. M., Vega N. M. (2024). Ecological drift during colonization drives within-host and between-host heterogeneity in an animal-associated symbiont. PloS Biol. 22, e3002304. doi: 10.1371/journal.pbio.3002304
Coleman G. A., Davín A. A., Mahendrarajah T. A., Szánthó L. L., Spang A., Hugenholtz P., et al. (2021). A rooted phylogeny resolves early bacterial evolution. Science 372, eabe0511. doi: 10.1126/science.abe0511
Cooper R. O., Cressler C. E. (2020). Characterization of key bacterial species in the Daphnia magna microbiota using shotgun metagenomics. Sci. Rep. 10, 652. doi: 10.1038/s41598-019-57367-x
Corbari L., Zbinden M., Cambon-Bonavita M.-A., Gaill F., Compère P. (2008). Bacterial symbionts and mineral deposits in the branchial chamber of the hydrothermal vent shrimp Rimicaris exoculata: relationship to moult cycle. Aquat. Biol. 1, 225–238. doi: 10.3354/ab00024
Cornejo-Granados F., Gallardo-Becerra L., Leonardo-Reza M., Ochoa-Romo J. P., Ochoa-Leyva A. (2018). A meta-analysis reveals the environmental and host factors shaping the structure and function of the shrimp microbiota. PeerJ 6, e5382. doi: 10.7717/peerj.5382
Costello E. K., Lauber C. L., Hamady M., Fierer N., Gordon J. I., Knight R. (2009). Bacterial community variation in human body habitats across space and time. Science 326, 1694–1697. doi: 10.1126/science.1177486
Couch C. E., Epps C. W. (2022). Host, microbiome, and complex space: applying population and landscape genetic approaches to gut microbiome research in wild populations. J. Heredity 113, 221–234. doi: 10.1093/jhered/esab078
Daborn G. R. (1975). Life history and energy relations of the giant fairy shrimp Branchinecta gigas Lynch 1937 (Crustacea: Anostraca). Ecology 56, 1025–1039. doi: 10.2307/1936144
Daday J. (1910). Quelques Phyllopodes Anostracés nouveaux. Annales des Sciences Naturelles 12, 241–264. Available online at: https://scholar.google.com/scholar_lookup?&title=Quelques%20Phyllopodes%20Anostracés%20nouveaux&journal=Annales%20des%20Sciences%20Naturelles&volume=12&pages=241-264&publication_year=1910&author=Daday%2CJ
Darcy J. L., Lynch R. C., King A. J., Robeson M. S., Schmidt S. K. (2011). Global distribution of Polaromonas phylotypes-evidence for a highly successful dispersal capacity. PloS One 6, e23742. doi: 10.1371/journal.pone.0023742
Dekaboruah E., Suryavanshi M. V., Chettri D., Verma A. K. (2020). Human microbiome: an academic update on human body site specific surveillance and its possible role. Arch. Microbiol. 202, 2147–2167. doi: 10.1007/s00203-020-01931-x
Delleuze M., Schwob G., Orlando J., Gérard K., Saucède T., Brickle P., et al. (2024). Habitat specificity modulates the bacterial biogeographic patterns in the Southern Ocean. FEMS Microbiology Ecol, fiae134. doi: 10.1093/femsec/fiae134
Dinno A. (2017). dunn. test: Dunn’s test of multiple comparisons using rank sums. R Package version 1, 1.
Doane M. P., Morris M. M., Papudeshi B., Allen L., Pande D., Haggerty J. M., et al. (2020). The skin microbiome of elasmobranchs follows phylosymbiosis, but in teleost fishes, the microbiomes converge. Microbiome 8, 93. doi: 10.1186/s40168-020-00840-x
Eckert E. M., Anicic N., Fontaneto D. (2021). Freshwater zooplankton microbiome composition is highly flexible and strongly influenced by the environment. Mol. Ecol. 30, 1545–1558. doi: 10.1111/mec.15815
Eckert E. M., Cancellario T., Bodelier P. L. E., Declerck S. A. J., Diwen L., Samad S., et al. (2022). A combination of host ecology and habitat but not evolutionary history explains differences in the microbiomes associated with rotifers. Hydrobiologia 850, 3813–3821. doi: 10.1007/s10750-022-04958-x
Edgar R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Edgar R. C., Haas B. J., Clemente J. C., Quince C., Knight R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Farkas J. (1985). Filamentous Flavobacterium sp. isolated from fish with gill diseases in cold water. Aquaculture 44, 1–10. doi: 10.1016/0044-8486(85)90037-7
Finn D. R., Yu J., Ilhan Z. E., Fernandes V. M. C., Penton C. R., Krajmalnik-Brown R., et al. (2020). MicroNiche: an R package for assessing microbial niche breadth and overlap from amplicon sequencing data. FEMS Microbiol. Ecol. 96. doi: 10.1093/femsec/fiaa131
Folmer O., Black M., Hoeh W., Lutz R., Vrijenhoek R. (1994). DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 3, 294–299.
Fusi M., Ngugi D. K., Marasco R., Booth J. M., Cardinale M., Sacchi L., et al. (2023). Gill-associated bacteria are homogeneously selected in amphibious mangrove crabs to sustain host intertidal adaptation. Microbiome 11, 189. doi: 10.1186/s40168-023-01629-4
Gao G., Li G., Liu M., Li P., Liu J., Ma S., et al. (2023). Changes in soil stoichiometry, soil organic carbon mineralization and bacterial community assembly processes across soil profiles. Sci. Total Environ. 903, 166408. doi: 10.1016/j.scitotenv.2023.166408
Ge Y., Jing Z., Diao Q., He J.-Z., Liu Y.-J. (2021). Host species and geography differentiate honeybee gut bacterial communities by changing the relative contribution of community assembly processes. MBio 12, 00751–00721. doi: 10.1128/mBio.00751-21
Giudice A. L., Bruni V., Michaud L. (2007). Characterization of Antarctic psychrotrophic bacteria with antibacterial activities against terrestrial microorganisms. J. basic Microbiol. 47, 496–505. doi: 10.1002/jobm.200700227
Groussin M., Mazel F., Alm E. J. (2020). Co-evolution and co-speciation of host-gut bacteria systems. Cell Host Microbe 28, 12–22. doi: 10.1016/j.chom.2020.06.013
Groussin M., Mazel F., Sanders J. G., Smillie C. S., Lavergne S., Thuiller W., et al. (2017). Unraveling the processes shaping mammalian gut microbiomes over evolutionary time. Nat. Commun. 8, 14319. doi: 10.1038/ncomms14319
Guivier E., Martin J. F., Pech N., Ungaro A., Chappaz R., Gilles A. (2018). Microbiota diversity within and between the tissues of two wild interbreeding species. Microbial Ecol. 75, 799–810. doi: 10.1007/s00248-017-1077-9
Hagen M., Hamrick J. (1996). Population level processes in Rhizobium leguminosarum bv. trifolii: the role of founder effects. Mol. Ecol. 5, 707–714. doi: 10.1111/j.1365-294X.1996.tb00367.x
Hammer T. J., Dickerson J. C., McMillan W. O., Fierer N. (2020). Heliconius butterflies host characteristic and phylogenetically structured adult-stage microbiomes. Appl. Environ. Microbiol. 86, e02007–e02020. doi: 10.1128/AEM.02007-20
Hammer T. J., Janzen D. H., Hallwachs W., Jaffe S. P., Fierer N. (2017). Caterpillars lack a resident gut microbiome. Proc. Natl. Acad. Sci. 114, 9641–9646. doi: 10.1073/pnas.1707186114
Hammer T. J., Sanders J. G., Fierer N. (2019). Not all animals need a microbiome. FEMS Microbiol. Lett. 366. doi: 10.1093/femsle/fnz117
Hawes T. (2009). Origins and dispersal of the Antarctic fairy shrimp. Antarctic Sci. 21, 477–482. doi: 10.1017/S095410200900203X
Hayward A., Poulin R., Nakagawa S. (2021). A broadscale analysis of host-symbiont cophylogeny reveals the drivers of phylogenetic congruence. Ecol. Lett. 24, 1681–1696. doi: 10.1111/ele.13757
Huot C., Clerissi C., Gourbal B., Galinier R., Duval D., Toulza E. (2019). Schistosomiasis vector snails and their microbiota display a phylosymbiosis pattern. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.03092
Jiao S., Yang Y., Xu Y., Zhang J., Lu Y. (2020). Balance between community assembly processes mediates species coexistence in agricultural soil microbiomes across eastern China. ISME J. 14, 202–216. doi: 10.1038/s41396-019-0522-9
Kapardar R. K., Ranjan R., Grover A., Puri M., Sharma R. (2010). Identification and characterization of genes conferring salt tolerance to Escherichia coli from pond water metagenome. Bioresource Technol. 101, 3917–3924. doi: 10.1016/j.biortech.2010.01.017
Karger D. N., Conrad O., Böhner J., Kawohl T., Kreft H., Soria-Auza R. W., et al. (2017). Climatologies at high resolution for the earth’s land surface areas. Sci. Data 4, 1–20. doi: 10.1038/sdata.2017.122
Kassambara A. (2020). ggplot2” based publication ready plots [R package ggpubr version 0.4.0. Compr. R Arch. Network. 25, 2022.
Kearse M., Moir R., Wilson A., Stones-Havas S., Cheung M., Sturrock S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Klindworth A., Pruesse E., Schweer T., Peplies J., Quast C., Horn M., et al. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41, e1. doi: 10.1093/nar/gks808
Kohl K. D. (2020). Ecological and evolutionary mechanisms underlying patterns of phylosymbiosis in host-associated microbial communities. Philos. Trans. R. Soc. B 375, 20190251. doi: 10.1098/rstb.2019.0251
Kohl K. D., Dearing M. D., Bordenstein S. R. (2018). Microbial communities exhibit host species distinguishability and phylosymbiosis along the length of the gastrointestinal tract. Mol. Ecol. 27, 1874–1883. doi: 10.1111/mec.14460
Kurtz Z. D., Muller C. L., Miraldi E. R., Littman D. R., Blaser M. J., Bonneau R. A. (2015). Sparse and compositionally robust inference of microbial ecological networks. PloS Comput. Biol. 11, e1004226. doi: 10.1371/journal.pcbi.1004226
Lahti L., Shetty S., Blake T., Salojarvi J. (2017). Tools for microbiome analysis in R. Version, Vol. 1. 28. Available online at: http://microbiome.github.iohttps://scholar.google.com/scholar?hl=fr&as_sdt=0%2C5&q=Lahti%2C+L.%2C+Shetty%2C+S.%2C+Blake%2C+T.%2C+and+Salojarvi%2C+J.+%282017%29.+Tools+for+microbiome+analysis+in+R.+Version+1%2C+28.&btnG=#d=gs_cit&t=1729863017070&u=%2Fscholar%3Fq%3Dinfo%3AOxH9VzHXxTAJ%3Ascholar.google.com%2F%26output%3Dcite%26scirp%3D0%26hl%3Dfr
Lange C., Boyer S., Bezemer T. M., Lefort M. C., Dhami M. K., Biggs E., et al. (2023). Impact of intraspecific variation in insect microbiomes on host phenotype and evolution. ISME J. 17, 1798–1807. doi: 10.1038/s41396-023-01500-2
Leasi F., Eckert E., Norenburg J., Thomas W. K., Sevigny J., Hall J., et al. (2023). Microbiota associated with meiofaunal nemerteans reveals evidence of phylosymbiosis. Authorea Preprints. doi: 10.22541/au.169339864.44365926/v1
Lee E., Lee K. W., Park Y., Choi A., Kwon K. K., Kang H. M. (2024). Comparative microbiome analysis of artemia spp. and potential role of microbiota in cyst hatching. Mar. Biotechnol. 26, 50–59. doi: 10.1007/s10126-023-10276-7
Lemoine F., Correia D., Lefort V., Doppelt-Azeroual O., Mareuil F., Cohen-Boulakia S., et al. (2019). NGPhylogeny. fr: new generation phylogenetic services for non-specialists. Nucleic Acids Res. 47, W260–W265. doi: 10.1093/nar/gkz303
Li H., Li T., Li X., Wang G., Lin Q., Qu J. (2018). Gut microbiota in Tibetan herdsmen reflects the degree of urbanization. Front. Microbiol. 9, 1745. doi: 10.3389/fmicb.2018.01745
Li H., Zhou R., Zhu J., Huang X., Qu J. (2019). Environmental filtering increases with elevation for the assembly of gut microbiota in wild pikas. Microbial Biotechnol. 12, 976–992. doi: 10.1111/1751-7915.13450
Lim S. J., Bordenstein S. R. (2020). An introduction to phylosymbiosis. Proc. R. Soc. B 287, 20192900. doi: 10.1098/rspb.2019.2900
Liu P., Zha T., Xia S.-Q., Sun P., Wu Y.-F., Zhao H., et al. (2023). Repeated digestion by earthworms on tetracycline degrading and biological responses in tetracycline-contaminated sewage sludge. Water Air Soil pollut. 234. doi: 10.1007/s11270-023-06366-w
Ludington W. B. (2024). The importance of host physical niches for the stability of gut microbiome composition. Philos. Trans. R. Soc. B 379, 20230066. doi: 10.1098/rstb.2023.0066
Lukic D., Pormehr N., Beladjal L., Vad C. F., Ptacnik R., Van Stappen G., et al. (2023). Life-history omnivory in the fairy shrimp Branchinecta orientalis (Branchiopoda: Anostraca). Hydrobiologia 850, 901–909. doi: 10.1007/s10750-022-05132-z
Lynch J. E. (1964). Packard’s and Pearse’s species of Branchinecta: analysis of a nomenclatural involvement. Am. Midland Nat. 71 (2), 466–488. doi: 10.2307/2423306
Macke E., Callens M., Massol F., Vanoverberghe I., De Meester L., Decaestecker E. (2020). Diet and genotype of an aquatic invertebrate affect the composition of free-living microbial communities. Front. Microbiol. 11, 503595. doi: 10.3389/fmicb.2020.00380
Mallott E. K. (2024). Disentangling the mechanisms underlying phylosymbiosis in mammals. Mol. Ecol. 33, e17193. doi: 10.1111/mec.17193
Mallott E. K., Amato K. R. (2021). Host specificity of the gut microbiome. Nat. Rev. Microbiol. 19, 639–653. doi: 10.1038/s41579-021-00562-3
Martin G. G., Natha Z., Henderson N., Bang S., Hendry H., Loera Y. (2020). Absence of a microbiome in the midgut trunk of six representative Crustacea. J. Crustacean Biol. 40, 122–130. doi: 10.1093/jcbiol/ruz087
Martiny J. B., Bohannan B. J., Brown J. H., Colwell R. K., Fuhrman J. A., Green J. L., et al. (2006). Microbial biogeography: putting microorganisms on the map. Nat. Rev. Microbiol. 4, 102–112. doi: 10.1038/nrmicro1341
Maturana C. S., Segovia N. I., González-Wevar C. A., Díaz A., Rosenfeld S., Poulin E., et al. (2020). Evidence of strong small-scale population structure in the Antarctic freshwater copepod Boeckella poppei in lakes on Signy Island, South Orkney Islands. Limnology Oceanography 65, 2024–2040. doi: 10.1002/lno.11435
Mazel F., Davis K. M., Loudon A., Kwong W. K., Groussin M., Parfrey L. W. (2018). Is host filtering the main driver of phylosymbiosis across the tree of life? Msystems 3, e00097–e00018. doi: 10.1128/msystems.00097-18
Mazel F., Guisan A., Parfrey L. W. (2024). Transmission mode and dispersal traits correlate with host specificity in mammalian gut microbes. Mol. Ecol. 33, e16862. doi: 10.1111/mec.16862
Mazel F., Knowles S. C. L., Videvall E., Sweeny A. R. (2023). Evolutionary patterns and processes in animal microbiomes. J. evolutionary Biol. 36, 1653–1658. doi: 10.1111/jeb.14248
McFall-Ngai M., Hadfield M. G., Bosch T. C., Carey H. V., Domazet-Lošo T., Douglas A. E., et al. (2013). Animals in a bacterial world, a new imperative for the life sciences. Proc. Natl. Acad. Sci. 110, 3229–3236. doi: 10.1073/pnas.1218525110
McMurdie P. J., Holmes S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PloS One 8, e61217. doi: 10.1371/journal.pone.0061217
Middlemiss K. L., Urbina M. A., Wilson R. W. (2015). Microbial proliferation on gill structures of juvenile European lobster (Homarus gammarus) during a moult cycle. Helgoland Mar. Res. 69, 401–410. doi: 10.1007/s10152-015-0445-4
Minich J. J., Härer A., Vechinski J., Frable B. W., Skelton Z. R., Kunselman E., et al. (2022). Host biology, ecology and the environment influence microbial biomass and diversity in 101 marine fish species. Nat. Commun. 13, 6978. doi: 10.1038/s41467-022-34557-2
Moeller A. H., Suzuki T. A., Lin D., Lacey E. A., Wasser S. K., Nachman M. W. (2017). Dispersal limitation promotes the diversification of the mammalian gut microbiota. Proc. Natl. Acad. Sci. 114, 13768–13773. doi: 10.1073/pnas.1700122114
Moran N. A., Ochman H., Hammer T. J. (2019). Evolutionary and ecological consequences of gut microbial communities. Annu. Rev. Ecol. Systematics 50, 451–475. doi: 10.1146/annurev-ecolsys-110617-062453
Moran N. A., Sloan D. B. (2015). The hologenome concept: helpful or hollow? PloS Biol. 13, e1002311. doi: 10.1371/journal.pbio.1002311
Motiei A., Brindefalk B., Ogonowski M., El-Shehawy R., Pastuszek P., Ek K., et al. (2020). Disparate effects of antibiotic-induced microbiome change and enhanced fitness in Daphnia magna. PloS One 15, e0214833. doi: 10.1371/journal.pone.0214833
Nougué O., Gallet R., Chevin L.-M., Lenormand T. (2015). Niche limits of symbiotic gut microbiota constrain the salinity tolerance of brine shrimp. Am. Nat. 186, 390–403. doi: 10.1086/682370
O’Brien P. A., Tan S., Yang C., Frade P. R., Andreakis N., Smith H. A., et al. (2020). Diverse coral reef invertebrates exhibit patterns of phylosymbiosis. ISME J. 14, 2211–2222. doi: 10.1038/s41396-020-0671-x
Oksanen J., Blanchet F. G., Kindt R., Legendre P., O’Hara R., Simpson G. L., et al. (2011). Vegan: Community ecology packag. R Package version 1, 17–18.
Olsen M., Blix A., Utsi T., Sørmo W., Mathiesen S. (1999). Chitinolytic bacteria in the minke whale forestomach. Can. J. Microbiol. 46, 85–94. doi: 10.1139/w99-112
Paggi J. (1996). Feeding ecology of Branchinecta gaini (Crustacea: Anostraca) in ponds of south Shetland Islands, Antarctica. Polar Biol. 16, 13–18. doi: 10.1007/BF02388730
Pan B., Han X., Yu K., Sun H., Mu R., Lian C. A. (2023). Geographical distance, host evolutionary history and diet drive gut microbiome diversity of fish across the Yellow River. Mol. Ecol. 32, 1183–1196. doi: 10.1111/mec.16812
Pankey S. M., Plachetzki D. C., Macartney K. J., Gastaldi M., Slattery M., Gochfeld D. J., et al. (2022). Cophylogeny and convergence shape holobiont evolution in sponge-microbe symbioses. Nat. Ecol. Evol. 6, 750–762. doi: 10.1038/s41559-022-01712-3
Paradis E., Schliep K. (2019). ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528. doi: 10.1093/bioinformatics/bty633
Peck L. S. (2004). Physiological flexibility: the key to success and survival for Antarctic fairy shrimps in highly fluctuating extreme environments. Freshw. Biol. 49, 1195–1205. doi: 10.1111/j.1365-2427.2004.01264.x
Pollock F. J., McMinds R., Smith S., Bourne D. G., Willis B. L., Medina M., et al. (2018). Coral-associated bacteria demonstrate phylosymbiosis and cophylogeny. Nat. Commun. 9, 4921. doi: 10.1038/s41467-018-07275-x
Polz M. F., Hunt D. E., Preheim S. P., Weinreich D. M. (2006). Patterns and mechanisms of genetic and phenotypic differentiation in marine microbes. Philos. Trans. R. Soc. B: Biol. Sci. 361, 2009–2021. doi: 10.1098/rstb.2006.1928
Qi W., Nong G., Preston J. F., Ben-Ami F., Ebert D. (2009). Comparative metagenomics of Daphnia symbionts. BMC Genomics 10, 1–21. doi: 10.1186/1471-2164-10-172
Qin M., Jiang L., Qiao G., Chen J. (2023). Phylosymbiosis: the eco-evolutionary pattern of insect-symbiont interactions. Int. J. Mol. Sci. 24. doi: 10.3390/ijms242115836
Risely A. (2020). Applying the core microbiome to understand host-microbe systems. J. Anim. Ecol. 89, 1549–1558. doi: 10.1111/1365-2656.13229
Rosa M. T., Loreto E. L. (2023). Stenostomum leucops (Catenulida, Platyhelminthes) has a flexible microbiome in time and space. Hydrobiologia 850, 3675–3683. doi: 10.1007/s10750-022-04931-8
Rosenberg E., Zilber-Rosenberg I. (2018). The hologenome concept of evolution after 10 years. Microbiome 6, 78. doi: 10.1186/s40168-018-0457-9
Rosenberg E., Zilber-Rosenberg I. (2021). Reconstitution and transmission of gut microbiomes and their genes between generations. Microorganisms 10, 70. doi: 10.3390/microorganisms10010070
Ross A. A., Müller K. M., Weese J. S., Neufeld J. D. (2018). Comprehensive skin microbiome analysis reveals the uniqueness of human skin and evidence for phylosymbiosis within the class Mammalia. Proc. Natl. Acad. Sci. 115, E5786–E5795. doi: 10.18653/v1/K18-2
Sadeghi J., Chaganti S. R., Johnson T. B., Heath D. D. (2023). Host species and habitat shape fish-associated bacterial communities: phylosymbiosis between fish and their microbiome. Microbiome 11, 258. doi: 10.1186/s40168-023-01697-6
Samad M. S., Lee H. J., Cerbin S., Meima-Franke M., Bodelier P. L. E. (2020). Niche differentiation of host-associated pelagic microbes and their potential contribution to biogeochemical cycling in artificially warmed lakes. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.00582
Savard P., Fernandes T., Dao A., McMeans B., Lazar C. S. (2023). Seasons influence the native gut microbiome of lake trout salvelinus namaycush. Appl. Microbiol. 3, 276–287. doi: 10.3390/applmicrobiol3010019
Schloss P. D., Westcott S. L., Ryabin T., Hall J. R., Hartmann M., Hollister E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Schwob G., Cabrol L., Poulin E., Orlando J. (2020). Characterization of the gut microbiota of the Antarctic heart urchin (Spatangoida) Abatus agassizii. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.00308
Schwob G., Cabrol L., Saucède T., Gérard K., Poulin E., Orlando J. (2024a). Unveiling the co-phylogeny signal between plunderfish Harpagifer spp. and their gut microbiomes across the Southern Ocean. Microbiol. Spectr. 12 (4), e03830–e03823. doi: 10.1101/2023.04.18.537398
Schwob G., Rosenfeld S., González-Wevar C., Orlando J. (2024b). Ecological filtering and phylogeographic structuring of Psychrilyobacter within two closely related limpet species from the Southern Ocean. Ann. Microbiol. 74 (1), 8. doi: 10.1186/s13213-024-01751-0
Schwob G., Segovia N. I., González-Wevar C., Cabrol L., Orlando J., Poulin E. (2021). Exploring the microdiversity within marine bacterial taxa: toward an integrated biogeography in the southern ocean. Front. Microbiol. 12. doi: 10.3389/fmicb.2021.703792
Soininen J., Korhonen J. J., Karhu J., Vetterli A. (2011). Disentangling the spatial patterns in community composition of prokaryotic and eukaryotic lake plankton. Limnology Oceanography 56, 508–520. doi: 10.4319/lo.2011.56.2.0508
Stegen J. C., Lin X., Fredrickson J. K., Chen X., Kennedy D. W., Murray C. J., et al. (2013). Quantifying community assembly processes and identifying features that impose them. ISME J. 7, 2069–2079. doi: 10.1038/ismej.2013.93
Stegen J. C., Lin X., Fredrickson J. K., Konopka A. E. (2015). Estimating and mapping ecological processes influencing microbial community assembly. Front. Microbiol. 6. doi: 10.3389/fmicb.2015.00370
Trevelline B. K., Sosa J., Hartup B. K., Kohl K. D. (2020). A bird’s-eye view of phylosymbiosis: weak signatures of phylosymbiosis among all 15 species of cranes. Proc. R. Soc. B 287, 20192988. doi: 10.1098/rspb.2019.2988
Turgay E., Steinum T. M., Eryalçın K. M., Yardımcı R. E., Karataş S. (2020). The influence of diet on the microbiota of live-feed rotifers (Brachionus plicatilis) used in commercial fish larviculture. FEMS Microbiol. Lett. 367, fnaa020. doi: 10.1093/femsle/fnaa020
Vavrek M. J. (2011). Fossil: palaeoecological and palaeogeographical analysis tools. Palaeontologia Electronica 14, 16.
Vega N. M., Gore J. (2017). Stochastic assembly produces heterogeneous communities in the Caenorhabditis elegans intestine. PloS Biol. 15, e2000633. doi: 10.1371/journal.pbio.2000633
Wang J., Pan Z., Yu J., Zhang Z., Li Y.-z. (2023). Global assembly of microbial communities. Msystems 8 (3), e01289–e01222. doi: 10.1128/msystems.01289-22
Weingarten E. A., Atkinson C. L., Jackson C. R. (2019). The gut microbiome of freshwater Unionidae mussels is determined by host species and is selectively retained from filtered seston. PloS One 14, e0224796. doi: 10.1371/journal.pone.0224796
Wickham H. (2016). ggplot2: Elegant Graphics for Data Analysis (Springer-Verlag New York: Springer).
Wilson A. C., Duncan R. P. (2015). Signatures of host/symbiont genome coevolution in insect nutritional endosymbioses. Proc. Natl. Acad. Sci. 112, 10255–10261. doi: 10.1073/pnas.1423305112
Woodhams D. C., Bletz M. C., Becker C. G., Bender H. A., Buitrago-Rosas D., Diebboll H., et al. (2020). Host-associated microbiomes are predicted by immune system complexity and climate. Genome Biol. 21, 23. doi: 10.1186/s13059-019-1908-8
Xiong J., Dai W., Zhu J., Liu K., Dong C., Qiu Q. (2017). The underlying ecological processes of gut microbiota among cohabitating retarded, overgrown and normal shrimp. Microbial Ecol. 73, 988–999. doi: 10.1007/s00248-016-0910-x
Yagi J. M., Sims D., Brettin T., Bruce D., Madsen E. L. (2009). The genome of Polaromonas naphthalenivorans strain CJ2, isolated from coal tar-contaminated sediment, reveals physiological and metabolic versatility and evolution through extensive horizontal gene transfer. Environ. Microbiol. 11, 2253–2270. doi: 10.1111/j.1462-2920.2009.01947.x
Yan Q., Li J., Yu Y., Wang J., He Z., Van Nostrand J. D., et al. (2016). Environmental filtering decreases with fish development for the assembly of gut microbiota. Environ. Microbiol. 18, 4739–4754. doi: 10.1111/1462-2920.13365
Youngblut N. D., Reischer G. H., Walters W., Schuster N., Walzer C., Stalder G., et al. (2019). Host diet and evolutionary history explain different aspects of gut microbiome diversity among vertebrate clades. Nat. Commun. 10, 2200. doi: 10.1038/s41467-019-10191-3
Zawierucha K., Trzebny A., Buda J., Bagshaw E., Franzetti A., Dabert M., et al. (2022). Trophic and symbiotic links between obligate-glacier water bears (Tardigrada) and cryoconite microorganisms. PloS One 17, e0262039. doi: 10.1371/journal.pone.0262039
Zhang C., Li H., Zeng Y., Ding H., Wang B., Li Y., et al. (2022). Diversity and assembly processes of microbial eukaryotic communities in Fildes Peninsula Lakes (West Antarctica). Biogeosciences 19, 4639–4654. doi: 10.5194/bg-19-4639-2022
Zhang M., Sun Y., Chen L., Cai C., Qiao F., Du Z., et al. (2016). Symbiotic bacteria in gills and guts of Chinese mitten crab (Eriocheir sinensis) differ from the free-living bacteria in water. PloS One 11, e0148135. doi: 10.1371/journal.pone.0148135
Zhang M., Zhang X., Tran N. T., Sun Z., Zhang X., Ye H., et al. (2021). Molting alters the microbiome, immune response, and digestive enzyme activity in mud crab (Scylla paramamosain). Msystems 6 (5), 10–1128. doi: 10.1128/msystems.00917-00921
Zhao Y., Duan C., Zhang X., Chen H., Ren H., Yin Y., et al. (2018). Insights into the gut microbiota of freshwater shrimp and its associations with the surrounding microbiota and environmental factors. Journal of Microbiology and Biotechnology 28, 946–956. doi: 10.4014/jmb.1709.09070
Zhou J., Ning D. (2017). Stochastic community assembly: does it matter in microbial ecology? Microbiol. Mol. Biol. Rev. 81 (4), 10–1128. doi: 10.1128/MMBR.00002-17
Keywords: invertebrate microbiome, gill, intestine, ecological filtering, ecological assembly processes, Branchinecta, West Antarctic Peninsula, freshwater ecosystems
Citation: Schwob G, Cabrol L, Vidal PM, Tapia YC, Moya F, Contador T, Orlando J and Maturana CS (2024) Which microbiome are we talking about? Contrasted diversity patterns and eco-evolutionary processes between gill and intestinal microbiomes of Antarctic fairy shrimps. Front. Ecol. Evol. 12:1438057. doi: 10.3389/fevo.2024.1438057
Received: 24 May 2024; Accepted: 17 October 2024;
Published: 07 November 2024.
Edited by:
Thomas George Bornman, South African Environmental Observation Network (SAEON), South AfricaReviewed by:
Marco Fusi, Newcastle University, United KingdomLara Schmittmann, Helmholtz Association of German Research Centers (HZ), Germany
Copyright © 2024 Schwob, Cabrol, Vidal, Tapia, Moya, Contador, Orlando and Maturana. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guillaume Schwob, Z3NjaHdvYkBpbnN0aXR1dG9iYXNlLmNs; Julieta Orlando, am9ybGFuZG9AdWNoaWxlLmNs