
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol., 25 September 2024
Sec. Evolutionary and Population Genetics
Volume 12 - 2024 | https://doi.org/10.3389/fevo.2024.1416876
 Fatmah Ahmed Safhi1
Fatmah Ahmed Safhi1 Areej Saud Jalal1*
Areej Saud Jalal1* Rana M. Alshegaihi2
Rana M. Alshegaihi2 Rahma Alshamrani3
Rahma Alshamrani3 Amnah M. Alamri3
Amnah M. Alamri3 Wessam Felemban3,4
Wessam Felemban3,4 Amani Omar Abuzaid3Mohammed A. A. Hussein5
Amani Omar Abuzaid3Mohammed A. A. Hussein5 Nora M. Al Aboud6
Nora M. Al Aboud6 Mahmoud Magdy7,8
Mahmoud Magdy7,8 Diaa Abd El-Moneim9
Diaa Abd El-Moneim9Introduction: This study presents the first complete plastome (cpDNA) sequence of Psydrax latifolia, a member of the Rubiaceae family, which includes small trees, smooth shrubs, and occasionally lianas. This specimen was collected near the Red Sea coast in Jazan province, Saudi Arabia, specifically in the paleotropical Fifa Mountains. The research aimed to characterize and compare the plastome of Psydrax latifolia with other species in the Rubiaceae family to enhance our understanding of its evolutionary dynamics and phylogenetic relationships.
Methods: The plastome of P. latifolia was sequenced and reconstructed using whole-genome next-generation sequencing (NGS) techniques. Comparative analyses were performed between the plastome of P. latifolia and 16 other species within the Rubiaceae family to identify genomic features and evolutionary patterns. The plastome structure, gene content, and codon usage were analyzed, with a focus on the Relative Synonymous Codon Usage (RSCU) in different regions of the plastome.
Results: The plastome of P. latifolia was found to be 153,242 base pairs (bp) in length, including a large single copy (LSC) region of 83,603 bp, a small single copy (SSC) region of 18,115 bp, and a pair of inverted repeats (IRs) of 25,762 bp each. It contained 87 protein-coding genes, 8 rRNA genes, and 33 tRNA genes, with an overall GC content of 37.30%. The RSCU analysis revealed regional variation, with the protein-coding region being more conserved than the intergenic spacer.
Discussion: This study provides the first complete plastome sequence of Psydrax latifolia, offering insights into its genomic structure and phylogenetic position within the Rubiaceae family. Comparative analyses with 16 Rubiaceae species highlighted distinct genomic features and evolutionary patterns. These findings contribute to the understanding of plastome evolution in the Rubiaceae family and provide a valuable resource for future phylogenetic and evolutionary studies.
Flowering plants belonging to the Rubiaceae family, one of the largest families of flowering plants, encompasses over 13,000 species distributed across tropical and subtropical regions. Recent classifications have provided insights into the evolutionary relationships within this family, but the genetic relationships among closely related species, particularly within the Psydrax genus, remain unclear (Ly et al., 2020). Among the Rubiaceae family, the Vanguerieae tribe forms a monophyletic group characterized by the following identifying features include the absence of raphides, axillary inflorescences, stylar knobs that serve as pollen collectors in secondary pollination, pendulous ovules, and drupaceous fruits, and an aestivated valvate corolla (Razafimandimbison et al., 2009). Canthium was the first genus of the tribe to be described, followed by Psydrax, containing the species Psydrax dicoccos. Despite this fact, the tribe is primarily found in Africa; the first described species were Asian (Lamarck and Poiret, 1783; Gaertner, 1788). According to Robyns (Robyns, 1928), the first monograph of the tribe was performed, in which a total of 17 genera were allocated to the tribe. From the Canthium genera, the Psydrax genus was reinstated in 1985; the Psydrax genus was reported with over 37 species of various ethnomedicinal uses (Bridson, 1985; Patro et al., 2014).
As a result of recent nomenclatural changes within the Philippine Vanguerieae (Lantz et al., 2002; Lantz and Bremer, 2004, 2005; Razafimandimbison et al., 2009), Psydrax puberula was described as a new species of this genus (Arriola and Alejandro, 2013). The current study focuses on Psydrax latifolia (F. Muell. ex Benth.) S.T. Reynolds & R.J.F. Hend. The inflorescences of P. latifolia bear petite, fragrant white flowers, each possessing a tubular structure with an aesthetically appealing arrangement. The species’ distinct morphology, including its arboreal stature, robust foliage, and aesthetically appealing flowers, contributes to its botanical significance within the Rubiaceae family (Ruhsam et al., 2008). Exploring P. latifolia is not only important for its botanical value but also relevant in the context of species identification challenges.
While specific references to Psydrax latifolia are limited, exploring related species within the Psydrax genus and their medicinal attributes can provide valuable insights. Psydrax species have notable medicinal uses, underscoring their pharmacological significance for different purposes (Feenna et al., 2020). Studies have linked bioactive extracts and compounds from Psydrax species to various beneficial effects, including anti-hyperglycemia, anti-inflammatory, anticonvulsant, and antimicrobial activities (Chukwudulue et al., 2022). Additionally, Psydrax latifolia, belonging to the Ixoroideae subfamily, exhibits morphological similarities with other Psydrax species, complicating species identification and phylogenetic classification. The overlap of medicinal use and morphological resemblance among Psydrax species further amplifies the challenge in distinguishing between species for accurate identification and utilization.
Among most plants, chloroplasts perform vital functions in photosynthesis, amino acid synthesis, and lipid synthesis, as well as being maternally inherited, semi-autonomous, and containing independent DNA from the nucleus (Daniell et al., 2016). Since the advent of high-throughput sequencing techniques and the reduction in costs, to date, over 12,994 plastome sequences have been deposited in the National Center for Biotechnology Information (Organelle genome database, NCBI). Higher plants have plastome (cp) composed of a double-stranded circular DNA molecule of a conserved quadripartite structure divided into a large single copy region (LSC) and a small single copy region (SSC), separated by two inverted repeats (IRs) (Kolodner and Tewari, 1979; Daniell et al., 2016; Meng et al., 2018). Depending on the plant species, most plastomes range from 120–160 kb in length, mainly affected by IR expansion/contraction or loss, and encode 120–130 genes (Palmer, 1985; Giardi and Piletska, 2006; Dong et al., 2012; Ruhlman and Jansen, 2014). Considering these unique features, plastome analysis provides a promising tool for resolving the identification challenges in species such as P. latifolia, where conventional morphological distinctions are often insufficient.
Due to its characteristics of parthenogenetic inheritance, a relatively small genome, a slower evolutionary rate than the nuclear genome, and a slow genome mutation rate (Birky, 1995; Song et al., 2017; Sun et al., 2020), the plastome has been widely used to develop molecular markers to classify medicinal plants, plant species identification, population genetics, genome evolution, and phylogenetics (Huo et al., 2019; Song et al., 2019; Guo et al., 2020; Tyagi et al., 2020). This method becomes particularly valuable in regions like the Fifa Mountains, where biodiversity is both rich and largely unexplored.
The Fifa mountains, located in the Asir region of Jazan Province located in southwestern Saudi Arabia, are a largely unexplored area despite their rugged and varied terrain. Ranging in elevation from 400 to about 2000 meters, these mountains have been recognized as a biodiversity hotspot and designated as an important plant area in the Arabian Peninsula. Despite its relatively limited size, the Fifa Mountains are rich in flora diversity, containing approximately 63% of Jazan’s floral species (Safhi et al., 2023). This exceptional biodiversity, combined with the challenges of species identification, highlights the importance of using advanced genetic tools like plastome analysis to better understand and classify the plant species found in these mountains.
This study aimed to address species identification and phylogenetic classification challenges through plastome assembly and annotation, which offers a powerful approach for resolving taxonomic ambiguities and understanding phylogenetic relationships. The complete plastome of P. latifolia was sequenced, annotated, and compared with 16 other Rubiaceae plastomes to identify genomic features and clarify phylogenetic relationships within this family. This approach not only helps to resolve the identification of Psydrax latifolia but also contributes to broader insights into the evolutionary relationships among members of the Rubiaceae family.
Psydrax latifolia, a small tree or smooth shrub with a right-angled branching pattern; and occasionally manifests as a few lianas, their stems are white to light or dark grey and finely fissured, while the branchlets are light brown or reddish brown in color. This flowering plant, characterized by its broad leaves (hence the epithet ‘latifolia’), exhibits an erect growth habit and is known for its sturdy yet supple branches. The leaves, typically arranged opposite each other along the stem, display a glossy, deep green hue and possess an ovate to elliptic shape (Bridson, 1985; Patro et al., 2014). Leaf shapes are oppositely lobed, broad-ovate to suborbicular, orbicular to elliptic, 3.6–8.5 cm long, and 3.0–8.7 cm wide. Petioles are 0.5–1.2 cm long, stout, flattened, and slightly winged by the leaf blades. Flowers are numerous, turbinate calyx tubes, campanulate corolla, 5.5 to 8.0 mm in length. Fruits are dark and shiny when ripe, have a slightly compressed shape, and are 5–9 mm long (Reynolds and Henderson, 2004).
Fresh samples of P. latifolia were collected from a single plant in the Fifa Mountains, located in the Asir region of Jazan Province, southwestern Saudi Arabia (17°15′ N 43°06′ E; Figure 1). Healthy leaves were collected and stored immediately in sterile bags containing 5g silica gel for DNA extraction. According to the manufacturer’s manual, total genomic DNA was extracted using WizPrep™ gDNA Mini Kit (Cell/Tissue, WIZBIO, Seoul, South Korea). DNA quality was assessed using 1% TBE agarose gels and measured using Quantus™ Fluorometer (Promega, USA) dsDNA Quantification Kit. After extraction, gDNA was stored at 20°C until further processing. The plant material has been deposited in the herbarium of King Abdulaziz University (code: KAU00008991).
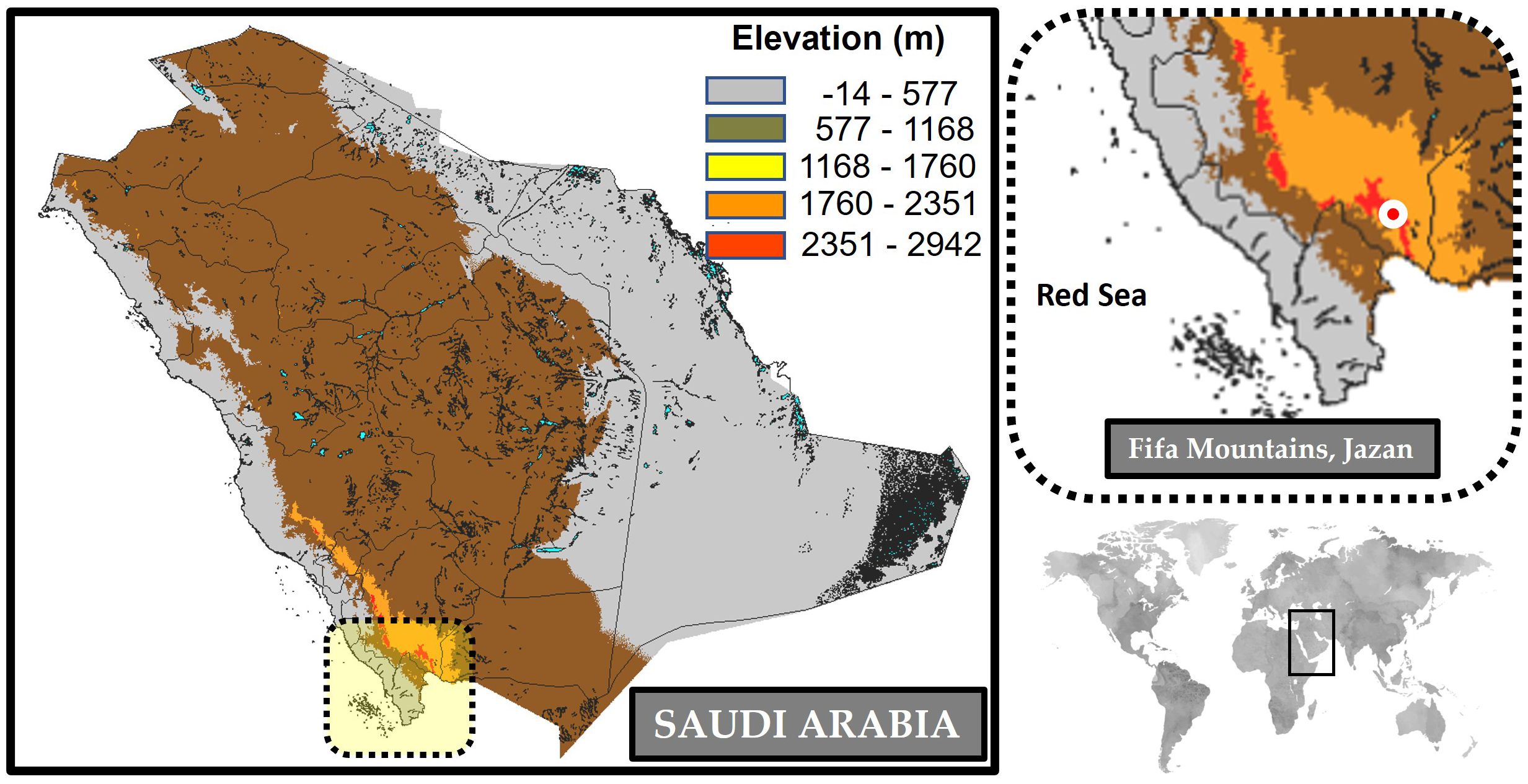
Figure 1. Geographical map of the Fifa Mountain located in Jazan province, Saudi Arabia.
To construct the DNA library, a 35 bp fragmented short insert was used according to the standard procedure for the TruSeq library preparation kit (Illumina, San Diego, California, USA). Using Il-lumina HiSeq 4000 platform (Novogene, China), the library was sequenced in pair-end mode with 150 bp length per read. High-quality clean reads with a Phred score of ≥20 obtained after trimming by BBDuk were used for de novo assembly using the single-contig method (Magdy et al., 2019, 2022; Magdy and Ouyang, 2020). Geneious Prime was used for P. latifolia plastome assembly, confirmation, and correction of coding sequences (Kearse et al., 2012). GeSeq was used to annotate the plastome based on the curated and refined organellar database of Chlorobox, producing graphical representation of the annotated plastome using the integrated OGDRAW tol (Tillich et al., 2017). Further, the anticodon sequences and typical cloverleaf secondary structures of all tRNAs have been confirmed by the tRNA scan-SE 2.0 search server (Lowe and Chan, 2016). In addition, P. latifolia plastome was deposited on the Banklt platform (NCBI, accession number OQ843458).
The long repeat sequences modeling was performed using a minimum repeat size of 30 and Hamming distance by REPuter software version 2.74 (Darling et al., 2004). Simple sequence repeats (SSRs) were detected using microsatellite identification (MISA) software version 1.0. (Beier et al., 2017) searching for SSR motif with length ranged between 1 to 6 with minimum number of repetition equal 10, 6, 5, 5, 5, and 5, respectively. The codon preference, including RSCU value, relative synonymous, and codon bias of the sequences encoded protein by P. latifolia was determined by MEGA X software version 10.2 (Kumar et al., 2018). The online tool Heatmapper (Babicki et al., 2016). The online tool Heatmapper was used to transform the codon frequencies using Euclidean distance (www.heatmapper.ca, accessed on the 5th of January 2023).
The basic characteristics of P. latifolia plastome and the other Rubiaceae family species (Table 1) were compared using the Irscope online tool (Amiryousefi et al., 2018) for IR/SSC and IR/LSC boundaries and junctions. (https://irscope.shinyapps.io/irapp/, accessed on 12th of January 2023). The plastomes were aligned by LAGAN alignment tool and compared using the mVISTA online tool (Frazer et al., 2004) (https://genome.lbl.gov/vista/mvista/submit.shtml, accessed on the 12th of January 2023).
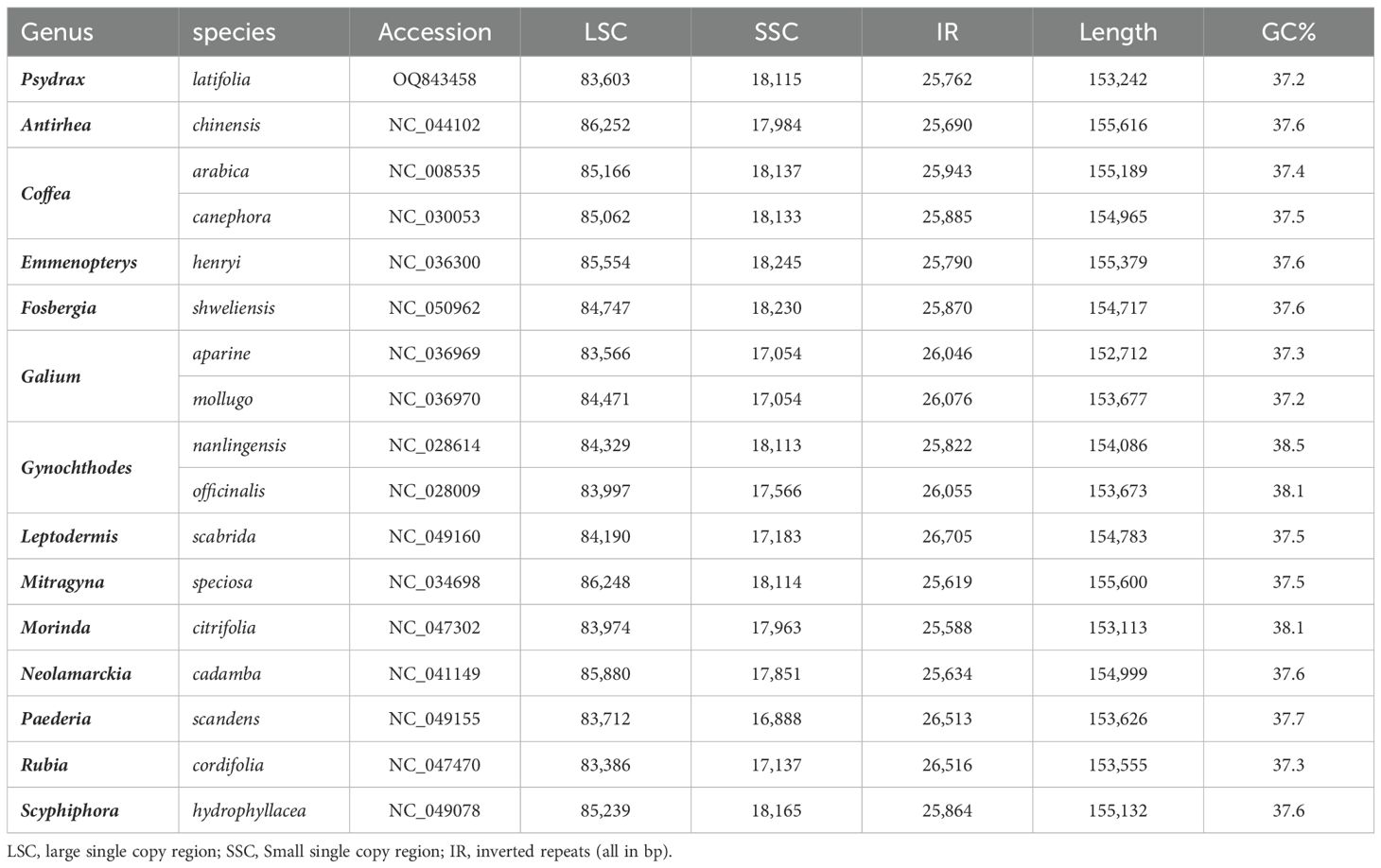
Table 1. The basic characteristics of P. latifolia chloroplast genome and the other Rubiaceae family species (obtained from NCBI).
To calculate the rates of synonymous and nonsynonymous substitution (dS/dN) in the protein-coding sequences of five Rubiaceae species, the protein coding sequence alignment tool MAFFT edition v7.3 (Katoh and Standley, 2013). After removing stop codons and gaps between the comparative sequences, the sequences were aligned. Ultimately, PAML’s CODEML software version 4.9 (Yang, 2007) was used to calculate dS/dN ratios. It was indicated that positive, neutral, or purifying selections would occur if ω > 1, ω = 1, and ω < 1, respectively (Nielsen, 2005).
To explore the phylogenetic relationships, the total chloroplasts, including the IR, SSC, and LSC regions, were aligned using Mauve genome aligner (Automatic seed weighting and calculation of the minimum Local Collinear Blocks “LCB” to perform a full alignment while assume collinear genomes) (Darling et al., 2004). Phylogenetic reconstruction was performed using FastTree V2 (Beier et al., 2017) following generalized time-reversible (GTR) models of nucleotide evolution and 1000 bootstrap iterations for reliability checks (Price et al., 2010), the full parameters and details can be found at http://www.microbesonline.org/fasttree/. The tree was graphed unrooted among the studied accessions.
The cp characteristics of P. latifolia have a conventional quadripartite structure characteristic typical of most land plants (Figure 2). The length of the plastome of P. latifolia was about 154,372 bp, long with a GC content of 38.2%, consisting of 52% in the protein-coding regions. Consisting of a pair of inverted repeats (IR) regions (25,762 bp), with a large single copy (LSC) region of 83,603 bp and a small single copy (SSC) region of 18,115 bp. The plastome of P. latifolia had a total of 128 genes, including 87 protein-coding genes (PCGs), 33 transfer RNA (tRNA) genes, and eight ribosomal RNA (rRNA) genes (Figure 2). rRNA and tRNA, the introns size, and the intergenic sequences had a total of 52%, 8%, 18%, and 40%, respectively (Table 2).
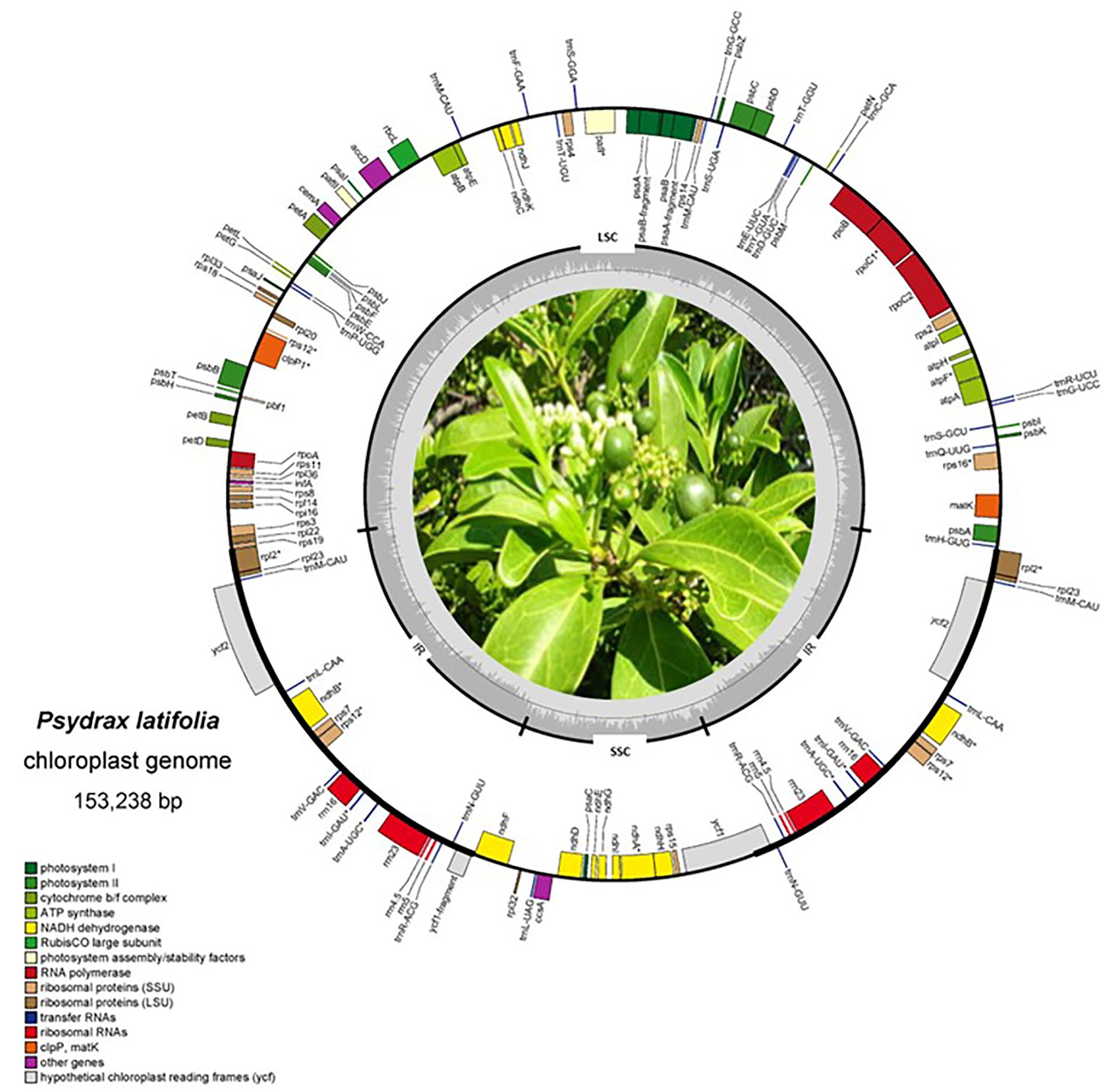
Figure 2. The complete chloroplast genome map of P. latifolia with a total length of 153,242 bp. The inner circle represents the GC content and is defined by cp regions (LSC, SSC, IRA, and IRB). The outer circle represents the complete cp sequence, with the genes annotated outside the circle to represent forward genes and the genes annotated inside the circle to represent reversed genes. All genes are colored according to their functional groups. (*) represent genes with introns.
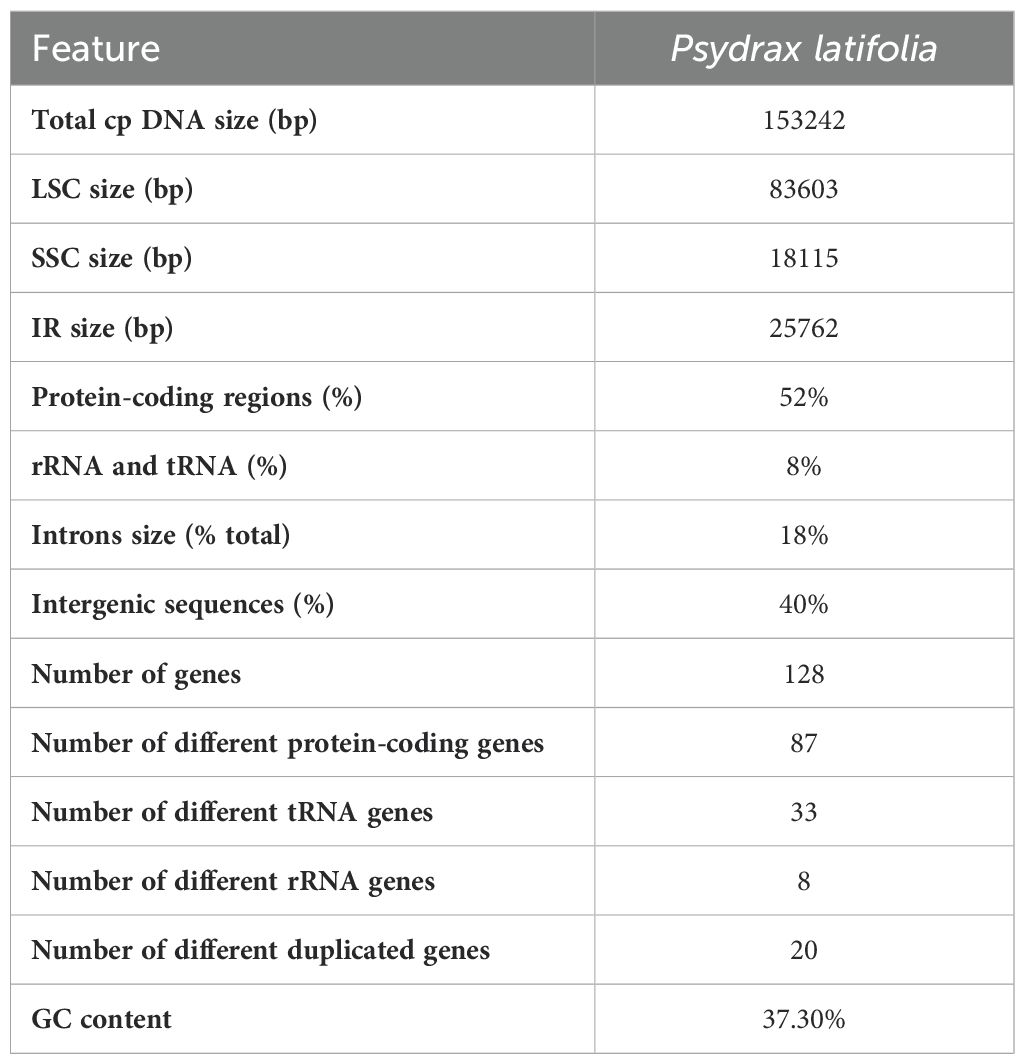
Table 2. The main basic features of P. latifolia chloroplast genome.
Analysis of repeat regions identified a total of 486 tandem repeats in the noncoding areas of the P. atifolia plastome. These repeats ranged from 6 to 142 base pairs in length and included 40 di-nucleotide, 78 tri-nucleotide, 72 tetra-nucleotide, 95 penta-nucleotide, and 142 hexa-nucleotide chloroplast simple sequence repeats (cpSSRs) (Table 3). Based on repeated class, the lowest number of cpSSRs was for 9-nucleotide repeats of 1%, the highest number of cpSSRs was for the hexanucleotides with the most repeated type (29%).
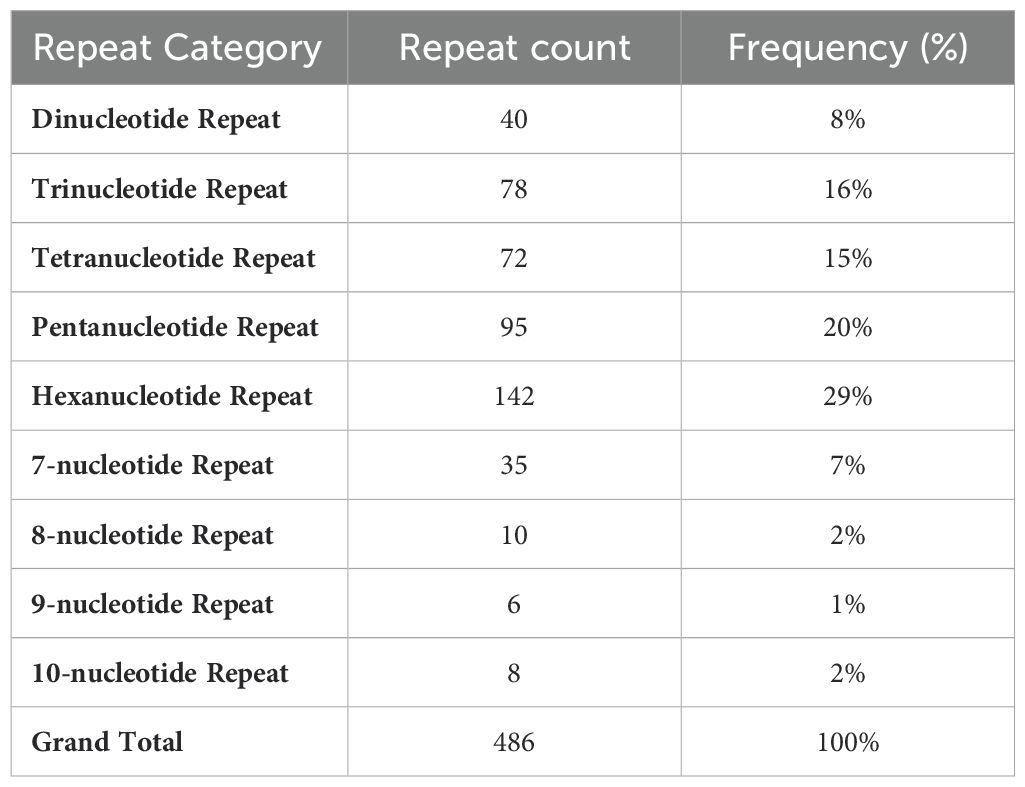
Table 3. Repeated sequences in the P. latifolia chloroplast genome, including repeat categories, repeat count and frequency percentage (%).
Among the protein-coding genes of P. latifolia, there are approximately 31,088 codons. The codon distribution is shown in Table 4. On average, UUU (Phenylalanine: F) encoding codons were less frequent, and CGC (Arginine: R) encoding codons were least frequent. The relative synonymous codon usage (RSCU) values for the 20 amino acids were estimated. According to the value of RSCU, synonymous codon preference falls into three patterns: (1) high preference (RSCU > 1); (2) no preference (RSCU = 1); and (3) low preference (RSCU < 1). Codon bias was observed for most amino acids in P. latifolia, except for AUG (Methionine: M) and UGG (Tryptophan: W). Among these 28 codons, there was a high preference for codon bias for AGA (Arginine:R) and GUU (Valine: V), while the lowest for CGC (Arginine: R) and CUG (Lucine: L). As a result of the analysis of codon usage bias, the genome of P. latifolia cp was found to be relatively conserved, as their evolution is quite similar.
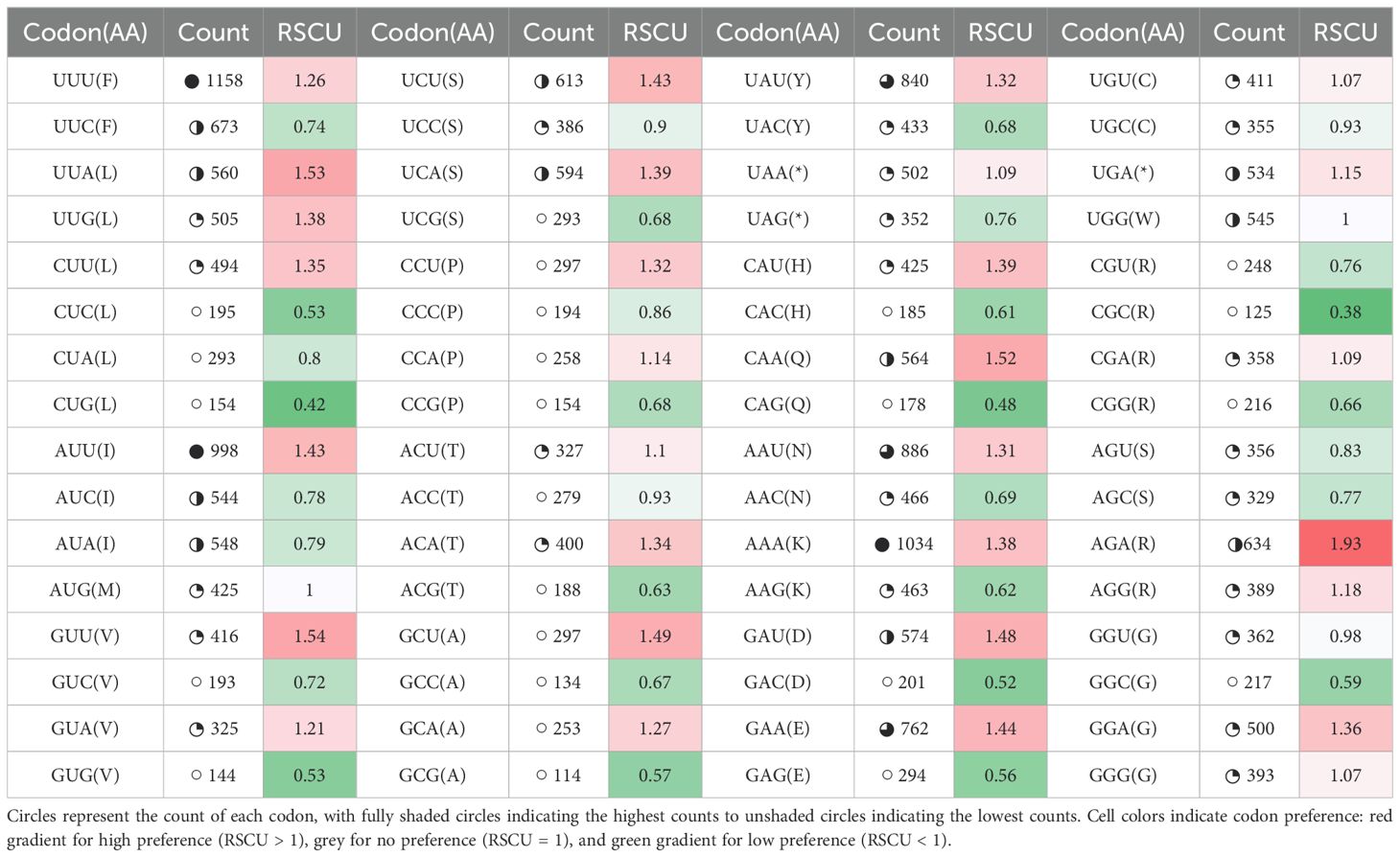
Table 4. Codon usage bias and Relative Synonymous Codon Usage (RSCU) values in the chloroplast genomes of P. latifolia.
Even though the IR region of the plastome is the most conserved in evolution, plastome length varies due to contractions and expansions in the border regions. Further, the expansion diversity and contraction diversity of the connected IR/SSC and IR/LSC regions have been examined. A comprehensive assessment of the four junctions between P. latifolia and nine representative species from the Rubiaceae family was conducted in the current study (Figure 3).Variations in the size of the plastome dynamically change the boundaries of the IR. Because of the LSC/IRb (JLB) boundary based on its position and gene synteny, its location between rpl22, rps19, and rpl2 was affected. The JSB boundary is located within the ndhF gene in all the species. In contrast, the ycf1 gene is located crossed all the JSA boundaries and across the JSB boundary except for Fosbergia shweliensis. The JLA boundary is located between rpl2 and trnH in all species. It was found that differences in the length of the four regions and whole genome sequences of plastomes were caused by variations in the IR/SC boundary region.

Figure 3. Comparison of LSC, SSC, and IR junctions between Psydrax latifolia and other Rubiaceae spices. Upper colored boxes represent genes, while arrows indicate their positions near junctions, while colored sections represent the three major chloroplast compartments (LSC: clear blue, SSC: green, and Ira/b: orange). The junction sites of the plastid genome are indicated by the abbreviations JLA (IRa/LSC), JLB (IRb/LSC), and JSA (SSC/IRa).
Among Rubiacea species, DNA polymorphism was estimated for the genetic group (i.e., CDS, rRNA, and tRNA) separately. In the case of coding sequences (CDS), the total number of recorded mutations was 79,615, with 13,680 (17.18%) segregating sites. The average number of mutations was 1,020 ± 1,230 sites, with a maximum value of 6,897 recorded for ycf2, and a minimum value of 90 recorded for petN gene. The average number of segregating sites was 175 ± 250 sites, with a maximum value of 1,733 recorded for ycf1 and a minimum value of 8 recorded for psbL gene. The haplotype diversity ranged between 1.00 to 0.825 (petN) among all genes (Figure 4).
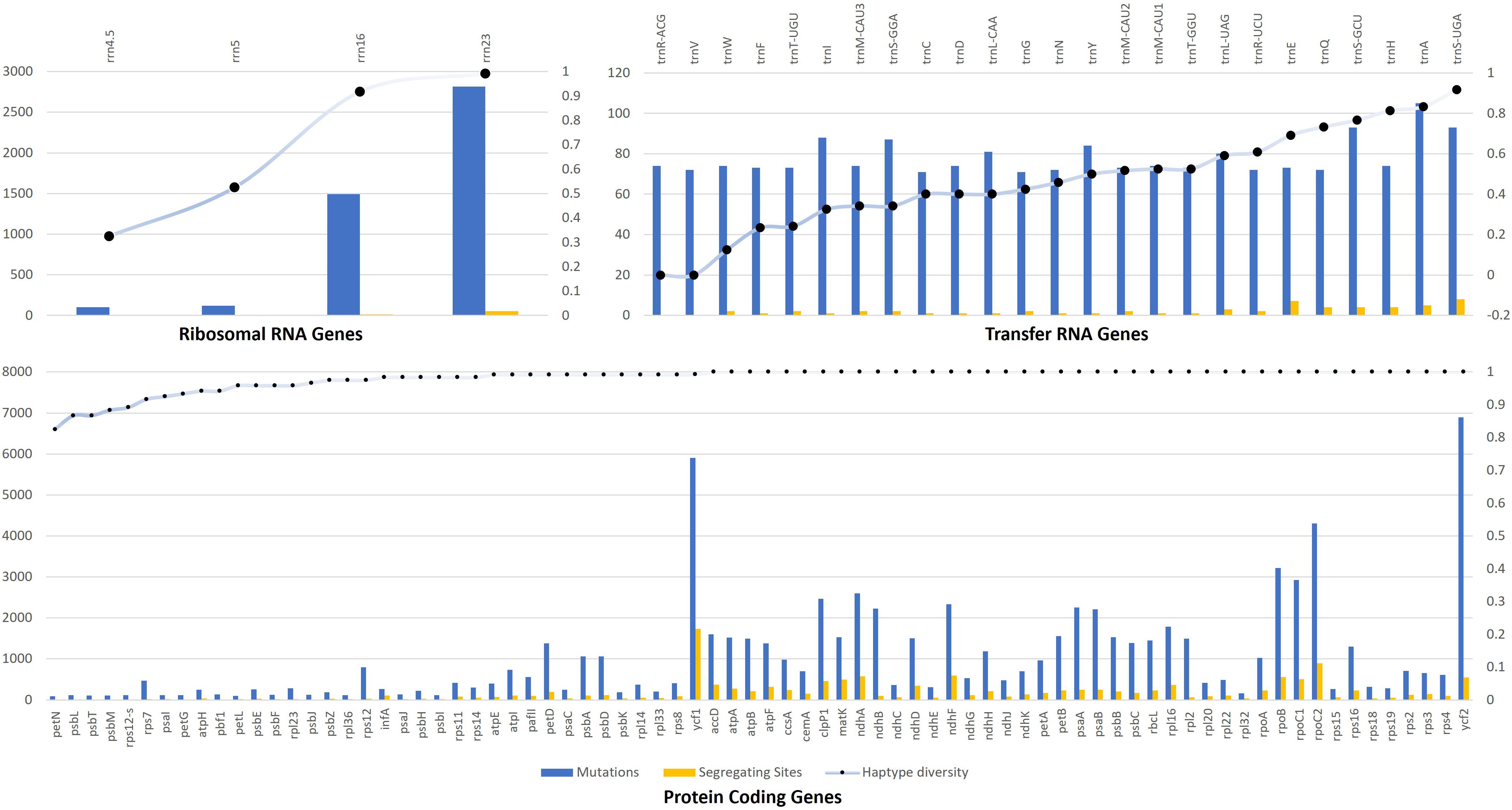
Figure 4. Genetic variability among P. latifolia and other Rubiaceae chloroplast genomes for ribosomal RNA, transfer RNA and protein-coding genes. Mutations are presented with blue columns, segregating sites with orange columns and haplotype diversity with dotted blue line.
In the case of rRNA, the total number of recorded mutations was 4,529, with 68 (1.5%) segregating sites. The average number of mutations was 1,132 ± 1,296 sites with a maximum value of 2,814 recorded for rrn23, and a minimum value of 103 recorded for rrn4.5 gene. The average number of segregating sites was 17 ± 24 sites, with a maximum value of 51 recorded for rrn23 and a minimum value of 1 recorded for rrn4.5 and rrn5 genes. The haplotype diversity ranged between 0.991 to 0.325 (rrn4.5) among all analyzed species (Figure 4). In the case of tRNA, the total number of recorded mutations was 1,950, with 58 (2.97%) segregating sites. The average number of mutations was 78 ± 9 sites, with a maximum value of 105 recorded for trnA, and a minimum value of 71 recorded for trnC and trnG genes. The average number of segregating sites was 2 ± 2 sites with a maximum value of 8 recorded for trnSUGA, and a minimum value of 0 recorded for trnRACG and trnV genes. The haplotype diversity ranged between 0.916 to 0 (trnRACG and trnV) among all tRNAs (Figure 4).
When genes were grouped by function, the average number of mutations was 4,423 ± 4191 (including 760 ± 745 segregating) sites with a maximum value of 12,801 (including 2,278 segregating) sites recorded for the ycf gene group of unknown function and a minimum value of 132 (including 12 segregating) recorded for infA (Photosystem biogenesis factor 1) gene. The most variable functional groups were the ycf group of unknown function (12,801 mutations include 2,278 segregating sites), DNA-dependent RNA polymerase (11,472 mutations include 2,180 segregating sites), and NADH oxidoreductase (12,228 mutations include 2,265 segregating sites; Figure 5A).
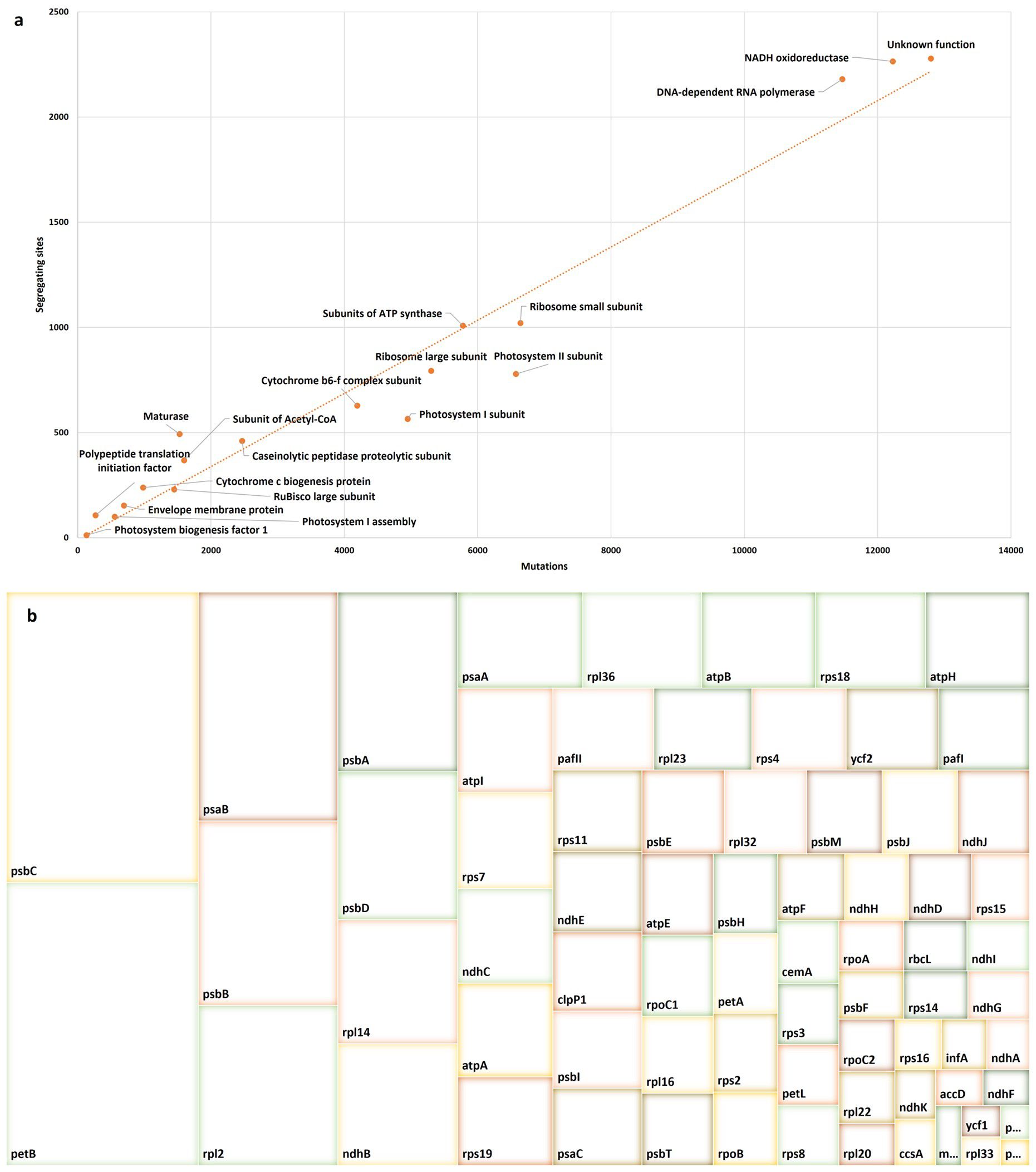
Figure 5. Genetic variability among P. latifolia and other Rubiaceae chloroplast genomes for each functional group (A) and the dN/dS ratio was estimated for each protein-coding gene (B). Colors are for improving visualization and reflect no specific value.
The protein-coding genes of P. latifolia were screened for single nucleotide polymorphisms (SNPs) and indels (insertions and deletions). These results showed 15,822 variations, including 13,192 SNPs and 653 indels (Supplementary Table S1). In contrast to the IR region, the SSC and LSC regions exhibit the greatest density of indel sites, discovered on ycf1, rpoC2, accD, ndhF, ccsA, infA and ndhK. All SNPs were classified into two types: synonymous (dS) and nonsynonymous (dN). Within the protein-coding genes, there were 11,199 synonymous and 1,994 nonsynonymous SNPs. The latter recorded an average of 24.9 ± 71 dN SNPs, with 15 genes recording dN SNPs above average. The most substitutions occurred in the ycf1 gene, followed by the ndhF, rpoC2, matK, and accD, respectively.
The synonymous (dS) and nonsynonymous (dN) substitution rates among the Rubiaceae species were used to detect selective pressure on PCGs by calculating the dN/dS ratio. Genes named pbf1, petD, petG, psaI, psaJ, psbL, psbZ, and rps12 recorded zero non-synonymous substitutions, and thus were excluded from the comparison. For the remaining genes, we did not observe purifying selection since most dN/dS ratios were greater than 1 The dN/dS ratios ranged from 1.3 (petN) to 97.5 (psbC) with an average of 14.44 ± 17.35, where psbC, petB, psaB, psbB, rpl2, psbA, psbD, rpl14, ndhB, psaA, rpl36, atpB, rps18, atpH, atpI, rps7, ndhC, and atpA recorded ratios above average, respectively (Figure 5B).
The complete plastome was analyzed of P. latifolia, in addition, 16 other complete plastome of Rubiaceae species were downloaded from GenBank for phylogenetic analysis. As a result of the phylogenetic analysis, the subfamily classification was divided into three (Rubioideae, Cinchonoideae, and Ixoroideae). The results showed a close relationship between P. latifolia and Scyphiphora hydrophyllacea. Furthermore, it was found that most nodes in the chloroplast ML tree were highly supported (Figure 6). The current study will provide valuable information for further phylogenetic analyses and evolutionary studies on the Ixoroideae.
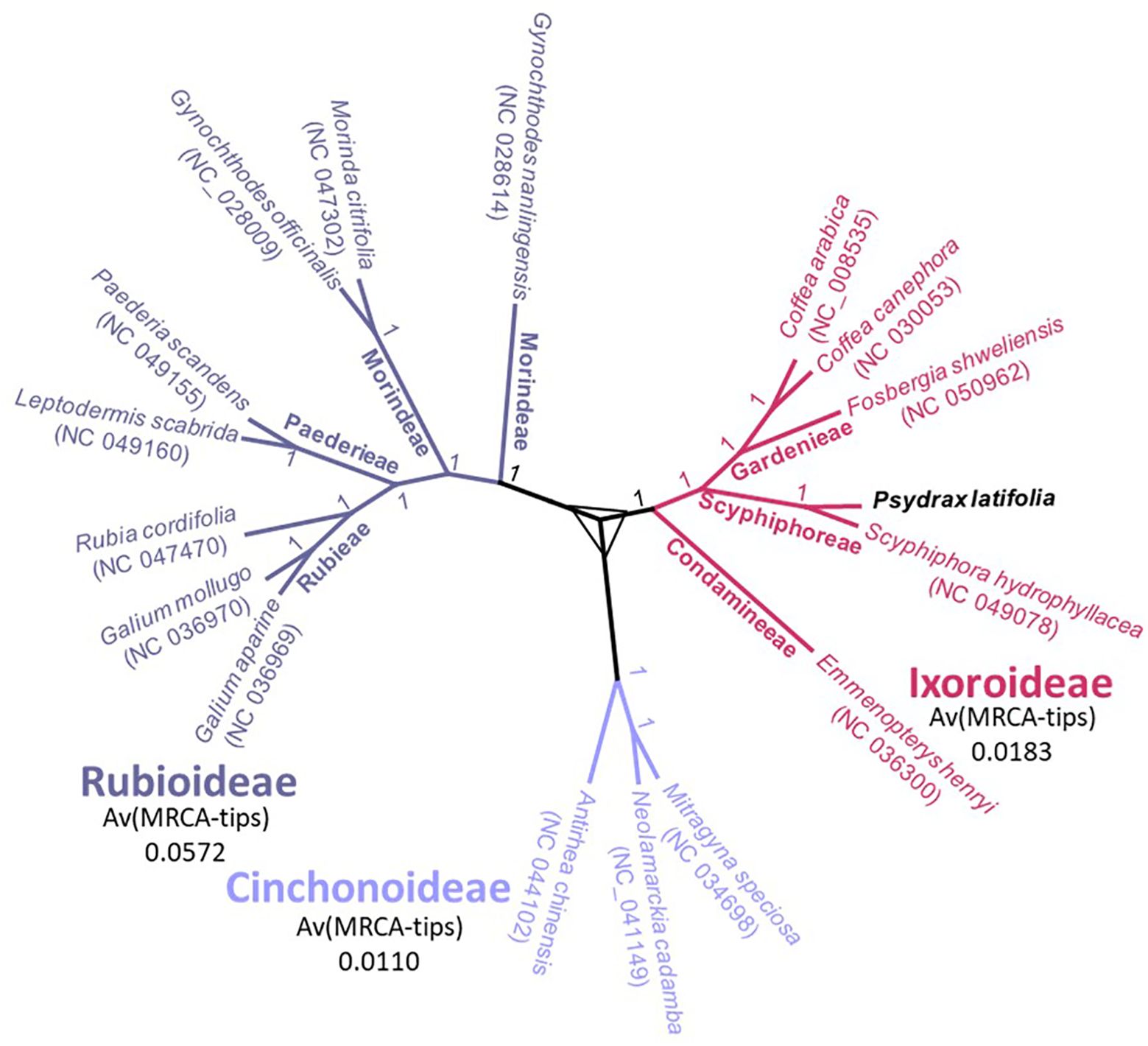
Figure 6. Phylogenetic trees of P. latifolia and other species belong to the Rubiaceae family using maximum likelihood based on the LSC region, SSC region, and IR region together. The bootstrap values of ML are written on each node. Each subfamily (distinct clade) is colored differently.
Using NGS technology in the current study, P. latifolia complete plastome was sequenced for the first time. A comprehensive comparison of published sixteen other species belonging to the Rubiaceae family from the NCBI genomic database after splicing, assembly, and gap-filling was conducted. Like most angiosperm genomes, P. latifolia plastomes contain a pair of IR regions (IRa and IRb) and a pair of SSC and LSC regions. As reported before, most land plants’ molecular analysis indicated that their plastomes consist of two identical IR regions with lower nucleotide substitution rates and fewer indels than their SSC and LSC counterparts (Li et al., 2017). According to our study, the plastome of P. latifolia and most land plants was conservative. Additionally, the plastome GC content were quite similar to those of other species studied previously (Zhang et al., 2019b; Geng et al., 2020; Niu and Liu, 2020; Zhang et al., 2020). Based on the structure of the plastome, GC content, and gene composition of the species studied in the present study, we observed that the plastome has a stable structure and a low evolution rate in these species. Since the plastome has a highly conserved nature and a low evolutionary rate, it is a hotspot for phylogenetic studies (Daniell et al., 2016); thus, it is a valuable resource for research in molecular phylogenetics and ecology (Li et al., 2017; Guan et al., 2022).
Two types of the identified SSRs in the plastome of P. latifolia were most prevalent in the plastid genome: Hexa-nucleotide repeats (29%), and Penta-nucleotide repeats (20%). The structural and comparative analysis of the complete plastome of Scyphiphora hydrophyllacea identified a total of 52 microsatellites, with A/T mononucleotides being the predominant SSR across nine related Rubiaceae species (Zhang et al., 2019c). Chloroplast simple sequence repeats (cpSSRs) are usually used as critical molecular markers for species identification (Yang et al., 2011; Jiao et al., 2012). The majority of these cpSSRs were found in LSC regions, followed by SSC regions, which is consistent with the plastid genome. There are many advantages to using SSRs as molecular markers, including abundance, high reproducibility, codominant inheritance, uniparental inheritance, and relative conservation, making them ideal for identifying species and assessing genetic variation at both the population and individual levels (Moore et al., 2007).
In P. latifolia, UUU (Phenylalanine: F) had the highest number of codons, while CGC (Arginine: R) had the lowest number of codons. A codon bias occurs when synonymous codons encode the same amino acid and are not used randomly, a common biological phenomenon (Hershberg and Petrov, 2008). It has been suggested that species with close phylogenetic relationships select codons similarly (Romero, 2000). During long-term evolution, organisms that encode the same amino acid prefer different codons encoding the amino acid (Daniell et al., 2016).
The RSCU results from a combination of natural selection, genetic drift, and mutation (Fu et al., 2022). The current study observed that most amino acids in P. latifolia exhibited codon bias except for AUG (Methionine: M) and UGG (Tryptophan: W). The highest preference for codon bias was found for (AGA) Arginine (R) and GUU (Valine: V), while the lowest preference was found for CGC (Arginine: R) and CUG (Lucine: L). Codon usage patterns are similar to those reported for related species, supporting the hypothesis of shared evolutionary pressures and has been extensively used to investigate the evolution and phylogeny of land plants (Wang et al., 2022). Gene expression and cellular function are majorly influenced by synonymous codon usage bias (Yang et al., 2021).
In P. latifolia, most of the variations in the plastome structure were caused by contraction and expansion in the IR region. In addition, the expansion diversity and contraction diversity of the connected IR/SSC and IR/LSC regions have been examined. In the current study, four junctions between P. latifolia and nine Rubiaceae species were comprehensively examined. In accordance with other plant species, IR regions are more conserved than LSC and SSC regions (Tang et al., 2004; Jiang et al., 2017; Cui et al., 2019). In the sequenced plastome, the longest and shortest IR regions differed, suggesting that the expansion/contraction of IR regions is not a major factor in cp evolution. The SC/IR boundaries were examined more closely; there was no difference in gene order. In contrast, the location of genes had some slight differences. Most Rubiaceae plants, for instance, have the tRNA trnH-GUG gene completely located in the LSC region (for example, Dunnia sinensis, Hedyotis ovata, and Trailliaedoxa gracilis) (Zhang et al., 2019a; Wikström et al., 2020; Zhang et al., 2020). A truncated copy of the ycf1 gene was present in the border of IRb/SSC and stretched over the boundary of SSC/IRa. On the other hand, the ycf1 gene is located across the JSA boundary and across the JSB boundary, except for Fosbergia shweliensis. In all species, the JLA boundary is located between rpl2 and trnH. This may provide a novel pattern for understanding the evolution of the plastome’s structure and evolutionary dynamics (Wicke and Naumann, 2018; Mehmood et al., 2020).
According to the dN/dS analysis, several genes had the highest ratio of dN and dS. This suggests they were selected for their sequence diversity and effect on the P. latifolia plastome sequence evolution rate. Despite the highly conserved genome, SNPs cluster in hotspots resulting in highly variable loci. Furthermore, variable hotspots containing indels have also been reported (Aldrich et al., 1988; Zhou et al., 2018). In contrast to commonly used molecular markers, the plastome can vary between closely related species thanks to its conserved sequence length of 110 to 160 kb (Li et al., 2015). Therefore, indels and SNPs are found in significant numbers across plastomes. This allowed identifying mutation hotspot regions specific to Rubiaceae families (for example, the ycf1 gene, followed by the ndhF, rpoC2, matK, and accD genes). There is one DNA barcode sequence in this list: matK. The genetic variation within these regions might also be sufficient to resolve the phylogenetic relationship of the Rubiaceae species.
By estimating the dN/dS ratio, we can gain insight into the constraints on organisms imposed by natural selection (Shi et al., 2019). To understand how the substitution rate affects the structure and function of genes, it is important to analyze the adaptive evolution of genes (Zhang et al., 2019a). There is a positive selection effect on the sequence divergence of most of the protein-coding genes investigated in this study. For example, the genes rpl and rps encode ribosomal proteins that have more divergent sequences compared to those related to photosynthesis such as psbB and psbC, which are associated with photosystem II (Souza et al., 2019). The matK is responsible for cutting and splicing group II RNA transcriptional introns (Hertel et al., 2013). In contrast, the rpoC2 gene encodes transcription proteins (Tyagi et al., 2020). In addition, psaA and ndhF genes play roles in photosynthesis and carbon fixation (Zhou et al., 2018; Huo et al., 2019). In terms of size, ycf1 and ycf2 encode two of the largest membrane proteins, which are unsuitable for routine DNA barcoding techniques (Drescher et al., 2000; Kikuchi et al., 2013). The survival and adaptation of plants depend upon all these genes, thus suggesting that all the detected DNA polymorphism is occurring with an adaptive evolutionary nature (Wang et al., 2022).
Plastomes are useful for phylogenetic studies, especially at low taxonomic levels, where they allow for resolving evolutionary relationships (Du et al., 2017). The availability of some of the complete plastomes allowed us to confirm the phylogenetic position of forest tree species among the Rubiaceae family, suggesting that the plastome sequences can effectively resolve species relationships, as demonstrated by (Spooner et al., 2017; Bedoya et al., 2019). The phylogenetic tree analysis showed that all species formed three significant clusters, which corresponded to the three subfamilies (Rubioideae, Cinchonoideae, and Ixoroideae) (Saldaña et al., 2022). The plastomes analysis of Rubiaceae provided a highly supported topology in accordance with previous reports (Bremer and Eriksson, 2009). The results of previous phylogenetic studies of the subfamily Ixoroideae were similar, but the support values were relatively low (Zhang et al., 2019a). Further evidence could be obtained by employing other members -whenever available- of the subfamily Ixoroideae and examining the plastome in order to accurately reconstruct the evolutionary history of P. latifolia. In addition to shedding light on P. latifolia evolutionary position, these findings will also provide valuable chloroplast genomic data for further study of Rubiaceae genesis and diversification.
The current study successfully sequenced the complete plastome of P. latifolia using next-generation sequencing technology. A comparison with 16 other Rubiaceae species revealed conserved regions (IR, SSC, and LSC) and similar GC content and gene composition, highlighting the plastome’s stability and low evolutionary rate. In P. latifolia, hexa-nucleotide and penta-nucleotide SSRs were prevalent, mostly located in the LSC region, serving as potential molecular markers after proper validation. Codon usage bias was observed, with UUU (Phenylalanine) being the most frequent and CGC (Arginine) the least, reflecting evolutionary pressures consistent with related species. Structural variations were primarily due to IR region contractions and expansions, with minimal impact on genome evolution. SC/IR boundary examination showed consistent gene order but slight variations in gene locations, providing insights into plastome evolution. The dN/dS analysis indicated selective pressures on several genes, identifying mutation hotspots crucial for understanding phylogenetic relationships within the Rubiaceae family. Phylogenetic analysis confirmed species clustering into Rubioideae, Cinchonoideae, and Ixoroideae subfamilies, aligning with previous studies. This study elucidates the evolutionary relationships of P. latifolia within the Rubiaceae family and underscores the plastome’s potential for phylogenetic and ecological research, species identification, and genetic variation assessment.
The original contributions presented in the study are publicly available. This data can be found here: NCBI GenBank, accession OQ843458.
FS: Conceptualization, Data curation, Formal analysis, Funding acquisition, Writing – review & editing, Writing – original draft. AJ: Formal analysis, Funding acquisition, Resources, Software, Writing – review & editing. RMA: Methodology, Project administration, Resources, Software, Validation, Writing – review & editing. RA: Project administration, Resources, Software, Validation, Visualization, Writing – review & editing. AA: Investigation, Methodology, Project administration, Resources, Software, Writing – review & editing. WF: Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Writing – review & editing. AA: Conceptualization, Data curation, Formal analysis, Resources, Software, Writing – review & editing. MH: Methodology, Resources, Software, Visualization, Writing – review & editing, Validation. NA: Conceptualization, Methodology, Project administration, Resources, Software, Writing – review & editing. MM: Investigation, Methodology, Project administration, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. DA: Data curation, Project administration, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2024R366), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
The authors extend their appreciation to Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2024R366), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2024.1416876/full#supplementary-material
Supplementary Table 1 | DNA polymorphism and dN/dS ratios of Psydrax latifolia chloroplast genes.
Aldrich J., Cherney B. W., Merlin E. (1988). The role of insertions/deletions in the evolution of the intergenic region betweenpsbA andtrnH in the chloroplast genome. Curr. Genet. 14, 137–146. doi: 10.1007/BF00569337
Amiryousefi A., Hyvönen J., Poczai P. (2018). IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031. doi: 10.1093/bioinformatics/bty220
Arriola A. H., Alejandro G. J. (2013). A new species of Psydrax (Vanguerieae, Rubiaceae) from Luzon, Philippines including its conservation status. Phytotaxa 149, 27. doi: 10.11646/phytotaxa.149.1.4
Babicki S., Arndt D., Marcu A., Liang Y., Grant J. R., Maciejewski A., et al. (2016). Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res. 44, W147–W153. doi: 10.1093/nar/gkw419
Bedoya A. M., Ruhfel B. R., Philbrick C. T., Madriñán S., Bove C. P., Mesterházy A., et al. (2019). Plastid genomes of five species of riverweeds (Podostemaceae): structural organization and comparative analysis in malpighiales. Front. Plant Sci. 10. doi: 10.3389/fpls.2019.01035
Beier S., Thiel T., Münch T., Scholz U., Mascher M. (2017). MISA-web: a web server for microsatellite prediction. Bioinformatics 33, 2583–2585. doi: 10.1093/bioinformatics/btx198
Birky C. W. (1995). Uniparental inheritance of mitochondrial and chloroplast genes: mechanisms and evolution. Proc. Natl. Acad. Sci. U.S.A. 92, 11331–11338. doi: 10.1073/pnas.92.25.11331
Bremer B., Eriksson T. (2009). Time tree of rubiaceae: phylogeny and dating the family, subfamilies, and tribes. Int. J. Plant Sci. 170, 766–793. doi: 10.1086/599077
Bridson D. M. (1985). The reinstatement of psydrax (Rubiaceae, subfam. Cinchonoideae tribe vanguerieae) and a revision of the African species. Kew Bull. 40, 687. doi: 10.2307/4109853
Chukwudulue U. M., Attah A. F., Okoye F. B. C. (2022). Linking phytochemistry to traditional uses and pharmacology of an underexplored genus - Psydrax: a review. Phytochem Rev. 21 (5), 1577–1604. doi: 10.1007/s11101-021-09798-6
Cui Y., Chen X., Nie L., Sun W., Hu H., Lin Y., et al. (2019). Comparison and phylogenetic analysis of chloroplast genomes of three medicinal and edible amomum species. IJMS 20, 4040. doi: 10.3390/ijms20164040
Daniell H., Lin C.-S., Yu M., Chang W.-J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17, 1–29. doi: 10.1186/s13059-016-1004-2
Darling A. C. E., Mau B., Blattner F. R., Perna N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394. doi: 10.1101/gr.2289704
Dong W., Liu J., Yu J., Wang L., Zhou S. (2012). Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PloS One 7, e35071. doi: 10.1371/journal.pone.0035071
Drescher A., Ruf S., Calsa T., Carrer H., Bock R. (2000). The two largest chloroplast genome-encoded open reading frames of higher plants are essential genes. Plant J. 22, 97–104. doi: 10.1046/j.1365-313x.2000.00722.x
Du Y., Bi Y., Yang F., Zhang M., Chen X., Xue J., et al. (2017). Complete chloroplast genome sequences of Lilium: insights into evolutionary dynamics and phylogenetic analyses. Sci. Rep. 7, 5751. doi: 10.1038/s41598-017-06210-2
Feenna O. P., Estella O. U., Chioma Obianuju P.-O., Ifeanyi N. F., Obodike E. C. (2020). Pharmacognostic and phytochemical studies of leaves of psydrax horizontalis schum. and thonn (Rubiaceae). PJ 12, 541–550. doi: 10.5530/pj.2020.12.82
Frazer K. A., Pachter L., Poliakov A., Rubin E. M., Dubchak I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. doi: 10.1093/nar/gkh458
Fu N., Ji M., Rouard M., Yan H.-F., Ge X.-J. (2022). Comparative plastome analysis of Musaceae and new insights into phylogenetic relationships. BMC Genomics 23, 223. doi: 10.1186/s12864-022-08454-3
Gaertner J. (1788). De fructibus et seminibus plantarum (Stutgardiae: typis Academiae Carolinae). doi: 10.5962/bhl.title.102753
Geng Y., Li Y., Yuan X., Luo T., Wang Y. (2020). The complete chloroplast genome sequence of Fosbergia shweliensis, an endemic species to Yunnan of China. Mitochondrial DNA Part B 5, 1796–1797. doi: 10.1080/23802359.2020.1750322
Giardi M. T., Piletska E. V. (2006). Biotechnological Applications of Photosynthetic Proteins: Biochips, Biosensors and Biodevices (Boston, MA: Springer US). doi: 10.1007/978-0-387-36672-2
Guan Y., Liu W., Duan B., Zhang H., Chen X., Wang Y., et al. (2022). The first complete chloroplast genome of Vicatia thibetica de Boiss.: genome features, comparative analysis, and phylogenetic relationships. Physiol. Mol. Biol. Plants 28, 439–454. doi: 10.1007/s12298-022-01154-y
Guo X.-L., Zheng H.-Y., Price M., Zhou S.-D., He X.-J. (2020). Phylogeny and comparative analysis of chinese chamaesium species revealed by the complete plastid genome. Plants 9, 965. doi: 10.3390/plants9080965
Hershberg R., Petrov D. A. (2008). Selection on codon bias. Annu. Rev. Genet. 42, 287–299. doi: 10.1146/annurev.genet.42.110807.091442
Hertel S., Zoschke R., Neumann L., Qu Y., Axmann I. M., Schmitz-Linneweber C. (2013). Multiple checkpoints for the expression of the chloroplast-encoded splicing factor MatK. Plant Physiol. 163, 1686–1698. doi: 10.1104/pp.113.227579
Huo Y., Gao L., Liu B., Yang Y., Kong S., Sun Y., et al. (2019). Complete chloroplast genome sequences of four Allium species: comparative and phylogenetic analyses. Sci. Rep. 9, 12250. doi: 10.1038/s41598-019-48708-x
Jiang D., Zhao Z., Zhang T., Zhong W., Liu C., Yuan Q., et al. (2017). The Chloroplast Genome Sequence of Scutellaria baicalensis Provides Insight into Intraspecific and Interspecific Chloroplast Genome Diversity in Scutellaria. Genes 8, 227. doi: 10.3390/genes8090227
Jiao Y., Jia H., Li X., Chai M., Jia H., Chen Z., et al. (2012). Development of simple sequence repeat (SSR) markers from a genome survey of Chinese bayberry (Myrica rubra). BMC Genomics 13, 201. doi: 10.1186/1471-2164-13-201
Katoh K., Standley D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kearse M., Moir R., Wilson A., Stones-Havas S., Cheung M., Sturrock S., et al. (2012). Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kikuchi S., Bédard J., Hirano M., Hirabayashi Y., Oishi M., Imai M., et al. (2013). Uncovering the protein translocon at the chloroplast inner envelope membrane. Science 339, 571–574. doi: 10.1126/science.1229262
Kolodner R., Tewari K. K. (1979). Inverted repeats in chloroplast DNA from higher plants. Proc. Natl. Acad. Sci. U.S.A. 76, 41–45. doi: 10.1073/pnas.76.1.41
Kumar S., Stecher G., Li M., Knyaz C., Tamura K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lamarck J. B., Poiret J. L. M. (1783). Encyclopédie méthodique: botanique /Par m. le chevalier de Lamarck (Paris, Liège: Panckoucke; Plomteux). doi: 10.5962/bhl.title.824
Lantz H., Andreasen K., Bremer B. (2002). Nuclear rDNA ITS sequence data used to construct the first phylogeny of Vanguerieae (Rubiaceae). Plant Systematics Evol. 230, 173–187. doi: 10.1007/s006060200003
Lantz H., Bremer B. (2004). Phylogeny inferred from morphology and DNA data: characterizing well-supported groups in Vanguerieae (Rubiaceae). Botanical J. Linn. Soc. 146, 257–283. doi: 10.1111/j.1095-8339.2004.00338.x
Lantz H., Bremer B. (2005). Phylogeny of the complex Vanguerieae (Rubiaceae) genera Fadogia, Rytigynia, and Vangueria with close relatives and a new circumscription of Vangueria. Plant Syst. Evol. 253, 159–183. doi: 10.1007/s00606-005-0313-9
Li X., Yang Y., Henry R. J., Rossetto M., Wang Y., Chen S. (2015). Plant DNA barcoding: from gene to genome: Plant identification using DNA barcodes. Biol. Rev. 90, 157–166. doi: 10.1111/brv.12104
Li Z., Long H., Zhang L., Liu Z., Cao H., Shi M., et al. (2017). The complete chloroplast genome sequence of tung tree (Vernicia fordii): Organization and phylogenetic relationships with other angiosperms. Sci. Rep. 7, 1869. doi: 10.1038/s41598-017-02076-6
Lowe T. M., Chan P. P. (2016). tRNAscan-SE On-line: integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 44, W54–W57. doi: 10.1093/nar/gkw413
Ly S. N., Garavito A., De Block P., Asselman P., Guyeux C., Charr J.-C., et al. (2020). Chloroplast genomes of Rubiaceae: Comparative genomics and molecular phylogeny in subfamily Ixoroideae. PloS One 15, e0232295. doi: 10.1371/journal.pone.0232295
Magdy M., El-Sherbeny E. A., Ramirez Sanchez A. (2022). The complete chloroplast genome of the Egyptian henbane (Hyoscyamus muticus L., Solanaceae). Mitochondrial DNA Part B 7, 1109–1111. doi: 10.1080/23802359.2022.2086493
Magdy M., Ou L., Yu H., Chen R., Zhou Y., Hassan H., et al. (2019). Pan-plastome approach empowers the assessment of genetic variation in cultivated Capsicum species. Hortic. Res. 6, 108. doi: 10.1038/s41438-019-0191-x
Magdy M., Ouyang B. (2020). The complete mitochondrial genome of the chiltepin pepper ( Capsicum annuum var. glabriusculum ), the wild progenitor of Capsicum annuum L. Mitochondrial DNA Part B 5, 683–684. doi: 10.1080/23802359.2020.1714496
Mehmood F., Abdullah, Shahzadi I., Ahmed I., Waheed M. T., Mirza B. (2020). Characterization of Withania somnifera chloroplast genome and its comparison with other selected species of Solanaceae. Genomics 112, 1522–1530. doi: 10.1016/j.ygeno.2019.08.024
Meng X.-X., Xian Y.-F., Xiang L., Zhang D., Shi Y.-H., Wu M.-L., et al. (2018). Complete chloroplast genomes from sanguisorba: identity and variation among four species. Molecules 23, 2137. doi: 10.3390/molecules23092137
Moore M. J., Bell C. D., Soltis P. S., Soltis D. E. (2007). Using plastid genome-scale data to resolve enigmatic relationships among basal angiosperms. Proc. Natl. Acad. Sci. U.S.A. 104, 19363–19368. doi: 10.1073/pnas.0708072104
Nielsen R. (2005). Molecular signatures of natural selection. Annu. Rev. Genet. 39, 197–218. doi: 10.1146/annurev.genet.39.073003.112420
Niu Y.-F., Liu J. (2020). The complete chloroplast genome of Morinda citrifolia (noni). Mitochondrial DNA Part B 5, 377–378. doi: 10.1080/23802359.2019.1703586
Palmer J. D. (1985). COMPARATIVE ORGANIZATION OF CHLOROPLAST GENOMES. Annu. Rev. Genet. 19, 325–354. doi: 10.1146/annurev.ge.19.120185.001545
Patro S. K., Sasmal D., Mazumndar P., Behera P., Lal U. R., Dash S., et al. (2014). Review on genus Canthium: Special reference to Canthium coromandelicum-an unexplored traditional medicinal plant of Indian Subcontinent. Am. J. Phytomed Clin. Therap 2, 796–813. Available at: https://www.academia.edu/download/35508323/PA-400383-_15_.pdf
Price M. N., Dehal P. S., Arkin A. P. (2010). FastTree 2 – approximately maximum-likelihood trees for large alignments. PloS One 5, e9490. doi: 10.1371/journal.pone.0009490
Razafimandimbison S. G., Lantz H., Mouly A., Bremer B. (2009). Evolutionary trends, major lineages, and new generic limits in the dioecious group of the tribe vanguerieae (Rubiaceae): insights into the evolution of functional dioecy 1. Ann. Missouri Botanical Garden 96, 161–181. doi: 10.3417/2006191
Reynolds S. T., Henderson R. J. (2004). Vanguerieae A. Rich. ex Dum.(Rubiaceae) in Australia, 3. Psydrax Gaertn. Austrobaileya, 817–889. Available at: https://www.jstor.org/stable/41739064
Robyns W. (1928). Tentamen monographiae vangueriae generumque affinium. Bull. du Jardin botanique l’État Bruxelles 11, 155. doi: 10.2307/3666455
Romero H. (2000). Codon usage in Chlamydia trachomatis is the result of strand-specific mutational biases and a complex pattern of selective forces. Nucleic Acids Res. 28, 2084–2090. doi: 10.1093/nar/28.10.2084
Ruhlman T. A., Jansen R. K. (2014). “The plastid genomes of flowering plants,” in Chloroplast Biotechnology. Ed. Maliga P. (Humana Press, Totowa, NJ), 3–38. doi: 10.1007/978-1-62703-995-6_1
Ruhsam M., Govaerts R., Davis A. P. (2008). Nomenclatural changes in preparation for a World Rubiaceae Checklist. Botan J. Linn Soc. 157, 115–124. doi: 10.1111/j.1095-8339.2008.00779.x
Safhi F. A., Alshamrani S. M., Bogmaza A. F. M., El-Moneim D. A. (2023). DNA barcoding of wild plants with potential medicinal properties from faifa mountains in Saudi Arabia. Genes 14, 469. doi: 10.3390/genes14020469
Saldaña C. L., Rodriguez-Grados P., Chávez-Galarza J. C., Feijoo S., Guerrero-Abad J. C., Vásquez H. V., et al. (2022). Unlocking the complete chloroplast genome of a native tree species from the amazon basin, capirona (Calycophyllum spruceanum, rubiaceae), and its comparative analysis with other ixoroideae species. Genes 13, 113. doi: 10.3390/genes13010113
Shi H., Yang M., Mo C., Xie W., Liu C., Wu B., et al. (2019). Complete chloroplast genomes of two Siraitia Merrill species: Comparative analysis, positive selection and novel molecular marker development. PloS One 14, e0226865. doi: 10.1371/journal.pone.0226865
Song Y., Chen Y., Lv J., Xu J., Zhu S., Li M. (2019). Comparative chloroplast genomes of sorghum species: sequence divergence and phylogenetic relationships. BioMed. Res. Int. 2019, 1–11. doi: 10.1155/2019/5046958
Song Y., Chen Y., Lv J., Xu J., Zhu S., Li M., et al. (2017). Development of chloroplast genomic resources for oryza species discrimination. Front. Plant Sci. 8. doi: 10.3389/fpls.2017.01854
Souza U. J. B. D., Nunes R., Targueta C. P., Diniz-Filho J. A. F., Telles M. P. D. C. (2019). The complete chloroplast genome of Stryphnodendron adstringens (Leguminosae - Caesalpinioideae): comparative analysis with related Mimosoid species. Sci. Rep. 9, 14206. doi: 10.1038/s41598-019-50620-3
Spooner D. M., Ruess H., Iorizzo M., Senalik D., Simon P. (2017). Entire plastid phylogeny of the carrot genus ( Daucus, Apiaceae): Concordance with nuclear data and mitochondrial and nuclear DNA insertions to the plastid. Am. J. Bot. 104, 296–312. doi: 10.3732/ajb.1600415
Sun J.-L., Han Y., Cui X.-M., Liu Y. (2020). Development and application of chloroplast molecular markers in Panax notoginseng. Zhongguo Zhong yao za zhi= Zhongguo Zhongyao Zazhi= China J. Chin. Materia Med. 45, 1342–1349. doi: 10.19540/j.cnki.cjcmm.20200104.105
Tang J., Xia H., Cao M., Zhang X., Zeng W., Hu S., et al. (2004). A comparison of rice chloroplast genomes. Plant Physiol. 135, 412–420. doi: 10.1104/pp.103.031245
Tillich M., Lehwark P., Pellizzer T., Ulbricht-Jones E. S., Fischer A., Bock R., et al. (2017). GeSeq – versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. doi: 10.1093/nar/gkx391
Tyagi S., Jung J.-A., Kim J. S., Won S. Y. (2020). A comparative analysis of the complete chloroplast genomes of three Chrysanthemum boreale strains. PeerJ 8, e9448. doi: 10.7717/peerj.9448
Wang Z., Cai Q., Wang Y., Li M., Wang C., Wang Z., et al. (2022). Comparative analysis of codon bias in the chloroplast genomes of theaceae species. Front. Genet. 13. doi: 10.3389/fgene.2022.824610
Wicke S., Naumann J. (2018). “Molecular evolution of plastid genomes in parasitic flowering plants,” in Advances in Botanical Research (Amsterdam: Elsevier), 315–347. doi: 10.1016/bs.abr.2017.11.014
Wikström N., Bremer B., Rydin C. (2020). Conflicting phylogenetic signals in genomic data of the coffee family (Rubiaceae). J. Syst. Evol. 58, 440–460. doi: 10.1111/jse.12566
Yang A.-H., Zhang J.-J., Yao X.-H., Huang H.-W. (2011). Chloroplast microsatellite markers in Liriodendron tulipifera (Magnoliaceae) and cross-species amplification in L. chinense. Am. J. Bot. 98, e123–e126. doi: 10.3732/ajb.1000532
Yang C., Zhao Q., Wang Y., Zhao J., Qiao L., Wu B., et al. (2021). Comparative analysis of genomic and transcriptome sequences reveals divergent patterns of codon bias in wheat and its ancestor species. Front. Genet. 12. doi: 10.3389/fgene.2021.732432
Yang Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Zhang Y., Chen S., Wang R. (2020). The complete chloroplast genome of Leptodermis scabrida (Rubiaceae): an endemic shrub in Himalaya-Hengduan Mountains. Mitochondrial DNA Part B 5, 169–170. doi: 10.1080/23802359.2019.1698371
Zhang X.-F., Wang J.-H., Zhao K.-K., Fan W.-W., Wang H.-X., Zhu Z.-X., et al. (2019b). Complete plastome sequence of Hedyotis ovata Thunb. ex Maxim (Rubiaceae): an endemic shrub in Hainan, China. Mitochondrial DNA Part B 4, 675–676. doi: 10.1080/23802359.2019.1572467
Zhang T., Xing Y., Xu L., Bao G., Zhan Z., Yang Y., et al. (2019a). Comparative analysis of the complete chloroplast genome sequences of six species of Pulsatilla Miller, Ranunculaceae. Chin. Med. 14, 53. doi: 10.1186/s13020-019-0274-5
Zhang Y., Zhang J.-W., Yang Y., Li X.-N. (2019c). Structural and Comparative Analysis of the Complete Chloroplast Genome of a Mangrove Plant: Scyphiphora hydrophyllacea Gaertn. f. and Related Rubiaceae Species. Forests 10, 1000. doi: 10.3390/f10111000
Keywords: Psydrax latifolia, wild plants, applied genomics, phylogenomic analysis, plastome
Citation: Safhi FA, Jalal AS, Alshegaihi RM, Alshamrani R, Alamri AM, Felemban W, Abuzaid AO, Hussein MAA, Al Aboud NM, Magdy M and Abd El-Moneim D (2024) The complete chloroplast genome of Psydrax latifolia: evolutionary dynamics, comparative genomics and phylogeny. Front. Ecol. Evol. 12:1416876. doi: 10.3389/fevo.2024.1416876
Received: 13 April 2024; Accepted: 02 September 2024;
Published: 25 September 2024.
Edited by:
Alison G. Nazareno, Federal University of Minas Gerais, BrazilReviewed by:
João Ricardo Bachega Feijó Rosa, RB Genetics & Statistics Consulting, BrazilCopyright © 2024 Safhi, Jalal, Alshegaihi, Alshamrani, Alamri, Felemban, Abuzaid, Hussein, Al Aboud, Magdy and Abd El-Moneim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Areej Saud Jalal, QXNqYWxhbEBwbnUuZWR1LnNh
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.