 Sai Jiang1†
Sai Jiang1† Ran Li1†
Ran Li1† Lina Jiang1Wei Wang1Yongcheng Liu1Yuewei Yang1
Lina Jiang1Wei Wang1Yongcheng Liu1Yuewei Yang1 Jichun Xing2*Zizhong Li2
Jichun Xing2*Zizhong Li2 Yujian Li1*
Yujian Li1*- 1School of Life Sciences, Qufu Normal University, Qufu, Shandong, China
- 2Institute of Entomology, Guizhou University, Guiyang, Guizhou, China
The mitochondrial genome is recognized for its utility in insect molecular research, due to its distinctive features, including fast evolutionary rate, low recombination, and maternal inheritance. In this study, we explored the mitochondrial genome in the context of the leafhopper subfamily Evacanthinae, which is significant in the agricultural pest sector due to the direct and indirect damage caused to plants by its species. We present complete mitochondrial genome sequences for three species: Evacanthus bivittatus, Carinata ganga, and Carinata recurvata. Comparative analyses of nucleotide composition, codon usage of protein coding genes (PCGs), nucleotide diversity, transfer RNA secondary structure, and gene overlap were conducted. To understand phylogenetic relationships among species, we constructed phylogenetic trees using nucleotide sequences from the 13 PCGs of the genomes newly sequenced in this study alongside other available leafhopper genome sequences. Phylogenetic analysis supported monophyly of the Evacanthinae subfamily and suggested a close relationship between the Evacanthini and Nirvanini tribes. Our research indicates that anticodon differences in trnS1 are insufficient to serve as taxonomic markers for distinguishing between these two tribes. This study contributes valuable genetic data supporting ongoing investigations into genetic diversity, molecular evolution, and species identification, while laying groundwork for future taxonomic and evolutionary endeavors within Cicadellidae.
1 Introduction
The mitochondrial genomes of insects are typically double-stranded circular molecules, comprising 37 genes (Wolstenholme, 1992) (13 protein-coding genes (PCGs), 22 transfer RNA (tRNA) genes, two ribosomal RNA (rRNA) genes) and a control region (Boore, 1999). Due to the characteristics of mitochondrial genomes, such as a relatively fast evolutionary rate, low recombination levels, and maternal inheritance, their sequences have been widely used for research into insect molecular evolution, population genetics, phylogenetics, and species identification (Gao et al., 2024; Li et al., 2023; Wilson et al., 1985).
The Evacanthinae are a subfamily of leafhoppers, widely distributed worldwide and consisting of 71 genera and 513 species (Dietrich, 2004). All Evacanthinae species feed on plants by piercing and sucking sap, which causes direct harm to the plants, and can also transmit plant viral diseases. The Evacanthinae are considered important pests in agriculture, forestry, orchards, and other commercially grown plants. For example, the species Sophonia orientalis Matsumura caused severe damage in Hawaii, particularly affecting fruit trees, vegetables, and ornamental plants (Aguin-Pombo et al., 2007). Previous studies have differed in the taxonomic status assigned to the Evacanthini. Oman et al. classified the Evacanthinae as a separate family, including only the Evacanthini (Oman et al., 1990), but Dietrich confirmed that the Nirvaninae are synonymous with the Evacanthinae and established a higher-level classification system for the Evacanthinae, comprising four tribes: Evacanthini, Nirvanini, Balbillini, and Pagaroniini (Dietrich, 2004). Further, Wang et al. conducted a phylogenetic analysis of the Evacanthinae using nuclear (28S) and mitochondrial (COI) gene sequences, and their findings supported the presence of four tribes within the Evacanthinae and indicated that the Evacanthini and Nirvanini were monophyletic. Moreover, they found that the Evacanthus genus was not monophyletic and clustered with other genera (Wang et al., 2017).
Most previous research on this topic has been based on insect morphological characteristics or partial mitochondrial sequences. Du et al. provided mitochondrial genome data for species related to the Evacanthini and Nirvanini, confirming that both tribes are monophyletic and exhibit significant differences in the trnS1 anticodon sequence (Du et al., 2021). There remains a lack of molecular data from many Evacanthinae species and more such data are needed to determine the relationship between the Evacanthinae and other subfamilies within the Cicadellidae.
Here, we present the complete mitochondrial genome sequences of three species: Evacanthus bivittatus, Carinata ganga, and Carinata recurvata, along with comparisons of their nucleotide composition, PCG codon usage frequency, nucleotide diversity, tRNA secondary structure, gene overlap, and non-coding regions. Additionally, we reconstructed the phylogenetic relationships among these three newly sequenced species together with other available leafhopper mitochondrial genome sequences, using concatenated nucleotide sequences of the 13 PCGs. The results of phylogenetic analysis support the monophyly of Evacanthinae, as proposed by Dietrich and suggest that the tribes, Evacanthini and Nirvanini, are sister groups (Dietrich, 2004). However, we found that the differences in trnS1 anticodons were not sufficiently significant to distinguish between the two tribes. These findings provide genetic information that will facilitate the study of genetic diversity, molecular evolution, and species identification, and will inform future research into classification and evolutionary history.
2 Materials and methods
2.1 Sample collection and DNA extraction
Specimens of Evacanthus bivittatus were collected in Cangyuan County, Yunnan Province, China (23°16′39.67″N, 99°11′21.45″E). Specimens of Carinata ganga and Carinata recurvata were collected from the Lijiang River in Guilin City, Guangxi Zhuang Autonomous Region, China (25°36′28.74″N, 109°57′53.29″E). The specimens were deposited in the College of Life Science, Qufu Normal University, Shandong Province. All specimens were preserved in 100% ethanol and subsequently transferred to the laboratory for storage at –80°C. Adult specimens were identified based on their morphological characteristics. Genomic DNA was extracted from the head and thorax of specimens using the SanPrep DNA Gel Extraction Kit (Sangon, Shanghai, China).
2.2 Mitogenome sequencing, assembly, annotation, and bioinformatic analyses
Genomic DNA samples were sent to Personalbio Inc. (Shanghai, China) for library construction and next-generation sequencing. Libraries with an insert size of 400 bp were prepared from each DNA sample using the TruSeq™ DNA Sample Prep Kit (Illumina, USA), then sequenced as 150 bp paired-end reads on the Illumina NovaSeq platform. Species library reads have been deposited in BioProject (PRJNA1059151, PRJNA1081200, PRJNA1081201) (https://www.ncbi.nlm.nih.gov/bioproject/). Paired-end reads were de novo assembled using Novoplasty 3.7 (Dierckxsens et al., 2017). After trimming the adapters and removing short and low-quality reads, more than 4 GB (30–40 million reads) of clean data were obtained for each sample. Circular mitogenome sequences were extracted using DNASTAR 7.1 (Lasergene version 5.0) (Burland, 2000) with the complete mitogenome of Chudania hellerina (GenBank accession: MN227164) as an initial seed. Mitogenomes were then annotated using MITOZ v.1.04 (Meng et al., 2019) and manually checked using Geneious v.8.1.3 (Kearse et al., 2012). Secondary structures of tRNA were drawn manually using Adobe Illustrator CC2017, based on predictions from the MITOS Web Server (Bernt et al., 2013). Mitogenome maps were generated using the Organellar Genome DRAW (OGDRAW) program (Lohse et al., 2013). The complete mitogenome sequences of E. bivittatus, C. ganga, and C. recurvata were submitted to the NCBI sequence archive (GenBank accession no: NC077613.1, NC082036.1, OR979469.1, respectively).Various bioinformatic analyses, such as nucleotide composition, composition skew, codon usage of PCGs, relative synonymous codon usage (RSCU), and mitogenomic organization tables, were conducted using PhyloSuite v.1.2.2 (Zhang et al., 2020).
2.3 Molecular phylogenetic analysis
A total of 37 mitogenomes from the Cicadellidae were used in phylogenetic analyses, with two Fulgoridae family species serving as outgroups. Nucleotide sequences of the 13 PCGs, excluding stop codons, were aligned using the G-INS-i (accurate) strategy and codon alignment mode in MAFFT v.7 (Katoh et al., 2002). The rRNA gene sequences, rrnL and rrnS, were aligned using the Q-INS-I algorithm in MAFFT v.7, taking into account rRNA gene secondary structure.
Regions of ambiguous alignment were removed using Gblocks 0.91b (Talavera and Castresana, 2007). Gene alignments were concatenated using PhyloSuite v.1.2.2. For Maximum Likelihood (ML) and Bayesian Inference (BI) phylogenetic analyses, partitioning scheme and nucleotide substitution models were chosen using ModelFinder (Kalyaanamoorthy et al., 2017) with Bayesian Information Criterion. ML analyses were performed using IQ-TREE v.1.6.3 (von Haeseler et al., 2015) by applying the ultrafast bootstrap approximation approach with 5,000 replicates. BI analysis was conducted using MrBayes v.3.2.7a (Ronquist et al., 2012) in the CIPRES Science Gateway, using four chains (one cold and three hot chains). Two independent runs of 2,000,000 generations were carried out, with sampling every 1,000 generations. The first 25% of trees were removed as burn-in. On reaching an average standard deviation of split frequencies < 0.01, stationarity was assumed. The resulting phylogenetic trees were visualized using the online site: https://www.chiplot.online/ (Xie et al., 2023).
3 Results and discussion
3.1 Mitogenome organization and nucleotide composition
The mitochondrial genomes of E. bivittatus, C. ganga, and C. recurvata consisted of double-stranded circular DNA molecules, containing 37 classic mitochondrial genes, including 13 PCGs, 22 tRNA genes, and 2 rRNA genes, as well as a single control region. Among these genes, 23 were encoded on the heavy strand (H-strand), while the remaining 14 were transcribed from the light strand (L-strand) (Figure 1).
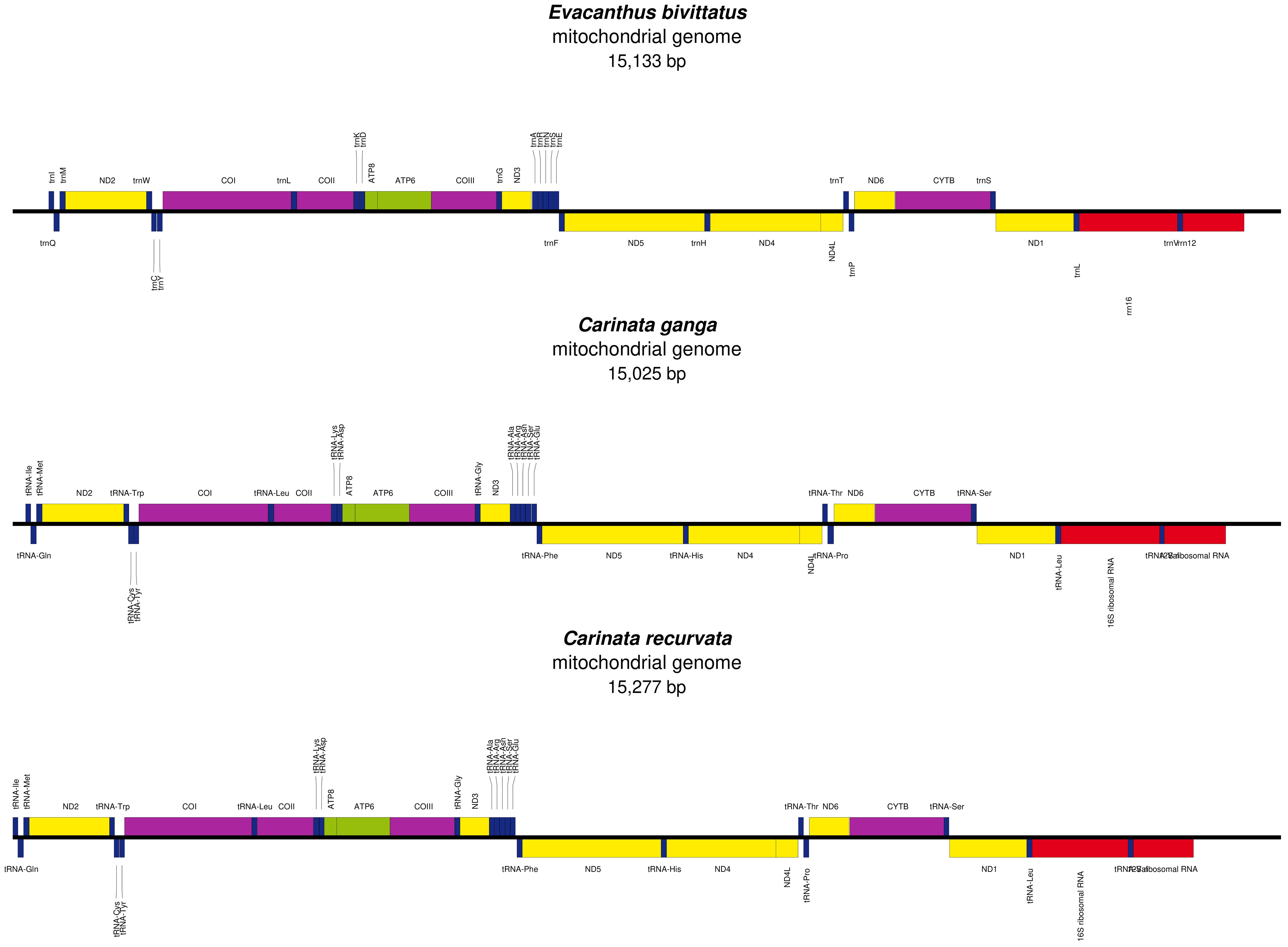
Figure 1. Linear maps of three mitochondrial genomes (E. bivittatus, C. ganga, C. recurvata). The upper half of the gene map indicates genes are coded on the major strand (J-strand), while the lower half indicates genes are coded on the minor strand (N-strand). Genes are represented by different color blocks.
The sizes of the E. bivittatus, C. ganga, and C. recurvata mitochondrial genomes were 15,133, 15,025, and 15,277 bp, respectively, attributable to differences in control region length (Fernández-Silva et al., 2003). The linear arrangements of the 37 genes in these three mitochondrial genomes were similar to those of other Cicadellidae species (Du et al., 2021; Wang et al., 2021a, 2021b). Excluding the control region, these mitochondrial genomes contained 9, 6, and 8 intergenic regions, respectively, as well as 9, 19, and 15 gene overlap regions. The mitochondrial genomes of all three species were highly condensed, with only a small amount of base overlap between neighboring genes, which suggests efficient RNA transcription and protein translation. The composition of the three mitochondrial genomes exhibited a strong bias toward A (adenine) + T (thymine), accounting for more than 70% of bases, consistent with the A-T bias observed in the majority of published Cicadellidae species (Wang et al., 2021a; 2019). This nucleotide composition asymmetry has often been considered an indicator of gene orientation and replication direction during replication and transcription processes (Perna and Kocher, 1995). The base skewness ([A-T]/[A+T]) values of E. bivittatus, C. ganga, and C. recurvata were 0.016, 0.057, and 0.055, respectively, with corresponding G-C skew values of –0.055, –0.105, and –0.12 (Table 1).
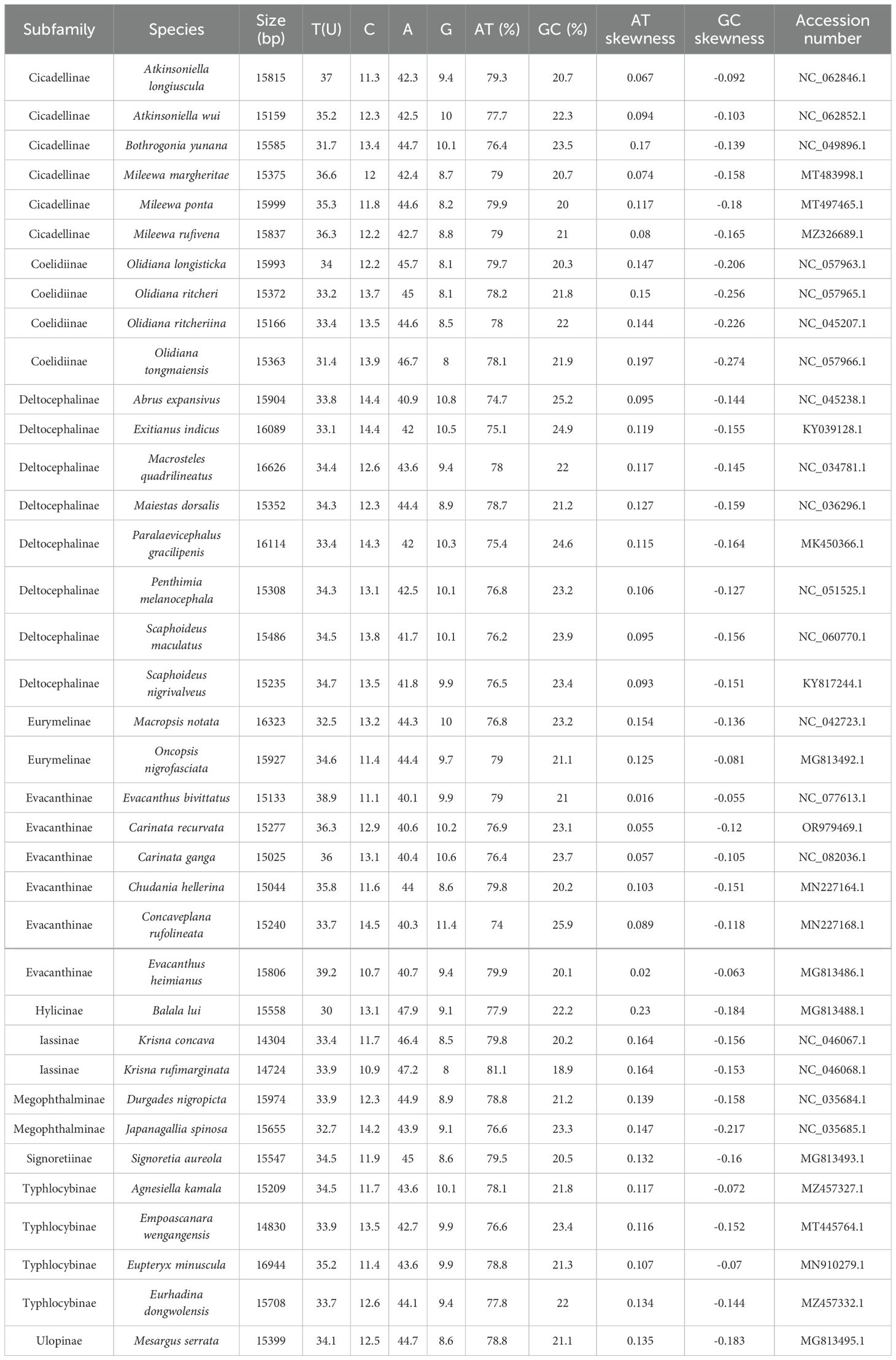
Table 1. Base composition and skewness of mitogenomes of 37 species.
3.2 Transfer RNA genes
The secondary structures of all tRNA molecules were identified using tRNAscan-SE 2.0 (Lowe and Eddy, 1997). The mitochondrial genomes of these three species each contained 22 tRNA genes, distributed throughout the entire mitochondrial genome, ranging in size from 61 to 73 nucleotides. Each mitochondrial genome had 14 and 8 tRNA genes encoded on the H- and L-strands, respectively (Figure 1); this pattern is common in Cicadellidae mitochondrial genomes (Du et al., 2021; Wang et al., 2021a, 2021b). Additionally, two serine tRNA genes (tRNASer(UCN) and tRNASer(AGN)) and two leucine tRNA genes (tRNALeu(UUR) and tRNALeu(CUN)) were identified. The trnS1 gene lacked the dihydrouridine loop, preventing it from folding into the typical cloverleaf structure, while the other 22 tRNAs could fold into the typical cloverleaf structure. Furthermore, noncanonical base pairs or mismatched base pairs were common in the relatively conserved tRNAs. Secondary structure prediction identified 20, 15, and 21 noncanonical base pairs in E. bivittatus, C. ganga and C. recurvata, respectively. The three mitochondrial genomes had 30, 2, 4, and 17 cases of G-U, A-C, A-A, and U-U base pair mismatches, respectively. The majority of noncanonical nucleotides were G-U pairs, which are known to form weak bonds in tRNA and contribute to the formation of noncanonical base pairs in tRNA secondary structures (Supplementary Figure S1). Furthermore, since the mitochondrial genome is not affected by recombination processes, these base pair mismatches may help eliminate detrimental mutations (Lynch, 1997).
3.3 Ribosomal RNAs and A+T-rich regions
The mitochondrial genomes of E. bivittatus, C. ganga, and C. recurvata all contained genes encoding the 12S and 16S rRNA subunits, which are located on the L-strand and separated by trnV (Figure 1). The total lengths of the rRNAs in these three mitochondrial genomes differed, measuring 1905, 1886, and 1901 bp, respectively (Supplementary Tables S1–S3). The A-T skew values of the two rRNAs in these leafhopper species were negative, while the G-C skew values were positive, indicating a higher AT content. The control region contains the initiation and control elements for mitochondrial genome DNA transcription and replication, and is the largest non-coding region in the mitochondrial genome (Taanman, 1999). The AT-rich region, which is the largest non-coding region in insect mitochondrial genomes, varies in size across diverse taxa, and even among individuals within the same species. Given the high variability of the insect AT control region, it is challenging to analyze its function and role in insect evolution through sequence similarity alignment. Through comparative analysis of insect AT control region sequences, Zhang et al. identified five conserved elements related to mitochondrial genome transcription and replication (Zhang et al., 1995). Taanman further proposed that the control region was the main regulatory region for mitochondrial genome replication and transcription (Taanman, 1999); however, in recent years, with the increasing numbers of sequenced and analyzed insect mitochondrial genomes, the AT control region has been found not to be a conserved structure in insects, and these regions do not contain conserved elements in many species (Dueñas et al., 2006).
3.4 Protein-coding genes
The lengths of the 13 PCGs in E. bivittatus, C. ganga, and C. recurvata all totaled 10,929 bp, encoding 3,633, 3,634, and 3,634 amino acids, respectively. Among the 13 PCGs, 9 (ND2, COI, COII, ATP8, ATP6, COIII, ND3, ND6, and CYTB) were encoded on the H-strand, while the remaining 4 (ND5, ND4, ND4L, and ND1) were encoded on the L-strand. As in other leafhopper species, the ATP8 and ND5 genes in E. bivittatus and C. recurvata feature TTG start codons, whereas the other PCGs had traditional ATN (A/T/C/G) start codons. The stop codons of the 13 PCGs in these three species included TAA, TAG, and T-. In E. bivittatus and C. recurvata, incomplete stop codons (T-) were identified in the COI, COII, COIII, and ND1 genes. Furthermore, in E. bivittatus and C. recurvata, the CYTB gene harbored a TAG stop codon. The remaining genes concluded with TAA stop codons (Supplementary Tables S1–S3). The existence of incomplete stop codons (T-) has been implicated in post-transcriptional mRNA regulation, potentially occurring subsequent to polyadenylation following transcription, a phenomenon commonly found in insect mitochondrial genomes (Ojala et al., 1981).
The RSCU was found to be very similar among the sequenced mitochondrial PCGs (Figure 2); codons ending with A and T were more frequently utilized, with the three most prevalent codons being UUA (Leu2), UCU (Ser2), and UCA (Ser2). Collectively, our findings suggest that the length, orientation, and codon usage frequencies of PCGs in Evacanthini leafhopper species are highly conserved.
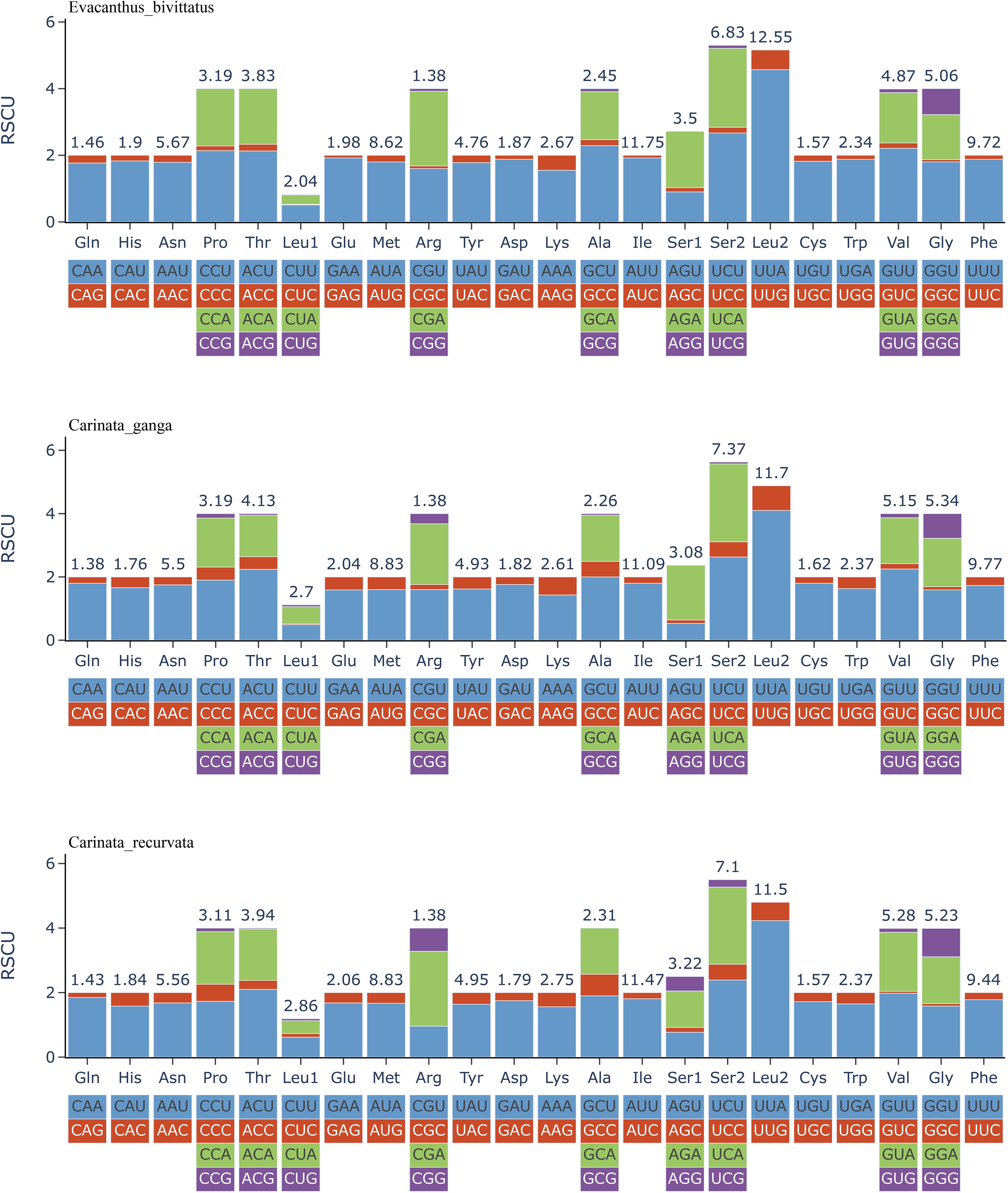
Figure 2. Relative synonymous codon usage (RSCU) of mitogenomes of three species. The stop codons are not shown.
Next, we analyzed nucleotide polymorphism in the 13 PCGs using a sliding window approach. Among the 13 genes, COI exhibited significantly lower polymorphism (Figure 3). Genetic distance analysis revealed that ATP8 (0.29) and ND2 (0.26) evolved relatively more rapidly, while COI (0.17) showed a slower rate of evolution (Figure 4). Ka/Ks ratio was used to estimate gene evolution rate, which ranged from 0.1 (COI) to 0.44 (ATP8). These findings indicate that all the PCGs underwent purifying selection throughout the lengthy process of evolution. ATP8 exhibited relatively less stringent purifying selection, while COI experienced the most intense selection pressure (Figure 4).
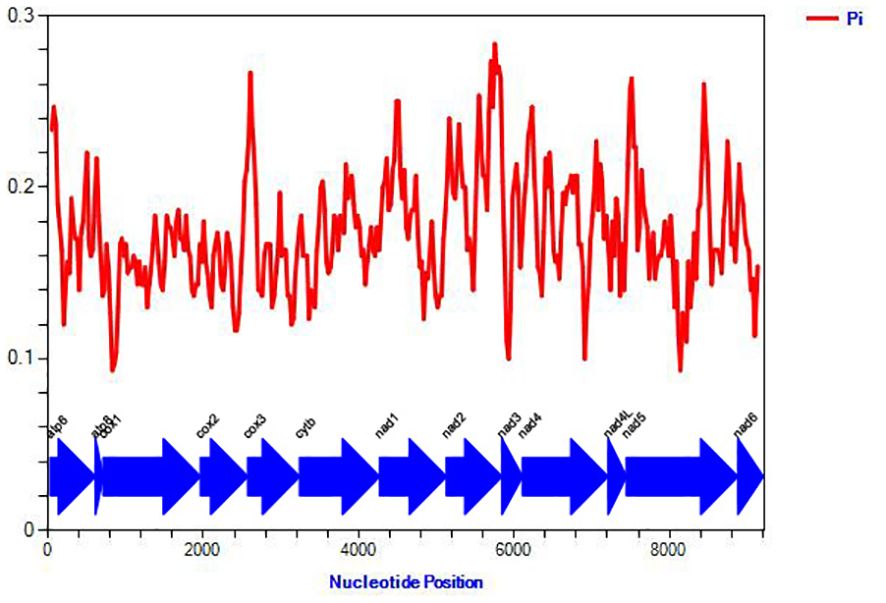
Figure 3. Sliding window analysis based on 13 aligned PCGs. The red line shows the value of nucleotide diversity Pi. The gene names are shown in the graph.
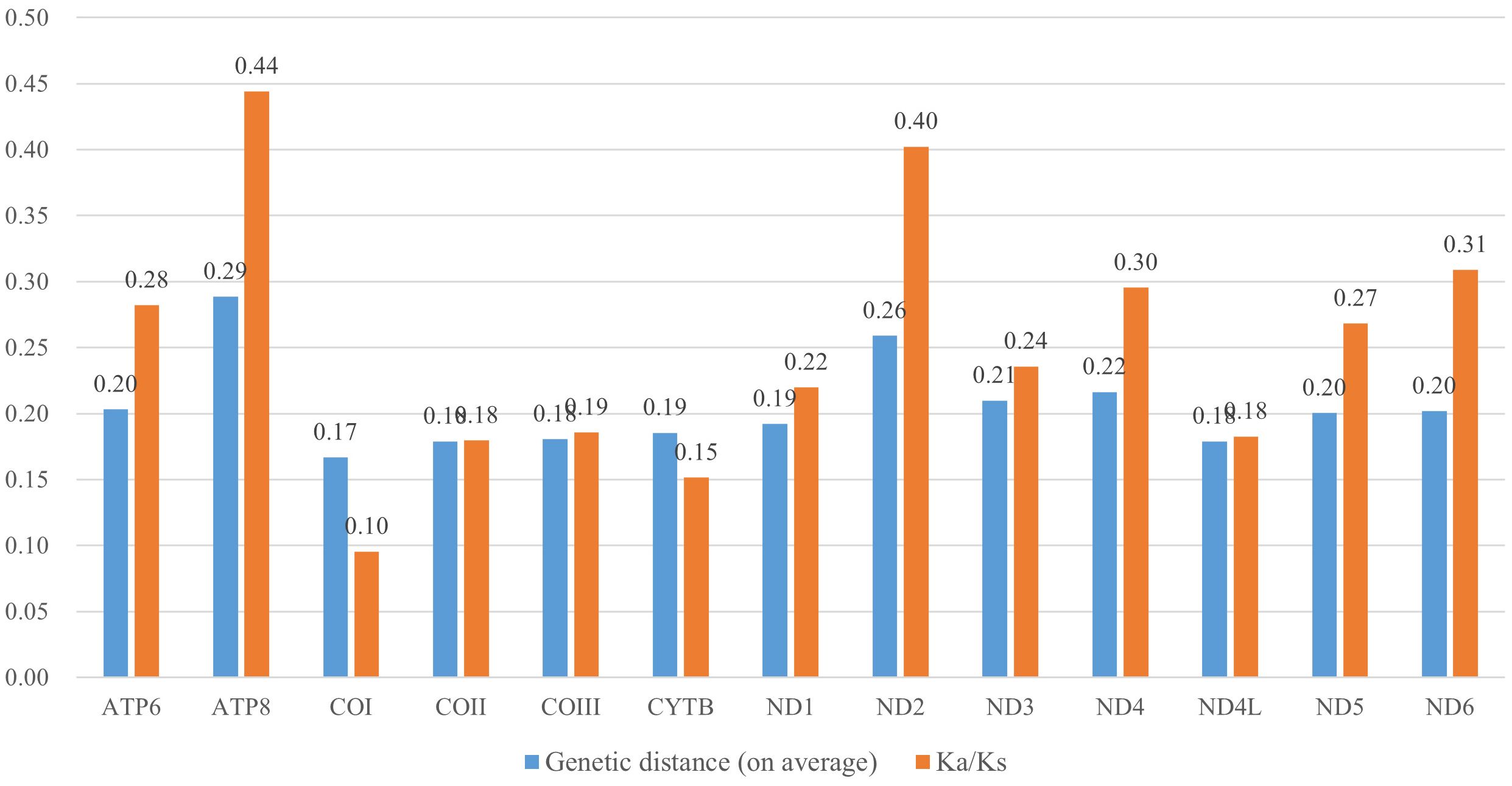
Figure 4. Genetic distance (average) and nonsynonymous (Ka) to synonymous (Ks) substitution rates of 13 protein coding genes among three species (Evacanthus bivittatus, Carinata ganga, and Carinata recurvata).
3.5 Phylogenetic analysis
To elucidate the phylogenetic relationships within the Evacanthini of the Cicadellidae, a phylogenetic analysis was conducted on 37 Cicadellidae species, including E. bivittatus, C. ganga, and C. recurvata. Additionally, two species from the Fulgoroidea were selected as outgroups. Bayesian Inference (BI) (Figure 5) and Maximum Likelihood (ML) (Figure 6) phylogenetic trees were constructed using concatenated sequences of the 13 PCGs.
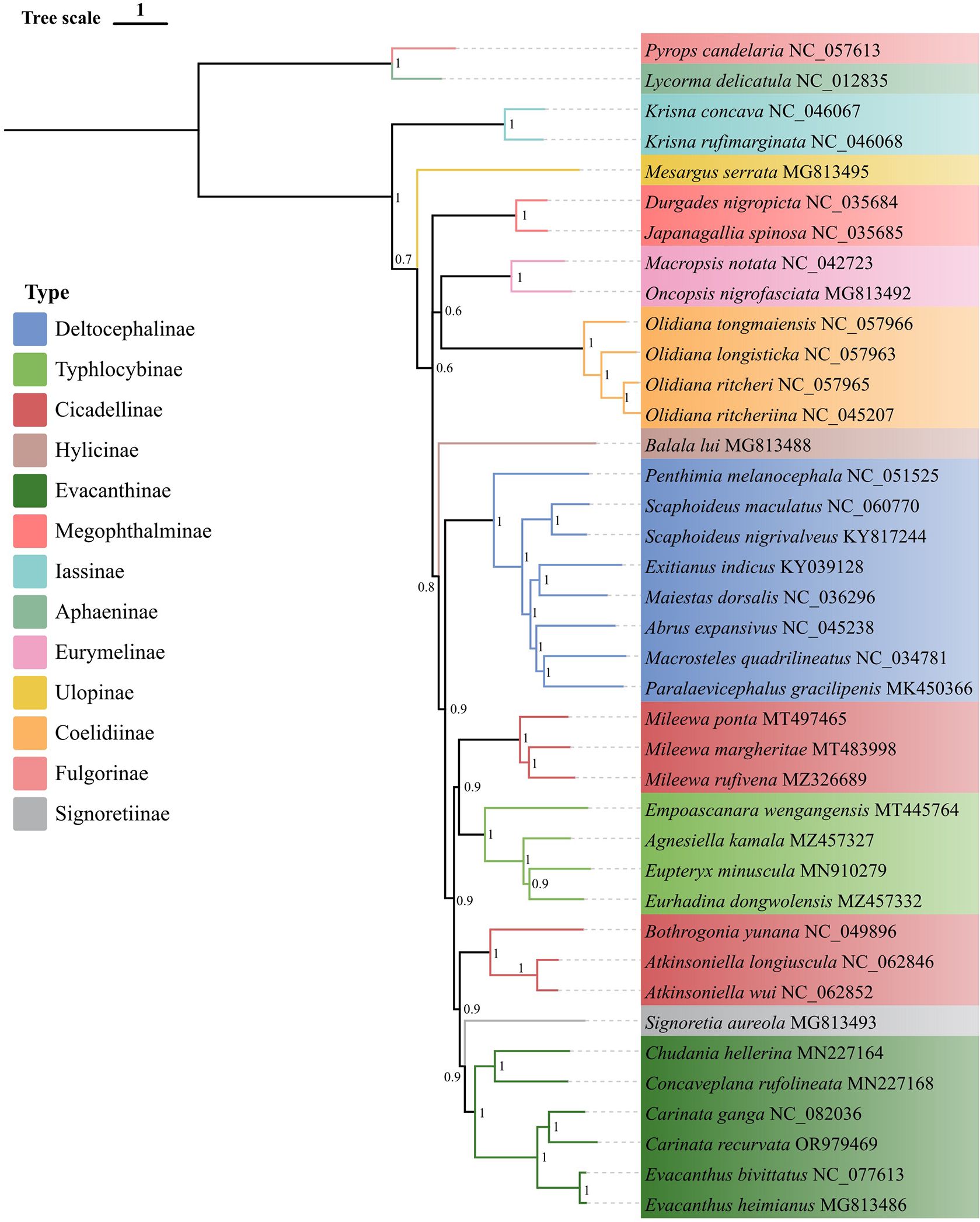
Figure 5. Phylogenetic tree obtained from Bayesian interference analysis, based on the protein coding gene dataset. Numbers on nodes represent posterior probability (PP).
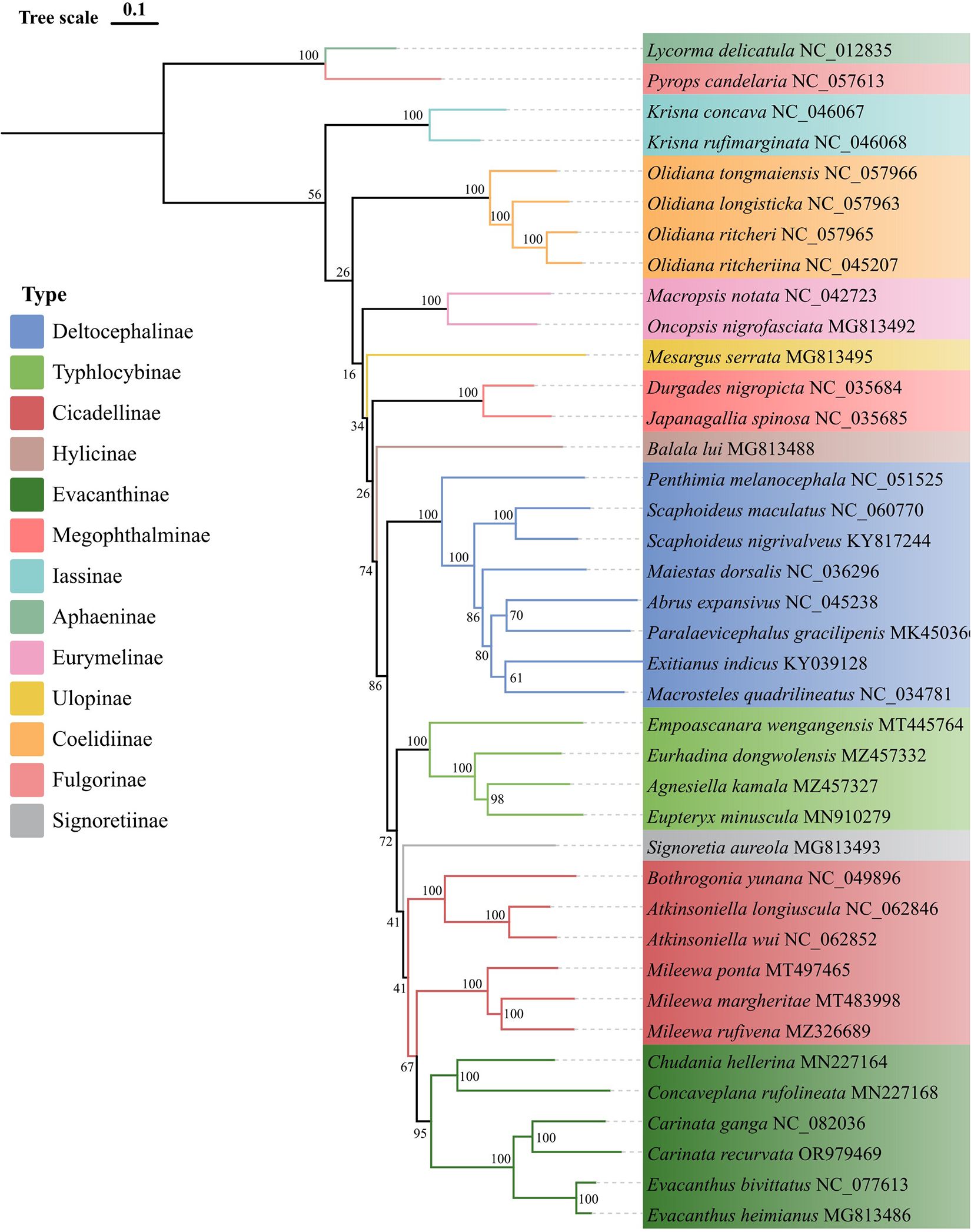
Figure 6. Phylogenetic tree obtained from maximum likelihood analysis, based on the protein coding gene dataset. Numbers on nodes represent bootstrap values (BV).
The resulting phylogenetic trees included 27 genera from 11 subfamilies of the Cicadellidae. They exhibited similar topologies and had high support values in both Maximum Likelihood bootstrap values and Bayesian posterior probabilities. The Evacanthini formed a distinct clade. Two species from the Carinata grouped together, as did two Evacanthus species. Two species formed a separate branch within the Nirvanini. The phylogenetic results also suggested that Evacanthini and Nirvanini are sister groups. Previous studies have supported the monophyly of Evacanthinae, which consists of four tribes: Evacanthini, Pagaroniini, Nirvanini, and Balbillini (Dietrich, 2004). Before the morphological phylogenetic analysis conducted by Dietrich (2004), Nirvaninae were regarded as a separate subfamily, while Evacanthini were included in Cicadellinae (Oman et al., 1990). Our phylogenetic studies corroborate Dietrich’ s conclusions, which are based on morphological traits related to the taxonomy of Evacanthinae and its tribes. These findings enhance understanding of the evolutionary relationships within this subfamily and further validate the use of morphological characteristics in insect taxonomy. The findings of this study also support the classification of Evacanthini and Nirvanini as two distinct branches within the Evacanthinae subfamily. A previous study reported a significant difference in the use of the anticodon (AGN) in trnS1 between representatives of Evacanthini and Nirvanini (Du et al., 2021); however, we found that both E. bivittatus and C. ganga used GCT as their anticodon, while C. recurvata had the TCT anticodon (Supplementary Figure S1). Additionally, our phylogenetic analysis showed that C. recurvata belongs to the Evacanthini, suggesting that trnS1 (AGN) anticodon use may not be a major distinguishing factor between Evacanthini and Nirvanini. Our findings also reveal that the Evacanthini tribe is monophyletic.
Although the other subfamilies were clearly clustered, the phylogenetic interconnections among them were inconsistent. The Cicadellinae could be divided into two groups, with Mileewa species forming one group and species from other genera forming another. Previous research has not clarified the relationship between Evacanthinae and Cicadellinae (Dietrich, 2004, 2005; Wang et al., 2017). Further research and additional data are needed to determine the sister groups of Evacanthinae.
Overall, the topology of the phylogenetic trees was largely consistent with the evolutionary relationships among the leafhoppers. Our analysis confirmed the monophyly of the Evacanthini and provided insights into its taxonomic status within the Evacanthinae. Furthermore, we present comprehensive data from three Evacanthini mitochondrial genomes, contributing to clarification of phylogenetic relationships within the Evacanthini.
4 Conclusion
In this study, we compared and analyzed the start codons, stop codons, and base usage of 13 PCGs in E. bivittatus, C. ganga, and C. recurvata. Furthermore, we predicted the secondary structures of tRNAs and found that the trnS1 anticodons in C. recurvata could not be used to differentiate the Evacanthini from the Nirvanini. Additionally, we described the number of codons and the relative synonymous codon usage of the PCGs in three Evacanthini species. Finally, we constructed BI and IQ trees using the 13 PCGs to analyze interspecific phylogenetic relationships within the Evacanthinae. Our research strongly supports the monophyly of the Evacanthinae subfamily, with the Evacanthini and Nirvanini tribes being sister taxa. Our findings enhance the understanding of mitochondrial evolution and phylogenetic relationships among different groups within the tribe Evacanthini.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The animal study was approved by Animal Care and Use Committee of Qufu normal University. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
SJ: Writing – original draft, Writing – review & editing. RL: Writing – review & editing. LJ: Writing – original draft, Writing – review & editing. WW: Data curation, Writing – review & editing. YCL: Writing – review & editing, Data curation, Software. YY: Writing – original draft. JX: Writing – original draft, Writing – review & editing. ZL: Visualization, Writing – review & editing. YJL: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This project was supported by the National Natural Science Foundation of China (Grant No. 32200359) and the Young Talents Invitation Program of Shandong Provincial Colleges and Universities (Grant No. 20190601).
Acknowledgments
We sincerely thank Professor Bin Zhang of Inner Mongolia Normal University and Dr. Xun Bian of Guangxi Normal University for their assistance in sampling.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2024.1410546/full#supplementary-material
References
Aguin-Pombo D., Aguiar A. M. F., Kuznetsova V. G. (2007). Bionomics and taxonomy of leafhopper Sophonia orientalis (Homoptera: Cicadellidae), a pacific pest species in the macaronesian archipelagos. Ann. Entomol. Soc Am. 100, 19–26. doi: 10.1603/0013-8746(2007)100[19:BATOLS]2.0.CO;2
Bernt M., Donath A., Jühling F., Externbrink F., Florentz C., Fritzsch G., et al. (2013). MITOS: improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319. doi: 10.1016/j.ympev.2012.08.023
Boore J. L. (1999). Animal mitochondrial genomes. Nucleic Acids Res. 27, 1767–1780. doi: 10.1093/nar/27.8.1767
Burland T. G. (2000). DNAStar’s Lasergene sequence analysis software. Methods Mol. Biol. 132, 71–91. doi: 10.1385/1-59259-192-2:71
Dierckxsens N., Mardulyn P., Smits G. (2017). NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45, e18. doi: 10.1093/nar/gkw955
Dietrich C. H. (2004). Phylogeny of the leafhopper subfamily Evacanthinae with a review of Neotropical species and notes on related groups (Hemiptera: Membracoidea: Cicadellidae). Syst. Entomol. 29, 455–487. doi: 10.1111/j.0307-6970.2004.00250.x
Dietrich C. H. (2005). Keys to the families of Cicadomoorpha and subfamilies and tribes of CicadellidaeI (Hemiptera: Auchenorrhyncha). Fla. Entomol. 88, 502–517. doi: 10.1653/0015-4040(2005)88[502:KTTFOC]2.0.CO;2
Du Y., Liang Z., Dietrich C. H., Dai W. (2021). Comparative analysis of mitochondrial genomes of Nirvanini and Evacanthini (Hemiptera: Cicadellidae) reveals an explicit evolutionary relationship. Genomics 113, 1378–1385. doi: 10.1016/j.ygeno.2021.03.017
Dueñas J. C. R., Gardenal C. N., Llinás G. A., Panzetta-Dutari G. M. (2006). Structural organization of the mitochondrial DNA control region in Aedes aEgypti. Genome 49, 931–937. doi: 10.1139/g06-053
Fernández-Silva P., Enriquez J. A., Montoya J. (2003). Replication and transcription of mammalian mitochondrial DNA. Exp. Physiol. 88, 41–56. doi: 10.1113/eph8802514
Gao W., Chen X., He J., Sha A., Luo Y., Xiao W., et al. (2024). Intraspecific and interspecific variations in the synonymous codon usage in mitochondrial genomes of 8 Pleurotus strains. BMC Genomics 25, 456. doi: 10.1186/s12864-024-10374-3
Kalyaanamoorthy S., Bui Quang M., Wong T. K. F., von Haeseler A., Jermiin L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Katoh K., Misawa K., Kuma K. I., Miyata T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Kearse M., Moir R., Wilson A., Stones-Havas S., Cheung M., Sturrock S., et al. (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Li Q., Xiao W., Wu P., Zhang T., Xiang P., Wu Q., et al. (2023). The first two mitochondrial genomes from Apiotrichum reveal mitochondrial evolution and different taxonomic assignment of Trichosporonales. IMA Fungus 14, 7. doi: 10.1186/s43008-023-00112-x
Lohse M., Drechsel O., Kahlau S., Bock R. (2013). OrganellarGenomeDRAW-a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41, W575–W581. doi: 10.1093/nar/gkt289
Lowe T. M., Eddy S. R. (1997). TRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964. doi: 10.1093/nar/25.5.955
Lynch M. (1997). Mutation accumulation in nuclear, organelle, and prokaryotic transfer RNA genes. Mol. Biol. Evol. 14, 914–925. doi: 10.1093/oxfordjournals.molbev.a025834
Meng G., Li Y., Yang C., Liu S. (2019). MitoZ: a toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 47, e63. doi: 10.1093/nar/gkz173
Ojala D., Montoya J., Attardi G. (1981). tRNA punctuation model of RNA processing in human mitochondria. Nature 290, 470–474. doi: 10.1038/290470a0
Oman P. W., Knight W. J., Nielson M. W. (1990). Leafhoppers (Cicadellidae): a bibliography, generic check-list and index to the world literature. The Journal of Agricultural Science, 116, (2): 1956–1985. doi: 10.1017/S0021859600077868
Perna N. T., Kocher T. D. (1995). Patterns of nucleotide composition at 4-fold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 41, 353–358. doi: 10.1007/BF00186547
Ronquist F., Teslenko M., van der Mark P., Ayres D. L., Darling A., Höhna S., et al. (2012). MrBayes 3.2: efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Taanman J. W. (1999). The mitochondrial genome: structure, transcription, translation and replication. Biochim. Biophys. Acta 1410, 103–123. doi: 10.1016/s0005-2728(98)00161-3
Talavera G., Castresana J. (2007). Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577. doi: 10.1080/10635150701472164
von Haeseler A., Schmidt H. A., Bui M. Q., Nguyen L. T. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Wang Y., Dietrich C. H., Zhang Y. (2017). Phylogeny and historical biogeography of leafhopper subfamily Evacanthinae (Hemiptera: Cicadellidae) based on morphological and molecular data. Sci. Rep. 7, 45387. doi: 10.1038/srep45387
Wang X. Y., Wang J. J., Dai R. H. (2021b). Mitogenomics of five Olidiana leafhoppers (Hemiptera: Cicadellidae: Coelidiinae) and their phylogenetic implications. PeerJ 9, e11086. doi: 10.7717/peerj.11086
Wang X. Y., Wang J. J., Fan Z. H., Dai R. H. (2019). Complete mitogenome of Olidiana ritcheriina (Hemiptera: Cicadellidae) and phylogeny of Cicadellidae. PeerJ 7, e8072. doi: 10.7717/peerj.8072
Wang J., Zhang Y. J., Yang L., Chen X. S. (2021a). The complete mitochondrial genome of Trifida elongate and comparative analysis of 43 leafhoppers. Comp. Biochem. Physiol. D 39, 100843. doi: 10.1016/j.cbd.2021.100843
Wilson A. C., Cann R. L., Carr S. M., George M., Gyllensten U. B., Helm-Bychowski K. M., et al. (1985). Mitochondrial DNA and two perspectives on evolutionary genetics. Biol. J. Linn. Soc 26, 375–400. doi: 10.1111/j.1095-8312.1985.tb02048.x
Wolstenholme D. R. (1992). Animal mitochondrial DNA: structure and evolution. Int. Rev. Cytol. 141, 173–216. doi: 10.1016/s0074-7696(08)62066-5
Xie J., Chen Y., Cai G., Cai R., Hu Z., Wang H. (2023). Tree visualization by one table (tvBOT): a web application for visualizing, modifying and annotating phylogenetic trees. Nucleic Acids Res. 51, W587–W592. doi: 10.1093/nar/gkad359
Zhang D., Gao F., Jakovlić I., Zou H., Zhang J., Li W. X., et al. (2020). PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20, 348–355. doi: 10.1111/1755-0998.13096
Keywords: Evacanthini, mitogenome, phylogeny, Evacanthus bivittatus, Carinata ganga, Carinata recurvata
Citation: Jiang S, Li R, Jiang L, Wang W, Liu Y, Yang Y, Xing J, Li Z and Li Y (2024) The complete mitochondrial genomes of three Evacanthini species, Evacanthus bivittatus, Carinata ganga, and Carinata recurvata, and phylogenomic analysis of the Evacanthini. Front. Ecol. Evol. 12:1410546. doi: 10.3389/fevo.2024.1410546
Received: 01 April 2024; Accepted: 08 August 2024;
Published: 29 August 2024.
Edited by:
Liang Liu, University of Georgia, United StatesReviewed by:
Lin Zhang, Hubei University of Chinese Medicine, ChinaQiang Li, Chengdu University, China
Copyright © 2024 Jiang, Li, Jiang, Wang, Liu, Yang, Xing, Li and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yujian Li, eXVqaWFuNTI4QDE2My5jb20=; Jichun Xing, eGluZ2ppY2h1bkAxMjYuY29t
†These authors have contributed equally to this work and share first authorship