 Elena V. Korneenko1,2*
Elena V. Korneenko1,2* Andrei E. Samoilov3
Andrei E. Samoilov3 Ivan K. Chudinov1,4
Ivan K. Chudinov1,4 Ivan O. Butenko1
Ivan O. Butenko1 Ignat V. Sonets3,5
Ignat V. Sonets3,5 Ilya V. Artyushin6Alexander P. Yusefovich6Sergei V. Kruskop6Sergei O. Sinitsyn7Ekaterina O. Klyuchnikova2Anna S. Gladkikh2Vladimir G. Dedkov2,8Marina V. Safonova9Peter Daszak10
Ilya V. Artyushin6Alexander P. Yusefovich6Sergei V. Kruskop6Sergei O. Sinitsyn7Ekaterina O. Klyuchnikova2Anna S. Gladkikh2Vladimir G. Dedkov2,8Marina V. Safonova9Peter Daszak10 Anna S. Speranskaya1
Anna S. Speranskaya1- 1Laboratory of Multiomics Research, Scientific Research Institute for Systems Biology and Medicine, Federal Service on Consumer Rights Protection and Human Well-Being Surveillance, Moscow, Russia
- 2Department of Epidemiology, Saint Petersburg Pasteur Institute, Federal Service on Consumer Rights Protection and Human Well-Being Surveillance, Saint Petersburg, Russia
- 3Laboratory of Mathematical Biology, Scientific Research Institute for Systems Biology and Medicine, Federal Service on Consumer Rights Protection and Human Well-Being Surveillance, Moscow, Russia
- 4Phystech School of Biological and Medical Physics, Moscow Institute of Physics and Technology (National Research University), Moscow, Russia
- 5Group of Bioinformatics, Institute of Gene Biology, Russian Academy of Sciences, Moscow, Russia
- 6Biological Faculty, Lomonosov Moscow State University, Moscow, Russia
- 7School of Life Sciences, Technical University of Munich (TUM), Freising, Germany
- 8Tropical and Vector Borne Diseases, Martsinovsky Institute of Medical Parasitology, Sechenov First Moscow State Medical University, Moscow, Russia
- 9Virology Laboratory, Anti-Plague Center, Federal Service on Consumer Rights Protection and Human Well-Being Surveillance, Moscow, Russia
- 10EcoHealth Alliance, New York, NY, United States
Introduction: Bats are considered the natural reservoirs of several viruses including coronaviruses (CoVs), two genera of which, alpha- and betacoronaviruses, infect humans. Despite widespread screening of bat samples for novel viruses, information on the diversity of coronaviruses in bats inhabiting Russian territory remains scarce. Here, we analyzed the presence and diversity of alphacoronaviruses (Alpha-CoVs) in bats from European Russia.
Methods: Fourty three fecal samples from bats of 8 species: P. nathusii, P. kuhlii, M. brandtii, M. daubentonii, N. noctula, V. murinus, M. dasycneme, and P. auritus were taken to study.
Results and discussion: We detected Alpha-CoV RdRp gene fragments in 30% (13/43) of samples examined in 75% (6/8) of species sampled. Phylogenetic analysis of RdRp showed that most of the identified Alpha-CoV sequences fall into clades within the Pedacovirus subgenus, with minor clusters of nyctacoviruses or myotacoviruses. We assert that closely related pedacoviruses have been circulating for a long time (from 2015 to 2021) in a large region from European Russia to Northern Europe. We propose that closely related pedacoviruses collected from common areas represent a separate species, which we name NE-Alpha coronavirus, with its host being bats of the genus Pipistrellus that inhabit a region from the European part of Russia to Northern Europe. Among the animals sampled, 4.6% (2/43) carried two Alpha-CoVs related to different subgenera (pedacovirus/myotacovirus or pedacovirus/nyctacovirus) simultaneously. We confirmed the presence of two different Alpha-CoV subgenera related to pedacovirus and nyctacovirus in P. kuhlii captured in 2021 and kept in captivity using whole genome sequencing of these viruses. The presence of two or more coronaviruses in one individual animal host is an essential prerequisite for recombination to occur. We also obtained two Alpha-CoV whole genomes from two specimens of P. nathusii captured in 2015. The genomic organization of BatCoV/MOW15-21 and BatCoV/MOW15-23 was similar to other Alpha-CoVs, but the assembled genomes contained a long insertion in the ORF1ab gene which has not been described in other Alpha-CoVs, except for a single sequence from P. nathusii captured in the Netherlands. We propose that the insertion encodes a previously undescribed domain of unknown function, probably related to the SEA domain superfamily.
Introduction
Coronaviruses (CoVs – order Nidovirales, family Coronaviridae, subfamily Coronavirinae) are enveloped viruses characterized by a positive-sense, single-stranded RNA genome of approximately 26–32 kilobases. They are classified into four genera: Alpha- (Alpha-CoV), Beta- (Beta-CoV), Gamma- (Gamma-CoV), and Deltacoronavirus (Delta-CoV) (Woo et al., 2009). A wide diversity of CoVs has been reported from bat host species, including dozens of strains from bats in Asia, Africa, the Americas, and Europe. Bats are also the presumed reservoir hosts of the closest relatives of the zoonotic viruses SARS-CoV and SARS-CoV-2 (Hu et al., 2015; Wacharapluesadee et al., 2015; Anthony et al., 2017; Wong et al., 2019).
Two genera of CoVs infect mammals: Alpha-CoVs and Beta-CoVs (King et al., 2011; Woo et al., 2012; Lelli et al., 2013). Alpha-CoVs include 15 subgenera with 26 species, while Beta-CoVs include 5 subgenera with 14 species (https://ictv.global/report_9th/RNApos/Nidovirales/Coronaviridae). Human and animal coronavirus infections mainly result in respiratory and enteric diseases (V’kovski et al., 2021). To date, seven coronaviruses which infect humans have been identified. Two of them are Alpha-CoVs (HCoV-NL63, HCoV-229E), while the other five are Beta-CoVs (HCoV-OC43, HCoV-HKU1, SARS-CoV, MERS-CoV, SARS-CoV-2). These Alpha-CoVs and two Beta-CoVs (HCoV-OC43, HCoV-HKU1) cause mild respiratory diseases with a seasonal pattern in humans and phylogenetic evidence suggests that some of these may have originated in bats centuries before present (Kesheh et al., 2022).
Bats are the natural reservoirs of several other viruses, including emerging viruses with high pathogenic potential for humans or livestock (Olival et al., 2017). Largescale environmental, demographic and ecological changes are thought to be the drivers of increasingly frequent zoonotic diseases that have emerged into people over the last few decades (Allen et al., 2017). For bat coronaviruses, these changes likely drive increasing human contact with bats for food, guano collection, competition over agricultural resources (e.g. fruit trees), exposure of domestic animals, and other factors, leading to increased opportunity for viral spillover (Holmes, 2013; Tao et al., 2017; Wang and Anderson, 2019; El-Sayed and Kamel, 2021). The risk of spillover is likely heightened for CoVs because of their capacity for recombination, facilitated by overlap of host species ranges, co-roosting by different bat species, and the presence of multiple CoV strains in individual bats (Anthony et al., 2017; Boni et al., 2020; Irving et al., 2021).
Currently, information on the geographic distribution, as well as geographic relationships of known species/subspecies of coronaviruses, is fragmentary. Over the past few decades, studies of European bats have shown that Alpha-CoVs and Beta-CoVs are present in bats in Italy, Germany, Spain, Luxembourg, the Netherlands, the United Kingdom, France, Hungary, Denmark, and Russia (August et al., 2012; Lazov et al., 2021; Tan et al., 2023). Data on SARS-like coronaviruses in horseshoe bats in the southern regions of Russia has been published (Alkhovsky et al., 2022). as well as a MERS-like coronavirus in Pipistrellus nathusii inhabiting Moscow region (Speranskaya et al., 2023). However, there were no published studies on the presence and characteristics of Alpha-CoVs in Russian populations of bats.
Fifty-seven species from 4 families of bats (Vespertilionidae, Rhinolophidae, Miniopteridae, Molossidae) have been recorded in Russia (Russian Working Group on Bats, https://zmmu.msu.ru/bats/rbgrhp/ewelc.html), 16 vespertilionids considered the commonest (https://zmmu.msu.ru/bats/rbgrhp/ewelc.html).
The aim of this study is to expand our knowledge of the diversity and distribution of Alpha-CoVs in bats inhabiting European Russia. describe the phylogeny of these and other viruses, and elucidate information on their likely geographic distribution in the region.
Materials and methods
Sample collection
In 2021 fecal samples were collected from 17 bats of 5 species: Vespertilio murinus (n=9) captured in Moscow and Moscow region; Myotis brandtii (n=3) captured in Kaluga, Moscow region (Solnechnogorsk) and Joshkar-Ola; Pipistrellus nathusii (n=1) and Nyctalus noctula (n=1) captured in Moscow; and Pipistrellus kuhlii (n=3) captured in Rostov-on-Don (Figure 1). Bat capture and sampling were conducted by professionally trained field staff of the biology department of Lomonosov Moscow State University. Samples collected in 2015 (n=26) were also tested in this study. Detailed description of sample collection of animals captured in 2015 is described in our previous study (Speranskaya et al., 2023). All bats captured in 2015 (n=26) were released after capture. In total 43 fecal samples were analyzed.
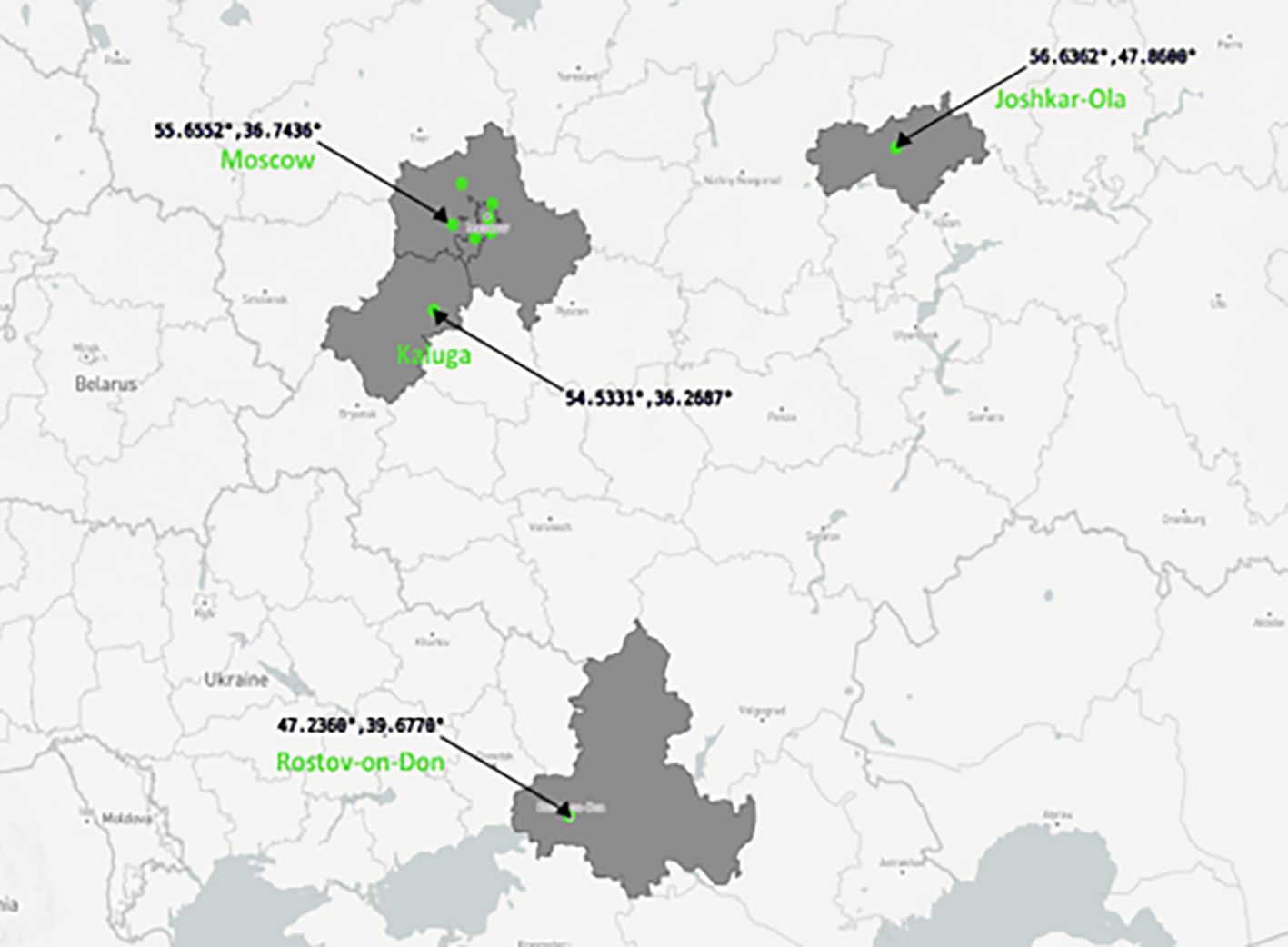
Figure 1 Russian regions where bats were captured in 2015 and 2021. Figure is built with plot.ly (https://plot.ly).
Animals captured in Rostov-on-Don, Joshkar-Ola, Moscow and Moscow region in 2021 were kept in captivity during several weeks of rehabilitation. Bats were housed in individual boxes for each species and relevant region. Fecal samples, rectal swabs, and ectoparasites were collected from individual animals using manual restraint without anesthesia. Species, sex, reproductive and health status were determined visually by trained field biologists. Fecal swabs were collected and kept in a transport media with mucolytic agent (AmpliSens, Russia) for transportation and stored at 4°C during transportation to the laboratory. Samples were then stored at −80°C before processing. The animals were released after the housing period.
Library preparation and high-throughput sequencing
Total RNA sequencing was performed for collected fecal samples. RNA was extracted using the QIAamp Viral RNA Mini Kit (Qiagen, Germany). RNA carrier was dissolved in AVE buffer and added to AVL buffer according to manufacturer’s recommendations before extraction. Fecal samples (140 μL) were added to the prepared AVL buffer with carrier RNA–Buffer AVE. Further steps were performed according to the original protocol. RNA was eluted with 60 μL of AVE buffer and stored at -70°C until analysis. First strand cDNA was obtained using the NEBNext Ultra II RNA First Strand Synthesis Module (NEB, England). Second strand cDNA was obtained using NEBNext Ultra™ II Non-Directional RNA Second Strand Synthesis Module (NEB, England). End preparation was performed with NEBNext End Repair Module which is a part of NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB, England). MGIEasy DNA Adapters (MGI, China) in the amount of 1.5 ul were ligated to double stranded cDNA using a ligation module which is also a part of NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB, England). Libraries were then amplified with PCR Primer Mix and PCR Enzyme Mix from the MGIEasy FS DNA Library Prep Kit. High throughput sequencing was performed using DNBSEQ-G400 (MGI, China) with DNBSEQ-G400RS High-throughput Rapid Sequencing Kit (FCS PE100) (MGI, China) and DNBSEQ-G400RS Rapid Sequencing Flow Cell (FCS) (MGI, China).
Analytical method: bat Alpha-CoV diversity and prevalence in European Russia
To estimate diversity and prevalence of Alpha-CoVs in bat fecal samples we performed amplification of RdRp gene fragments for CoVs, followed by NGS of the amplified fragments as described previously in (Speranskaya et al., 2023).
Partial genome assembly of Alpha-CoVs
MGI PE100 raw data obtained from Pipistrellus kuhlii (sample №132) collected in 2021 was used to assemble partial genomes of two coronaviruses. The quality of the raw reads was assessed with FastQC (Andrews, 2010). Trimming was performed using fastp with default parameters (Chen, 2023). Reads aligned with hisat2 (Kim et al., 2019) to the human genome hg38p105 were excluded from analysis, and reads aligned to the RdRp palm fragment base (palmDB) (Edgar et al., 2022) with DIAMOND (Buchfink et al., 2021) were further assembled with metaSPAdes (Nurk et al., 2017). Contigs shorter than 350 bp were discarded from further analysis. Taxonomic classification of resulting partial assemblies was carried out Kraken2 (Wood et al., 2019) and its pluspf database, CAT (https://github.com/dutilh/CAT), the latest version of its database and blastn (Altschul et al., 1990). Contigs belonging to Coronaviridae genome were further analyzed. Read mapping was performed using bowtie2 (Langmead and Salzberg, 2012). The result of read mapping was visualized using IGV genome browser (Robinson et al., 2011). Genome annotation of assembled Alpha-CoV contigs was performed using ZCURVE_CoV 2.1 (http://tubic.tju.edu.cn/sars/) and edited manually.
Complete genome assembly of Alpha-CoVs
In addition to data from bats captured in 2021, we used Illumina PE250 raw data obtained during our previous work (Speranskaya et al., 2023) to assemble complete genomes of Alpha-CoVs from bats captured in 2015. The quality of the raw reads was assessed with FastQC (Andrews, 2010) Paired reads were filtered with Trimmomatic (Bolger et al., 2014) using parameters SLIDINGWINDOW:4:25 MINLEN:40. Genome assembly was completed by metaSPAdes (Nurk et al., 2017). After trimming, genome assembly and selection of Coronaviridae sequences we obtained two contigs with lengths of 20,098 and 10,135 bp. Coronaviridae sequences were selected by BLASTn (Altschul et al., 1990) of assembled contigs using all of the available Coronaviridae genomes as a reference. Read mapping was performed using bowtie2 (Langmead and Salzberg, 2012). The result of read mapping was visualized using IGV genome browser (Robinson et al., 2011). Genome annotation was performed using two programs, Geneious Prime 2022.1.1 (https://www.geneious.com) and ZCURVE_CoV 2.1 (http://tubic.tju.edu.cn/sars/), and edited manually. TRS-L and TRS-B alignment was performed manually. Prediction of protein domains and functional sites of the ORF1ab was performed using the online tool InterPro (https://www.ebi.ac.uk/interpro/).
Alignment of the NSP3 region of assembled genomes was performed using the online program MAFFT version 7 (https://mafft.cbrc.jp/alignment/server/). The genome with a similar insertion in the NSP3 region (OQ405400.1) and tree sequences without insertion (OQ401253.1, ON457561.1, MZ218060.10 obtained from GenBank) were aligned and manually reviewed for the comparison.
Phylogenetic analysis of partial Alpha-CoV genomes
Maximum likelihood (ML) phylogenetic trees were built for partial RdRp gene fragments (326-563 bp), full RdRp genes (2700 bp), and full S gene (4100 bp) of assembled contigs and full genomes. For phylogenetic analysis, we downloaded a dataset of complete genome references containing 27 Alpha-CoV sequences related to 12 subgenera presented in the ICTV taxonomy browser (https://ictv.global/taxonomy) and 6 sequences closest to obtained RdRp gene fragments (acc. num. MG923572.2, MN535734.1, EU375869.1, MN535733.1, MZ218060.1). Thirteen partial RdRp fragments were aligned to 27 Alpha-CoVs from ICTV and 5 closest sequences. Four full RdRp genes, four S gene of assembled contigs and full genomes were aligned to 27 downloaded sequences using the alignment program MAFFT version 7 (https://mafft.cbrc.jp/alignment/server/index.html) by default. Three phylogenetic trees were built for aligned regions using IQ-TREE multicore version 2.0.7 with bootstrap (1000 replicates), best-fit model: GTR+F+I+G4 chosen according to BIC. Phylogenetic trees were visualized using the online website (https://itol.embl.de).
Phylogenetic analysis of complete Alpha-CoV genomes
For phylogenetic analysis of complete genomes, all sequences belonging to genus Alphacoronavirus with a genome size from 25,000 nt to 35,000 nt were downloaded from GenBank NCBI. We excluded sequences belonging to “Porcine epidemic diarrhea virus” or “Swine acute diarrhea syndrome coronavirus” in order to limit the number of genomes. Additionally, we filtered sequences containing “porcine”, “swine”, “transmissible”, or “unverified” in the title, but one genome of transmissible gastroenteritis virus was included to represent the Tegacovirus subgenus. Duplicate sequences have been removed using in-house scripts. Complete genomes including 155 bat-CoVs, 33 camel-CoVs, 26 canine-CoVs, 72 feline-CoVs, 6 ferret-Covs, 135 human-CoVs, 7 mink-CoVs, 10 rodent-CoVs, 1 alpaca-CoV, 2 shrew-CoVs, and 1 raccoon dog CoV sequences were taken for analysis. SARS-CoV-2 isolate Wuhan-Hu-1 was chosen as an outgroup.
The complete genomes, including the newly discovered Alpha-CoVs, were aligned using Mafft v7.505 (2022/Apr/10) (Katoh et al., 2018). The maximum likelihood (ML) phylogenetic tree was constructed using IQ-TREE multicore version 2.0.7 (Minh et al., 2020) with bootstrap (1000 replicates), best-fit model: GTR+F+I+G4 chosen according to BIC. The phylogenetic tree was visualized using the online website (https://itol.embl.de).
Results
Pedacoviruses, nyctacoviruses and myotacoviruses in bats from European Russia according to RdRp gene fragment analysis
We analyzed the presence and diversity of Alpha-CoVs in bats from the European part of Russia in total 43 fecal samples from bats of 8 species: P. nathusii (n=7), P. kuhlii (n=3), M. brandtii (n=6), M. daubentonii (n=5), N. noctula (n=5), V. murinus (n=10), M. dasycneme (n=5), and P. auritus (n=2). RdRp gene fragments of Alpha-CoVs were detected in 30% of the investigated samples (13 of 43), in six of eight species of investigated bats. Namely, Alpha-CoVs were detected in P. nathusii (in 4 of 7 specimens), M. brandtii (1 of 6), M. daubentonii (3 of 5), N. noctula (1 of 5), P. kuhlii (3 of 3), and V. murinus (1 of 10). In total, 13 Alpha-CoVs were detected using RdRp gene fragment sequences (Figure 2). In fecal samples of nine M. dasycneme bats, as well as in two P. auritus, Alpha-CoVs were not found.
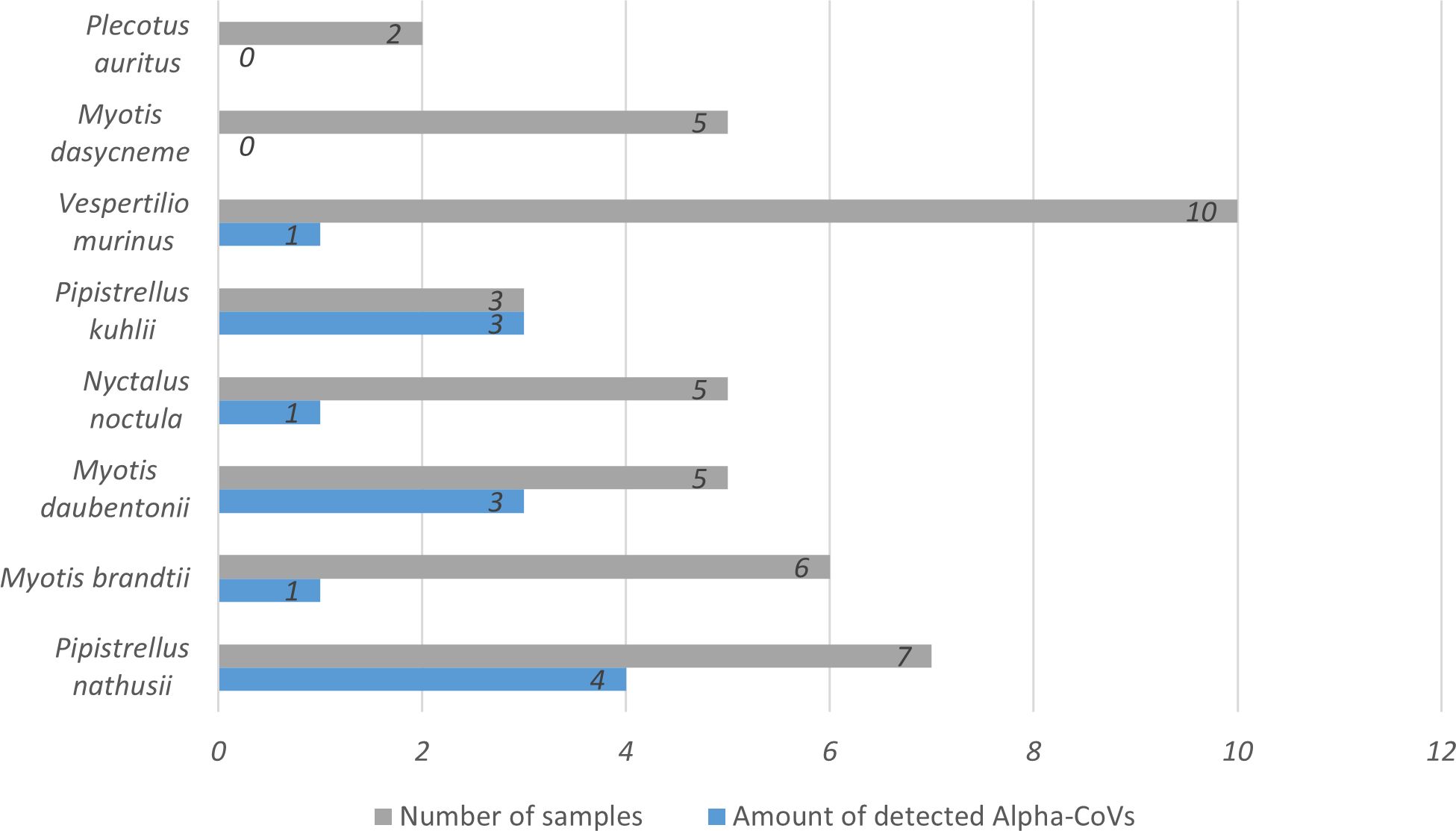
Figure 2 Amount of detected Alpha-CoVs among the bat samples of 8 species. Diagram was built with Microsoft Excel (https://office.microsoft.com/excel).
Two animals carried two different isolates of Alpha-CoVs. One of these animals, M. daubentonii (sample №30 collected in 2015), contained two RdRp sequences with highest similarity to BtCoV/18802-1/M.das/DK/2016 from M. dasycneme, Denmark and P.nat/Germany/D5.73/2007 from P. nathusii, Germany. While the other bat, P. kuhlii (sample №132 collected in 2021), contained a contig with identity to Alpha-CoVs from bats captured in Denmark and China (BtCoV/7542-55/P.pyg/DK/2014 from P. pygmaeus, Denmark and BtNv-AlphaCoV/SC2013 from N. velutinus, China).
Alpha-CoVs were found in bats captured in 2015 (n=8) as well as in bats captured in 2021 (n=5). RdRp obtained from bats captured in 2015 have >95% identity with RdRp of different Alpha-CoVs from bats captured in Northern Europe (in Denmark, Finland or Germany from 2014 to 2018, for details see Table 1). Four RdRps found in bats collected in 2021 have 98% identity to the same sequence (MZ218060.1) obtained from P. pygmaeus captured in 2014 in Denmark.
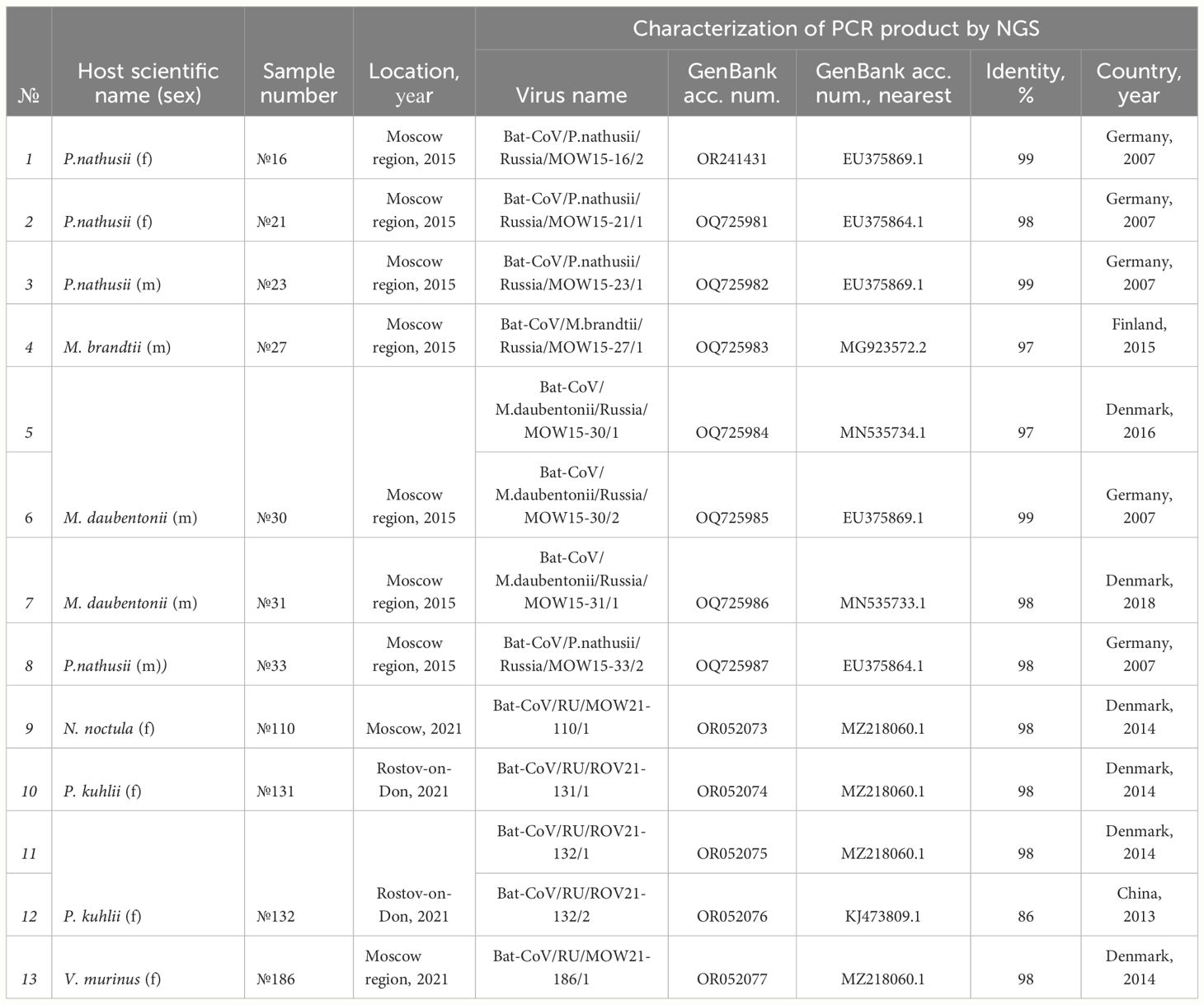
Table 1 Alpha-CoVs found in bats inhabiting European Russia and their similarity to Alpha-CoVs of bats captured in other countries.
In addition to the RdRp data presented in Table 1 firstly, we also analyzed four Alpha-CoVs from P. nathusii captured in 2015 mentioned in our published study (Speranskaya et al., 2023), namely OR241431, OQ725981, OQ725982, and OQ725987. All of these four RdRp sequences had >98% identity with RdRp of viruses found in bats from Germany collected in 2007 (EU375869.1), bat coronavirus P.nat/Germany/D5.73/2007 (EU375864.1). Phylogenetic analysis of RdRp gene fragments showed that most sequences from bats inhabiting Russia (marked as red triangle) fall into clades with pedacoviruses (brown, n=10), with minor clusters of nyctacoviruses (light green, n=1) or myotacovirus (green, n=2) (Figure 3). Low bootstrap value of the clades can probably be explained by the high similarity of analyzed RdRp fragments; we are not able to accurately relate the obtained RdRp fragments to a specific subgenus.
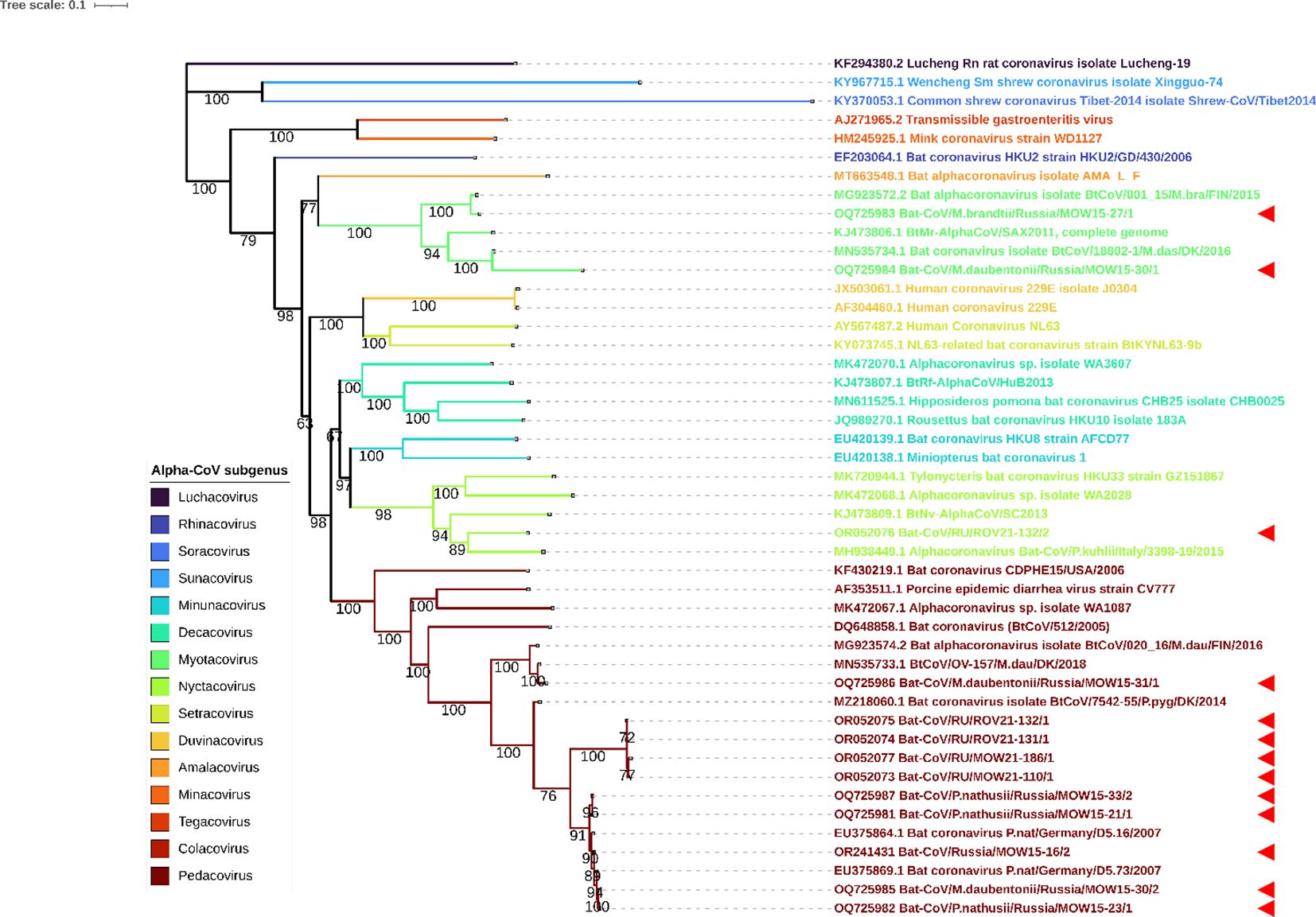
Figure 3 ML phylogenetic tree on 400-bp RdRp gene fragments of 13 Alpha-CoVs from Russia, 27 Alpha-CoV sequences downloaded from ICTV and 6 closest sequences from GenBank NCBI. Best-fit model of substitution according to BIC: GTR + F + I + G4. Numbers show bootstrap values. The red arrows represent RdRps obtained in current study.
Complete genomes of pedacoviruses from bats captured in European Russia in 2015
We obtained two complete genomes of Alpha-CoVs from fecal samples of two bats of P. nathusii captured in 2015 near Moscow (specimens №21 and №23). For genome assembly, we used sequence raw data SRA (GenBank acc. num. SRR15525307, SRR15540905) with 11.7 million and 14.9 million paired reads for samples №21 and №23, respectively. After trimming and quality filtration, 11.4 million and 12.0 million reads remained. Further de novo assembly revealed contigs of 28,245 bp in samples №21 and 28,387 bp in samples №23, with both viral sequences associated with family Coronaviridae. The fraction of reads mapped on the resulting contig consisted 0.15% (17.836 reads of 11.4 million) and 0.12% (13.767 of 12.0 million reads) for samples №21 and №23, respectively. The obtained sequences were named: Bat-CoV/P.nathusii/Russia/MOW15-21/2015, short name BatCoV/MOW15-21 (acc. num. OP919651.1); and Bat-CoV/P.nathusii/Russia/MOW15-23/2015, short name BatCoV/MOW15-23 (acc. num. OQ230639). Pairwise alignment of BatCoV/MOW15-21 to BatCoV/MOW15-23 showed 99% identity to each other according to blastn. The nearest complete genome from GenBank showed 91% identity with pedacovirus PpiGB01 (acc. num. OQ401253) from P. pipistrellus from the UK for the both obtained genomes in our study.
The genomic organization of BatCoV/MOW15-21 and BatCoV/MOW15-23 is similar to other alphacoronaviruses and encodes six ORFs: ORF1ab (putative mature nonstructural proteins, including RNA-dependent RNA polymerase (RdRp); ORF1a; S (Spike); ORF3; E (envelope); M (membrane glycoprotein); N (nucleocapsid phosphoprotein); and two non-translated 5′ terminus and 3′ terminus. In the ORF1ab of BatCoV/MOW15-21, the predicted slippery sequence “UUUAAAC”, which is involved in the synthesis of the replicase pp1ab polyprotein by ribosomal frameshift, is localized at the 12834–12840 nt position. In the ORF1ab of BatCoV/MOW15-23, it is localized at 12809-12815 n.t. (Table 2).
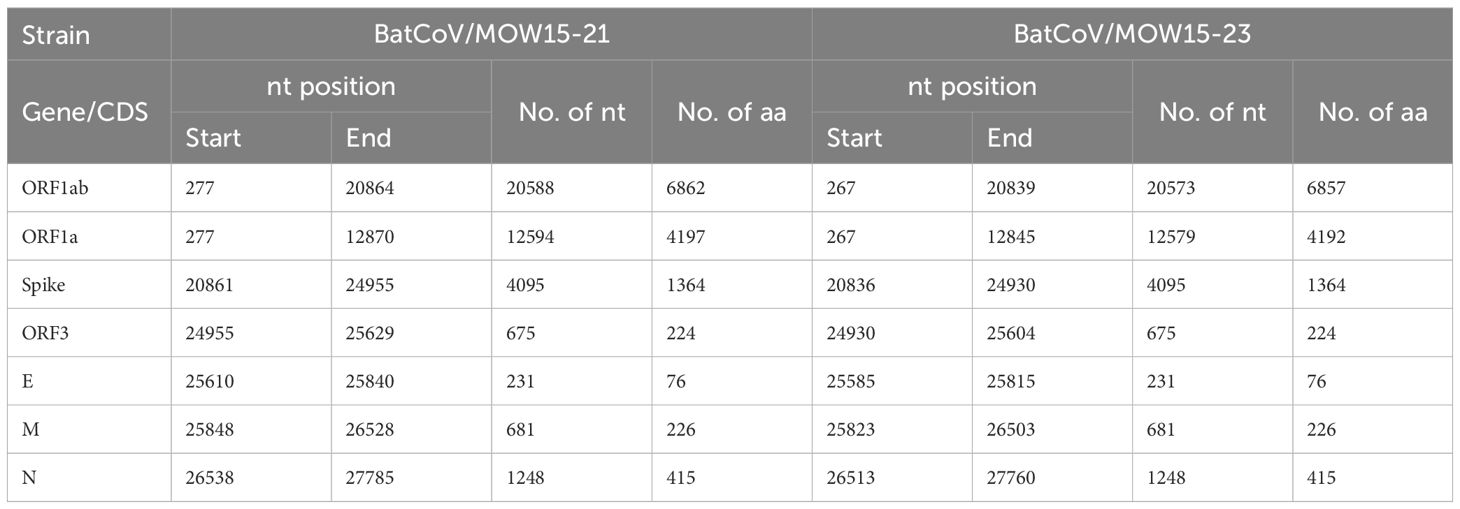
Table 2 Locations of predicted ORFs, protein sequences of BatCoV/MOW15-21 and BatCoV/MOW15-23.
The positions of the 15 putative cleavage sites of nonstructural protein (NSP1–16) are presented in Table 3. The locations and protein sequences of putative leader transcriptional regulatory sequence (TRS-L) and body transcriptional regulatory sequence (TRS-B) of predicted ORFs are described in Table 4. The core sequences of the TRS-L (5′-CTAAAC-3′) were identified in the 5′ untranslated sequences of BatCoV/MOW15-21 and BatCoV/MOW15-23 which proceeded ORF1ab and N gene, but the TRS motifs of S, ORF3, E, and M genes differed from the core sequences of the leader TRS. They were as follows: the TRS motif of S was identified as 5′-GTCAAC-3′; the TRS motif of ORF3 was identified as 5′-CTAAAG-3′; the TRS motif of E was identified as 5′-CTAGAC-3′; and the TRS motif of M was identified as 5′-CGAAAT-3′.
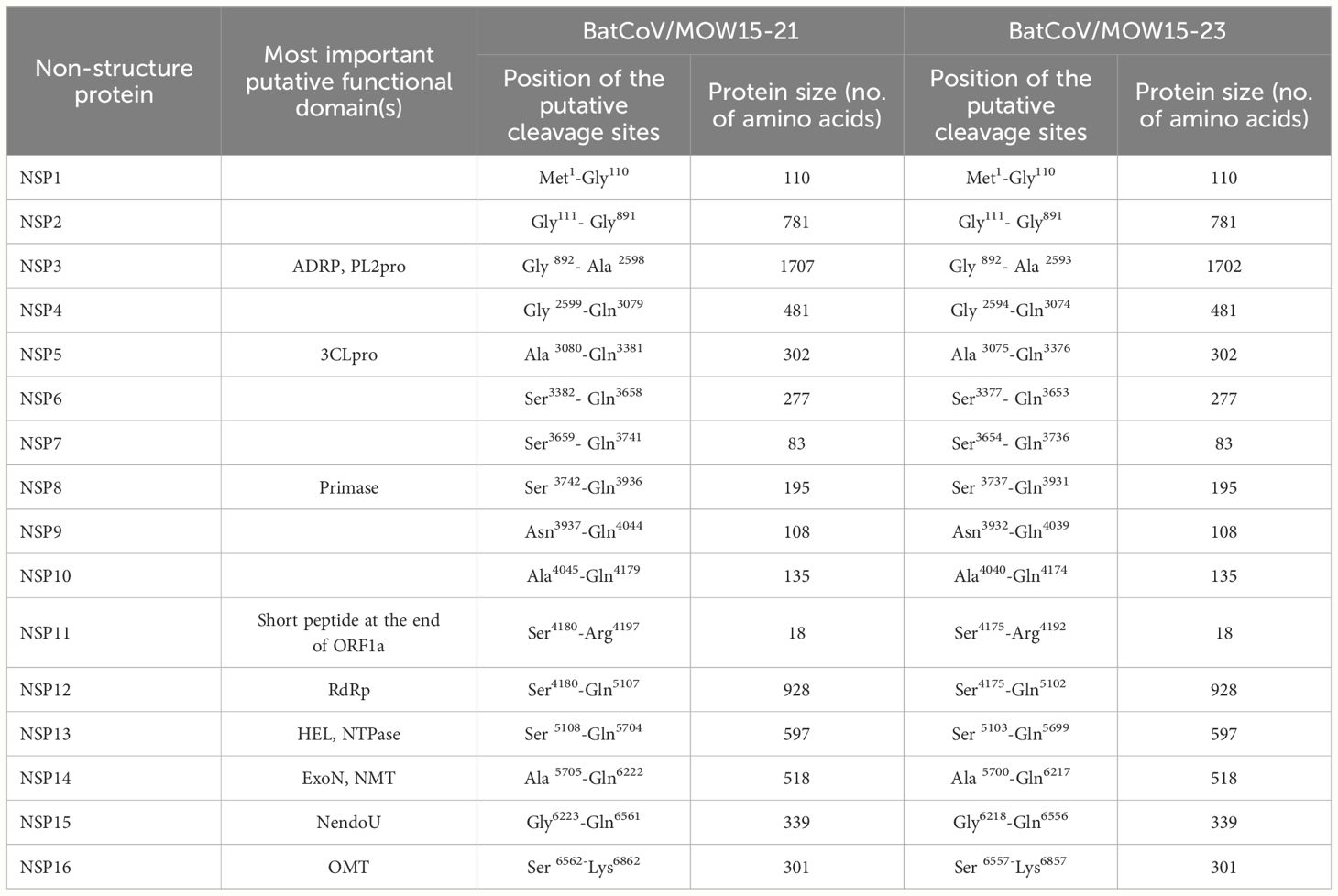
Table 3 Predicted putative pp1ab cleavage sites of BatCoV/MOW15-21 and BatCoV/MOW15-23.
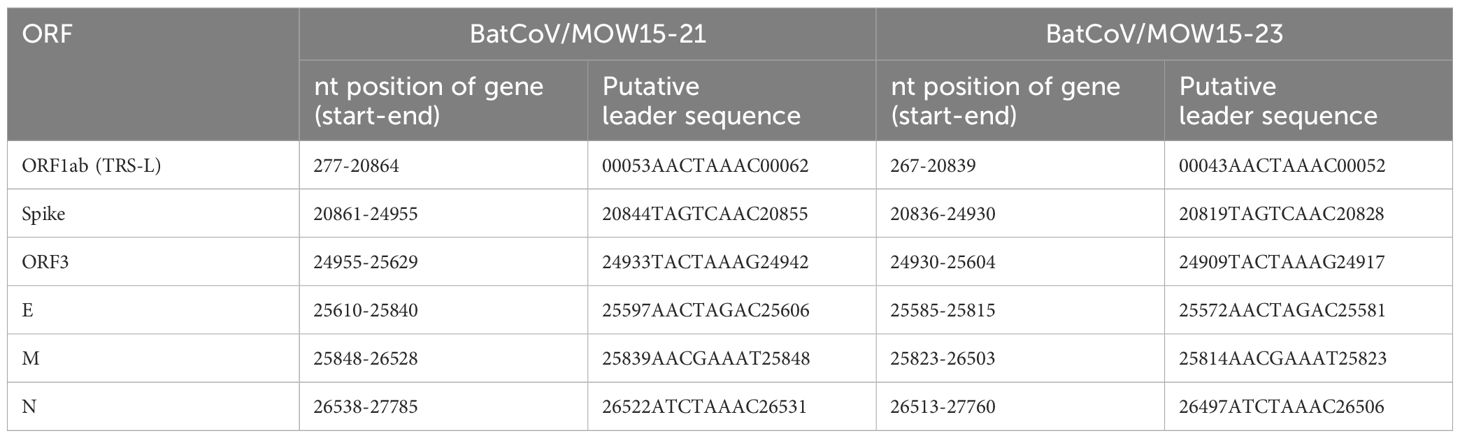
Table 4 Locations of predicted ORFs, protein sequences, putative leader TRS-L and TRS-B of BatCoV/MOW15-21 and BatCoV/MOW15-23.
Both genomes BatCoV/MOW15-21 and BatCoV/MOW15-23 contain long insertions in ORF1ab (396 bp and 382 bp, correspondingly) not found in other genomes of Alpha-CoVs, except Alpha-CoV from bats of P. nathusii captured in 2018 from Netherlands (OQ405400.1, alignment in Figure 4).
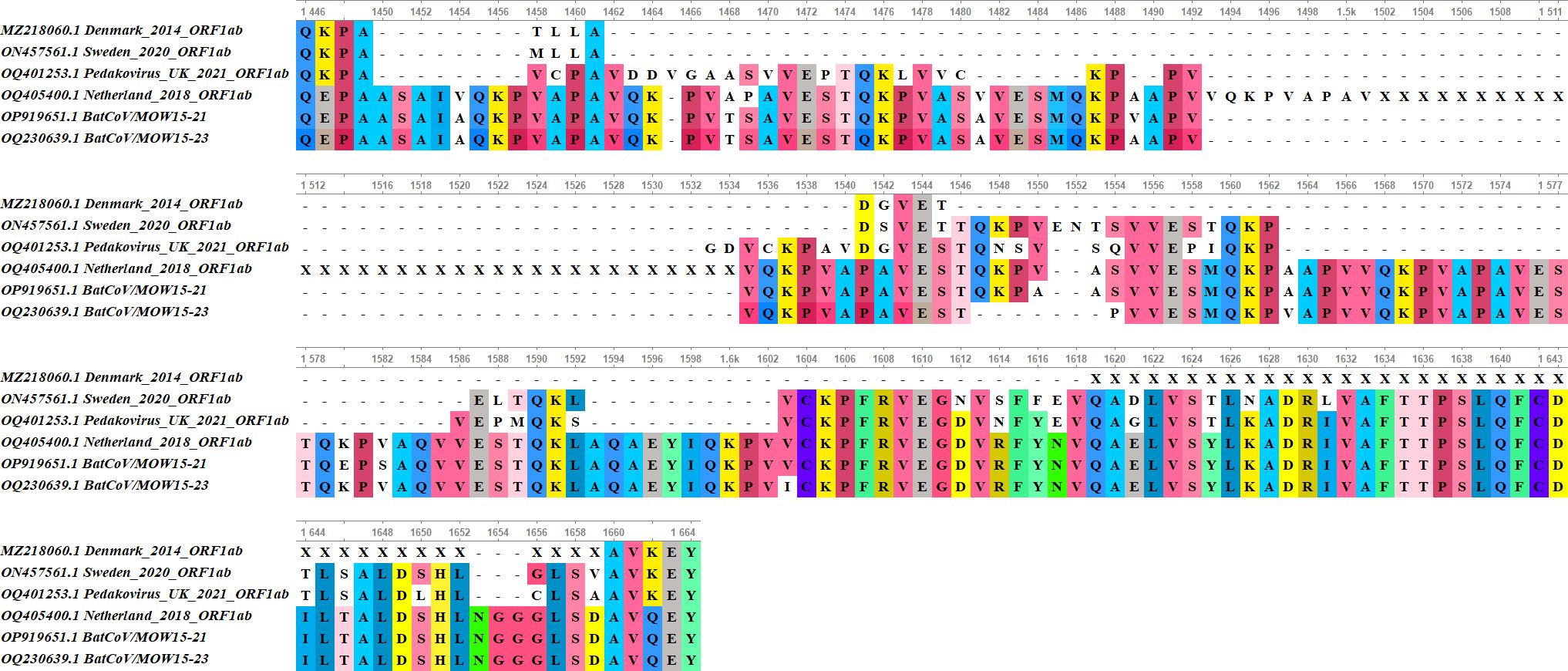
Figure 4 Alignment of the NSP3 region with insertion in BatCoV/MOW15-21, BatCoV/MOW15-23, AlphaCoV/P.nathusii/NL/2018-403.3 genome with similar insertion (OQ405400.1) and related Alpha‐CoVs genomes without insertion (OQ401253.1, ON457561.1, MZ218060.1).
Insertions in the genomes are located in the NSP3 protein encoding region. In BatCoV/MOW15-21, a 396 n.t. insertion is located at 4537-4933 bp (1512-1644 a.a. of polyprotein encoding by ORF1ab). In BatCoV/MOW15-23, a 382 n.t. insertion is located at 4527-4908 (1509-1636 a.a.). Prediction of protein domain with InterProScan revealed in BatCoV/MOW15-21 and BatCoV/MOW15-23: two ubiquitin-like domains (1 and 2), macrodomain, two papain-like proteases (1 and 2), C-terminal domain, and Y3 domain (Table 5). The region of insertion in both genomes corresponds to the region between Macrodomain (ADRP, “X-domain”) and Ubiquitin-like domain 2 (Ubl2). Multiple searches have shown that in most databases information on potential function of similar domains is absent. However, it is possible that insertion is related to an SEA domain, according to an InterPro ‘phmmer search tool’ result (biosequence analysis using profile hidden Markov Models, protein sequence vs protein sequence database).
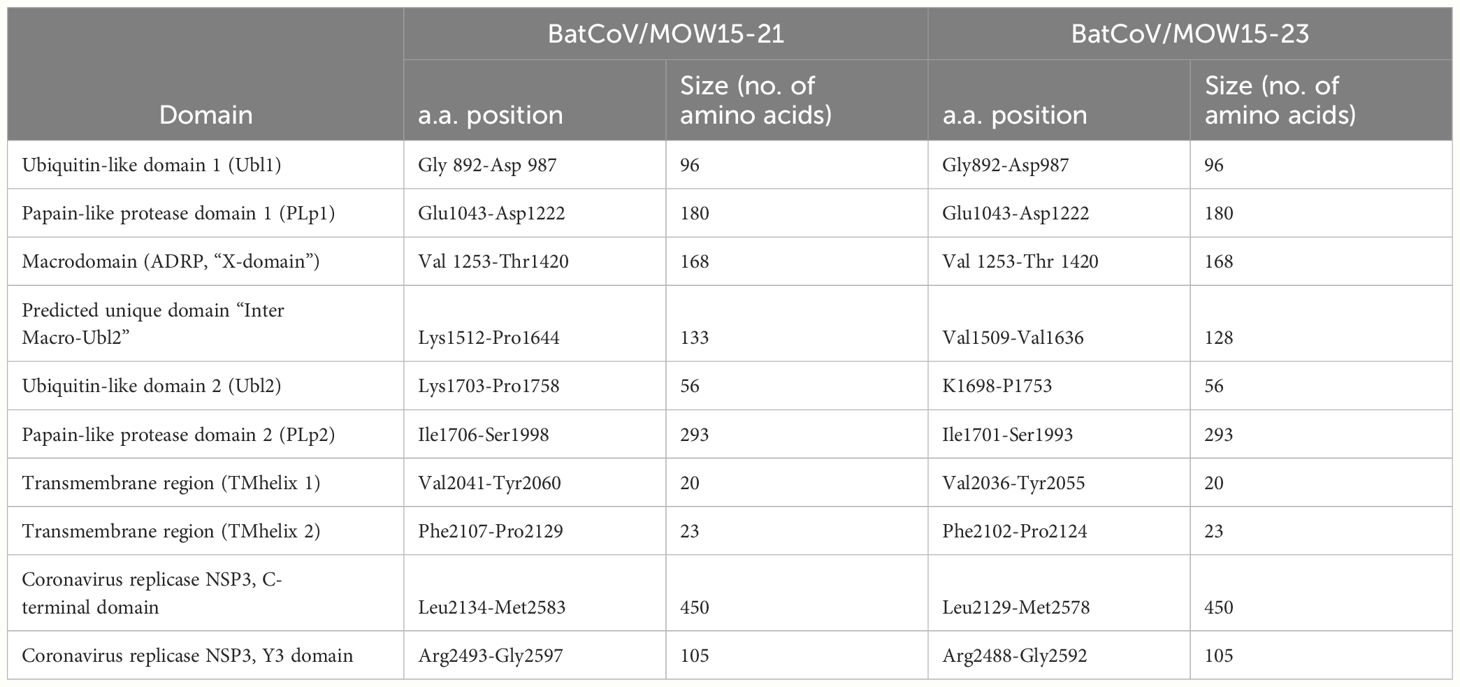
Table 5 Prediction of NSP3 domains.
Amino acid sequence alignment of the ORF1ab gene showed that a similar insertion was observed in a bat fecal sample from P. nathusii from Netherlands (OQ405400.1) captured in 2018, but this sequence contains unassembled regions and is marked as unverified in GenBank. Visualization of raw reads mapping to genome assembly demonstrates that the region of insertion is well covered (>80× coverage), hence this sequence is not an artifact of assembly (Supplementary Figures 1, 2).
According to ICTV demarcation criteria for genera and species of viruses that share more than 90% aa sequence identity in the conserved replicase domains nsp3 (ADRP, X-domain), nsp5 (3CLpro), nsp12 (RdRp), nsp13 (Hel), nsp14 (ExoN), nsp15 (NendoU) and nsp16 (O-MT)) are considered to belong to the same species (https://ictv.global/report_9th/RNApos/Nidovirales/Coronaviridae). A separate comparison of the amino acid sequences of 7 domains of ORF1ab showed >90% identity with isolates AlphaCoV/P.nathusii/NL/2018-403.3 from the Netherlands, BtCoV/F-MV2/P.pyg/SWE/2020 (acc. number ON457561.1) from Sweden, and PpiGB01 from the UK (acc. number OQ401253).
We performed phylogenetic analysis to determine the taxonomical position of Alpha-CoVs with assembled genomes. Phylogenetic analysis of 445 complete genomes of Alpha-CoVs demonstrates that BatCoV/MOW15-21 and BatCoV/MOW15-23 fall into the clade with pedacoviruses (brown). This clade forms three subclades: from China and Australia; from China and Vietnam; and from Northern Europe. BatCoV/MOW15-21 and BatCoV/MOW15-23 fall into subclades of Northern European viruses. Both viruses from Russia show >80% nt. identity with pedacovirus PpiGB01 (acc. number OQ401253) from P. pipistrellus from UK and Alpha-CoVs from M. daubentonii inhabiting Denmark and Finland (Figure 5).
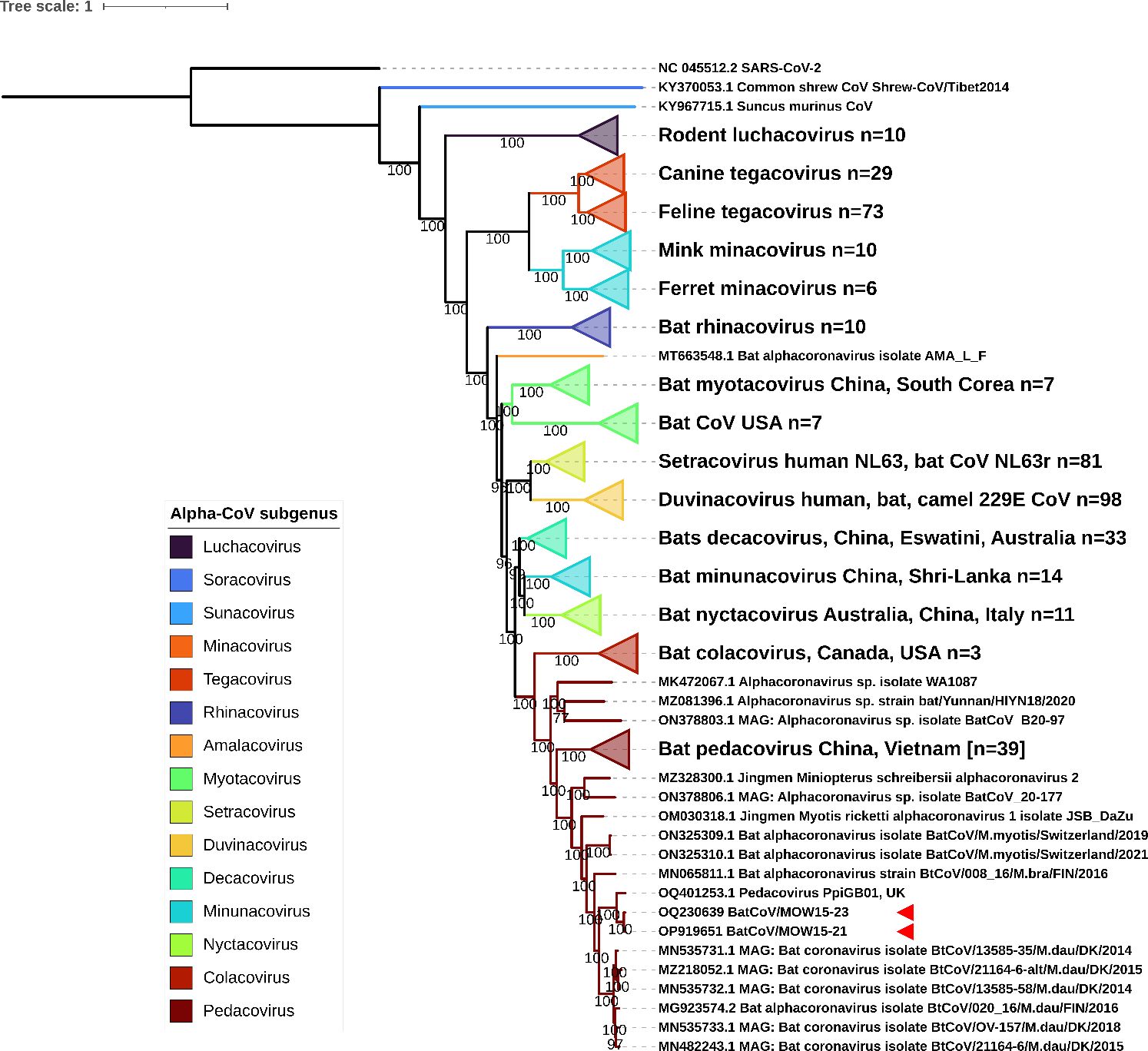
Figure 5 ML phylogenetic tree built for 445 Alpha-CoV complete genomes. Best-fit model of substitution according to BIC: GTR + F + I + G4. Numbers show bootstrap values. The red arrows mark BatCoV/MOW15-21 and BatCoV/MOW15-23.
Pedacoviruses and nyctacoviruses found in P. kuhlii captured in south Russia in 2021
As mentioned above, P. kuhlii (bat №132 collected in 2021) contained two different contigs of RdRp. We performed metavirome analysis and genome assembly of Alpha-CoVs from the specimen of P. kuhlii (bat №132). The analysis revealed two contigs of 21,184 bp and 24,456 bp fragment size. The assembled sequences were named Bat-CoV/RU/ROV21-132/4-Ped (acc. num. OR147948) and Bat-CoV/RU/ROV21-132/3-Nyct (acc. num. OR147947). The first of them, ROV21-132/4-Ped, featured 99.78% identity to BatCoV/MOW15-21 described in this work. The second, ROV21-132/3-Nyct, had 82% identity to Alpha-CoV Bat-CoV/P.kuhlii/Italy/206679-3/2010 (GenBank MH938450.1) from P. kuhlii captured in Italy in 2010.
We performed phylogenetic analysis using the RdRp and S genes to determine the taxonomical position of ROV21-132/4-Ped and ROV21-132/3-Nyct. ML phylogenetic trees constructed on 31 RdRp gene fragments and 31 S gene fragments showed similar results. ROV21-132/4-Ped clusters with BatCoV/MOW15-21, BatCoV/MOW15-23, and with other pedacoviruses from Northern Europe (Figures 6, 7). The second of the two investigated Alpha-CoVs from P. kuhlii, ROV21-132/3-Nyct, clusters with nyctacoviruses in the RdRp gene tree as well as in S gene tree (Figures 6, 7).
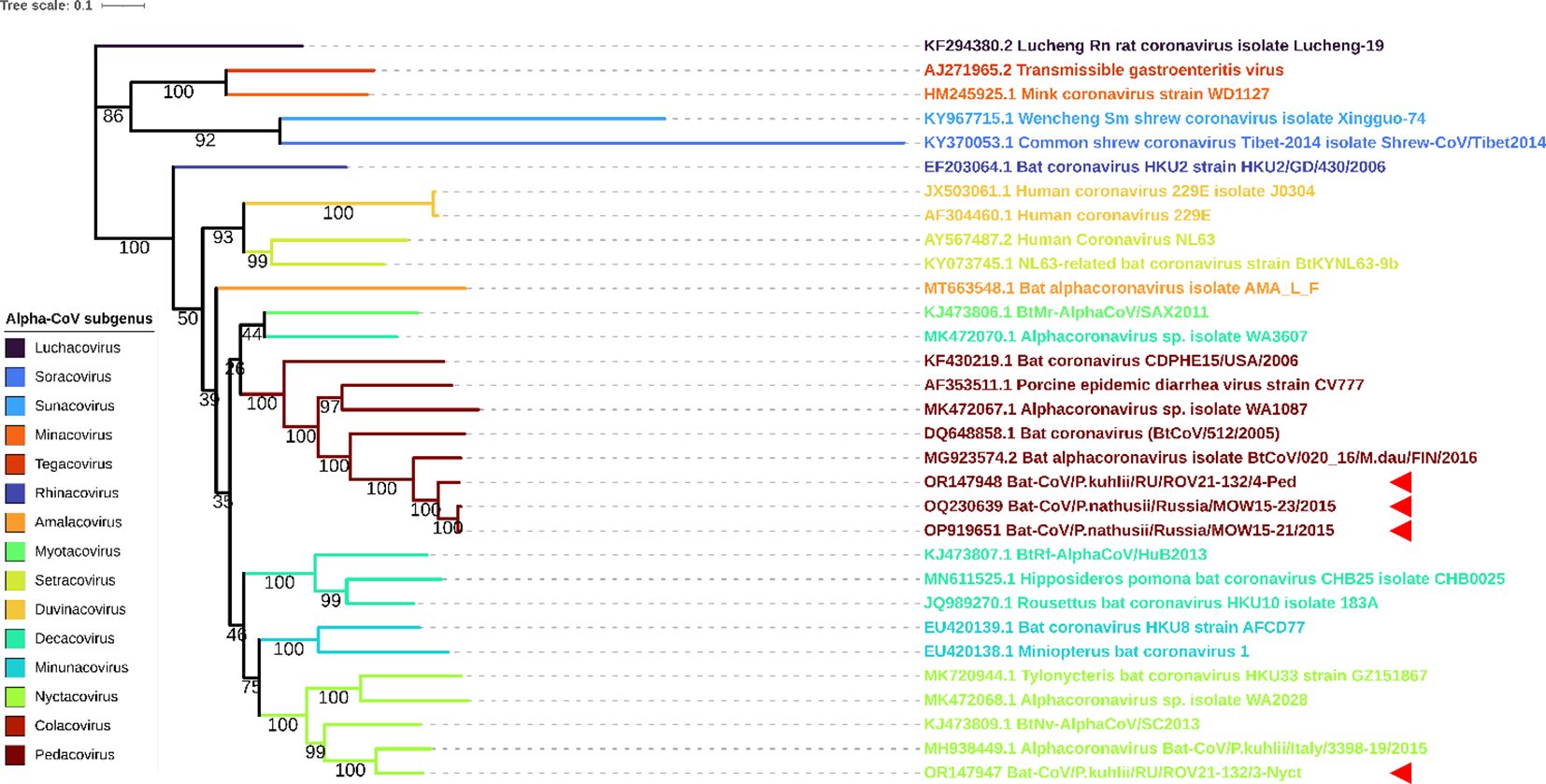
Figure 6 ML phylogenetic tree built for full RdRp genes of 4 Alpha-CoVs from Russia (OP919651, OQ230639, OR147947, OR147948) and 27 Alpha-CoV sequences downloaded from ICTV. Best-fit model of substitution according to BIC: GTR + F + I + G4. Numbers show bootstrap values. Stars present Alpha-CoVs from Russia. The red arrows represent full RdRp sequences obtained in current study.
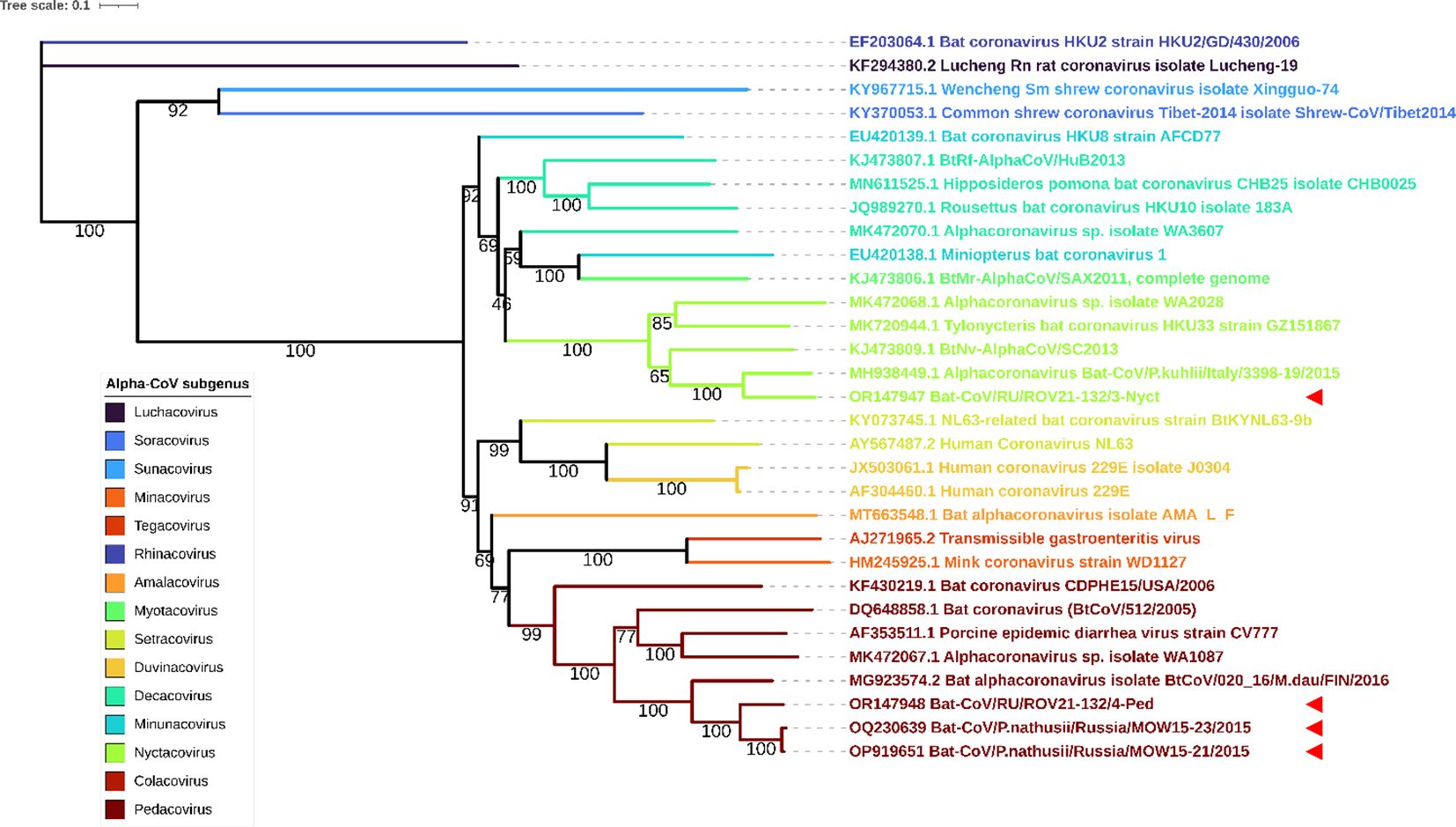
Figure 7 ML phylogenetic tree built for S genes of 4 Alpha-CoVs from Russia (OP919651, OQ230639, OR147947, OR147948) and 27 Alpha-CoV sequences downloaded from ICTV. Best-fit model of substitution according to BIC: GTR + F + I + G4. Numbers show bootstrap values. The red arrows represent full S gene sequences obtained in current study.
Discussion
Novel bat CoV host and geographic distribution
In this study thirteen Alpha-CoVs were observed in 8 bat species inhabiting European Russia. Two of forty-three investigated animals (4.6%) carried two different Alpha-CoVs: M. daubentonii collected in 2015 and P. kuhlii, collected in 2021. M. daubentonii carried two different Alpha-CoVs, that are related to Pedacovirus and Myotacovirus subgenera. In P. kuhlii (bat №132), two Alpha-CoVs from different subgenera, Pedacovirus and Nyctacovirus, were found.
Previous studies have also reported co-infection with two coronaviruses (Tong et al., 2009; Drexler et al., 2010; Lau et al., 2010; Wacharapluesadee et al., 2015; Ge et al., 2016), and the presence of two different Alpha-CoVs in one specimen (Chu et al., 2008). Like those of many negative stranded RNA viruses, CoV genomes are characterized by high frequency of mutation and recombination events, and low error correction capacity. The simultaneous presence of two or more coronaviruses in one animal is an essential prerequisite for recombination events. Further work on the longitudinal frequency of co-infections may shed light on the likelihood of recombination events (Chu et al., 2008).
We found that the closest relatives of the Alpha-CoVs we found in European-Russian bats were from Scandinavia, the United Kingdom, and Germany (Figure 6). We found two Alpha-CoVs (BatCoV/MOW15-21, BatCoV/MOW15-23) whose complete genomes contained long insertions in the NSP3 encoding region of ORF1ab not found in other genomes of Alpha-CoVs, except Alpha-CoV from P. nathusii captured in the Netherlands in 2018 (Mols et al., 2023). Both BatCoV/MOW15-21 and BatCoV/MOW15-23 are related to pedacoviruses, which is one of fifteen Alpha-CoV subgenera.
Phylogenetic analysis shows that pedacoviruses form three subclades: (a) from China; (b) from China and Vietnam; and (c) from Northern Europe. BatCoV/MOW15-21 and BatCoV/MOW15-23 fall into subclades of Northern European viruses. RdRp and S sequences from the partial genome of pedacovirus from P. khulii, ROV21-132/4-Ped which was obtained during this investigation, also fall into the “Northern European” clade. It clusters closely with BatCoV/MOW15-21, BatCoV/MOW15-23, and other pedacoviruses from Northern Europe. Interestingly, all viruses from this subclade were isolated from different Pipistrellus species (P. nathusii, P. pygmaeus, P. pipistrellus, P. khulii) which were captured in different countries of Northern or Northeastern Europe, including Russia.
The results of the phylogenetic analysis based on the short RdRp sequences indicate that detected Alpha-CoVs belong to the “Northern European”. RdRps obtained from bats captured in 2015 have >95% identity with RdRps of different Alpha-CoVs from bats captured in Northern Europe: Dеnmark, Finland or Germany from 2014 to 2018. Four RdRps found in bats collected in 2021 have 98% identity to the same sequence (MZ218060.1) from the bat captured in 2014 in Denmark. Thus, it appears that related pedacoviruses have been circulating for a long time (at least 7 years, from 2015 to 2021) in from the region covered by European Russia and Northern Europe. We propose that pedacoviruses from this subclade are likely a separate subspecies of Alpha-CoV (Northern Europe) (Figure 8).
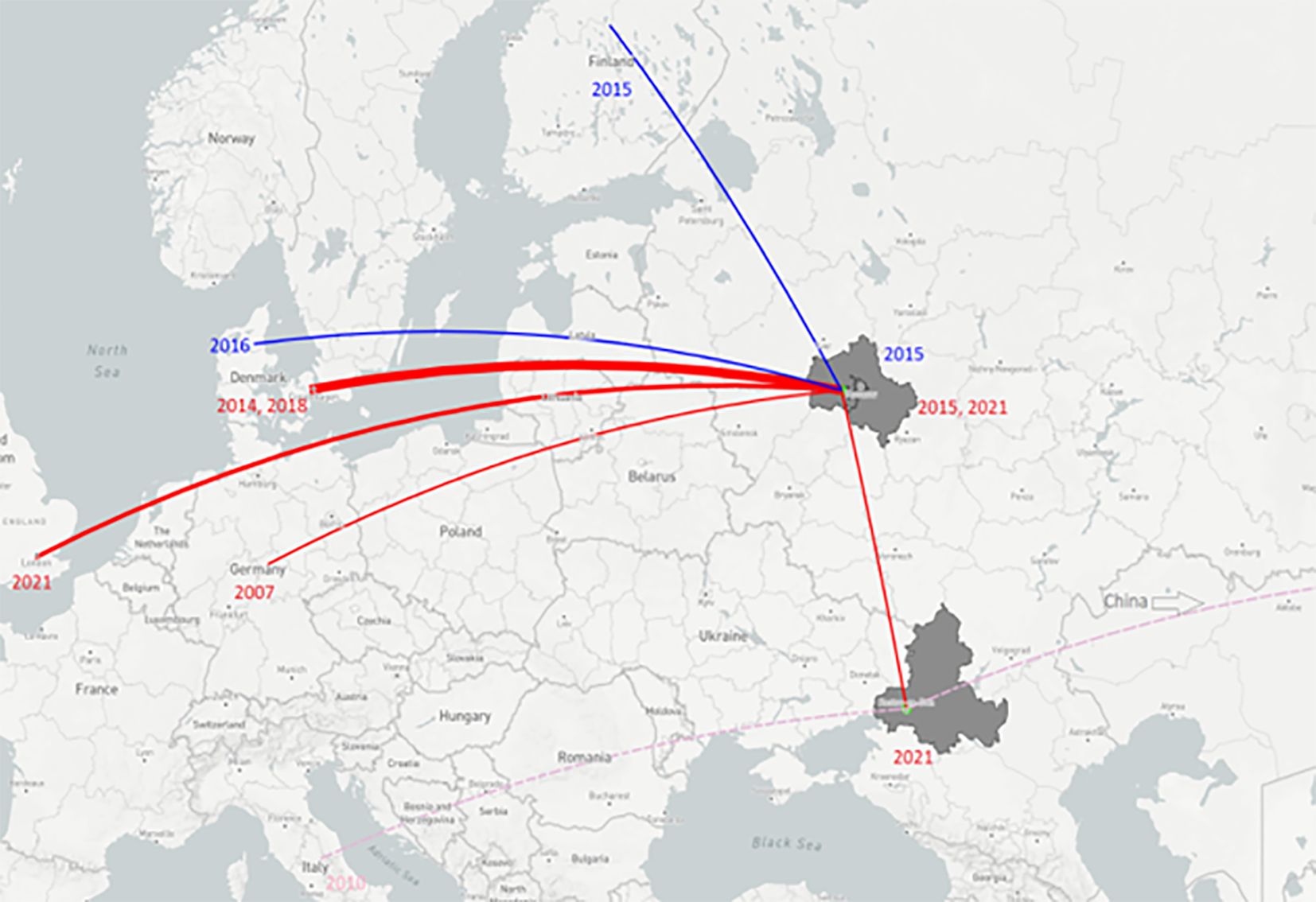
Figure 8 Map of relationships between Alpha-CoVs extracted from bats in different regions. Key: light green dots represent places where Alpha-CoV-positive samples were captured; red lines – the relationship between pedacoviruses from bats captured in European Russia and the closest pedacoviruses of Northern European bats (>90% identity according blastn); blue lines – relationship between myotacoviruses found in Moscow region and the closest of bats from Finland and Denmark (>90% identity according blastn); pink dashed lines – relationship between nyctacoviruses found in Rostov-on-Don and the closest from bats of China/Italy (<90% identity according blastn). Line width represents the number of sample hits with high identity. Figure built with plot.ly (https://plot.ly).
According to ICTV demarcation criteria for Coronaviridae family viruses sharing >90% amino acid sequence identity in the conserved replicase domains should be considered conspecific (https://ictv.global/report_9th/RNApos/Nidovirales/Coronaviridae). We compared amino acid sequences of seven conserved ORF1ab domains, and none of them fell below the 90% threshold value. It therefore seems that these viruses represent a separate taxonomic unit (species or subspecies), which we named “Northern-European Bat Alpha-CoV” (NE-Alpha-CoV). We expect that further studies of coronaviruses from Pipistrellus will find sequences representing this species. P. nathusii, P. pygmaeus, and P. pipistrellus are among the most widespread bat species in western Palaearctic, inhabiting bulk part of Europe from the North Sea coasts to the southern Ural Mountain, Caspian sea and the latter species also widely distributed in Western and Central Asia (Russo, 2023). P. kuhlii represents more “southern” species with main range in Mediterranean region. Just half a century ago it was recorded mostly south of 47° latitude, however since 1980s it spread widely northward reaching Central Europe and Central regions of the European part of Russia (Il’in and Smirnov, 2000; Sachanowicz et al., 2006; Bobrov et al., 2008; Ancillotto et al., 2016). Moreover, solitary individuals are also found far beyond the edge of the range (e.g. in Moscow region: see Bobrov et al., 2008). Reports from UK (e.g. Hutson, 1992) could actually be transported specimens, but also may represent beginning of expansion to the British Isles (Sachanowicz et al., 2006). Such distant flights may well be accompanied by contact with other species of Pipistrellus, although this issue remains unexplored. All west Palaearctic Pipistrellus species are quite closely related belonging to so-called “western clade” of the genus (Zhukova et al., 2022). P. nathusii, P. pipistrellus, and P. pygmaeus have broadly similar physiology. These are small aerial hawkers feeding at a height of several meters along the linear elements of the landscape (Dietz and Kiefer, 2016; Russo, 2023). All three species are to some extent associated with forest habitats (at least in Europe; in Asia, P. pipistrellus also inhabits arid open landscapes: see e.g. Benda et al., 2012). These species prefer natural tree hollows or slit-like shelters in wooden buildings as summer roosts used for rising offspring (Dietz and Kiefer, 2016). In sufficiently large hollows and, moreover, in buildings, pipistrelle colonies can be located side by side with other species with similar preferences: with other species of Pipistrellus, as well as with V. murinus, Myotis spp., Nyctalus spp. etc. (Orlova and Zapart, 2012; Dietz and Kiefer, 2016; Vasenkov et al., 2017). During seasonal migration, young animals of different species are also able to occupy common daytime roosts (Hutterer et al., 2005). The interaction of individuals in such mixed colonies is not well studied; however, there could be an exchange of ectoparasites and, accordingly, pathogens (Lagerveld et al., 2021).
Phylogenetic analysis of Alpha-CoV complete genomes performed in current study have suggested that Alpha-CoVs found in Russia clusters with other Alpha-CoVs from bats and form distinct clade with Alpha-CoVs found in human or other mammals. Similar conclusions were given by the authors of other studies (Osborne et al., 2011; Tan et al., 2023). Experiments on binding of the CoVs from bats with human cell receptors performed in study Tan et al. demonstrated that the bat Alpha-CoVs (Pedacovirus subgenus) could not enter human cell overexpressing ACE2 or transmembrane serine protease 2 (Tan et al., 2023).
Long distance spread of virus may also occur due to migration activity of some bat species. The migratory rate of P. pipistrellus and P. pygmaeus apparently differs in various areas (Hutterer et al., 2005): they are sedentary or make only local movements in the western and southern parts of their range, but from the forest zone of Eastern Europe they make long-distance migration of hundreds of kilometers (directed from northeast to southwest). However, mtDNA and microsatellite data suggest that a distance of 1000 km (in the direction from west to east) completely excludes gene flow between populations in these species (Hulva et al., 2010). On the contrary, P. nathusii is the most distant migrant among bats in general, capable of seasonal movements over distances of more than 2000 km. Recently migration from northwest Russia to the French Alps via a straight-line distance of 2486 km has been reported for this species (Vasenkov et al., 2022). This pipistrelle regularly migrates over the North Sea (Lagerveld et al., 2021). The National Nathusius’ Pipistrelle Project (NNPP) has also identified long-distance movement of P. nathusii between the south of England and mainland Europe, including the coast of the Netherlands, Latvia and Lithuania. It can be expected that this species may involuntarily transfer pathogens over similar distances in the direction of its migration routes (northeast–southwest). Detailed studies of migration routes are still required, but analysis of viral sequences suggest that the same viruses are circulating in a particular area.
Possible function of a unique domain in the Nsp3 region of BatCoV/MOW15-21 and BatCoV/MOW15-23
Previous studies have shown that NSP3 is the largest protein encoded by the CoV genome, with an average molecular mass of about 200 kD. In SARS-CoV-2, Nsp3 contains 1945 residues (~212 kDa) (Osipiuk et al., 2021). Nsp3 comprises various domains (from 10 to 16), the organization of which differs in CoV genera due to duplication or absence of some domains. Unique domains exist in Nsp3 of some coronaviruses, for example (SUD/HVR) of SARS [PMC7854729]. The following domains and two transmembrane regions of Nsp3 are conserved in Alpha-CoVs: (1) the ubiquitin-like domain 1 (Ubl1); (2) the Glu-rich acidic domain (also called “hypervariable region”); (3) the papain-like protease 1 (PL1pro); (4) a macrodomain (also named “X domain”); (5) the ubiquitin-like domain 2 (Ubl2); (6) the papain-like protease 2 (PL2pro); (7) the Nsp3 ectodomain (3Ecto, also called “zinc-finger domain”), flanked on two sides by transmembrane regions (TMhelix 1 and 2); and (8) the domains Y1 and CoV-Y of unknown functions (Lei et al., 2018). PLpro is highly conserved and found in all coronaviruses, usually in two copies, denoted as PL1pro and PL2pro. PLpro of SARS-CoV-2 is a slightly basic, 315 residue protein. In SARS, PLpro is located in Nsp3 between a unique domain (SUD/HVR) and a nucleic acid-binding domain (NAB) (Osipiuk et al., 2021).
Protein domain prediction with InterProScan revealed typical Alpha-CoV Nsp3 structure (BatCoV/MOW15-21, BatCoV/MOW15-23): two ubiquitin-like domains (1 and 2), macrodomain, two papain-like proteases (PL1pro and PL2pro), C-terminal domain + Y domain. We found large insertions of 130+ a.a. between macrodomain and PL2pro in BatCoV/MOW15-21 and BatCoV/MOW15-23, which distinguish these viruses from other bat pedacoviruses (with the exception of AlphaCoV/P.nathusii/NL/2018-403.3).
Conclusion
Thus, this is the first report of Alpha-CoVs detected in bats inhabiting European Russia. Our results demonstrate that closely related pedacoviruses have been circulating for a long time (from 2015 to 2021) in a large region from European Russia to Northern Europe and represent a separate species, which we named NE-Alpha coronavirus. We showed that newly discovered complete genomes of Alpha-CoV from P. nathusii had typical genome structure, but contained an insertion located in a region described in the literature as “a region of unknown function and structure”. Further research and characterization of this region is required. It is also necessary to obtain more viral genomes for study circulation and evolution of Alpha-CoVs among bats inhabiting Northern Europe.
Data availability statement
The original contributions presented in the study are publicly available. The raw fastq files associated with this study have been deposited in the NCBI SRA database under accession numbers SRX24496538–SRX24496547, SRX24496555–SRX24496587, SRX11807218, SRX11807474, SRX11807359, SRX11823289, SRX11822922, SRX11823236, SRX11824039, SRX11824943, SRX11831395, SRX11831451, SRX11832353, SRX11839038, and SRX11839039. Partial and complete genomes of Alpha-CoVs have been deposed in NCBI GenBank under accession numbers OR147948, OR147947, OP919651, and OQ230639. RdRp gene fragment sequences have been deposited in NCBI GenBank under accession numbers OR241431, OQ725981, OQ725982, OQ725983, OQ725984, OQ725985, OQ725986, OQ725987, OR052073, OR052074, OR052075, OR052076, and OR052077.
Ethics statement
The animal study was approved by local ethics committee of Saint Petersburg Pasteur Institute, Federal Service on Consumer Rights Protection and Human Well-Being Surveillance, Saint Petersburg, Russia. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
EvK: Conceptualization, Data curation, Methodology, Writing – original draft, Writing – review & editing. AES: Data curation, Formal analysis, Methodology, Writing – review & editing. IC: Data curation, Methodology, Writing – review & editing. IB: Data curation, Writing – review & editing. IS: Data curation, Writing – review & editing. IA: Data curation, Methodology, Writing – review & editing. AY: Methodology, Writing – review & editing. SK: Writing – review & editing. MS: Writing – review & editing. SS: Writing – review & editing. EoK: Methodology, Writing – review & editing. AG: Methodology, Writing – review & editing. VD: Writing – original draft, Writing – review & editing. AS: Conceptualization, Data curation, Methodology, Writing – original draft, Writing – review & editing. PD: Conceptualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The molecular virology and bioinformatic works were supported by state project (No. 12203090069-4) (AS, AES, EK); The sequencing work was supported by Russian Foundation for Basic Research Grant (20-04-60561) (AS, IA, AY, AES, EK); Biosampling, Zoology and phylogeography data interpretation were supported by Russian Science Foundation Grant 22-24-00017 (IA and SK), and was made in line with the ZMMU state theme (No. 121032300105-0) (IA and SK).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2024.1324605/full#supplementary-material
References
Alkhovsky S., Lenshin S., Romashin A., Vishnevskaya T., Vyshemirsky O., Bulycheva Y., et al. (2022). SARS-like coronaviruses in horseshoe bats (Rhinolophus spp.) in Russia 2020. Viruses 14, 113. doi: 10.3390/v14010113
Allen T., Murray K. A., Zambrana-Torrelio C., Morse S. S., Rondinini C., Di Marco M., et al. (2017). Global hotspots and correlates of emerging zoonotic diseases. Nat. Commun. 8, 1–10. doi: 10.1038/s41467-017-00923-8
Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Ancillotto L., Santini L., Ranc N., Maiorano L., Russo D. (2016). Extraordinary range expansion in a common bat:the potential roles of climate change and urbanisation. Sci. Nat. 103, 15. doi: 10.1007/s00114-016-1334-7
Andrews S. (2010). FastQC - A quality control tool for high throughput sequence data (Babraham Bioinformatics). Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
Anthony S. J., Gilardi K., Menachery V. D., Goldstein T., Ssebide B., Mbabazi R., et al. (2017). Further evidence for bats as the evolutionary source of middle east respiratory syndrome coronavirus. mBio 8. doi: 10.1128/mBio.00373-17
August T. A., Mathews F., Nunn M. A. (2012). Alphacoronavirus detected in bats in the United Kingdom. Vector-Borne Zoonotic Dis. 12, 530–533. doi: 10.1089/vbz.2011.0829
Bobrov V., Varshavsky A., Hlyap L. (2008). Alien mammals in the ecosystems of Russia (Moscow: KMK Scientific press Ltd). 232 pp.
Bolger A. M., Lohse M., Usadel B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Boni M. F., Lemey P., Jiang X., Lam T. T. Y., Perry B. W., Castoe T. A., et al. (2020). Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat. Microbiol. 5, 1408–1417. doi: 10.1038/s41564-020-0771-4
Buchfink B., Reuter K., Drost H. G. (2021). Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368. doi: 10.1038/s41592-021-01101-x
Chen S. (2023). Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using fastp. iMeta 2, e107. doi: 10.1002/imt2.107
Chu D. K. W., Peiris J. S. M., Chen H., Guan Y., Poon L. L. M. (2008). Genomic characterizations of bat coronaviruses (1A, 1B and HKU8) and evidence for co-infections in Miniopterus bats. J. Gen. Virol. 89, 1282–1287. doi: 10.1099/vir.0.83605-0
Drexler J. F., Gloza-Rausch F., Glende J., Corman V. M., Muth D., Goettsche M., et al. (2010). Genomic characterization of severe acute respiratory syndrome-related coronavirus in European bats and classification of coronaviruses based on partial RNA-dependent RNA polymerase gene sequences. J. Virol. 84, 11336–11349. doi: 10.1128/JVI.00650-10
Edgar R. C., Taylor J., Lin V., Altman T., Barbera P., Meleshko D., et al. (2022). Petabase-scale sequence alignment catalyses viral discovery. Nature 602, 142–147. doi: 10.1038/s41586-021-04332-2
El-Sayed A., Kamel M. (2021). Coronaviruses in humans and animals: the role of bats in viral evolution. Environ. Sci. pollut. Res. 28, 19589–19600. doi: 10.1007/s11356-021-12553-1
Ge X. Y., Wang N., Zhang W., Hu B., Li B., Zhang Y. Z., et al. (2016). Coexistence of multiple coronaviruses in several bat colonies in an abandoned mineshaft. Virol. Sin. 31, 31–40. doi: 10.1007/s12250-016-3713-9
Holmes E. C. (2013). What can we predict about viral evolution and emergence? Curr. Opin. Virol. 3, 180–184. doi: 10.1016/j.coviro.2012.12.003
Hu B., Ge X., Wang L.-F., Shi Z. (2015). Bat origin of human coronaviruses. Virol. J. 12, 1–10. doi: 10.1186/s12985-015-0422-1
Hulva P., Fornůsková A., Chudárková A., Evin A., Allegrini B., Benda P., et al. (2010). Mechanisms of radiation in a bat group from the genus Pipistrellus inferred by phylogeography, demography and population genetics. Mol. Ecol. 19, 5417–5431. doi: 10.1111/mec.2010.19.issue-24
Hutterer R., Ivanova T., Meyer-Cords C. H., Rodrigues L. (2005). Bat migrations in Europe: a review of banding data and literature. Naturschutz und Biologische Vielfalt 28, 3–162.
Il’in V. Y., Smirnov D. G. (2000). Specific features of distribution of resident bat species (Chiroptera: Vespertilionidae) in the eastern East European plain and adjacent regions. Russian J. Ecol. 31, 101–107. doi: 10.1007/BF02828363
Irving A. T., Ahn M., Goh G., Anderson D. E., Wang L. F. (2021). Lessons from the host defences of bats, a unique viral reservoir. Nature 589, 363–370. doi: 10.1038/s41586-020-03128-0
Katoh K., Rozewicki J., Yamada K. D. (2018). MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 20, 1160–1166. doi: 10.1093/bib/bbx108
Kesheh M. M., Hosseini P., Soltani S., Zandi M. (2022). An overview on the seven pathogenic human coronaviruses. Rev. Med. Virol. 32, e2282. doi: 10.1002/rmv.2282
Kim D., Paggi J. M., Park C., Bennett C., Salzberg S. L. (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915. doi: 10.1038/s41587-019-0201-4
King A. M. Q., Adams M. J., Carstens E. B., Lefkowitz E. J. (2011). Virus Taxonomy Classification and Nomenclature of Viruses., 1st ed. (Vienna: Springer)
Lagerveld S., Jonge Poerink B., Geelhoed S. C. V. (2021). Offshore occurrence of a migratory bat, Pipistrellus nathusii, depends on seasonality and weather conditions. Animals 11, 3442. doi: 10.3390/ani11123442
Langmead B., Salzberg S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lau S. K. P., Li K. S. M., Huang Y., Shek C.-T., Tse H., Wang M., et al. (2010). Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome-related rhinolophus bat coronavirus in China reveal bats as a reservoir for acute, self-limiting infection that allows recombination events. J. Virol. 84, 2808–2819. doi: 10.1128/JVI.02219-09
Lazov C. M., Belsham G. J., Bøtner A., Rasmussen T. B. (2021). Full-genome sequences of alphacoronaviruses and astroviruses from myotis and pipistrelle bats in Denmark. Viruses 13, 1073. doi: 10.3390/v13061073
Lei J., Kusov Y., Hilgenfeld R. (2018). Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antiviral Res. 149, 58–74. doi: 10.1016/j.antiviral.2017.11.001
Lelli D., Papetti A., Sabelli C., Rosti E., Moreno A., Boniotti M. B. (2013). Detection of coronaviruses in bats of various species in Italy. Viruses 5, 2679–2689. doi: 10.3390/v5112679
Minh B. Q., Schmidt H. A., Chernomor O., Schrempf D., Woodhams M. D., Von Haeseler A., et al. (2020). IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. doi: 10.1093/molbev/msaa015
Mols V. C., Lamers M. M., Leijten L. M., Breugem T. I., van de Bildt M. W., van den Doel P. B., et al. (2023). Intestinal tropism of a betacoronavirus (Merbecovirus) in nathusius’s pipistrelle bat (Pipistrellus nathusii), its natural host. J. Virol. 97, e00099-23. doi: 10.1128/jvi.00099-23
Nurk S., Meleshko D., Korobeynikov A., Pevzner P. A. (2017). MetaSPAdes: A new versatile metagenomic assembler. Genome Res. 27, 824–834. doi: 10.1101/gr.213959.116
Olival K. J., Hosseini P. R., Zambrana-Torrelio C., Ross N., Bogich T. L., Daszak P. (2017). Host and viral traits predict zoonotic spillover from mammals. Nature 546, 646–650. doi: 10.1038/nature22975
Orlova M. V., Zapart A. (2012). Interaction of ectoparasites in cohabitating colonies of pond bats Myotis dasycneme (Boie 1825) and species of genus Pipistrellus from northern Poland. Ann. Parasitol. 58, 211–215.
Osborne C., Cryan P. M., O’Shea T. J., Oko L. M., Ndaluka C., Calisher C. H., et al. (2011). Alphacoronaviruses in new World bats: Prevalence, persistence, phylogeny, and potential for interaction with humans. PLoS One 6, e19156. doi: 10.1371/journal.pone.0019156
Osipiuk J., Azizi S. A., Dvorkin S., Endres M., Jedrzejczak R., Jones K. A., et al. (2021). Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat. Commun. 12, 743. doi: 10.1038/s41467-021-21060-3
Robinson J. T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E. S., Getz G., et al. (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. doi: 10.1038/nbt.1754
Russo D. (Ed.) (2023). Handbook of the Mammals of Europe (Cham, Switzerland: Springer Nature). Chiroptera963 pp. doi: 10.1007/978-3-030-44029-9
Sachanowicz K., Wower A., Bashta A. T. (2006). Further range extension of Pipistrellus kuhlii (Kuhl 1817) in central and eastern Europe. Acta Chiropt 8, 543–548. doi: 10.3161/1733-5329(2006)8[543:FREOPK]2.0.CO;2
Speranskaya A. S., Artiushin I. V., Samoilov A. E., Korneenko E. V., Khabudaev K. V., Ilina E. N., et al. (2023). Identification and Genetic Characterization of MERS-Related Coronavirus Isolated from Nathusius’ Pipistrelle (Pipistrellus nathusii) near Zvenigorod (Moscow Region, Russia). Int. J. Environ. Res. Public Health 20, 3702. doi: 10.3390/ijerph20043702
Tan C. C. S., Trew J., Peacock T. P., Mok K. Y., Hart C., Lau K., et al. (2023). Genomic screening of 16 UK native bat species through conservationist networks uncovers coronaviruses with zoonotic potential. Nat. Commun. 14, 3322. doi: 10.1038/s41467-023-38717-w
Tao Y., Shi M., Chommanard C., Queen K., Zhang J., Markotter W., et al. (2017). Surveillance of bat coronaviruses in Kenya identifies relatives of human coronaviruses NL63 and 229E and their recombination history. J. Virol. 91, 10–1128. doi: 10.1128/JVI.01953-16
Tong S., Conrardy C., Ruone S., Kuzmin I. V., Guo X., Tao Y., et al. (2009). Detection of novel SARS-like and other coronaviruses in bats from Kenya. Emerg. Infect. Dis. 15, 482. doi: 10.3201/eid1503.081013
Vasenkov D., Desmet J., Popov I., Sidorchuk N. (2022). Bats can migrate farther than it was previously known: a new longest migration record by Nathusius’ pipistrelle Pipistrellus nathusii (Chiroptera: Vespertilionidae). Mammalia 86, 524–526. doi: 10.1515/mammalia-2021-0139
Vasenkov D. A., Sidorchuk N. V., Khokhryakov V. R. (2017). Sedentary” and “migrating” population of bats in the “Smolenskoye Poozerye” National Park. Plecotus 20, 54–67.
V’kovski P., Kratzel A., Steiner S., Stalder H., Thiel V. (2021). Coronavirus biology and replication: implications for SARS-CoV-2. Nat. Rev. Microbiol. 19, 155–170. doi: 10.1038/s41579-020-00468-6
Wacharapluesadee S., Duengkae P., Rodpan A., Kaewpom T., Maneeorn P., Kanchanasaka B., et al. (2015). Diversity of coronavirus in bats from Eastern Thailand Emerging viruses. Virol. J. 12, 1–7. doi: 10.1186/s12985-015-0289-1
Wang L. F., Anderson D. E. (2019). Viruses in bats and potential spillover to animals and humans. Curr. Opin. Virol. 34, 79–89. doi: 10.1016/j.coviro.2018.12.007
Wong A. C. P., Li X., Lau S. K. P., Woo P. C. Y. (2019). Global epidemiology of bat coronaviruses. Viruses 11, 174. doi: 10.3390/v11020174
Woo P. C. Y., Lau S. K. P., Lam C. S. F., Lai K. K. Y., Huang Y., Lee P., et al. (2009). Comparative analysis of complete genome sequences of three avian coronaviruses reveals a novel group 3c coronavirus. J. Virol. 83, 908–917. doi: 10.1128/JVI.01977-08
Woo P. C. Y., Lau S. K. P., Lam C. S. F., Lau C. C. Y., Tsang A. K. L., Lau J. H. N., et al. (2012). Discovery of seven novel mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J. Virol. 86, 3995–4008. doi: 10.1128/JVI.06540-11
Wood D. E., Lu J., Langmead B. (2019). Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 1–13. doi: 10.1186/s13059-019-1891-0
Keywords: alphacoronaviruses, bats, European Russia, coronaviruses, high-throughput sequencing
Citation: Korneenko EV, Samoilov AE, Chudinov IK, Butenko IO, Sonets IV, Artyushin IV, Yusefovich AP, Kruskop SV, Sinitsyn SO, Klyuchnikova EO, Gladkikh AS, Dedkov VG, Safonova MV, Daszak P and Speranskaya AS (2024) Alphacoronaviruses from bats captured in European Russia in 2015 and 2021 are closely related to those of Northern Europe. Front. Ecol. Evol. 12:1324605. doi: 10.3389/fevo.2024.1324605
Received: 19 October 2023; Accepted: 18 June 2024;
Published: 09 July 2024.
Edited by:
Xi Huang, Beijing Normal University, ChinaReviewed by:
Beza Ramasindrazana, Institut Pasteur de Madagascar, MadagascarIgor Popov, Maastricht University, Netherlands
Copyright © 2024 Korneenko, Samoilov, Chudinov, Butenko, Sonets, Artyushin, Yusefovich, Kruskop, Sinitsyn, Klyuchnikova, Gladkikh, Dedkov, Safonova, Daszak and Speranskaya. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elena V. Korneenko, bGVuYWtvcm5lZW5rbzBAZ21haWwuY29t