 Jason M. Wood1†
Jason M. Wood1† Camilla Urbaniak1
Camilla Urbaniak1 Ceth Parker1†
Ceth Parker1† Nitin Kumar Singh1†
Nitin Kumar Singh1† Season Wong2
Season Wong2 Arunkumar Arumugam2
Arunkumar Arumugam2 Dana J. Skorupa3,4Ashlyn Hemmah3Phoebe Laaguiby5
Dana J. Skorupa3,4Ashlyn Hemmah3Phoebe Laaguiby5 Fathi Karouia6,7,8†
Fathi Karouia6,7,8† Brent M. Peyton3,4,9†
Brent M. Peyton3,4,9† Scott Tighe5*†
Scott Tighe5*† Kasthuri Venkateswaran1*†
Kasthuri Venkateswaran1*†- 1Jet Propulsion Laboratory, Biotechnology and Planetary Protection Group, California Institute of Technology, Pasadena, CA, United States
- 2AI Biosciences, Inc., College Station, TX, United States
- 3Center for Biofilm Engineering, Montana State University, Bozeman, MT, United States
- 4Department of Chemical and Biological Engineering, Montana State University, Bozeman, MT, United States
- 5Advanced Genomics Lab, University of Vermont Cancer Center, Burlington, VT, United States
- 6Exobiology Branch, NASA Ames Research Center, Moffett Field, CA, United States
- 7Department of Pharmaceutical Chemistry, University of California San Francisco, San Francisco, CA, United States
- 8Space Research Within Reach, San Francisco, CA, United States
- 9Thermal Biology Institute, Montana State University, Bozeman, MT, United States
Microbial diversity estimation involves extracting nucleic acids from intricate sample matrices. Preparing nucleic acid samples is time-consuming, necessitating effective cell lysis and obtaining pure, inhibitor-free nucleic acid purifications before further use. An automated system offers advantages for field deployment due to its ease of use and quick autonomous results. This is especially beneficial for rapid measurement of in situ microbial diversity in remote areas. Our study aimed to assess microbial diversity of Yellowstone hot springs using a field-deployable lab in a resource-limited remote setting and demonstrate on-site nucleic acid sample processing and sequencing. We collected microbial mat and sediment samples from several Yellowstone National Park hot springs, focusing on the Five Sister Springs (FSS), spring LNN010, and Octopus Spring (OS). The samples were processed for DNA extraction on-site and further sequenced in the lab for microbial diversity. In addition, DNA extracted from one sample was sequenced and analyzed on-site as proof-of-concept. Using either Illumina or Oxford Nanopore Technology sequencing, we found similar microbial diversities. Bacteria (over 90%) were predominant at the FSS and OS sites, with archaea accounting for less than 10%. Metagenomic results were taxonomically categorized based on the closest known organism with a sequenced genome. The dominant archaeal community member was Candidatus Caldiarchaeum subterraneum, and among bacteria, Roseiflexus sp. RS-1 was abundant in mat samples. Interestingly, Bacterium HR17 was also frequently found, suggesting the need for more research on this newly recognized bacterial community member. The presence of Bacterium HR17 in these hot springs suggests its potential role in nitrogen cycling, informing both ecological understanding and industrial potential. This pioneering study assessed the microbiome of Yellowstone hot springs in about 8-9 hours using an automated system for nucleic acid extraction. By its deployment, the system’s value in elucidating the microbial diversity of extreme environments without the need to bring samples to the lab for processing had been highlighted. Sample processing and sequencing had been included in the benefits of the field-deployable lab, and the Nanopore platform had been utilized.
Introduction
Monitoring microbial activity is vital for probing extreme Earth environments. At present, in situ microbial testing systems face challenges, including size, mass, power constraints, and lack of portability. Although molecular detection systems that have overcome these constraints such as Oxford Nanopore Technology (ONT) sequencers are at hand, a primary hurdle is laborious nucleic acid (NA) sample preparation, centering on DNA/RNA isolation and purification. This step is crucial for precise microbial community analysis using portable sequencers in extreme environments.
Bio-detection via DNA/RNA analysis hinges on successful NA extraction from intricate sample matrices, irrespective of laboratory or field detection (Daar et al., 2002; Tang et al., 2005; Chin et al., 2007; Dundas et al., 2008; Yager et al., 2008). These upstream steps necessitate effective cell lysis, NA recovery, and purification from potential contaminants. Manual DNA extraction kits, while available, are prone to contamination, inconsistencies, and user error (Kwok and Higuchi, 1989; Jungkind, 2001). To address this, we developed a compact prototype, the µTitan, showcasing its prowess in swift, medium-throughput automated sample processing. The µTitan’s architecture, detailed earlier (Urbaniak et al., 2020), features an enclosed NA extraction cartridge, ensuring no direct sample contact. This cartridge facilitates magnetic particle-based DNA/RNA extraction but retains the adaptability to various extraction chemistries.
The nucleic acid extraction efficiency of the µTitan system had been previously compared with a commercial bench-top automated DNA extraction system (Promega Maxwell-16) and a manual extraction procedure using the QIAamp DNA Micro Kit on a whole cell microbial reference comprised of 9 bacterial strains (Urbaniak et al., 2020). Following such lab-based validation analysis yielding superior recovery, the µTitan was field-tested at Yellowstone National Park (YNP) during this study. Commercially available automated NA extraction systems falter in field deployment due to size, power demands, and reagent stability issues. In contrast, µTitan is battery powered, portable, and operated efficiently in YNP’s challenging hot spring settings.
These hot springs, notably within YNP’s lower geyser basin, have been extensively studied after bringing the samples to the lab and processed (Brock and Brock, 1968; Klatt et al., 2011; Becraft et al., 2020), but sample processing on site and subsequent sequencing has not been attempted. The consensus has been that microbial diversity varies between samples extracted for DNA on-site and those transported to a lab for subsequent processing. Factors such as storage conditions, temperature, and transportation methods were cited as reasons for differential estimation of microbial diversity. Immediate on-site processing was thought to offer a truer depiction of the microbial community structure than processing after storage and transportation, as previously reported (Holzhausen et al., 2021). Consequently, the primary emphasis of this study was placed on extracting DNA from environmental samples on site and subsequent measurement of the microbial diversity of the YNP hot springs.
This study aimed to assess the microbial diversity of YNP hot springs using a portable lab designed for use in remote areas with scarce resources, with an emphasis on in-field DNA extraction. To demonstrate its practicality, ONT sequencing was performed in the field on a single sample. In addition, a more comprehensive sequencing of all kinds of samples (n= 48; including those high and low biomass) was conducted in a laboratory setting, employing both ONT and Illumina sequencing for a thorough data comparison; such a comparison was not carried out in the past. We incorporated enzymatic pretreatment with Metapolyzyme into the µTitan system since these cell wall degrading enzymes were reported to yield better DNA from hard to lyse samples such as urine (Zhang et al., 2022). Subsequent to this pretreatment, shotgun sequencing was performed utilizing both ONT and Illumina platforms. The comparable sequencing performance of these methods affirms the potential of Nanopore sequencing for field-based biological research.
Materials and methods
Site description
All field experiments were conducted in YNP at alkaline geothermal features in the White Creek thermal area located in Lower Geyser Basin (Figure 1) on June 18, 2019. The White Creek thermal area was selected for this study because of the variety of source pools with chemosynthetic zones above the upper temperature limit for photosynthesis as well as several robust outflow channels with prominent photosynthetic zones containing microbial mats. To ensure the safety of on-site personnel, sampling locations away from known thin-crust zones were initially identified by experienced personnel, and sample collection was performed at a safe distance from the edge. Spring water, microbial mat, and sediment were collected from the Five Sisters Springs (44.5325°N, 110.7971°W), spring LNN010 (44.5325°N, 110.7974°W), Octopus Spring (44.5340°N, 110.7978°W), and effluent channels from these features (see Table 1 for a list of sampling locations).

Figure 1 Locations of the Yellowstone National Park hot spring samples collected.
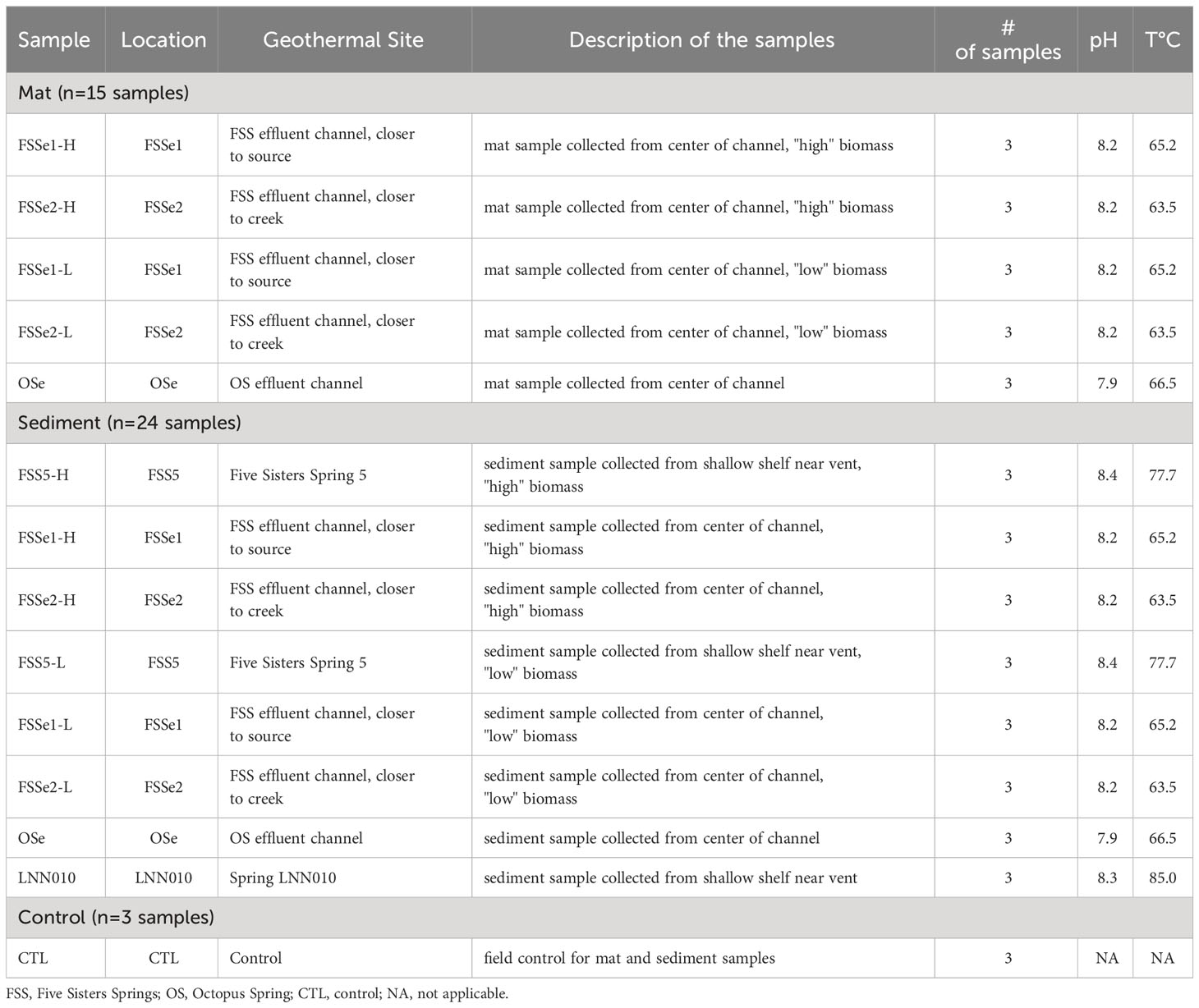
Table 1 Yellowstone National Park hot spring samples collected during this study.
Measurement of environmental conditions
To document environmental conditions, measurements of water temperature and aqueous chemistry were either measured in situ with portable equipment or preserved on-site and analyzed back at the lab. The temperature and pH of each site was measured in situ using a HQ30d combined pH-temperature probe (HACH, Loveland, CO, USA), with dissolved concentrations of oxygen also measured in the field using either a high or low-range dissolved oxygen method and a portable colorimeter (Hach DR900, Hach Co., Loveland, CO). Aqueous geochemistry was assessed for each hot spring by filter (0.22 µm) sterilizing site water (5 mL) directly into sterile 15 mL conical tubes. Some filtered site water was acidified in the field with 5% trace metal grade nitric acid prior to transport and used for total dissolved metals analysis. Concentrations of total metals were measured using an Agilent 7500ce ICP-MS by comparing to certified standards (Environmental Calibration Standard 5183-4688, Agilent Technologies, Santa Clara, CA, USA) at the Environmental and Biofilm Mass Spectrometry Facility at Montana State University - Bozeman. Ion chromatography was used to determine concentrations of dominant anions. For this, non-acidified filtered samples were analyzed using a Dionex ICS-1100 chromatography System (Dionex Corp., Sunnyvale, CA, USA) equipped with a 25 μL injection loop and an AS22-4x250 mm anion exchange column, using an eluent concentration of 4.5 mmol·L-1 sodium carbonate and 1.4 mmol·L-1 sodium bicarbonate flowing at a rate of 1.2 mL·min-1. Filtered samples collected in closed headspace scintillation vials were used for total carbon, total nitrogen, dissolved inorganic carbon, and non-purgeable organic carbon measurements with a Shimadzu total organic carbon analyzer (TOC-VCSH, Shimadzu, Japan) coupled to a chemiluminescence detector (TNM-1 total nitrogen unit) in the Environmental Analytical Laboratory at Montana State University. An overview of the aqueous geochemistry is given in Supplementary Table S1; the following dissolved constituents were not detected: Be, Mg, Al, V, Cr, Mn, Fe, Co, Ni, Cu, Se, Ag, Cd, Ba, U, and Bi (lowest detection limit ranges from 0.5-10.0 µg L-1).
Sample collection
Microbial mat (n=15) and sediment samples (n = 24) were aseptically collected using a small autoclaved metal scoop. Microbial mat and sediment samples collected from Five Sisters Springs, spring LNN010, and Octopus Spring (Figure 1) were taken from sites determined to be safe within reach (10-60 cm) of the shore of the spring. Detailed descriptions of the temperature and pH of each sampling location are given in Table 1. In total, 42 samples were collected, including 3 sampling device controls for all of the assays. Samples collected from effluent channels of these springs were taken from the approximate center of the flow path of water. The top approximately 1 cm of sediment or microbial mat was collected from water depths ranging from 2-10 cm. Small metal spatulas used to collect these samples were rinsed with spring water between replicate samplings. Collected samples were placed into a 50 mL Falcon tube (Thermo Fisher Scientific, Waltham, MA, USA) until further processing. Spatulas similar to those used to collect mat or sediment samples that were left exposed to the environment but were otherwise unused were washed with sterile phosphate buffered saline (PBS) solution and resulting microbial communities were referred to as “kitome” and served as the field control in subsequent analyses.
Triplicate mat samples were collected from several high biomass and low biomass hot spring locations, and primarily focused on the Five Sisters Spring (FSSe1 and FSSe2) and Octopus Spring (OSe). In total, 15 mat samples were analyzed and in addition, three “kitome” controls were included. Sediment samples in triplicate from high biomass (n=9) and low biomass (n=9) of three locations from the Five Sisters Spring (FSS5, FSSe1, and FSSe2) and one location from the Octopus Spring (OSe; n=3) were collected. Sediment samples collected from three Five Sisters Spring locations (FSS5, FSSe1, and FSSe2) were divided into high biomass (H) and low biomass (L) categories based on visual inspection. The temperature of sediment samples of FSS5 was hot (77.7°C; but above 72°C, the approximate upper limit for photosynthesis at this pH) (Allewalt et al., 2006), though not much warmer than FSSe1 (65.2°C) and FSSe2 (63.5°C) locations. Sediments from Five Sisters Spring 5 was designated as FSS5-H and FSS5-L; from the same site as where FSSe1 mat sample collected was named FSSe1-H and FSSe1-L; and from the same site as where FSSe2 mat sample was collected was called as FSSe2-H and FSSe2-L (Figure 1). In addition, sediment samples from Octopus Spring (OSe; 66.5°C; n=3) and the hottest area spring sampled, LNN010 (85°C), were also collected in triplicate (LNN010; n=3). Furthermore, three field controls were considered here. In total there were 24 sediment samples analyzed (Table 1).
Mat samples collected from two of the Five Sisters Spring locations (FSSe1 and FSSe2) were divided into high biomass (H) and low biomass (L) categories based on visual inspection to ensure that high biomass did not overwhelm the chemistry used by the µTitan. As shown in Figure 1, FSSe1 was just after the Five Sisters Spring 5 water run-off whereas FSSe2 was situated closer to the creek. Importantly, the temperature profiles of FSSe1 (65.2°C) and FSSe2 (63.5°C) locations were similar. Of note, the µTitan was able to measure microbial diversity in both high and low biomass samples, with only minor differences in composition detected.
Sample processing
The sample preparation process involved adding 1.2 mL of sterile PBS to each microbial mat and sediment sample. These samples were then individually transferred into 2 mL Matrix Lysing E bead tubes (MP Biomedicals, located in Irvine, California, USA). The tubes were subjected to mechanical disruption using a battery-operated oscillating power tool, specifically the JobPLUS ONE 18V multi-tool, which was equipped with a P246 console and a P570 head attachment from Ryobi (Anderson, South Carolina, USA). To secure each tube during the process, blue painter’s tape (ScotchBlue brand from 3M) was used to fasten the tube firmly to the flat end of the tool’s blade. The device was then operated at its maximum power setting, delivering 20,000 oscillations per minute for a duration of one minute, to effectively lyse the samples. 25 µL of 10 µg µL-1 MetaPolyzyme (MAC4L, Millipore Sigma, St. Louis, MO, USA) was added to each tube and incubated for 1 hr at 35°C, followed by another round of bead beating for 15 s. Tubes were then left to settle for 15 min, before aliquoting 100 µL of the supernatant into the µTitan system for DNA extraction.
µTitan DNA extraction
The µTitan system was operated in a vehicle in the parking lot using a laptop computer equipped with open-source software (Repetier-Hot-World GmbH & Co., Willich, Germany) that uses G-code based programing. Full operations of the µTitan system have been previously described (Urbaniak et al., 2020).
A commercially available magnetic particle–based nucleic acid isolation kit (NucliSENS Magnetic Particle Extraction Kit, bioMerieux, Durham, NC, USA) was purchased, and the reagents from this kit were used to pre-fill the µTitan cartridges. These reagents included lysis buffer, magnetic particle solution, wash buffer #1, wash buffer #2, wash buffer #3, and an elution buffer. The µTitan extraction protocol (e.g., volume, time of incubation, and number of repeated washing steps) is as follows: 100 µL of the pre-processed sample was added to the 2nd well of a µTitan cartridge containing 400 µL of lysis buffer and incubated for 10 min. 8 µL of NucliSENS magnetic particles, for capturing NAs, were then added to the lysed sample solution in the 2nd well. The magnetic particles were intermittently mixed for 10 min, allowing for NA to bind to the magnetic particles. The cartridge was then secured on the µTitan system. An extraction tip mounted on the follower magnet set was placed on the 1st well of the cartridge. Wells # 3–7 contained wash buffer # 1 (600 µL and 250 µL), wash buffer # 2 (600 µL and 250 µL), and wash buffer # 3 (250 µL). Well # 8 contained Tris elution buffer (60 µL). Each pre-processed sample was run in triplicate. To ensure the quality of the work and to check for contamination, molecular-grade water was used for extraction during each run of the machine instead of a sample, and is considered the µTitan instrument control.
Sequencing
Field sequencing was performed on a single sample of microbial mat collected from the effluent channel of Five Sisters Spring (FSSe2). DNA was immediately extracted in the field using the μTitan automated field sample extraction system (Urbaniak et al., 2020) and quantified using a Qubit spectrofluorometer using DNA high sensitivity reagents (Q32851, Thermo Fisher Scientific, Waltham, MA, USA). The concentration of recovered DNA was 0.5 ng µL-1 and a total of 8 µL (4 ng) was used for the Rapid Sequencing Kit (SQK-RAD004, Oxford Nanopore Technologies, Oxford, United Kingdom). While this is much lower than the recommended 200-400 ng input, the expectation was that fewer sequences would be generated. Library synthesis was carried out in a parking area at Yellowstone National Park that we termed Mobile Research Sequence Laboratory (MRSL; Figure 1) and a portable heat block was used for the two incubation steps of 35°C and 80°C. Sequencing was conducted using a standalone version of the MinKNOW software specially arranged for NASA field sequencing with the ONT MinION MK1B (Urbaniak et al., 2020). After flow cell QC was complete, the RAD004 library was added to the R9.4 flow cell and sequencing was initiated. Sequencing was performed in the vehicle while driving back to Bozeman, Montana with intermediate data analysis performed via a cell phone hotspot connection and the One Codex online software (https://www.onecodex.com/). Sequencing was allowed to continue until no additional data was generated.
Laboratory-based Oxford Nanopore sequencing was performed using the GridION-X5 sequencer equipped with Standard flow cells (R9.4). Libraries were synthesized using the rapid PCR Barcoding kit (RPB004) with 3 µL of adjusted input volume. PCR cycles were adjusted for total input amounts as follows; 16 cycles for 1-3 ng, 18 cycles for 0.25-1 ng, and 22 cycles for 0.0-0.25 ng. Read depth varied for each sample and low reads were obtained for samples that had DNA at or below detection limits, but not for all cases. Low read depth was noted for some samples that had high inputs and may be explained by possible microbial content, biofilm interference, or other possible inhibitors such as excessive extracellular polysaccharides or organic acids.
Laboratory-based Illumina sequencing was also performed on all samples for comparison. Whole genome shotgun library synthesis was performed on 1 ng input or maximum input volume using the Nextera XT whole genome kit, cleaned using AMPure XP beads to purify the library DNA to remove short library fragments as per the Illumina protocol (Illumina Inc., San Diego, CA, USA). These libraries were further quantified using the Qubit spectrofluorometer (Thermo Fisher Scientific, Waltham, MA, USA) and analyzed for quality using the Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA). Libraries were pooled equimolar and sequenced using single-end 150 bp chemistry on a HiSeq 1500/2500 system (Illumina, San Diego, CA, USA). Samples were demultiplexed and fastq files were used for analysis.
Sequence quality control and annotation
Quality control of short-read Illumina sequence data was performed using Trimmomatic (v0.32) (Bolger et al., 2014) to remove adapter sequences and low-quality reads, using a quality cutoff value set at minimum Phred score of 20 along the length of the read. Quality control of long-read Nanopore sequence data was performed using Nanofilt (v2.6.0) (De Coster et al., 2018) and default parameters. Reads shorter than 80 bp were removed from the data. The DIAMOND aligner (v2.0.4) (Buchfink et al., 2015) was used to query the NCBI-NR protein database, and the MEGAN 6 (Huson et al., 2016) lowest common ancestor (LCA) algorithm was used to bin high quality reads to their respective taxonomy. Additionally, the eggnog, KEGG, and SEED databases were queried for functional analysis (results not shown). For long-read Nanopore sequence annotation, the DIAMOND frameshift and range curling mode was used. The frameshift option performs frameshift-alignment of DNA sequences against a protein reference database. The parameter used with the frameshift mode was -F 15, leading to a dynamic programming penalty of 15. Range-culling estimates an alignment locally; this option was used with the parameter as follows –range-culling and –top 10.
A read threshold was set to understand the abundance of microbial diversity in these samples. For ONT long reads, a minimum of 10 reads was considered and for Illumina, >500 reads was considered for the microbial abundance calculations. When computed, >99.9% of total reads were accounted for with these thresholds. The construction of metagenomic trees for dominant species was facilitated by the ‘Tree’ function in the MEGAN6 software suite (Huson et al., 2016). This tool permitted an in-depth examination and depiction of genus-level information extracted from metagenomic samples.
Statistical analyses
To determine whether long-read ONT sequencing could supplement or replace short-read Illumina sequencing for more rapid analyses in remote locations, multiple statistical analyses were performed. Analysis of Similarities (ANOSIM) using the R vegan package (Oksanen et al., 2013) was used to compare similarities in community composition among similar samples (e.g., mat samples, sediment samples) as sequenced using either the Illumina or Oxford Nanopore platforms. Alpha diversity among similar samples was measured with Chao1, Shannon, and Simpson indices as calculated by the R vegan package (Oksanen et al., 2013) and a custom R script (https://github.com/sandain/R/blob/main/diversity.R) that performed rarefaction based on the size of the smallest library. The R vegan package (Oksanen et al., 2013) was also used to visualize beta diversity using nonmetric multidimensional scaling (NMDS) of Bray-Curtis dissimilarity.
Results
Validation of the µTitan instrument
The µTitan system was effectively used for DNA extraction, and its performance was found to be comparable to or even better than a similar commercially available laboratory-based automated nucleic acid extraction device (Urbaniak et al., 2020). In a challenging environment like YNP and on a moving vehicle at speeds up to 85 miles per hour, the µTitan system was tested with the whole cell microbial reference community to simulate remote settings with limited resources. Under these controlled conditions, the performance of the µTitan was found to be consistent with its operation in a standard laboratory setting (Urbaniak et al., 2020). It was highlighted that the instrument had the capability to process samples in remote settings, delivering near real-time information from environmental and human samples (Chan et al., 2016a). This study confirmed the effectiveness of the µTitan system for DNA isolation, and it was previously reported that the device had also been validated for successful RNA extraction (Chan et al., 2016c). The versatility of the µTitan instrument was acknowledged, allowing for analyses of microbiomes, metagenomes, transcriptomes, and viromes in remote environments (Chan et al., 2016b). This is the first time the µTitan system has been deployed at YNP to process samples on-site and extract DNA instead of transporting samples to the lab for processing.
Chemistry
Vent fluid temperatures ranged between 68°C to 85°C, with the effluent channels of Octopus Spring and Five Sisters Springs measuring between 62°C to 64°C (Supplementary Table S1). Aqueous chemistry was measured from water samples taken either directly from the vent fluid or above the photosynthetic mats. The White Creek thermal area is generally categorized as a water-dominated alkaline-chloride system (Fournier, 1989), which is fed from subsurface boiling in deep-heated reservoirs. Collectively, the sampling results all feature high chloride (309 ± 10 mg L-1), high arsenic (716 ± 29 µg L-1) and low sulfate (18 ± 1 mg L-1) concentrations (Supplementary Table S1). In addition, the sampled springs are alkaline (pH 7.99–8.85) and contain high concentrations of sodium (140 ± 6 mg L-1 Na+), which is typical of the sodium and chloride-rich waters that form alkaline-chloride hot spring fluids.
Microbial diversity
Read counts for each detected species were summed for replicate samples collected from the same hot spring location before relative abundances were calculated. In general, >3 kbp long reads were yielded by Nanopore data, and 150 bp reads were yielded by Illumina (Supplementary Table S2). Although microbial diversity was measured equally by both the Illumina and ONT sequencing platforms, the microbial profile composition was detailed based on ONT results. However, the microbial diversity profile based on Illumina was also provided in several Supplementary Files.
Phylogenetic trees representing the predominant archaeal and bacterial species of mat samples were constructed, allowing for a detailed analysis and graphical representation of species-specific data obtained, as illustrated in Supplementary Figure S1. Similarly, the construction of phylogenetic trees for key archaeal and bacterial species facilitated a visual display of data pertinent to these species, drawn from sediment samples, as depicted in Supplementary Figure S2.
Archaeal diversity of mat samples
Differential archaeal diversity of the microbial mats was assayed using the Illumina (Supplementary Table S3) and ONT sequencing platforms (Supplementary Table S4; Figure 2, Table 2). Approximately 6.7x104 of the Illumina short reads and 2x103 of the ONT long reads were identified as Archaea after high quality trimming.
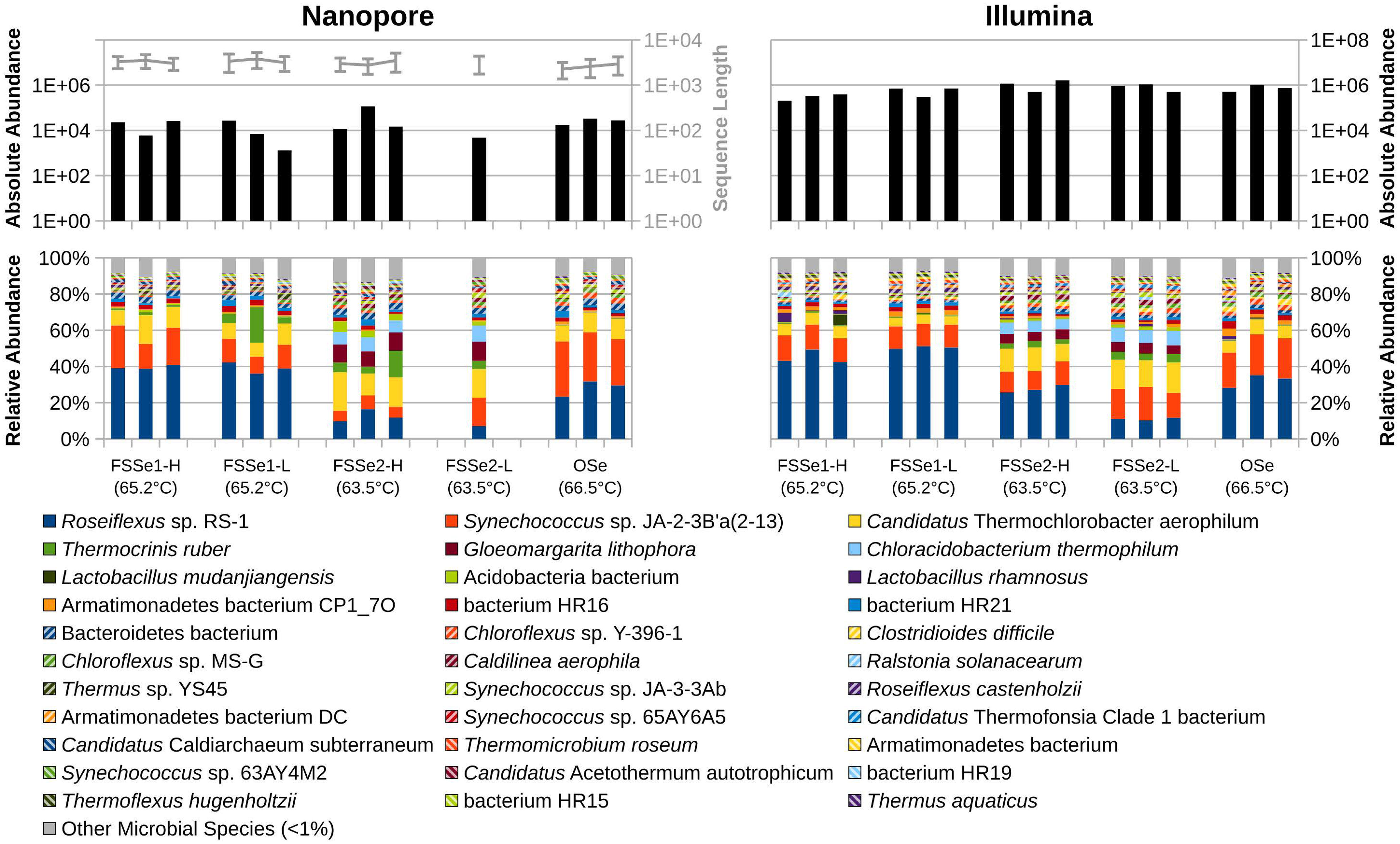
Figure 2 Absolute abundance of sequences sampled (top) and relative abundance of bacterial and archaeal diversity (bottom) of YNP hot spring mat samples as sampled by Oxford Nanopore (left) and Illumina (right) sequencing. Sequence lengths for Nanopore data represented in light-gray (top-left).
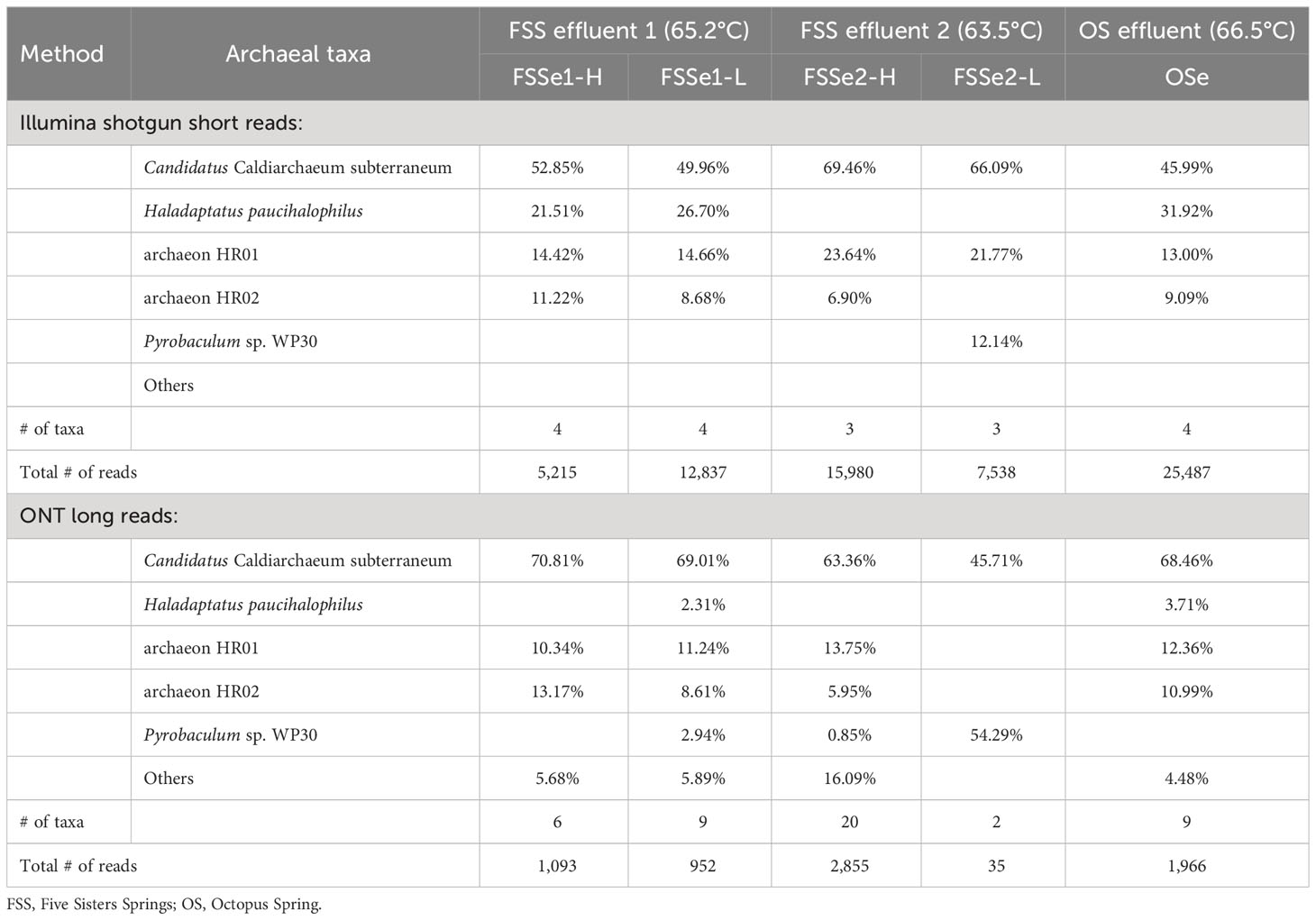
Table 2 Dominant archaeal sequences retrieved from YNP Springs mat samples.
In both the low-biomass and high-biomass microbial mat samples Candidatus Caldiarchaeum subterraneum was overwhelmingly present. It is interesting to note that a close relative of Haladaptatus paucihalophilus (a halophilic archaeon) was detected at high relative abundance in FSSe1-H, FSSe1-L, and OSe, but not in FSSe2, by both the Illumina short read and ONT long read platforms. Whereas Pyrobaculum sp. WP30 (~12%) was found in high numbers at FSSe2-L mat samples. The community composition of the Octopus Spring mat resembled that of FSSe1 with respect to archaeal abundance and taxa profile.
The ONT long reads of Five Sisters Spring high biomass mat samples exhibited six archaeal taxa from FSSe1 location (FSSe1-H) that were closer to the hot water run off (81.3°C, FSS3 and 78.5°C, FSS1), whereas a high number of Archaea (20 taxa) were observed in FSSe2 high biomass (FSSe2-H) samples that were collected from the cooler geothermal outflow channel (63.5°C; FSSe2). When the low biomass samples were analyzed, FSSe1 location (FSSe1-L) had 9 archaeal taxa whereas in FSSe2 location (FSSe2-L) samples only 2 archaea were retrieved. In FSSe1-H and FSSe2-H samples, the archaeal diversity was similar, with 64% to 71% of the archaeal diversity comprised of Candidatus C. subterraneum. It is also interesting to note that the archaeal taxa detected in the FSSe1 mats were all present at >1% abundance, while in the FSSe2-H mat, only 8 of the 20 archaeal taxa displayed a >1% abundance. The two archaeal taxa retrieved from the FSSe2-L mat samples belonged to Candidatus C. subterraneum (~46%) or Pyrobaculum sp. WP30 (~54%). The archaeal diversity profile of the Octopus Spring mat samples resembled the FSSe1-H and FSSe2-H archaeal diversity which included Candidatus C. subterraneum (~68%), archaeon HR02 (~12%), and HR01 (~11%) sequences constituting >91% of total archaeal abundance.
Bacterial diversity of mat samples
Differential bacterial diversity of the microbial mats (Figure 2, Table 3) was assayed using the Illumina (Supplementary Table S5) and ONT sequencing platforms (Supplementary Table S6). The ONT long reads of Five Sisters Spring high biomass mat samples exhibited 89 bacterial taxa from FSSe1 location (FSSe1-H) that were closer to the hot water run off (81.3°C, FSS3 and 78.5°C, FSS1), whereas high number of bacteria (187 taxa) were observed in FSSe2 high biomass (FSSe2-H) samples that were collected from the relatively warm area (63.5°C; FSSe2) (Table 3; Supplementary Table S6). When low biomass samples were analyzed, FSSe1 location (FSSe1-L) had 74 bacterial taxa whereas in FSSe2-L samples only 38 bacterial taxa were retrieved. Even though the bacterial diversity of FSSe1 mat samples resembles that of FSSe2 mat samples, their proportions were different. Similar to the Illumina dataset, Roseiflexus sp. RS-1 was most abundant in all four mat samples analyzed via ONT, but the abundance levels of this community member varied, ranging between 5-40%. As well, lower abundance levels of T. ruber was detected in FSSe1-H (1%) mat samples when compared to FSSe1-L (8%), FSSe2-H (5%), and FSSe2-L (~34%). The two bacterial taxa retrieved from both FSSe1 mat samples belonged to Synechococcus sp. JA-2-3B’a(2-13) (22% in FSSe1-H and 13% in FSSe1-L) or Candidatus T. aerophilum (11% in FSSe1-H and 9% in FSSe1-L samples). Similar incidences of these bacterial taxa at about 10% or more was noticed in FSSe2 mat samples as well. The bacterial diversity profile of the Octopus Spring mat samples resembled the FSSe1-H and FSSe2-H bacterial diversity which included Roseiflexus sp. RS-1 (~30%), Synechococcus sp. JA-2-3B’a(2-13) (~29%), and Candidatus T. aerophilum (~11%) sequences constituting ~70% of total bacterial abundance. Even though 107 bacterial taxa were found from Octopus Spring mat samples, only 12 taxa exhibited >1% of the total bacterial reads.
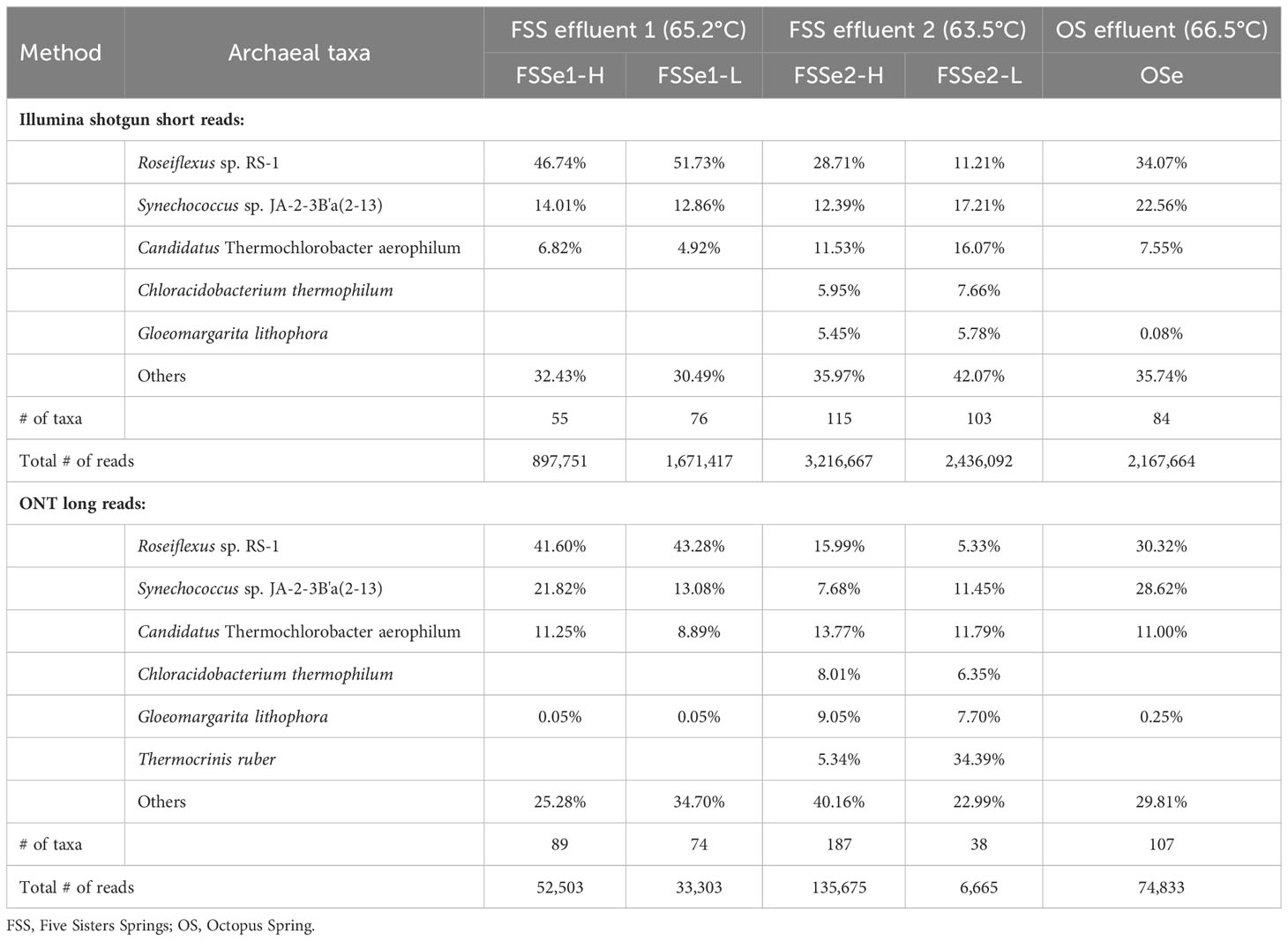
Table 3 Dominant bacterial sequences retrieved from YNP Springs mat samples.
Archaeal diversity of sediment samples
Differential archaeal diversity of sediment samples assayed using Illumina (Supplementary Table S7) and ONT sequencing (Supplementary Table S8; Figure 3, Table 4) are presented. Approximately 1.2x106 Illumina short reads and 5.6x104 ONT long reads associated with archaea after high quality trimming from the sediment samples were retrieved.
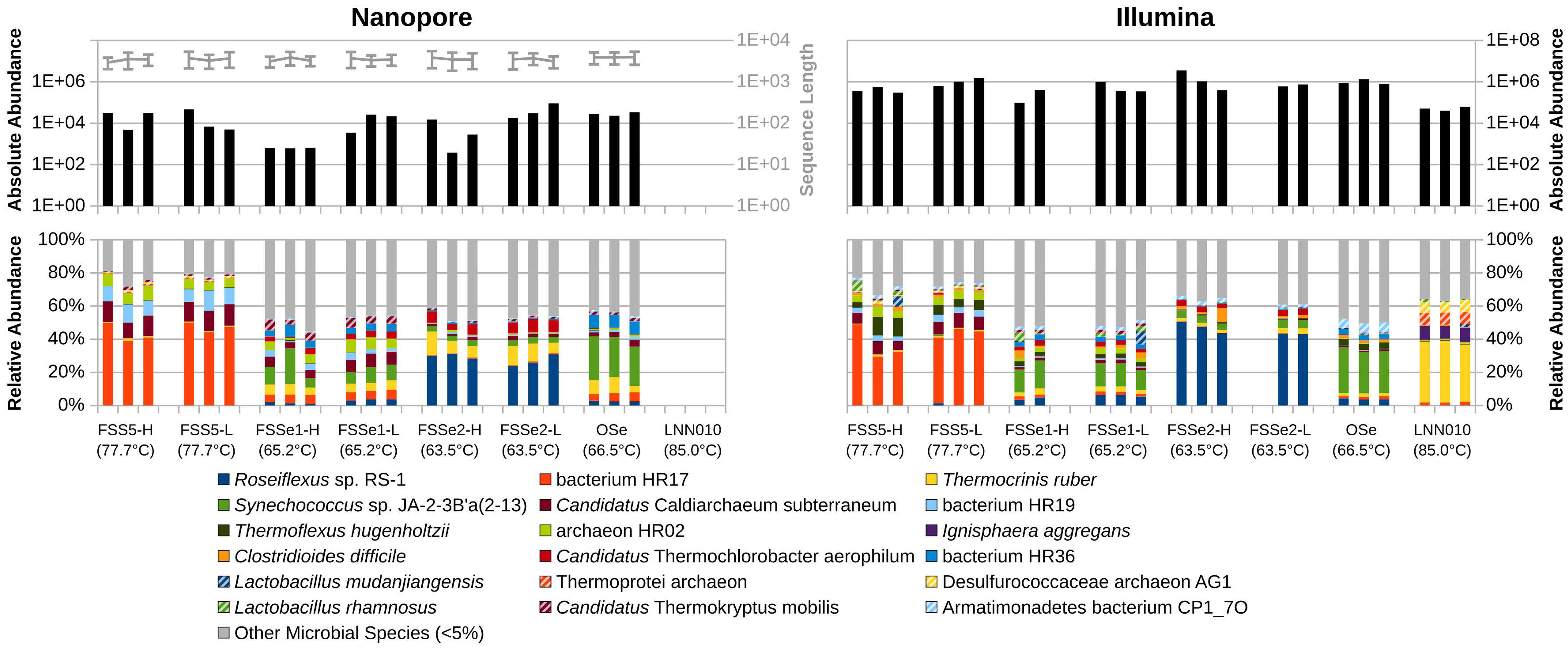
Figure 3 Absolute abundance of sequences sampled (top) and relative abundance of bacterial and archaeal diversity (bottom) of YNP hot spring sediment samples as sampled by Oxford Nanopore (left) and Illumina (right) sequencing. Sequence lengths for Nanopore data represented in light-gray (top-left).
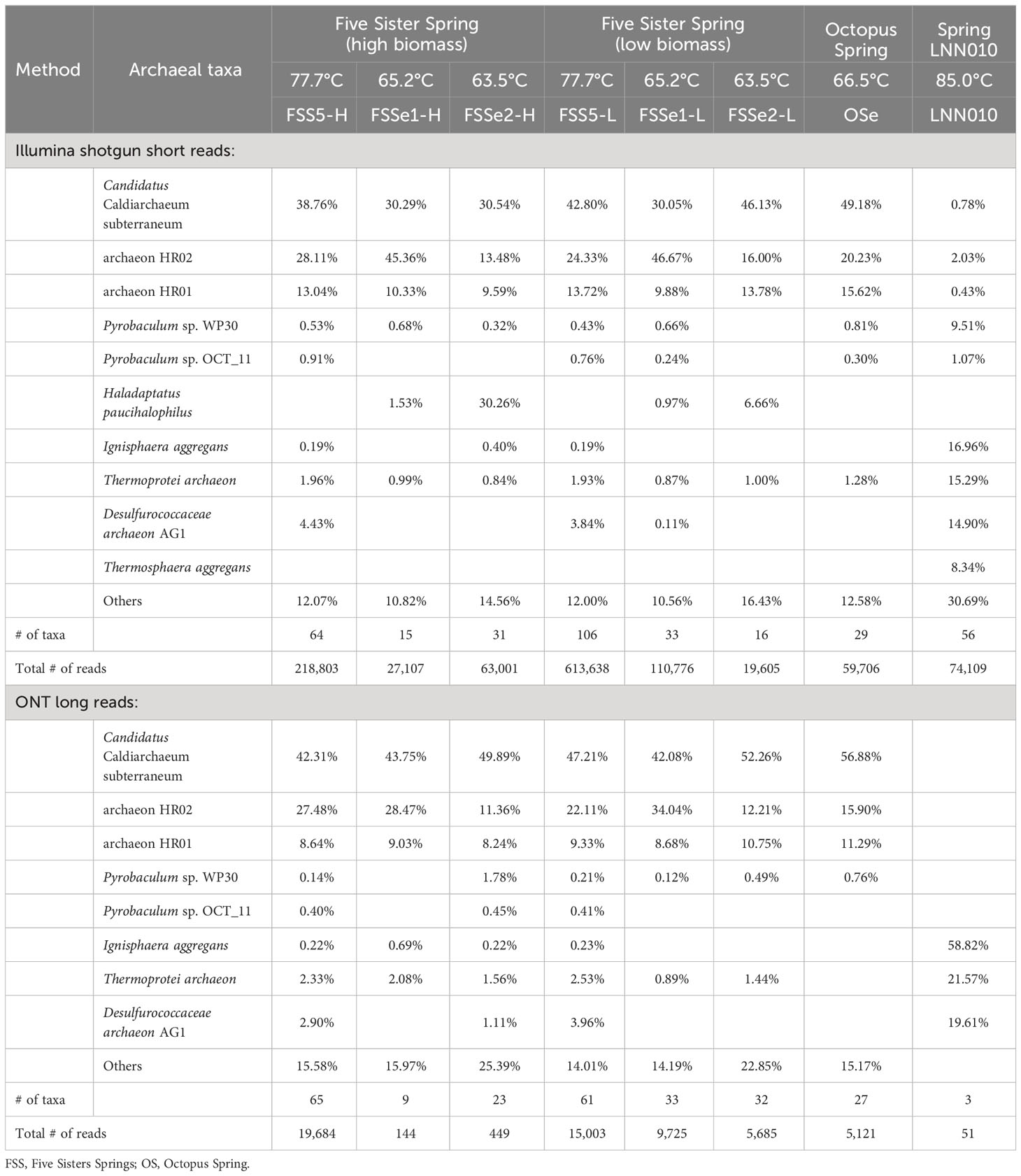
Table 4 Dominant archaeal sequences retrieved from YNP Springs sediment samples.
ONT long reads of Five Sisters Spring high biomass mat samples exhibited 65 archaeal taxa from the FSS5 location (FSS5-H) that were closer to the hot water run off (81.3°C, FSS3 and 78.5°C, FSS1), whereas only 9 and 23 taxa were observed in FSSe1 and FSSe2 high biomass sediment samples that were collected from the relatively warm area (65-63°C). In high biomass sediment (FSS5, FSSe1, and FSSe2) samples, the archaeal diversity was similar and high abundance of Candidatus C. subterraneum (42% to 70%) was noted. The second and third dominant taxa were archaeon HR02 and HR01 with ~10% of total archaeal abundance. As noticed in mat samples Pyrobaculum sp. WP30 was absent in sediment samples but this archaeon was dominant in the upstream water (~55% to 64%). The archaeal diversity profile of the Octopus Spring sediment samples resembled some Five Sister Spring results from FSS5, FSSe1, and FSSe2 archaeal diversity which included Candidatus C. subterraneum (~57%), archaeon HR02 (~15%), and HR01 (~11%) sequences; constituting >84% of total archaeal abundance. In contrast, the hottest sediments from the LNN010 possessed extremely different archaea (Ignisphaera aggregans, Thermoprotei archaeon, and Desulfurococcaceae archaeon AG1) but in total, only 51 reads of ONT long reads were retrieved.
Bacterial diversity of sediment samples
Differential bacterial diversity of sediment samples assayed using Illumina (Supplementary Table S9) and ONT sequencing (Supplementary Table S10; Figure 3, Table 5) are presented. ONT long reads of Five Sisters Spring high biomass sediment samples exhibited 96 bacterial taxa from FSS5 location (FSS5-H) that were closer to the hot water run off (81.3°C, FSS3 and 78.5°C, FSS1), whereas FSSe1-H and FSSe2-H sediment samples had only 6 and 23 bacterial taxa whose locations were relatively in warm area (66.5°C and 63.5°C). In contrast, when low biomass samples were analyzed, the bacterial taxa incidence was reversed where FSSe2-L (286 taxa) and FSSe1-L (142 taxa) had more compared to FSS5-L (85 taxa) sediment samples. The dominant bacteria present in these sediment samples were distinct in which FSS5-H and FSS5-L possessed Bacterium HR17 (>66%) and HR19 (>12%) in high numbers but FSSe1 (3% to 7%) and FSSe2 (<1%) sediment samples had much lower abundance. On the contrary, FSSe1 sediments harbored a high number of Synechococcus sp. JA-2-3B’a(2-13) (12% to 23%) and in FSS5 sediments Roseiflexus sp. RS-1 was abundant (31% to 32%). The sediment sample bacterial diversity was not similar to the mat diversity. Unlike mat samples, the bacterial diversity profile of the Octopus Spring sediment samples was different and had Synechococcus sp. JA-2-3B’a(2-13) (27%) in abundance followed by bacterium HR36 (8.5%), T. ruber (7.8%), and bacterium HR17 (5.1%). Even though 155 bacterial taxa were found from Octopus Spring sediments (Supplementary Table S5), only 20 taxa exhibited >1% of the total bacterial reads. The LNN010 sediments showed only 43 ONT long reads, of which only the recognizable member was T. ruber while others were mostly attributed to “kitome” contaminants.
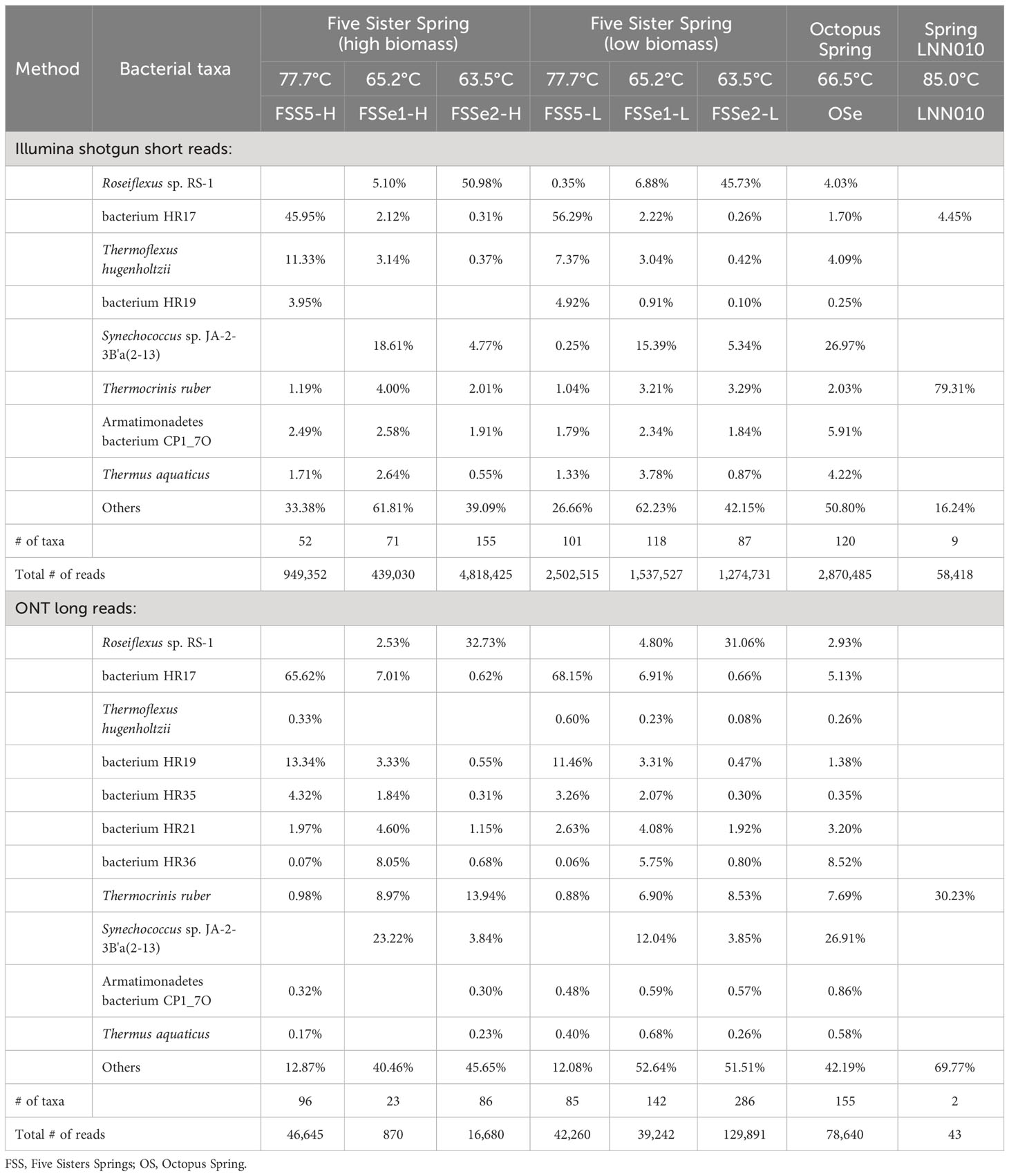
Table 5 Dominant bacterial sequences retrieved from YNP Springs sediment samples.
The three negative sampling device control samples were evaluated for exogenous DNA often referred to as “kitome” and resulted from 2.5x104 Illumina reads consisting of Stenotrophomonas (55%), Clostridioides (19%), Lacticaseibacillus (12%), Ralstonia (12%), and Clostridium sp. C105KSO13 (2%). The ONT data revealed only one bacterial taxon, Roseiflexus sp, which may be incidental contamination from the air by the sampling device (spatula) since this bacterial taxon was found in abundant quantities in most of the hot spring samples.
Illumina vs ONT methods
Relative abundance of Illumina and Oxford Nanopore sequence data was similar among samples collected from the same location with some differences. In general, both sequencing platforms retrieved sequences associated with dominant bacterial and archaeal taxa (>95% of the reads).
Analysis of similarities (ANOSIM) among similar sample types sequenced by either Illumina or ONT demonstrate a high degree of similarity in relative sequence abundance. Mat samples collected from the effluent channels of Octopus Spring and Five Sisters Springs resulted in a similar set of microbial constituents sequenced (R=0.1652; P=0.015) by these two platforms. Likewise, sediment samples collected from various sites, including Five Sisters Springs and their effluent channel, the effluent channel of Octopus Spring, and LNN010 contained a similar set of microbes (R=0.1258; P=0.007). However, NMDS analyses indicated differential microbial diversity profiles of the examined hot spring samples where mat, sediment, and control samples grouped themselves and exhibited differential profiles between the groups (Figure 4). In addition, Illumina sequence data indicated higher alpha diversity using the Chao1, Shannon, and Simpson indices for all sample types (Figure 5).
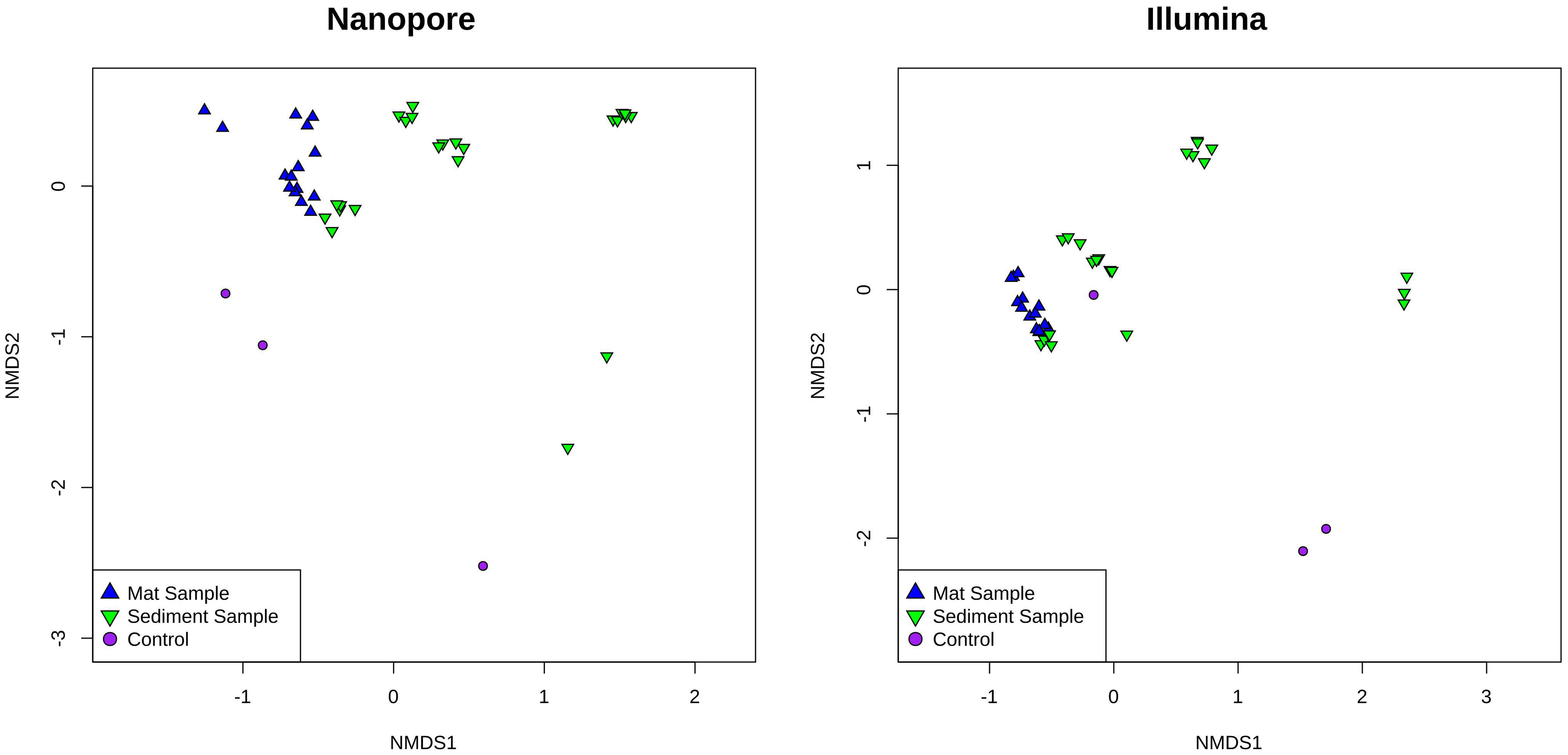
Figure 4 Nonmetric multidimensional scaling (NMDS) analysis of YNP hot spring mat and sediment samples as sampled by Oxford Nanopore (left) and Illumina (right) sequencing. Mat samples are represented as blue up-facing triangles, sediment samples are green down-facing triangles, and control samples are purple circles.
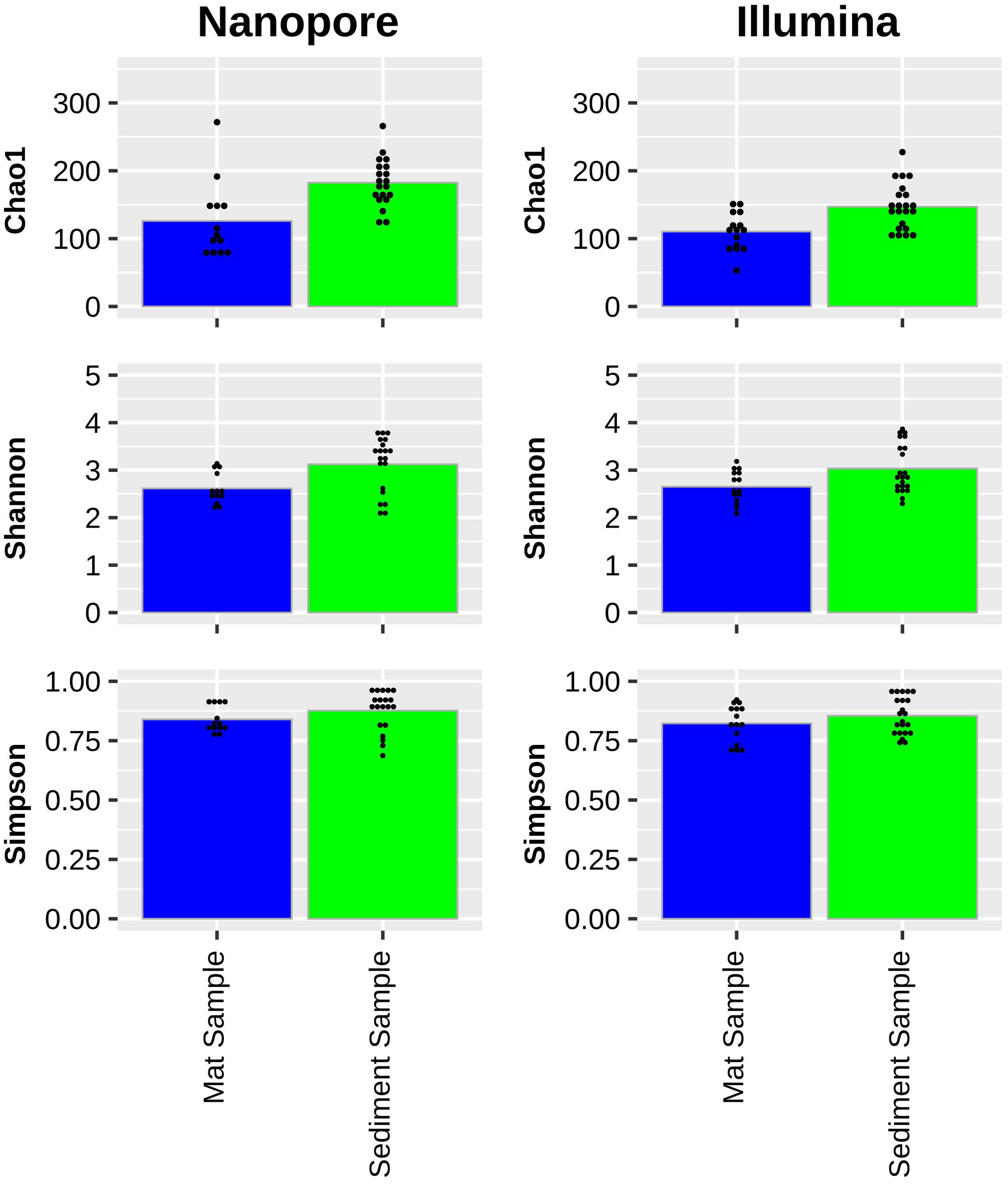
Figure 5 Chao1 (top), Shannon (middle), and Simpson (bottom) microbial diversity indices of YNP hot spring samples as sampled by Oxford Nanopore (left) and Illumina (right) sequencing. Average indices of mat samples (blue) and sediment samples (green) are depicted with small-black dots representing index values of individual samples.
Field measurement of microbial diversity using Nanopore
In order to demonstrate the portability of the µTitan platform and the ONT sequencer, a field measurement of microbial diversity of a single microbial mat sample collected from the effluent channel of Five Sisters Spring (FSSe2) was performed while on site. During transport back to Bozeman, MT, sequence data were generated using the MinION sequencer linked to a computer and connected via a cellular hot-spot. A total of 4.6 Mbp were generated from 3843 reads with an average length of 1.2 kbp. Data analysis was performed using the One Codex microbiome analysis software. These immediate data (Supplementary Table S11) found Roseiflexus, Synechococcus, Candidatus Thermochlorobacter, and Thermomicrobium, all expected of this sample.
Mobile research sequencing laboratory
These results demonstrate that it is possible to set up a mobile research sequencing laboratory (MRSL) in a remote setting to rapidly (~8-9 hrs for sample collection, processing, sequencing, and bioinformatic characterization) attain measurements of microbial diversity. This is the first study of the field deployable µTitan system for extracting nucleic acids from complex materials, including hot water, microbial mats, and sediments in a remote setting like Yellowstone National Park. Although Metapolyzyme was used to process urine samples in the past (Zhang et al., 2022), this is the first reported use of this specialized hydrolase cell wall degrading enzyme mixture, to better lyse microbial organisms for a more complete microbiome analysis from environmental samples. Importantly, we demonstrated that the µTitan system can be used with either Illumina or ONT sequencing platforms, resulting in very similar perspectives of the microbial community. The portability of the µTitan and ONT platforms open more avenues for studying microbial diversity in remote locations.
Discussion
NASA’s interest in deploying miniaturized, low-powered, portable instruments for future space missions is due to the practicality of such devices in field conditions. The µTitan system, with its low power and stability needs, enables the analysis of microbial diversity in the challenging environments of YNP’s extreme hot springs while using limited resources. Although groundbreaking for on-site microbial analysis, automated NA extraction systems for field deployment might encounter limitations. Dependence on uninterrupted power supply is problematic in isolated locations. Environmental elements like severe temperatures and moisture levels can impact the systems’ function and the integrity of samples. The complexity of these systems necessitates expert handling, while ensuring sterility to avert contamination. Contamination, which could skew results, is difficult to avoid outdoors. The systems’ restricted sample processing capacity and the requirement for detailed, often complex, subsequent data analysis may limit their use in extensive environmental assessments. Nevertheless, integrating the µTitan with advanced shotgun metagenome analysis facilitated a comprehensive evaluation of the microbial ecology within these extreme thermal environments, providing species- or strain-level resolution (Singh et al., 2018).
This is the first study using an automated nucleic acid extraction system deployed in a remote field such as YNP to rapidly measure an environmental microbiome. This workflow included various methods associated with sample collection, processing, extracting nucleic acids with, for the first time, the inclusion of the enzyme cocktail, Metapolyzyme. DNA sequencing operations were also included in the field using the MinION ONT platform, and remote data analysis performed via a cell phone hotspot. To our knowledge, this is the first study to compare shotgun microbiome data collected using Metapolyzyme assisted automated DNA extraction using both Illumina and ONT sequencing to benchmark the microbiome data results of each platform in measuring the microbial diversity of the YNP hot spring systems.
Previous studies that used only the Illumina platform have revealed that when low-biomass samples were extracted using the µTitan system, the negative control “kitomes” of the reagents were distinct from the microbial diversity of the samples (Urbaniak et al., 2020), and replicate extractions (n=3) were consistent and reproducible with little difference between field or lab performance. In this study, DNA extractions were performed in the open field surrounded by hot springs and steam clouds in the air (aerosols) during sample collection, which can cause ubiquitous contamination of sampling gear with shorter fragments of aerosolized eDNA. Unlike the previous study (Urbaniak et al., 2020), both Illumina and ONT platforms were utilized during this study. These short aerosolized eDNA (<500-bp) may not be detected by long read sequencers (ONT platform), but may be picked up by short-read sequencers (Illumina platform). This may explain the differences seen in the “kitomes” detected by the two platforms in this study. Further research into the amount of aerosolized eDNA around hot springs and differences in sequencing ability for these short fragment eDNA using long- and short-read sequencers is warranted.
The ability of Illumina and ONT to sequence small amplicons originating from clinical samples was previously performed and showed a genus-level concordance of 96.7% and 90.3% on the Illumina and ONT platforms, respectively (Stefan et al., 2022). Similarly, these two platforms were used to measure microbial diversity associated with bivalve communities, in Italy and Portugal, and it was found that agreement between sequencing methods was only 69% (Illumina detected higher diversity) and the difference between methods was non-significant (Egeter et al., 2022). In contrast, when indoor samples were assayed for 16S rRNA amplicons using Illumina and ONT platforms, there was no significant difference between the microbial compositions at the family level. However, at the genus, and particularly at the species level, the ONT MinION reported greater taxonomic resolution than Illumina MiSeq (Nygaard et al., 2020). Differences between the ONT and Illumina platforms noted in these previous studies informed the decision to sequence samples from this study of YNP hot springs using both platforms. However, as shown in Figure 5, no major differences were observed in the microbial diversity of YNP mat and sediment samples when these two platforms were used.
The waters of the FSS and OS hot springs are alkaline (~7.9-8.9 pH) in nature and high in chloride (300-340 mg L-1), fluoride (27-30 mg L-1), sulfate (14-18 mg L-1), and arsenic (676-748 µg L-1). In such an extreme environment, dominant microbes of certain microbial populations were documented based on targeted gene amplification/sequencing (Barns et al., 1994; Blank et al., 2002; Podar et al., 2020) and shotgun metagenome sequencing methods (Colman et al., 2022). Though previous metagenomic studies have been conducted at OS (Klatt et al., 2011), to date only 16S rRNA gene targeted amplicon sequencing has been used to discern the microbial community composition in the FSS hot spring system (Peach et al., 2022). To our knowledge, work here is the first to generate shotgun metagenomics sequencing from the FSS system hot spring complex.
While detection of Candidatus C. subterraneum, an uncultivated Aigarchaeota has been reported in other microbial mats from geothermal sites including water stream of a sub-surface gold mine in Japan (Takami et al., 2015), Lobios hot spring water (76°C, pH 8.2), Spain (López-López et al., 2015), Mid-Cayman Rise Vent Fluids (Zhou et al., 2020), and in a Jinze hot spring in China (GenBank accession #: DTCM00000000.1), it has not previously been reported in the hot springs studied during this study, until now. This may be attributed to the use of Metapolyzyme combined with deep shotgun sequencing. Since it is well known that this enzyme cocktail has remarkable cell wall degradation characteristics and that shotgun sequencing is far more agnostic and sensitive than 16S, it can potentially allow for more comprehensive lysis and detection, revealing entirely new microbial diversity. As previously noted, the metabolic and physiological features mined from the metagenome assembled genome of this uncultivated Aigarchaeota appeared to support aerobic conditions for growth with a heterotrophic lifestyle. Because of the reference database used here, metagenomic sequences detected from the microbial mats from the YNP system binned to Candidatus C. subterraneum in the microbial mats from the YNP system, however are likely more related to a different novel Aigarchaeota, Candidatus Calditenuis aerorheumensis previously isolated from OS (Beam et al., 2016). In sediments, Candidatus C. subterraneum was overwhelmingly present as noted in mat samples. The LNN010 sediments contained 56 archaeal taxa as observed by short read Illumina sequencing but only 3 archaea were retrieved by the ONT platform. This discrepancy could be attributed to the fact that these archaeal taxa might not be viable and the genetic sequences retrieved by the Illumina system might be fragmented hence the long read ONT platform did not retrieve them.
It is interesting that Haladaptatus paucihalophilus was detected in the effluent channels of FSS and OS as these systems do not have the necessary salt concentration (0.8-5.1M; Supplementary Table S1), pH (5-7.5; Table 1), or temperature (25-45°C; Table 1) to grow this archaeon (Savage et al., 2007). While the detection of this microorganism may simply be an artifact of inadequate reference sequences in the database used for annotation, there is the possibility that there is a closely related non-halophilic, thermophilic archaeon that has heretofore been undetected in these hot spring systems. Halophiles are known to be difficult organisms to lyse using the standard bead-beating method (Leuko et al., 2008), and as such have likely been overlooked in prior studies. Metapolyzyme used in this study creates spheroplasts quite easily, allowing for the detection of this potentially novel archaeon. More studies are warranted.
Among bacterial members, sequences similar to Roseiflexus sp. RS-1 were found to be high in FSS/OS mat samples during this study which was also reported in OS mats (Madigan et al., 2005). The members of the Chloroflexota phylum, thermophiles capable of chlorophototrophy, were the major components of photosynthetic microbial mats in sulfidic and non-sulfidic hot springs in Yellowstone National Park (Nübel et al., 2002). It was reported that Roseiflexus dominated in non-sulfidic hot springs and Chloroflexus sequences were retrieved from sulfidic springs on the basis of genetic and lipid biomarker evidence (Nübel et al., 2002; Meer et al., 2005). However, sulfate content was high (14-18 mg L-1) in FSS/OS systems where Roseiflexus dominated when compared to Chloroflexus, which begged the question of whether the FSS and OS systems were non-sulfidic or not.
Attempts to isolate Aigarchaeota and Candidatus C. subterraneum have thus far been unsuccessful (Beam et al., 2016; Malygina et al., 2023). Abundance of sequences related to Candidatus C. subterraneum in sediment samples was similar to mat samples indicating that this archaeon might accumulate and thrive in mat or sediments where they may depend on other microbial community members for growth. However, differential bacterial abundance in sediments (dominated by Bacterium HR17 instead of Roseiflexus) in some FSS/OS mat samples need more research. Originally detected in samples from a subsurface geothermal stream in Japan, Bacterium HR17 is thought to play a key role in denitrification (Kato et al., 2018). The detection of Bacterium HR17 in YNP hot springs helps to unlock the mysteries of nitrogen cycling in these extreme environments. Cultivation of Bacterium HR17 in the laboratory and/or the creation of a metagenome-assembled genome would help inform both ecological understanding and its industrial potential.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA935810.
Author contributions
JW: Investigation, Validation, Writing – original draft, Writing – review & editing, Data curation, Formal Analysis, Visualization, Conceptualization, Methodology, Supervision. CU: Investigation, Writing – original draft, Writing – review & editing, Conceptualization, Methodology. CP: Investigation, Writing – review & editing, Methodology, Visualization. NS: Investigation, Methodology, Validation, Writing – review & editing, Data curation, Formal Analysis. SW: Investigation, Methodology, Writing – review & editing, Resources, Supervision, Writing – original draft. AA: Formal Analysis, Investigation, Methodology, Writing – review & editing. DS: Investigation, Methodology, Writing – review & editing, Data curation. AH: Investigation, Methodology, Writing – review & editing. PL: Investigation, Methodology, Writing – review & editing. FK: Investigation, Writing – review & editing. BP: Investigation, Writing – review & editing, Methodology, Resources, Supervision, Conceptualization. ST: Formal Analysis, Investigation, Methodology, Resources, Supervision, Writing – review & editing, Conceptualization. KV: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Resources, Supervision, Validation, Writing – review & editing, Funding acquisition, Project administration, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was supported by the TRISH through Cooperative Agreement NNX16AO69A awarded to KV. The W. M. Keck Foundation provided support for DJS and BMP. The NASA postdoctoral fellowship supported part of JW, CP, and CU time. Preliminary work of this research was supported by a NASA SBIR Contract (NNX17CP21P) awarded to SW. The funders had no role in the study design, data collection, and interpretation; the writing of the manuscript; or the decision to submit the work for publication.
Acknowledgments
This study was conducted under Yellowstone National Park research permit YELL-2018-SCI-5480 held by BMP. Adriana Gonzalez (student) and Bo Liu (AI Biosciences) are acknowledged for their assistance in organizing supplies for the work. We acknowledged the Jet Propulsion Laboratory supercomputing facility staff, notably Narendra J. Patel (Jimmy) and Edward Villanueva, for their continuous support in providing the best possible infrastructure for BIGDATA analysis.
Conflict of interest
SW is currently employed at AI Biosciences, while AA was formerly associated with the same institution.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
This manuscript was prepared as an account of work sponsored by NASA, an agency of the US Government. The US Government, NASA, California Institute of Technology, Jet Propulsion Laboratory, and their employees make no warranty, expressed or implied, or assume any liability or responsibility for the accuracy, completeness, or usefulness of information, apparatus, product, or process disclosed in this manuscript, or represents that its use would not infringe upon privately held rights. The use of, and references to any commercial product, process, or service does not necessarily constitute or imply endorsement, recommendation, or favoring by the US Government, NASA, California Institute of Technology, or Jet Propulsion Laboratory. Views and opinions presented herein by the authors of this manuscript do not necessarily reflect those of the US Government, NASA, California Institute of Technology, or Jet Propulsion Laboratory, and shall not be used for advertisements or product endorsements.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2024.1306008/full#supplementary-material
Supplementary Figure 1 | Phylogeny of dominant Archaea and Bacteria detected in microbial mat samples.
Supplementary Figure 2 | Phylogeny of dominant Archaea and Bacteria detected in sediment samples.
References
Allewalt J. P., Bateson M. M., Revsbech N. P., Slack K., Ward D. M. (2006). Effect of temperature and light on growth of and photosynthesis by synechococcus isolates typical of those predominating in the octopus spring microbial mat community of yellowstone national park. Appl. Environ. Microbiol. 72, 544–550. doi: 10.1128/AEM.72.1.544-550.2006
Barns S. M., Fundyga R. E., Jeffries M. W., Pace N. R. (1994). Remarkable archaeal diversity detected in a Yellowstone National Park hot spring environment. Proc. Natl. Acad. Sci. 91, 1609–1613. doi: 10.1073/pnas.91.5.1609
Beam J. P., Jay Z. J., Schmid M. C., Rusch D. B., Romine M. F., Jennings Rde M., et al. (2016). Ecophysiology of an uncultivated lineage of Aigarchaeota from an oxic, hot spring filamentous ‘streamer’ community. ISME J. 10, 210–224. doi: 10.1038/ismej.2015.83
Becraft E. D., Wood J. M., Cohan F. M., Ward D. M. (2020). Biogeography of american northwest hot spring A/B′-lineage synechococcus populations. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.00077
Blank C. E., Cady S. L., Pace N. R. (2002). Microbial composition of near-boiling silica-depositing thermal springs throughout Yellowstone National Park. Appl. Environ. Microbiol. 68, 5123–5135. doi: 10.1128/AEM.68.10.5123-5135.2002
Bolger A. M., Lohse M., Usadel B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brock T. D., Brock M. L. (1968). Measurement of steady-state growth rates of a thermophilic alga directly in nature. J. Bacteriology 95, 811–815. doi: 10.1128/jb.95.3.811-815.1968
Buchfink B., Xie C., Huson D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Chan K., Coen M., Hardick J., Gaydos C. A., Wong K.-Y., Smith C., et al. (2016a). Low-cost 3D printers enable high-quality and automated sample preparation and molecular detection. PloS One 11, e0158502. doi: 10.1371/journal.pone.0158502
Chan K., Weaver S. C., Wong P.-Y., Lie S., Wang E., Guerbois M., et al. (2016b). Rapid, affordable and portable medium-throughput molecular device for zika virus. Sci. Rep. 6, 38223. doi: 10.1038/srep38223
Chan K., Wong P.-Y., Yu P., Hardick J., Wong K.-Y., Wilson S. A., et al. (2016c). A rapid and low-cost PCR thermal cycler for infectious disease diagnostics. PloS One 11, e0149150. doi: 10.1371/journal.pone.0149150
Chin C. D., Linder V., Sia S. K. (2007). Lab-on-a-chip devices for global health: past studies and future opportunities. Lab. Chip 7, 41–57. doi: 10.1039/B611455E
Colman D. R., Amenabar M. J., Fernandes-Martins M. C., Boyd E. S. (2022). Subsurface Archaea associated with rapid geobiological change in a model Yellowstone hot spring. Commun. Earth Environ. 3, 205. doi: 10.1038/s43247-022-00542-2
Daar A. S., Thorsteinsdóttir H., Martin D. K., Smith A. C., Nast S., Singer P. A. (2002). Top ten biotechnologies for improving health in developing countries. Nat. Genet. 32, 229–232. doi: 10.1038/ng1002-229
De Coster W., D’hert S., Schultz D. T., Cruts M., Van Broeckhoven C. (2018). NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34, 2666–2669. doi: 10.1093/bioinformatics/bty149
Dundas N., Leos N. K., Mitui M., Revell P., Rogers B. B. (2008). Comparison of automated nucleic acid extraction methods with manual extraction. J. Mol. Diagn. 10, 311–316. doi: 10.2353/jmoldx.2008.070149
Egeter B., Veríssimo J., Lopes-Lima M., Chaves C., Pinto J., Riccardi N., et al. (2022). Speeding up the detection of invasive bivalve species using environmental DNA: A Nanopore and Illumina sequencing comparison. Mol. Ecol. Resour. 22, 2232–2247. doi: 10.1111/1755-0998.13610
Fournier R. O. (1989). Geochemistry and dynamics of the yellowstone national park hydrothermal system. Annu. Rev. Earth Planetary Sci. 17, 13–53. doi: 10.1146/annurev.ea.17.050189.000305
Holzhausen E. A., Nikodemova M., Deblois C. L., Barnet J. H., Peppard P. E., Suen G., et al. (2021). Assessing the impact of storage time on the stability of stool microbiota richness, diversity, and composition. Gut Pathog. 13, 75. doi: 10.1186/s13099-021-00470-0
Huson D. H., Beier S., Flade I., Górska A., El-Hadidi M., Mitra S., et al. (2016). MEGAN community edition - interactive exploration and analysis of large-scale microbiome sequencing data. PloS Comput. Biol. 12, e1004957. doi: 10.1371/journal.pcbi.1004957
Jungkind D. (2001). Automation of laboratory testing for infectious diseases using the polymerase chain reaction– our past, our present, our future. J. Clin. Virol. 20, 1–6. doi: 10.1016/S1386-6532(00)00148-7
Kato S., Sakai S., Hirai M., Tasumi E., Nishizawa M., Suzuki K., et al. (2018). Long-term cultivation and metagenomics reveal ecophysiology of previously uncultivated thermophiles involved in biogeochemical nitrogen cycle. Microbes Environ. 33, 107–110. doi: 10.1264/jsme2.ME17165
Klatt C. G., Wood J. M., Rusch D. B., Bateson M. M., Hamamura N., Heidelberg J. F., et al. (2011). Community ecology of hot spring cyanobacterial mats: predominant populations and their functional potential. ISME J. 5, 1262–1278. doi: 10.1038/ismej.2011.73
Kwok S., Higuchi R. (1989). Avoiding false positives with PCR. Nature 339, 237–238. doi: 10.1038/339237a0
Leuko S., Goh F., Ibáñez-Peral R., Burns B. P., Walter M. R., Neilan B. A. (2008). Lysis efficiency of standard DNA extraction methods for Halococcus spp. in an organic rich environment. Extremophiles 12, 301–308. doi: 10.1007/s00792-007-0124-8
López-López O., Knapik K., Cerdán M. E., González-Siso M. I. (2015). Metagenomics of an alkaline hot spring in galicia (Spain): microbial diversity analysis and screening for novel lipolytic enzymes. Front. Microbiol. 6 1291. doi: 10.3389/fmicb.2015.01291
Madigan M. T., Jung D. O., Karr E. A., Sattley W. M., Achenbach L. A., van der Meer M. T. J. (2005). “Diversity of anoxygenic phototrophs in contrasting extreme environments,” in Geothermal biology and geochemistry in Yellowstone National Park. Eds. Inskeep W. P., Mcdermott T. R. (Bozeman, Montana: Montana State University Publications).
Malygina A., Balkin A., Polyakova E., Stefanov S., Potekhin A., Gogoleva N. (2023). Taxonomic diversity of the microbial biofilms collected along the thermal streams on kunashir island. Ecologies 4, 106–123. doi: 10.3390/ecologies4010009
Meer M.T.J.V.D., Schouten S., Bateson M. M., Nübel U., Wieland A., Kühl M., et al. (2005). Diel variations in carbon metabolism by green nonsulfur-like bacteria in alkaline siliceous hot spring microbial mats from yellowstone national park. Appl. Environ. Microbiol. 71, 3978–3986. doi: 10.1128/AEM.71.7.3978-3986.2005
Nübel U., Bateson M. M., Vandieken V., Wieland A., Kühl M., Ward D. M. (2002). Microscopic examination of distribution and phenotypic properties of phylogenetically diverse chloroflexaceae-related bacteria in hot spring microbial mats. Appl. Environ. Microbiol. 68, 4593–4603. doi: 10.1128/AEM.68.9.4593-4603.2002
Nygaard A. B., Tunsjø H. S., Meisal R., Charnock C. (2020). A preliminary study on the potential of Nanopore MinION and Illumina MiSeq 16S rRNA gene sequencing to characterize building-dust microbiomes. Sci. Rep. 10, 3209. doi: 10.1038/s41598-020-59771-0
Oksanen J., Blanchet F. G., Kindt R., Legendre P., Minchin P. R., O’hara R. B., et al. (2013). Vegan: community ecology package”. 2.0-10. Available at: https://CRAN.R-project.org/package=vegan.
Peach J. T., Mueller R. C., Skorupa D. J., Mesle M. M., Kanta S., Boltinghouse E., et al. (2022). Longitudinal analysis of the Five Sisters hot springs in Yellowstone National Park reveals a dynamic thermoalkaline environment. Sci. Rep. 12, 18707. doi: 10.1038/s41598-022-22047-w
Podar P. T., Yang Z., Björnsdóttir S. H., Podar M. (2020). Comparative analysis of microbial diversity across temperature gradients in hot springs from yellowstone and Iceland. Front. Microbiol. 11. doi: 10.3389/fmicb.2020.01625
Savage K. N., Krumholz L. R., Oren A., Elshahed M. S. (2007). Haladaptatus paucihalophilus gen. nov., sp. nov., a halophilic archaeon isolated from a low-salt, sulfide-rich spring. Int. J. Systematic Evolutionary Microbiol. 57, 19–24. doi: 10.1099/ijs.0.64464-0
Singh N. K., Wood J. M., Karouia F., Venkateswaran K. (2018). Succession and persistence of microbial communities and antimicrobial resistance genes associated with International Space Station environmental surfaces. Microbiome 6, 214. doi: 10.1186/s40168-018-0609-y
Stefan C. P., Hall A. T., Graham A. S., Minogue T. D. (2022). Comparison of illumina and oxford nanopore sequencing technologies for pathogen detection from clinical matrices using molecular inversion probes. J. Mol. Diagnostics 24, 395–405. doi: 10.1016/j.jmoldx.2021.12.005
Takami H., Arai W., Takemoto K., Uchiyama I., Taniguchi T. (2015). Functional classification of uncultured “Candidatus caldiarchaeum subterraneum” Using the maple system. PloS One 10, e0132994. doi: 10.1371/journal.pone.0132994
Tang Y. W., Sefers S. E., Li H., Kohn D. J., Procop G. W. (2005). Comparative evaluation of three commercial systems for nucleic acid extraction from urine specimens. J. Clin. Microbiol. 43, 4830–4833. doi: 10.1128/JCM.43.9.4830-4833.2005
Urbaniak C., Wong S., Tighe S., Arumugam A., Liu B., Parker C. W., et al. (2020). Validating an automated nucleic acid extraction device for omics in space using whole cell microbial reference standards. Front. Microbiol. 11, 1909. doi: 10.3389/fmicb.2020.01909
Yager P., Domingo G. J., Gerdes J. (2008). Point-of-care diagnostics for global health. Annu. Rev. BioMed. Eng. 10, 107–144. doi: 10.1146/annurev.bioeng.10.061807.160524
Zhang L., Chen T., Wang Y., Zhang S., Lv Q., Kong D., et al. (2022). Comparison analysis of different DNA extraction methods on suitability for long-read metagenomic nanopore sequencing. Front. Cell Infect. Microbiol. 12, 919903. doi: 10.3389/fcimb.2022.919903
Keywords: automated DNA extraction system, µTitan, environmental microbiome, microbial diversity, Yellowstone National Park
Citation: Wood JM, Urbaniak C, Parker C, Singh NK, Wong S, Arumugam A, Skorupa DJ, Hemmah A, Laaguiby P, Karouia F, Peyton BM, Tighe S and Venkateswaran K (2024) Assessing microbial diversity in Yellowstone National Park hot springs using a field deployable automated nucleic acid extraction system. Front. Ecol. Evol. 12:1306008. doi: 10.3389/fevo.2024.1306008
Received: 03 October 2023; Accepted: 23 January 2024;
Published: 09 February 2024.
Edited by:
Siyan Zhao, National University of Singapore, SingaporeReviewed by:
William C. Nelson, Pacific Northwest National Laboratory (DOE), United StatesGuofang Xu, National University of Singapore, Singapore
Copyright © 2024 Wood, Urbaniak, Parker, Singh, Wong, Arumugam, Skorupa, Hemmah, Laaguiby, Karouia, Peyton, Tighe and Venkateswaran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kasthuri Venkateswaran, a2p2ZW5rYXRAanBsLm5hc2EuZ292; Scott Tighe, c2NvdHQudGlnaGVAdXZtLmVkdQ==
†ORCID: Jason M. Wood, orcid.org/0000-0002-0847-4695
Ceth Parker, orcid.org/0000-0002-4328-490X
Nitin Kumar Singh, orcid.org/0000-0001-5344-1190
Fathi Karouia, orcid.org/0000-0002-1142-9684
Brent M. Peyton, orcid.org/0000-0003-0033-0651
Scott Tighe, orcid.org/0000-0002-3988-0741
Kasthuri Venkateswaran, orcid.org/0000-0002-6742-0873