
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
DATA REPORT article
Front. Ecol. Evol., 22 January 2024
Sec. Phylogenetics, Phylogenomics, and Systematics
Volume 12 - 2024 | https://doi.org/10.3389/fevo.2024.1291268
 Autumn Gray1,2*†
Autumn Gray1,2*† Anna S. Zhuk3,4†
Anna S. Zhuk3,4† Jan F. Kamler5†
Jan F. Kamler5† Cody W. Edwards2,6†
Cody W. Edwards2,6† Henrique V. Figueiró2,7†
Henrique V. Figueiró2,7† Klaus-Peter Koepfli2,8†
Klaus-Peter Koepfli2,8†The Cape fox, Vulpes chama, (Smith, 1833) is one of the 12 true fox species (genus Vulpes) found in the Canidae and the only Vulpes species found in sub-Saharan Africa (Kamler et al., 2016; Loveridge et al., 2019). They are distributed from southwestern Angola and Namibia to Botswana, southwestern Zimbabwe, and South Africa, where they occupy arid to semi-arid grasslands and savannahs (Kamler et al., 2016; Loveridge et al., 2019). Among canid species that occur in this region, which also includes the bat-eared fox, Otocyon megalotis (Desmarest, 1822), black-backed jackal, Lupulella mesomelas (Schreber, 1775), side-striped jackal, Lupulella adusta (Sundevall, 1847), and African painted dog, Lycaon pictus (Temminck, 1820), Cape foxes are the smallest in terms of body size, averaging 2.5-4.5 kg (Lavoie et al., 2019). The Cape fox, shown in Figure 1, is identifiable by its dark silver-gray and light pale fur coat and black-tipped tail, along with a short muzzle and large ears compared to its body size (Lavoie et al., 2019). Their diet typically includes small vertebrate prey, insects, leporids, and carrion from large mammal carcasses (Klare et al., 2014). Cape foxes are socially monogamous and territorial, and they exhibit male-biased dispersal with young females sometimes remaining as helpers (Kamler and Macdonald, 2014). They are not uncommon in their range and are currently listed as ‘Least Concern’ on the IUCN Red List of Threatened Species with a stable population (Hoffmann, 2014). These mesocarnivores do not have any immediate threats to their population although they face mortality through animal control and the pelt trade, with the latter being insignificant to their population trend (Lavoie et al., 2019). Their density can change due to the presence of black-backed jackals, but the two species do not compete for food resources (Kamler et al., 2012; Kamler et al., 2013). The Cape fox has been able to expand its range, despite these threats, due to changing agricultural practices in recent years (Kamler et al., 2016).

Figure 1 Photograph of a Cape fox, Vulpes chama (A) taken in Kgalagadi Transfrontier Park, South Africa, by Mark Anderson© and (B) taken in Benfontein Nature Reserve, South Africa, by Alex Sliwa©. (A) Showcases the Cape fox’s large ears compared to its head and short muzzle while (B) shows the identifiable smaller slender body, black tipped tail, and silver-gray back with a pale body.
While the genetics and genomics of more well-known fox species such as the red fox, Vulpes vulpes (Linnaeus, 1758), have been investigated in depth (e.g., Sacks et al., 2018; Quinn et al., 2022), much is still unknown about the genetics of the Cape fox. For example, the karyotype and thus chromosome number of this species has not yet been characterized. Past studies have shown that V. chama is morphologically similar to the Bengal fox, Vulpes bengalensis, (Shaw, 1800), but Cape foxes have larger ears and wider skulls (Ellerman and Morrison-Scott, 1966; Stains, 1975) and one of the first molecular phylogenetic analyses suggested they diverged early within the Vulpes group (Geffen et al., 1992). Complete mitochondrial genomes (hereafter, mitogenomes) are commonly used to analyze phylogeographical relationships within species or phylogenetic relationships among species (DeSalle and Hadrys, 2017). These applications are facilitated by the small size, near absence of recombination, and high substitution rate of mitogenomes that result in a high phylogenetic information content whose evolution is relatively straightforward to interpret (Brown et al., 1979; Moritz et al., 1987). To date, complete mitogenomes have been sequenced or assembled in only eight species in the genus Vulpes: red fox, V. vulpes (Arnason et al., 2006), Arctic fox, V. lagopus (Yan et al., 2016), fennec fox, V. zerda (Yang et al., 2016), corsac fox, V. corsac (Zhao et al., 2016a), Bengal fox, V. bengalensis (Gaur et al., 2019), Pale fox, V. pallida (Rocha et al., 2023), Rüppell’s fox, V. rueppellii (Rocha et al., 2023), and Tibetan fox, V. ferrilata (Zhao et al., 2016c). Therefore, to better understand its evolutionary relationship with other species in the Canidae, we aimed to sequence the first complete mitogenome for the Cape fox.
We employed genome skimming to sequence and assemble the complete mitogenome of the Cape fox. In this approach, relatively shallow sequencing (low sequencing depth) of the overall genome using high-throughput sequencing methods yields deep sequencing of the high-copy portions of a genome, such as ribosomal DNA, mitogenomes, and, in the case of plants, chloroplast genomes (Straub et al., 2012; Dodsworth, 2015). Genome skimming provides a relatively efficient and inexpensive method to generate mitogenomes from either fresh or low-quality tissues such as those obtained from road-killed or preserved specimens (e.g., Trevisan et al., 2019; Gray et al., 2022). Furthermore, genome skimming is being increasingly used to generate mitogenome and other high-copy number datasets for a variety of investigations, such as phylogenetic relationships (e.g., Ren et al., 2017; Taite et al., 2023), parasite detection (Papaiakovou et al., 2023), and taxonomic identification (Bohmann et al., 2020).
A tissue sample was collected from the ear of a dead female Cape fox on 22 of May 2006 in the Benfontein Nature Reserve, Northern Cape Province, South Africa (28°51’S 24°48’E). This animal had been radio collared as part of an ecological study (Kamler et al., 2013) and was subsequently killed by a black-backed jackal. The sample was preserved in 100% ethanol and maintained in the personal collection of J.F. Kamler, until it was transferred to the collection of K-P. Koepfli (https://smconservation.gmu.edu/people/klaus-koepfli/, Klaus-Peter. Koepfli,a2tvZXBmbGlAZ211LmVkdQ==) at the Smithsonian-Mason School of Conservation, George Mason University, under the catalog ID VCH_ID_15.
DNA extraction, library preparation, and sequencing was conducted at Psomagen Inc. (Rockville, Maryland, USA). A Mag-Bind Blood and Tissue Kit (Omega Bio-Tek Inc., Norcross, GA) was used for genomic DNA extraction from a ~10 mg piece of tissue, and concentration and quality were assessed with Picogreen and Victor X2 fluorometry (Life Technologies, Carlsbad, CA), an Agilent 4200 Tapestation (Agilent Technologies, Santa Clara, CA), and 1% gel electrophoresis. A Covaris S220 Ultrasonicator (Covaris, Woburn, MA) was used to generate fragments of 350 base pairs (bp), which were then incorporated into a genomic library prepared using the TruSeq DNA PCR-free library kit (Illumina, San Diego, CA). The library was quality checked and quantified using an Agilent 4200 Tapestation and a Lightcycler qPCR assay (Roche Life Science, St. Louis, MO) and then paired-end sequenced (2 x 150 bp) to a depth of 10x on an Illumina NovaSeq 6000 instrument. This resulted in 87,056,472 reads (42.2% GC and 57.8% AT), with 91.81% having a >30 quality score.
Raw reads were evaluated for quality using FastQC (Andrews, 2010), and then imported into Geneious Prime version 2023.1.2 (http://www.geneious.com). BBDuk version 38.84 (Bushnell, 2014) was used to trim low-quality reads (<Q20) and reads <30 bp, resulting in 79,989,082 reads. The processed reads were initially mapped to the reference genome of the red fox, Vulpes vulpes (NCBI accession number AM181037) using the Geneious mapper (settings: medium sensitivity and 10 iterations of fine-tuning), resulting in a consensus sequence of 16,807 bp with a mean coverage of 296.3x. However, we observed 22 ambiguous bases in this sequence, most of which were located within the control region. We then utilized GetOrganelle version 1.7.7.0 to de novo assemble the Cape fox mitogenome using the full read set, employing the red fox (AM181037) and fennec fox, Vulpes zerda (KJ603240) as seeds and the -F animal_mt parameter, while the remaining parameters were set to their default values (Jin et al., 2020). From this we obtained a 16,772 bp sequence with no ambiguous bases and with a mean coverage of 275.7x as estimated with Mosdepth (Pedersen and Quinlan, 2018). This sequence was imported into MITOS2 (Donath et al., 2019) for annotation.
We performed phylogenetic analyses to assess the relationship of the Cape fox relative to other species within the Canidae based on mitogenome sequences, which represent a single non-recombining locus and that is maternally inherited (Harrison, 1989). First, MAFFT version 7.490 (Katoh and Standley, 2013) (settings: AUTO algorithm, 200PAM/k=2 scoring matrix, 1.53 gap open penalty, and an offset value of 0.123) was used to align the mitogenome of the Cape fox (GenBank accession number ON756054) with those from 23 other canid species and two species of Ursus (bears), which were used as outgroups. Mitogenome sequences were downloaded from the National Center for Biotechnology Information’s GenBank database for the following species: Vulpes zerda KJ603240 (Yang et al., 2016), V. vulpes AM181037 (Arnason et al., 2006), V. corsac KJ140137 (Zhao et al., 2016a), V. ferrilata KT033906 (Zhao et al., 2016c), V. lagopus KP200876 (Yan et al., 2016), raccoon dog, Nyctereutes procyonoides koreensis KF709435 (Kim et al., 2015), Otocyon megalotis MW257223 (Hassanin et al., 2021), gray fox, Urocyon cinereoargenteus KP129096 (Hofman et al., 2015), island fox, U. littoralis catalinae KP129018 (Hofman et al., 2015), bush dog, Speothos venaticus MW257226 (Hassanin et al., 2021), maned wolf, Chrysocyon brachyurus KJ508409 (Zhao et al., 2016b), Lycaon pictus KT598692 (Hwang et al., 2015), Lupulella mesomelas KT448280 (Koepfli et al., 2015), African wolf, Canis lupaster KT378606 (Urios et al., 2015), gray wolf, C. lupus chanco EU442884 (Chen et al., 2010), golden jackal, C. aureus cruesemanni ON986207 (Sosale et al., 2023), Ethiopian wolf, C. simensis MT793779 (Jie, 2022b), red wolf, C. rufus MT793780 (Jie, 2022a), coyote, C. latrans DQ480509 (Björnerfeldt et al., 2006), dhole, Cuon alpinus GU063864 (Zhang and Chen, 2011), American black bear, Ursus americanus JX196366 (Miller et al., 2012), and brown bear, U. arctos isabellinus MW257206 (Hassanin et al., 2021). For three species, we downloaded whole genome short reads or mitogenome sequence from the European Nucleotide Archive: Vulpes pallida SRR24041004 (Rocha et al., 2023), V. rueppellii SRR24040945 (Rocha et al., 2023), and V. bengalensis MG200266 (Gaur et al., 2019). Before MAFFT alignment, the mitogenomes of V. pallida and V. rueppellii were assembled with the Geneious mapper, using the same settings as for the Cape fox, and then annotated using MITOS2 (Donath et al., 2019). We obtained an initial alignment of 17,317 bps. The part of the alignment corresponding to the control region was trimmed due to alignment uncertainty, resulting in a final alignment of 15,568 bps. A maximum-likelihood tree was estimated from the alignment using RAxML version 8.2.11 (Stamatakis, 2014) (settings: nucleotide model = GTRGAMMA, algorithm = rapid hill-climbing, with starting trees = 20). Only four nucleotide substitution models are available for phylogenetic analysis using RAxML: GTR+GAMMA, GTR+GAMMA+I, GTR+CAT, and GTR+CAT+I, with the I parameter accounting for the proportion of invariant sites. The GTR+GAMMA model was selected because our alignment only included 26 sequences (Stamatakis, 2016) and the alpha parameter for the gamma distribution itself accounts for sites with low rates of substitution, which thereby precludes the need to use the GTR+GAMMA+I model (Yang, 2006). 1000 replicates of rapid bootstrapping were used to estimate node support. Alignment, phylogenetic analyses, and visualization of the Cape fox mitogenome map and phylogenetic tree were conducted using program plugins available in the Geneious Prime version 2023.1.2 software package (http://www.geneious.com).
The resulting complete mitogenome for the Cape fox consists of 16,772 bp (ON756054) which has 37 genes that includes 2 rRNAs, 22 tRNAs, and 13 protein-coding regions (Figure 2A). An origin of replication and a control region were also identified. This sequence had a nucleotide composition of: A = 31.0% (5,196 bp), C = 27.6% (4,632 bp), G = 14.9% (2,505 bp), and T = 26.5% (4,439 bp). The overall GC-content was 42.6% (7,137 bp) of the nucleotides.
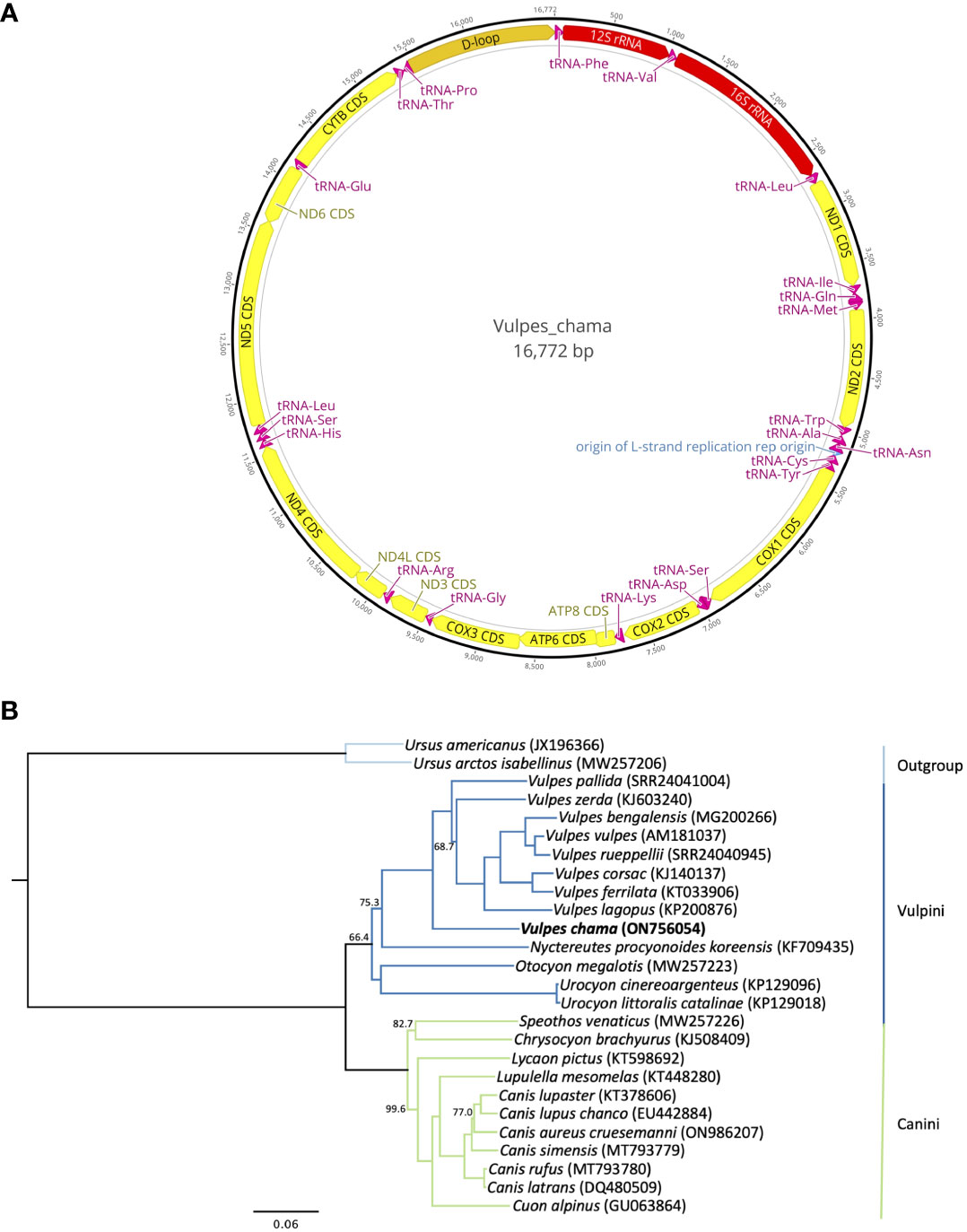
Figure 2 (A) Annotated mitogenome of the Cape Fox, Vulpes chama. Annotated features include protein-coding genes (yellow bars), rRNA genes (red bars), tRNA genes (magenta bars), origin of replication (blue bar), and the control region (orange bar). Arrowed bars indicate direction of transcription on either the plus strand (arrows oriented to the right) or minus strand (arrows oriented to the left). The black outer ring shows the relative nucleotide position of each gene in the mitogenome. Image of the annotated mitogenome was created in Geneious Prime 2023.1.2 (http://www.geneious.com). (B) Maximum-likelihood phylogenetic tree reconstructed from a 15,568 bp alignment of mitogenomes, with the control regions trimmed, showing the relationship of Vulpes chama to other species within the Canidae. The tree was rooted using two species of bears (Ursidae). All nodes in the tree were supported by 100% bootstrap support except for those shown with <100% support. The clade joining Otocyon megalotis and the two species of Urocyon received <50% bootstrap support. Branch colors represent the tribes within Canidae. GenBank accession numbers, or European Nucleotide Archive accession numbers for Vulpes bengalensis, V. pallida, and V. rueppellii, are included next to the species name in parentheses.
This assembled sequence of 16,772 bp is comparable to the mitogenomes of the eight other Vulpes species that have been sequenced thus far (16,656-16,813 bp). The Cape fox mitogenome, including the control region, differs by 11.81%-13.13% from the other species in the genus Vulpes. The Cape fox also has a slightly larger control region, 1,311 bp, than most of the other Vulpes species (1,196-1,354 bp). The only species with a larger control region are V. pallida (1,335 bp), V. rueppellii (1,351 bp), and V. vulpes (1,354 bp). The maximum likelihood tree (Figure 2B) shows V. chama as the earliest diverging species in the monophyletic group of Vulpes species. The relationships among the Vulpes species were resolved with 100% bootstrap support, except for the placement of V. zerda with 68.7% bootstrap support. The phylogenetic tree also supports the division of Canidae into two major tribes, the Vulpini, with a bootstrap support of 66.4% and which includes the fox-like canids, and the Canini, with 100% support and which includes the South American canids (here represented only by Chrysocyon and Speothos), and the wolf-like canids in genera Lupulella, Lycaon, Cuon, and Canis. This fundamental split within Canidae was also found by Hassanin et al. (2021) based on Bayesian analyses of mitogenomes, by Bardeleben et al. (2005) based on a Bayesian analysis of six nuclear and two mitochondrial DNA segments, and by Zrzavý and Řičánková (2004) based on a combined dataset of three mitochondrial gene segments and 188 morphological, development, behavioral, and cytogenetic characters. The Vulpini-Canini division was also reconstructed for the most part in Lindblad-Toh et al. (2005) using nuclear DNA sequences, and in Perini et al. (2010) using a dataset of combined nuclear and mitochondrial sequences, with the exception of Urocyon, which was placed as the earliest branching lineage with the family.
With the sequenced mitogenome of the Cape fox, the mitogenomes of 9 of the 12 species of Vulpes and 24 of the 35 species of Canidae have been sequenced and characterized to date. The topology estimated using maximum likelihood is consistent with that inferred from parsimony analysis of shared mitochondrial restriction fragments and a 402 bp segment of the cytochrome b gene in a study that included eight species of Vulpes, with the Cape fox as the earliest branching lineage, followed by a lineage containing Blanford’s fox (V. cana) and fennec fox as sister species, and then a clade that included Arctic, swift (V. velox), Corsac, red, and Rüppell’s foxes (Geffen et al., 1992). However, maximum likelihood analysis of the 402 bp cytochrome b gene segment switched the positions of the Blanford’s fox + fennec fox and Cape fox lineages, with the former as the earlier branching lineage (Geffen et al., 1992). The relationship among species of Vulpes inferred from the mitogenome sequences also differ from the topology reported by Lindblad-Toh et al. (2005) based on parsimony and Bayesian analysis of 14,948 bp of 12 exon and 10 intron loci sampled from the nuclear genome (14,948 bps). In the latter tree, Blanford’s fox and fennec fox again comprise a clade that is the earliest branching lineage among the eight Vulpes species included, followed by the Cape fox, and then a clade containing the Arctic, kit (V. macrotis), Corsac, Rüppell’s, and red foxes. More recently, Rocha et al. (2023) investigated the influence of introgression on adaptation to arid environments in several species of foxes found in North Africa, including fennec, red, and Rüppell’s foxes, but their phylogenomic analyses (including analysis of partial and complete mitogenomes) did not include a Cape fox, so no direct comparison can be made with this study. However, among the Vulpes species included in Rocha et al. (2023), the pale fox was the earliest branching lineage in the mitochondrial phylogeny, followed by the fennec fox, and then a clade compromising Arctic, Corsac + Tibetan, Bengal, and Rüppell’s + red foxes. Based on the phylogeny presented here and the topology reported by Lindblad-Toh et al. (2005), there appears to be mito-nuclear discordance in the early branching patterns among Vulpes species, which is not uncommon given the different processes influencing the inheritance of these two genomic compartments (Toews and Brelsford, 2012).
Our results also provide some insights into the biogeographic history of Vulpes species found in Africa. The Cape fox has the most disjunct distribution of all living species of true foxes (not including the red foxes introduced to and currently found in Australia). The mitogenome phylogenetic tree suggests that the Cape fox was the earliest lineage to diverge among the extant species of Vulpes (Figure 2B) followed by the pale fox and fennec fox, respectively. As the Cape fox is found in sub-Saharan Africa far south of the equator (Kamler et al., 2016), and the pale and fennec foxes are both found north of the equator, in the Sahel or Sahara Desert respectively (Leite, 2012), the divergence patterns and current ranges suggest that the ancestors of these lineages colonized Africa independently, with subsequent independent colonization of the continent by Rüppell’s and red foxes. Species within Vulpes have high-dispersal and the earliest fossil evidence for the genus in Africa is recorded from Late Miocene deposits in Chad (7 million years ago, mya), ascribed to V. riffautae (Bonis et al., 2007). This taxon is slightly preceded by fossils of V. kernensis and V. stenognathus from North America, also of Late Miocene age (~8 mya) (Bartolini-Lucenti & Madurell-Malapeira, 2020). The phylogenetic results and fossil evidence therefore raise the possibility that Africa was one of the earliest cradles for the evolution of true foxes, with subsequent diversification occurring in Eurasia and North America. Comprehensive phylogenomic analyses that include data from all extant species are needed to better understand the evolution and biogeography of true foxes.
In conclusion, using a genome skimming approach, we have assembled and annotated the first complete mitogenome of the Cape fox, which was used to elucidate its phylogenetic position within the speciose Canidae. The phylogenetic results are congruent with past studies in that species of Vulpes constitute a monophyletic group and are distinct from other “fox-like” species such as the African bat-eared fox (Otocyon megalotis) and the North American gray fox (Urocyon cinereoargenteus) (Lindblad-Toh et al., 2005). Analysis suggests that the Cape fox is an early diverging lineage within the Vulpes clade, although mitogenomes of other species of true foxes (Vulpes), such as the Blanford’s fox (V. cana), kit fox (V. macrotis), and swift fox (V. velox), need to be sequenced to further verify this hypothesis. Our findings can be used to further the genomic understanding of the Cape fox and other Vulpes species.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, Nucleotide : ON756054, SRA : SRR25541219, BioSample ID : SAMN36861376, BioProject : PRJNA847318.
The animal study was approved by the Department of Tourism, Environment and Conservation, Kimberley, South Africa. The sample used to generate the Cape fox mitogenome sequence was obtained opportunistically from a predated specimen for which ethics approval was non-applicable. Capture and handling methods followed the animal care and use guidelines of the American Society of Mammalogists (Sikes and The Animal Care and Use Committee of the American Society of Mammalogists, 2016), and our research protocol (#0401/05) was approved by the Department of Tourism, Environment and Conservation, Kimberley, South Africa. The study was conducted in accordance with the local legislation and institutional requirements.
AG: Investigation, Writing – original draft, Formal Analysis, Visualization, Writing – review & editing. AZ: Formal Analysis, Investigation, Writing – review & editing, Writing – original draft. JK: Conceptualization, Resources, Writing – review & editing. CE: Conceptualization, Writing – review & editing. HF: Conceptualization, Writing – review & editing. KPK: Conceptualization, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. AZ was funded by Saint-Petersburg State University project #95444727.
We would like to thank Mark Anderson and Alex Sliwa for providing images of the Cape fox. We would also like to thank Kristal Miller, Spencer Harman, Alison Meredith, Piper Thacher, and Medhini Sosale for their immense intellectual thoughts, support, and encouragement throughout the project.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Andrews S. (2010). FastQC: A quality control tool for high throughput sequence data. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
Arnason U., Gullberg A., Janke A., Kullberg M., Lehman N., Petrov E. A., et al. (2006). Pinniped phylogeny and a new hypothesis for their origin and dispersal. Mol. Phylogenet. Evol. 41 (2), 345–354. doi: 10.1016/j.ympev.2006.05.022
Bardeleben C., Moore R. L., Wayne R. K. (2005). A molecular phylogeny of the Canidae based on six nuclear loci. Mol. Phylogenet. Evol. 37 (3), 815–831. doi: 10.1016/j.ympev.2005.07.019
Bartolini-Lucenti S., Madurell-Malapeira J. (2020). Unraveling the fossil record of foxes: An updated review on the Plio-Pleistocene Vulpes spp. from Europe. Quat. Sci. Rev. 236, 106296. doi: 10.1016/j.quascirev.2020.106296
Björnerfeldt S., Webster M. T., Vilà C. (2006). Relaxation of selective constraint on dog mitochondrial DNA following domestication. Genome Res. 16 (8), 990–994. doi: 10.1101/gr.5117706
Bohmann K., Mirarab S., Bafna V., Gilbert M. T. P. (2020). Beyond DNA barcoding: The unrealized potential of genome skim data in sample identification. Mol. Ecol. 29 (14), 2521–2534. doi: 10.1111/mec.15507
Bonis L., de, Peigné S., Likius A., Mackaye H. T., Vignaud P., Brunet M. (2007). The oldest African fox (Vulpes riffautae n. Sp., Canidae, Carnivora) recovered in late Miocene deposits of the Djurab desert, Chad. Naturwissenschaften 94 (7), 575–580. doi: 10.1007/s00114-007-0230-6
Brown W. M., George M., Wilson A. C. (1979). Rapid evolution of animal mitochondrial DNA. Proc. Natl. Acad. Sci. United States America 76 (4), 1967–1971. doi: 10.1073/pnas.76.4.1967
Chen L., Zhang H., Ma J. (2010). The mitochondrial genome of the Mongolian wolf Canis lupus chanco and a phylogenetic analysis of Canis. Shengtai Xuebao/Acta Ecol. Sin. 30, 1463–1471.
DeSalle R., Hadrys H. (2017). Evolutionary biology and mitochondrial genomics: 50 000 mitochondrial DNA genomes and counting. eLS, 1–35. doi: 10.1002/9780470015902.a002
Desmarest A. G. (1822). “Mammalogie ou description des especes de mammiferes. Seconde partie, contenant les ordres de Rongeurs des Edentes, des Pachydermes, des ruminans et des Cetaces,” in En encyclopedie methodique, 196 vols, vol. 7. (Veuve Agasse, Paris, France), 277–555.
Dodsworth S. (2015). Genome skimming for next-generation biodiversity analysis. Trends Plant Sci. 20 (9), 525–527. doi: 10.1016/j.tplants.2015.06.012
Donath A., Jühling F., Al-Arab M., Bernhart S. H., Reinhardt F., Stadler P. F., et al. (2019). Improved annotation of protein-coding genes boundaries in metazoan mitochondrial genomes. Nucleic Acids Res. 47 (20), 10543–10552. doi: 10.1093/nar/gkz833
Ellerman J. R., Morrison-Scott T. C. S. (1966). Checklist of Palearctic and INDIAn mammals 1758–1946. 2nd ed (London, United Kingdom: British Museum of Natural History).
Gaur A., Sreenivas A., Vaishnavi K., Tabasum W. (2019). Complete mitogenome of Indian Fox (Vulpes bengalensis) (European Nucleotide Archive). Available at: https://www.ebi.ac.uk/ena/browser/view/MG200266.1.
Geffen E., Mercure A., Girman D. J., Macdonald D. W., Wayne R. K. (1992). Phylogenetic relationships of the fox-like canids: mitochondrial DNA restriction fragment, site and cytochrome b sequence analysis. J. Zool. (London) 228, 27–39. doi: 10.1111/j.1469-7998.1992.tb04430.x
Gray A., Brito J. C., Edwards C. W., Figueiró H. V., Koepfli K.-P. (2022). First complete mitochondrial genome of the Saharan striped polecat (Ictonyx libycus). Mitochondrial DNA Part B 7 (11), 1957–1960. doi: 10.1080/23802359.2022.2141080
Harrison R. G. (1989). Animal mitochondrial DNA as a genetic marker in population and evolutionary biology. Trends Ecol. Evol. 4 (1), 6–11. doi: 10.1016/0169-5347(89)90006-2
Hassanin A., Veron G., Ropiquet A., Vuuren B. J., van, Lécu A., Goodman S. M., et al. (2021). Evolutionary history of Carnivora (Mammalia, Laurasiatheria) inferred from mitochondrial genomes. PloS One 16 (2), e0240770. doi: 10.1371/journal.pone.0240770
Hoffmann M. (2014). “Vulpes chama,” in IUCN Red List of Threatened Species (The IUCN Red List of Threatened Species 2014). Available at: https://www.iucnredlist.org/en.
Hofman C. A., Rick T. C., Hawkins M. T. R., Funk W. C., Ralls K., Boser C. L., et al. (2015). Mitochondrial genomes suggest rapid evolution of dwarf California channel islands foxes (Urocyon littoralis). PloS One 10 (2), e0118240. doi: 10.1371/journal.pone.0118240
Hwang K. C., Jeong Y. W., Kim J. J., Jeong J. H., Lee J., Hyun S. H., et al. (2015). Lycaon pictus mitochondrion, complete genome, (944203056) (NCBI Nucleotide Database). Available at: http://www.ncbi.nlm.nih.gov/nuccore/KT598692.1.
Jie Z. (2022a). Canis rufus mitochondrion, complete genome, (2220274603) (NCBI Nucleotide Database). Available at: http://www.ncbi.nlm.nih.gov/nuccore/MT793780.1.
Jie Z. (2022b). Canis simensis mitochondrion, complete genome, (2220274589) (NCBI Nucleotide Database). Available at: http://www.ncbi.nlm.nih.gov/nuccore/MT793779.1.
Jin J.-J., Yu W.-B., Yang J.-B., Song Y., dePamphilis C. W., Yi T.-S., et al. (2020). GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21 (1), 241. doi: 10.1186/s13059-020-02154-5
Kamler J. F., Macdonald D. W. (2014). Social organization, survival, and dispersal of cape foxes (Vulpes chama) in South Africa. Mamm. Biol. 79, 64–70. doi: 10.1016/j.mambio.2013.09.004
Kamler J. F., Palmer G., Cowell C., Mills M., Stuart C., Stuart M., et al. (2016). “A conservation assessment of Vulpes chama,” in The Red List of Mammals of South Africa, Swaziland and Lesotho. Eds. Child M., Roxburgh L., Do Lihn San E., Raimondo D., Davies-Mostert H. (South Africa: South African National Biodiversity Institute and Endangered Wildlife Trust). Available at: https://www.ewt.org.za/wp-content/uploads/2019/02/39.-Cape-Fox-Vulpes-chama_LC.pdf.
Kamler J. F., Stenkewitz U., Klare U., Jacobsen N. F., Macdonald D. W. (2012). Resource partitioning among cape foxes, bat-eared foxes, and black-backed jackals in South Africa. J. Wildlife Manage. 76 (6), 1241–1253. doi: 10.1002/jwmg.354
Kamler J. F., Stenkewitz U., Macdonald D. W. (2013). Lethal and sublethal effects of black-backed jackals on cape foxes and bat-eared foxes. J. Wildl. Manag. 94, 2, 295–306. doi: 10.1644/12-MAMM-A-122.1
Katoh K., Standley D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30 (4), 772–780. doi: 10.1093/molbev/mst010
Kim H. R., Cho J. Y., Park Y. C. (2015). Intraspecific comparison of complete mitogenome sequences from two Asian raccoon dogs (Canidae: Nyctereutes procyonoides). Mitochondrial DNA 26 (6), 827–828. doi: 10.3109/19401736.2013.855913
Klare U., Kamler J. F., Macdonald D. W. (2014). Seasonal diet and numbers of prey consumed by Cape foxes Vulpes chama in South Africa. Wildlife Biol. 20 (3), 190–195. doi: 10.2981/wlb.00006
Koepfli K.-P., Pollinger J., Godinho R., Robinson J., Lea A., Hendricks S., et al. (2015). Genome-wide evidence reveals that African and Eurasian golden jackals are distinct species. Curr. Biol. 25 (16), 2158–2165. doi: 10.1016/j.cub.2015.06.060
Lavoie M., Renard A., Pitt J. A., Larivière S. (2019). Vulpes chama (Carnivora: canidae). Mamm. Species (Online) 51 (972), 11–17. doi: 10.1093/mspecies/sez002
Leite J. V. (2012). Evolution and Biogeography of Canids (Canis and Vulpes) in North-West Africa (Portugal: Master’s, Universidade do Porto). Available at: https://www.proquest.com/docview/2689289835/abstract/B8E291E7BD404CC2PQ/1.
Lindblad-Toh K., Wade C. M., Mikkelsen T. S., Karlsson E. K., Jaffe D. B., Kamal M., et al. (2005). Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 438 (7069), 803–819. doi: 10.1038/nature04338
Loveridge A. J., Seymour-Smith J. L., Sibanda A. L., Mabika C., Ngosi E., Macdonald D. W. (2019). First confirmed record of a Cape fox, Vulpes chama, in Zimbabwe. Afr. J. Ecol. 57 (3), 411–414. doi: 10.1111/aje.12605
Miller W., Schuster S. C., Welch A. J., Ratan A., Bedoya-Reina O. C., Zhao F., et al. (2012). Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change. Proc. Natl. Acad. Sci. 109 (36), E2382–E2390. doi: 10.1073/pnas.1210506109
Moritz C., Dowling T. E., Brown W. M. (1987). Evolution of animal mitochondrial DNA: relevance for population biology and systematics. Annu. Rev. Ecol. Systematics 18 (1), 269–292. doi: 10.1146/annurev.es.18.110187
Papaiakovou M., Fraija-Fernández N., James K., Briscoe A. G., Hall A., Jenkins T. P., et al. (2023). Evaluation of genome skimming to detect and characterise human and livestock helminths. Int. J. Parasitol. 53 (2), 69–79. doi: 10.1016/j.ijpara.2022.12.002
Pedersen B. S., Quinlan A. R. (2018). Mosdepth: Quick coverage calculation for genomes and exomes. Bioinformatics 34 (5), 867–868. doi: 10.1093/bioinformatics/btx699
Perini F., Russo C., Schrago C. (2010). The evolution of South American endemic canids: A history of rapid diversification and morphological parallelism. J. Evolution. Biol. 23, 311–322. doi: 10.1111/j.1420-9101.2009.01901.x
Quinn C. B., Preckler-Quisquater S., Akins J. R., Cross P. R., Alden P. B., Vanderzwan S. L., et al. (2022). Contrasting genetic trajectories of endangered and expanding red fox populations in the western U.S. Heredity 129 (2), 123–136. doi: 10.1038/s41437-022-00522-4. Article 2.
Ren Z., Harris A. J., Dikow R. B., Ma E., Zhong Y., Wen J. (2017). Another look at the phylogenetic relationships and intercontinental biogeography of eastern Asian – North American Rhus gall aphids (Hemiptera: Aphididae: Eriosomatinae): Evidence from mitogenome sequences via genome skimming. Mol. Phylogenet. Evol. 117, 102–110. doi: 10.1016/j.ympev.2017.05.017
Rocha J. L., Silva P., Santos N., Nakamura M., Afonso S., Qninba A., et al. (2023). North African fox genomes show signatures of repeated introgression and adaptation to life in deserts. Nat. Ecol. Evol. 7 (8), 1267–1286. doi: 10.1038/s41559-023-02094-w. Article 8.
Sacks B. N., Lounsberry Z. T., Statham M. J. (2018). Nuclear genetic analysis of the red fox across its trans-pacific range. J. Hered. 109 (5), 573–584. doi: 10.1093/jhered/esy028
Schreber J. C. D. (1775). Die Säugthiere in Abbildungen nach der Natur mit Beschreibungen (Erster Theil): Der Mensch (Verlag Wolfgang Walther: Der Affe. Der Maki. Die Fledermaus. Erlangen).
Shaw G. (1800). General zoology or systematic natural history. Volume 1. Part 2 (London, England: Mammalia. G. Kearsley).
Sikes R. S., and The Animal Care and Use Committee of the American Society of Mammalogists (2016). 2016 Guidelines of the American Society of Mammalogists for the use of wild mammals in research and education. J. Mamma. 97 (3), 663–688. doi: 10.1093/jmammal/gyw078
Sosale M. S., Songsasen N., İbiş O., Edwards C. W., Figueiró H. V., Koepfli K.-P. (2023). The complete mitochondrial genome and phylogenetic characterization of two putative subspecies of golden jackal (Canis aureus cruesemanni and Canis aureus moreotica). Gene 866, 147303. doi: 10.1016/j.gene.2023.147303
Stains H. J. (1975). “Distribution and taxomony of the canidae,” in The wild canids: their systematics, behavioral ecology and evolution. Ed. Fox M. W. (Malabar, Florida: Robert E. Krieger Publishing Company), Pp. 3–Pp.26.
Stamatakis A. (2014). RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30 (9), 1312–1313. doi: 10.1093/bioinformatics/btu033
Stamatakis A. (2016) The RAxML v8.2.X Manual. Available at: https://cme.h-its.org/exelixis/web/software/raxml/.
Straub S. C. K., Parks M., Weitemier K., Fishbein M., Cronn R. C., Liston A. (2012). Navigating the tip of the genomic iceberg: Next-generation sequencing for plant systematics. Am. J. Bot. 99 (2), 349–364. doi: 10.3732/ajb.1100335
Sundevall C. J. (1847). Nya mammalia från Sydafrika. Öfversigt af Kongl. Vetenskapsakademiens Förhandlingar. 3, 118–121.
Taite M., Fernández-Álvarez F. Á., Braid H. E., Bush S. L., Bolstad K., Drewery J., et al. (2023). Genome skimming elucidates the evolutionary history of Octopoda. Mol. Phylogenet. Evol. 182, 107729. doi: 10.1016/j.ympev.2023.107729
Temminck C. J. (1820). Sur le genre hyena, et description d’une espece nouvelle, decouverte en Afrique. Annales generals des sciences physiques, Bruxelles De l’impr. Weissenbruch 3, 46–57.
Toews D. P. L., Brelsford A. (2012). The biogeography of mitochondrial and nuclear discordance in animals. Mol. Ecol. 21 (16), 3907–3930. doi: 10.1111/j.1365-294X.2012.05664.x
Trevisan B., Alcantara D. M. C., MaChado D. J., Marques F. P. L., Lahr D. J. G. (2019). Genome skimming is a low-cost and robust strategy to assemble complete mitochondrial genomes from ethanol preserved specimens in biodiversity studies. PeerJ 7, e7543. doi: 10.7717/peerj.7543
Urios V., Donat-Torres M. P., Ramirez C., Monroy-Vilchis O., Idrissi H. R. (2015). El análisis del genoma mitocondrial del cánido estudiado en Marruecos manifiesta que no es ni lobo (Canis lupus) ni chacal euroasiático (Canis aureus).
Yan S.-Q., Guo P.-C., Yue Y., Li W.-H., Bai C.-Y., Li Y.-M., et al. (2016). The complete sequence of the mitochondrial genome of Arctic fox (Alopex lagopus). Mitochondrial DNA Part A 27 (6), 4095–4096. doi: 10.3109/19401736.2014.1003860
Yang X., Zhao C., Zhang H., Zhang J., Chen L., Sha W., et al. (2016). Complete mitochondrial genome of the fennec fox (Vulpes zerda). Mitochondrial DNA Part A 27 (1), 637–638. doi: 10.3109/19401736.2014.908467
Zhang H., Chen L. (2011). The complete mitochondrial genome of dhole Cuon alpinus: Phylogenetic analysis and dating evolutionary divergence within canidae. Mol. Biol. Rep. 38 (3), 1651–1660. doi: 10.1007/s11033-010-0276-y
Zhao C., Yang X., Zhang H., Zhang J., Chen L., Sha W., et al. (2016b). The complete mitochondrial genome sequence of the maned wolf (Chrysocyon brachyurus). Mitochondrial DNA Part A 27 (1), 539–540. doi: 10.3109/19401736.2014.905847
Zhao C., Zhang H., Liu G., Yang X., Zhang J. (2016c). The complete mitochondrial genome of the Tibetan fox (Vulpes ferrilata) and implications for the phylogeny of Canidae. Comptes Rendus Biol. 339 (2), 68–77. doi: 10.1016/j.crvi.2015.11.005
Zhao C., Zhang J., Zhang H., Yang X., Chen L., Sha W., et al. (2016a). The complete mitochondrial genome sequence of the corsac fox (Vulpes corsac). Mitochondrial DNA Part A 27 (1), 304–305. doi: 10.3109/19401736.2014.892088
Keywords: Vulpes chama, Cape fox, Vulpini, mesocarnivore, mitochondrial genome (mitogenome), genome skimming, phylogeny
Citation: Gray A, Zhuk AS, Kamler JF, Edwards CW, Figueiró HV and Koepfli K-P (2024) The complete mitogenome of the Cape fox, Vulpes chama (Canidae: Mammalia). Front. Ecol. Evol. 12:1291268. doi: 10.3389/fevo.2024.1291268
Received: 08 September 2023; Accepted: 08 January 2024;
Published: 22 January 2024.
Edited by:
Mark A. Elgar, The University of Melbourne, AustraliaReviewed by:
Chuan Ma, Chinese Academy of Agricultural Sciences, ChinaCopyright © 2024 Gray, Zhuk, Kamler, Edwards, Figueiró and Koepfli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Autumn Gray, YWdyYXkyMkBnbXUuZWR1; YXV0aWVncmF5QGdtYWlsLmNvbQ==
†ORCID: Autumn Gray, orcid.org/0000-0002-2445-2832
Anna S. Zhuk, orcid.org/0000-0001-8683-9533
Jan F. Kamler, orcid.org/0000-0003-4148-2088
Cody W. Edwards, orcid.org/0000-0001-5912-6693
Henrique V. Figueiró, orcid.org/0000-0002-8368-7156
Klaus-Peter Koepfli, orcid.org/0000-0001-7281-0676
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.