 Xue Li Zhu
Xue Li Zhu Jiao Jiao Yuan1†
Jiao Jiao Yuan1† Luca Bartolozzi
Luca Bartolozzi Xia Wan
Xia Wan
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol. , 09 February 2023
Sec. Biogeography and Macroecology
Volume 11 - 2023 | https://doi.org/10.3389/fevo.2023.974315
Cyclommatus stag beetles (Coleoptera, Lucanidae) are very interesting insects, because of their striking allometry (mandibles can be longer that the whole body in large males of some species) and sexual dimorphism. They mainly inhabit tropical and subtropical forests in Asia. To date, there has been no molecular phylogenetic research on how these stag beetles evolved and diversified. In this study, we constructed the first phylogenetic relationship for Cyclommatus using multi-locus datasets. Analyses showed that Cyclommatus is monophyletic, being subdivided into two well-supported clades (A and B). The clade A includes the island species from Southeast Asia, and the clade B is formed by the continental species. The divergent time estimates showed these beetles split from the outgroup around 43.10 million years ago (Mya) in the late Eocene, divided during the late Oligocene (around 24.90 Mya) and diversified further during the early and middle Miocene (around 18.19 Mya, around 15.17 Mya). RASP analysis suggested that these beetles likely originated in the Philippine archipelago, then dispersed to the other Southeast Asian archipelagoes, Indochina Peninsula, Southeast Himalayas, and Southern China. Moreover, relatively large genetic distance and stable morphological variations signified that the two clades reach the level of inter-generic differences, i.e., the current Cyclommatus should be separated in two genera: Cyclommatus Parry, 1863 including the clade A species, and Cyclommatinus Didier, 1927 covering the clade B species. In addition, the evidence we generated indicated these beetles’ diversification was promoted probably by both long-distance dispersal and colonization, supporting an “Upstream” colonization hypothesis. Our study provides insights into the classification, genetics and evolution of stag beetles in the Oriental region.
Cyclommatus Parry (1863) is one of the most interesting genera in the Lucanidae family, with more than 60 species, distributed across the continental area of the Indochina Peninsula, Sino-Himalaya region, Southeast Asia archipelago, and parts of New Guinea (Fujita, 2010). These stag beetles exhibit highly intraspecific variability owing to male polymorphism with impressive mandibles, sexual dimorphism and color varieties. So far, some of them have been good models for the studies of behavior and functional morphology, or popular pets and valuably private collections (Kojima, 1996; New, 2010; Gotoh et al., 2014, 2016; Goyens et al., 2015; Liu et al., 2017). These saproxylic beetles also play an important role in forest ecosystems (Araya, 1993; Tanahashi et al., 2009; Songvorawit et al., 2017). However, the taxonomic position of the genus has been a-controversial issue. Their phylogenetic relationships are lacking studies. The origin, dispersal and diversification of these beetles remain poorly known.
Historically, Cyclommatus taxonomy has been revised several times mostly basing on external morphology. The genus Cyclommatus was created by Parry in 1863. Two generic names before Parry’s proposition, Lucanus (Cyclophthalmus) Hope and Westwood (1845) and Megaloprepes Thomson (1862), were both preoccupied names. Didier (1927) divided Cyclommatus Parry into two generic taxa, basing on both morphological characters and distinct geographical distributions: Cyclommatus (s. s.) Parry (1863), containing the species with well-developed mandibles mainly from the insular area of Southeast Asia, and Cyclommatinus Didier (1927) in which the species show rather uniform male mandibles and that are distributed in the continental region of Sino-Himalaya. Shortly afterwards, Nagel (1936) designated the new genus Cyclommatellus consisting of three species, with a peculiar sixth-antennal joint in larger male. Arrow (1938, 1950) did not agree with Didier’s and Nagel’s classifications that was mainly based on charactyers of large males, being often obsolete or absent in smaller males and females. The opinion of Arrow, i.e., Cyclommatinus and Cyclommatellus were synonyms of Cyclommatus Parry, was followed by most of the taxonomists (Benesh, 1960; Mizunuma and Nagai, 1994; Krajčík, 2001; Fujita, 2010; Huang and Chen, 2017). Meanwhile, Didier and Séguy (1953) did not follow Arrow’s opinion in their catalogue. Maes (1992) listed the two synonymized genera as subgenera of Cyclommatus without any taxonomic discussion. Thus, the molecular phylogeny analysis could be very useful to resolve the morphology-based controversy. Unfortunately, only a handful of studies provided a few availably molecular data about this genus (Kim and Farrell, 2015; Liu et al., 2017; Yuan et al., 2021). To provide robustly phylogenetic framework and taxonomic utility of Cyclommatus stag beetles, we performed a multi-locus analysis.
Moreover, all collected data show that Cyclommatus stag beetles are endemic or restricted to the tropical and subtropical forests, either in continental areas or islands (Krajčík, 2001; Fujita, 2010; Huang and Chen, 2017), which seems to indicate a low dispersal capacity of these beetles. Similarly, low dispersal abilities were observed in other stag beetle species, such as Lucanus cervus (Thomaes, 2009) and Lucanus datunensis (Lin et al., 2011); they also inhabit tropical and subtropical forests. Therefore, Cyclommatus stag beetles could be an excellent model for testing specific biogeographic hypotheses. There is an interesting topic about the origin of this genus and how it spreads from Southeast Asia islands to Southern China. To test whether the establishment of Cyclommatus satisfies the “Upstream” hypothesis (the colonization of continents by island fauna) or the “Downstream” hypothesis (the colonization of islands by continental fauna) (Filardi and Moyle, 2005; Jonsson et al., 2011), we also made a spatio-temporal analysis.
Considering these issues, we used a combination of six DNA regions (COI, 16S rDNA, 28S rDNA, Cytb, Wingless, ITS) of 25 representative Cyclommatus stag beetles in order to: (1) discuss the phylogenetic relationship between the two clades of the Cyclommatus stag beetles; (2) explain the current geographical distribution pattern of Cyclommatus stag beetles during evolution.
The sample collection in this study was approved by the Anhui University Academic Ethics Committee, Anhui Province, China.
A total of 115 specimens of 25 taxa of Cyclommatus stag beetles were used in this study. Several were collected in the field by members of our research team, other specimens were donated by universities or research institutes, and some by private collectors; they were preserved in 100% ethanol at −20°C for molecular analyses. Six species closely related to Cyclommatus were selected: Lucanus swinhoei Parry, 1874, L. maculifemoratus taiwanus Miwa, 1936, Noseolucanus denticulus Boucher, 1996, Prismognathus davidis Deyrolle, 1878, P. prossi Bartolozzi and Wan, 2006 and P. triapicalis Houlbert, 1915; they were used for outgroup comparisons in phylogenetic analyses. According to the collecting data of the samples, we drew a distribution map of the species (Figure 1). The voucher specimens and their extracted genomic DNA are deposited at Anhui University, Hefei, China, and in addition, details of the specimen vouchers and specimen locations are listed in Supplementary Table S1.
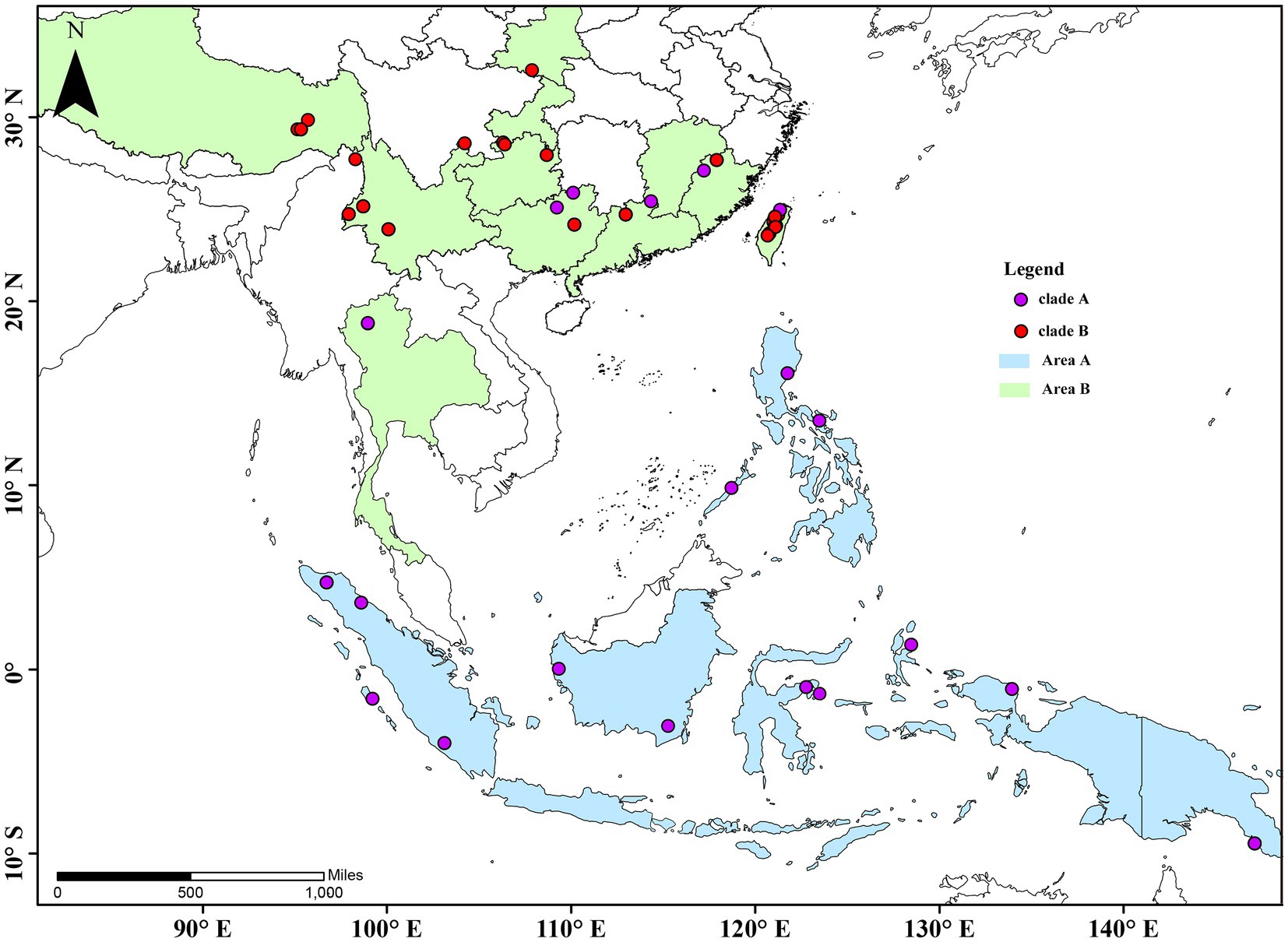
Figure 1. Collection sites and species distribution map of Cyclommatus stag beetles. The map is drew using ARCGIS v10.2 (http://www.esri.com/sofware/arcgis) based on the geospatial data from the National Geomatics Center of China.
Genomic DNA was extracted from the thoracic muscle tissues of adult beetles preserved in absolute ethanol using the Blood and Tissue Kit (Qiagen, Germany) according to the manufacturer’s protocol. Extracted DNA was kept in a freezer at −20°C until needed for the Polymerase chain reaction (PCR). The primer sets in the present study used to amplify three mitochondrial genes 16S rDNA, COI, Cytb, and three nuclear genes 28S rDNA, ITS, and Wingless are shown in Table 1 (Simon et al., 1994; Hosoya et al., 2001; Marcilla et al., 2001; Abouheif and Wray, 2002; Balke et al., 2004; Ward and Downie, 2005; Monaghan et al., 2007; Wild and Maddison, 2008).
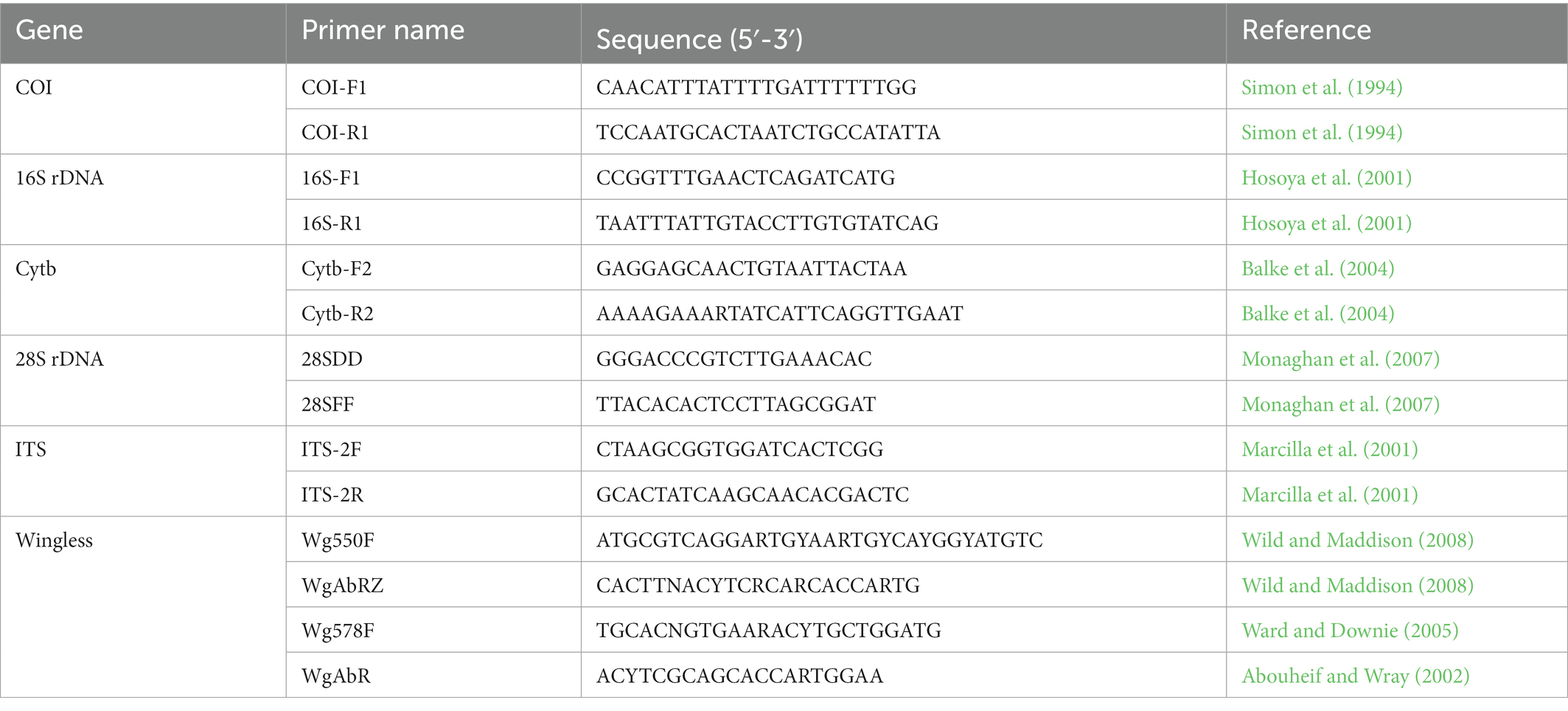
Table 1. Information on the primers used in this study.
PCR amplification reaction was conducted in 25 μl volumes reactions, containing 12.5 μl 2× EasyTaq SuperMix (dye), 10 μM of each primer 1 μl, 2 μl template DNA, and 8.5 μl sterile double-distilled water (ddH2O). The PCR amplification conditions were as follows: initial denaturation at 94°C for 2 min, followed by 35–37 cycles of denaturation at 94°C for 40 s, annealing at 52–58°C for 50 s, and elongation at 70°C for 1 min, and finally 7 min extension step at 72°C. Amplifications were purified using the Template DNA Amplify Kit (Ensure Biologicals). Sequencing the successful PCR products, using the ABI PRISM Big Dye terminator v3.1 sequencing kit (Life Technologies, United States) and cycling on the ABI PRISM 3730xl automated sequencers (Life Technologies, United States) at Sangon Biotech Company, China. The sequences were deposited in GenBank with the accession number (Supplementary Table S1).
All sequences were viewed, assembled, and edited in Geneious Prime v2019.1.1.1 Sequences for each of the six DNA regions were aligned and then inspected by eye for accuracy and trimmed to minimize missing characters in MEGA v7.0 (Kumar et al., 2016). We use PartitionFinder v2.1.1 (Lanfear et al., 2017), a small program in PhyloSuite v1.2.1 (Zhang et al., 2020), to select the best-fitting model selection and best data partition scheme. The total of six partitions were determined to be optimal using the greedy algorithm under the corrected Akaike Information Criterion (AICc): (1) 16S, (2) COI, (3) Cytb, (4) 28S, (5) ITS, (6) Wingless. Finally, best-fitting models were selected for each partition under the AICc: the GTR + I + G model for (1), (2), (3), (4), (5), and the GTR + G model for (6).
Phylogenetic analyses were conducted based on maximum likelihood (ML) and Bayesian inference (BI). An ML tree was constructed using the IQ-TREE webserver2 (Nguyen et al., 2015). In the ML analysis, the “automatic” option was set under the optimal evolutionary model, and the ultrafast Bootstrap approximation method with 10,000 replicates was used to construct the phylogenetic tree. We also implemented a Bayesian analysis as performed in MrBayes v3.2 (Ronquist et al., 2012; Trifinopoulos et al., 2016). Four Markov chains were run simultaneously and sampled once every 1,000 generations, for a total of 10.5 × 108 generations. The initial 25% of trees were discarded as burn-in. Analysis was settled based on the average standard deviation of frequency division being less than 0.01. Phylogenetic trees were viewed and edited in FIGTREE v1.4.4.3
BEAST v2.6.0 (Bouckaert et al., 2014) software was used to perform the analysis for time divergence estimation, which combined two mitochondrial genes (COI and 16S rDNA). A strict molecular clock was applied in a Markov chain Monte Carlo (MCMC) simulation running for 10.5 × 108 generations with samplings every 1 × 104 generations using BEAST. For the beetles, substitution rates of 0.017 per lineage in a million years (Myr) for COI and 0.0054/lineage/Myr for 16S rDNA were found to be optimal (Papadopoulou et al., 2010). TRACER v1.7 (Rambaut et al., 2018) was used to analyze the output results of the Effective Sample Size (ESS) for the posterior distribution of estimated parameter values. Next, the first 25% of the run was discarded as burn-in, all post-burn-in trees from the four independent runs were combined using the software LOG COMBINER v2.6.0 (Bouckaert et al., 2014). Used TREE ANNOTATOR v2.5.1 (Bouckaert et al., 2014) to summarize information from the individual post-burn-in trees onto a single maximum clade credibility (MCC) tree. The final tree was described using FIGTREE v1.4.4.
To further explore the origins and dispersal routes of Cyclommatus, the ancestral distribution of the genus sequences was reconstructed based on the combination of COI and 16S rDNA. Ancestral range reconstruction analysis was conducted with the program RASP v4.2 (Yu et al., 2020) based on the MCC tree from the BEAST, which incorporates the following models:
Dispersal-Extinction-Cladogenesis (DEC, Ree and Smith, 2008; Massana et al., 2015), DEC + Jump parameter (DEC + J, Matzke, 2014a), Dispersal Vicariance Analysis with Likelihood implementation (DIVA LIKE, Ronquist, 1997; Matzke, 2014a), DIVALIKE+ Jump parameter (DIVALIKE + J, Matzke, 2014b), Bayesian inference for discrete Areas with Likelihood implementation (BAYAREALIKE, Landis et al., 2013; Matzke, 2014a), BAYAREALIKE + Jump parameter (BAYAREALIKE + J, Matzke, 2014b), as implemented in BioGeoBEARS (Matzke, 2013, 2014a,b; R Core Team, 2021). Choose the best model according to the corrected AICc and Akaike weights from the previously mentioned six models. The distribution range of Cyclommatus, was based on specimens collected information and fieldwork. Then, the areas were encoded as: (A) the main islands in Southeast Asia; (B) the mainland from the Indochina Peninsula to the southeast of the Himalayas.
Phylogenetic inferences using maximum likelihood (ML) and Bayesian inference (BI) showed a highly similar topology, which was divided into two major clades within the Cyclommatus stag beetles, indicated by us as clade A and clade B (Figure 2). The outgroups formed an independent clade, separated from other high support specimens. The clade A includes 36 specimens representing 19 taxa and is primarily found in the Southeast Asian Islands (Figure 2). However, C. mniszechi Thomson, 1856 is also found in Southeast China, and not only on Taiwan Island. There are several species from New Guinea, such as C. eximius Möllenkamp, 1909, C. imperator Boileau, 1905, C. pulchellus Möllenkamp, 1900, C. margaritae Gestro, 1877 and C. weinreichi Lacroix, 1972. The species located in the Philippines include C. alagari De Lisle, 1968, C. suzumurai Mizumura and Nagai, 1991, and C. zuberi Parry, 1876. The remaining species, for example, C. elaphus Gestro, 1881, C. truncatus Schenk, 2000, C. dehaani Westwood, 1842, C. canaliculatus Ritsema, 1891, C. cupreonitens Boileau, 1901, C. tarandus Thunberg, 1806, and C. metallifer Boisduval, 1835 are mainly distributed in Indonesia. The clade B includes 79 samples representing 10 taxa distributed mainly in the continental area from Indochina to the Himalayan area. C. bicolor Bomans, 1991 is from Thailand and C. scutellaris Möllenkamp, 1912 is broadly distributed in southern China. C. vitalisi Pouillaude, 1913, C. assamensis assamensis Séguy, 1955, C. assamensis yingjiangensis Huang and Chen (2017), C. nagaii Fujita (2010) and C. albersi Kraatz, 1894 are all distributed in the southwestern regions of China. The remaining taxa C. saltini Bomans, 1991, C. asahinai asahinai Kurosawa, 1974 and C. asahinai nanlingensis Schenk, 2016 are mainly located in southeastern China (Figures 1, 2).
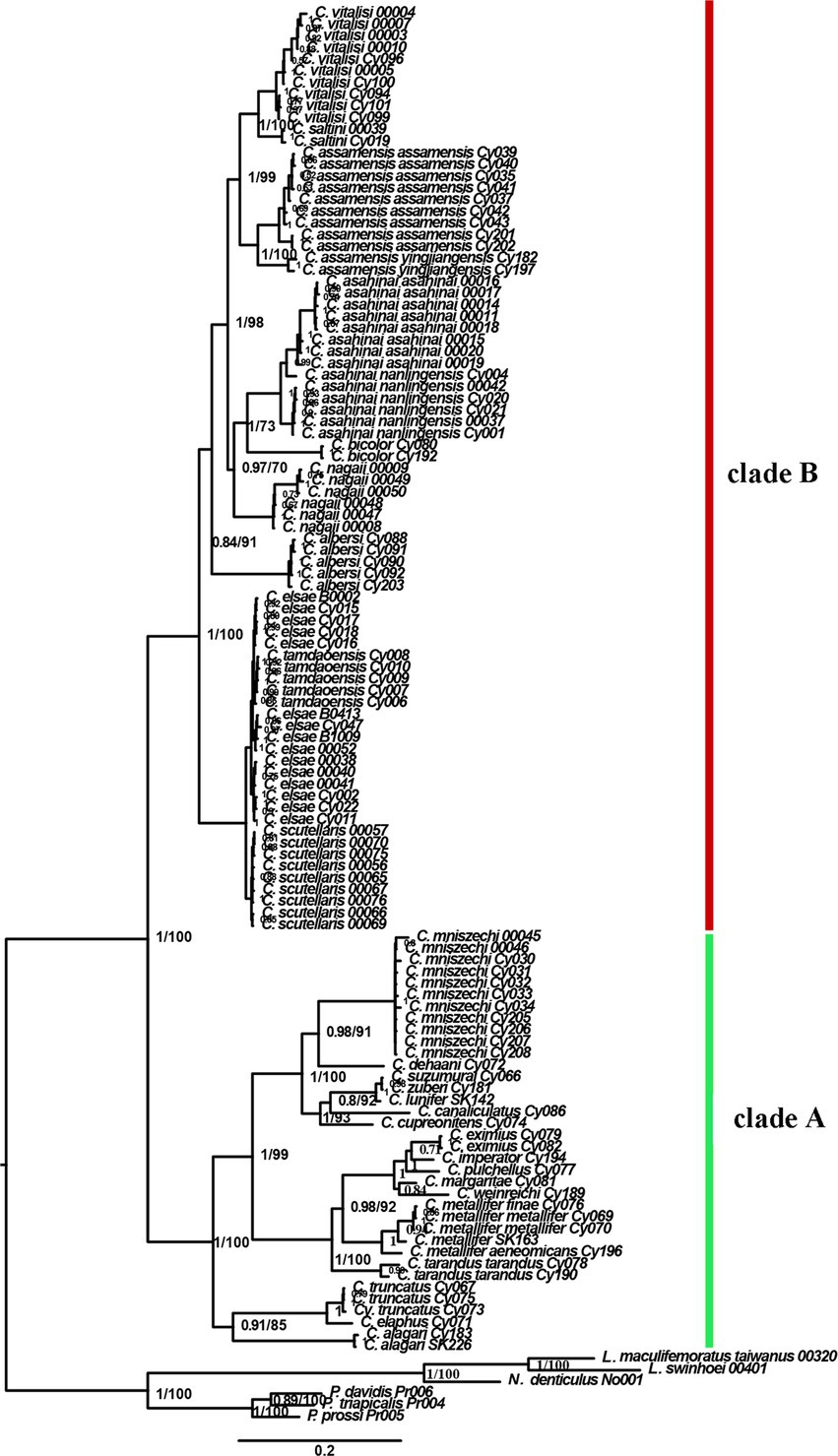
Figure 2. Bayesian topology showing the relationships within Cyclommatus stag beetles and outgroups. Numbers next to each node represent Bayesian posterior probabilities (first number) and partitioned maximum likelihood bootstrap support (second number). The phylogenetic tree is based on Bayesian inference analysis of concatenated DNA sequence data from 16S rDNA, COI, 28S rDNA, ITS, Cytb, and Wingless.
Genetic distances (K2P-distances) using the COI gene were calculated between the clade A and clade B at 0.176 (Table 2), while the average mean genetic distance within the genus is 0.160.

Table 2. The genetic distance for the Cyclommatus stag beetles (Kimura 2-parameter).
The estimation of divergence time showed that Cyclommatus stag beetles (Figure 3) date back to 43.10 million years ago (Mya) in the Late Eocene [95% highest posterior density (HPD) confidence interval: 27.61–66.39 Mya]. Then, in the Late Oligocene (circa 24.90 Mya) [95% HPD, 16.24–36.55 Mya, node 1], Cyclommatus stag beetles were divided into clade A and clade B. Clade A showed interspecific divergence during the Early Miocene circa (ca.) 18.19 Mya [95% HPD, 12.43–25.95 Mya, node 2], while clade B experienced divergence during the Middle Miocene around 15.17 Mya [95% HPD, 9.58–22.60 Mya, node 3].
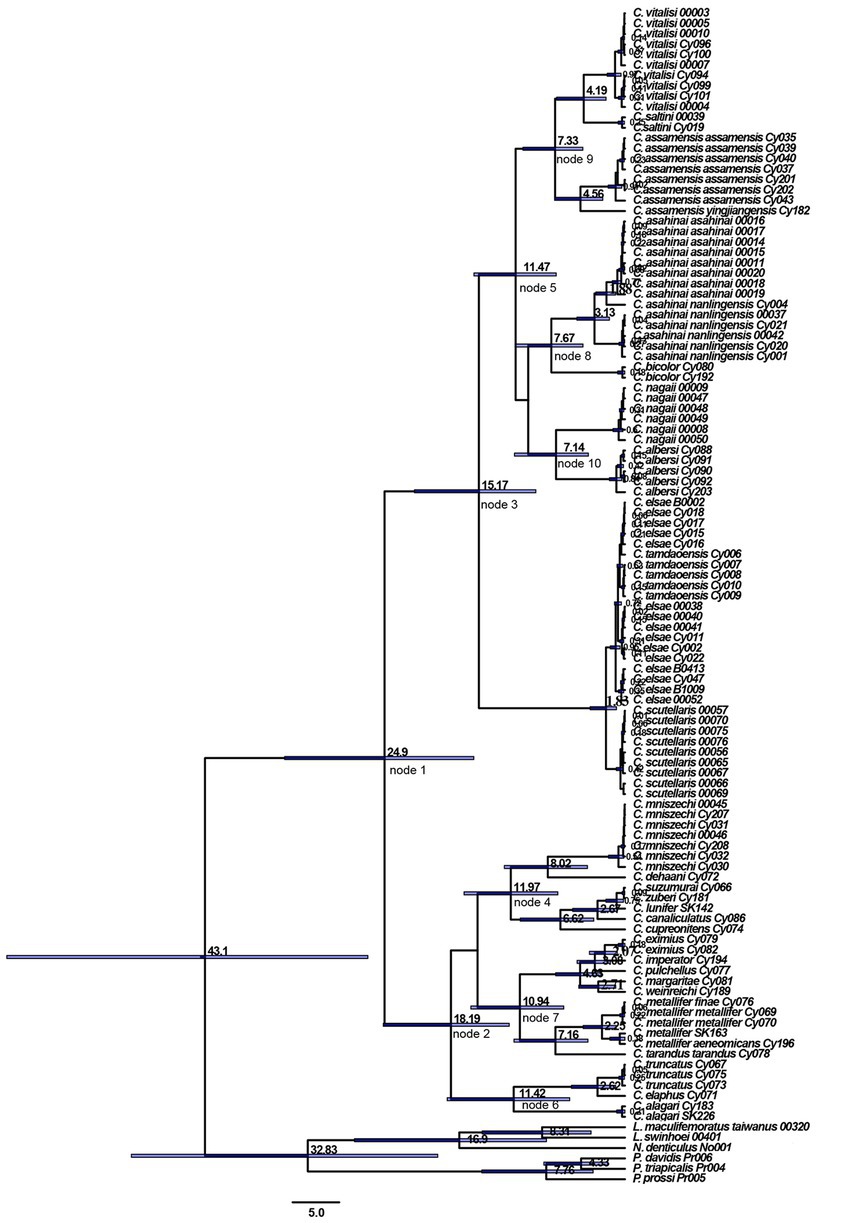
Figure 3. Divergence time estimation of Cyclommatus stag beetles based on COI and 16S rDNA gene. Divergence time estimates are represented next to the nodes (in millions of years) with horizontal bars indicating 95% highest posterior density intervals.
We used the RASP to analyze the ancestral distribution area of Cyclommatus stag beetles AICc scores showed that the best model for this stag beetle dataset in this analysis was the DIVALIKE model (Table 3). Based on this model analysis, at least two geographically isolated events (v) and one dispersal event could explain the genus distribution pattern (Figure 4). Most of the other events were presumed to be speciation within the distribution range, and the distribution range of descendent species are the same as that of their ancestor (y).
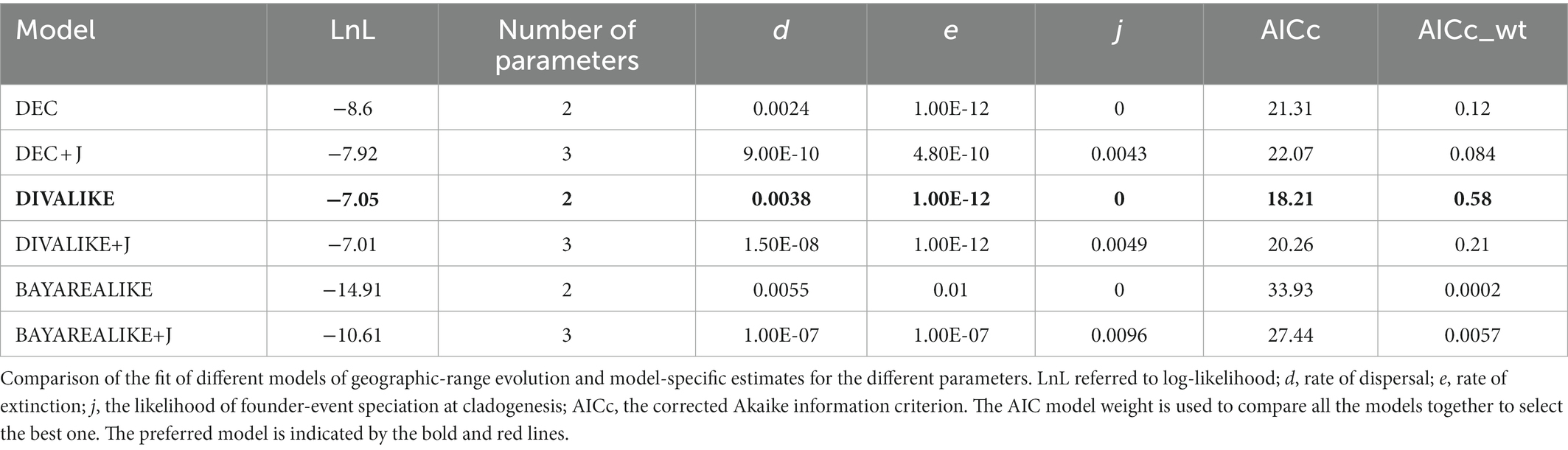
Table 3. Statistical results from RASP of Cyclommatus stag beetles.
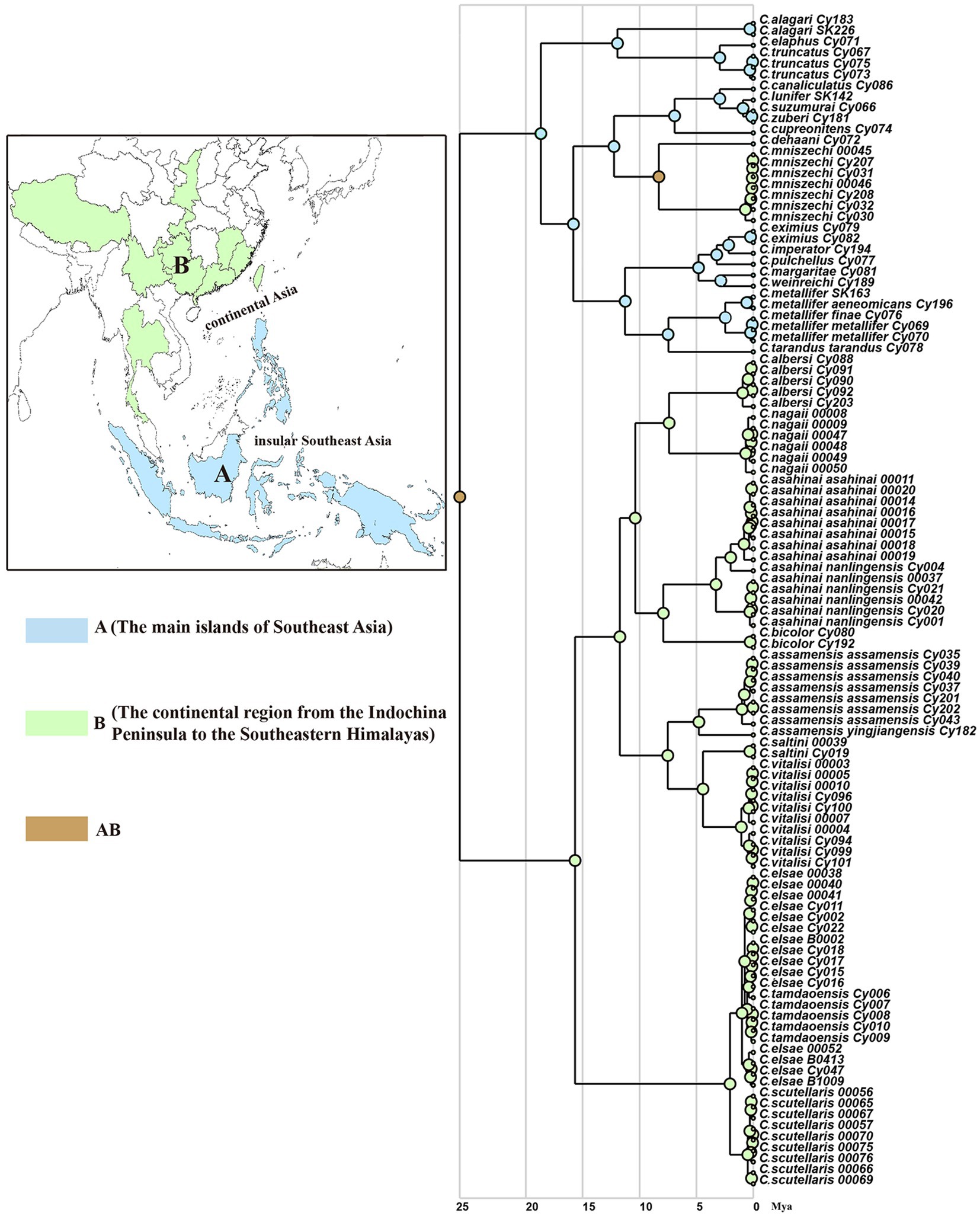
Figure 4. Spatiotemporal reconstruction of Cyclommatus stag beetles based on the best-fitting model (DIVALIKE) inferred using RASP and the maximum clade credibility tree from BEAST analyses. Color circles reflect biogeographic designations (for species at tips), and different colors within the pie charts at the nodes represent the probabilities of different geographical areas. A, the main islands of Southeast Asia; B, the continental region from Indochina Peninsula to the Southeast Himalaya region.
According to this model the ancestor of Cyclommatus stag beetles most probably inhabited the Philippine Islands (Southeast Asian islands), then dispersed to other Southeast Asian islands, the Indochinese peninsula, the southeastern Himalayas, and finally to southern China (Figure 4). Initially, a geographic isolation event divided this population into two groups: clade A (Southeast Asia) and clade B (Southern China). Later, clade A diversified by expanding their range to the main island area of Southeast Asia, and subsequently, experienced isolation events such as the distribution of C. mniszechi in southeastern China (area B). Finally, the species of clade B diversified gradually through the colonization of the mainland from the Indochina Peninsula to the Southeast Himalayas.
Phylogenetic inferences using ML and BI showed a highly similar topology, which confirmed that Cyclommatus stag beetles belong into a monophyletic group. The results are consistent with previous studies (Kim and Farrell, 2015; Tsai and Yeh, 2016; Wu, 2016), although their biological samples were not fully sufficient. The phylogenetic analysis also showed that the samples formed two genetically distinct population clusters (i.e., clade A and B). Clade A species are mainly distributed in Southeast Asian islands, while clade B covers mainly the southern part of China. Two well supported genetic separations implied that species of clade A and clade B could belong to two different taxa, respectively. Furthermore, the K2P-distance between clade A and clade B provided informative sign to assess their generic relationship. The average inter-genetic K2P distance is 0.220 (range 0.174–0.259) in Lucanidae (Wu, 2016). Within a few of closely related lucanid genera, such as between Falcicornis Planet, 1894 and Dorcus MacLeay, 1819; Rhaetus Parry, 1864 and Rhaetulus Westwood, 1871, the K2P-distances values are 0.173 and 0.174, respectively (Wu, 2016; Chen, 2018). The K2P-distance value between the clade A and clade B (=0.176) in our study suggests that the two clades could represent different genera. Additionally, morphological comparison (the terminology used in this study from Holloway (2007) of the two clades indicated some variability, and their differences were supported by Wan (2007), Fujita (2010), Huang and Chen (2017). In species of clade A, the males usually have well-developed mandibles, which can be even 2–3 times their body length (total length of head, pronotum and elytra), and in their genitalia the parameres are long and pointed; their permanently everted internal sac are relatively short, usually no more than twice the length of aedeagus; the female spermathecal gland and spermatheca are clearly separated, and the bursa copulatrix is short (e.g., in C. miniszechi). In reverse, the male mandibles are less developed in the clade B species, usually not exceeding the total length of the head and pronotum; the parameres are rounded and blunt, the permanently everted internal sac relatively long, at least twice the length of aedeagus; the spermathecal gland and spermatheca of female are almost attached to the base of bursa copulatrix, and the bursa copulatrix is long (e.g., in C. assamensis).
In summary, the well-separated topology, relatively high genetic differences and stable morphological variations between the clade A and clade B showed that they could reach the level of inter-generic differences. Consequently, the current Cyclommatus stag beetles could be divided in the typical genus Cyclommatus Parry (1863) and the resurrected genus Cyclommatinus Didier (1927). Despite having no molecular data for Cyclommatellus Nagel (1936) in this study, we suggest to include it in Cyclommatus Parry (1863) basing on the morphological characters. Taxonomic notes on the genera Cyclommatus Parry (1863) and Cyclommatinus Didier (1927) are presented, which include new and revised status for Cyclommatinus. Type species, synonimies, generic diagnosis, updated species checklist with distribution data of the two genera are also given.
Our results are consistent with the “Upstream” colonization hypothesis in Cyclommatus stag beetles, which a general colonization from Southeast Asia islands to mainland Asia, and from tropical archipelagos to subtropical continents, with Southeast Asian being the source of mainland Asian lineages. This diversification pattern is similar to those previously found in other taxa (van der Kaars, 1991; Grismer et al., 2017; Williams et al., 2017; Chen et al., 2018; Liang et al., 2018; Poyarkov et al., 2022). Therefore, islands can be a source and a sink for continental biodiversity (Bellemain and Ricklefs, 2008; Weinell et al., 2021). The direction of species dispersal from continents to islands has also been recorded in Asia (Matuszak et al., 2016), which is the “Downstream” colonization hypothesis. However, it is necessary to synthesize the diversity patterns of other endemic genera within this large geographic range to produce a comprehensive understanding of regional source-sink dynamics between continents and islands.
Our biogeographic analysis shows that Cyclommatus stag beetles appear to have originated from the Late Eocene to the Early Oligocene transition (ca. 43.10 Mya). During this period, the global climate shifted from warm and humid to cold and dry, especially in Southeast Asia (Zachos et al., 2005). At the same time, the continuously humid forests shrunk and fragmented (Bain and Hurley, 2011; Buerki et al., 2013), which may have promoted the initial diversification of Cyclommatus stag beetles.
At the beginning of the Oligocene, the global climate began to cool sharply and sea levels dropped, leaving more islands in Southeast Asia exposed and connected, allowing species to spread southward to the islands in Southeast Asia (Tan et al., 2020). At this time, the Qinghai-Tibet Plateau and its tectonic morphology had been initially formed, and the arid areas began to retreat inland, while most of the inland arid areas began to gradually diverge from the humid areas affected by the East Asian monsoon (Harrison et al., 1992; Kutzbach and Ruddiman, 1993; Zhisheng et al., 2001; Qiang et al., 2011). These factors may have facilitated the differentiation of Cyclommatus stag beetles to form two clades corresponding to clade A and clade B during the late Oligocene (ca. 24.90 Mya).
In the Late Jurassic, western Sulawesi, Java, and Borneo separated from Australia and united with the Sunda lands during the Cretaceous (Hall, 2013). During the Tertiary period, Sulawesi separated from Borneo, forming the Makassar Strait, which became a barrier for species exchange between Borneo and Sulawesi with the central and northern Borneo and Palawan land surfaces in the Early Miocene (Hall, 2013). Thus, we speculated that it promoted the species diversity of the clade A through geographical isolation due to islands and straits, in the Early Miocene (ca. 18.19 Mya). After, it was the main uplift stage of the Qinghai-Tibet Plateau, the arid inland areas of Asia began to expand further (Harrison et al., 1992; Kutzbach and Ruddiman, 1993), and the monsoon gradually became stronger in the period of the Middle Miocene. Subsequently, the global temperature began to drop (Zachos et al., 2005). These events are believed to have accelerated the prosperity of most groups in the clade B of Cyclommatus stag beetles. In previous studies, there are several Asian taxa with distributions which are similar to our clade B group (e.g., Che et al., 2010; Gao et al., 2013; Klaus et al., 2016; Chen et al., 2018; Yan et al., 2020).
In the late Miocene, affected by the impact of the Indian Asian plate, Cyclommatus stag beetles differentiated many times. C. mniszechi, which is an clade A taxon, gradually spread from Southeast Asian islands to the Asian continent (8.02 Mya). The southern China populations of this species resemble the clade B species in external morphology, indicating that this species adapted its phenotype to the continental habitat environment after spreading from the Taiwan island to the mainland. This species could be used as an example of the spread to the continent from the islands. In general, our study strengthens the idea that Borneo and Indochina are evolutionary hotspots for the biodiversity in Southeast Asia (de Bruyn et al., 2014).
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
The animal study was reviewed and approved by Anhui University.
XZ and JY contributed to data curation, formal analysis, methodology, and writing of the original draft. XW supervised the manuscript writing and helped with data analysis. LZ contributed to the visualization, methodology, and investigation. LB contributed to sample collection, review and editing. All authors reviewed, edited, and approved the manuscript.
This work is supported by the National Natural Science Foundation of China (Nos. 31872276 and 31572311) and included study design, fieldwork, sample collection, laboratory work, data analysis, writing, and publication.
We are grateful to Eunjoong Kim, Jian-Yue Qiu, Hao Xu, Yun-Fei Wu, Ai-Min Shi, and Chang-Chin Chen for their kind donations of specimens.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2023.974315/full#supplementary-material
Abouheif, E., and Wray, G. A. (2002). Evolution of the gene network underlying wing Polyphenism in ants. Science 297, 249–252. doi: 10.1126/science.1071468
Araya, K. (1993). Relationship between the decay types of dead wood and occurrence of lucanid beetles (Coleoptera: Lucanidae). Appl. Entomol. Zool. 28, 27–33. doi: 10.1303/aez.28.27
Arrow, G. J. (1938). Some notes on stag-beetles and descriptions of a few new species. Ann. Mag. Nat. Hist. 2, 49–63. doi: 10.1080/03745481.1938.9755437
Arrow, G. J. (1950). The Fauna of India including Pakistan, Ceylon, Burma and Malaya. Coleoptera, Lamellicornia, Lucanidae and Passalidae. IV. London, Taylor and Francis Ltd.
Bain, R. H., and Hurley, M. M. (2011). A biogeographic synthesis of the amphibians and reptiles of Indochina. Bull. Amer. Mus. Nat. Hist. 360, 1–138. doi: 10.1206/360.1
Balke, M., Ribera, I., and Vogler, A. P. (2004). MtDNA phylogeny and biogeography of Copelatinae, a highly diverse group of tropical diving beetles (Dytiscidae). Mol. Phylogenet. Evol. 32, 866–880. doi: 10.1016/j.ympev.2004.03.014
Bellemain, E., and Ricklefs, R. E. (2008). Are islands the end of the colonization road? Trends Ecol. Evol. 23, 461–468. doi: 10.1016/j.tree.2008.05.001
Bouckaert, R., Heled, J., Kuehnert, D., Vaughan, T., Wu, C.-H., Xie, D., et al. (2014). BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 10:e1003537. doi: 10.1371/journal.pcbi.1003537
Buerki, S., Forest, F., Stadler, T., and Alvarez, N. (2013). The abrupt climate change at the Eocene-Oligocene boundary and the emergence of South-East Asia triggered the spread of sapindaceous lineages. Ann. Bot. 112, 151–160. doi: 10.1093/aob/mct106
Che, J., Zhou, W.-W., Hu, J.-S., Yan, F., Papenfuss, T. J., Wake, D. B., et al. (2010). Spiny frogs (Paini) illuminate the history of the Himalayan region and Southeast Asia. Proc. Natl. Acad. Sci. U. S. A. 107, 13765–13770. doi: 10.1073/pnas.1008415107
Chen, Y. J. (2018). Studies on the comparative mitochondrial genomics and phylogeny of Dorcus s. 1. (Coleoptera: Lucanidae). Master thesis. Hefei: Anhui University.
Chen, J.-M., Poyarkov, N. A., Suwannapoom, C., Lathrop, A., Wu, Y.-H., Zhou, W.-W., et al. (2018). Large-scale phylogenetic analyses provide insights into unrecognized diversity and historical biogeography of Asian leaf-litter frogs, genus Leptolalax (Anura: Megophryidae). Mol. Phylogenet. Evol. 124, 162–171. doi: 10.1016/j.ympev.2018.02.020
de Bruyn, M., Stelbrink, B., Morley, R. J., Hall, R., Carvalho, G. R., Cannon, C. H., et al. (2014). Borneo and Indochina are major evolutionary hotspots for southeast Asian biodiversity. Syst. Biol. 63, 879–901. doi: 10.1093/sysbio/syu047
Didier, R. (1927). Quelques modifications à la classification des Lucanides [Col.]. A propos du genre Cyclommatus Parry. Bul. Soc. Entomol. Fr. 32, 101–103. doi: 10.3406/bsef.1927.27797
Didier, R., and Séguy, E. (1953). Catalogue Illustré des Lucanides du Globe. Encyclopédie Entomologique Série A 27, 1–223.
Filardi, C. E., and Moyle, R. G. (2005). Single origin of a pan-Pacific bird group and upstream colonization of Australasia. Nature 438, 216–219. doi: 10.1038/nature04057
Gao, Y.-D., Harris, A., Zhou, S.-D., and He, X.-J. (2013). Evolutionary events in Lilium (including Nomocharis, Liliaceae) are temporally correlated with orogenies of the Q–T plateau and the Hengduan Mountains. Mol. Phylogenet. Evol. 68, 443–460. doi: 10.1016/j.ympev.2013.04.026
Gotoh, H., Miyakawa, H., Ishikawa, A., Ishikawa, Y., Sugime, Y., Emlen, D. J., et al. (2014). Developmental link between sex and nutrition; doublesex regulates sex-specific mandible growth via juvenile hormone signaling in stag beetles. PLoS Genet. 10:e1004098. doi: 10.1371/journal.pgen.1004098
Gotoh, H., Zinna, R. A., Warren, I., DeNieu, M., Niimi, T., Dworkin, I., et al. (2016). Identification and functional analyses of sex determination genes in the sexually dimorphic stag beetle Cyclommatus metallifer. BMC Genom. 17:250. doi: 10.1186/s12864-016-2522-8
Goyens, J., Dirckx, J., and Aerts, P. (2015). Costly sexual dimorphism in Cyclommatus metallifer stag beetles. Funct. Ecol. 29, 35–43. doi: 10.1111/1365-2435.12294
Grismer, L. L., Wood, P. L., Aowphol, A., Cota, M., Grismer, M. S., Murdoch, M. L., et al. (2017). Out of Borneo, again and again: biogeography of the stream toad genus Ansonia Stoliczka (Anura: Bufonidae) and the discovery of the first limestone cave-dwelling species. Biol. J. Linnean Soc. 120, 371–395. doi: 10.1111/bij.12886
Hall, R. (2013). The palaeogeography of Sundaland and Wallacea since the late Jurassic. J. Limnol. 72, 1–17. doi: 10.4081/jlimnol.2013.s2.e1
Harrison, T. M., Copeland, P., Kidd, W. S., and Yin, A. (1992). Raising tibet. Science 255, 1663–1670. doi: 10.1126/science.255.5052.1663
Holloway, B. A. (2007). Fauna of New Zealand, Lucanidae (Insecta: Coleoptera). Number 61, Lincoln: Manaaki Whenua.
Hope, F. W., and Westwood, J. O. (1845). A catalogue of the Lucanoid Coleoptera in the collection of the rev. F.W. Hope, M.a., F.R.S., etc., together with descriptions of the New species therein contained. London: J.C. Bridgewater.
Hosoya, T., Honda, M., and Araya, K. (2001). Genetic variations of 16S rRNA gene observed in Ceruchus lignarius and Dorcus rectus rectus (Coleoptera: Lucanidae). Entomol. Sci. 4, 335–344.
Huang, H., and Chen, C. C. (2017). Stag beetles of China III. New Taipei City: Formosa Ecological Company.
Jonsson, K. A., Fabre, P.-H., Ricklefs, R. E., and Fjeldsa, J. (2011). Major global radiation of corvoid birds originated in the proto-Papuan archipelago. Proc. Natl. Acad. Sci. U. S. A. 108, 2328–2333. doi: 10.1073/pnas.1018956108
Kim, S. I., and Farrell, B. D. (2015). Phylogeny of world stag beetles (Coleoptera: Lucanidae) reveals a Gondwanan origin of Darwin’s stag beetle. Mol. Phylogenet. Evol. 86, 35–48. doi: 10.1016/j.ympev.2015.02.015
Klaus, S., Morley, R. J., Plath, M., Zhang, Y.-P., and Li, J.-T. (2016). Biotic interchange between the Indian subcontinent and mainland Asia through time. Nat. Commun. 7:12132. doi: 10.1038/ncomms12132
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Kutzbach, J. E., and Ruddiman, P. (1993). Sensivitivity of Eurasian climate to surface uplift of the Tibetian plateau. J. Geol. 101, 177–190. doi: 10.1086/648215
Landis, M. J., Matzke, N. J., Moore, B. R., and Huelsenbeck, J. P. (2013). Bayesian analysis of biogeography when the number of areas is large. Syst. Biol. 62, 789–804. doi: 10.1093/sysbio/syt040
Lanfear, R., Frandsen, P. B., Wright, A. M., Senfeld, T., and Calcott, B. (2017). PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 34, msw260–msw773. doi: 10.1093/molbev/msw260
Liang, B., Zhou, R.-B., Liu, Y.-L., Chen, B., Grismer, L., and Wang, N. (2018). Renewed classification within Goniurosaurus (Squamata: Eublepharidae) uncovers the dual roles of a continental island (Hainan) in species evolution. Mol. Phylogenet. Evol. 127, 646–654. doi: 10.1016/j.ympev.2018.06.011
Lin, C. P., Huang, J. P., Lee, Y. H., and Chen, M. Y. (2011). Phylogenetic position of a threatened stag beetle, Lucanus datunensis (Coleoptera: Lucanidae) in Taiwan and implications for conservation. Conserv. Genet. 12, 337–341. doi: 10.1007/s10592-009-9996-8
Liu, J., Chenggong, L. I., You, S., Wan, X., and Ecology, D. O. (2017). The first complete mitogenome of Cyclommatus stag beetles (Coleoptera: Lucanidae) with the phylogenetic implications. Entomotaxonomia 39, 294–299. doi: 10.11680/entomotax.2017035
Maes, J. M. (1992). Lista de los Lucanidae (Coleoptera) del Mundo. Rev. Nicaragua. Entomol. 22A, 1–60.
Marcilla, A., Bargues, M. D., Ramsey, J. M., Magallon-Gastelum, E., Salazar-Schettino, P. M., Abad-Franch, F., et al. (2001). The ITS-2 of the nuclear rDNA as a molecular marker for populations, species, and phylogenetic relationships in Triatominae (Hemiptera: Reduviidae), vectors of Chagas disease. Mol. Phylogenet. Evol. 18, 136–142. doi: 10.1006/mpev.2000.0864
Massana, K. A., Beaulieu, J. M., Matzke, N. J., and O’Meara, B. C. (2015). Non-null effects of the null range in biogeographic models: exploring parameter estimation in the DEC model. bioRxiv 2015:026914. doi: 10.1101/026914
Matuszak, S., Muellner-Riehl, A. N., Sun, H., and Favre, A. (2016). Dispersal routes between biodiversity hotspots in Asia: the case of the mountain genus Tripterospermum (Gentianinae, Gentianaceae) and its close relatives. J. Biogeogr. 43, 580–590. doi: 10.1111/jbi.12617
Matzke, N. J. (2013). Probabilistic historical biogeography: new models for founder-event speciation, imperfect detection, and fossils allow improved accuracy and model-testing. Front. Biogeogr. 5, 242–248. doi: 10.5901/ajis.2015.v4n2p137
Matzke, N. J. (2014a). Model selection in historical biogeography reveals that founder-event speciation is a crucial process in island clades. Syst. Biol. 63, 951–970. doi: 10.1093/sysbio/syu056
Matzke, N. J. (2014b). BioGeography with Bayesian (and likelihood) evolutionary analysis in R scripts. R package BioGeoBEARS version 0.2.1.
Monaghan, M. T., Inward, D. J. G., Hunt, T., and Vogler, A. P. (2007). A molecular phylogenetic analysis of the Scarabaeinae (dung beetles). Mol. Phylogenet. Evol. 45, 674–692. doi: 10.1016/j.ympev.2007.06.009
Nagel, P. (1936). Neues über Hirschkäfer (Col. Fam. Lucanidae). Stett. Entomol. Zeitung. 97, 289–302.
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Papadopoulou, A., Anastasiou, I., and Vogler, A. P. (2010). Revisiting the insect mitochondrial molecular clock: the mid-Aegean trench calibration. Mol. Biol. Evol. 27, 1659–1672. doi: 10.1093/molbev/msq051
Parry, F. J. S. (1863). A few remarks upon Mr. James Thomson’s catalogue of Lucanidae, published in the Annales de la Société Entomologique de France, 1862. Trans. R. Entomol. Soc. Lond. 1, 442–452. doi: 10.1111/j.1365-2311.1863.tb01291.x
Poyarkov, N. A., Nguyen, T. V., Pawangkhanant, P., Yushchenko, P., Brakels, P., Nguyen, L. H., et al. (2022). An integrative taxonomic revision of slug-eating snakes (Squamata: Pareidae: Pareineae) reveals unprecedented diversity in Indochina. PeerJ 10, e12713–e12799. doi: 10.7717/peerj.12713
Qiang, X. K., An, Z. S., Song, Y. G., Chang, H., Sun, Y. B., Liu, W. G., et al. (2011). New eolian red clay sequence on the western Chinese loess plateau linked to onset of Asian desertification about 25 ma ago. Sci. China Earth Sci. 54, 136–144. doi: 10.1007/s11430-010-4126-5
R Core Team. (2021). R: A language and environment for statistical computing. R foundation for statistical computing. Vienna, Austria. Available at: http://www.Rproject.org/
Rambaut, A., Drummond, A. J., Xie, D., Baele, G., and Suchard, M. A. (2018). Posterior summarization in Bayesian Phylogenetics using Tracer 1.7. Syst. Biol. 67, 901–904. doi: 10.1093/sysbio/syy032
Ree, R. H., and Smith, S. A. (2008). Maximum likelihood inference of geographic range evolution by dispersal, local extinction, and cladogenesis. Syst. Biol. 57, 4–14. doi: 10.1080/10635150701883881
Ronquist, F. (1997). Dispersal-vicariance analysis: a new approach to the quantification of historical biogeography. Syst. Biol. 46, 195–203. doi: 10.1093/sysbio/46.1.195
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Hohna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Simon, C., Frati, F., Beckenbach, A., Crespi, B., Liu, H., and Flook, P. (1994). Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 87, 651–701. doi: 10.1093/aesa/87.6.651
Songvorawit, N., Butcher, B. A., and Chaisuekul, C. (2017). Decaying Wood preference of stag beetles (Coleoptera: Lucanidae) in a tropical dry-Evergreen Forest. Environ. Entomol. 46, 1322–1328. doi: 10.1093/ee/nvx143
Tan, K., Malabrigo, P. L., and Ren, M. X. (2020). Origin and evolution of biodiversity hotspots in Southeast Asia. Acta Ecol. Sin. 40, 3866–3877. doi: 10.5846/stxb201904160762
Tanahashi, M., Matsushita, N., and Togashi, K. (2009). Are stag beetles fungivorous? J. Insect Physiol. 55, 983–988. doi: 10.1016/j.jinsphys.2009.07.002
Thomaes, A. (2009). “A protection strategy for the stag beetle (Lucanus cervus, (L., 1758), Lucanidae) based on habitat requirements.” in Proceedings of the 5th Symposium and Workshop on the Conservation of Saproxylic Beetles. 149–160.
Thomson, J. (1862). Catalogue des Lucanides de la collection de M. James Thomson, suivi d'un appendix renfermant la description des coupes génériques et spécifiques nouvelles. Ann. Soc. Entomol. Fr. 2, 389–436.
Trifinopoulos, J., Nguyen, L.-T., von Haeseler, A., and Minh, B. Q. (2016). W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44, W232–W235. doi: 10.1093/nar/gkw256
Tsai, C.-L., and Yeh, W.-B. (2016). Subspecific differentiation events of montane stag beetles (Coleoptera, Lucanidae) endemic to Formosa Island. PLoS One 11:e0156600. doi: 10.1371/journal.pone.0156600
van der Kaars, W. A. (1991). Palynological aspects of site 767 in the Celebes Sea. In Proceedings of the ocean drilling program, scientific results. College Station, TX 124, 369–374.
Wan, X. (2007). Study on the systematics of Lucanidae from China (Coleoptera: Scarabaeoidea). Ph.D. thesis. Beijing: Institute of Zoology, Chinese Academy of Sciences.
Ward, P. S., and Downie, D. A. (2005). The ant subfamily Pseudomyrmecinae (hymenoptera: Formicidae): phylogeny and evolution of big-eyed arboreal ants. Syst. Entomol. 30, 310–335. doi: 10.1111/j.1365-3113.2004.00281.x
Weinell, J. L., Barley, A. J., Siler, C. D., Orlov, N. L., Ananjeva, N. B., Oaks, J. R., et al. (2021). Phylogenetic relationships and biogeographic range evolution in cat-eyed snakes, Boiga (Serpentes: Colubridae). Zool. J. Linnean Soc. 192, 169–184. doi: 10.1093/zoolinnean/zlaa090
Wild, A. L., and Maddison, D. R. (2008). Evaluating nuclear protein-coding genes for phylogenetic utility in beetles. Mol. Phylogenet. Evol. 48, 877–891. doi: 10.1016/j.ympev.2008.05.023
Williams, E. W., Gardner, E. M., Harris, R., Chaveerach, A., Pereira, J. T., and Zerega, N. J. C. (2017). Out of Borneo: biogeography, phylogeny and divergence date estimates of Artocarpus (Moraceae). Ann. Bot. 119, 611–627. doi: 10.1093/aob/mcw249
Wu, Y.Y. (2016). Molecular Phylogeny of Lucanidae from China (Coleoptera: Lucanidae). Master thesis. Hefei: Anhui University.
Yan, P., Pan, T., Wu, G., Kang, X., Ali, I., Zhou, W., et al. (2020). Species delimitation and evolutionary history of tree frogs in the Hyla chinensis group (Hylidae, amphibian). Front. Ecol. Evol. 8:234. doi: 10.3389/fevo.2020.00234
Yu, Y., Blair, C., and He, X. (2020). RASP 4: ancestral state reconstruction tool for multiple genes and characters. Mol. Biol. Evol. 37, 604–606. doi: 10.1093/molbev/msz257
Yuan, J. J., Chen, D., and Wan, X. (2021). A multilocus assessment reveals two new synonymies for east Asian Cyclommatus stag beetles (Coleoptera, Lucanidae). ZooKeys 1021, 65–79. doi: 10.3897/zookeys.1021.58832
Zachos, J. C., Rohl, U., Schellenberg, S. A., Sluijs, A., Hodell, D. A., Kelly, D. C., et al. (2005). Rapid acidification of the ocean during the Paleocene-Eocene thermal maximum. Science 308, 1611–1615. doi: 10.1126/science.1109004
Zhang, D., Gao, F., Jakovlic, I., Zou, H., Zhang, J., Li, W. X., et al. (2020). PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20, 348–355. doi: 10.1111/1755-0998.13096
Keywords: Lucanidae, Cyclommatus, molecular phylogeny, divergence time, “upstream” colonization hypothesis
Citation: Zhu XL, Yuan JJ, Zhou LY, Bartolozzi L and Wan X (2023) Molecular phylogeny and historical biogeography of Cyclommatus stag beetles (Coleoptera: Lucanidae): Insights into their evolution and diversification in tropical and subtropical Asia. Front. Ecol. Evol. 11:974315. doi: 10.3389/fevo.2023.974315
Edited by:
Oana Moldovan, Emil Racovita Institute of Speleology, Romanian Academy, RomaniaCopyright © 2023 Zhu, Yuan, Zhou, Bartolozzi and Wan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xia Wan, ✉ d2FueGlhQGFodS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.