 Junxia Mao
Junxia Mao Ying Tian
Ying Tian Qian Liu
Qian Liu Danyang Li
Danyang Li Xinfan Ge
Xinfan Ge Xubo Wang
Xubo Wang Zhenlin Hao
Zhenlin Hao- Key Laboratory of Mariculture & Stock Enhancement in North China’s Sea, Ministry of Agriculture and Rural Affairs, Dalian Ocean University, Dalian, China
Introduction: The Pacific oyster (Crassostrea gigas), one of the major aquaculture shellfish worldwide, has strong environmental adaptability. However, genetic diversity and population structure of the Pacific oysters in Dalian Sea, the major natural and farming area of the species in China, has not been systematically investigated, especially at genome-wide level, limiting the conservation and management of the species.
Methods: In this study, whole-genome resequencing of 105 individuals from seven Pacific oyster populations, including five wild and one cultured populations in Dalian and one wild population in Qingdao relatively distant from others, were first performed.
Results: A total of 2,363,318 single nucleotide polymorphisms (SNPs) were identified. Based on all these SNPs, similar but relatively low genetic diversity (0.2352~0.2527) was found in the seven populations. The principal component analysis (PCA), phylogenetic and population structure analysis consistently revealed weak differentiation among the seven populations. Frequent migration events were detected among the studied populations by TreeMix, which probably led to a high genetic similarity of these populations. Rapid linkage disequilibrium (LD) decay was observed in the genome of the Pacific oyster. Investigation of genome-wide selection signatures of these populations identified many selected genes involved in the biological processes related to DNA metabolism and stability, shell formation, and environmental stress response, which may be critical for oysters to adapt to the stressful environments.
Discussion: This study laid theoretical basis for the subsequent germplasm conservation, management and genetic breeding of the indigenous Pacific oysters, and provided novel insights for the adaptive evolutionary mechanism of oysters.
1 Introduction
The Pacific oyster (Crassostrea gigas), a sessile and filter-feeding bivalve mollusc, is native to the coast of East Asia, including China, Japan, and Korea. Its high fecundity and broad environmental adaptability, such as high tolerance to a variety of different salinities and temperatures, in conjunction with its rapid growth rate and robust disease resistance, have made it an economically important species produced throughout the world (Miossec et al., 2009; Zhang et al., 2012; Li et al., 2018). Due to its ecological and economic importance, the Pacific oyster has also been a model marine organism in research of biology, marine ecology, genetics, and genomics (Li et al., 2018). Recently, much attention has also been paid to this species in terms of their adaptive mechanism to the changing environments (Zhang et al., 2012; Dineshram et al., 2016; Zhang et al., 2016; Li et al., 2018; Wang et al., 2023). However, the wild Pacific oysters are facing a crisis of population decline in East Asia in recent years because of anthropogenic and natural causes, such as global warming, over-fishing, expansion of oyster aquaculture, and deterioration of seawater environment caused by human activities (Aranishi, 2006; Guo et al., 2006; Li et al., 2015). The ancestral genetic diversity of different wild populations probably has been disturbed or lost, which would negatively affect the adaptation of populations to their habitats (Aranishi, 2006).
China is the major producing country of the Pacific oyster, and the production in Liaoning and Shandong provinces accounts for over 90% of the total in China (Zhong et al., 2017). Dalian, a city lying on the south side of Liaoning province, the northeast of China (E120°58′–E123°31′, N38°43′–N40°10′), spans the Yellow Sea and the Bohai Sea and is the major farming area of Pacific oysters in Liaoning Province. Additionally, almost the entire coast of Dalian has a natural distribution of the Pacific oyster. However, the Pacific oyster industry in Dalian is facing challenges due to a lack of excellent varieties, a high mortality rate, a slow growth rate, and poor stress and disease resistance of cultured Pacific oyster (Teng et al., 2022). Understanding the genetic diversity and differentiation of the Pacific oysters distributed in Dalian, though probably a relatively narrow spatial distribution, is critical for the conservation and development of the local genetic resources, which could help select fine germplasms for genetic breeding programs for different breeding purposes. Nevertheless, related research has rarely been performed. It was reported that current aquaculture practices had led to serious genetic effects on wild populations due to the mixture of farmed individuals into wild populations by accidental escape or human intentional releasing of farmed individuals (Meistertzheim et al., 2013; Semeraro et al., 2015; Šegvić-Bubić et al., 2020). Taking into account the large farming scale of the Pacific oyster in Dalian, the genetic impacts of cultured individuals on the wild should be evaluated, but the situation is also completely unclear. Therefore, the population genetics of the Pacific oysters in Dalian needs a systematic study.
Though some studies have been carried out to investigate the genetic diversity and population structures of different native Pacific oyster populations, most studies used a very limited number of markers, and even inconsistent conclusions were obtained due to different types and numbers of markers or sequences used (Aranishi, 2006; Rohfritsch et al., 2013; Li et al., 2015; Guo et al., 2016; Zhong et al., 2017), which may confuse scientists during genetic resource management. With the advent of next-generation sequencing and the availability of genomes of many species, whole-genome sequencing has been a powerful tool for studying the population genetics of the species on a fine scale using genome-wide single-nucleotide polymorphisms (SNPs). It also has the potential to identify genomic selection signatures and genes related to economic or adaptive important traits (Liu et al., 2017; Li et al., 2018; Wang et al., 2021; Li et al., 2022; Chen et al., 2023; Zheng et al., 2023).
In the present study, whole-genome resequencing was first performed for different geographical populations of the Pacific oyster in Dalian, and genetic variation was surveyed. A comprehensive analysis of genetic diversity, population structure, and phylogenetic relationships was conducted among the wild populations together with one local cultured population. Then, gene flow, linkage disequilibrium (LD), and demographic history of these populations were inferred. In addition, potentially selected genomic regions and genes were identified, which would be helpful for elucidating the adaptive mechanism of the Pacific oyster to the local environment. The study provided novel insights into subsequent germplasm conservation, management, and development of genetic breeding strategies for indigenous Pacific oysters.
2 Materials and methods
2.1 Sample collection
To investigate the genetic diversity and architecture of Pacific oysters distributed in the Dalian Sea area (Liaoning, China), a total of 105 Pacific oysters from the seven different populations (15 individuals for each population) were collected in July 2022. As shown in Figure 1, six populations were collected along the coast of Dalian, including five wild populations of Lvshun (LS), Laohutan (LHT), Sanshan island (SSD), Guanglu island (GLD), and Xingshutun (XST) and one cultured population that was selected for years by the farm in Guanglu island (cultured (CT)). Additionally, a relatively distant wild population distributed in the Qingdao Sea area (QD) was also collected. The fresh muscle tissues of 105 individuals were sampled, immediately frozen in liquid nitrogen, and stored at −80°C for DNA extraction. All experiments were carried out according to the animal ethics guidelines approved by the Ethics Committee of Dalian Ocean University.
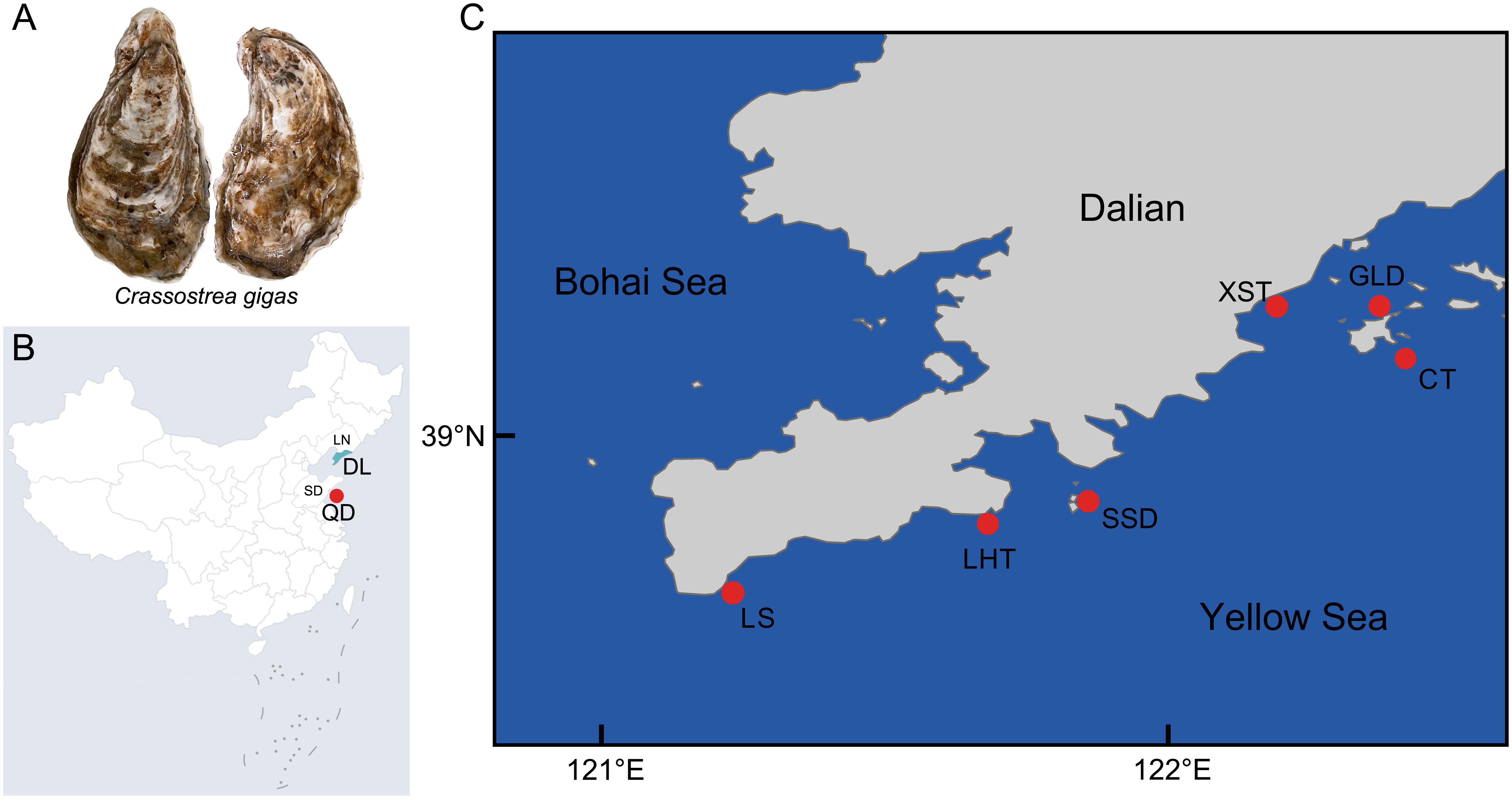
Figure 1 Geographical distribution of the studied Pacific oyster (Crassostrea gigas) populations. (A) The picture of the Pacific oyster. (B) The Chinese map. (C) The map of Dalian. LS, Lvshun; LHT, Laohutan; SSD, Sanshan island; GLD, Guanglu island; XST, Xingshutun; CT, cultured population; QD, Qingdao; DL, Dalian; SD, Shandong province; LN, Liaoning province.
2.2 DNA extraction, library construction, and resequencing
Genomic DNA samples were prepared following the standard phenol–chloroform DNA isolation method. The DNA quality and concentration were assessed by agarose gel electrophoresis and Nanodrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). The DNA samples were interrupted with a length of ∼350 bp using a Covaris ultrasonic crusher. Then, the sequence library for each sample was constructed using a NEB Next® Ultra™ II DNA Library Prep Kit, following the manufacturer’s protocol. Finally, all the libraries were subjected to 150-bp paired-end sequencing on the Illumina NovaSeq 6000 platform. All the sequencing data are deposited at the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) platform with accession no. PRJNA999033.
2.3 SNP calling and annotation
Raw sequencing data were processed to remove adapters and low-quality reads, which contained unidentified nucleotides (N) >10% or >50% bases of a read with quality value ≤5. The high-quality clean reads of each individual were mapped to the genome of the Pacific oyster (https://www.ncbi.nlm.nih.gov/genome/?term=txid29159) using the BWA software (Li and Durbin, 2009) with the parameters “mem -t 4 -k 32 -M”. Duplicate read data were excluded from the mapping results by SAMTOOLS software (Li et al., 2009) with the parameter “rmdup”. SAMTOOLS software (Li et al., 2009) was used to perform SNP calling for all samples on a population scale with the Bayesian model. The SNPs were screened using the following criteria: Dp (minimum sequencing depth for a single sample) ≥3; miss (miss rate for one SNP site) ≤0.2, and maf (minimum allele frequency) ≥0.05. The screened SNPs were annotated using ANNOVAR software (Wang et al., 2010).
2.4 Genetic diversity and population structure analysis
The nucleotide diversity (Pi), observed heterozygosity (Ho), expected heterozygosity (He), inbreeding coefficients (Fis), and genetic differentiation (Fst) were statistically analyzed with generic formulas using the R package to estimate the genetic diversity and differentiation levels of the seven Pacific oyster populations. A phylogenetic tree was constructed to reveal the phylogenetic relationships between individuals of the seven populations. The TreeBest software (http://treesoft.sourceforge.net/treebest.shtml) was used to calculate the genetic distance matrix, and then the phylogenetic tree was inferred through the neighbor-joining (NJ) method using 1,000 bootstraps. The principal component analysis (PCA) was performed using the GCTA software (Yang et al., 2011), and the first three principal components were calculated. The population structure was analyzed using the ADMIXTURE software (Alexander et al., 2009) with the genetic clusters (K value) ranging from 2 to 6, and the optimal K value was determined using the minimum value of the cross-validation (CV) error rate.
2.5 Migration events between the populations
Migration events between the populations and the directionality of gene flow were inferred using the TreeMix software (Pickrell and Pritchard, 2012). First, a maximum-likelihood tree was constructed based on the population allele frequency covariance matrix. Then, migration edges were added to the phylogenetic tree to improve the fit by comparing the estimated covariance modeled by this tree to the real observed covariance between populations.
2.6 Linkage disequilibrium and demographic history analysis
The PopLDdecay software (Zhang et al., 2019) was used to calculate the squared correlation (r2) between any two loci to evaluate the LD decay of each population. The demographic history of the seven populations was detected using the pairwise sequentially Markovian coalescent (PSMC) method (Li and Durbin, 2011), and the parameters of g = 2 and a rate of 1.39 × 10−9 mutations per generation (Zhao et al., 2014) were used to estimate the distribution time.
2.7 Selective sweep analysis
To identify genomic regions with signatures of selection, both Fst values and θπ ratios were calculated between population pairs (Li et al., 2018). The overlapping windows simultaneously with significantly high Fst values (top 5% regions) and significantly high and low θπ ratios (log2-transformed; top 5% regions) distributions were considered as regions with selection signals along the genome. Selected regions for one population were merged, and genes within or overlapped by these selective regions were defined as candidate selective genes. To further investigate the biological significance of the selective genes of each population, Gene Ontology (GO; http://www.geneontology.org/) and Kyoto Encyclopedia of Genes and Genomes (KEGG; http://www.genome.jp/kegg/) enrichment analyses were implemented based on the hypergeometric distribution test in R package. GO terms and KEGG pathways with a p-value <0.05 and gene number ≥2 were considered significantly enriched.
3 Results
3.1 Whole-genome resequencing and genetic variations
Whole-genome resequencing of 105 individuals of Pacific oysters from the seven populations (XST, GLD, QD, LS, SSD, LHT, and CT) generated a total of 4.5 billion high-quality reads (~667.93 Gb of sequences) with an average of 42.4 million reads (~10X) per individual (Supplementary Table S1). Reads were aligned to the Pacific oyster genome using BWA with an average mapping rate of 91.42% (Supplementary Table S1). After variant calling and filtering, a total of 2,363,318 SNPs were identified, of which 573,572 (~24.3%) were exonic, 1,105,751 (~46.8%) were intronic, and 531,806 (~22.5%) were intergenic (Supplementary Table S2). In addition, a large number of SNPs (380,861, ~16.1%) were found located in coding regions with 68.7% synonymous and 31.2% non-synonymous (Supplementary Table S2). The ts/tv (transition/transversion) ratio for all the detected SNPs was 1.30.
3.2 Genetic diversity of the seven populations
Genetic diversity analysis revealed that the nucleotide diversity (Pi), observed heterozygosity (Ho), and expected heterozygosity (He) varied among the seven Pacific oyster populations, which ranged from 0.2352 to 0.2527, 0.2441 to 0.2853, and 0.2268 to 0.2437, respectively (Table 1). The inbreeding coefficients (Fis) for all the seven populations were positive and small (ranging from 0.0249 to 0.0843), which indicated a relatively low degree of inbreeding effect in these populations (Table 1). The overall assessment showed that the LHT population had the highest genetic diversity and that the QD population had the lowest genetic diversity.

Table 1 Genetic diversity analysis of the seven Pacific oyster populations.
3.3 Phylogenetic relationship and population structure
Low values of genetic differentiation were detected between the studied populations, which ranged from 0.01787 (XST and GLD) to 0.02428 (XST and CT) (Table 2). The genetic differentiation degree between the CT and the other six wild populations was a little higher (Table 2). An NJ phylogenetic tree was constructed to analyze the genetic relationship among individuals of different populations. Individuals from CT, SSD, and LHT were closely related and clustered together, and individuals from QT, GLD, and XST grouped into another cluster (Figure 2A), while individuals from the LS population were scattered in the tree. The PCA plot showed similar results. Though individuals from the same populations were not clearly separate from the other populations, which indicated the low genetic differentiation of these populations, closer genetic relationships were also found among the CT, SSD, and LHT populations and the QD, GLD, and XST populations (Figure 2B).
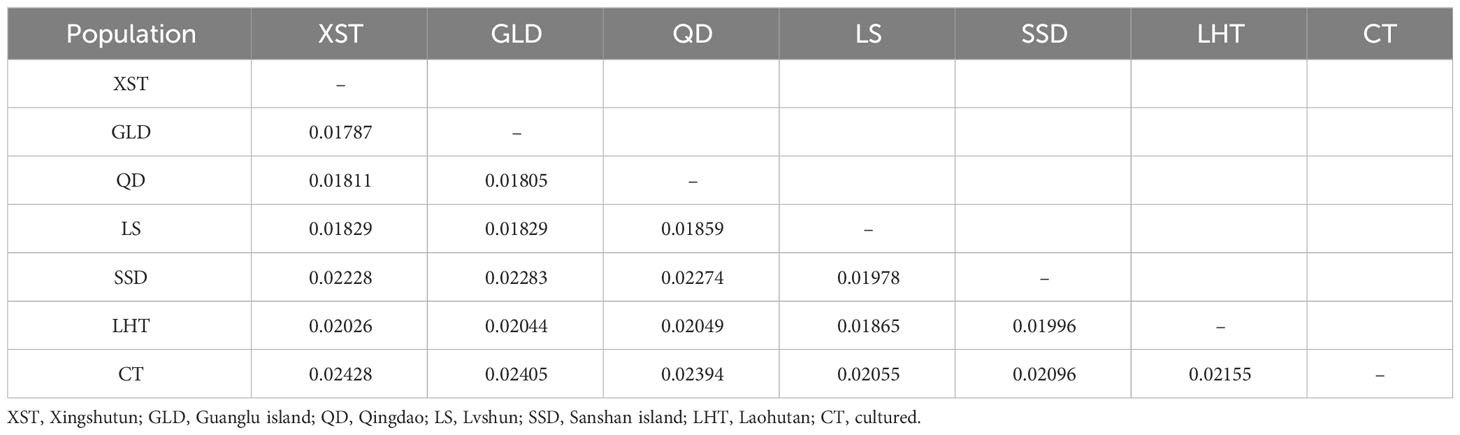
Table 2 Fst values between the seven Pacific oyster populations.
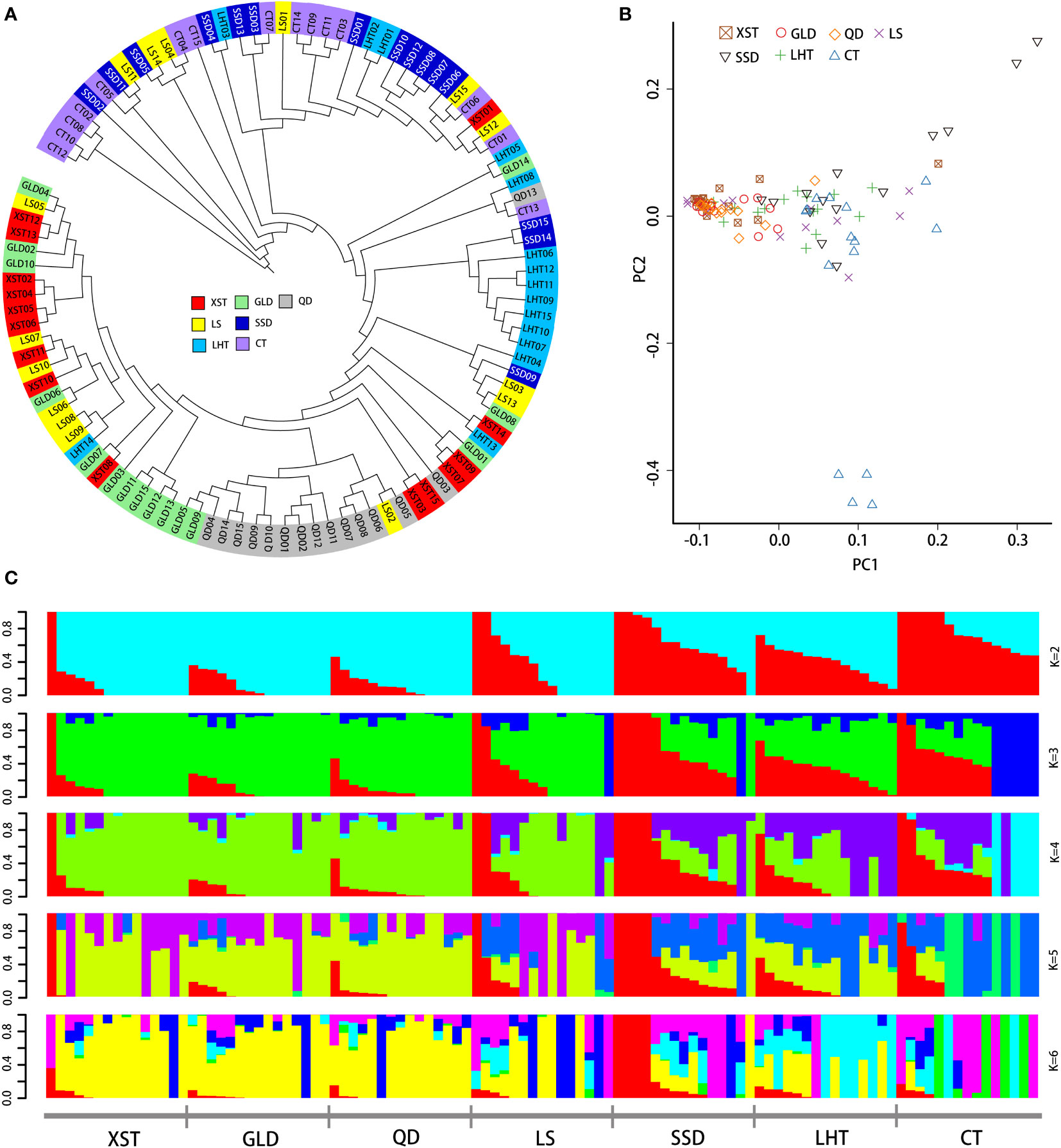
Figure 2 Phylogenetic relationship and population structure of the seven Pacific oyster populations. (A) Neighbor-joining phylogenetic tree of the seven populations. (B) Principal component analysis for the first two PCs of 105 Pacific oysters. (C) Admixture analysis with five assumed ancestral numbers (K = 2 to 6). PCs, principal components.
Admixture analysis was performed to investigate admixture degrees among the seven populations with the assumed ancestral number K from 2 to 8. K = 2 was suggested as the most plausible ancestral number by cross-validation error test (Supplementary Figure S1), reflecting the sampled populations, which could be divided into two groups. In this scenario, group 1 mainly contained individuals from CT, SSD, and LHT, indicating that the three populations probably originated from the same ancestor. Group 2 including the QT, GLD, and XST populations probably descended from another ancestor (Figure 2C). Population admixture based on K = 3 to 6 showed a similar result (Figure 2C).
3.4 Potential gene flows between the populations
TreeMix analysis was conducted to build a maximum-likelihood tree and infer migration events between the studied populations, and the model was estimated using the residuals from the tree. The results suggested that the genetic relationship showed in the maximum-likelihood tree with four migration events was consistent with that shown by the NJ tree, PCA, and admixture analyses (Figure 3A), and the model could fit the data well (Figure 3B). Maximum-likelihood trees with other number migration events were inconsistent with the NJ tree and had higher residuals (Supplementary Figure S2). Based on this, gene flows were detected from CT to LS, CT to QD, and SSD to LS and the common ancestor of XST, GLD, QD, and LS to LHT (Figure 3A).
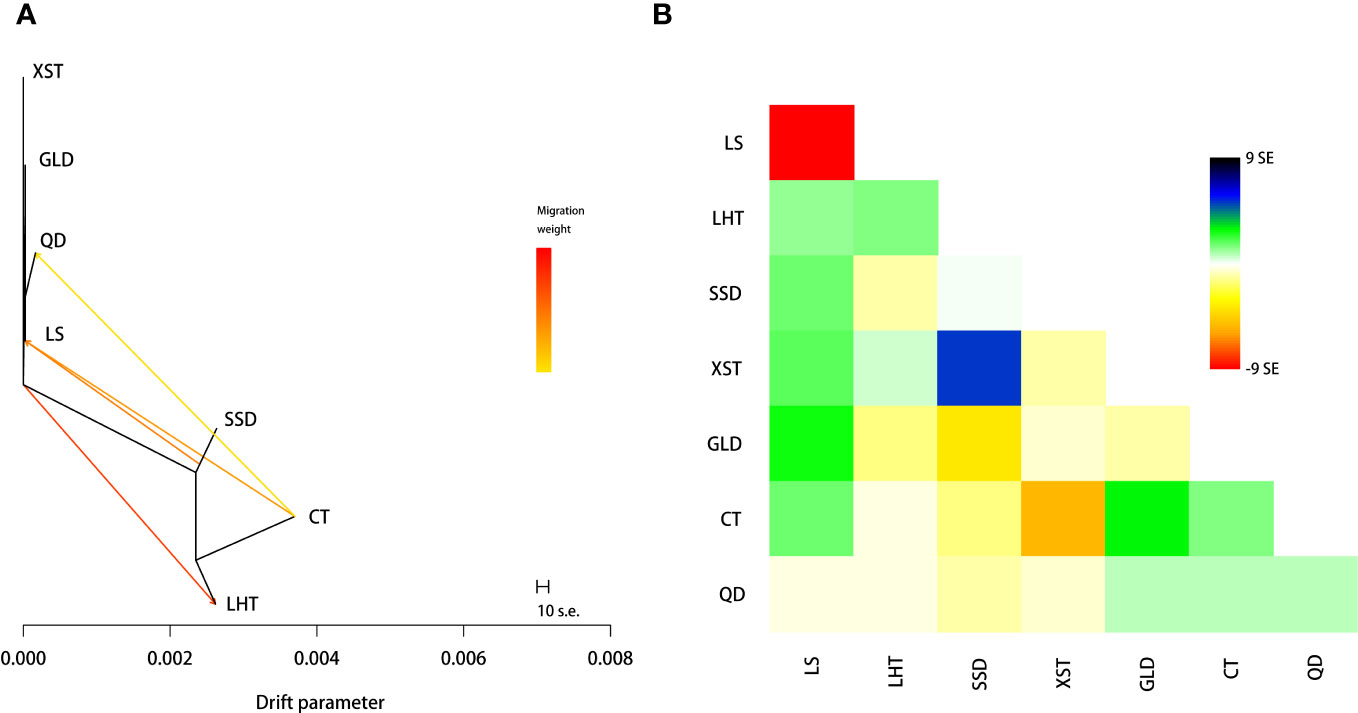
Figure 3 Maximum-likelihood tree and gene flow inferred among the seven Pacific oyster populations. (A) Maximum-likelihood tree with four migration events. Arrows represent the direction of gene glow, and the line colors indicate the migration weight. (B) Residual fit from the maximum-likelihood tree in panel (A).
3.5 Linkage disequilibrium and demographic history
LD analysis showed that the LD decayed rapidly with genetic distance in the Pacific oyster and that the r2 decreased to 0.15 within 500 bp (Figure 4A). A similar finding was also reported in the previous study by Li et al. (2018). The extent of LD of one species can be affected by many factors, such as genomic history, demographic history, and selection (Ardlie et al., 2002; Li and Merilä, 2011). The Pacific oyster is an outcrossing heterozygote species having a high mutation rate, which will cause a decrease in LD (Gupta et al., 2005; Guo et al., 2016). The fastest decay rate was detected in the LS population, while the slowest was in the CT population (Figure 4A), suggesting a low level of LD in wild oyster populations. Artificial selection will reduce allele numbers in cultured populations, thus increasing LD levels (Guo et al., 2016; Zhong et al., 2017). PSMC was utilized to infer the historically effective population size to explore the demographic histories of the seven populations. As shown in Figures 4B–H, individuals from the same population represented similar historical fluctuations in effective population size, and all populations displayed a decline in population size approximately 105 years ago.
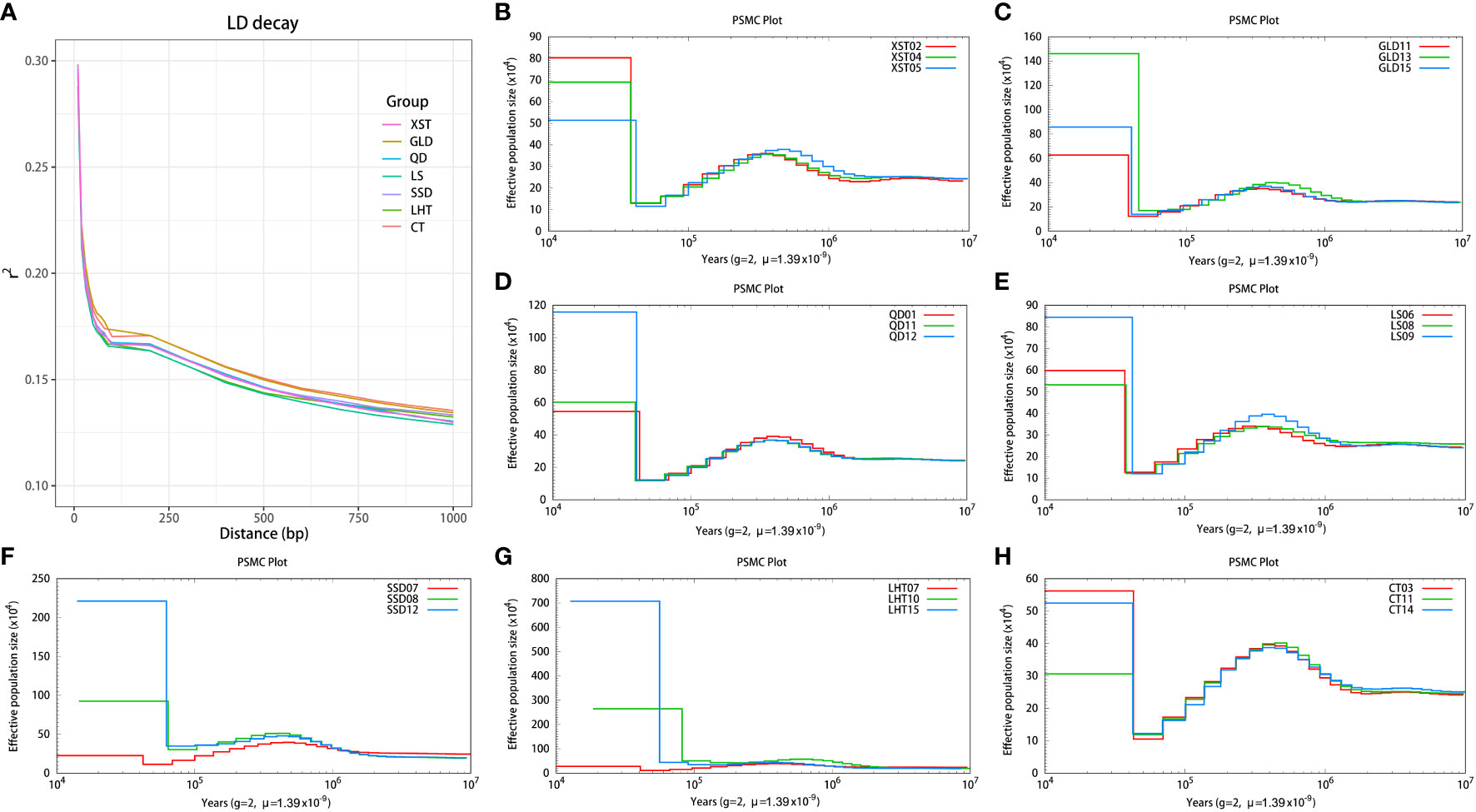
Figure 4 Linkage disequilibrium (LD) decay and demographic history of the seven Pacific oyster populations. (A) LD decay in different populations. (B–H) The effective population size and history of the seven populations.
3.6 Selective regions of the seven populations
Genome-wide scan of selective sweeps has been a powerful method to detect potential regions and genes involved in local adaptation. To search for the genomic evidence of the Pacific oyster adapting to environment changes, both θπ ratios and Fst values were used to identify genomic selection signals of the studied populations. The regions within the top 5% were defined as the selected regions. Pairwise comparisons of the seven groups identified 348 genomic regions harboring 1,157 genes in XST, 366 genomic regions harboring 1,269 genes in GLD, 368 genomic regions harboring 1,278 genes in QD, 276 genomic regions harboring 975 genes in LS, 172 genomic regions harboring 601 genes in SSD, 192 genomic regions harboring 588 genes in LHT, and 209 genomic regions harboring 710 genes in CT showing significant selection signals (Figure 5, Supplementary Figure S3).
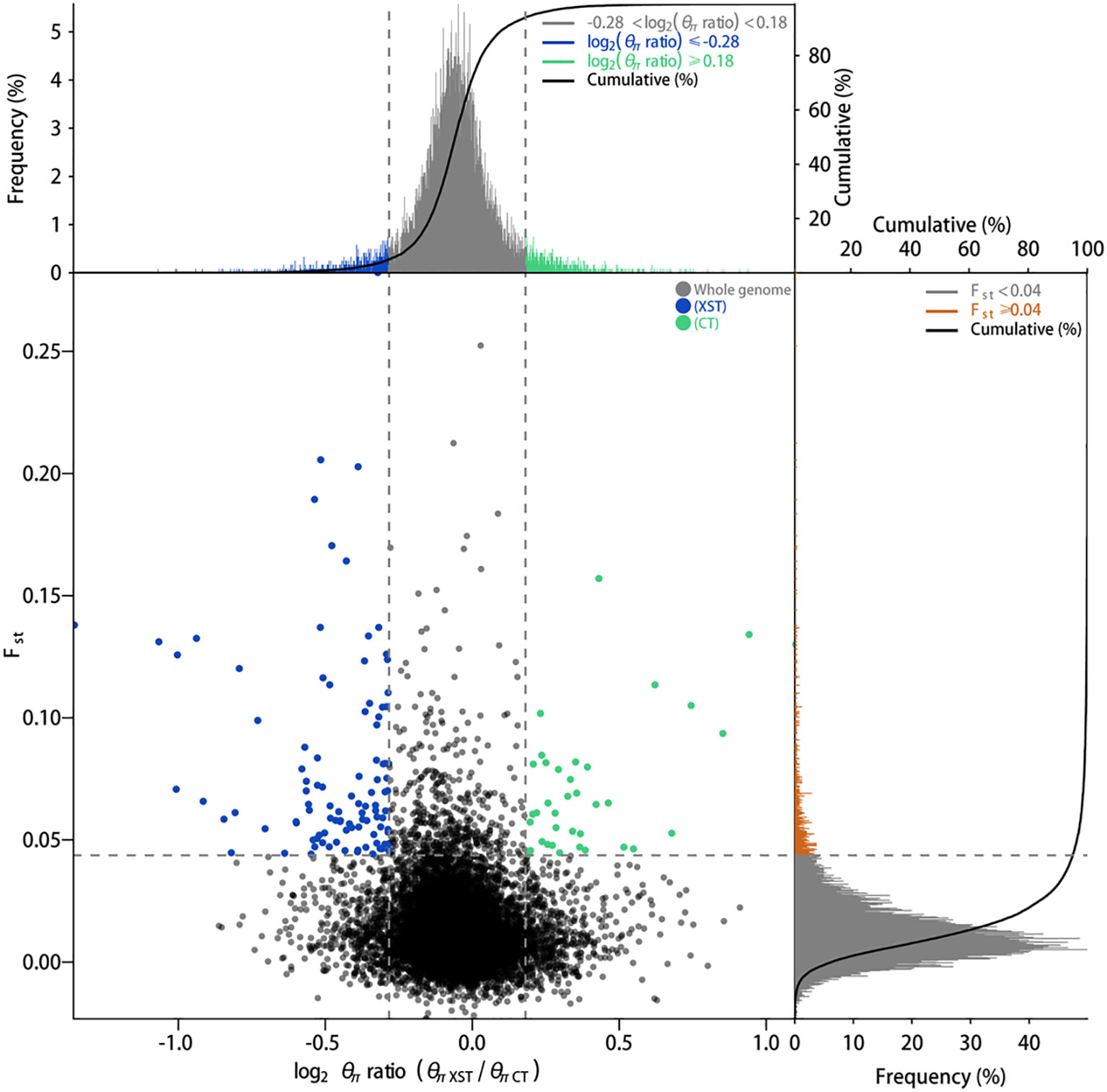
Figure 5 Selective region identification based on both θπ ratios and Fst values. Data points located to the left and right of the vertical dashed lines (top 5%) and above the horizontal dashed line (top 5%) were identified as regions under significant selection in XST and CT populations, respectively. Distribution of θπ ratios and Fst values between other paired populations presented in Supplementary Figure S3. XST, Xingshutun; CT, cultured.
GO and KEGG enrichment analyses were performed for the selected genes of each population. There were separately 121, 54, 137, 114, 103, 77, and 104 GO terms significantly enriched in the XST, GLD, QD, LS, SSD, LHT, and CT populations, respectively. Those GO terms related to DNA metabolism and stability, such as DNA metabolic process, DNA integration, DNA replication, DNA recombination DNA binding, DNA modification, nuclease activity, and transposition, were found mostly significantly enriched in all the seven populations (Figure 6, and highlighted in yellow in Supplementary Tables S3–S9). Additionally, some apoptosis-related GO terms were found significantly enriched in the QD and LS populations (highlighted in blue in Supplementary Tables S5, S6), and some glycoprotein-related GO terms were significantly enriched in the CT population (highlighted in gray in Supplementary Table S9).
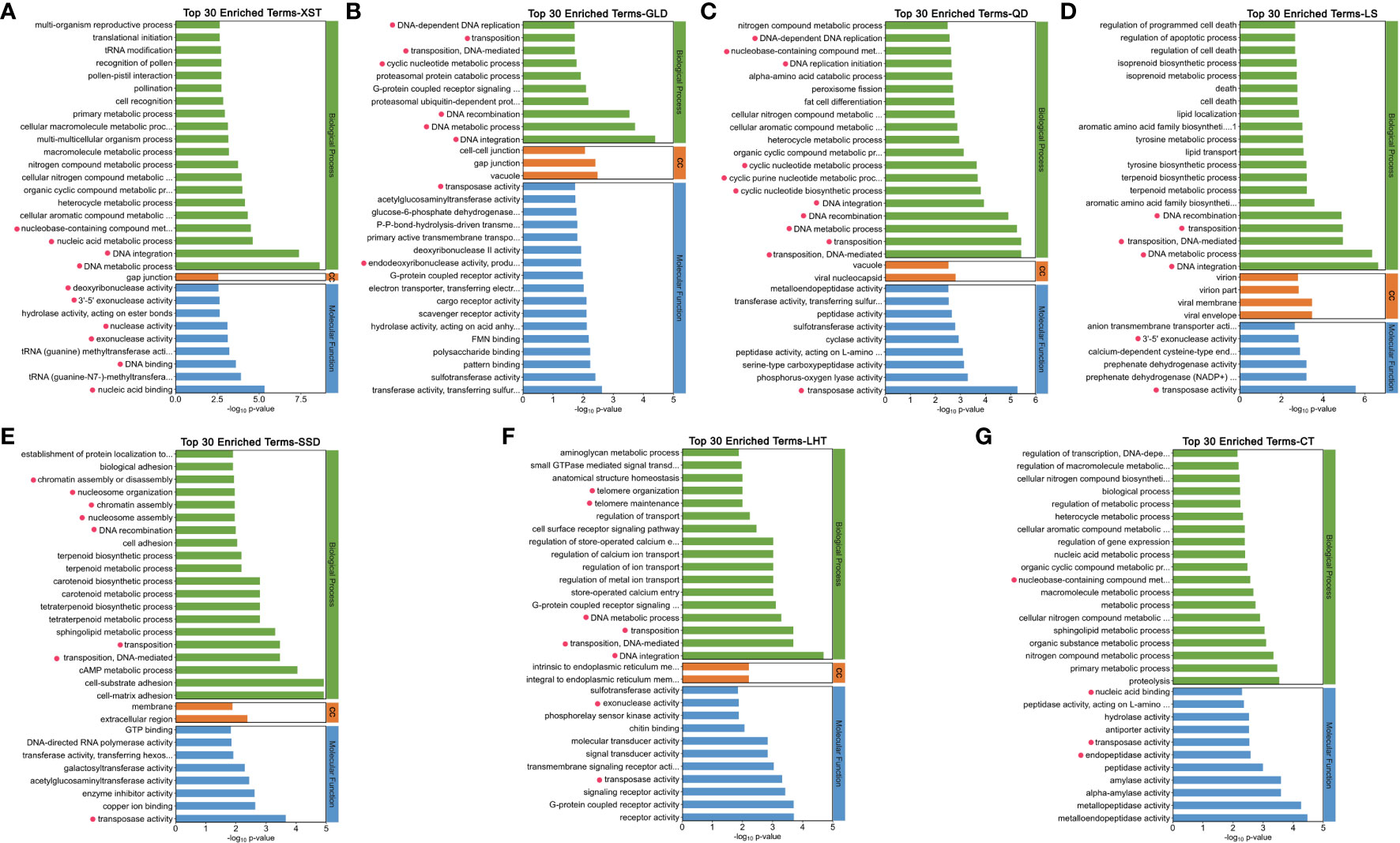
Figure 6 GO enrichment analysis for selected genes in the seven populations, respectively. (A) XST. (B) GLD. (C) QD. (D) LS. (E) SSD. (F) LHT. (G) CT. Top 30 significantly enriched GO termed were showed. Termed related to DNA metabolism and stability are highlighted with red dots. CC, cellular component.
There were separately 10, 7, 9, 10, 9, 5, and 11 pathways significantly enriched in the XST, GLD, QD, LS, SSD, LHT, and CT populations (Figure 7). Pathways of “Notch signaling pathway” (in the XST, GLD, QD, LS, and CT populations), “Glycosaminoglycan biosynthesis-chondroitin sulfate/dermatan sulfate” (in the XST, GLD, QD, LS, SSD, and CT populations), and “Glycosaminoglycan biosynthesis-heparan sulfate/heparin” (in the GLD, QD, and SSD populations), which were related to shell formation, were found significantly enriched. Additionally, some environmental stress-related pathways, such as “Protein processing in endoplasmic reticulum” (in the QD and LHT populations) and “Ubiquitin mediated proteolysis” (in the GLD, QD, LS, LHT, and CT populations), were also significantly enriched. Moreover, some important selected genes involved in these pathways, such as Notch1, Notch2, HSP40, HSP70, HSP90, Bip, ATF6, ERP57, SAR1, TRAP, IAP, BIRC2, BIRC3, Apc7, and Apc10, may play critical roles in adaptive evolution in the Pacific oyster (Table 3).
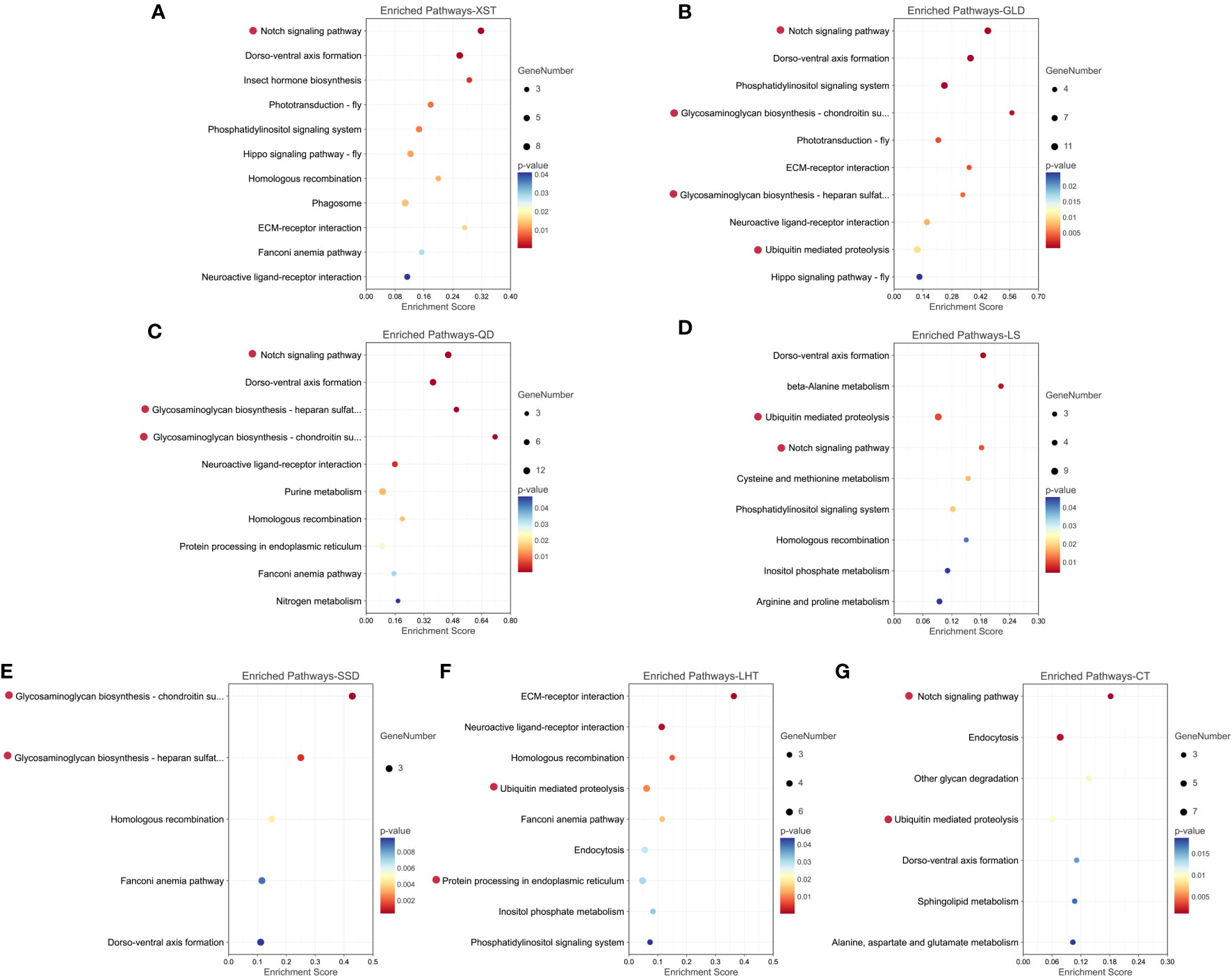
Figure 7 Significantly enriched KEGG pathways for selected genes in the seven populations, respectively. (A) XST. (B) GLD. (C) QD. (D) LS. (E) SSD. (F) LHT. (G) CT. Key candidate pathways are highlighted with red dots.
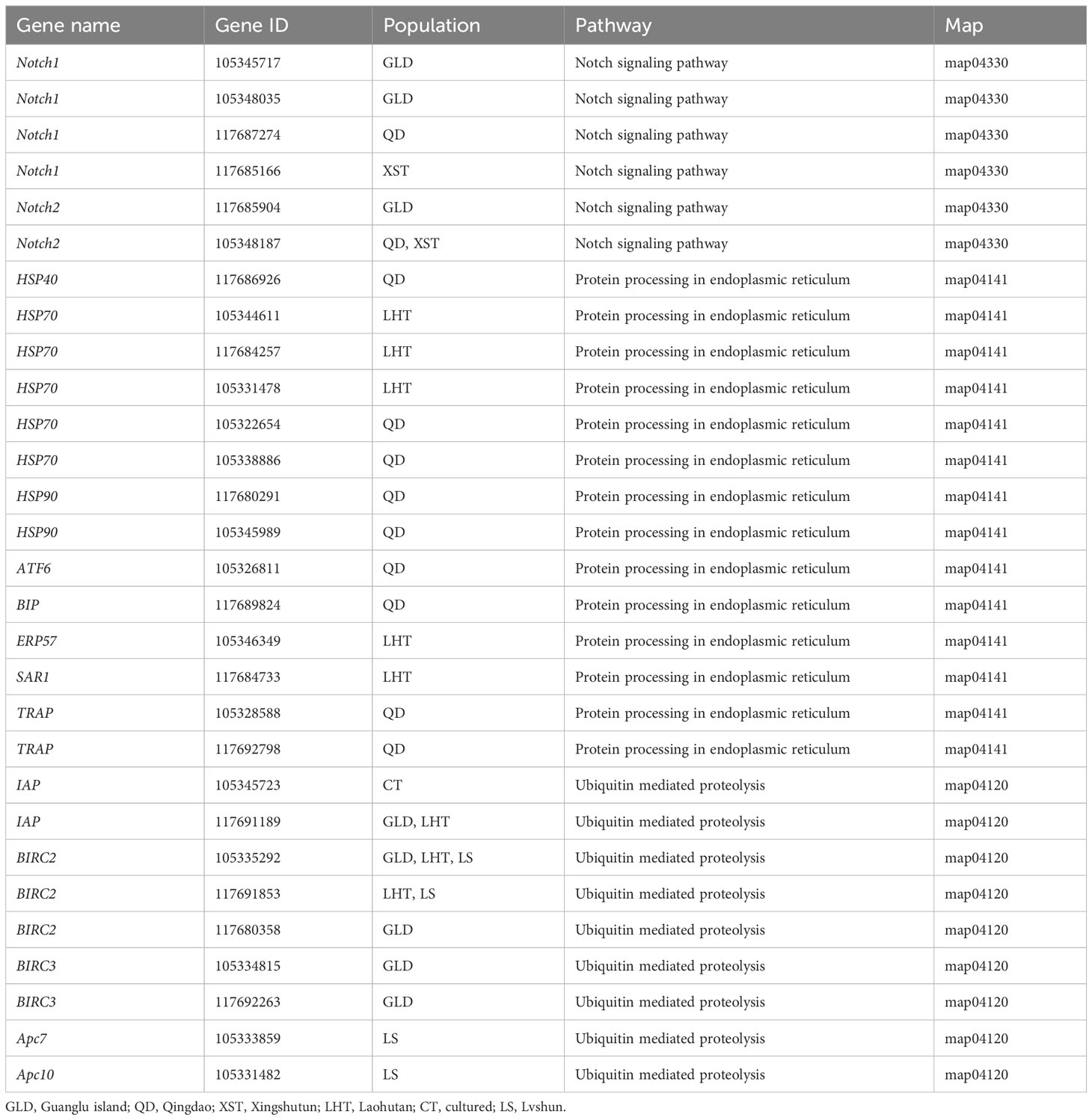
Table 3 Key candidate selected genes in the seven Pacific oyster populations.
4 Discussion
4.1 Low levels of genetic diversity and differentiation of the Pacific oysters in Dalian
To reveal the genetic diversity and structures of the Pacific oyster populations in the Dalian Sea area, one of the main natural distribution and aquaculture areas for the Pacific oyster, and evaluate the effects of farming on the genetic diversity of wild populations, genome-wide SNPs were first detected for natural and cultured populations in Dalian by whole-genome resequencing in the present study. Similar and relatively low genetic diversity (0.2352–0.2527) was found in all the studied populations of Dalian, together with the population in Qingdao, with genome-wide SNPs, which was higher than the levels detected in different geographical populations (0.0142–0.0147), separately distributed in Bohai Sea (BS), Bohai/Yellow Sea transition area (B&YT), northern Yellow Sea (NYS), and southern Yellow Sea (SYS) by Li et al. (2018). Nevertheless, in the study by Li et al. (2015), extremely low nucleotide diversity (0.000322–0.001432) was found in 12 Pacific oyster populations, distributed in China, Korea, and Japan, using partial mitochondrial cytochrome oxidase I (mtCOI) sequences. The Ho of the wild populations in the present study ranged from 0.2441 to 0.2853, with an average of 0.2593, which was very close to that of the cultured population (0.2577). Similar values have also been reported in the selected lines of Pacific oysters (0.2479–0.2733) studied by Zhong et al. (2017) with 103 SNPs, but higher Ho (0.2703–0.2939) was detected in the wild populations (separately located in Inchon of Korea, Miyagi Prefecture of Japan, and Rushan and Dongying of Shandong province in China) in their study. However, a much higher Ho has also been found in other selected lines of Pacific oysters. For example, the Ho detected using microsatellites and mtDNA for the generations of black shell strain Pacific oyster ranged from 0.583 to 0.731 (Xu et al., 2019). The different results among studies were partially due to the different marker types and numbers utilized (Rohfritsch et al., 2013).
Weak differentiation among populations in Dalian was revealed by Fst, PCA, phylogenetic analysis, and population structure analysis. The QD population (southern Yellow Sea) was intended to be used as a group with a relatively long distance from these Dalian populations (northern Yellow Sea), but no significant differentiation was found between the QD and Dalian populations here. Relatively low paired Fst values (0.01787–0.02428) were detected between the studied populations, and PCA and phylogenetic analysis could not clearly divide each population in this work. In the study by Li et al. (2018), slightly higher Fst values (0.036–0.042) were found between different populations in northern China (BS, B&YT, NYS, and SYS), which also showed a clear clustering of different populations by phylogenetic analysis. However, individuals from a larger spanning (China, Korea, and Japan) were clustered together with mtCOI (Li et al., 2015), which were probably limited by the very few genetic variations used. Additionally, frequent migration events have also been observed among the studied populations by TreeMix analysis, possibly leading to a high genetic similarity of these populations. Despite that the adult Pacific oyster is sessile without migration, a 2- to 3-week planktonic larval stage allows a long-distance migration of larvae as a result of high dispersal capabilities (Shanks, 2009; Selkoe and Toonen, 2011; Li et al., 2015). These life history characteristics promote gene exchange between populations during the larval stage, which is probably the main reason limiting genetic differentiation between different Pacific oyster populations with insufficient geographical barriers (Li et al., 2015; Li et al., 2018). At the same time, the ocean current system along the coast of Dalian may further influence the genetic connectivity of the Pacific oyster by causing large-scale oceanic mixture and providing high randomness of diffusivity of eggs and larvae, thus increasing gene exchange and genetic homogeneity of marine organisms (Li et al., 2022). Though there is weak differentiation among the studied populations, closer genetic relationships could be found among the CT, SSD, and LHT populations and the QD, GLD, and XST populations.
It should be noted that the CT population in this study also showed similar genetic diversity and low differentiation with other wild populations, and it especially showed a closer genetic relationship and more similar population structure with the SSD and LHT populations. A similar result was also found in the study by Zhong et al. (2017) in which no clear division was between selected and wild Pacific oysters. In the study of European populations of Pacific oysters, cultured individuals from the Bay of Arcachon genetically resembled the naturalized populations from Texel to southern France (Meistertzheim et al., 2013). In the present study, gene flows from the cultured population to wild populations, such as CT to LS and CT to QD, and then gene flows between wild populations probably resulted in a genetic mixture of wild and cultured Pacific oysters. The cultured population possibly has greatly affected the genetic diversity of neighboring wild populations, which was also found in other marine species, such as pikeperch (Sander lucioperca) (Tsaparis et al., 2022), European flat oyster (Ostrea edulis) (Šegvić-Bubić et al., 2020), and other Pacific oyster populations (Meistertzheim et al., 2013). This was probably caused by the individuals’ escaping, pelagic larval dispersal, and human-made stocking (Meistertzheim et al., 2013; Semeraro et al., 2015; Šegvić-Bubić et al., 2020). It has been proved that current aquaculture practices result in serious genetic diversity reductions (Semeraro et al., 2015). Cultured populations are usually strengthened in some economic traits through artificial selection, while their genetic diversity is commonly declined due to insufficient number of parents, non-random mating, or genetic drift (Evans et al., 2004; Gaffney, 2006; Xu et al., 2019). Therefore, the mix of the cultured individuals probably caused the genetic diversity of wild populations reduced or lost, which would badly influence the adaptive ability of the species to the changing environment (Meistertzheim et al., 2013; Šegvić-Bubić et al., 2020). Reduced diversity was also observed in farm-associated populations (natural populations located in the vicinity of oyster-farm installations) of European flat oyster due to gene flows between farmed and farm-associated populations (Šegvić-Bubić et al., 2020). Hence, protection and restoration of the wild germplasm resources of the Pacific oyster in Dalian and reasonable management of aquaculture of the species are presently necessary.
4.2 Selected signatures related to habitat adaptation of the Pacific oysters in Dalian
Natural selection can improve species’ habitat adaptation ability by altering their phenotypic and genomic characteristics (Li et al., 2022). Detection of selection signatures in the whole genome is necessary for understanding the adaptive genetic mechanisms of marine organisms under changing climates. For instance, genes associated with material metabolism and ion transfer were found selected in wild pomfret (Pampus echinogaster) populations, which were critical for the adaptation of the species to spatially heterogeneous temperatures (Li et al., 2022). In spotted sea bass (Lateolabrax maculatus), genes encoding essential structural components or regulating striated muscle fibers were identified as positively selected in the northern population, which altered the swimming ability of the fish to cope with environmental temperature changes (Chen et al., 2023). As sessile bivalves, the Pacific oyster lives in intertidal and estuarine regions, where the environments are harsh and dramatically fluctuating, making the species face serious environmental challenges. The Pacific oyster has strong environmental adaptability, and its adaptive mechanism has drawn much attention recently due to global climate changes. Genome-wide scan of selection signatures of the Pacific oyster has only been studied by Li et al. (2018), and genes involved in metabolic processes, protein phosphorylation, autophagy, protein folding, ubiquitination, and ion channels were detected under selection, which may play roles in local adaptation. In the present study, genome-wide selection signals and genes were first identified in different Dalian populations. GO and KEGG enrichment analyses revealed biological processes related to DNA metabolism and stability, shell formation, and environmental stress response significantly enriched, and some similar selected processes have also been detected in other bivalves, such as the pearl oyster (Pinctada fucata martensii) (Zheng et al., 2023) and the bay scallop (Argopecten irradians) (Wang et al., 2021), which may be key processes in bivalves to adapt to environmental changes.
A growing body of evidence proves that environmental stress can influence the genome stability of organisms (Galhardo et al., 2007). For example, environmental stress increases mutagenesis in mammalian cells by suppressing DNA repair pathways (Yuan et al., 2000; Mihaylova et al., 2003). In bivalves, genes involved in DNA replication and/or repair processes were also found regulated under different environmental stresses (Artigaud et al., 2015; Shen et al., 2019; Mao et al., 2022), which may be one of the main reasons for the high levels of polymorphism of many bivalve species and then be the evolved mechanism of the group to adapt to highly variable environments (Guo et al., 2015). In the present study, many functions related to DNA metabolism and stability were significantly enriched for the selected genes of all the seven Pacific oyster populations, which would modulate mutagenesis of genes involved in environmental adaptation. A similar result has also been found in the bay scallop recently in which genes involved in the function of base excision repair underwent selection through selective sweep analysis, indicating it may be a common adaptive mechanism of bivalves to environmental changes.
The calcified external shells, one major evolutionary innovation contributing to the success of molluscs, are considered to serve as solid support for the molluscan soft body and possess an adaptive potential for ecological diversification (Marin and Luquet, 2004; Zhang et al., 2012). The very thick shells in sessile oysters do provide a critical physical line for the species to defend against predation and desiccation (Zhang et al., 2012). Glycosaminoglycan, which has high calcium affinity, was suggested to play a key part in crystal nucleation in the process of the nacreous layer formation in shelled molluscs (Lopes-Lima et al., 2005; Du et al., 2017; Zheng et al., 2023). A study of selective sweep in different pearl oyster populations discovered that typical signaling pathways for biomineralization of shell formation, such as glycosaminoglycan biosynthesis, were under selection pressure (Zheng et al., 2023). Consistent with the findings in the pearl oyster, pathways of glycosaminoglycan biosynthesis were also significantly enriched for the selected genes in the Pacific oyster populations in the present study. The Notch signaling pathway, an evolutionarily conserved cell signaling system, functions in a great diversity of biological processes such as cell proliferation and growth, cell fate determination, differentiation, and stem cell maintenance (Artavanis-Tsakonas et al., 1999; Bray, 2006). Recently, the Notch pathway has been proposed to play a crucial role in shell formation and pigmentation in molluscs, including pearl oysters (Pinctada margaritifera and P. fucata martensii) (Jiao et al., 2019; Auffret et al., 2020), the clam (Meretrix meretrix) (Yue et al., 2015), and the Pacific oyster (Feng et al., 2015). Here, we first detected that this pathway underwent significant selection in different Pacific oyster populations, and some core genes of the pathway, i.e., Notch1 (two in GLD, one in XST, and one in QD) and Notch2 (one in GLD and one in QD and XST), were involved. The Notch pathway is activated by binding of the ligands, such as Delta and Serrate, to the Notch receptors, leading to a concerted two-step proteolysis of the Notch proteins (Krämer, 2002; Liu et al., 2015). Notch1 and Notch2 are the two closest related Notch paralogs but usually have apparent different biological functions in mammals (Liu et al., 2015). Notch-related genes in both clams and Pacific oysters were predicted to function as upstream components of the shell color determination by combining with calcium signaling pathway (Feng et al., 2015; Yue et al., 2015), and the white-shelled Pacific oyster possibly utilized endocytosis to downregulate the Notch expression level and resulted in the apoptosis of melanoblasts, hindering pigmentation (Feng et al., 2015). Therefore, the present study provided important candidate loci for the research on shell biomineralization in the Pacific oyster, and the genetic divergence of the above genomic regions controlling shell formation was probably of great importance for molluscs to adapt to changing environments.
Multiple lines of evidence suggest that environmental stresses can induce endoplasmic reticulum (ER) stress, which refers to a physiological or pathological state leading to the accumulation of misfolded or unfolded proteins in the ER lumen (Bánhegyi et al., 2007; Cybulsky, 2010; Zhao et al., 2022). The ER has serious quality control systems, i.e., the unfolded protein response (UPR) and ER-associated degradation (ERAD), to rescue misfolded or unfolded proteins, which are vital in cellular homeostasis under stress (Ellgaard et al., 1999; Yoshida, 2007; Cybulsky, 2010). Protein processing in the endoplasmic reticulum is the key pathway for these processes to recognize and selectively direct abnormal proteins to be either correctly refolded or degraded (Yoshida, 2007; Cybulsky, 2010). Meanwhile, the pathway of ubiquitin mediated proteolysis plays an important role in degrading irreparable proteins (Hampton, 2002). The two pathways have been reported to be regulated by different environmental stresses in diverse molluscs, such as the Yesso scallop (Patinopecten yessoensis) (Mao et al., 2022), the Zhikong scallop (Chlamys farreri) (Mao et al., 2022), and the hard clam (Mercenaria mercenaria) (Hu et al., 2022). In this study, both the two pathways were found under significant selection in the Pacific oyster. Some key genes involved in different processes of the pathway of protein processing in the endoplasmic reticulum were found selected. HSPs are classes of chaperone proteins that play important roles in protein folding and quality control (Georgopoulos and Welch, 1993). A great expansion of HSP genes was detected in the oyster genomes, such as 88 HSP70 genes found in the Pacific oyster genome, which are probably central for the oyster to defend against all stresses (Zhang et al., 2012). There were one HSP40 (in QD), five HSP70 (three in LHT and two in QD), and two HSP90 (in QD) genes significantly selected in the Pacific oyster genome of different populations. HSP40, HSP70, and HSP90, ancient and highly conserved chaperone families, are critical to maintaining ER homeostasis (Kotler and Street, 2023). They can work together as a unified system so that HSP40 chaperones can transfer their clients to HSP70 and HSP70 can transfer clients to HSP90 (Kotler and Street, 2023). Bip, a member of the HSP70 family, is one of the most abundant ER chaperones, which facilitates protein translocation into the ER and protein folding within the ER and modulates the UPR and ERAD (Yoshida, 2007; Kotler and Street, 2023). Bip gene was found selected in the QD population. Genomic evidence of selection signatures in the HSP gene family implied their important role for the Pacific oyster in environmental adaption. Strong selection signals were also discovered in the genes of the inhibitor of apoptosis (IAP) family, including two IAP (one in CT and one in GLD and LHT), three BIRC2 (one in GLD, LHT, and LS; one in LHT and LS; and one in GLD), and two BIRC3 (in GLD). IAPs function primarily by suppressing the activity of caspases to inhibit apoptosis (Richter and Duckett, 2000). The general expansion of IAPs in many molluscan genomes, including the Pacific oyster, indicates a powerful anti-apoptosis system in molluscs (Zhang et al., 2012; Song et al., 2021). Additionally, the ubiquitin ligase activity of IAPs can contribute to ubiquitin mediated proteolysis (McDonald and El-Deiry, 2004). Genetic divergence of IAPs probably led to the different abilities of different Pacific oyster populations to tolerate diverse environmental stresses by regulating programmed cell death or protein degradation.
The present work provided many important selected loci that may play key roles in adapting to the stressful and changing environment in the Pacific oyster and were valuable for understanding the evolutionary adaptive mechanism of oysters. However, the study has its limitations in that phenotypic and environmental values of the different populations were not collected, which limited linking phenotypic and genomic features related to local adaptation to specific environmental conditions, and further research is needed.
5 Conclusions
In this study, a novel catalog of population genomic data was generated in the Pacific oyster by whole-genome resequencing. Population genetic analysis has comprehensively revealed the genomic variation, genetic diversity, population structure, phylogenetic relationships, and migration events of wild and cultured populations in Dalian. Additionally, linkage disequilibrium and demographic history of these populations were inferred. Moreover, potential genomic selection signatures associated with environmental adaptation were discovered, with selected genes involved in DNA metabolism and stability, shell formation, and environmental stress response. The study laid the theoretical basis for the subsequent germplasm conservation, management, and genetic breeding of the indigenous Pacific oysters and provided novel insights into the adaptive evolutionary mechanism of oysters.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Files, further inquiries can be directed to the corresponding author/s.
Ethics statement
The animal study was approved by the Ethics Committee of Dalian Ocean University. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
JM: Conceptualization, Data curation, Formal Analysis, Investigation, Resources, Supervision, Visualization, Writing – original draft, Writing – review & editing. YT: Conceptualization, Data curation, Formal Analysis, Investigation, Resources, Supervision, Writing – review & editing. QL: Data curation, Formal Analysis, Investigation, Resources, Writing – review & editing. DL: Investigation, Resources, Writing – review & editing. XG: Investigation, Resources, Writing – review & editing. XW: Formal Analysis, Writing – review & editing. ZH: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This project was supported by National Key Research and Development Program of China (2021YFB2600200), the Dalian Science and Technology Innovation Fund (2021JJ12SN34), the Central Government Subsidy Project for Liaoning Fisheries (2023), the National Natural Science Foundation of China (42076101).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2023.1337980/full#supplementary-material
References
Alexander D. H., Novembre J., Lange K. (2009). Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19 (9), 1655–1664. doi: 10.1101/gr.094052.109
Aranishi F. (2006). A novel mitochondrial intergenic spacer reflecting population structure of Pacific oyster. J. Appl. Genet. 47, 119–123. doi: 10.1007/BF03194610
Ardlie K. G., Kruglyak L., Seielstad M. (2002). Patterns of linkage disequilibrium in the human genome. Nat. Rev. Genet. 3 (4), 299–309. doi: 10.1038/nrg777
Artavanis-Tsakonas S., Rand M. D., Lake R. J. (1999). Notch signaling: cell fate control and signal integration in development. Science 284 (5415), 770–776. doi: 10.1126/science.284.5415.77
Artigaud S., Richard J., Thorne M. A., Lavaud R., Flye-Sainte-Marie J., Jean F., et al. (2015). Deciphering the molecular adaptation of the king scallop (Pecten maximus) to heat stress using transcriptomics and proteomics. BMC Genomics 16 (1), 1–14. doi: 10.1186/s12864-015-2132-x
Auffret P., Le Luyer J., Sham Koua M., Quillien V., Ky C. L. (2020). Tracing key genes associated with the Pinctada margaritifera albino phenotype from juvenile to cultured pearl harvest stages using multiple whole transcriptome sequencing. BMC Genomics 21 (1), 1–13. doi: 10.1186/s12864-020-07015-w
Bánhegyi G., Baumeister P., Benedetti A., Dong D., Fu Y., Lee A. S., et al. (2007). Endoplasmic reticulum stress. Ann. New York Acad. Sci. 1113 (1), 58–71. doi: 10.1196/annals.1391.007
Bray S. J. (2006). Notch signalling: a simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 7 (9), 678–689. doi: 10.1038/nrm2009
Chen B., Zhou Z., Shi Y., Gong J., Li C., Zhou T., et al. (2023). Genome-wide evolutionary signatures of climate adaptation in spotted sea bass inhabiting different latitudinal regions. Evolutionary Appl. 16, 1029–1043. doi: 10.1111/eva.13551
Cybulsky A. V. (2010). Endoplasmic reticulum stress in proteinuric kidney disease. Kidney Int. 77 (3), 187–193. doi: 10.1038/ki.2009.389
Dineshram R., Chandramouli K., Ko G. W. K., Zhang H., Qian P. Y., Ravasi T., et al. (2016). Quantitative analysis of oyster larval proteome provides new insights into the effects of multiple climate change stressors. Global Change Biol. 22 (6), 2054–2068. doi: 10.1111/gcb.13249
Du X., Fan G., Jiao Y., Zhang H., Guo X., Huang R., et al. (2017). The pearl oyster Pinctada fucata martensii genome and multi-omic analyses provide insights into biomineralization. Gigascience 6 (8), gix059. doi: 10.1093/gigascience/gix059
Ellgaard L., Molinari M., Helenius A. (1999). Setting the standards: quality control in the secretory pathway. Science 286 (5446), 1882–1888. doi: 10.1126/science.286.5446.1882
Evans F., Matson S., Brake J., Langdon C. (2004). The effects of inbreeding on performance traits of adult Pacific oysters (Crassostrea gigas). Aquaculture 230 (1-4), 89–98. doi: 10.1016/j.aquaculture.2003.09.023
Feng D., Li Q., Yu H., Zhao X., Kong L. (2015). Comparative transcriptome analysis of the Pacific oyster Crassostrea gigas characterized by shell colors: identification of genetic bases potentially involved in pigmentation. PLoS One 10 (12), e0145257. doi: 10.1371/journal.pone.0145257
Gaffney P. M. (2006). The role of genetics in shellfish restoration. Aquat. Living Resour. 19 (3), 277–282. doi: 10.1051/alr:2006028
Galhardo R. S., Hastings P. J., Rosenberg S. M. (2007). Mutation as a stress response and the regulation of evolvability. Crit. Rev. Biochem. Mol. Biol. 42 (5), 399–435. doi: 10.1080/10409230701648502
Georgopoulos C., Welch W. J. (1993). Role of the major heat shock proteins as molecular chaperones. Annu. Rev. Cell Biol. 9 (1), 601–634. doi: 10.1146/annurev.cb.09.110193.003125
Guo X., He Y., Zhang L., Lelong C., Jouaux A. (2015). Immune and stress responses in oysters with insights on adaptation. Fish shellfish Immunol. 46 (1), 107–119. doi: 10.1016/j.fsi.2015.05.018
Guo X., Li Q., Kong L., Yu H. (2016). Linkage disequilibrium in wild and cultured populations of Pacific oyster (Crassostrea gigas). J. Ocean Univ. China 15, 327–333. doi: 10.1007/s11802-016-2832-0
Guo X., Zhang G., Qian L., Wang H., Liu X., Wang A. (2006). Oysters and oyster farming in China: a review. J. Shellfish Res. 25, 734.
Gupta P. K., Rustgi S., Kulwal P. L. (2005). Linkage disequilibrium and association studies in higher plants: present status and future prospects. Plant Mol. Biol. 57, 461–485. doi: 10.1007/s11103-005-0257-z
Hampton R. Y. (2002). ER-associated degradation in protein quality control and cellular regulation. Curr. Opin. Cell Biol. 14 (4), 476–482. doi: 10.1016/S0955-0674(02)00358-7
Hu Z., Feng J., Song H., Zhou C., Yu Z., Yang M., et al. (2022). Mechanisms of heat and hypoxia defense in hard clam: insights from transcriptome analysis. Aquaculture 549, 737792. doi: 10.1016/j.aquaculture.2021.737792
Jiao Y., Yang S., Cao Y., Zheng Z., Deng Y., Wang Q., et al. (2019). Genome and transcriptome analyses providing insight into the immune response of pearl oysters after allograft and xenograft transplantations. Fish Shellfish Immunol. 90, 109–117. doi: 10.1016/j.fsi.2019.04.061
Kotler J. L., Street T. O. (2023). Mechanisms of protein quality control in the endoplasmic reticulum by a coordinated Hsp40-Hsp70-Hsp90 system. Annu. Rev. biophysics 52, 509–524. doi: 10.1146/annurev-biophys-111622-091309
Krämer H. (2002). Sorting out signals in fly endosomes. Traffic 3 (2), 87–91. doi: 10.1034/j.1600-0854.2002.030201.x
Li H., Durbin R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25 (14), 1754–1760. doi: 10.1093/bioinformatics/btp324
Li H., Durbin R. (2011). Inference of human population history from individual whole-genome sequences. Nature 475 (7357), 493–496. doi: 10.1038/nature10231
Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25 (16), 2078–2079. doi: 10.1093/bioinformatics/btp352
Li L., Li A., Song K., Meng J., Guo X., Li S., et al. (2018). Divergence and plasticity shape adaptive potential of the Pacific oyster. Nat. Ecol. Evol. 2, 1751–1760. doi: 10.1038/s41559-018-0668-2
Li S., Li Q., Yu H., Kong L., Liu S. (2015). Genetic variation and population structure of the Pacific oyster Crassostrea gigas in the northwestern Pacific inferred from mitochondrial COI sequences. Fisheries Sci. 81, 1071–1082. doi: 10.1007/s12562-015-0928-x
Li Y., Lou F., Liu S., Li H., Xiang J., Shan B., et al. (2022). Differentiation and temperature adaptation of Pampus echinogaster based on genome-wide SNPs. Front. Mar. Sci. 9, 936217. doi: 10.3389/fmars.2022.936217
Li M. H., Merilä J. (2011). Population differences in levels of linkage disequilibrium in the wild. Mol. Ecol. 20 (14), 2916–2928. doi: 10.1111/j.1365-294X.2011.05154.x
Liu L., Ang K. P., Elliott J. A., Kent M. P., Lien S., MacDonald D., et al. (2017). A genome scan for selection signatures comparing farmed Atlantic salmon with two wild populations: Testing colocalization among outlier markers, candidate genes, and quantitative trait loci for production traits. Evolutionary Appl. 10 (3), 276–296. doi: 10.1111/eva.12450
Liu Z., Brunskill E., Varnum-Finney B., Zhang C., Zhang A., Jay P. Y., et al. (2015). The intracellular domains of Notch1 and Notch2 are functionally equivalent during development and carcinogenesis. Development 142 (14), 2452–2463. doi: 10.1242/dev.125492
Lopes-Lima M., Ribeiro I., Pinto R. A., MaChado J. (2005). Isolation, purification and characterization of glycosaminoglycans in the fluids of the mollusc Anodonta cygnea. Comp. Biochem. Physiol. Part A: Mol. Integr. Physiol. 141 (3), 319–326. doi: 10.1016/j.cbpb.2005.06.007
Mao J., Huang X., Sun H., Jin X., Guan W., Xie J., et al. (2022). Transcriptome analysis provides insight into adaptive mechanisms of scallops under environmental stress. Front. Mar. Sci. 9, 971796. doi: 10.3389/fmars.2022.971796
Marin F., Luquet G. (2004). Molluscan shell proteins. Comptes Rendus Palevol 3 (6-7), 469–492. doi: 10.1016/j.crpv.2004.07.009
McDonald E. R., El-Deiry W. S. (2004). Suppression of caspase-8- and -10-associated RING proteins results in sensitization to death ligands and inhibition of tumor cell growth. PNAS 101 (16), 6170–6175. doi: 10.1073/pnas.0307459101
Meistertzheim A. L., Arnaud-Haond S., Boudry P., Thébault M. T. (2013). Genetic structure of wild European populations of the invasive Pacific oyster Crassostrea gigas due to aquaculture practices. Mar. Biol. 160, 453–463. doi: 10.1007/s00227-012-2102-7
Mihaylova V. T., Bindra R. S., Yuan J., Campisi D., Narayanan L., Jensen R., et al. (2003). Decreased expression of the DNA mismatch repair gene Mlh1 under hypoxic stress in mammalian cells. Mol. Cell. Biol. 23 (9), 3265–3273. doi: 10.1128/MCB.23.9.3265-3273.2003
Miossec L., Le Deuff R. M., Goulletquer P. (2009). Alien species alert: Crassostrea gigas (Pacific oyster). ICES Cooperative Res. Rep. 299, 1017–6195. doi: 10.17895/ices.pub.5417
Pickrell J., Pritchard J. (2012). Inference of population splits and mixtures from genome-wide allele frequency data. PloS Genet. 8, e1002967. doi: 10.1371/journal.pgen.1002967
Richter B. W., Duckett C. S. (2000). The IAP proteins: caspase inhibitors and beyond. Science's STKE 2000 (44), 1–4. doi: 10.1126/stke.2000.44.pe1
Rohfritsch A., Bierne N., Boudry P., Heurtebise S., Cornette F., Lapegue S. (2013). Population genomics shed light on the demographic and adaptive histories of European invasion in the Pacific oyster, Crassostrea gigas. Evolutionary Appl. 6 (7), 1064–1078. doi: 10.1111/eva.12086
Šegvić-Bubić T., Žužul I., Talijančić I., Ugrin N., Lepen Pleić I., Stagličić N., et al. (2020). Translocation and aquaculture impact on genetic diversity and composition of wild self-sustainable Ostrea edulis populations in the Adriatic Sea. Front. Mar. Sci. 7, 84. doi: 10.3389/fmars.2020.00084
Selkoe K. A., Toonen R. J. (2011). Marine connectivity: a new look at pelagic larval duration and genetic metrics of dispersal. Mar. Ecol. Prog. Ser. 436, 291–305. doi: 10.3354/meps09238
Semeraro A., Mohammed-Geba K., Arias A., Anadon N., García-Vázquez E., Borrell Y. J. (2015). Genetic diversity and connectivity patterns of harvested and aquacultured molluscs in estuaries from Asturias (northern Spain). Implications Manage. strategies. Aquaculture Res. 47 (9), 2937–2950. doi: 10.1111/are.12745
Shanks A. L. (2009). Pelagic larval duration and dispersal distance revisited. Biol. Bull. 216 (3), 373–385. doi: 10.1086/BBLv216n3p373
Shen Y., Huang Z., Liu G., Ke C., You W. (2019). Hemolymph and transcriptome analysis to understand innate immune responses to hypoxia in Pacific abalone. Comp. Biochem. Physiol. Part D: Genomics Proteomics 30, 102–112. doi: 10.1016/j.cbd.2019.02.001
Song H., Guo X., Sun L., Wang Q., Han F., Wang H., et al. (2021). The hard clam genome reveals massive expansion and diversification of inhibitors of apoptosis in Bivalvia. BMC Biol. 19, 1–20. doi: 10.1186/s12915-020-00943-9
Teng W., Zheng J., Xie X., Liu X., Li Q., Wang Q. (2022). The current situation and suggestions on development of oyster industry in Dalian. Chin. Fisheries Economics 40 (2), 84–90.
Tsaparis D., Lecocq T., Kyriakis D., Oikonomaki K., Fontaine P., Tsigenopoulos C. S. (2022). Assessing genetic variation in wild and domesticated pikeperch populations: implications for conservation and fish farming. Animals 12 (9), 1178. doi: 10.3390/ani12091178
Wang X., Cong R., Li A., Wang W., Zhang G., Li L. (2023). Transgenerational effects of intertidal environment on physiological phenotypes and DNA methylation in Pacific oysters. Sci. Total Environ. 871, 162112. doi: 10.1016/j.scitotenv.2023.162112
Wang K., Li M., Hakonarson H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38 (16), e164. doi: 10.1093/nar/gkq603
Wang H., Lv J., Zeng Q., Liu Y., Xing Q., Wang S., et al. (2021). Genetic differentiation and selection signatures in two bay scallop (Argopecten irradians) breeds revealed by whole-genome resequencing analysis. Aquaculture 543, 736944. doi: 10.1016/j.aquaculture.2021.736944
Xu L., Li Q., Xu C., Yu H., Kong L. (2019). Genetic diversity and effective population size in successive mass selected generations of black shell strain Pacific oyster (Crassostrea gigas) based on microsatellites and mtDNA data. Aquaculture 500, 338–346. doi: 10.1016/j.aquaculture.2018.10.007
Yang J., Lee S. H., Goddard M. E., Visscher P. M. (2011). GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88 (1), 76–82. doi: 10.1016/j.ajhg.2010.11.011
Yoshida H. (2007). ER stress and diseases. FEBS J. 274 (3), 630–658. doi: 10.1111/j.1742-4658.2007.05639.x
Yuan J., Narayanan L., Rockwell S., Glazer P. M. (2000). Diminished DNA repair and elevated mutagenesis in mammalian cells exposed to hypoxia and low pH. Cancer Res. 60 (16), 4372–4376.
Yue X., Nie Q., Xiao G., Liu B. (2015). Transcriptome analysis of shell color-related genes in the clam Meretrix meretrix. Mar. Biotechnol. 17, 364–374. doi: 10.1007/s10126-015-9625-0
Zhang C., Dong S. S., Xu J. Y., He W. M., Yang T. L. (2019). PopLDdecay: a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 35 (10), 1786–1788. doi: 10.1093/bioinformatics/bty875
Zhang G., Fang X., Guo X., Li L., Luo R., Xu F., et al. (2012). The oyster genome reveals stress adaptation and complexity of shell formation. Nature 490, 49–54. doi: 10.1038/nature11413
Zhang G., Li L., Meng J., Qi H., Qu T., Xu F., et al. (2016). Molecular basis for adaptation of oysters to stressful marine intertidal environments. Annu. Rev. Anim. Biosci. 4, 357–381. doi: 10.1146/annurev-animal-022114-110903
Zhao X., Li L., Li C., Liu E., Zhu H., Ling Q. (2022). Heat stress-induced endoplasmic reticulum stress promotes liver apoptosis in largemouth bass (Micropterus salmoides). Aquaculture 546, 737401. doi: 10.1016/j.aquaculture.2021.737401
Zhao X., Yu H., Kong L., Liu S., Li Q. (2014). Comparative transcriptome analysis of two oysters, Crassostrea gigas and Crassostrea hongkongensis provides insights into adaptation to hypo-osmotic conditions. PLoS One 9 (11), e111915. doi: 10.1371/journal.pone.0111915
Zheng Z., Hao R., Yang C., Jiao Y., Wang Q., Huang R., et al. (2023). Genome-wide association study analysis to resolve the key regulatory mechanism of biomineralization in Pinctada fucata martensii. Mol. Ecol. Resour. 23 (3), 680–693. doi: 10.1111/1755-0998.13743
Keywords: Pacific oyster, genetic diversity, population structure, selection signature, whole-genome resequencing
Citation: Mao J, Tian Y, Liu Q, Li D, Ge X, Wang X and Hao Z (2024) Revealing genetic diversity, population structure, and selection signatures of the Pacific oyster in Dalian by whole-genome resequencing. Front. Ecol. Evol. 11:1337980. doi: 10.3389/fevo.2023.1337980
Received: 14 November 2023; Accepted: 15 December 2023;
Published: 09 January 2024.
Edited by:
Hui Zhang, Chinese Academy of Sciences (CAS), ChinaCopyright © 2024 Mao, Tian, Liu, Li, Ge, Wang and Hao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhenlin Hao, aGFvemhlbmxpbkBkbG91LmVkdS5jbg==
†These authors have contributed equally to this work