 Carolina Martínez-Eraso
Carolina Martínez-Eraso Heiber Cárdenas1
Heiber Cárdenas1 Mario A. Cerón-Romero
Mario A. Cerón-Romero- 1Department of Biology, Universidad del Valle, Cali, Colombia
- 2Department of Biology, Case Western Reserve University, Cleveland, OH, United States
Antigenic diversity is critical for parasites to coevolve with their hosts. Plasmodium falciparum generates antigenic diversity through ectopic recombination of their antigenic gene-rich subtelomeres, a mechanism that takes place after chromosomal ends anchor in clusters near the nuclear periphery. A study mapping the phylogenomic history of genes across the chromosomes of P. falciparum showed that this mechanism to generate antigenic diversity extends to all chromosomes. Yet, its existence, importance, and evolutionary history in other Plasmodium species remain largely unknown. In this study, we survey and compare genomic features associated with the mechanism to generate antigenic diversity through ectopic recombination of subtelomeres in 19 species widely distributed in the genus Plasmodium. By comparing these features across species using a phylogenomic framework, we assess the existence and intensity of this mechanism, as well as propose different hypotheses for its evolution. Our results suggest that ectopic recombination of subtelomeres is more critical for the diversification of pir or rif/stevor genes than other antigenic gene families. Furthermore, its intensity varies among subgenera and was likely acquired and lost multiple times in the phylogeny of Plasmodium. These results demonstrate, for the first time, the genomic and evolutionary complexity of this mechanism for generating antigenic diversity in the genus Plasmodium.
1 Introduction
The genus Plasmodium belongs to the clade Apicomplexa and includes more than 200 species of protozoan hemoparasites that use dipterans as vectors to infect a great diversity of vertebrate hosts. Phylogenetic analyses of these parasites show conflicts with the phylogeny of their hosts (Rich and Xu, 2011; Böhme et al., 2018; Galen et al., 2018). For instance, the two most common species infecting humans, P. falciparum and P.vivax, belong to two distinct subgenera that infect primates, Laverania and Plasmodium, respectively (Sharp et al., 2020). In addition to Plasmodium and Laverania, the next most studied subgenera are Haemamoeba and Vinckeia, which infect birds and rodents, respectively (Pacheco et al., 2011; Perkins, 2014; Sharp et al., 2020). The most popular proposal about the phylogenetic order of these subgenera suggests that Haemamoeba diverges first, followed by Laverania, and finally, by the sister clades Vinckeia and Plasmodium (Borner et al., 2016; Galen et al., 2018; Escalante et al., 2022). However, both the root of the species tree and the monophyly of the subgenus Plasmodium are still a matter of debate (Rutledge et al., 2017; Böhme et al., 2018; Galen et al., 2018).
The discordance between the phylogenies of Plasmodium species and those of their hosts suggests that these parasites have highly dynamic genomes and their infection mechanisms have allowed them to frequently change and diversify their hosts (Rich and Xu, 2011; Böhme et al., 2018; Galen et al., 2018). Hence, comparative analyses of their genomes can reveal evolutionary patterns such as those related to their infection mechanisms. However, although there are more than 30 annotated genomes and over 200 described species of Plasmodium, most molecular and genomic studies have focused on P. falciparum due to its high virulence, and on P. vivax due to its wide global distribution (WHO, 2014). In P. falciparum, a 23 Mb genome is organized into 14 linear chromosomes ranging from 0.7 to 3.4 Mb (Kemp et al., 1987; Hernández-Rivas et al., 2013), and other Plasmodium species, mainly those infecting mammals, show similar chromosomal organization and size (Carlton et al., 1999; Carlton et al., 2008; Pain et al., 2008). Moreover, subtelomeres of P. falciparum were found to be significantly less conserved than the chromosomic internal regions, a feature that is strongly linked to its virulence (Hernández-Rivas et al., 2013; Reed et al., 2021), which has also been documented in P. vivax and P. knowlesi (del Portillo et al., 2001; Pain et al., 2008). Notably, the high subtelomeric variation of P. falciparum lies in sequences encoding virulence factors, while the rest of the subtelomeres are composed of repeats that tend to be conserved (Scherf et al., 2001; Hernández-Rivas et al., 2013). These more specific details about subtelomeric structures have been less explored in other Plasmodium species.
The importance of the chromosome structure in promoting antigenic diversity, proposed in P. falciparum and P. vivax (del Portillo et al., 2001; Figueiredo et al., 2002), has also been described in other eukaryotic taxa such as excavate parasites. However, both the mechanisms and the chromosomal regions involved are very variable (Arkhipova and Morrison, 2001; Silva Pereira et al., 2020). In P. falciparum as well as in Trypanosoma cruzi, an important mechanism for generating antigenic diversity is ectopic recombination of subtelomeres (Freitas-Junior et al., 2000; Ramirez, 2020). This mechanism in P. falciparum occurs through the anchoring of chromosomes at the nuclear periphery, bringing the subtelomeres of non-homologous chromosomes closer (Freitas-Junior et al., 2000). Thus, subtelomeric repetitive sequences facilitate the occurrence of gene conversion events and the production of new gene variants (Freitas-Junior et al., 2000; Barry et al., 2003), resulting in large antigenic gene families that tend to be species-specific (Kooij et al., 2005; Frank et al., 2008; Otto et al., 2018). To date, this process has not been described in T. cruzi, and therefore, homology between their mechanisms of antigenic diversity production cannot be assumed (Ramirez, 2020).
The evolutionary history of the mechanism of subtelomeric ectopic recombination to generate antigenic diversity remains unknown. However, describing the presence and intensity of this mechanism in each species and for each antigenic gene family can provide important clues to reconstruct such history. Intensity can be defined as the significance of this mechanism in generating antigenic diversity for a species, including whether it is widely employed across all chromosomes and if most or only a few antigenic gene families rely on it. In P. falciparum, at least three subtelomeric multigene families have been documented, var, rif, and stevor (Su et al., 1995; Cheng et al., 1998). A study mapping of the phylogenomic history of genes across the chromosomes of P. falciparum reported recombination hotspots of rif and stevor in the subtelomeres of the 14 chromosomes, likely due to intense ectopic recombination of subtelomeres to generate diversity in these two gene families (Cerón-Romero et al., 2018). Other antigenic gene families have been described in other Plasmodium species such as vir in P. vivax (del Portillo et al., 2001; Carlton et al., 2008), sicavar and kir in P. knowlesi (Al-Khedery et al., 1999; Janssen et al., 2004), cir in P. chabaudi, bir in P. berghei, and yir in P. yoelii (Janssen et al., 2002). However, the presence and intensity of ectopic recombination of subtelomeres to generate diversity in these gene families remain largely undetermined. Deciphering the diversifying mechanisms for these gene families can also help to understand their evolutionary history. For instance, it would help to resolve the dilemma regarding the homology of the superfamily pir with rif/stevor (Janssen et al., 2004; Cunningham et al., 2010; Harrison et al., 2020), given that phylogenetics and sequence similarity analyses support the homology (Janssen et al., 2004), but protein structure analyses reject it (Harrison et al., 2020).
Given that the importance of the mechanism of ectopic recombination of subtelomeres to generate antigenic diversity in Plasmodium is still widely unexplored, this study aims to determine the presence of this mechanism in 19 Plasmodium species and to compare its intensity across their phylogeny. To achieve this, we produced chromosome maps of gene conservation that allowed us to identify expected genomic evidence of this mechanism, for instance, young subtelomeric regions (i.e., containing genes that are present in a few species) with a high density of antigenic gene families. A higher prevalence of these features in a genome might be the result of more intense ectopic recombination of subtelomeres to produce antigenic diversity. Contrasting the presence and the importance of this mechanism across the phylogeny of Plasmodium, we proposed hypothetical events in the evolutionary history of this mechanism in Plasmodium parasites. The results of this study demonstrate, for the first time, the genomic and evolutionary complexity of this mechanism in Plasmodium. Furthermore, they reveal that its importance, which seems to have been acquired and lost multiple times in the history of Plasmodium, is clade-specific, and is more closely associated with the pir and rif/stevor genes.
2 Materials and methods
2.1 Database for phylogenetic reconstruction
Given the contention on different aspects of the current phylogeny of Plasmodium, especially on the root of the tree that is critical to interpreting the directionality of the evolution of the subgenera, we aimed to reconstruct a more robust phylogeny with a richer database and more contrasted approaches. To achieve this, a database of 40 complete genomes was constructed (Supplementary Dataset 1). The sequences were obtained from PlasmoDB (Aurrecoechea et al., 2009), PiroplasmaDB (Amos et al., 2022), and GenBank (Benson et al., 2013). The genomes for this database were chosen based on the quality of the sequence annotations, the maximization of the diversity currently described for the genus, and the keeping of a certain level of evenness across taxa. To ensure a balance between maximizing diversity and maintaining evenness across taxa, one to four genomes were selected per species (based on the available isolates per species), including each described and available subgenus of Plasmodium. Since previous studies have shown that Plasmodium is either paraphyletic or polyphyletic (Martinsen et al., 2008; Schaer et al., 2013; Borner et al., 2016; Galen et al., 2018), we sought to include the sister taxa Hepatocystis and Nycteria in the phylogenetic analysis. However, we found only 1 genome of Hepatocystis and none of Nycteria. Therefore, this database includes 35 Plasmodium species and 1 Hepatocystis species, a parasite of the red colobus monkey Piliocolobus tephrosceles (Aunin et al., 2020). Plasmodium species are distributed into four distinct subgenera (Perkins, 2014; Escalante et al., 2022): Laverania (12), Haemamoeba (2), Plasmodium (14), and Vinckeia (7). On the other hand, the remaining four species of the database (i.e., Babesia bovis, Babesia bigemina, Theileria equi, and Theileria annulata) comprised the outgroup and were chosen based on previous phylogenetic studies about Plasmodium (Perkins and Schall, 2002; Pick et al., 2011; Borner et al., 2016).
2.2 Reconstruction of the phylogeny of Plasmodium
Six different phylogenetic approaches were used to reconstruct the species tree used as a phylogenetic framework to compare conservation profiles among Plasmodium species. The first approach involved using OrthoFinder v2.5.4 to identify gene families, infer orthologs, and construct a species tree based on those orthologs (Emms and Kelly, 2015; Emms and Kelly, 2017; Emms and Kelly, 2018). While certain rapidly evolving regions of the chromosomes, such as the subtelomeres, may be susceptible to missing data due to challenges in sequencing and mapping, it is highly unlikely that this issue will impact our analysis. This is because we specifically chose gene families that are more conserved and present in all taxa. Even if these gene families happen to be subtelomeric, it should not pose a problem for reconstructing the phylogeny, as they still need to meet our taxa inclusion criteria.
The gene families present in all taxa were aligned with the einsi method of MAFFT v7.505 (Katoh et al., 2005). Then, PAL2NAL v14.0 (Suyama et al., 2006) was used to get codon alignments from the amino acid alignments. Subsequently, the codon alignments were used to reconstruct the phylogeny for each gene family with IQ-TREE v1.6.9 (Nguyen et al., 2015). The parameters -B 1000 -alrt 1000 were applied to obtain branch support (i.e., UFBoot and SH-aLRT) in all trees (Guindon et al., 2010; Hoang et al., 2018) and ModelFinder to determine the most appropriate substitution model for each gene family according to the BIC score (Kalyaanamoorthy et al., 2017).
The phylogenetic gene trees obtained with IQ-TREE were used to infer a species tree with the five remaining phylogenetic approaches. They include a supermatrix analysis by alignment concatenation (de Queiroz and Gatesy, 2007) and four summary gene tree methods, three of which used multi-copy gene families as input: ASTRAL-Pro v1.8.1.3 (Zhang et al., 2020), ASTRAL v5.7.8-DISCO v1.3 (Willson et al., 2022), and SpeciesRax v2.0.4 (Morel et al., 2022); and one that required single-copy gene trees: ASTRAL v5.7.8 (Rabiee et al., 2019). For the supermatrix analysis, the alignments of single-copy gene families were concatenated using Mega X v10.2.6 (Stecher et al., 2020), and the resulting supermatrix was used to infer another species tree with IQ-TREE (Nguyen et al., 2015) using ModelFinder to determine the best-fit model according to the BIC score (Kalyaanamoorthy et al., 2017).
The six obtained species trees were compared to each other and against other previously published versions (e.g., Borner et al., 2016; Galen et al., 2018; Escalante et al., 2022). To evaluate the quality of these phylogenetic reconstructions, in addition to analyzing the branch support of the species trees, we analyzed the median node support values of the gene family trees used as input for the species tree inference. Given previous evidence that the GC content on Plasmodium genomes can affect the topology of their species tree (Galen et al., 2018), we removed the third base position on all the codon alignments with a custom Python script and reconstructed another set of four species trees using ASTRAL-Pro v1.8.1.3 (Zhang et al., 2020), ASTRAL v5.7.8 (Rabiee et al., 2019), ASTRAL v5.7.8-DISCO v1.3 (Willson et al., 2022) and SpeciesRax v2.0.4 (Morel et al., 2022). Finally, a majority rule consensus tree of the ten species trees was constructed using PAUP* v4.0a168 (Swofford, 2002; full trees in Supplementary Dataset 2) and used as the phylogenetic framework for further analyses. All custom scripts can be found at https://github.com/camae2246/Plasmodium_ERS_2023.git.
2.3 Database for chromosome mapping of gene conservation
Out of the 35 genomes of Plasmodium used for the species tree reconstruction, 19 are reference genomes with necessary annotations for comparing chromosomal conservation profiles. These 19 genomes represent the four Plasmodium subgenera: 7 from Laverania, 1 from Haemamoeba, 7 from Plasmodium, and 4 from Vinckeia (Supplementary Dataset 1). The maps of gene conservation require the assessment of conservation on every coding gene using a diverse genome database as a reference. We leveraged our diverse genome database with 134 species distributed across the SAR clade (Stramenopiles, Alveolata, Rhizaria) and Excavata (Supplementary Table S1, Supplementary Dataset 3). Given the focus taxa of our maps (i.e., Plasmodium species), we sampled more Alveolates (Apicomplexa, Ciliophora, and Dinozoa) to have a higher resolution on this clade. The rest of the sampling was evenly distributed between Stramenopila and Rhizaria, and we also included 26 Excavata species (15 Discoba and 11 Metamonada). When selecting the genomes, we aimed to maximize the phylogenetic diversity in every clade. For each of the 134 species, including the Plasmodium species, we collected their protein sequences and, whenever possible, their coding sequences as well. All the sequences were collected from different public databases such as PlasmoDB (Aurrecoechea et al., 2009), ToxoDB (Harb and Roos, 2020), GenBank (Benson et al., 2013), PiroplasmaDB (Amos et al., 2022), CryptoDB (Heiges et al., 2006), FungiDB (Basenko et al., 2018) and TriTrypDB (Aslett et al., 2010).
Assessing gene conservation of the coding genes in each Plasmodium species requires the reconstruction of a phylogenetic tree per gene. To achieve this, we used the genomic database to infer gene families and reconstructed their phylogenies using OrthoFinder v.2.5.4 (Emms and Kelly, 2015) with the “only-trees” configuration (-ot). Subsequently, a single-copy gene tree was obtained for each gene tree using the -s option of DISCO v1.3 (Willson et al., 2022). The resulting single-copy gene trees were used as input for producing the phylogenomic maps of the chromosomes with PhyloChromoMap v1.2 (Cerón-Romero et al., 2018).
2.4 Construction of chromosome maps of gene conservation
We used PhyloChromoMap v1.2 to construct the chromosomal conservation profiles of the 19 Plasmodium species. Apart from the single-copy gene trees required to run PhyloChromoMap, we also needed to create a gene family mapping file with information from PlasmoDB and a centromere mapping file. For the latter, we created a custom Python script with the sliding window method to locate centromeres as the largest chromosomal region with the highest AT content, a feature that has been reported previously in some Plasmodium species (Gardner et al., 2002; Hoeijmakers et al., 2012). Furthermore, we analyzed the distribution of AT content along the chromosomes and compared the obtained centromeric regions with the available records in PlasmoDB.
2.5 Identification and analysis of young chromosomic regions
Young regions were defined as distinctive portions of the phylogenomic chromosome maps exhibiting low gene conservation. This is because rapidly evolving sequences are prone to frequent rearrangements, resulting in high variability. These young regions were determined using a custom Ruby script and visual inspection. Using the script, we identified candidate young regions that include a maximum of one conserved gene (present in three or more major clades). Initially, a standard minimum (80 kb) and maximum (200 kb) size were determined for young regions in all species, based on what was described in P. falciparum (Cerón-Romero et al., 2018). However, after visually inspecting the chromosomal maps, we modified the size of some regions located outside this range and reviewed manually each young region to ensure the presence of species-specific (young) genes. According to previous reports on different species of Plasmodium, telomeric regions are too small to be considered for this analysis (960–6700 bp; Bottius et al., 1998; Figueiredo et al., 2002). On the other hand, there is no consensus size for the subtelomeres. Since we were only interested in subtelomeres undergoing ectopic recombination for the purposes of this study, we only accounted for the young subtelomeric regions, which we defined as continuous regions from the chromosomal end with only species-specific genes and genes in less than three major clades.
2.6 Analysis of the distribution of antigenic genes
We searched for antigenic genes in young regions since this is an expected outcome of antigenic diversity production through ectopic recombination. To accomplish this, we created Python scripts to identify the top ten gene families per species with the highest number of sequences in young regions and compare the frequency of these families in subtelomeric young regions, internal young regions, and conserved regions. As a final step, we verified if these gene families were antigenic by using BLASTN v2.14.1+ (Zhang et al., 2000; Morgulis et al., 2008) to search for similar sequences with antigenic properties and by reviewing the literature on their products based on the information included in the genome annotations (Supplementary Datasets 4, 5). Subsequently, the antigenic genes were located on the chromosome maps to analyze their distribution across the karyotypes (Supplementary Dataset 6).
2.7 Analysis of ectopic recombination intensity to generate antigenic diversity
Based on the identified young regions and antigenic genes, we established criteria to classify species according to their signals of intensity in antigenic diversity production through ectopic recombination of subtelomeres (Supplementary Table S2). These criteria, together with the presence of antigenic sequences (≥3) in young regions, allowed us to determine candidate regions to undergo ectopic recombination to generate antigenic diversity (hereinafter referred to as “CERAD regions”). Then, we contrasted this information with the inferred consensus species tree to find patterns of evolution of this mechanism of antigenic diversity production in the evolutionary history of Plasmodium.
2.8 Statistical analysis
We assessed various traits for each species, including features of the gene conservation profiles, the distribution of antigenic genes, and the presence of CERAD regions along the chromosomes based on our observations in the chromosome maps. To estimate statistically significant differences among the subgenera (Plasmodium, Vinckeia, and Laverania), Welch’s test (Welch, 1947) was used after confirming the assumption of normality (Shapiro Wilk’s test for small sample sizes; Shapiro and Wilk, 1965), irrespective of homoscedasticity (Zimmerman, 2004). When the data did not follow a normal distribution, we used the Mann-Whitney U test (Mann and Whitney, 1947) after evaluating the homogeneity of variances with Levene’s test (Levene, 1960). All pairwise comparison tests were performed as one-tailed tests to determine which group had a significantly higher/lower value for each trait than another group. A p-value < 0.05 was considered statistically significant. We used R software (R Core Team, 2022) to conduct the statistical analyses.
3 Results
3.1 Phylogenetic reconstruction of Plasmodium
OrthoFinder detected 7,415 gene families, of which 823 are present in all taxa and generated an alignment and a phylogenetic tree. Of these 823 gene families, 597 are single-copy and 226 are multi-copy. Laverania, Vinckeia, and Plasmodium have a mean ratio of 1.132, 1.136, and 1.132 sequences per gene family respectively, and Haemamoeba (P. relictum) has 1.075 sequences per gene family. The quality of the 823 phylogenetic trees generated with IQ-TREE was good since 99% of the gene families had a median bootstrap (UFBoot) value between 80 and 100.
Phylogenomic analysis of Plasmodium showed a highly consistent topology among the species tree reconstruction approaches (normalized consensus fork = 1.0; Figures 1C-G) and suggests that the subgenus Plasmodium is a non-monophyletic group (Figure 1A). Only the tree generated by OrthoFinder shows Plasmodium as a monophyletic group but with low branch support (<0.50, Figure 1B). In contrast, the remaining five trees showed Plasmodium as non-monophyletic and at the base of the tree, with high branch support (>0.80, Figures 1C-G). This topology also has important implications for Hepatocystis and Vinckeia, which appear in the early bifurcations of the OrthoFinder tree (Figure 1B), but share a most recent common ancestor and form the sister clade of Laverania-Haemamoeba in the other five phylogenetic trees (Figures 1C-G). Finally, although the monophyly of Plasmodium is not supported by these results, seven of its taxa (P. coatneyi, P. inui, P. fragile, P. knowlesi, P. cynomolgi, P. vivax and P. vivax-like) form a recurrent monophyletic clade in the species trees, except in the tree generated by the supermatrix method (Figure 1G). Removing the third base of the codons, generated the same major result: the subgenus Plasmodium as non-monophyletic and at the base of the tree. The only notable difference is in the monophyletic subgroup inside the subgenus Plasmodium, which also contains P. gonderi in the trees generated with the codon alignments without the third base (Supplementary Figure S1).
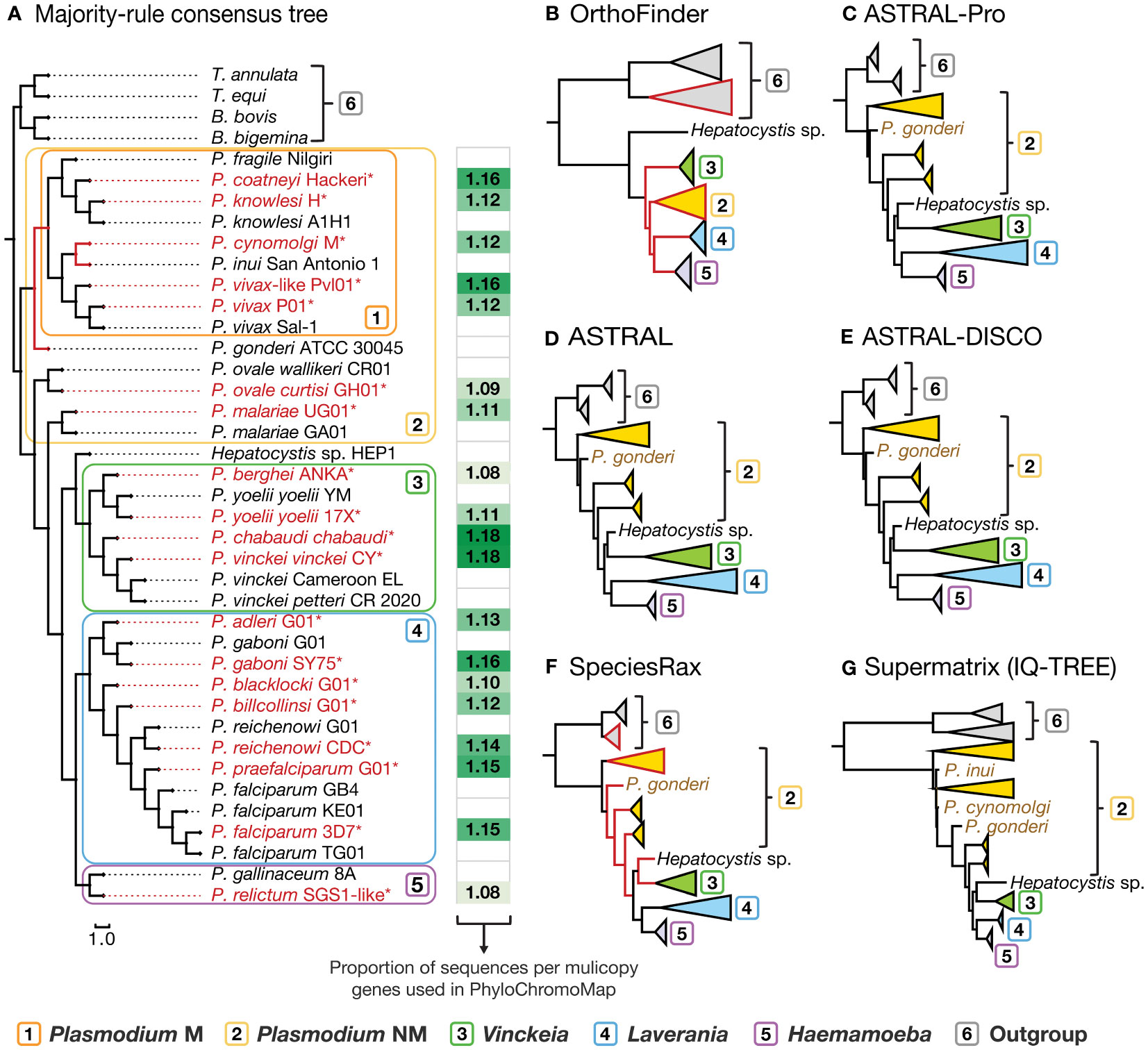
Figure 1 Phylogenetic reconstruction of the genus Plasmodium. (A) Majority-rule (50%) consensus tree generated with PAUP*, suggesting that the subgenus Plasmodium is not monophyletic. The proportion of sequences per gene family obtained with OrthoFinder, used in PhyloChromoMap, is shown next to each species. (B) OrthoFinder, (C) ASTRAL-Pro, (D) ASTRAL, (E) ASTRAL-DISCO, (F) SpeciesRax, (G) Supermatrix-(IQ-TREE). Red branches indicate low statistical support: Consensus (<70%), OrthoFinder (STAG consensus <70%), SpeciesRax (EQPIC<0,2). For the ASTRAL-Pro, ASTRAL, and ASTRAL-DISCO trees, all branches showed 100% support (LPP), except between P. falciparum strains 3D7 and TG01 (LPP=95-97%). All the branches in the supermatrix tree showed high support (SH-aLRT≥99%, UFBoot≥90%). The methods ASTRAL-Pro, ASTRAL, ASTRAL-DISCO, and SpeciesRax, were repeated with alignments with the third base of the codons removed with the results only showing one notable change: P. gonderi as part of the monophyletic subgroup of Plasmodium. A complementary figure with these last four trees can be found in Supplementary Figure S1. Complete trees can be found in the Supplementary Dataset 2. The ten species trees were used for the consensus tree. B bigemina, B bovis, T. annulata, and T. equi were used as an outgroup to estimate the root of the trees. (*) = Species whose gene conservation profile was analyzed. Plasmodium NM, non-monophyletic (all species); Plasmodium M, monophyletic (monophyletic subgroup excluding P. gonderi, P. ovale and P. malariae).
3.2 Gene conservation profiles
OrthoFinder generated 63,661 multi-copy gene trees, and these were subsequently used by DISCO to create a database consisting of 31,260 single-copy gene trees. To achieve this, DISCO decomposed the multi-copy gene trees by choosing only one leaf per species in each gene tree. Therefore, the 32,401 gene trees that were discarded by DISCO were those that finished with less than 4 taxa after the decomposition and could not produce a gene tree (Supplementary Figure S2). This reduction of the gene tree database did not have a significant impact on further analyses because the ratio of number of trees per species remained high: Apicomplexa 98%, Other alveolates 72%, Stramenopila 94%, Rhizaria 90%, Discoba 91%, and Metamonada 77% (Supplementary Figure S2). Overall, between 25-50% of their phylogenetic trees were discarded for less than 13% of the species, and between 40-50% of the gene families were discarded for less than 5% of the species. Moreover, the missing (discarded) genes in the conservation maps would also be interpreted as young. Finally, for the construction of the phylogenetic maps, a centromere was detected in all chromosomes of each species except for chromosome 2 of P. cynomolgi and P. coatneyi, chromosome 12 of P. relictum, and chromosomes 2 and 6 of P. vivax-like.
Phylogenetic chromosome maps showed that Vinckeia exhibits a significantly different gene conservation pattern compared to Plasmodium and Laverania (Figures 2A-C). This pattern was characterized by young subtelomeres at almost all chromosome ends, and a few young internal regions, which do not exceed 85 kb. Accordingly, the proportion of young subtelomeres in Vinckeia is significantly higher than in Laverania (One-tailed Wilcoxon-Mann-Whitney, W = 25, p = 0.0224) and Plasmodium (One-tailed Wilcoxon-Mann-Whitney, W = 25, p = 0.0182). Likewise, the proportion of chromosomes with young internal regions and the average size of these regions was significantly lower in Vinckeia than in Laverania (One-tailed Wilcoxon-Mann-Whitney, W = 4.5, p = 0.0401; One-tailed Welch, t = -4.4409, p = 0.0012, respectively), and Plasmodium (One-tailed Welch, t = -3.3365, p = 0.0054; One-tailed Welch, t = -2.3642, p = 0.0225, respectively). Unlike Vinckeia, the subgenus Plasmodium showed high chromosomal structural variation among its species, even in those that are part of its monophyletic clade, and Laverania exhibited an intermediate pattern of variation compared to what was observed in Vinckeia and Plasmodium (Figure 2D). On the other hand, Haemamoeba (P. relictum) exhibits less than 20% of chromosome ends as young subtelomeres and less than 25% of chromosomes with young internal regions whose size is less than 85 kb (Figures 2A-C). The results of all statistical tests were summarized in the Supplementary Tables S3-5.
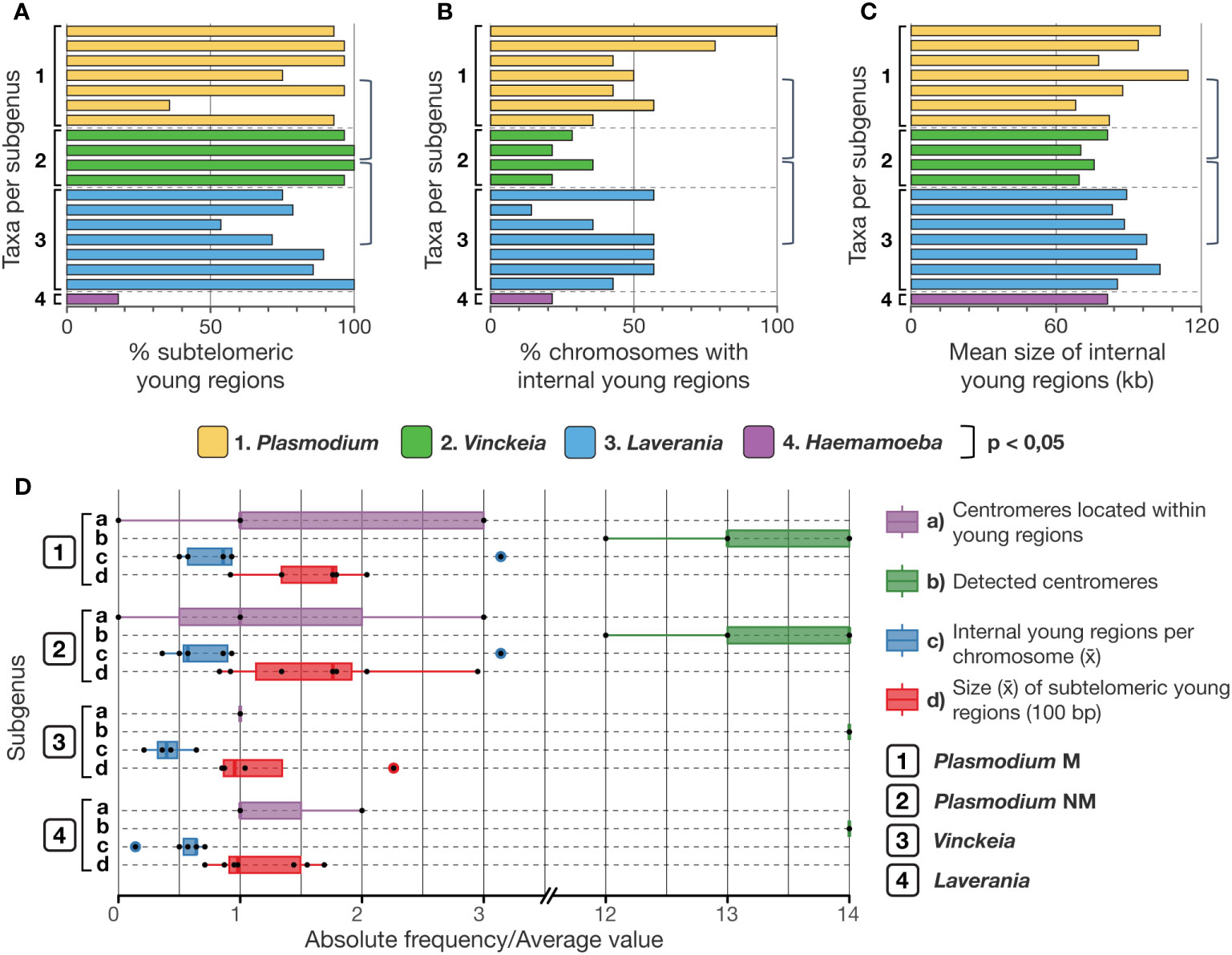
Figure 2 Comparison of features derived from gene conservation profiles among Plasmodium subgenera. (A) Percentage of young subtelomeres. Vinckeia shows a significantly higher percentage than Laverania (One-tailed Wilcoxon-Mann-Whitney, W = 25, p = 0.0224) and Plasmodium (One-tailed Wilcoxon-Mann-Whitney, W = 25, p = 0.0182). (B) Percentage of chromosomes with young internal regions. Vinckeia shows significantly lower values than Laverania (One-tailed Wilcoxon-Mann-Whitney, W = 4.5, p = 0.0401) and Plasmodium (One-tailed Welch, t = -3.3365, p = 0.0054). (C) Average size of young internal regions (kb). Vinckeia shows significantly smaller values than Laverania (One-tailed Welch, t = -4.4409, p = 0.0012) and Plasmodium (One-tailed Welch, t = -2.3642, p = 0.0225). (D) Features with higher variation in Plasmodium than in Vinckeia and Laverania, where even the monophyletic clade of Plasmodium shows a higher variation. Plasmodium NM, non-monophyletic (all species); Plasmodium M, monophyletic (monophyletic subgroup).
3.3 Distribution of antigenic gene families
The search for the ten predominant gene families in young regions per species resulted in a total of 133 gene families (Supplementary Dataset 5). In this process, P. relictum was the only species with less than ten families found (Figure 3A). Out of the total number of gene families obtained, 11 were excluded from the analysis because BLAST searches of these sequences do not retrieve an implicit antigenic gene and it was not possible to verify whether they were antigenic based on the genome annotations or the reviewed literature on their products. In addition, 14 gene families were classified as candidate antigenic gene families since their sequences seem to play a key role in the virulence of these parasites, but their antigenic role could not be confirmed. Following this classification, most of the gene families per species (>80%; Supplementary Dataset 4) were suitable for the analysis of distribution on the chromosome maps, except in Haemamoeba (P. relictum) where 57% of its gene families were discarded (Figure 3A).
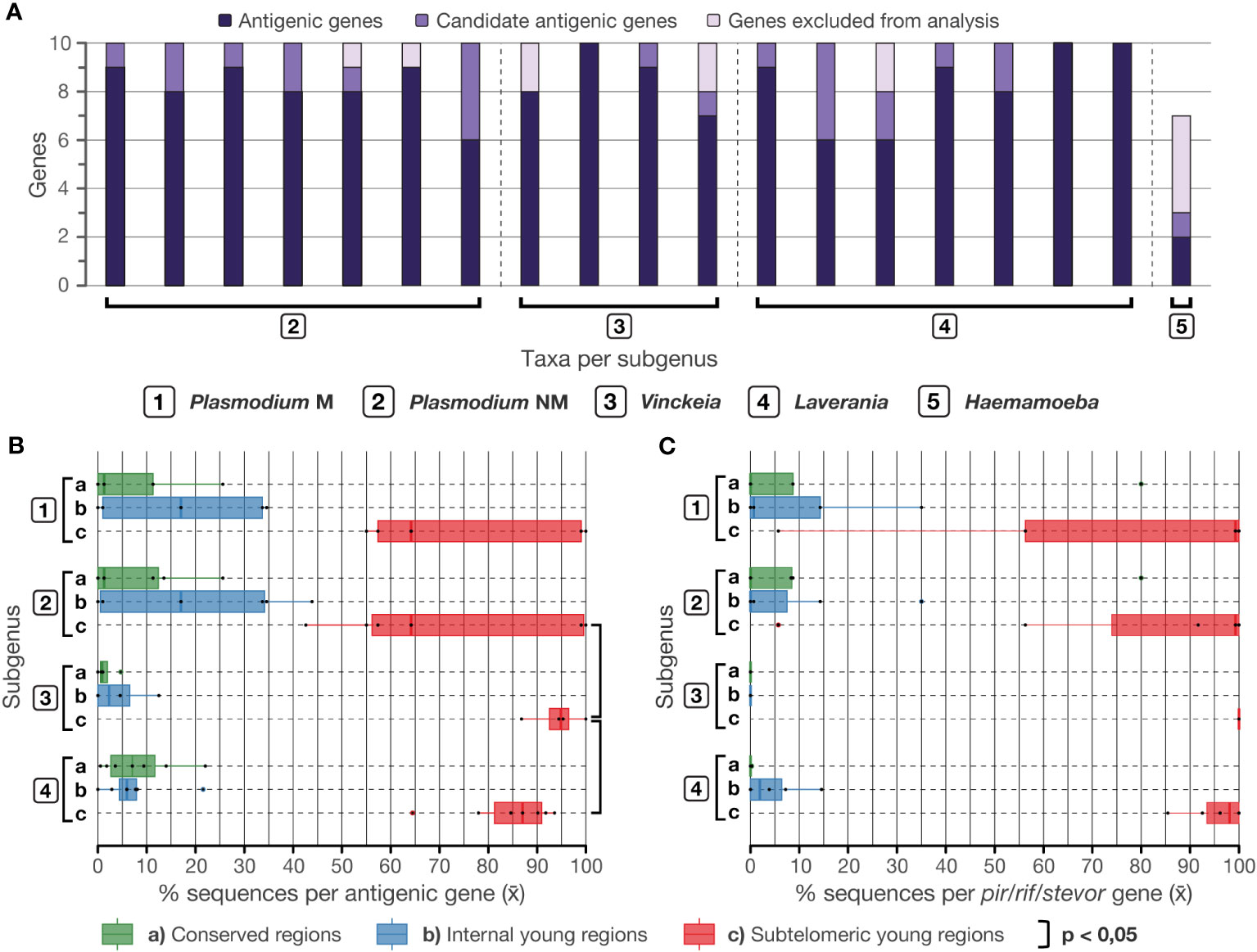
Figure 3 Analysis of the distribution of antigenic genes on chromosomes. (A) Classification of predominant genes in young regions according to the literature (Supplementary Dataset 5). (B) Average percentage of sequences per antigenic gene family in each chromosome region. Genes in Vinckeia exhibited a significantly higher percentage of subtelomeric sequences compared to Plasmodium (One-tailed Welch, t = 2.0581, p = 0.0394) and Laverania (One-tailed Welch, t = 2.1049, p = 0.0323). (C) Average percentage of sequences per pir/rif/stevor gene family (pir in Vinckeia and Plasmodium, rif/stevor in Laverania) in each chromosome region. A higher percentage of the sequences of these genes tend to locate preferentially in subtelomeric young regions. Plasmodium NM, non-monophyletic (all species); Plasmodium M, monophyletic (monophyletic subgroup).
The distribution of antigenic genes on chromosome maps (Figure 4; complete maps per species in Supplementary Dataset 6) revealed that these genes tend to prefer subtelomeric regions (Figure 3B). Vinckeia species exhibit the highest averages (>85%) of the number of sequences per gene family in subtelomeric young regions, and thus, a low variation is observed in the distribution of this trait. As a result, this subgenus exhibits a significantly higher average of this trait than Plasmodium (One-tailed Welch, t = 2.0581, p = 0.0394) and Laverania (One-tailed Welch, t = 2.1049, p = 0.0323). In contrast, Plasmodium is the subgenus (even when evaluating only its monophyletic subgroup) that shows the highest variation in the averages of the number of sequences per gene family in the different chromosomal regions, while Laverania presents an intermediate pattern of variation (Figure 3B). In the case of Haemamoeba (P. relictum), there is no clear location preference in the few antigenic genes detected (Supplementary Dataset 4).
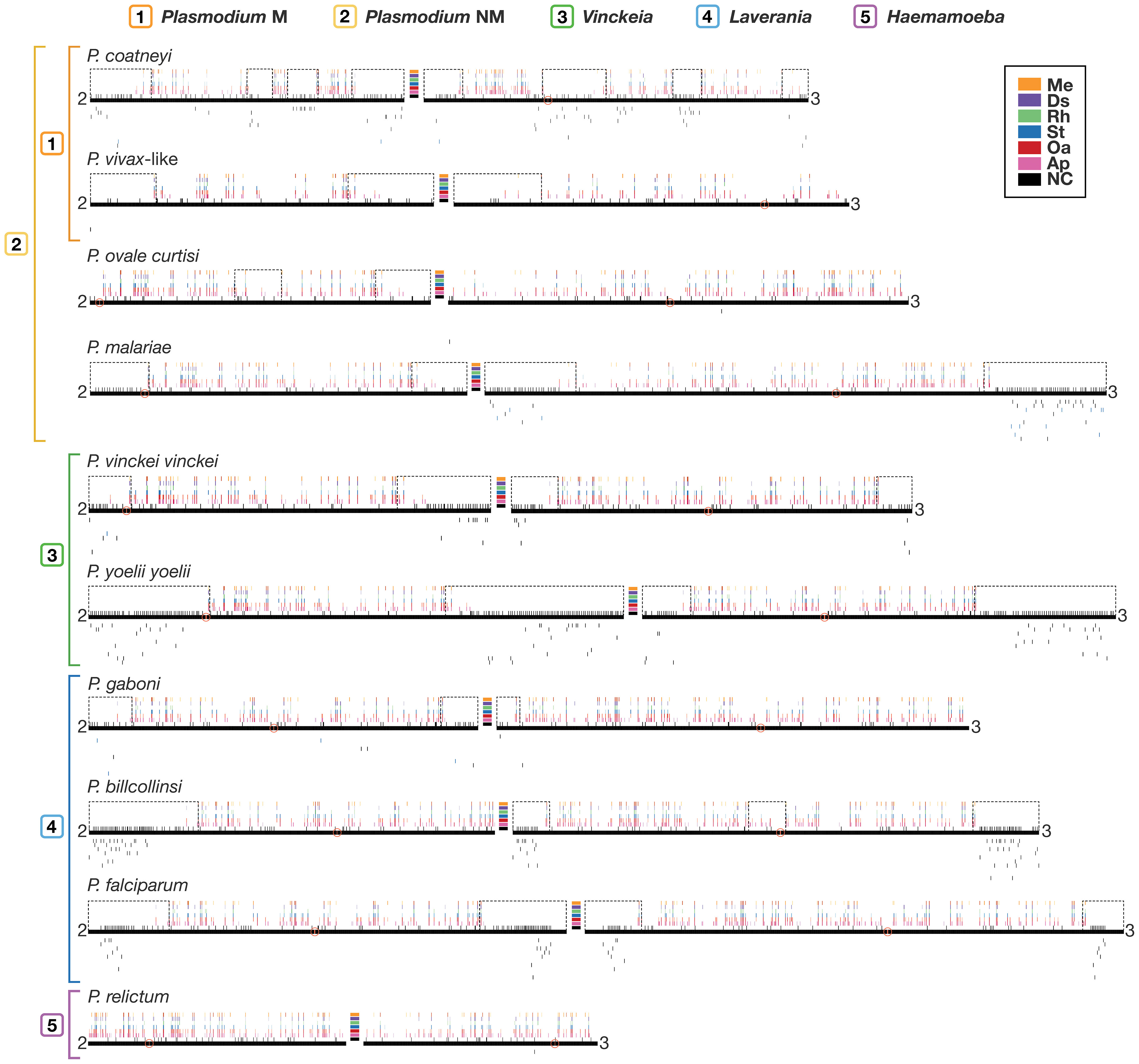
Figure 4 Examples of chromosome maps showing the gene conservation profile, presence of young regions, and distribution of antigenic genes in ten Plasmodium species distributed in Plasmodium, Vinckeia, Laverania, and Haemamoeba. Black lines represent chromosomes and bars above reflect levels of conservation, with dashed boxes around “young” regions. Detected centromeres are indicated by a red circle. Above the black line, the first row (NC) indicates genes whose phylogenetic trees do not meet the criteria of having more than ten taxa. The remaining rows (bottom to top) are heatmaps reflecting the proportion of lineages of Apicomplexa (Ap), Other Alveolates (Oa), Stramenopila (St), Rhizaria (Rh), Discoba (Ds), and Metamonada (Me) that contain the indicated gene. Lines below the chromosomes show the location of sequences belonging to antigenic gene families (black) or candidate antigenic gene families (blue), one per row, found in each species. Plasmodium NM, non-monophyletic (all species); Plasmodium M, monophyletic (monophyletic subgroup). The complete chromosome maps of the 19 Plasmodium species can be found at Supplementary Dataset 6.
All Plasmodium and Vinckeia species sampled presented pir genes, while 86% of Laverania species exhibited rif/stevor genes. When analyzing the distribution of sequences from these families, it was found that they tend to be located preferentially in the subtelomeres (Figure 3C). This preference is most evident in Vinckeia where all the species exhibit a consistent pattern of having pir sequences in subtelomeric regions. In contrast, Plasmodium shows a high variation in the average percentage of pir sequences in each chromosomal region, and Laverania shows an intermediate variation for its rif/stevor sequences.
3.4 Intensity of ectopic recombination of subtelomeres to produce antigenic diversity
Analysis of CERAD region distribution shows a tendency to concentrate these regions in the subtelomeres, and not in internal regions, in Vinckeia, Laverania, and Plasmodium (Figure 5). This tendency is less clear in Laverania than in Vinckeia, and even less clear in Plasmodium (the complete group and the monophyletic subgroup) where there is a high variation of this pattern among its species. Accordingly, Vinckeia showed a higher distribution of CERAD regions in subtelomeres (Figure 5A) than Laverania (One-tailed Welch, t = 3.0488, p = 0.0069), Plasmodium (One-tailed Wilcoxon-Mann-Whitney, W = 25, p = 0.0224), and even the monophyletic subgroup of Plasmodium (One-tailed Welch, t = 2.3540, p = 0.0299); and a significantly lower distribution of the percentage of chromosomes with internal CERAD regions (Figure 5B) than Laverania (One-tailed Welch, t = -2.3959, p = 0.0202). On the other hand, no CERAD regions were detected in P. relictum (Haemamoeba) considering the low number of antigenic genes and the minority of young regions found.
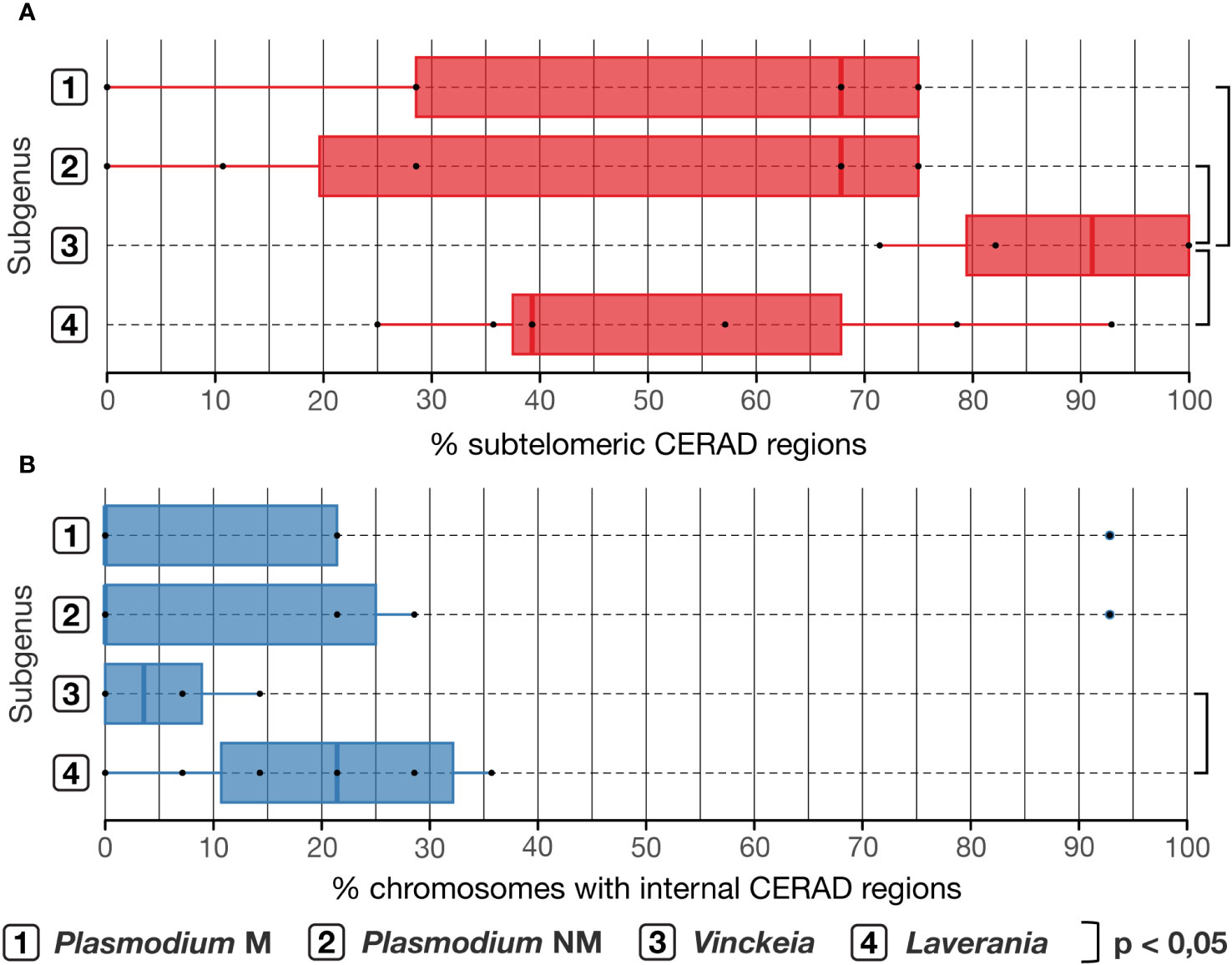
Figure 5 Analysis of presence of chromosomal CERAD regions. (A) Percentage of subtelomeric CERAD regions. Vinckeia has significantly higher percentages than Laverania (One-tailed Welch, t = 3.0488, p = 0.0069), Plasmodium NM (One-tailed Wilcoxon-Mann-Whitney, W = 25, p = 0.0224), and Plasmodium M (One-tailed Welch, t = 2.3540, p = 0.0299). (B) Percentage of chromosomes with internal CERAD regions. Vinckeia has significantly lower percentages than Laverania (One-tailed Welch, t = -2.3959, p = 0.0202). CERAD regions, Candidate regions to undergo Ectopic Recombination to generate Antigenic Diversity; Plasmodium NM, non-monophyletic (all species); Plasmodium M, monophyletic (monophyletic subgroup).
The evaluation of the intensity of ectopic recombination to produce antigenic diversity across the phylogeny shows a greater intensity of this mechanism in Vinckeia, an intermediate intensity in Laverania, high variation in Plasmodium, and zero intensity in Haemamoeba (Figure 6A). These results are consistent with the presence of antigenic genes (Figure 6B), particularly pir/rif/stevor (Figure 6C), and the distribution of CERAD regions on the chromosomes (Figures 6D, E). Taken together, these features mark a distinct pattern in each subgenus. In Vinckeia, this pattern is characterized by a high intensity of this mechanism to produce antigenic diversity, an accumulation of CERAD regions in subtelomeres rather than in internal parts of the chromosomes, and a high number of pir genes. Meanwhile, in Laverania, an intermediate level of this mechanism is observed, which gradually increases as it approaches the clade of P. falciparum and P. praefalciparum, occurring in conjunction with the increase in the percentage of CERAD regions and the number of rif/stevor genes. On the other hand, Plasmodium exhibits abrupt changes in the intensity levels of the mechanism and other evaluated features, reflecting the high variation that characterizes this subgenus, which is also evident in its monophyletic subgroup. Additionally, a hundred comparisons across equally sized subsamples of Plasmodium, Laverania, and Vinckeia (n=4) consistently reveal the same patterns among clades, indicating that these observations are not influenced by clade size differences (Supplementary Dataset 7).
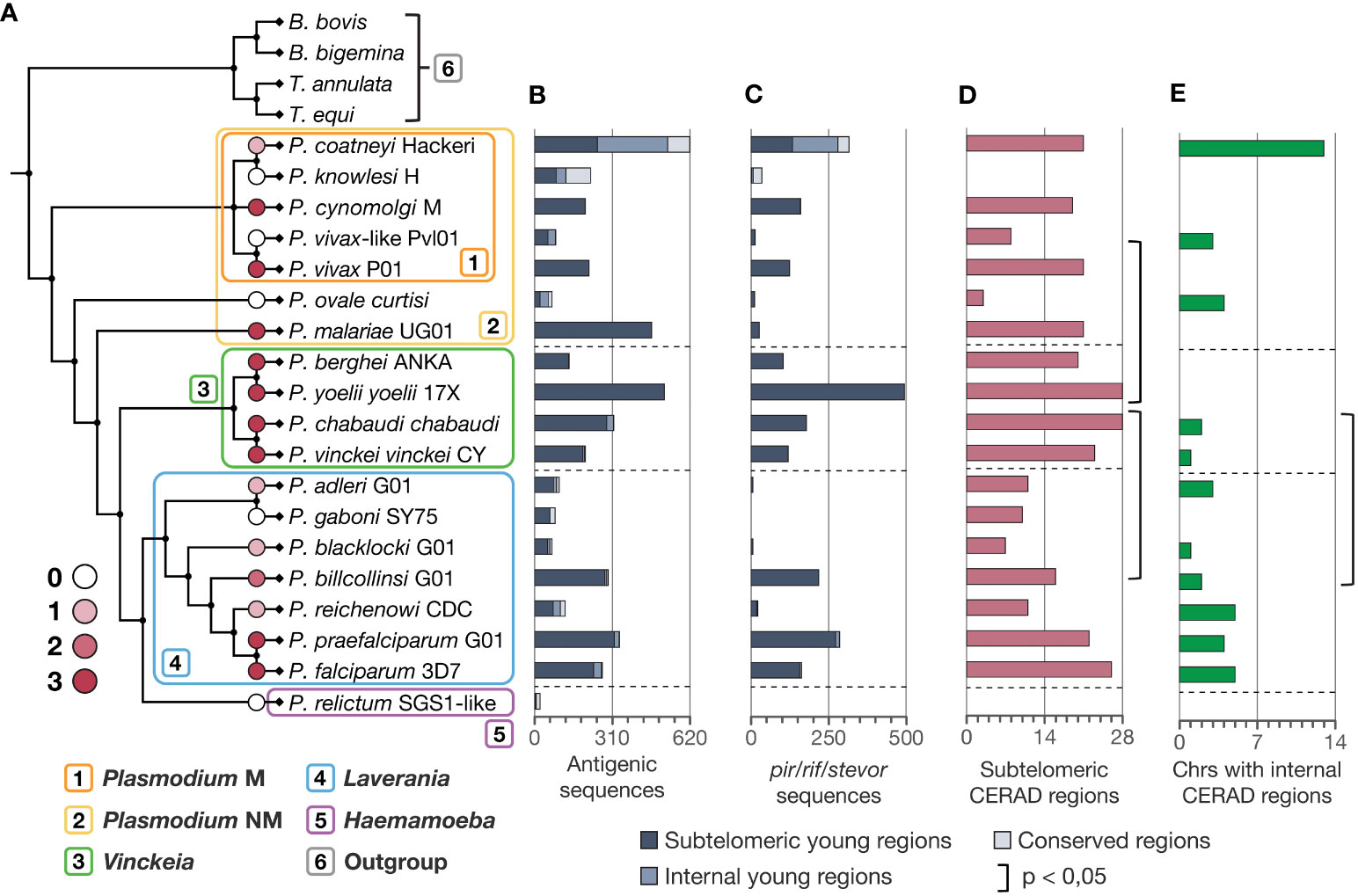
Figure 6 Comparison of genomic features associated with ectopic recombination of subtelomeres to generate antigenic diversity in Plasmodium species. (A) Intensity levels of ectopic recombination to produce antigenic diversity placed in the phylogeny of Plasmodium. (B) Number of antigenic sequences in each chromosomal region. (C) Number of pir/rif/stevor sequences (pir in Vinckeia and Plasmodium, rif/stevor in Laverania) in each chromosomal region. (D) Number of CERAD subtelomeres per species. Vinckeia exhibits a significantly higher number of CERAD subtelomeric regions than Laverania (One-tailed Welch, t = 3.0488, p = 0.0069) and Plasmodium (One-tailed Wilcoxon-Mann-Whitney, W = 25, p = 0.0224). (E) Number of chromosomes with internal CERAD regions per species. Vinckeia has a significantly lower number of chromosomes with internal CERAD regions than Laverania (One-tailed Welch, t = -2.3959, p = 0.0202). CERAD regions, Candidate regions to undergo Ectopic Recombination to generate Antigenic Diversity; Plasmodium NM, non-monophyletic (all species); Plasmodium M, monophyletic (monophyletic subgroup).
4 Discussion
This study presents, for the first time, a distribution of the mechanism to generate antigenic diversity through ectopic recombination of subtelomeres in the genus Plasmodium, represented by 19 species distributed among the subgenera Plasmodium, Vinckeia, Laverania and Haemamoeba. This mechanism, previously described only in P. falciparum, occurs after chromosome ends anchor in clusters near the nuclear periphery (Freitas-Junior et al., 2000) and can be inferred by analyzing the distribution of associated genomic features, such as the presence of young subtelomeres and antigenic genes concentrated towards the subtelomeres (Cerón-Romero et al., 2018). We applied this approach to 19 species of Plasmodium (Figure 6). Furthermore, contrasting the presence of the associated genomic features with the phylogeny of the group allowed us to establish hypotheses about the origin and evolution of this molecular mechanism to generate antigenic diversity in the evolutionary history of Plasmodium. Based on this, the results of this work provide three important findings: 1) The phylogeny of Plasmodium does not support the subgenus Plasmodium as monophyletic; 2) Regardless of the discordance of the phylogeny in this study and others previously published (Galen et al., 2018; Pacheco et al., 2018; Escalante et al., 2022), Vinckeia shows a consistent pattern of high levels of intensity of this molecular mechanism in all its species, whereas Laverania exhibits a pattern of intermediate intensity and Plasmodium shows a high variation in intensity levels; 3) This molecular mechanism has been evolutionarily more associated with pir and rif/stevor genes, which fuels the debate about the homology of these gene families (Cunningham et al., 2010; Harrison et al., 2020).
The phylogeny of Plasmodium reconstructed in this study (Figure 1) contrasts with the most widely accepted proposal, in which the subgenus Plasmodium is monophyletic, and avian and reptilian parasites diverge first (i.e., closer to the root). However, it is important to keep in mind that this proposal arose from early studies based on analyses with mitochondrial DNA and/or few nuclear loci (Escalante et al., 1998; Perkins and Schall, 2002; Hayakawa et al., 2008; Martinsen et al., 2008; Krief et al., 2010). More recent studies with genomic data have reported mixed results. Some studies support the monophyly of Plasmodium (Pacheco et al., 2011; Loy et al., 2017; Pacheco et al., 2018; Escalante et al., 2022), while others reject it (Rutledge et al., 2017; Böhme et al., 2018). On the other hand, the pattern observed in this study with the subgenus Plasmodium at the base of the phylogeny was also obtained in a recent study, but it was explained as a phylogenetic artifact caused by the attraction between this subgenus and the outgroup due to their similarity in GC content (Galen et al., 2018). Although we saw significant differences in GC content among groups (Kruskal-Wallis, chi-squared = 29.454, df = 3, p-value = 1.798e-06), our results demonstrated that removing the third base of the codons in the alignments, a proxy to reduce base composition bias, did not affect the major finding of the phylogenetic analysis - the sugbenus Plasmodium as the earliest divergent and non-monophyletic group.
The lack of consensus among phylogenetic studies may be largely due to differences in database size (genes and species) (Martinsen et al., 2008; Krief et al., 2010; Galen et al., 2018; Pacheco et al., 2018), the lack of comparison between different phylogenetic approaches (Martinsen et al., 2008; Pacheco et al., 2011; Pick et al., 2011), and the assumption of a root for the phylogeny instead of inferring it (Pacheco et al., 2011; Escalante et al., 2022). Considering the above, the phylogenetic analysis performed in this study is the most robust to date. However, future efforts that provide more data from taxa related to Haemamoeba, P. ovale, and P. malariae, and their inclusion in phylogenetic studies can have important changes in the topology of this species tree. Therefore, the interpretations made for the rest of the analyses were done considering different evolutionary scenarios (e.g., the subgenus Plasmodium as a monophyletic and non-monophyletic group).
Ectopic recombination of subtelomeres to produce antigenic diversity shows different levels of significance in Vinckeia, Laverania, Haemamoeba, and Plasmodium, proving to be clade-specific. Our results demonstrate that Vinckeia is the subgenus with the most uniform pattern among species (Figures 2–6), characterized by the presence of ectopic recombination of subtelomeres at high levels, suggesting that this feature may have been crucial for the evolution of this group. In contrast, this mechanism seems to be important in Laverania but no more so than in Vinckeia (intermediate intensity) and its importance increases as one progresses toward the clade of P. falciparum and P. praefalciparum (Figure 6). Consistent with this intermediate intensity in Laverania and in contrast to what was observed in Vinckeia, the results suggest that in some cases, internal chromosomal regions of Laverania may ectopically recombine with the subtelomeres, as has been proposed for the var genes of P. falciparum (Marty et al., 2006; Claessens et al., 2014). In the case of P. relictum (Haemamoeba), this mechanism does not seem to be important to generate antigenic diversity (Figures 4, 6, Supplementary Dataset 6). If present, this mechanism could have acquired another function, and antigenic diversity is then promoted by other means (Pain et al., 2008; Zhang et al., 2019). On the other hand, the subgenus Plasmodium exhibits a high variation among its species, even within its monophyletic clade. This variation suggests that this mechanism is important only for half of its species (Figure 6A) and implies that its significance was either lost or acquired multiple times independently within this subgenus.
Considering the differences among the subgenera in their patterns of intensity of ectopic recombination to generate antigenic diversity, we can propose different evolutionary scenarios to explain the significance of this mechanism for each of them. Based on our consensus phylogeny (Figure 1A), we can infer that this mechanism emerged and gained importance independently on several occasions in Plasmodium, whereas two scenarios may have occurred in the Vinckeia-Laverania-Haemamoeba clade. The first scenario is an independent acquisition in the ancestors of Vinckeia and Laverania, with different levels of importance in both clades. The other scenario is the acquisition of this mechanism in the ancestor of the Vinckeia-Laverania-Haemamoeba clade, with an independent loss in Haemamoeba (P. relictum) and one Laverania species (P. gaboni). The likelihood of both scenarios depends largely on whether future studies provide evidence of this mechanism in other Haemamoeba species. On the other hand, according to the phylogeny with avian clades as the first divergent groups (Galen et al., 2018; Escalante et al., 2022), the most parsimonious scenario is that this trait appeared after the divergence of the avian groups, with different consequences for each clade: intermediate and gradual importance in Laverania, absolute importance in Vinckeia, and independent losses in Plasmodium. However, if other Haemamoeba species have this trait, it is also possible that it is an ancestral trait of the four subgenera with multiple independent losses.
The mechanism of ectopic recombination of subtelomeres is more linked to the generation of diversity of the pir and rif/stevor gene families than to other gene families, reigniting the debate over whether these families are part of the same superfamily (Cunningham et al., 2010). Although studies based on the comparison of their protein structures, which are more conserved and useful to detect homology than sequences, have determined significant differences between pir and rif/stevor (Harrison et al., 2020), the values to establish significant differences can be arbitrary and debatable, especially when talking about proteins with a high evolutionary rate (Hernandez-Rivas et al., 1996; Rich and Ayala, 2000; Claessens et al., 2014). In fact, according to our analysis, rif and pir are among the most recombinant gene families (Supplementary Dataset 4). Therefore, our results suggest one more feature in common between these families that may contribute to future studies aiming to establish homology among them. Likewise, further studies clarifying whether there is homology between these gene families would also be useful to establish whether the association between the mechanism of ectopic recombination of subtelomeres and the diversity of these antigenic gene families is of ancestral nature.
In conclusion, we can infer from this study that ectopic recombination of subtelomeres is the primary mechanism for generating diversity in pir and rif/stevor genes, which explains the difference in the intensity of this mechanism among different clades of Plasmodium and suggests that other gene families probably prefer alternative mechanisms to generate antigenic diversity. However, it is important to mention some of the limitations that we encountered during the execution of the analyses. For example, although this study improves several aspects of previous phylogenetic studies, the available genomic sequences for some groups in Plasmodium, especially Haemamoeba, are still very scarce. Future efforts to sequence more of those taxa and include them in phylogenetic studies could alter the phylogenetic topology proposed here. Anticipating this limitation, the evolutionary scenarios we discussed also consider alternate phylogenetic topologies. Moreover, complementary studies on how some of the genomic features analyzed here vary with traits of the immune system of the hosts and vectors can offer valuable insights to understand further the evolutionary history of this molecular mechanism in Plasmodium. Finally, it is worth noting that the inferences we made here about the presence of this molecular mechanism depend on its expected consequences in the genome, such as the presence of subtelomeric young regions with a high density of antigenic genes. Therefore, future studies focused on analyzing the presence of the protein machinery, still unknown, involved in this process at the cellular level (Figueiredo and Scherf, 2005; Hernández-Rivas et al., 2013), would be crucial to validate the propositions presented in this study.
Data availability statement
The raw data used for this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author contributions
MC-R and CM-E conceived of the study and broad approach, and designed the experiments in collaboration with HC, CM-E performed the analyses. CM-E and MC-R wrote the manuscript with input from HC. All authors contributed to the article and approved the submitted version.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We thank the Scientific Computing Laboratory (LACCo) of CIBioFi for providing computational resources. Further, we thank Oscar E. Ospina (Moffitt Cancer Center), Abdel H. Halloway (Case Western Reserve University), and our referees for their valuable insights and comments on the manuscript. Finally, we extend our thanks to Andrea C. Niño Castro, head of the Department of Biology of Universidad del Valle, for her support during the early stages of the project.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2023.1177350/full#supplementary-material
References
Al-Khedery B., Barnwell J. W., Galinski M. R. (1999). Antigenic variation in malaria: a 3′ Genomic alteration associated with the expression of a P. knowlesi variant antigen. Mol. Cell 3 (2), 131–141. doi: 10.1016/S1097-2765(00)80304-4
Amos B., Aurrecoechea C., Barba M., Barreto A., Basenko E. Y., Bażant W., et al. (2022). VEuPathDB: the eukaryotic pathogen, vector and host bioinformatics resource center. Nucleic Acids Res. 50 (D1), D898–D911. doi: 10.1093/nar/gkab929
Arkhipova I. R., Morrison H. G. (2001). Three retrotransposon families in the genome of Giardia lamblia: Two telomeric, one dead. Proc. Natl. Acad. Sci. 98 (25), 14497–14502. doi: 10.1073/pnas.231494798
Aslett M., Aurrecoechea C., Berriman M., Brestelli J., Brunk B. P., Carrington M., et al. (2010). TriTrypDB: a functional genomic resource for the Trypanosomatidae. Nucleic Acids Res. 38 (suppl_1), D457–D462. doi: 10.1093/nar/gkp851
Aunin E., Böhme U., Sanderson T., Simons N. D., Goldberg T. L., Ting N., et al. (2020). Genomic and transcriptomic evidence for descent from Plasmodium and loss of blood schizogony in Hepatocystis parasites from naturally infected red colobus monkeys. PloS Pathog. 16 (8), e1008717. doi: 10.1371/journal.ppat.1008717
Aurrecoechea C., Brestelli J., Brunk B.P., Dommer J., Fischer S., Gajria B., et al. (2009). PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Res. 37 (suppl_1), D539–D543. doi: 10.1093/nar/gkn814
Barry J. D., Ginger M. L., Burton P., McCulloch R. (2003). Why are parasite contingency genes often associated with telomeres? Int. J. Parasitol. 33 (1), 29–45. doi: 10.1016/S0020-7519(02)00247-3
Basenko E. Y., Pulman J. A., Shanmugasundram A., Harb O. S., Crouch K., Starns D., et al. (2018). FungiDB: an integrated bioinformatic resource for fungi and oomycetes. J. Fungi 4 (1), p.39. doi: 10.3390/jof4010039
Benson D. A., Cavanaugh M., Clark K., Karsch-Mizrachi I., Lipman D. J., Ostell J., et al. (2013). GenBank. Nucleic Acids Res. 41 (Database issue), D36–D42. doi: 10.1093/nar/gks1195
Böhme U., Otto T. D., Cotton J. A., Steinbiss S., Sanders M., Oyola S. O., et al. (2018). Complete avian malaria parasite genomes reveal features associated with lineage-specific evolution in birds and mammals. Genome Res. 28 (4), 547–560. doi: 10.1101/gr.218123.116
Borner J., Pick C., Thiede J., Kolawole O. M., Kingsley M. T., Schulze J., et al. (2016). Phylogeny of haemosporidian blood parasites revealed by a multi-gene approach. Mol. Phylogenet. Evol. 94, 221–231. doi: 10.1016/j.ympev.2015.09.003
Bottius E., Bakhsis N., Scherf A. (1998). Plasmodium falciparum telomerase: de novo telomere addition to telomeric and nontelomeric sequences and role in chromosome healing. Mol. Cell. Biol. 18 (2), 919–925. doi: 10.1128/MCB.18.2.919
Carlton J. M., Adams J. H., Silva J. C., Bidwell S. L., Lorenzi H., Caler E., et al. (2008). Comparative genomics of the neglected human malaria parasite Plasmodium vivax. Nature 455 (7214), 757–763. doi: 10.1038/nature07327
Carlton J. M.-R., Galinski M. R., Barnwell J. W., Dame J. B. (1999). Karyotype and synteny among the chromosomes of all four species of human malaria parasite. Mol. Biochem. Parasitol. 101 (1), 23–32. doi: 10.1016/S0166-6851(99)00045-6
Cerón-Romero M. A., Nwaka E., Owoade Z., Katz L. A. (2018). PhyloChromoMap, a Tool for Mapping Phylogenomic History along Chromosomes, Reveals the Dynamic Nature of Karyotype Evolution in Plasmodium falciparum. Genome Biol. Evol. 10 (2), 553–561. doi: 10.1093/gbe/evy017
Cheng Q., Cloonan N., Fischer K., Thompson J., Waine G., Lanzer M., et al. (1998). stevor and rif are Plasmodium falciparum multicopy gene families which potentially encode variant antigens. Mol. Biochem. Parasitol. 97 (1), 161–176. doi: 10.1016/S0166-6851(98)00144-3
Claessens A., Hamilton W. L., Kekre M., Otto T. D., Faizullabhoy A., Rayner J. C., et al. (2014). Generation of antigenic diversity in Plasmodium falciparum by structured rearrangement of var genes during mitosis. PloS Genet. 10 (12), e1004812. doi: 10.1371/journal.pgen.1004812
Cunningham D., Lawton J., Preiser P., Langhorne J. (2010). The pir multigene family of Plasmodium: Antigenic variation and beyond. Mol. Biochem. Parasitol. 170 (2), 65–73. doi: 10.1016/j.molbiopara.2009.12.010
del Portillo H. A., Fernandez-Becerra C., Bowman S., Oliver K., Preuss M., Sanchez C. P., et al. (2001). A superfamily of variant genes encoded in the subtelomeric region of Plasmodium vivax. Nature 410 (6830), 839–842. doi: 10.1038/35071118
de Queiroz A., Gatesy J. (2007). The supermatrix approach to systematics. Trends Ecol. Evol. 22 (1), 34–41. doi: 10.1016/j.tree.2006.10.002
Emms D. M., Kelly S. (2015). OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16 (1), 157. doi: 10.1186/s13059-015-0721-2
Emms D. M., Kelly S. (2017). STRIDE: species tree root inference from gene duplication events. Mol. Biol. Evol. 34 (12), 3267–3278. doi: 10.1093/molbev/msx259
Emms D. M., Kelly S. (2018). STAG: species tree inference from all genes. bioRxiv. doi: 10.1101/267914
Escalante A. A., Cepeda A. S., Pacheco M. A. (2022). Why Plasmodium vivax and Plasmodium falciparum are so different? A tale of two clades and their species diversities. Malaria J. 21 (1), 139. doi: 10.1186/s12936-022-04130-9
Escalante A. A., Freeland D. E., Collins W. E., Lal A. A. (1998). The evolution of primate malaria parasites based on the gene encoding cytochrome b from the linear mitochondrial genome. Proc. Natl. Acad. Sci. 95 (14), 8124–8129. doi: 10.1073/pnas.95.14.8124
Figueiredo L. M., Freitas-Junior L. H., Bottius E., Olivo-Marin J.-C., Scherf A. (2002). A central role for Plasmodium falciparum subtelomeric regions in spatial positioning and telomere length regulation. EMBO J. 21 (4), 815–824. doi: 10.1093/emboj/21.4.815
Figueiredo L., Scherf A. (2005). Plasmodium telomeres and telomerase: the usual actors in an unusual scenario. Chromosome Res. 13 (5), 517–524. doi: 10.1007/s10577-005-0996-3
Frank M., Kirkman L., Costantini D., Sanyal S., Lavazec C., Templeton T. J., et al. (2008). Frequent recombination events generate diversity within the multi-copy variant antigen gene families of Plasmodium falciparum. Int. J. Parasitol. 38 (10), 1099–1109. doi: 10.1016/j.ijpara.2008.01.010
Freitas-Junior L. H., Bottius E., Pirrit L. A., Deitsch K. W., Scheidig C., Guinet F., et al. (2000). Frequent ectopic recombination of virulence factor genes in telomeric chromosome clusters of P. falciparum. Nature 407 (6807), 1018–1022. doi: 10.1038/35039531
Galen S. C., Borner J., Martinsen E. S., Schaer J., Austin C. C., West C. J., et al. (2018). The polyphyly of Plasmodium: comprehensive phylogenetic analyses of the malaria parasites (order Haemosporida) reveal widespread taxonomic conflict. R. Soc. Open Sci. 5 (5), 171780. doi: 10.1098/rsos.171780
Gardner M. J., Hall N., Fung E., White O., Berriman M., Hyman R. W., et al. (2002). Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419 (6906), pp.498–pp.511. doi: 10.1038/nature01097
Guindon S., Dufayard J.-F., Lefort V., Anisimova M., Hordijk W., Gascuel O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of phyML 3.0. System. Biol. 59 (3), 307–321. doi: 10.1093/sysbio/syq010
Harb O. S., Roos D. S. (2020). “ToxoDB: Functional Genomics Resource for Toxoplasma and Related Organisms,” in Toxoplasma gondii: Methods and Protocols, Methods in Molecular Biology. Ed. Tonkin C. J. (New York, NY: Springer US), 27–47. doi: 10.1007/978-1-4939-9857-9_2
Harrison T. E., Reid A. J., Cunningham D., Langhorne J., Higgins M. K. (2020). Structure of the Plasmodium-interspersed repeat proteins of the malaria parasite. Proc. Natl. Acad. Sci. 117 (50), 32098–32104. doi: 10.1073/pnas.2016775117
Hayakawa T., Culleton R., Otani H., Horii T., Tanabe K.. (2008). Big bang in the evolution of extant malaria parasites. Mol. Biol. Evol. 25 (10), 2233–2239. doi: 10.1093/molbev/msn171
Heiges M., Wang H., Robinson E., Aurrecoechea C., Gao X., Kaluskar N., et al. (2006). CryptoDB: a Cryptosporidium bioinformatics resource update. Nucleic Acids Res. 34 (suppl_1), D419–D422. doi: 10.1093/nar/gkj078
Hernández-Rivas R., Herrera-Solorio A. M., Sierra-Miranda M., Delgadillo D. M., Vargas M. (2013). Impact of chromosome ends on the biology and virulence of Plasmodium falciparum. Mol. Biochem. Parasitol. 187 (2), pp.121–pp.128. doi: 10.1016/j.molbiopara.2013.01.003
Hernandez-Rivas R., Hinterberg K., Scherf A. (1996). Compartmentalization of genes coding for immunodominant antigens to fragile chromosome ends leads to dispersed subtelomeric gene families and rapid gene evolution in Plasmodium falciparum. Mol. Biochem. Parasitol. 78 (1), 137–148. doi: 10.1016/S0166-6851(96)02618-7
Hoang D. T., Chernomor O., von Haeseler A., Minh B. Q., Vinh L. S. (2018). UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35 (2), 518–522. doi: 10.1093/molbev/msx281
Hoeijmakers W. A. M., Flueck C., Françoijs K.-J., Smits A. H., Wetzel J., Volz J. C., et al. (2012). Plasmodium falciparum centromeres display a unique epigenetic makeup and cluster prior to and during schizogony. Cell. Microbiol. 14 (9), 1391–1401. doi: 10.1111/j.1462-5822.2012.01803.x
Janssen C. S., Barrett M. P., Turner C. M. R., Phillips R. S. (2002). A large gene family for putative variant antigens shared by human and rodent malaria parasites. Proc. R. Soc. Lond. B 269(1489), 431–436. doi: 10.1098/rspb.2001.1903
Janssen C. S., Phillips R. S., Turner C. M. R., Barrett M. P. (2004). Plasmodium interspersed repeats: the major multigene superfamily of malaria parasites. Nucleic Acids Res. 32 (19), 5712–5720. doi: 10.1093/nar/gkh907
Kalyaanamoorthy S., Minh B. Q., Wong T. K. F., von Haeseler A., Jermiin L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14 (6), 587–589. doi: 10.1038/nmeth.4285
Katoh K., Kuma K., Toh H., Miyata T. (2005). MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33 (2), 511–518. doi: 10.1093/nar/gki198
Kemp D. J., Thompson J. K., Walliker D., Corcoran L. M. (1987). Molecular karyotype of Plasmodium falciparum: conserved linkage groups and expendable histidine-rich protein genes. Proc. Natl. Acad. Sci. 84 (21), 7672–7676. doi: 10.1073/pnas.84.21.7672
Kooij T. W. A., Carlton J. M., Bidwell S. L., Hall N., Ramesar J., Janse C. J., et al. (2005). A Plasmodium whole-genome synteny map: indels and synteny breakpoints as foci for species-specific genes. PloS Pathog. 1 (4), e44. doi: 10.1371/journal.ppat.0010044
Krief S., Escalante A. A., Pacheco M. A., Mugisha L., André C., Halbwax M., et al. (2010). On the diversity of malaria parasites in African apes and the origin of Plasmodium falciparum from bonobos. PloS Pathog. 6 (2), p.e1000765. doi: 10.1371/journal.ppat.1000765
Levene H. (1960). “Robust tests for equality of variances,” in Contributions to Probability and Statistics: Essays in Honor of Harold Hotelling. Eds. Olkin I., Hotelling H. (Palo Alto, California, USA: Stanford University Press), 278–292.
Loy D. E., Liu W., Li Y., Learn G. H., Plenderleith L. J., Sundararaman S. A., et al. (2017). Out of Africa: origins and evolution of the human malaria parasites Plasmodium falciparum and Plasmodium vivax. Int. J. Parasitol. 47 (2–3), 87–97. doi: 10.1016/j.ijpara.2016.05.008
Mann H. B., Whitney D. R. (1947). On a Test of Whether one of Two Random Variables is Stochastically Larger than the Other. Ann. Math. Stat 18 (1), 50–60. doi: 10.1214/aoms/1177730491
Martinsen E. S., Perkins S. L., Schall J. J. (2008). A three-genome phylogeny of malaria parasites (Plasmodium and closely related genera): Evolution of life-history traits and host switches. Mol. Phylogenet. Evol. 47 (1), 261–273. doi: 10.1016/j.ympev.2007.11.012
Marty A. J., Thompson J. K., Duffy M. F., Voss T. S., Cowman A. F., Crabb B. S. (2006). Evidence that Plasmodium falciparum chromosome end clusters are cross-linked by protein and are the sites of both virulence gene silencing and activation. Mol. Microbiol. 62 (1), pp.72–pp.83. doi: 10.1111/j.1365-2958.2006.05364.x
Morel B., Schade P., Lutteropp S., Williams T. A., Szöllősi G. J., Stamatakis A. (2022). SpeciesRax: A tool for maximum likelihood species tree inference from gene family trees under duplication, transfer, and loss. Mol. Biol. Evol. 39 (2), msab365. doi: 10.1093/molbev/msab365
Morgulis A., Coulouris G., Raytselis Y., Madden T. L., Agarwala R., Schäffer A. A. (2008). Database indexing for production MegaBLAST searches. Bioinformatics 24, 1757–1764. doi: 10.1093/bioinformatics/btn322
Nguyen L.-T., Schmidt H. A., von Haeseler A., Minh B. Q. (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32 (1), 268–274. doi: 10.1093/molbev/msu300
Otto T. D., Böhme U., Sanders M., Reid A., Bruske E. I., Duffy C. W., et al. (2018). Long read assemblies of geographically dispersed Plasmodium falciparum isolates reveal highly structured subtelomeres. Wellcome Open Res. 3, 52. doi: 10.12688/wellcomeopenres.14571.1
Pacheco M. A., Battistuzzi F. U., Junge R. E., Cornejo O. E., Williams C. V., Landau I., et al. (2011). Timing the origin of human malarias: the lemur puzzle. BMC Evolution. Biol. 11 (1), 299. doi: 10.1186/1471-2148-11-299
Pacheco M. A., Matta N. E., Valkiūnas G., Parker P. G., Mello B., Stanley C. E., et al. (2018). Mode and rate of evolution of haemosporidian mitochondrial genomes: timing the radiation of avian parasites. Mol. Biol. Evol. 35 (2), 383–403. doi: 10.1093/molbev/msx285
Pain A., Böhme U., Berry A. E., Mungall K., Finn R. D., Jackson A. P., et al. (2008). The genome of the simian and human malaria parasite Plasmodium knowlesi. Nature 455 (7214), 799–803. doi: 10.1038/nature07306
Perkins S. L. (2014). Malaria’s many mates: past, present, and future of the systematics of the order haemosporida. J. Parasitol. 100 (1), pp.11–pp.25. doi: 10.1645/13-362.1
Perkins S. L., Schall J. (2002). A molecular phylogeny of malarial parasites recovered from cytochrome b gene sequences. J. Parasitol. 88 (5), 972–978. doi: 10.1645/0022-3395(2002)088[0972:AMPOMP]2.0.CO;2
Pick C., Ebersberger I., Spielmann T., Bruchhaus I., Burmester T. (2011). Phylogenomic analyses of malaria parasites and evolution of their exported proteins. BMC Evolution. Biol. 11 (1), 167. doi: 10.1186/1471-2148-11-167
Rabiee M., Sayyari E., Mirarab S. (2019). Multi-allele species reconstruction using ASTRAL. Mol. Phylogenet. Evol. 130, 286–296. doi: 10.1016/j.ympev.2018.10.033
Ramirez J. L. (2020). An evolutionary view of Trypanosoma cruzi telomeres. Front. Cell. Infect. Microbiol. 9. doi: 10.3389/fcimb.2019.00439
R Core Team (2022). R: A language and environment for statistical computing (Vienna, Austria: R Foundation for Statistical Computing). Available at: https://www.R-project.org/.
Reed J., Kirkman L. A., Kafsack B. F., Mason C. E., Deitsch K. W. (2021). Telomere length dynamics in response to DNA damage in malaria parasites. iScience 24 (2). doi: 10.1016/j.isci.2021.102082
Rich S. M., Ayala F. J. (2000). Population structure and recent evolution of Plasmodium falciparum. Proc. Natl. Acad. Sci. 97 (13), 6994–7001. doi: 10.1073/pnas.97.13.6994
Rich S. M., Xu G. (2011). Resolving the phylogeny of malaria parasites. Proc. Natl. Acad. Sci. 108 (32), 12973–12974. doi: 10.1073/pnas.1110141108
Rutledge G. G., Böhme U., Sanders M., Reid A. J., Cotton J. A., Maiga-Ascofare O., et al. (2017). Plasmodium malariae and P. ovale genomes provide insights into malaria parasite evolution. Nature 542 (7639), 101–104. doi: 10.1038/nature21038
Schaer J., Perkins S. L., Decher J., Leendertz F. H., Fahr J., Weber N., et al. (2013). High diversity of West African bat malaria parasites and a tight link with rodent Plasmodium taxa. Proc. Natl. Acad. Sci. 110 (43), 17415–17419. doi: 10.1073/pnas.1311016110
Scherf A., Figueiredo L. M., Freitas-Junior L. H. (2001). Plasmodium telomeres: a pathogen’s perspective. Curr. Opin. Microbiol. 4 (4), 409–414. doi: 10.1016/S1369-5274(00)00227-7
Shapiro S. S., Wilk M. B. (1965). An analysis of variance test for normality (complete samples). Biometrika 52 (3–4), 591–611. doi: 10.1093/biomet/52.3-4.591
Sharp P. M., Plenderleith L. J., Hahn B. H. (2020). Ape origins of human malaria. Annu. Rev. Microbiol. 74 (1), 39–63. doi: 10.1146/annurev-micro-020518-115628
Silva Pereira S., de Almeida Castilho Neto K. J. G., Duffy C. W., Richards P., Noyes H., Ogugo M., et al. (2020). Variant antigen diversity in Trypanosoma vivax is not driven by recombination. Nat. Commun. 11 (1), 844. doi: 10.1038/s41467-020-14575-8
Stecher G., Tamura K., Kumar S. (2020). Molecular evolutionary genetics analysis (MEGA) for macOS. Mol. Biol. Evol. 37 (4), 1237–1239. doi: 10.1093/molbev/msz312
Su X., Heatwole V. M., Wertheimer S. P., Guinet F., Herrfeldt J. A., Peterson D. S., et al. (1995). The large diverse gene family var encodes proteins involved in cytoadherence and antigenic variation of Plasmodium falciparum-infected erythrocytes. Cell 82 (1), 89–100. doi: 10.1016/0092-8674(95)90055-1
Suyama M., Torrents D., Bork P. (2006). PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34 (suppl_2), W609–W612. doi: 10.1093/nar/gkl315
Swofford D. (2002) PAUP*: Phylogenetic Analysis Using Parsimony (*and Other Methods). Available at: https://paup.phylosolutions.com/.
Welch B. L. (1947). The generalization of ‘Student’s’ problem when several different population variances are involved. Biometrika 34 (1–2), 28–35. doi: 10.1093/biomet/34.1-2.28
WHO (2014) World malaria report 2014 (World Health Organization). Available at: https://www.who.int/publications/i/item/9789241564830 (Accessed 10 February 2023).
Willson J., Roddur M. S., Liu B., Zaharias P., Warnow T (2022). DISCO: species tree inference using multicopy gene family tree decomposition. System. Biol. 71 (3), 610–629. doi: 10.1093/sysbio/syab070
Zhang Z., Schwartz S., Wagner L., Miller W. (2000). A greedy algorithm for aligning DNA sequences. J. Comput. Biol.: A J. Comput. Mol. Cell Biol. 7 (1–2), 203–214. doi: 10.1089/10665270050081478
Zhang X., Alexander N., Leonardi I., Mason C., Kirkman L. A., Deitsch K. W. (2019). Rapid antigen diversification through mitotic recombination in the human malaria parasite Plasmodium falciparum. PloS Biol. 17 (5), e3000271. doi: 10.1371/journal.pbio.3000271
Zhang C., Scornavacca C., Molloy E. K., Mirarab S. (2020). ASTRAL-pro: quartet-based species-tree inference despite paralogy. Mol. Biol. Evol. 37 (11), 3292–3307. doi: 10.1093/molbev/msaa139
Keywords: antigenic genes, chromosome maps, gene conservation profiles, subtelomeres, Plasmodium, species tree, ectopic recombination, antigenic diversity
Citation: Martínez-Eraso C, Cárdenas H and Cerón-Romero MA (2024) Phylogenomics and chromosome mapping show that ectopic recombination of subtelomeres is critical for antigenic diversity and has a complex evolutionary history in Plasmodium parasites. Front. Ecol. Evol. 11:1177350. doi: 10.3389/fevo.2023.1177350
Received: 01 March 2023; Accepted: 18 December 2023;
Published: 25 January 2024.
Edited by:
Monica Medina, The Pennsylvania State University (PSU), United StatesReviewed by:
Kristan Alexander Schneider, Hochschule Mittweida, GermanyRaúl A. González-Pech, The Pennsylvania State University (PSU), United States
Copyright © 2024 Martínez-Eraso, Cárdenas and Cerón-Romero. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mario A. Cerón-Romero, bWFyaW8uY2Vyb25yb21lcm9AY2FzZS5lZHU=