 Swellan Luciann Pinto1,2*
Swellan Luciann Pinto1,2* Megan Carvalho Henriquez1,2,3,4Saul Cheves Hernandez5Gwen Duytschaever1
Megan Carvalho Henriquez1,2,3,4Saul Cheves Hernandez5Gwen Duytschaever1 Janneke Wit2,6Russell William Avramenko2,6
Janneke Wit2,6Russell William Avramenko2,6 John Stuart Gilleard2,6Joseph Daniel Orkin7,8
John Stuart Gilleard2,6Joseph Daniel Orkin7,8 Amanda Dawn Melin1,2,9,10*
Amanda Dawn Melin1,2,9,10*- 1Department of Anthropology and Archaeology, University of Calgary Faculty of Arts, Calgary, AB, Canada
- 2Host Parasite Interactions Network, University of Calgary, Calgary, AB, Canada
- 3Department of Anthropology, The Graduate Center, City University of New York, New York, NY, United States
- 4The New York Consortium in Evolutionary Primatology (NYCEP), The Graduate Center, CUNY, New York, NY, United States
- 5Sector Santa Rosa Monkey Project, Wildlife Research, Área de Conservación, Guanacaste, Costa Rica
- 6Department of Comparative Biology and Experimental Medicine, University of Calgary Faculty of Veterinary Medicine, Calgary, AB, Canada
- 7Department of Medicine and Life Sciences (MELIS), Institut de Biologia Evolutiva, Universitat Pompeu Fabra-Consejo Superior de Investigaciones Científicas (CSIC), Barcelona, Spain
- 8Département d’anthropologie, Université de Montréal, Montréal, QC, Canada
- 9Alberta Children’s Hospital Research Institute, University of Calgary, Calgary, AB, Canada
- 10Department of Medical Genetics, University of Calgary, Calgary, AB, Canada
Genomic screening of fecal DNA provides insight into diet, parasite infection dynamics, and other aspects of the ecology and pathogens of wild populations. Here, we amplify and sequence the V4/V5 regions of the eukaryotic 18S ribosomal RNA gene from fecal DNA of wild capuchin monkeys (Cebus imitator). We collected 94 fecal samples from 26 individuals, each sampled 1-4 times across a 19-month period and examined the eukaryotic diversity in 63 of these samples which had sufficient numbers and quality of reads during downstream analyses. We found a total of 234 distinct amplicon sequence variants (ASVs) classified as Eukaryotes in our samples. Of these, 66 were assigned to the phylum Nematoda. 64 ASVs are from taxa that possibly parasitize monkeys or their food items: 33 were assigned to lungworms (Superfamily Metastrongyloidae; genus Angiostrongylus), two to the genus Strongyloides, and one to the genus Austrostrongylus. The remaining 28 ASVs were assigned to nematodes that likely parasitize plants and/or invertebrates that the monkeys consume. Taken together with past dietary and coprological study of the same primate population, our results suggest that invertebrate consumption and parasitic infection, especially by lungworms, is common and widespread among this population of wild monkeys. We also discuss limitations of our approach, including the amplification of off-target ASVs, and make suggestions for future research. Overall, 18S screening shows promise for identifying various components of the capuchin gastrointestinal eukaryotic ecosystem, including parasitic helminths, and its utility will increase with the improvement of genetic databases.
1 Introduction
Genomic screening of fecal DNA provides a non-invasive method for studying the dietary and disease ecology of wild animals (Bradley et al., 2007; Henry, 2012; Srivathsan et al., 2016). Molecular approaches, such as amplifying diagnostic genetic markers or metagenomic screening, can reveal taxonomic relationships that complement and enhance data obtained from traditional, coproscopic methods used in parasitology (Ross et al., 2010; Callejón et al., 2013; Perera et al., 2013; Tanaka et al., 2014; Srivathsan et al., 2016; Mann et al., 2020; Sharma et al., 2022). Genetic markers used to identify eukaryotic contents from vertebrate feces include hypervariable regions within mitochondrial genes, such as the cytochrome oxidase I (COI), as well as regions within ribosomal RNA (rRNA) genes such as the internal transcribed spacer 2 (ITS-2), and 18S (Aivelo and Medlar, 2018; Sharma et al., 2022). To achieve finer levels of taxonomic resolution in parasites, or to study the genetic population structure of those parasites, COI and ITS-2 are effective genetic markers to target (Criscione et al., 2007; Criscione et al., 2010; Blasco-Costa et al., 2016; Pafčo et al., 2018; Papaiakovou et al., 2022); however, documenting higher level (e.g. order, family, genus) taxonomy using the 18S rRNA gene, which is largely conserved across eukaryotes, is useful for broad scale surveys, and for first-pass identification of taxa of particular interest or importance (Blasco-Costa et al., 2016; Avramenko et al., 2017; Aivelo and Medlar, 2018; Stensvold, 2019; Mann et al., 2020).
To better understand the ecology and parasites of wild capuchin monkeys from a genetic perspective, and to contribute to growing eukaryotic genetic databases, we amplified and sequenced the V4/V5 regions of the 18S ribosomal RNA gene derived from the fecal DNA of a population of wild, Costa Rican capuchins (Cebus imitator; Rowe and Myers, 2016). White-faced capuchins are well suited for parasitological study because their omnivorous diet and behavior expose them to a wide variety of parasites. This population is of particular interest as the monkeys are individually identifiable and the habitat is highly seasonal (Melin et al., 2008; Melin et al., 2014). In particular, capuchins drink from small pools of groundwater in the dry season, which are shared by group members and other species (ex. tapirs, deer, peccaries) of the tropical forest community. The potential for acquiring parasitic infections in these situations is high because some gastrointestinal parasites, such as Strongyloides sp., deposit infective stages in the surrounding soil and sediment (Viney, 2017). Aggregation around sparse and stagnant water sources also facilitates the horizontal transfer of parasites between hosts through an accumulation of fecal matter (Titcomb et al., 2021). However, during the wet season, when both water and food resources are abundant, hosts can spread out and access fresh resources, potentially reducing the chances of parasite transmission between hosts (Griffin and Nunn, 2012). Our aim is to assess the broad-level identity, richness, and genetic variability of eukaryotes in our study population across distinct ecological seasons, with a focus on nematode parasites, complementing previous studies which examined seasonality in parasite composition in capuchins at this field site.
2 Materials and methods
2.1 Study site, species, and sample collection
This study was conducted using fecal samples collected from wild white-faced capuchin monkeys living in Sector Santa Rosa, Área de Conservación Guanacaste, in northwestern Costa Rica (10.82049 lat.; 85.62813 long.). This tropical dry forest biome experiences distinct dry (November – mid-May) and wet (mid-June – October) seasons (Figure 1), which impacts the resources, diet, and behavior of the resident primates (Campos and Fedigan, 2009; Melin et al., 2014; Orkin et al., 2018). We collected 94 fecal samples from 26 wild, habituated monkeys during both the wet and dry seasons during a 19-month period between January 2014 and July 2015. Freshly voided fecal samples were collected and stored in 15ml sterile tubes containing 8 ml of 96% ethanol. Sampled monkeys differed in age and sex and were distributed across five social groups: GN (N=8) LV (N=5), AD (N=5), RM (N=5), and EX (N=3).

Figure 1 (A) Wild white-faced capuchin monkeys inhabit the highly seasonal forests of Guanacaste Province in northwestern Costa Rica. (B, C) In the dry season, the forest is largely defoliated and monkeys drink from stan ding pools of water. (D) In the wet season, monkeys lick water from leaves and drink from holes and crevices of densely foliated trees. Photo credits: AD Melin.
Monkey feces were collected in Costa Rica under permission from CONEGABIO, approval no. R-025-2014-OT-CONEGABIO and exported under Área de Conservación Guanacaste permit no. DSVS-029-2014-ACG-PI-060-2014. Feces were imported to the University of Calgary with permission from the Public Health Agency of Canada (PHAC), permit no. P-15-6481. Our research protocols were approved by the Animal Care Committee of the University of Calgary, approval no. AC15-0161.
2.2 18S amplification and sequencing
We extracted fecal DNA using the NucleoSpin Tissue kit (Macherey-Nagel) following manufacturer recommendations and stored the DNA extracts at -80°C. To amplify the 18S region of eukaryotic rRNA, we used previously published primers: 563F (5’-GCCAGCAVCYGCGGTAAY-3’) and 1132R (5’-CCGTCAATTHCTTYAART-3’), which have been shown in in-silico experiments to amplify and discriminate between a broad range of eukaryotic rRNA, with paired-end reads as short as 150 bp, while minimizing prokaryotic amplification (Hadziavdic et al., 2014; Hugerth et al., 2014). While we did not include a mammal-blocking primer during our PCR steps, we would suggest future studies consider using one, as host DNA amplification occurred in this study (Mann et al., 2020). After successfully identifying and sequencing amplicons of the expected size and taxa in our primer validation (Supplementary Tables 1, 2), we proceeded with PCR on 94 fecal DNA extracts. 80 out of the 94 samples produced amplicons around the expected size of ~550 bp and were carried forward for further analyses.
We constructed 18S libraries following the two-step protocol used by Avramenko et al. (2015). In brief, we amplified the targeted region using a mixture that contained the primer sequences above, attached to adaptors with a variable number of Ns in between the primer and adaptor sequence. In the initial PCR (PCR1) the variable Ns introduced heterogeneity into the mixture and the adaptors were used to attach unique barcodes in preparation for PCR2, which was carried out to attach sample-specific barcodes (Supplementary Table 3). We purified PCR products after each PCR following the AMPure XP protocol, using a 1X homemade solid phase reversible immobilization (SPRI) mixture with carboxyl coated magnetic beads (Sera Mag Magnetic Speed-beads; Life Technologies) (DeAngelis et al., 1995; Rohland and Reich, 2012). Following a second purification, we measured our DNA libraries’ concentration using a Qubit dsDNA HS assay (Life Technologies) and a D1000 TapeStation assay (Agilent). We then added 40 ng of DNA from each sample to a pooled library that was sequenced on an Illumina Miseq using a 2x250 paired end v2 kit (Miseq Reagent Kits v2, MS-103-2003) with 20% phiX spike in.
2.3 Identification of eukaryotic taxa
We checked the quality of the short reads with fastqc v0.11.9c (Andrews, 2010). Illumina Nextera adaptors were trimmed, and primer sequences were removed using Cutadapt v4.1 (Martin, 2011). We identified amplicon sequence variants (ASVs) with DADA2 v1.14.1(Callahan et al., 2016) and made several modifications to the standard bioinformatic pipeline to accommodate non-overlapping reads. Specifically, we used the justConcatenate=TRUE option in the mergePairs step, following Hu et al. (2016), which concatenates each pair of forward and reverse-complemented reads with a 10 bp spacer sequence (all Ns) introduced between them. Subsequent alignment and taxonomic identification steps ignore the spacer and locally align the two regions with the introduction of gaps where necessary. To minimize the introduction of false diversity from primer sequences that were not completely removed, we trimmed the forward reads to a length of 227 bp and the reverse reads to 125 bp, with a maximum expected error of two. Subsequently, we merged paired end reads with the 10 bp spacer between forward and reverse reads, removed chimeras, and identified unique ASVs. Combined sequence reads were 362 bases long. We assigned taxonomy to the read pairs using the Protist Ribosomal Reference database (PR2) for 18S (version 4.14.0) (Guillou et al., 2013; Hu et al., 2016).
We constructed a eukaryotic phylogenetic neighbor joining tree that was optimized using a GTR model in phangorn 2.10.0 (Schliep, 2011). Downstream analyses were conducted in R using phyloseq 1.42.0 (Callahan et al., 2016). In total, we generated 1,651,332 reads from which we identified 1,809 ASVs, 234 of which remained after removing ASVs assigned to bacteria (1,567,428 raw reads), mitochondria (3,647 raw reads), plasmids (223 raw reads), unidentifiable kingdoms (86 raw reads), Mammalia (6,579 raw reads), and samples with fewer than 2000 raw reads. Of the 80 samples we sequenced, 70 samples generated sufficient numbers and quality of reads. 63 of the 70 samples, representing 25 individuals, had adequate amounts of metadata available to be included in downstream analyses (Supplementary Table 4).
Eukaryotic alpha diversity was characterized with the Chao1 index for each sample collected in the more intensively sampled year, 2014, which included 56 samples from 25 individuals. For individuals that were sampled multiple times within a single 2014 season (wet or dry), we calculated the mean Chao1 value across replicate samples. The limitations of our dataset (zero-inflated non-normal distribution, small sample sizes, and non-uniform sampling across seasons) hindered our ability to meet the statistical requirements for linear mixed effects models, so we tested seasonal alpha diversity for significance using non-parametric Wilcoxon tests.
To measure the relative abundance of the different taxa and construct TCS parsimony networks, we rarefied our remaining dataset of 56 samples to only include samples with exactly 500 reads to control for stochasticity in sequencing depth. After rarefaction, 24 samples from 16 individuals remained. To identify 18S sequences originating from potentially parasitic nematodes, we manually identified ASVs that were assigned to nematode genera likely to infect mammals based on literature review. To explore the genetic diversity of putatively parasitic nematodes within and between species, we used the rarified dataset of 24 samples to build TCS parsimony haplotype networks using R (RStudio Version 1.2.5001) and the packages ape v5.6-2 (Paradis and Schliep, 2019), pegas v1.2 (Paradis, 2010), adegenet v2.1.10 (Jombart, 2008), and Bios2cor v2.2.1 (Taddese et al., 2021). We set TCS node size to correspond to the prevalence of that ASV across all 24 samples and we represented ASV prevalence in samples by season across both sampling years within each ASV node.
3 Results
3.1 Prevalence, richness, and taxonomy of eukaryotes
Of the 234 eukaryotic ASVs that passed quality control, we found ASVs from 18 classes, 22 orders, and 25 families (Supplementary Table 5). ASVs assigned to the same genus and/or species differed, on average, by 10.9%, or 33 bases (range 1 – 123 bp; 0.3 – 41.5% dissimilarity). We group all putative parasitic worms, including an ASV mapping to an acanthocephalan, into the paraphyletic group “helminths’’ (Supplementary Table 6). In addition, we also found a large number (1511) of ASVs that were assigned to Bacteria, which likely reflects a lack of specificity in our primer design, and which we did not analyze further. Within the rarefied dataset, eukaryotic relative abundance was dominated by members of the Ascomycota fungi, including Penicillium sp., yeasts, molds, and Candida sp. (Maharachchikumbura et al., 2021; Figure 2A). Lesser contributions from members of the Apicomplexa, such as Adelina sp. may represent coccidian parasites of insects (Lange and Wittenstein, 2001) while ASVs mapping to Metamonada largely fall in the genus Hypotrichomonas, and are known to infect a variety of hosts, from insects to mammals (Céza et al., 2015; Supplementary Table 5). Within Metazoa we found ASVs assigned to taxa that capuchins likely consume such as Arthropoda (e.g., insects and arachnids) and Archaeplastida (e.g., Embryophyceae and Chlorellales; Figure 2A). Furthermore, we found the presence of putatively parasitic roundworms (class: Nematoda) in 54 out of 70 samples.
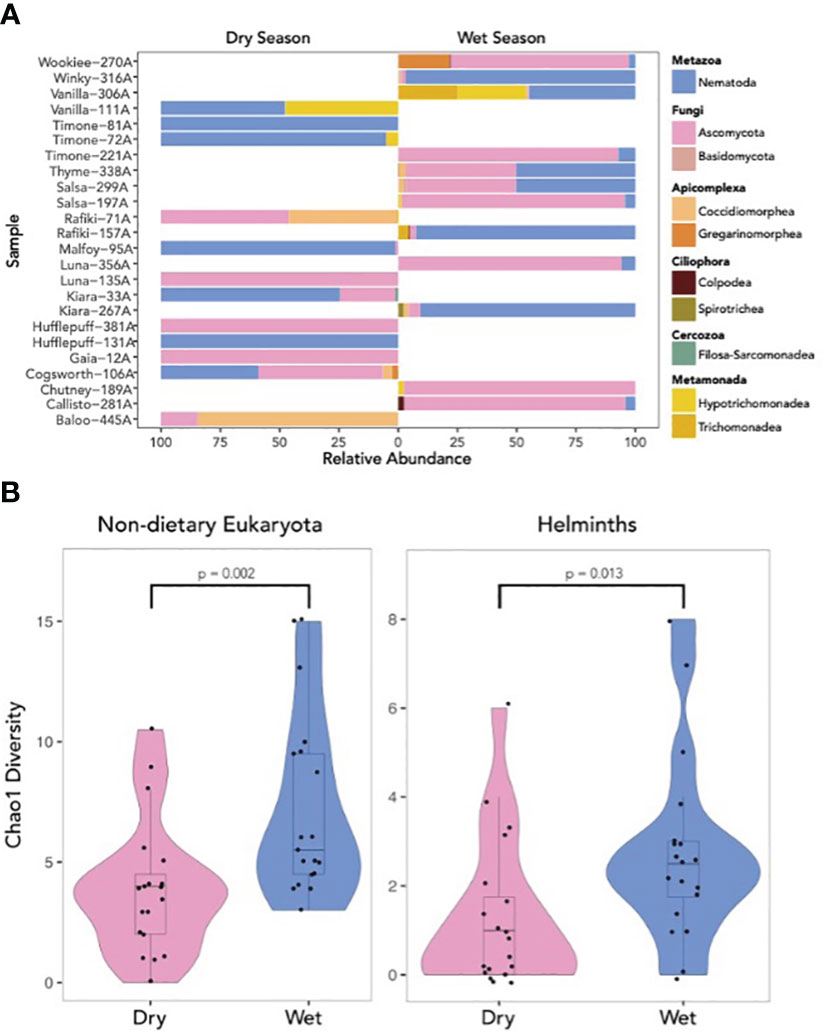
Figure 2 (A) Relative abundance of eukaryotic classes from filtered 18S samples rarefied to 500 reads. Most samples are dominated by reads from members of the Nematoda and Ascomycota. Samples are split between those collected in the wet and dry seasons. While all monkeys were initially sampled in both wet and dry seasons, not all samples survived filtration and rarefaction steps, limiting our ability to exhaustively include samples from the same monkeys in both seasons. (B) Violin plots of the Chao 1 diversity of 56 fecal samples collected from 25 wild capuchin monkeys (Cebus imitator) in the dry season and wet season of 2014 in the tropical dry forest of Área de Conservacion Guanacaste, Costa Rica. Taxa classified as helminths are plotted on the right and non-dietary eukaryotes on the left. Alpha diversity is significantly higher, with low effect sizes, in the wet season samples.
We examined the richness of each ASV across our population in 2014. Chao1 alpha diversity was significantly higher in the wet than dry season in both the non-dietary eukaryotic (W = 72.5, p=0.002, 95% CI = (-5.000, -1.000)) and helminth-only (W= 96, p = 0.013, 95% CI = (-2.500 - -0.500)) datasets (Figure 2B).
3.2 Diversity of abundant parasitic helminths
We found 64 ASVs that were assigned to five species of parasitic worms present in existing databases (Figure 3). 33 ASVs - including the four most prevalent - were assigned to Angiostrongylus vasorum, a clade V lungworm species. Two ASVs were assigned to Strongyloides cebus, a clade IV nematode species. Single ASVs were assigned to Austrostrongylus victoriensis, and Onicola sp. We also identified ASVs originating from taxa that are likely to have been incidentally ingested by capuchins via food items (fruit and insects), including ASVs that were assigned to Myctolaimus sp. (a parasite of beetles), Parasitodiplogaster sp., Schistonchus aureus (both parasites of fig wasps) (infraorder Tylenchomorpha), and the Schistonchus genus more generally (Giblin-Davis et al., 2006).
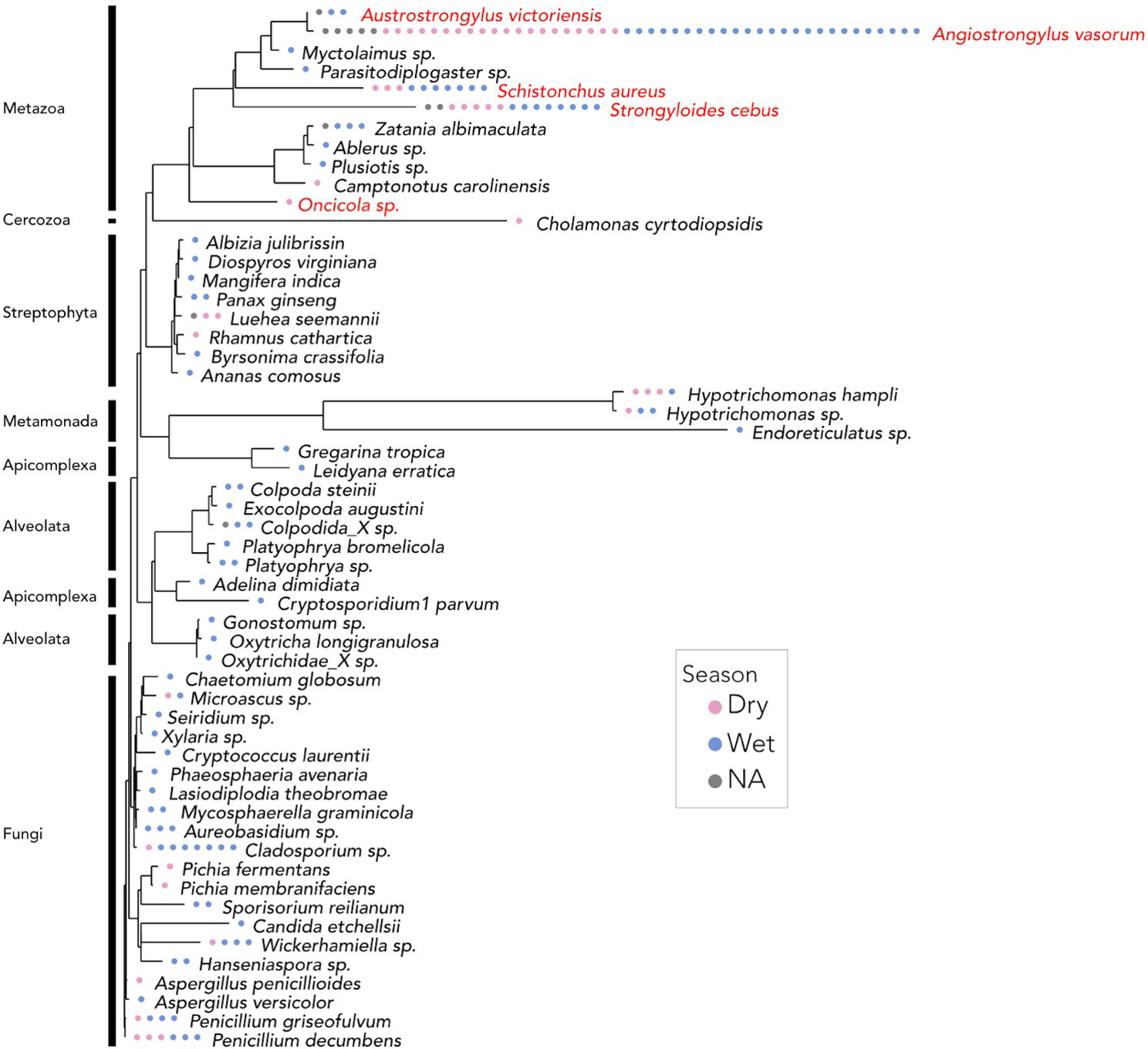
Figure 3 18S amplicon maximum likelihood tree of eukaryotic species identified in Cebus imitator samples. ASVs have been agglomerated to the species level, and ASVs that could not be confidently assigned to the level of genus or below are not included. Each point indicates the presence of a given species in a sample at least once and is color coded by sampling season. Putatively parasitic or commensal worms are labeled in red text. The widespread presence of nematode taxa likely results from our primer design. Plants and arthropods are presumed to be of dietary origin.
To explore general seasonal patterns in the genetic diversity of ASVs which mapped to putative parasitic taxa, which we grouped into “helminths”, we generated parsimony networks (TCS) using the rarified dataset of 24 samples (Figure 4). ASVs assigned to six helminth taxa were included in our TCS networks: Angiostrongylus vasorum, Austrostrongylus victoriensis, Parasitodiplogaster sp., Schistonchus aureus, Schistonchus sp., and Strongyloides cebus. The 19 ASVs mapping to Angiostrongylus vasorum and the 13 ASVs mapping to Schistonchus aureus were the most diverse and each formed their own cluster. ASVs mapping to Angiostrongylus vasorum and Schistonchus aureus were found in samples collected during both the wet and dry seasons. ASV 020, which mapped to Strongyloides cebus, was also detected in samples collected during both seasons, while ASV 067, which mapped to Austrostrongylus victoriensis, and ASV 082, which mapped to Parasitodiplogaster sp. were only identified in wet season samples.
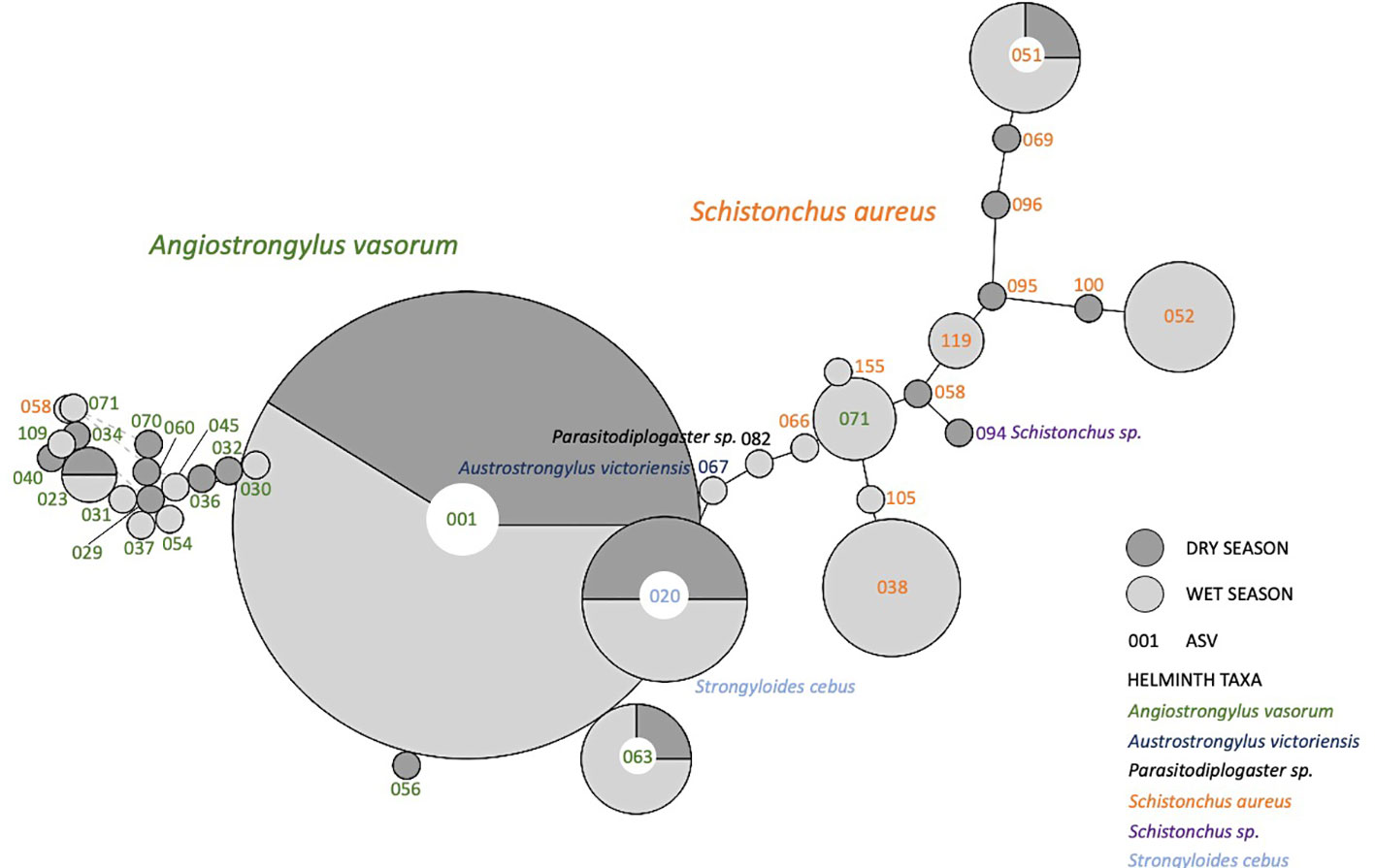
Figure 4 Parsimony haplotype network of capuchin parasite 18S rRNA gene ASVs from 24 filtered 18S samples rarefied to 500 reads. ASV numbers are located either within or next to haplotype nodes. The colors of the ASV numbers correspond to the helminth taxa that those ASVs mapped to. Node size corresponds to haplotype frequency (range 1 - 17), with smaller nodes representing rarer haplotypes and larger nodes representing more abundant haplotypes. Shading of the pie charts within ASV nodes corresponds to the number of samples in which that ASV was found in either the wet (light gray) or the dry (dark gray) season. Light gray linkages (represented with dashed lines) between ASV nodes represent ‘alternative paths’, or reticulations considering unsampled haplotypes, which may affect patterns of connectedness between the current ASVs.
4 Discussion
We used 18S metabarcoding to study eukaryotic diversity and prevalence of different taxa in a population of wild capuchin monkeys inhabiting a seasonal dry tropical forest. We identified 234 ASVs assigned to the Domain Eukaryota, including 66 ASVs assigned to nematodes, and 1 ASV assigned to an acanthocephalan parasite.
4.1 Prevalence, richness, and taxonomy of eukaryotes
Despite the limitations of 18S deep amplicon sequencing, we identified a wide range of eukaryotes in our samples that appear biologically meaningful to the ecology of capuchin monkeys and contribute to our understanding of communities present in the tropical dry forest. Many of the sequences were assigned to invertebrates that are capuchin prey items, such as arachnids (2 ASVs) and insects (18 ASVs) (Melin et al., 2008; Melin et al., 2010; Supplementary Table 6). Typically, 50%, and up to 80% of the capuchin diet consists of invertebrate prey (Melin et al., 2010; Mosdossy et al., 2015; Mallott et al., 2018) and capuchins eat an impressive variety of species (Fragaszy et al., 2004; Bergstrom et al., 2019). As the 18S databases improve, tracking the fecal DNA of invertebrate predators may be a promising way to study invertebrate biodiversity, natural food webs, and ecosystem natural history (Alberdi et al., 2018; Srivathsan et al., 2019; Bueno de Mesquita et al., 2021).
4.2 Diversity of abundant parasitic helminths
Given the potential fitness impacts and broader effects on ecosystem dynamics and conservation (Marcogliese, 2005), understanding the dynamics of helminth infection in wild animals is an enduring goal (Eley et al., 1989; Obanda et al., 2019). The rarified dataset revealed ASVs which mapped to 6 parasitic helminths that either infect capuchins or their prey items (Figure 4). The most prevalent parasitic ASVs mapped to the lungworm Angiostrongylus vasorum and were found in 17 of the 24 samples collected in both the wet and dry seasons. One ASV (ASV 020), mapping to the capuchin threadworm Strongyloides cebus, was found in 6 of the 24 samples and was also collected in both seasons from multiple individuals. These taxonomic assignments are the closest genetic matches to our ASVs, though several of our ASVs may represent one or more parasite species not represented in the PR2 database and so, our results should be treated conservatively.
When we compare our results to a coprological study of the same population, we gain additional context for our findings. Parr et al. (2013a) found lungworms, Filariopsis barretoi, to be very common in the Santa Rosa population. The genus Filariopsis is not represented in the PR2 database but belongs to the same superfamily (Metastrongyloidea; Rego and Schaeffer, 1988) as Angiostrongylus. Given the high prevalence of this lungworm in this population, one or more of the 34 ASVs assigned to Angiostrongylus vasorum (a canid lungworm) in this study may belong to Filariopsis barretoi. Other lungworms in this genus such as, Angiostrongylus cantonesi and A. dujardini, infect platyrrhine monkeys, i.e., tamarins and marmosets (Solórzano-García and de León, 2018). However, despite the PR2 database carrying sequences for A. cantonesi and A. dujardini, our ASVs were still assigned to lungworms that infect canids, suggesting that the lungworm from our samples may be genetically distinct from those that have been reported to infect other primates.
The second most prevalent nematode found by Parr et al. (2013a) was in the genus Strongyloides. We found two ASVs in total that were assigned to Strongyloides cebus, one from our rarified samples (ASV 020) and one ASV (ASV 219) from our larger sample set (Supplementary Table 6). Strongyloides cebus was first described in capuchin monkeys and is also reported to parasitize other platyrrhine monkeys (Mati et al., 2013; Solórzano-García and de León, 2018). In our rarified samples, Strongyloides cebus ASVs were represented equally in samples collected in both the wet and dry seasons. These findings are consistent with previous coprological studies of this population (Parr et al., 2013b), which also identified Strongyloides eggs and larvae in samples collected across seasons. However, Parr et al. ‘s (2013b) found a higher prevalence of Strongyloides during the dry season, suggesting that Strongyloides infections in this capuchin population may have a seasonal pattern. This study’s small sample size (N=24) may obscure any seasonal patterns of infection for this parasite, but future studies would benefit from exploring seasonal patterns of parasitic infection in this population using larger genetic datasets.
Finally, several of the putative helminth ASVs we discovered are not likely to be capuchin monkey parasites and rather were assigned to species that parasitize plants and/or invertebrates consumed by the capuchins. ASVs assigned to the genera Schistonchus, Parasitodiplogaster, and Myctolaimus are known to be parasites of the fig plants, fig wasps, and longhorn beetles of Central and South America, respectively (Decrappeo and Giblin-Davis, 2001; Giblin-Davis et al., 2006; Kanzaki and Giblin-Davis, 2014). The presence of these taxa are feasible dietary byproducts, as capuchin monkeys in SSR heavily consume figs from several species of Ficus, including the wasps living inside the fig as well as numerous invertebrates (Parr et al., 2011; Valenta and Melin, 2012).
4.3 Seasonality and 18S diversity
While our sample sizes in this study are preclusive of detailed seasonal analyses, we find evidence of greater Chao 1 diversity in both eukaryotes generally, and helminths more specifically, in the wet season than in the dry season. At our field site, a greater diversity of Eukaryotes, including parasites, in the wet season would be plausible, given the flush of invertebrates, fruits, water sources and general biodiversity that often accompany the start of the rains in seasonal tropical forests (Frankie et al., 2004; Orkin et al., 2019; Melin et al., 2020). However, this is somewhat inconsistent with a previous coprological study of parasites in the same population (Parr et al., 2013b), which found 7 helminth morphotypes in both the wet and dry seasons. The inconsistencies in the results of parasitological studies at the Santa Rosa field site are not unique as the primate parasitology literature is generally mixed on whether environmental conditions and seasonality strongly influence parasitic infections (e.g., Akinyi et al., 2019; de Winter et al., 2020; Blersch et al., 2021; Bueno de Mesquita et al., 2021; Bethge et al., 2022). However, conclusions are likely confounded by differences in study design, sampling, analyses, host-parasite relationships, and site-specific factors. While we refrain from making strong conclusions regarding seasonality in the present study, our results suggest the potential for fruitful discovery in future in-depth study of parasite dynamics in the seasonal dry forest.
4.4 Comments on the promise and limitations of 18S screening
The 18S region of Eukaryotic DNA has elements that are well conserved across eukaryotes, and we demonstrate the promise of 18S fecal screening to detect food items, monkey parasites, the parasites of their prey and other eukaryotes. However, as reviewed in Stensvold (2019), many factors, from primer selection to reference database selection, can affect the accuracy and efficacy of these amplicon sequencing techniques. Additionally, compared to other diagnostic genetic markers, such as ITS-2 and COI, 18S amplicon sequences often do not exhibit enough diversity to make confident assignments below the genus level (Kounosu et al., 2019). Furthermore, the classifications we present here are based on taxonomic assignment that uses publicly available reference databases. Due to the current incompleteness of these databases many ASVs were assigned to the same few reference species, and we cannot at present fully determine to what extent the ASVs are meaningfully different or if they represent more than one species or genus (Stensvold, 2019; Papaiakovou et al., 2022). As future studies continue to add to these databases, they will become increasingly powerful tools in genetic parasitology.
We also recovered a large number (1511) of ASVs that were assigned to Bacteria, suggesting that, despite selecting primers reported to minimize non-specific amplification, our primer choice was not sufficiently selective to exclude all bacteria. Indeed, recent in-silico studies have suggested that while the 563F/1132R primer pair targets a diagnostically variable gene region in a broad range of helminths, it may also target bacterial 16S genetic sequences (Kounosu et al., 2019). Other 18S gene region primer pairs, such as 616*F/1132R and 1183F/1631R, have yielded promising results in recent experiments, and may be more suitable for future studies, especially with the addition of PCR blocking primers to minimize off-target amplification (Kounosu et al., 2019; Mann et al., 2020).
Additionally, the amplicon-based method used in this study is not well suited to examine infection intensity due to genetic heterogeneity between and within individual worms, as well as biases in PCR amplification and sequencing (Avramenko et al., 2015; Viney, 2017; Pafčo et al., 2018). While parasite egg counts also have limitations when used to infer infection intensity, such as discordance between fecal egg counts and adult worm burdens in hosts and temporal stochasticity in egg shedding among different parasites, they offer a more biologically valid approach (Roberts, 2000; Romeo et al., 2014; Byrne et al., 2018). Ideally, researchers will work to validate approaches that integrate molecular and morphological methods (e.g., Avramenko et al., 2015) and consistently refine our abilities to estimate true infection intensity parameters.
Sequencing of fecal DNA has the potential to augment information gained by coprological analyses of parasites. Many parasites are morphologically indistinguishable in their egg and larval state, making misidentifications a likely common occurrence and making it difficult to identify parasites to the genus or species level (Frias et al., 2018; Solórzano-García and de León, 2018). For example, in coprological studies conducted in this host population, researchers described a single “Strongyloides morphotype”, based on egg morphology (Parr et al., 2013a), while our study was able to assign two ASVs to the Strongyloides cebus and detect genetic variability within those ASVs as well. The numerous ASVs found in our samples which were assigned to specific parasitic genera suggest that the genetic diversity of these parasites is higher than previously thought. These findings will allow future studies to investigate patterns of parasite genetic diversity in host populations, possibly resulting in new information on parasite transmission between hosts, novel infections acquired from the environment, temporal patterns of parasitic infections in host populations, and more.
As natural habitats continue to be altered by human development and climate change, changes in ecological community structure and the relationships between parasites and their hosts may be affected (Brooks and Hoberg, 2007). Developing simple, multifaceted approaches for monitoring parasite diversity in host populations is important for monitoring host health and detecting patterns of parasitic infections throughout host populations. 18S has proven its utility as a preliminary screening technique and as a valuable genetic marker used to detect a wide breadth of Eukaryotes in fecal samples. Future studies may build on the ASV data generated by this study to further our understanding the Eukaryotic diversity within host species, and within various ecosystems more generally.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found in GenBank, accession nos. SAMN28885587-SAMN28885663. The code used for the analyses described in this paper can be found in GitHub, https://github.com/swellanpinto/Capuchin_Parasites.
Ethics statement
The animal study was approved by Animal Care Committee of the University of Calgary. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
SLP and ADM conceived of the study. SLP, ADM, and JDO planned the overall research project and experiments. SCH collected samples and data from the field. SLP and GD conducted the laboratory procedures and analyses under the guidance and supervision of JW, RWA, JSG and ADM. JDO, SLP and MCH analyzed the data with input from ADM. SLP, ADM, JDO, and MCH wrote the manuscript. All authors edited the manuscript and approve its submission.
Funding
Research was supported by the National Sciences and Engineering Research Council of Canada (AM), the Canada Research Chairs Program (AM), the Alberta Innovates (Omics) Graduate Student Scholarship (SP), the National Science and Engineering Research Council of Canada Graduate Scholarship (SP), the Queen Elizabeth II Graduate Scholarship (SP), the O’Brien Centre Funding Summer Studentship (SP), the Alberta Children’s Hospital Research Institute (SP, AM), and the National Science Foundation Graduate Research Fellowship Program (MH). The project that gave rise to these results received the support of a fellowship from “la Caixa” Foundation (ID 100010434) and from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 847648. The fellowship code is LCF/BQ/PI20/11760004 (JO).
Acknowledgments
We thank Shasta Webb for helpful comments and suggestions on the data analyses and presentation. We thank Roger Blanco, Maria Marta Chavarria, Jeremy Hogan, Emily Walco, and Monica Myers for assistance in research access and data collection.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2023.1176681/full#supplementary-material
References
Aivelo T., Medlar A. (2018). Opportunities and challenges in metabarcoding approaches for helminth community identification in wild mammals. Parasitology. 145 (5), 608–621. doi: 10.1017/s0031182017000610
Akinyi M. Y., Jansen D., Habig B., Gesquiere L. R., Alberts S. C., Archie E. A. (2019). Costs and drivers of helminth parasite infection in wild female baboons. J. Anim. Ecol. 88 (7), 1029–1043. doi: 10.1111/1365-2656.12994
Alberdi A., Aizpurua O., Gilbert M. T. P., Bohmann K. (2018). Scrutinizing key steps for reliable metabarcoding of environmental samples. Methods Ecol. Evol. 9 (1), 134–147. doi: 10.1111/2041-210X.12849
Andrews S. (2010) FastQC: A quality control tool for high throughput sequence data. Available at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
Avramenko R. W., Redman E. M., Lewis R. R., Yazwinski T. A., Wasmuth J. D., Gilleard J. S. (2015). Exploring the gastrointestinal “nemabiome”: deep amplicon sequencing to quantify the species composition of parasitic nematode communities. PloS One 10 (12), e0143559. doi: 10.1371/journal.pone.0143559
Avramenko R. W., Redman E. M., Lewis R., Bichuette M. A., Palmeira B. M., Yazwinski T. A., et al. (2017). The use of nemabiome metabarcoding to explore gastro-intestinal nematode species diversity and anthelmintic treatment effectiveness in beef calves. Int. J. Parasitol. 47 (13), 893–902. doi: 10.1016/j.ijpara.2017.06.006
Bergstrom M. L., Hogan J. D., Melin A. D., Fedigan L. M. (2019). The nutritional importance of invertebrates to female Cebus capucinus imitator in a highly seasonal tropical dry forest. Am. J. Phys. Anthropology. 170 (2), 207–216. doi: 10.1002/ajpa.23913
Bethge J., Razafimampiandra J. C., Wulff A., Dausmann K. H. (2022). Seasonal changes in the parasite prevalence of a small Malagasy lemur species (Lepilemur edwardsi). Integr. Zoology. 00, 1–13. doi: 10.1111/1749-4877.12647
Blasco-Costa I., Cutmore S. C., Miller T. L., Nolan M. J. (2016). Molecular approaches to trematode systematics: ‘best practice’ and implications for future study. Systematic Parasitol. 93 (3), 295–306. doi: 10.1007/s11230-016-9631-2
Blersch R., Bonnell T. R., Barrett L., Henzi S. P. (2021). Seasonal effects in gastrointestinal parasite prevalence, richness and intensity in vervet monkeys living in a semi-arid environment. J. Zoology 314 (3), 163–173. doi: 10.1111/jzo.12877
Bradley B. J., Stiller M., Doran-Sheehy D. M., Harris T., Chapman C. A., Vigilant L., et al. (2007). Plant DNA sequences from feces: potential means for assessing diets of wild primates. Am. J. Primatology 69 (6), 699–705. doi: 10.1002/ajp.20384
Brooks D. R., Hoberg E. P. (2007). How will global climate change affect parasite–host assemblages? Trends Parasitol. 23 (12), 571–574. doi: 10.1016/j.pt.2007.08.016
Bueno de Mesquita C. P., Nichols L. M., Gebert M. J., Vanderburgh C., Bocksberger G., Lester J. D., et al. (2021). Structure of chimpanzee gut microbiomes across tropical Africa. Msystems 6 (3), e01269–e01220. doi: 10.1128/mSystems.01269-20
Byrne R. L., Fogarty U., Mooney A., Marples N. M., Holland C. V. (2018). A comparison of helminth infections as assessed through coprological analysis and adult worm burdens in a wild host. Int. J. Parasitology: Parasites Wildlife 7 (3), 439–444. doi: 10.1016/j.ijppaw.2018.11.003
Callahan B. J., McMurdie P. J., Rosen M. J., Han A. W., Johnson A. J. A., Holmes S. P. (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13 (7), 581–583. doi: 10.1038/nmeth.3869
Callejón R., Nadler S., De Rojas M., Zurita A., Petrášová J., Cutillas C. (2013). Molecular characterization and phylogeny of whipworm nematodes inferred from DNA sequences of cox1 mtDNA and 18S rDNA. Parasitol. Res. 112 (11), 3933–3949. doi: 10.1007/s00436-013-3584-z
Campos F. A., Fedigan L. M. (2009). Behavioral adaptations to heat stress and water scarcity in white-faced capuchins (Cebus capucinus) in Santa Rosa National Park, Costa Rica. Am. J. Phys. Anthropology 138 (1), 101–111. doi: 10.1002/ajpa.20908
Céza V., Pánek T., Smejkalová P., Čepička I. (2015). Molecular and morphological diversity of the genus Hypotrichomonas (Parabasalia: Hypotrichomonadida), with descriptions of six new species. Eur. J. protistology 51 (2), 158–172. doi: 10.1016/j.ejop.2015.02.003
Criscione C. D., Anderson J. D., Raby K., Sudimack D., Subedi J., Rai D. R. (2007). Microsatellite markers for the human nematode parasite Ascaris lumbricoides: development and assessment of utility. J. Parasitol. 93 (3), 704–708. doi: 10.1645/GE-1058R.1
Criscione C. D., Anderson J. D., Sudimack D., Subedi D., Upadhayay J., Jha R. P., et al. (2010). Landscape genetics reveals focal transmission of a human macroparasite. PloS Negl. Trop. Dis. 4 (4), e665. doi: 10.1371/journal.pntd.0000665
DeAngelis M. M., Wang D. G., Hawkins T. L. (1995). Solid-phase reversible immobilization for the isolation of PCR products. Nucleic Acids Res. 23 (22), 4742–4743. doi: 10.1093/nar/23.22.4742
Decrappeo N., Giblin-Davis R. M. (2001). Schistonchus aureus n. sp. and S. laevigatus n. sp. (Aphelenchoididae): Associates of Native Floridian Ficus spp. and Their Pegoscapus Pollinators (Agaonidae). J. Nematol. 33 (2-3), 91–103.
de Winter I. I., Umanets A., Gort G., Nieuwland W. H., van Hooft P., Heitkönig I. M. A., et al. (2020). Effects of seasonality and previous logging on faecal helminth-microbiota associations in wild lemurs. Sci. Rep. 10 (1), 16818. doi: 10.1038/s41598-020-73827-1
Eley R. M., Strum S. C., Muchemi G., Reid G. D. F. (1989). Nutrition, body condition, activity patterns, and parasitism of free-ranging troops of olive baboons (Papio anubis) in Kenya. Am. J. Primatology 18 (3), 209–219. doi: 10.1002/ajp.1350180304
Fragaszy D. M., Visalberghi E., Fedigan L. M. (2004). The complete capuchin: the biology of the Genus Cebus. (Cambridge, United Kingdom: Cambridge University Press).
Frankie G. W., Mata A., Vinson S. B. (2004). Biodiversity conservation in Costa Rica: Learning the lessons in a seasonal dry forest. (Berkeley and Los Angeles, California, United States: Univ of California Press).
Frias L., Stark D. J., Lynn M. S., Nathan S. K., Goossens B., Okamoto M., et al. (2018). Lurking in the dark: Cryptic Strongyloides in a Bornean slow loris. Int. J. Parasitology. Parasites Wildlife 7 (2), 141–146. doi: 10.1016/j.ijppaw.2018.03.003
Giblin-Davis R. M., Ye W., Kanzaki N., Williams D., Morris K., Thomas W. K. (2006). Stomatal Ultrastructure, Molecular Phylogeny, and Description of Parasitodiplogaster laevigata n. sp. (Nematoda: Diplogastridae), a Parasite of Fig Wasps. J. Nematol. 38 (1), 137–149.
Griffin R. H., Nunn C. L. (2012). Community structure and the spread of infectious disease in primate social networks. Evolutionary Ecol. 26, 779–800. doi: 10.1007/s10682-011-9526-2
Guillou L., Bachar D., Audic S., Bass D., Berney C., Bittner L., et al. (2013). The Protist Ribosomal Reference database (PR2): a catalog of unicellular eukaryote small sub-unit rRNA sequences with curated taxonomy. Nucleic Acids Res. 41 (Database issue), D597–D604. doi: 10.1093/nar/gks1160
Hadziavdic K., Lekang K., Lanzen A., Jonassen I., Thompson E. M., Troedsson C. (2014). Characterization of the 18S rRNA gene for designing universal eukaryote specific primers. PloS One 9 (2), e87624. doi: 10.1371/journal.pone.0087624
Henry A. G. (2012). Recovering dietary information from extant and extinct primates using plant microremains. Int. J. Primatology 33 (3), 702–715. doi: 10.1007/s10764-011-9556-1
Hu Y. O. O., Karlson B., Charvet S., Andersson A. F. (2016). Diversity of Pico- to Mesoplankton along the 2000 km Salinity Gradient of the Baltic Sea. Front. Microbiol. 7. doi: 10.3389/fmicb.2016.00679
Hugerth L. W., Muller E. E. L., Hu Y. O. O., Lebrun L. A., Roume H., Lundin D., et al. (2014). Systematic design of 18S rRNA gene primers for determining eukaryotic diversity in microbial consortia. PloS One 9 (4), e95567. doi: 10.1371/journal.pone.0095567
Jombart T. (2008). adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24 (11), 1403–1405. doi: 10.1093/bioinformatics/btn129
Kanzaki N., Giblin-Davis R. M. (2014). Phylogenetic status and morphological characters of Rhabditolaimus anoplophorae (Rhabditida: Diplogastridae). J. Nematol. 46 (1), 44–49.
Kounosu A., Murase K., Yoshida A., Maruyama H., Kikuchi T. (2019). Improved 18S and 28S rDNA primer sets for NGS-based parasite detection. Sci. Rep. 9 (1), 1–12. doi: 10.1038/s41598-019-52422-z
Lange C. E., Wittenstein E. (2001). An Adelina sp. (Apicomplexa: Coccidia) found in Anurogryllus muticus (De Geer) (Orthoptera: Gryllidae). J. Invertebrate Pathol. 77 (1), 83–84. doi: 10.1006/jipa.2000.4999
Maharachchikumbura S. S. N., Chen Y., Ariyawansa H. A., Hyde K. D., Haelewaters D., Perera R. H., et al. (2021). Integrativ approaches for species delimitation in Ascomycota. Fungal Diversity 109, 155–179. doi: 10.1007/s13225-021-00486-6
Mallott E. K., Amato K. R., Garber P. A., Malhi R. S. (2018). Influence of fruit and invertebrate consumption on the gut microbiota of wild white-faced capuchins (Cebus capucinus). Am. J. Phys. Anthropology 165 (3), 576–588. doi: 10.1002/ajpa.23395
Mann A. E., Mazel F., Lemay M. A., Morien E., Billy V., Kowalewski M., et al. (2020). Biodiversity of protists and nematodes in the wild nonhuman primate gut. ISME J. 14 (2), 609–622. doi: 10.1038/s41396-019-0551-4
Marcogliese D. J. (2005). Parasites of the superorganism: Are they indicators of ecosystem health? Int. J. Parasitol. 35 (7), 705–716. doi: 10.1016/j.ijpara.2005.01.015
Martin M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17 (1), 10–12. doi: 10.14806/ej.17.1.200
Mati V. L. T., Ferreira Junior F. C., Pinto H. A., De Melo A. L. (2013). Strongyloides cebus (Nematoda: Strongyloididae) in Lagothrix cana (Primates: Atelidae) from the Brazilian Amazon: aspects of clinical presentation, anatomopathology, treatment, and parasitic biology. J. Parasitol. 99 (6), 1009–1018. doi: 10.1645/13-288.1
Melin A. D., Fedigan L. M., Hiramatsu C., Kawamura S. (2008). Polymorphic color vision in white faced capuchins (Cebus capucinus): Is there foraging niche divergence among phenotypes? Behav. Ecol. Sociobiology 62 (5), 659–670. doi: 10.1007/s00265-007-0490-3
Melin A. D., Fedigan L. M., Young H. C., Kawamura S. (2010). Can color vision variation explain sex differences in invertebrate foraging by capuchin monkeys? Curr. Zoology. 56 (3), 300–312. doi: 10.1093/czoolo/56.3.300
Melin A. D., Hiramatsu C., Parr N. A., Matsushita Y., Kawamura S., Fedigan L. M. (2014). The behavioral ecology of color vision: Considering fruit conspicuity, detection distance and dietary importance. Int. J. Primatology 35 (1), 258–287. doi: 10.1007/s10764-013-9730-8
Melin A. D., Hogan J. D., Campos F. A., Wikberg E., King‐Bailey G., Webb S., et al. (2020). Primate life history, social dynamics, ecology, and conservation: Contributions from long-term research in Área de Conservación Guanacaste, Costa Rica. Biotropica 52 (6), 1041–1064. doi: 10.1111/btp.12867
Mosdossy K. N., Melin A. D., Fedigan L. M. (2015). Quantifying seasonal fallback on invertebrates, pith, and bromeliad leaves by white-faced capuchin monkeys (Cebus capucinus) in a tropical dry forest. Am. J. Phys. Anthropology 158 (1), 67–77. doi: 10.1002/ajpa.22767
Obanda V., Maingi N., Muchemi G., Ng’ang’a C. J., Angelone S., Archie E. A. (2019). Infection dynamics of gastrointestinal helminths in sympatric non-human primates, livestock and wild ruminants in Kenya. PloS One 14 (6), e0217929. doi: 10.1371/journal.pone.0217929
Orkin J. D., Campos F. A., Myers M. S., Cheves Hernandez S. E., Guadamuz A., Melin A. D. (2019). Seasonality of the gut microbiota of free-ranging white-faced capuchins in a tropical dry forest. ISME J. 13 (1), 183–196. doi: 10.1038/s41396-018-0256-0
Orkin J. D., Montague M. J., Tejada-Martinez D., de Manuel M., del Campo J., Hernandez S. C., et al. (2018). The evolution of ecological flexibility, large brains, and long lives: capuchin monkey genomics revealed with fecalFACS. Proceedings of the National Academy of Sciences 118 (7), e2010632118. doi: 10.1101/366112
Pafčo B., Čížková D., Kreisinger J., Hasegawa H., Vallo P., Shutt K., et al. (2018). Metabarcoding analysis of strongylid nematode diversity in two sympatric primate species. Sci. Rep. 8 (1), 1–11. doi: 10.1038/s41598-018-24126-3
Papaiakovou M., Littlewood D. T. J., Doyle S. R., Gasser R. B., Cantacessi C. (2022). Worms and bugs of the gut: the search for diagnostic signatures using barcoding, and metagenomics–metabolomics. Parasites Vectors 15 (1), 118. doi: 10.1186/s13071-022-05225-7
Paradis E. (2010). pegas: an R package for population genetics with an integrated–modular approach. Bioinformatics 26 (3), 419–420. doi: 10.1093/bioinformatics/btp696
Paradis E., Schliep K. (2019). ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35 (3), 526–528. doi: 10.1093/bioinformatics/bty633
Parr N. A., Fedigan L. M., Kutz S. J. (2013a). A coprological survey of parasites in white aced capuchins (Cebus capucinus) from Sector Santa Rosa, ACG, Costa Rica. Folia primatological 84 (2), 102–114. doi: 10.1159/000348287
Parr N. A., Fedigan L. M., Kutz S. J. (2013b). Predictors of parasitism in wild white-faced capuchins (Cebus capucinus). Int. J. Primatology 34 (6), 1137–1152. doi: 10.1007/s10764-013-9728-2
Parr N. A., Melin A. D., Fedigan L. M. (2011). Figs are more than fallback foods: The relationship between ficus and cebus in a tropical dry forest. Int. J. Zoology 2011, 967274. doi: 10.1155/2011/967274
Perera A., Maia J. P. M. C., Jorge F., Harris D. J. (2013). Molecular screening of nematodes in lacertid lizards from the Iberian Peninsula and Balearic Islands using 18S rRNA sequences. J. Helminthology 87 (2), 189–194. doi: 10.1017/S0022149X12000181
Rego A. A., Schaeffer G. (1988). Filariopsis barretoi (travassos 1921) (Nematoda: metastrongyloidea) lung parasite of primates from South America - taxonomy, synonyms and pathology. Memorias do Instituto Oswaldo Cruz 83 (2), 183–188. doi: 10.1590/S0074-02761988000200006
Roberts L. S. (2000). The crowding effect revisited. J. Parasitol. 86 (2), 209–211. doi: 10.1645/0022-3395(2000)086[0209:TCER]2.0.CO;2
Rohland N., Reich D. (2012). Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Res. 22 (5), 939–946. doi: 10.1101/gr.128124.111
Romeo C., Wauters L. A., Cauchie S., Martinoli A., Matthysen E., Saino N., et al. (2014). Faecal egg counts from field experiment reveal density dependence in helminth fecundity: Strongyloides robustus infecting grey squirrels (Sciurus carolinensis). Parasitol. Res. 113, 3403–3408. doi: 10.1007/s00436-014-4005-7
Ross J. L., Ivanova E. S., Spiridonov S. E., Waeyenberge L., Moens M., Nicol G. W., et al. (2010). Molecular phylogeny of slug-parasitic nematodes inferred from 18S rRNA gene sequences. Mol. Phylogenet. Evol. 55 (2), 738–743. doi: 10.1016/j.ympev.2010.01.026
Schliep K. P. (2011). phangorn: phylogenetic analysis in R. Bioinformatics 27 (4), 592–593. doi: 10.1093/bioinformatics/btq706
Sharma A. K., Davison S., Pafco B., Clayton J. B., Rothman J. M., McLennan M. R., et al. (2022). The primate gut mycobiome-bacteriome interface is impacted by environmental and subsistence factors. NPJ biofilms microbiomes 8 (1), 12. doi: 10.1038/s41522-022-00274-3
Solórzano-García B., de León G. P.-P. (2018). Parasites of neotropical primates: A review. Int. J. Primatology 39, 155–182. doi: 10.1007/s10764-018-0031-0
Srivathsan A., Ang A., Vogler A. P., Meier R. (2016). Fecal metagenomics for the simultaneous assessment of diet, parasites, and population genetics of an understudied primate. Front. Zoology 13, 17. doi: 10.1186/s12983-016-0150-4
Srivathsan A., Nagarajan N., Meier R. (2019). Boosting natural history research via metagenomic clean-up of crowdsourced feces. PloS Biol. 17 (11), e3000517. doi: 10.1371/journal.pbio.3000517
Stensvold C. R. (2019). Pinning down the role of common luminal intestinal parasitic protists in human health and disease–status and challenges. Parasitology 146 (6), 695–701. doi: 10.1017/S0031182019000039
Taddese B., Garnier A., Deniaud M., Henrion D., Chabbert M. (2021). Bios2cor: an R package integrating dynamic and evolutionary correlations to identify functionally important residues in proteins. Bioinformatics 37 (16), 2483–2484. doi: 10.1093/bioinformatics/btab002
Tanaka R., Hino A., Tsai I. J., Palomares-Rius J. E., Yoshida A., Ogura Y., et al. (2014). Assessment of helminth biodiversity in wild rats using 18S rDNA-based metagenomics. PloS One 9 (10), e110769. doi: 10.1371/journal.pone.0110769
Titcomb G., Mantas J. N., Hulke J., Rodriguez I., Branch D., Young H. (2021). Water sources aggregate parasites with increasing effects in more arid conditions. Nat. Commun. 12 (1), 7066. doi: 10.1038/s41467-021-27352-y
Valenta K., Melin A. D. (2012). Protein limitation explains variation in primate colour vision phenotypes: A unified model for the evolution of primate trichromatic vision. In García M. D., Zoology (IntechOpen) 23, 29–46.
Keywords: genetic parasitology, primate parasitology, deep amplicon sequencing, metabarcoding, parasite communities, platyrrhines, diet analyses, seasonality
Citation: Pinto SL, Henriquez MC, Cheves Hernandez S, Duytschaever G, Wit J, Avramenko RW, Gilleard JS, Orkin JD and Melin AD (2023) Promise and limitations of 18S genetic screening of extracted fecal DNA from wild capuchins. Front. Ecol. Evol. 11:1176681. doi: 10.3389/fevo.2023.1176681
Received: 28 February 2023; Accepted: 02 August 2023;
Published: 24 August 2023.
Edited by:
Elizabeth Mallott, Washington University in St. Louis, United StatesReviewed by:
Justin Wilcox, New York University Abu Dhabi, United Arab EmiratesAllison E. Mann, University of North Texas Health Science Center, United States
Copyright © 2023 Pinto, Henriquez, Cheves Hernandez, Duytschaever, Wit, Avramenko, Gilleard, Orkin and Melin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Swellan Luciann Pinto, c3dlbGxhbi5waW50b0B1Y2FsZ2FyeS5jYQ==; U3dlbGxhbnBpbnRvQGdtYWlsLmNvbQ==; Amanda Dawn Melin, YW1hbmRhLm1lbGluQHVjYWxnYXJ5LmNh