 Vadivelu Karuppaiah1*
Vadivelu Karuppaiah1* Ankush S. Gadge1
Ankush S. Gadge1 Dhananjay V. Shirsat1
Dhananjay V. Shirsat1 Parakkattu S. Soumia1
Parakkattu S. Soumia1 Pawan Mainkar1
Pawan Mainkar1 Satish Kumar1
Satish Kumar1 Durgesh K. Jaiswal2Vijay Mahajan1*
Durgesh K. Jaiswal2Vijay Mahajan1*- 1Crop Protection Section, Indian Council of Agricultural Research (ICAR) Directorate of Onion and Garlic Research, Pune, India
- 2Department of Botany, Savitribai Phule Pune University, Pune, India
The Indian stingless bee Tetragonula iridipennis (Hymenoptera: Apidae), popularly recognized as the Indian dammer bee, is an economically important and widely distributed non-Apis bee species in India. The taxonomic gaps, systematics, evolutionary puzzles, and structural motifs within the mitogenomes of this species have rarely been examined and are not fully understood. Next-generation sequencing was employed to decipher the complete mitochondrial genome of T. iridipennis (15,045 bp). De novo genome assembly revealed that it encompasses 34 genes: protein-coding genes (13), transfer RNA (tRNAs) genes (19), and ribosomal RNA (rRNA) genes (2). Additionally, genome organization, including gene content, nucleotide composition, codon usage, and gene rearrangement, was investigated to better comprehend, utilize, and conserve this germplasm resource. The average gene length was 400 bp; maximum and minimum lengths were 1,530 bp (cox1) and 57 bp (tRNA-S1), respectively. Phylogenetic analysis has suggested that T. iridipennis is mostly closely related to T. pagdeni and Lepidotrigona species. All the stingless bee species (Meliponini) formed a distinct clade that shared a closer relationship with bumble bees (Bombini) than honey bees (Apini). The nucleotide composition of T. iridipennis was biased toward A+T, which accounted for 75.95% of the whole mitogenome. Length and compositional differences between T. iridipennis and other bees were detected, and gene order was compared. The mitogenome of T. iridipennis showed the highest gene rearrangement score (78), suggesting this species has a hyperactive evolutionary history. The variations of gene positions and gene rearrangement in the mitogenome could also aid in resolving the phylogenetic relationships and evolutionary history in Meliponini. Additionally, this is the first report of a complete mitochondrial genome sequence of T. iridipennis.
1 Introduction
The stingless bees (Hymenoptera: Apidae: Meliponini) are an important group of non-Apis bees that visit many flowering plants in the tropics and subtropics (Roubik, 1993; Heard, 1999; Vossler, 2012; Layek and Karmakar, 2018). They are considered as a viable substitute for addressing the issue of pollination deficit in many agricultural, forest, and horticultural ecosystems (Garibaldi et al., 2013). Additionally, honey produced by stingless bees is very much in demand as it is rich in medicinal value (Al-Hatamleh et al., 2020). Furthermore, it is considered that uncovering the molecular features of this species is vital to understanding the molecular phylogeny and biodiversity of social insects (Wang et al., 2021). Wider adaptation to extreme environmental conditions and weedy types of reproduction in warmer conditions appears to be a superior trait of this species for their wide inhabitation. Among the corbiculate bees, enormous diversity is observed in stingless bees, which comprise over 600 distinct species (Gruter, 2020). The geospatial distribution of this species suggests that they are predominantly found in forests and agricultural ecosystems of tropical areas and urban realms of the Neotropical, Afrotropical, and Australasian regions (Gruter, 2020). The stingless bee species Tetragonula iridipennis, T. bengalensis, T. praeterita, T. laeviceps, T. ruficornis, and Lepidotrigona arcifera (Rahman et al., 2018) are widely distributed across India. Recently, two new species, T. kyrdemkulaiensis and T.srikantanathi, were identified in Northeast India (Viraktamath and Thangjam, 2021). Moreover, 17 diverse genera of stingless bees have reported in Indo-Malay/Australasia (Gruter, 2020; Rasmussen et al., 2017). Among Tetragonula, Moure is the most complex genus and alone comprises 34 species, predominantly inhabiting the Indian subcontinent. Tetragonula iridipennis (Smith) is an economically important stingless bee species natively known as the Indian stingless bee, Dammer bee, or Mosquito bee and is widespread in Asian countries, including Bangladesh, Bhutan, India, Indonesia, Myanmar, Nepal, Pakistan, and Sri Lanka (Rasmussen, 2013; Rahman et al., 2018; Engel et al., 2019; Bhatta et al., 2020; Nidup, 2021).
The smaller size of stingless bees means they are difficult to identify morphologically due to indistinctive differences and overlapping morphometric characteristics, leading to the misidentification of these species. Furthermore, the evolutionary histories of many taxa remain unexplored and are not obvious in the Meliponini tribe. As a result, there are many gaps in the taxonomic knowledge, which could have an impact on how efficiently stingless bee resources are protected and utilized (Wang et al., 2022). Again, morphology-based classification requires vast experience to effectively distinguish subtle differences between species. In this context, molecular features, such as complete mitochondrial DNA (mtDNA) sequences, are being efficiently utilized to distinguish the arthropod species and resolve contradictory evolutionary histories (Ohler and Delorme, 2006). In addition, the record of maternal inheritance obtained from mitochondrial genome is widely utilized in inferring phylogenetic relationships between diverse lineages. Furthermore, mtDNA poses several advantages as a biological marker, owing to its increased rate of mutation, nucleotide substitution rates, rare genetic recombination events, maternal inheritance, and easy availability (Howlader et al., 2015). Therefore, it has been extensively used in phylogenetic analysis, molecular evolution, genome structure analysis, and phylogeographic studies (Li et al., 2021). Additionally, mtDNA has been utilized to identify cryptic species, study population genetic structure, and evaluate micro-evolutionary processes (Zhang et al., 2018). Hence, the use of molecular datasets, especially mitogenomes, becomes essential to better understand stingless bee taxa and their molecular systematics, as well as conservation studies.
Although there have been numerous studies on arthropod mitochondrial genome sequencing, very few studies have been carried out with the Meliponini tribe. Six stingless bee species, Melipona fasciculata (MH680930.1), Melipona bicolor (AF466146.2), Melipona scutellaris (KP202303), Lepidotrigona terminata (MN737481.1), Lepidotrigona flavibasis (MN747147.1), and Tetragonula pagdeni (NC066054.1 and OK336459), have been characterized so far. Considering the distribution, diversity, and economic importance of T. iridipennis on the Indian subcontinent, molecular systematics of this species are vital for its characterization and conservation studies. Therefore, the complete mt genome of T. iridipennis was assembled using next-generation sequencing (NGS) for the first time. The sequence information and datasets generated will be helpful in uncovering the phylogenetics, and the evolutionary history of this species thereby aids conservation biology. Accordingly, the objectives of this study include: (i) characterization of the whole-mitochondrial genome of T. iridipennis; (ii) analyze the structural features of the mitochondrial genome, inherent nucleotide composition, codon usage bias, and the tRNA gene translocation; and (iii) establish the major phylogenetic relationship with the Apidae family and the gene order, as well as gene arrangement. The extensive genetic information generated from this study will aid in comparative genomics, molecular systematics, and population genetic studies of stingless bees.
2 Materials and methods
2.1 Sample collection
T. iridipennis-visited (workers bee) onion umbels were collected from onion seed production plots at the experimental farm of the Indian Council of Agricultural Research-Directorate of Onion and Garlic Research (ICAR-DOGR), located in Pune, Maharashtra, India (latitude of 18° 51’ 59.99” N, longitude of 73° 53’ 59.99” E, and 645 m above sea level). The insect sample was collected during the peak onion blooming period (February and March) and the voucher specimen was preserved (DOGR Voucher SHB01) following stipulated procedures. Species identity was confirmed by DNA barcoding at ICAR-DOGR, Pune, India, and the sequence information has been archived (NCBI GeneBank accession number: OQ103112).
2.2 Sample preparation and DNA extraction
Sample specimens collected from the onion field were initially preserved in 95% ethanol and stored at 4°C until experimentation. Total genomic DNA was extracted from the legs of a single worker bee of T. iridipennis using a DNeasy Blood and Tissue Kit (QIAGEN, Germany). The quantitative and qualitative features of extracted DNA were confirmed through 1% agarose gel electrophoresis and spectrophotometric measurement (at 260 and 280 nm) using a SmartSpec 3000 UV/Visible Spectrophotometer (Bio-Rad, Hercules, CA, USA). Mitochondrial DNA was isolated from the genomic DNA using a REPLI-g Mitochondrial DNA Kit (QIAGEN, Germany).
2.3 Sequence assembly, annotation, and analysis
The sequence features from the mitochondrial genome library were obtained using an Illumina Next Seq 500 platform, and primary FASTQ data were generated. The FASTQ data were analyzed for total number of bases, the number of reads, GC% value, Q30, and the percentage of fuzzy bases. Then, the high-quality reads were generated by removing adapter and junk sequences and ambiguous reads, using the fastp tool (v0.12.4) (Chen et al., 2018). The resultant high-quality reads were further aligned with the reference sequences using BWA MEM (version 0.7.17) (Li and Durbin, 2009). Protein-coding and other RNA genes were identified from the consensus sequence extracted using SAM tools mpileup (Li et al., 2009). Genes from the invertebrate’s mitochondrial genome were predicted using the MITOS web server (http://mitos.bioinf.uni-leipzig.de/index.py) (Bernt et al., 2013).
A de novo de-Brujin graph-based assembly was carried out to assemble the short reads into larger stretches of DNA contigs. These contigs were used for genome annotation, which assigns functions to various regions of the genome of the organism. The mitogenome was generated using the Megahit assembler (Li et al., 2015) as part of the MitoZ package (Meng et al., 2019). The assembly was further quality-checked using the Quast tool (Gurevich et al., 2013). Using MitoZ software (https://github.com/linzhi2013/MitoZ), the whole-mitochondrial genome was annotated (Meng et al., 2019) and subsequently verified with three mitogenome sequences of Melipona bicolor (AF466146.2), Tetragonula pagdeni (NC066054.1 and OK336459), and Lepidotrigona flavibasis (MN747147.1) available from the GeneBank database. The MITOS web server was used for assessing the assembled mitogenome and to predict the secondary structural aspects of tRNA genes (Bernt et al., 2013). A circular genome map, GC concentration, and GC skewness were generated using the molecular biology tool GCview server (http://stothard.afns.ualberta.ca/cgview_server/) (Grant and Stothard, 2008).
The base composition (A+T contents), codon usage, and relative synonymous codon usage (RSCU) of the protein-coding genes (PCGs) were analyzed using MEGA version XI (https://www.megasoftware.net/). The GC and AT skews were computed using the following formula: GC skew = [G-C]/[G+C] and AT skew = [A-T]/[A+T] (Perna and Kocher, 1995). The intergenic spacers and overlapping regions between the genes were manually computed. The complete mitogenome sequence of T. iridipennis following assembly and annotation was submitted to the NCBI sequence archive (GenBank accession no: OQ139639).
2.4 Phylogenetic analysis
The genetic relationships and taxonomy of T. iridipennis with other stingless bees (Meliponini) and hymenopteran bees (honey bees and bumble bees) were analyzed using 15 concatenated whole-mitogenome sequences of Hymenoptera species retrieved from the NCBI database.
Partition Finder 2.1.1 with the Bayesian information criterion (BIC) and greedy algorithm (www.phylo.org) was used to select the best partitioning schemes (Lanfear et al., 2017). The GTR+G model was preferred by the BIC for nucleotide alignment analysis. The nucleotide sequences from 15 different species were aligned using the MUSCLE tool implemented in MEGA 11 (https://www.megasoftware.net/) software. The phylogenetic tree was generated in MEGA 11 (Tamura et al., 2021) using the maximum likelihood (ML) method, general time reversible model with gamma distribution (GTR+G) default parameters, and bootstrap values from 500 iterations.
2.5 Gene rearrangement evaluation
The mitogenome of T. iridipennis, along with the mitogenomes of 15 chosen species, were examined for gene orders and potential gene rearrangement. This was carried out using the PhyloSuite software package (http://phylosuite.jushengwu.com/) (Zhang et al., 2020a). For quantitative analysis and comparison of the rearrangements among various mitogenomes, the rearrangement score (RS) of each individual gene was computed using qMGR software (http://qmgr.hnnu.edu.cn/) (Zhang et al., 2020b).
3 Results
3.1 Mitogenome organization and nucleotide composition
The circular mitogenome of the Indian stingless bee T. iridipennis was 15,045 bp in size (Figure 1). There were 34 sequence elements in total, comprising 13 protein-coding genes, 19 tRNA genes, and two rRNA genes. Analysis of the strand localization of 34 coding genes revealed that H-strand (+) harbors 28 genes whereas the L-strand (−) has six genes (Table 1). The T. iridipennis mitochondrion had an average gene length of 400 bp, while the maximum and minimum lengths were 1,530 bp (cox1) and 57 bp (tRNA-S1 genes), respectively. The base composition of the entire mitogenome of T. iridipennis was 40.66%, 35.29%, 12.65%, and 11.40% for A, T, C, and G nucleotides, respectively (Table 2). The nucleotide composition ranged from 64.94% to 85.47% for the 13 PCGs and two rRNAs, and it was 75.95% for the overall mitogenome. The AT skewness ranged between −0.050 to −0.212 for the 13 PCGs, which was relatively stronger than for the two rRNA genes, rrnS (0.047) and rrnL (0.032) (Table 2).
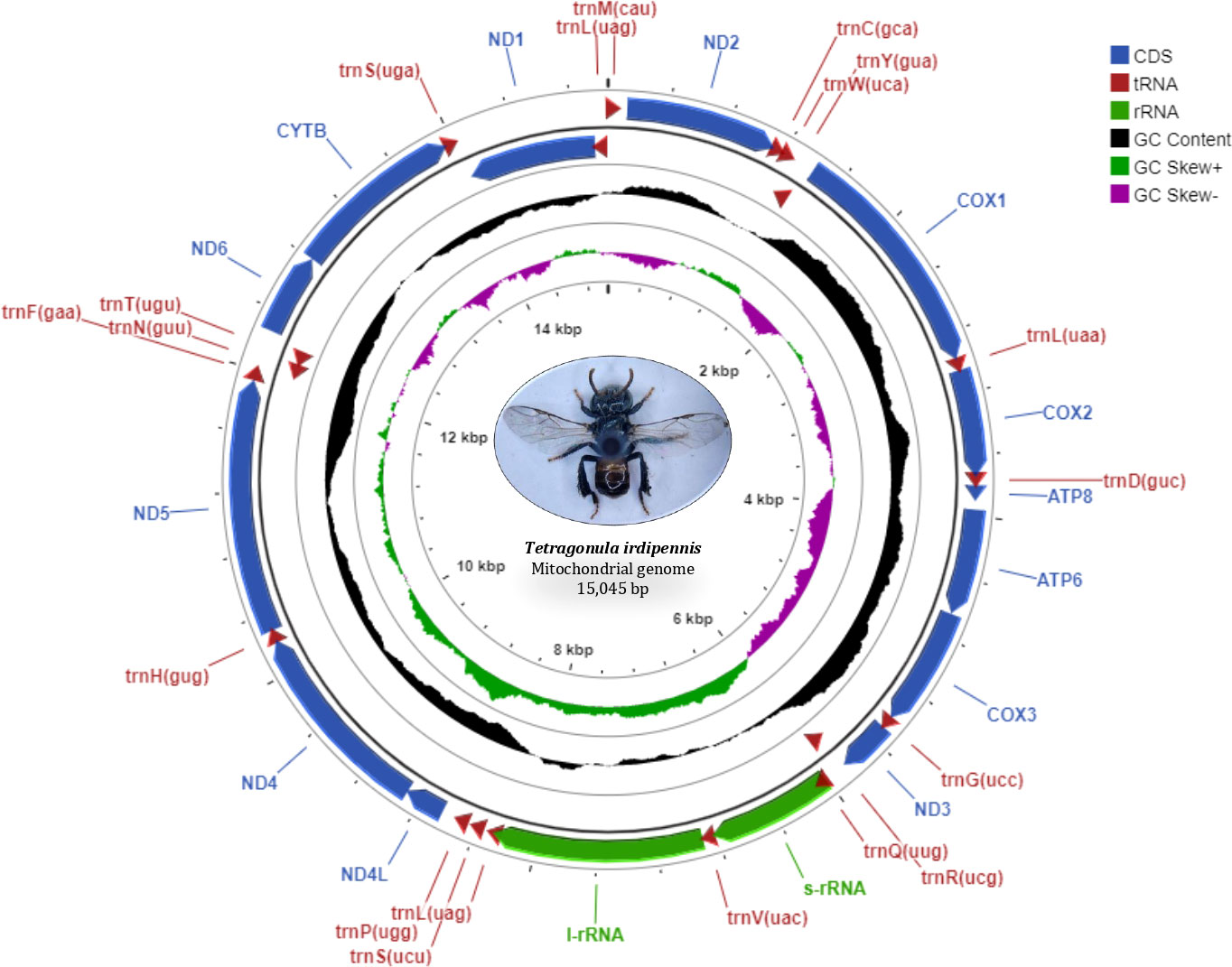
Figure 1 Mitochondrial genome map of T. iridipennis. From outer to inner, the first circle shows the gene map (PCGs, rRNA, tRNAs, and CR); tRNA genes are abbreviated by one letter symbols according to the IUPAC-IUB single-letter amino acid codes. The second circle shows the GC content and the third circle shows GC skew calculated as (G-C)/(G+C). GC content and GC skew are plotted as the deviation from the average value of the entire sequence.
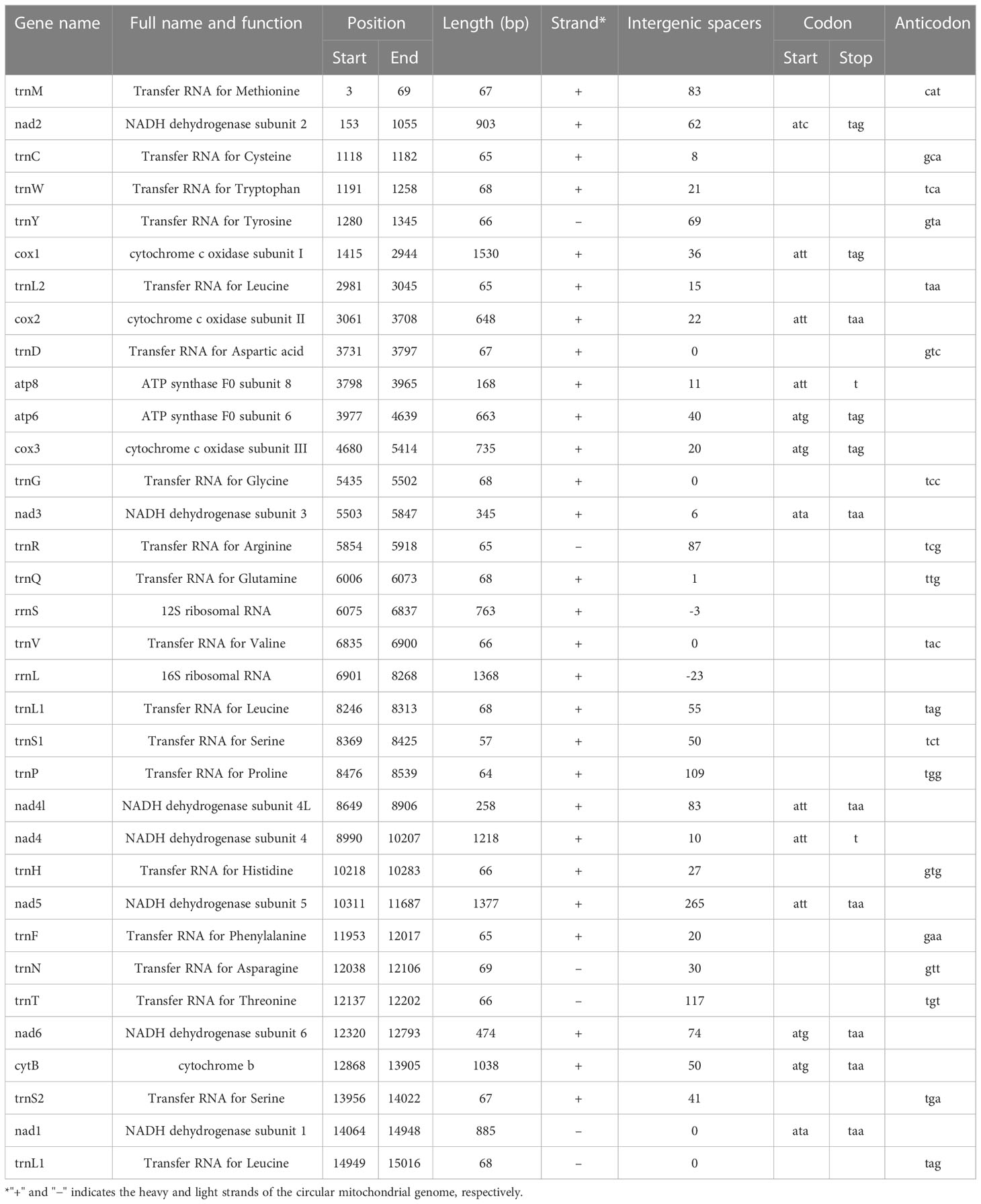
Table 1 Summary table of the characteristics of the complete mitogenome of T. iridipennis.
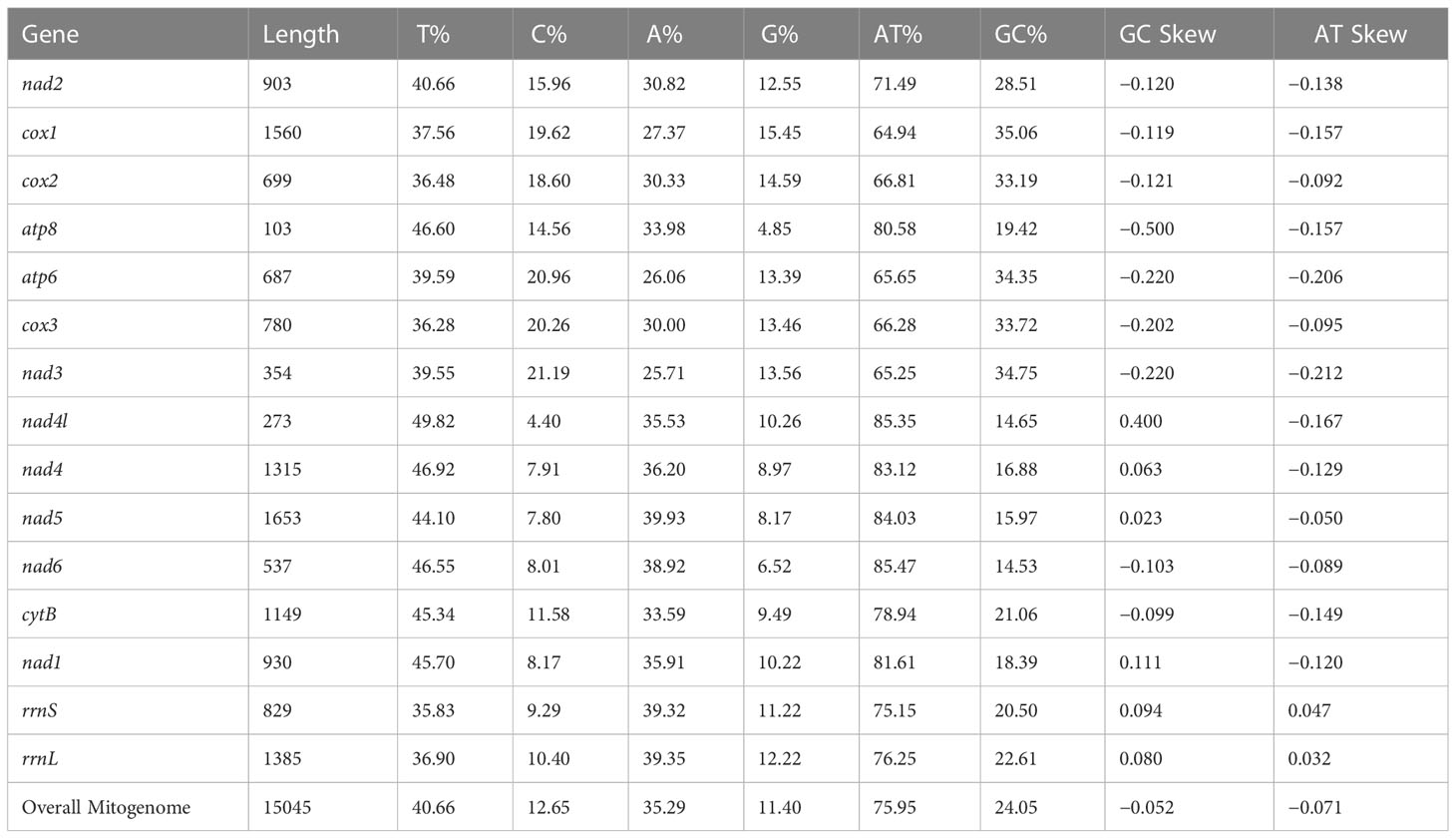
Table 2 AT/CG skews in the 13 PCGs and two rRNAs of the T. iridipennis mitogenome.
3.2 Protein-coding genes and codon usage bias
The total length of 13 PCGs of the mitogenome of T. iridipennis was 10,242 bp, which is nearly 68.08% of the genome. The length range of coding sequences varied from 168 bp (atp8) to 1,530 bp (cox1). Genes such as cox1, cox2, cox3, nad2, nad3, nad4, nad4l, nad5, nad6I, CytB, atp6, and atp8 were located on the H-strand (+), while nad1 alone was located on the L-strand (−). The four start codons, ATA, ATC, ATG, and ATT, were found in the T. iridipennis mitochondrial genome. Six genes (cox1, cox2, atp8, nad4, nad4l, and nad5) adopted ATT as their start codon, four genes (atp6, cox3, nad6, and CytB) used ATG, two genes (nad3 and nad1) used ATA, and ATC was only used by nad2. Seven PCGs (cox2, nad1, nad3, nad4l, nad5, nad6, and CytB) are characterized with TAA as their termination codon, while four mitochondrial genes (cox1, cox3, nad2, and atp6) use TAG, and the incomplete termination codon ‘T’ is used by two genes (atp8 and nad4).
The relative synonymous codon use (RSCU) and amino acid usage in the protein-coding genes of the T. iridipennis mitochondrion are summarized in Table 3. Leucine (L, Leu), isoleucine (I, Ile), phenylalanine (F, Phe), serine (S, Ser), tyrosine (Y, Tyr), and asparagine (N, Asn) were the most frequent amino acids, whereas tryptophan (W, Trp) and alanine (A, Ala) were the rarest (Figure 2). In general, UUU, AUU, UUA, UAU, and AAU were the most frequently used codons.
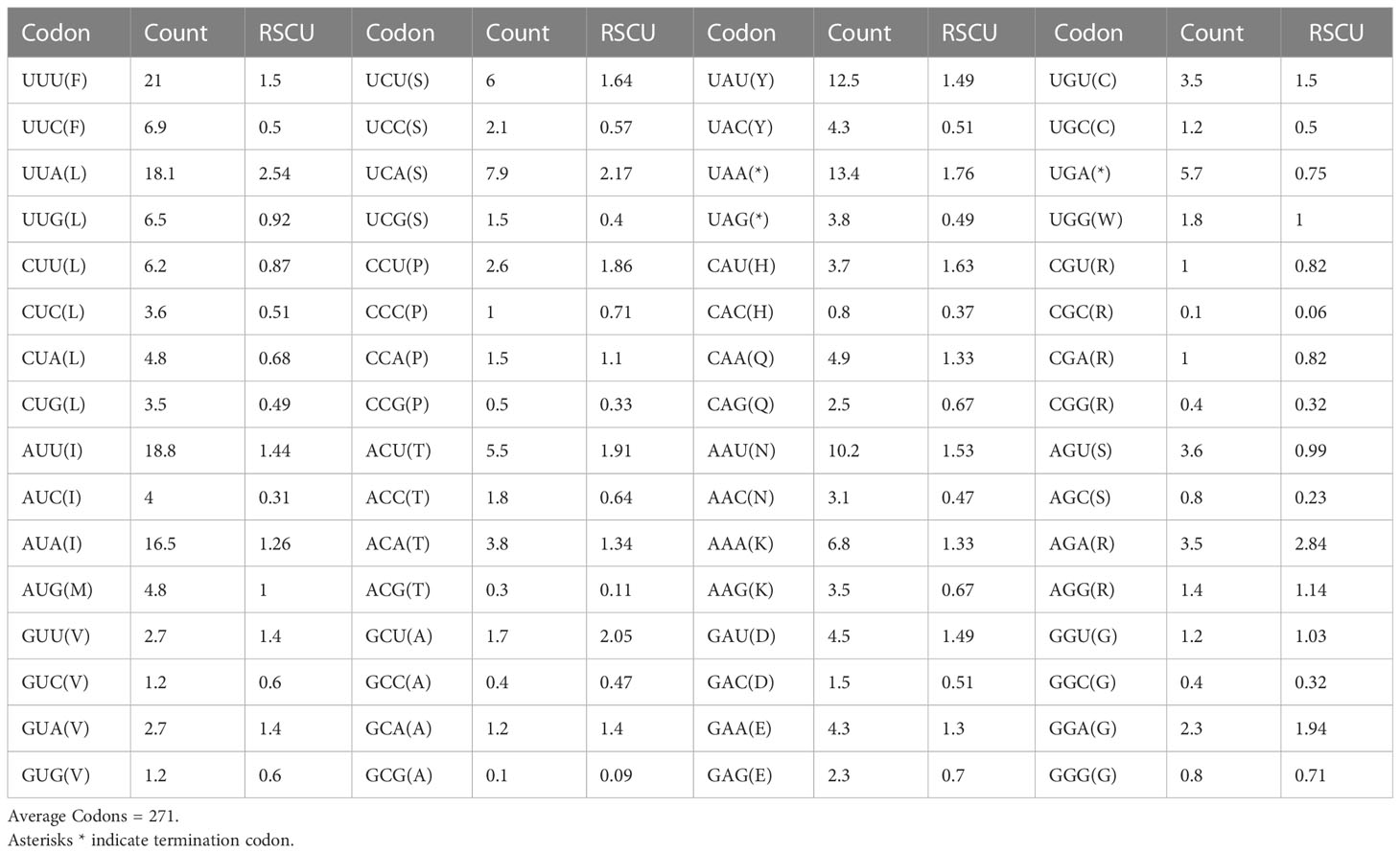
Table 3 Codon usage and relative synonymous codon usage (RSCU) pattern of the 13 mitochondrial protein-coding genes from T. iridipennis.
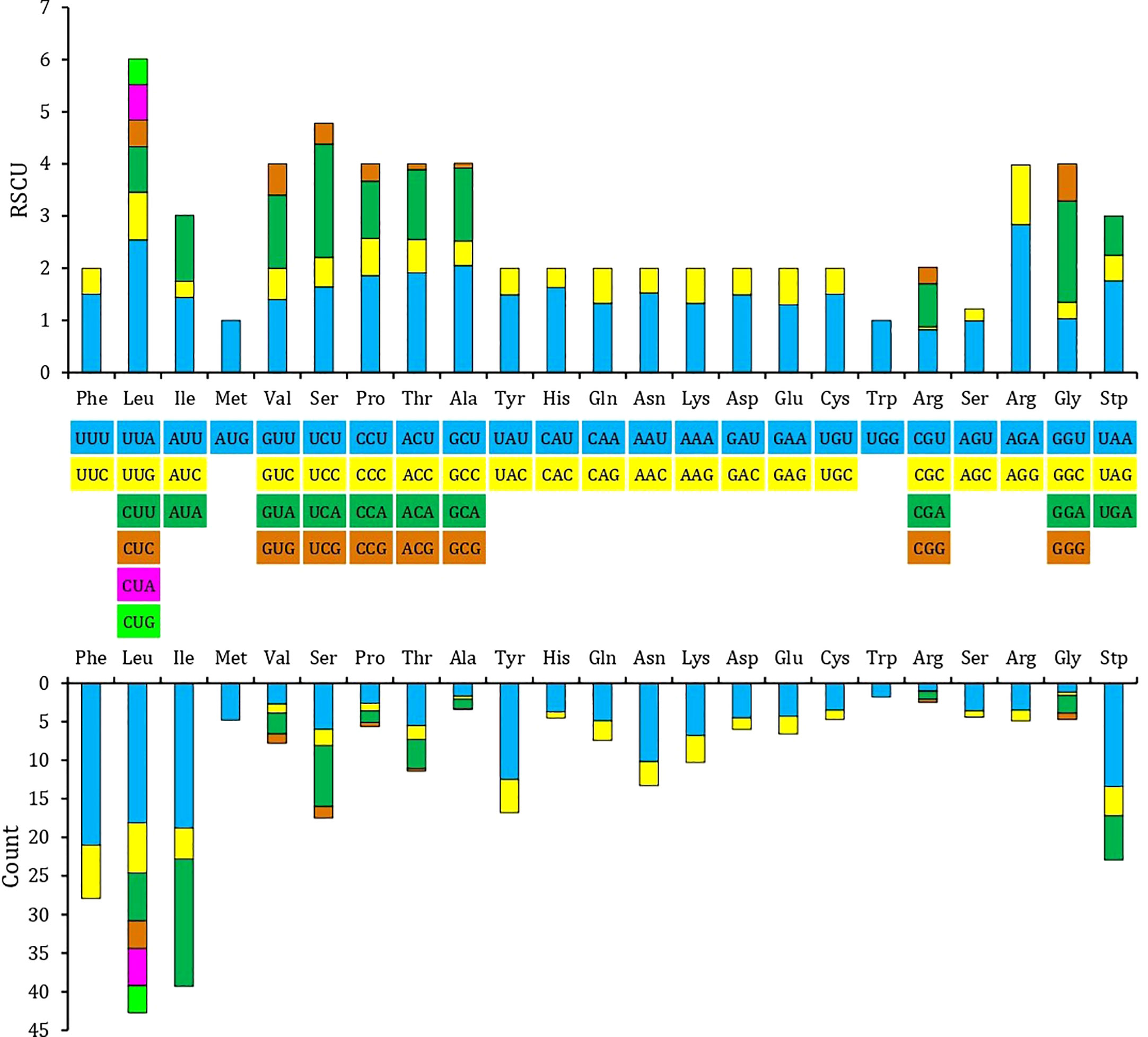
Figure 2 Relative synonymous codon usage (RSCU) in the mitogenome of T. iridipennis. The different colors in the column chart represent the codon families corresponding to the amino acids below and consistent colors are used to represent the same codon families.
3.3 Transfer RNAs and ribosomal RNAs
The 19 tRNA genes, which vary in size from 57 to 69 bp, were scattered over the circular mitochondrial genome of T. iridipennis. The H-strand (+) of the mitochondria contained 14 tRNA genes, whereas the remaining five tRNA genes were found on the L-strand (−) (Figure 3; Table 1). The 19 tRNAs collectively comprised 1,255 bp covering 8.34% of the whole mitogenome. The characteristic clover-leaf secondary structures of tRNA genes were found except for trnS1ser (UCU), which lacked a dihydrouridine (DHU) arm. Four genes encoding tRNAs for Ala, Glu, Ile, and Lys were absent in the mitochondrial genome of T. iridipennis, whereas two copies of trnL1 were found. A total of five mismatched base pairs (tRNACys, tRNAGly, tRNAGln, tRNAThr, tRNAVal) were present in the tRNAs of T. iridipennis.
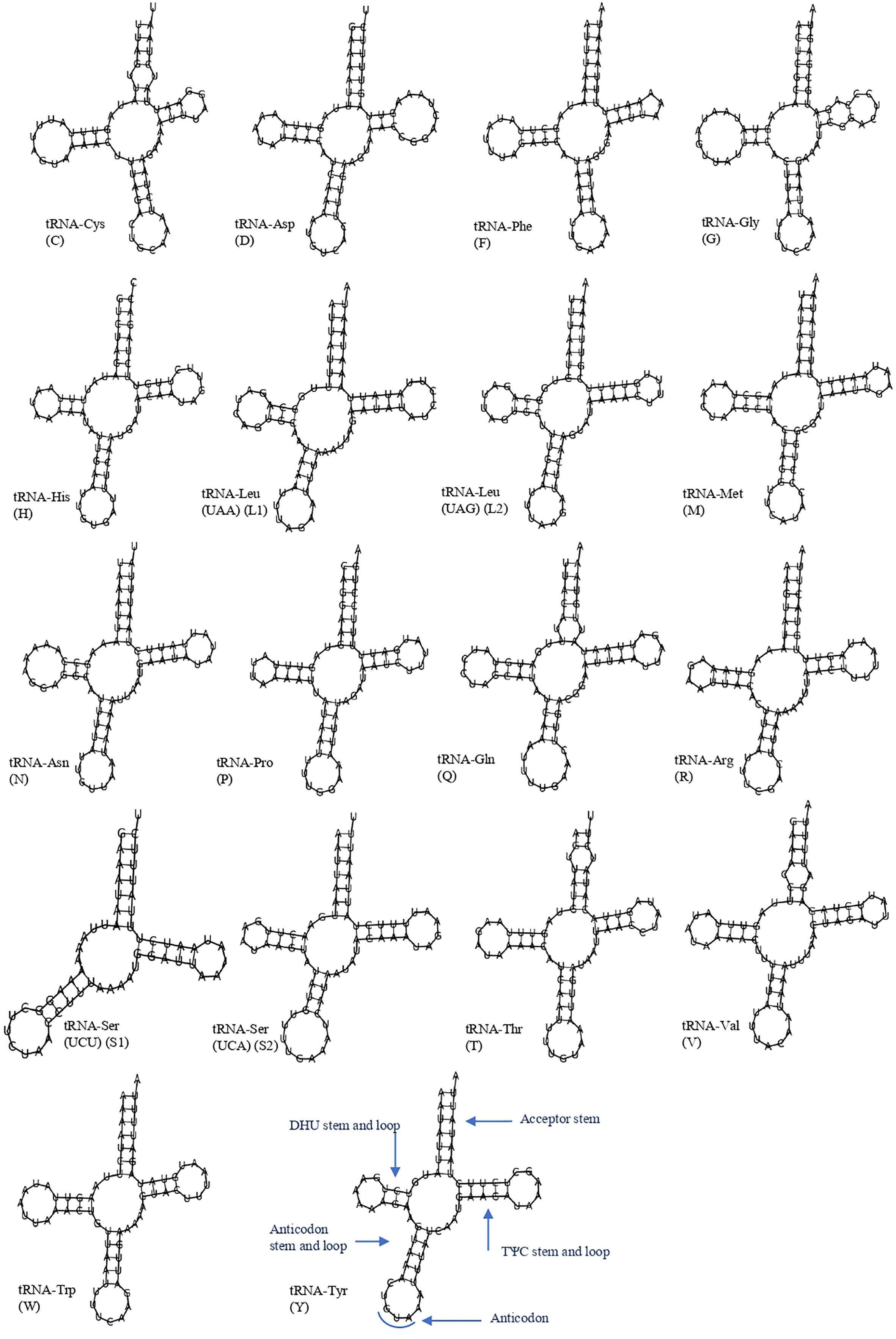
Figure 3 Putative secondary structures for 19 tRNA genes of T. iridipennis. The uppercase alphabet letters are the single letter abbreviation of the amino acids.
The two rRNA genes (rrnS-and rrnL) located on the H-strand (+) constitute 14.16% of the total mitochondrial genome. The rrnS gene (763 bp long and an A+T content of 75.15%) was located between tRNAGln and tRNAVal. The rrnL gene (1,368 bp in size and an A+T content of 76.25%) was found between tRNAVal and tRNALeu.
3.4 The overlapping and intergenic spacer region
Two overlapping regions (26 bp in length) were identified in the mitogenome of T. iridipennis. These regions varied in length from 3 bp between rrnS and trnV and 23 bp between rrnL and trnL1. The intergenic spacers varied in length from 1 to 265 bp in the T. iridipennis mitogenome, constituting 1,412 bp, and were found interspersed in 27 regions of the genome. The longest intergenic spacer was 265 bp and located between nad5 and trnF. Similarly sized intergenic spacer regions were also located between trnT and nad6, and trnP and nad4l, with lengths of 117 bp and 109 bp, respectively.
3.5 Phylogenetic analysis
Molecular phylogenomics were evaluated from the phylogenetic tree constructed using the concatenated complete mitogenome sequences of 15 Hymenopteran bees, including seven stingless bees (Figure 4). Phylogenetic analysis revealed three major clades based on the tribes (Meliponini, Bombini, and Apini) along with an outgroup comprising Megachile sculpturalis (KT223644) and Colletes gigas (KM978210). The Meliponini tribe was grouped together, forming a major monophyletic clade of stingless bees. T. iridipennis from the current study (OQ139639) was closely related to T. pagdeni (OK336459) and clustered together with two Lepidotrigona species (MN747147.1 and MN737481.1) under sub-clade Ia. Meanwhile, three Melipona species (MH680930, AF466146, and KP202303) were grouped under sub-clade Ib. At the genus level, Tetragonula exhibited greater molecular proximity to Lepidotrigona than to the Melipona taxa. The phylogenetic analysis also showed that stingless bees (Meliponini) have a closer relationship with the clade corresponding to bumble bees (Bombus) of the Bombini tribe than with the clade belonging to honey bees (Apis) of the Apini tribe in the Apidae family.
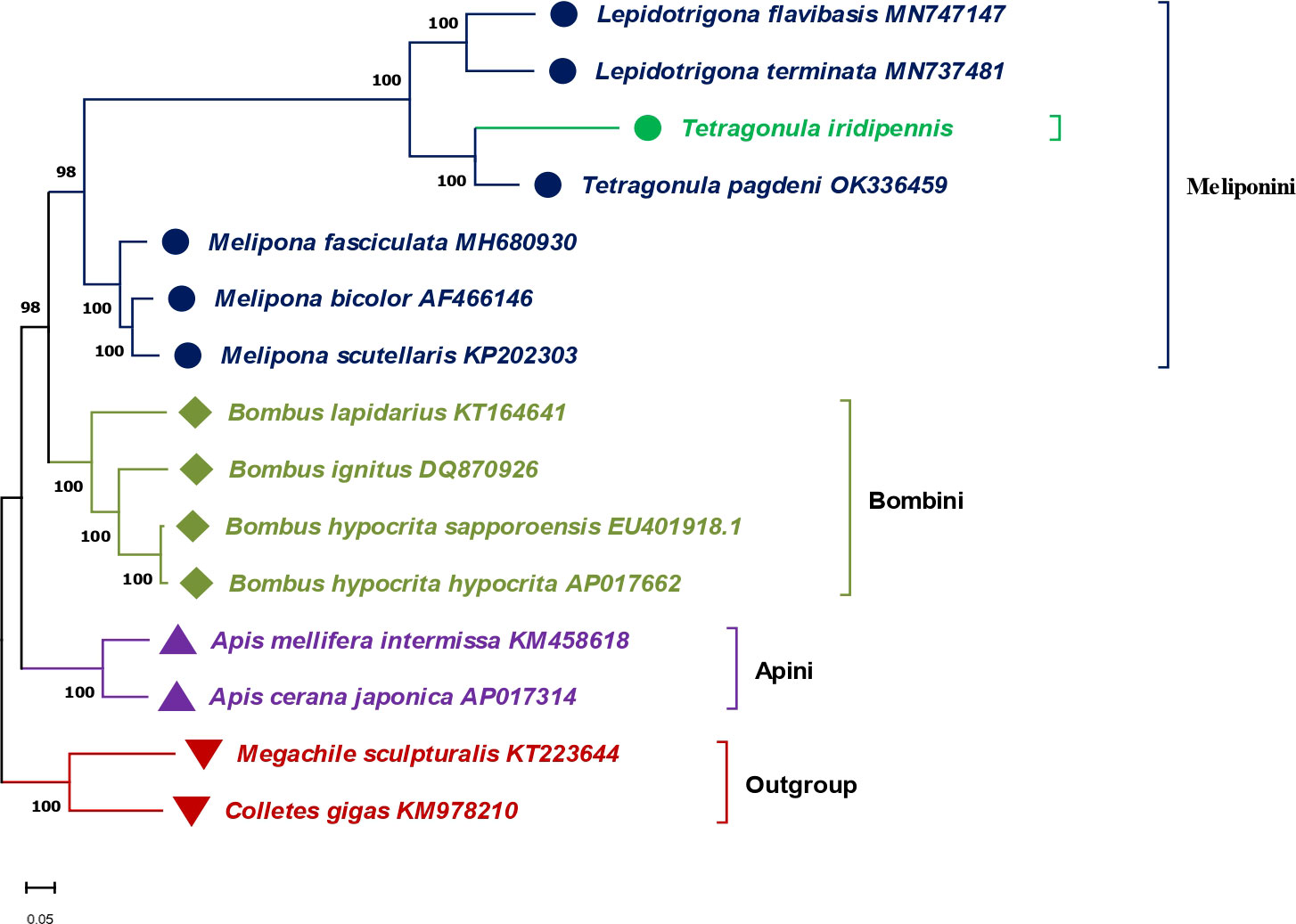
Figure 4 Phylogenetic tree showing the relationship between T. iridipennis and 14 other Hymenopteran species based on the concatenated nucleotide sequences of the whole mitogenome using maximum likelihood (ML) and Bayesian inference (BI) analysis. Megachilidae (Megachile sculpturalis) and Colletidae (Colletes gigas) were used as an outgroup. GenBank accession numbers of each sequence are displayed in the tree beside their corresponding species names.
3.6 Gene rearrangement evaluation
The order of genes of the mitogenome of T. iridipennis was quite distinct from the six previously reported stingless bee mitogenomes (OK336459, MN747147, MN737481, AF466146, MH680930, AF466146, and KP202303), and dramatic gene rearrangement was observed in T. iridipennis (Figure 5). Genetic rearrangement events were remarkably consistent with the output of the phylogenetic tree (Figures 5, 6), supporting the idea that the order of genes in the mitogenome may contain crucial evolutionary data for phylogenetic research. The RS value, which describes the extent of gene rearrangement of each mitogenome, was determined. T. iridipennis had the highest RS value (78) among all Apidae insects considered in the current study (Figure 6), indicating an active evolutionary past of T. iridipennis. The mitochondrial gene rearrangement score of T. iridipennis was relatively higher among the Hymenopteran insect species. Among the stingless bee (Meliponini) clade, the locations of tRNA-K differed. Gene rearrangement showed M-K-A-I block in Lepidotrigona, whereas the reversed order I-A-K-M block was present in Melipona, and K-A-I-M was present in T. pagdeni. By contrast, T. iridipennis had I-M block instead on the usual gene rearrangement in Hymenopteran orders due to missing tRNA-A and tRNA-K (Figure 5).
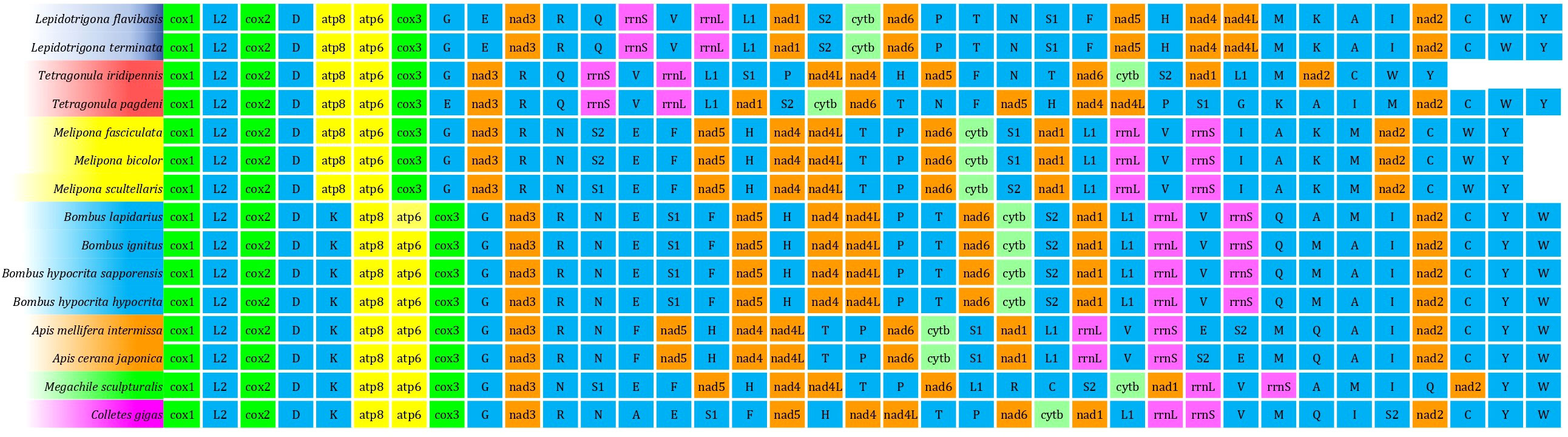
Figure 5 The gene order of each mitogenome for comparison of the mitochondrial gene orders between Tetragonula stingless bees and other Hymenopteran insects. The same colour indicates the sequence of the similar gene. The uppercase alphabet letters are the single letter abbreviation of the amino acids.
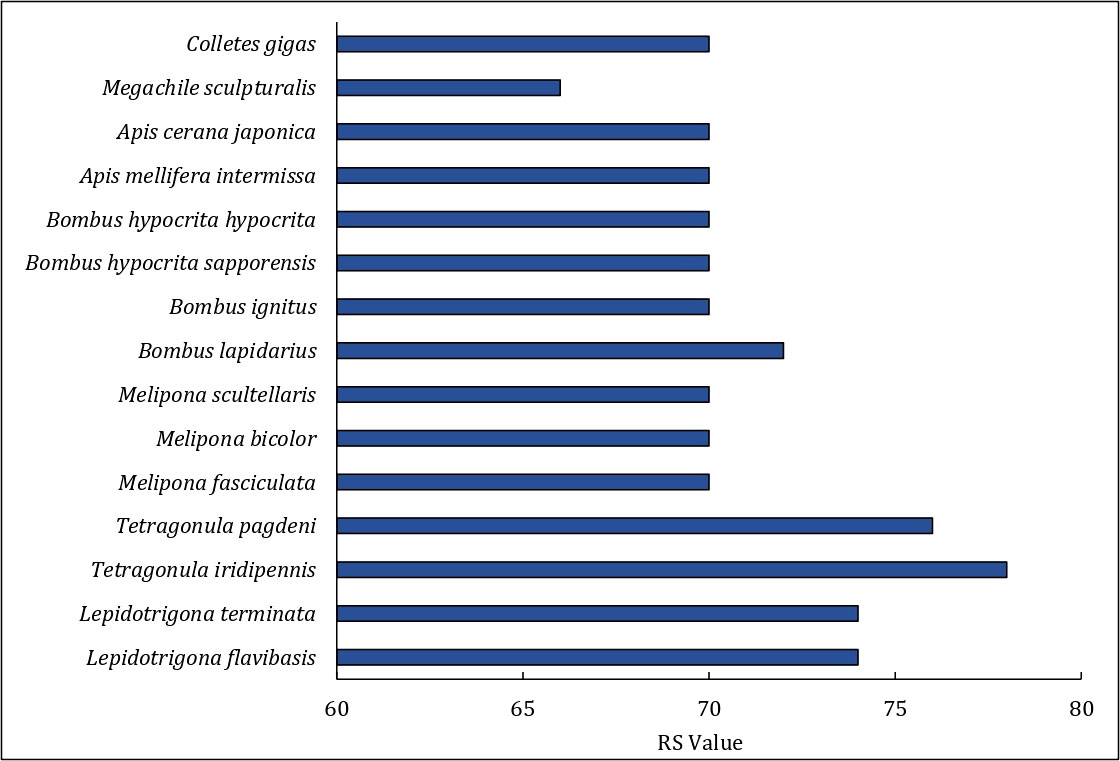
Figure 6 The Rearrangement Score (RS value) of each mitogenome for comparing Tetragonula stingless bees and other Hymenopteran insects.
4 Discussion
In general, the insect mitogenome is characterized as a circular double-stranded polymer 14–19 kb in length and encodes 37 genes, including 22 tRNA genes, two rRNA genes, and 13 PCGs (Boore, 1999; Cameron, 2014; De Mandal et al., 2014). The size of the whole mitogenome of T. iridipennis from the present study (15,045 bp) is in accordance with the mitogenome of other stingless bees, such as T. pagdeni (16,061 bp), L. flavibasis (15,408 bp), L. terminata (15,431 bp), M. bicolor (15,001 bp), M. fasciculata (14,753 bp), and M. scutellaris (14,862 bp), belonging to the same tribe, Meliponini, in the Apidae family.
The nucleotide base composition of T. iridipennis revealed an obvious bias toward A and T. Among the four nucleotides, T and G were the most and least favored, respectively. According to previous reports, the nucleotide composition of stingless bees belonging to different species was A+T rich (Wang et al., 2021; Wang et al., 2022). A similar trend was observed in the T. iridipennis mitogenome. The bias of the base composition of an individual strand can be described by skewness. The AT (−0.071) and GC (−0.052) skewness values were both negative, indicating a bias towards Ts and Cs rather than As and Gs, respectively, in the mitogenome of T. iridipennis. According to the hypothesis, biasness might be due to the active use of these bases (Ts and Cs) by DNA polymerase during mtDNA replication (Clary and Wolstenholme, 1985). Furthermore, less energy is required to break the A-T linkages, and the subsequent transcription will generate an AT bias in species that depend on mitochondrial efficiency to maintain a high metabolic rate (Xia, 1996).
The PCGs [three Cytochrome oxidases (cox1, cox2, and cox3), seven NADH dehydrogenases (nad1, nad2, nad3, nad4, nad4l, nad5, and nad6), one Cytochrome B gene (CytB), and two ATPases (atp6 and atp8)] of the mitochondrial genome of T. iridipennis are distributed throughout. Most species belonging to the Hymenoptera, such as T. pagdeni (Wang et al., 2022), L. flavibasis (Wang et al., 2021), L. terminata (Wang et al., 2020), M. bicolor (Silvestre et al., 2008), M. scutellaris (Pereira et al., 2016), and Apis dorsata (Chhakchhuak et al., 2016), have the same number of PCGs, with little variation in their size and location. Like most Hymenopterans, the initiation codon (ATN) and the incomplete stop codon (T) were observed in T. iridipennis. However, no anomalous codon was observed in T. iridipennis PCGs, as found in L. flavibasis (GTG start codon for nad2) and A. cerana (GTG start codon for nad2) (Wang et al., 2021). In T. iridipennis, the degenerate codons frequently used A and T rather than G and C. Similar results were reported by Wang et al. (2022) in T. pagdeni. In addition, the incomplete stop codon (T) can give rise to functional stop codons due to processes such as polyadenylation and polycistronic transcription cleavage (Ojala et al., 1981). RSCU analysis indicated a bias toward AT-rich codons, which mainly results from an AT mutation bias (Wang et al., 2022).
The predicted secondary structures of the tRNAs of T. iridipennis were very similar to T. pagdeni and L. flavibasis (Wang et al., 2021; Wang et al., 2022). Nonetheless, a few differences were present in the TΨC and D arms (Silvestre et al., 2008). The tRNA-Ser1 (UCU) lacks the dihydrouridine (DHU) arm and thereby a simple loop is formed rather than the typical clover-leaf shape. The mitogenomes of many stingless bees showed similar results (Wang et al., 2020; Wang et al., 2021; Wang et al., 2022). The anticodons were the same for all the stingless bee species studied by various researchers (Silvestre et al., 2008; Wang et al., 2021; Wang et al., 2022). In confirmation with Silvestre et al. (2008), the anticodon loop of 14 tRNAs of T. iridipennis had seven nucleotides and its arm had five base pairs, except for 5 tRNAs (tRNA-Leu1, tRNA-Gln, tRNA-Arg, tRNA-Ser2 and tRNA-Val) that only had four base pairs on the anticodon loop, similar to A. mellifera tRNA-Val (4 bp). Additionally, the acceptor arm had a conserved size (7 bp) in most Meliponini stingless bees, except in M. bicolor tRNA-Arg (6 bp) (Silvestre et al., 2008). The two copies of trnL1 reported in the mitochondrial genome of T. iridipennis might have occurred due to the partial duplication that commonly occurs in insect mtDNA (Zhang and Hewitt, 1997). Unlike the 22–23 tRNA genes commonly found in animal mitochondrial genomes, 19 tRNAs were identified in T. iridipennis mtDNA in the present study. The four missing tRNA genes coded Ala, Glu, Ile, and Lys amino acids. Similar results were observed by Silvestre et al., 2008 in M. bicolor with the absence of tRNA-Cys, tRNA-Gly, tRNA-Ile, and tRNA-Ser1. The mitochondrial tRNAs inferred for Hymenopteran insects T. pagdeni, L. flavibasis, M. bicolor, A. mellifera, A. cerana, and Bombus ignites were found to have mismatched base pairs (Silvestre et al., 2008; Tan et al., 2011; Wang et al., 2021; Wang et al., 2022). Mitochondrial tRNAs of Hymenopteran species are characterized by stem mismatches and sequences that are rectified later by a posttranscriptional editing mechanism (Tan et al., 2011). The relative locations of two genes encoding ribosomal RNAs in the T. iridipennis mitogenome were identical to those in T. pagdeni and L. flavibasis but different from Melipona species, thereby clustering them in different clades of the phylogenetic tree (Silvestre et al., 2008; Wang et al., 2021; Wang et al., 2022).
Two overlapping regions of 26 bp in length have been reported in the mitogenome of T. iridipennis. Similar overlapping regions have also been recorded in the mitogenomes of T. pagdeni and L. flavibasis, with lengths of 25 and 24 bp, respectively (Wang et al., 2021; Wang et al., 2022). These regions of T. iridipennis, T. pagdeni, and L. flavibasis ranged in length from 1–3 bp between rrnS and trnV and 23 bp between rrnL and trnL1, constituting a single clade in evolutionary history. Owing to the apparent lack of strong purifying selection and accumulation of multiple base changes and insertion/deletions, this region is referred to as a second origin of mtDNA replication and transcription (Cornuet et al., 1991).
According to Wang et al. (2019); Wang et al. (2021), complete mitochondrial genome sequences are usually preferred for phylogenetic analysis over partial or incomplete combination sequences and a few nuclear genes as they yield topologies with strong support values. The complete mitogenomes of T. iridipennis and 14 common Hymenopteran species retrieved from the NCBI database were used for the phylogenetic analysis. In accordance with other studies, the current phylogenetic analysis of stingless bees (Meliponini) revealed the following relationships: (Tetragonula + Lepidotrigona) + Melipona) + Bombus) + Apis) + (Megachile + Colletes) (Wang et al., 2020; Wang et al., 2021; Wang et al., 2022). At the genus level within the Meliponini tribe, Tetragonula is more closely linked to Lepidotrigona than to Melipona, and these findings closely align with some earlier reports by Wang et al. (2022); Wang et al. (2021); Wang et al. (2020). Additionally, Zhao et al. (2017) and Wang et al. (2020); Wang et al. (2021); Wang et al. (2022) have provided evidence that stingless bees are phylogenetically more closely related to bumble bees than honey bees. The phylogenetic relationships were reconstructed based on the complete mitogenome, supporting the inferences obtained from the conventional morphology-based phylogenetics of the Apidae family.
The gene order of most insect mitogenomes is the same; nevertheless, gene rearrangements in the mitogenomes of some species are very common. In general, rearrangements of genes involve gene inversion, genetic transposition, inverse transposition, and tandem duplication, causing loss of gene function (Cameron, 2014; Chen et al., 2018). The absence of the super conserved tRNA D-K block in the stingless bee group implies that tRNA-K movement could have occurred early in the progenitor of the stingless bee, thereby phylogenetically separating the stingless bee from other Hymenopteran insects (Figure 5). Similar findings were found in T. pagdeni and L. flavibasis (Wang et al., 2021; Wang et al., 2022). Furthermore, T. iridipennis was found to exhibit unique translocation, which broke the relatively frequent intact block Nad1-L1-rrnL-V-rrnS box in the mitogenomes of Hymenopteran insects, suggesting that T. iridipennis had a very active mitogenome. However, in Lepidotrigona and Tetragonula, this box had moved and turned upside down (Wang et al., 2021; Wang et al., 2022). The translocated Nad-1 moved to the position next to S-2 (Figure 5). The super-conserved atp8-atp6-cox3-G block in Hymenopteran mitogenomes was retained in T. iridipennis. However, Wang et al. (2022) found an uncommon tRNA-Gly translocation by substituting E for G in T. pagdeni. The rRNA, tRNA, and (PCG) genes of T. iridipennis experienced considerable rearrangement. These occurrences imply that the Tetragonula species’ mitogenomes have been very active. Most invertebrate mitogenomes have largely retained gene ordering. Natural selection may not completely eradicate mitogenome rearrangements, and they may reflect the dynamics and rates of evolution within a given species or genus (Shao et al., 2003).
Many cultivated crops are cross pollinated and mainly depend on insect pollinators, especially from the order Hymenoptera, for their ecosystem services. Among insect pollinators, honey bees are important pollinators belonging to the Apidae family. Nevertheless, non-Apis species of stingless bees are imperative in the pollination of agricultural, forest, and horticultural crops in the tropics and subtropics. Mitochondrial genomics has helped greatly in elucidating the evolutionary relationships of Hymenopteran insects at multiple taxonomic levels, especially for Apoidea. Hence, the scientific literature is replete with mitochondrial genomics pertaining to the Apidae family; however, very few reports are available pertaining to the Meliponini tribe. This study provides novel molecular insights into the genetic architecture of the mitogenomic and phylogenetic framework of T. iridipennis in Hymenoptera and facilitates future studies on the evolution of invertebrates. In this study, the entire T. iridipenni mitogenome sequence was made available for the first time. The circular mitogenome of T. iridipennis is 15,045 bp in length and contains 34 genes, comprising 13 protein-coding genes, NADH dehydrogenase genes, cytochrome c oxidase genes, ATPase genes, a Cytochrome B gene, two ribosomal RNA genes, and 19 transfer RNA genes. These findings provide crucial information for upcoming mtDNA analysis of other Meliponini species, helping to resolve the intraspecific and interspecific study of biological, ecological, and evolutionary issues. Because many stingless bee species are endangered, the application of molecular tools in deciphering the population genomics of the species is crucial for biodiversity conservation. Furthermore, the greater degree of gene rearrangements found in tRNA genes of the T. iridipennis mitochondrial genome underscores the genetic fluidity of the species. Phylogenetic analysis conducted using complete mitogenomes of 15 Hymenopteran species confirmed the phylogenetic position of T. iridipennis as a sister species to T. pagdeni. Furthermore, phylogenetic analysis of gene order could help in resolving the molecular evolutionary genomics of bees and could shed light on the emergence of eusociality among corbiculate bees (Silvestre et al., 2008). It is anticipated that the incorporation of more molecular data and genetic dissection of tRNA gene clusters in the mitogenomes of several species will provide novel insights into the evolution and biogeography of the Meliponini tribe of the Apidae family.
5 Conclusions
In summary, the complete mitochondrial genome of T. iridipennis was sequenced using next-generation sequencing for the first time. The circular mitogenome of T. iridipennis is 15,045 bp in length and contains 34 sequence elements, comprising 13 protein-coding genes, 19 tRNA genes, and two rRNA genes. Phylogenetic analysis revealed that T. iridipennis is closely related to T. page and Lepidotrigona species. The genetic information generated from this study will aid in comparative genomics, molecular systematics, and population genetic studies of many species of the Tetragonula genus in particular and hymenopterans bees in general.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Ethics statement
Ethical review and approval was not required for the study on the insect pollinator, Tetragonula iridipennis, in accordance with the local legislation and institutional requirements. The insect specimens were collected from the onion field by net sweeping, and no specific permissions were required for these locations/activities. The species in our study are common pollinators and did not involve endangered or protected species in India. The ICAR-Directorate of Onion and Garlic Research, Pune, Maharashtra, India, approves the work.
Author contributions
VK and PS conceptualized the study and designed the experiment. VK, AG, and DS carried out the methodology. AG, DS, PM, and SK analyzed and interpreted the data with inputs. VK, AG, DS, PS, and DJ wrote and edited the manuscript and prepared display elements. VM provided resources. All authors contributed to the article and approved the submitted version.
Funding
The research was funded by the Department of Science and Technology (DST) – Science and Engineering Research Board (SERB), New Delhi (CRG/2021/004267).
Acknowledgments
We are grateful to the Department of Science and Technology (DST) – Science and Engineering Research Board (SERB), New Delhi, India for financial grant (CRG/2021/004267) support and the Indian Council of Agricultural Research and Directorate of Onion and Garlic Research (ICAR-DOGR), Pune, Maharashtra, India, for providing lab facilities.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Al-Hatamleh M. A., Boer J. C., Wilson K. L., Plebanski M., Mohamud R., Mustafa M. Z. (2020). Antioxidant-based medicinal properties of stingless bee products: recent progress and future directions. Biomolecules 10, 923. doi: 10.3390/biom10060923
Bernt M., Donath A., Juhling F., Externbrink F., Florentz C., Fritzsch G., et al. (2013). MITOS: improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319. doi: 10.1016/j.ympev.2012.08.023
Bhatta C., Gonzalez V. H., Smith D. (2020). Traditional uses and relative cultural importance of Tetragonula iridipennis (Smith)(Hymenoptera: apidae: meliponini) in Nepal. J. Melittol. 97), 1–13. doi: 10.17161/jom.vi97.13620
Boore J. L. (1999). Animal mitochondrial genomes. Nucleic Acids Res. 27 (8), 1767–1780. doi: 10.1093/nar/27.8.1767
Cameron S. L. (2014). Insect mitochondrial genomics: implications for evolution and phylogeny. Annu. Rev. Entomol. 59, 95–117. doi: 10.1146/annurev-ento-011613-162007
Chen L., Chen P. Y., Xue X. F., Hua H. Q., Li Y. X., Zhang F., et al. (2018). Extensive gene rearrangements in the mitochondrial genomes of two egg parasitoids, Trichogramma japonicum and Trichogramma ostriniae (Hymenoptera: chalcidoidea: trichogrammatidae). Sci. Rep. 8, 1–11. doi: 10.1038/s41598-018-25338-3
Chhakchhuak L., De Mandal S., Gurusubramanian G., Nachimuthu S. K. (2016). The near complete mitochondrial genome of the giant honey bee, Apis dorsata (Hymenoptera: apidae: apinae) and its phylogenetic status. Mitochondr. DNA Part A 27, 3483–3484. doi: 10.3109/19401736.2015.1066359
Clary D. O., Wolstenholme D. R. (1985). The mitochondrial DNA molecule of Drosophila yakuba: nucleotide sequence, gene organization, and genetic code. J. Mol. Evol. 22, 252–271. doi: 10.1007/BF02099755
Cornuet J. M., Garnery L., Solignac M. (1991). Putative origin and function of the intergenic region between COI and COII of Apis mellifera l. mitochondrial DNA. Genetics 128, 393–403. doi: 10.1093/genetics/128.2.393
De Mandal S., Chhakchhuak L., Gurusubramanian G., Kumar N. S. (2014). Mitochondrial markers for identification and phylogenetic studies in insects- a review. DNA Barcodes 2, 1–9. doi: 10.2478/dna-2014-0001
Engel M. S., Kahono S., Peggie D. (2019). A key to the genera and subgenera of stingless bees in Indonesia (Hymenoptera: apidae). Treubia 45, 65–84. doi: 10.14203/treubia.v45i0.3687
Garibaldi L. A., Steffan-Dewenter I., Winfree R., Aizen M. A., Bommarco R., Cunningham S. A., et al. (2013). Wild pollinators enhance fruit set of crops regardless of honey bee abundance. -Science 339, 1608–1611. doi: 10.1126/science.1255213
Grant J. R., Stothard P. (2008). The CGView server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 36, 181–184. doi: 10.1093/nar/gkn179
Gruter C. (2020). STINGLESS BEES: their behaviour, ecology and evolution. (Cham, Switzerland: Springer). doi: 10.1007/978-3-030-60090-7
Gurevich A., Saveliev V., Vyahhi N., Tesler G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Heard T. A. (1999). The role of stingless bees in crop pollination. Annu. Rev. Entomol. 44 (1), 183–206. doi: 10.1146/annurev.ento.44.1.183
Howlader M. S. A., Nair A., Gopalan S. V., Merila J. (2015). A new species of Microhyla (Anura: microhylidae) from nilphamari, Bangladesh. PloS One 10, e0119825. doi: 10.1371/journal.pone.0119825
Lanfear R., Frandsen P. B., Wright A. M., Senfeld T., Calcott B. (2017). Partition finder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 34, 772–773. doi: 10.1093/molbev/msw260
Layek U., Karmakar P. (2018). Nesting characteristics, floral resources, and foraging activity of Trigona iridipennis smith in bankura district of West Bengal, India. Insectes Soc 65, 117–132. doi: 10.1007/s00040-017-0593-4
Li H., Durbin R. (2009). Fast and accurate short read alignment with burrows–wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li D., Liu C. M., Luo R., Sadakane K., Lam T. W. (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de bruijn graph. Bioinformatics 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Li Y., Zhang H., Wu X., Li D., Yan P., Wu X. (2021). The complete mitochondrial genome of Rhacophorus dennysi (Anura: rhacophoridae) with novel gene arrangements and its phylogenetic implications. Pakistan J. Zool. 53, 2013. doi: 10.17582/journal.pjz/20190901010935
Meng G., Li Y., Yang C., Liu S. (2019). MitoZ: a toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 47, e63–e63. doi: 10.1093/nar/gkz173
Nidup T. (2021). Report on the stingless bees of Bhutan (Hymenoptera: apidae: meliponini). J. Threatened Taxa 13 (5), 18344–18348. doi: 10.11609/jott.4504.13.5.18344-18348
Ohler A., Delorme M. (2006). Well known does not mean well studied: morphological and molecular support for existence of sibling species in the Javanese gliding frog Rhacophorus reinwardtii (Amphibia, anura). C. R. Biol. 329, 86–97. doi: 10.1016/j.crvi.2005.11.001
Ojala D., Montoya J., Attardi G. (1981). tRNA punctuation model of RNA processing in human mitochondria. Nature 290, 470–474. doi: 10.1038/290470a0
Pereira U. D. P., Bonetti A. M., Goulart L. R., Santos A. R. D., Oliveira G. C. D., Cuadros-Orellana S., et al. (2016). Complete mitochondrial genome sequence of Melipona scutellaris, a Brazilian stingless bee. Mitochondr. DNA Part A 27, 3387–3388. doi: 10.3109/19401736.2015.1018233
Perna N. T., Kocher T. D. (1995). Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 41, 353–358. doi: 10.1007/BF01215182
Rahman A., Das P. K., Rajkumari P., Saikia J., Sharmah D. (2018). Stingless bees (Hymenoptera: apidae: meliponini): diversity and distribution in India. Apidologie 39, 102–118. https://wgbis.ces.iisc.ac.in/biodiversity/sahyadri_enews/newsletter/issue46/bibliography/51_Stingless%20Bees%20(Hymenoptera.pdf.
Rasmussen C. (2013). Stingless bees (Hymenoptera: apidae: meliponini) of the Indian subcontinent: diversity, taxonomy and current status of knowledge. Zootaxa 3647 (3), 401–428. doi: 10.11646/zootaxa.3647.3.1
Rasmussen C., Thomas J. C., Engel M. S. (2017). A new genus of Eastern hemisphere stingless bees (Hymenoptera: apidae), with a key to the supraspecific groups of indomalayan and Australasian meliponini. Am. Mus. Novit. 2017, 1–33. doi: 10.1206/3888.1
Roubik D. W. (1993). Tropical pollinators in the canopy and understory: field data and theory for stratum “preferences”. J. Insect Behav. 6, 659–673. doi: 10.1007/BF01201668
Shao R., Dowton M., Murrell A., Barker S. C. (2003). Rates of gene rearrangement and nucleotide substitution are correlated in the mitochondrial genomes of insects. Mol. Biol. Evol. 20, 1612–1619. doi: 10.1093/molbev/msg176
Silvestre D., Dowton M., Arias M. C. (2008). The mitochondrial genome of the stingless bee melipona bicolor (Hymenoptera, apidae, meliponini): sequence, gene organization and a unique tRNA translocation event conserved across the tribe meliponini. Gen. Mol. Biol. 31, 451–460. doi: 10.1590/S1415-47572008000300010
Tamura K., Stecher G., Kumar S. (2021). MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027. doi: 10.1093/molbev/msab120
Tan H. W., Liu G. H., Dong X., Lin R. Q., Song H. Q., Huang S. Y., et al. (2011). The complete mitochondrial genome of the Asiatic cavity-nesting honeybee apis cerana (Hymenoptera: apidae). PloS One 6, e23008. doi: 10.1371/journal.pone.0023008
Viraktamath S., Thangjam R. (2021). Two new species of tetragonula (Hymenoptera: apidae: meliponini) from north-East India with notes on their nest structure. Biologia 76 (6), 1691–1704. doi: 10.2478/s11756-020-00662-0
Vossler F. G. (2012). Flower visits, nesting and nest defence behaviour of stingless bees (Apidae: meliponini): suitability of the bee species for meliponiculture in the argentinean chaco region. Apidologie 43, 139–161. doi: 10.1007/s13592-011-0097-6
Wang J. D., Wang C. Y., Zhao M., He Z., Feng Y. (2019). The complete mitochondrial genome of an edible aquatic insect Epophthalmia elegans (Odonata: corduliidae) and phylogenetic analysis. Mitochondr. DNA Part B 4, 1381–1382. doi: 10.1080/23802359.2019.1598303
Wang C. Y., Yang P. L., Zhao M., Xu H. L., Liu L. N., Feng Y., et al. (2022). Unusual mitochondrial tRNA rearrangements in stingless bee Tetragonula pagdeni and phylogenetic analysis. Entomol. Sci. 25, e12526. doi: 10.1111/ens.12526
Wang C. Y., Zhao M., Wang S. J., Xu H. L., Yang Y. M., Liu L. N., et al. (2021). The complete mitochondrial genome of Lepidotrigona flavibasis (Hymenoptera: meliponini) and high gene rearrangement in Lepidotrigona mitogenomes. J. Insect Sci. 21, 10. doi: 10.1093/jisesa/ieab038
Wang C. Y., Zhao M., Xu H. L., Zhang F. L., Zhong Y. H., Feng Y., et al. (2020). Complete mitochondrial genome of the stingless bee Lepidotrigona terminata (Hymenoptera: meliponinae) and phylogenetic analysis. Mitochondr. DNA Part B 5, 752–753. doi: 10.1080/23802359.2020.1715298
Xia X. (1996). Maximizing transcription efficiency causes codon usage bias. Genetics 144, 1309–1320. doi: 10.1093/genetics/144.3.1309
Zhang D., Gao F., Jakovlić I., Zou H., Zhang J., Li W. X., et al. (2020a). PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Res. 20, 348–355. doi: 10.1111/1755-0998.13096
Zhang D. X., Hewitt G. M. (1997). Insect mitochondrial control region: a review of its structure, evolution and usefulness in evolutionary studies. Biochem. Syst. Ecol. 25, 99–120. doi: 10.1016/S0305-1978(96)00042-7
Zhang J., Kan X., Miao G., Hu S., Sun Q., Tian W. (2020b). qMGR: a new approach for quantifying mitochondrial genome rearrangement. Mitochondrion 52, 20–23. doi: 10.1016/j.mito.2020.02.004
Zhang J. Y., Zhang L. P., Yu D. N., Storey K. B., Zheng R. Q. (2018). Complete mitochondrial genomes of Nanorana taihangnica and N. yunnanensis (Anura: dicroglossidae) with novel gene arrangements and phylogenetic relationship of dicroglossidae. BMC Evol. Biol. 18, 1–13. doi: 10.1186/s12862-018-1140-2
Keywords: Indian stingless bee, Meliponini, mitogenome, next-generation sequencing, non-apis, phylogeny, systematics
Citation: Karuppaiah V, Gadge AS, Shirsat DV, Soumia PS, Mainkar P, Kumar S, Jaiswal DK and Mahajan V (2023) The complete mitochondrial genome of the Indian dammer bee, Tetragonula iridipennis, and the phylogenomics of Meliponini. Front. Ecol. Evol. 11:1171242. doi: 10.3389/fevo.2023.1171242
Received: 21 February 2023; Accepted: 07 June 2023;
Published: 22 June 2023.
Edited by:
Cristina Grande, Autonomous University of Madrid, SpainCopyright © 2023 Karuppaiah, Gadge, Shirsat, Soumia, Mainkar, Kumar, Jaiswal and Mahajan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vadivelu Karuppaiah, a2FydXBwYWlhaHYyMDA4QGdtYWlsLmNvbQ==; Vijay Mahajan, dmlqYXkubWFoYWphbkBpY2FyLmdvdi5pbg==