 Agnès Vallier†Elisa Dell’Aglio†Mariana Galvão FerrariniOphélie HurtadoCarole Vincent-MonégatAbdelaziz HeddiRita Rebollo*Anna Zaidman-Rémy*
Agnès Vallier†Elisa Dell’Aglio†Mariana Galvão FerrariniOphélie HurtadoCarole Vincent-MonégatAbdelaziz HeddiRita Rebollo*Anna Zaidman-Rémy*- Université de Lyon, INRAE, INSA Lyon, BF2I, UMR 203, Villeurbanne, France
Reference genes are a fundamental tool for analyses of gene expression by real-time quantitative PCR (qRT-PCR), in that they ensure the correct comparison between conditions, stages, or treatments. Because of this, selection of appropriate genes to use as references is crucial for proper application of the technique. Nevertheless, efforts to find appropriate, stably expressed transcripts are still lacking, in particular in the field of insect science. Here, we took advantage of a massive transcriptomic high-throughput analysis of various developmental stages of the gut and associated-bacteriomes of the cereal weevil Sitophilus oryzae and identified a subset of stably expressed genes with the potential to be used as housekeeping genes from the larva to the adult stage. We employed several normalization techniques to select the most suitable genes among our subset. Our final selection includes two genes–TAO, and YTH3–which can also be used to compare transcript abundance at various developmental stages in symbiotic insects, and in insects devoid of endosymbionts (aposymbiotic). Since they are well conserved, these genes have the potential to be useful for many other insect species. This work confirms the interest in using large-scale, unbiased methods for reference gene selection.
1. Introduction
Although high-throughput transcriptomic analyses are becoming more affordable and accessible, analysis of a subset of transcripts with quantitative real-time PCR (qRT-PCR) (Higuchi et al., 1992) is still routinely required in laboratory practice, exploratory experiments, experimental validations as well as diagnostics protocols (Kubista et al., 2006). The instrumentation for qRT-PCR is common in molecular biology facilities, along with established guidelines [MIQE guidelines (Bustin et al., 2009)], making it a highly affordable and reliable technique. However, the qRT-PCR accuracy relies on the use of a control method for data normalization that is often achieved by comparing the expression level of target genes against stably expressed transcripts from so-called reference genes, housekeeping genes or constitutively expressed genes. These transcripts must be stably expressed in the biological samples and experimental conditions that are tested, in order to be used as a comparison to evaluate changes in the expression of the transcripts of interest.
Hence, an appropriate set of qRT-PCR reference genes must be established for each experimental condition, species and tissue examined (Bustin et al., 2009). However, historically, only a small group of highly stably expressed genes has been used and tested in various conditions and species. A review summarizing recent work on reference genes in 78 insect species from 2008 to 2017 has shown that the majority of reports focused on the same gene candidates, including Actin, Tubulin, Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and the ribosomal gene 18S (Lü et al., 2018). All these genes displayed great stability and high expression levels in various experimental conditions, tissues and species, but none appeared to be a universal “passe-partout,” as highlighted now that transcriptomic analyses are routinely employed in different tissues and at different developmental stages. For example, GAPDH (Dveksler et al., 1992) often shows stable expression between insect tissues and treatments but is not often adapted for comparisons between developmental stages (Fu et al., 2013; Pan et al., 2015).
Since the production of transcriptomic data is becoming more and more affordable, it is now possible to choose the best set of reference genes for each species, condition, and treatment, by comparing whole transcriptomes instead of limiting the choice to a set of potential candidates. Examples of this non-aprioristic process are already available for various organisms, including plants (Yim et al., 2015; Liang et al., 2020; Lopes et al., 2021), mammals (Zhang et al., 2020), and human tissues (Caracausi et al., 2017), but not yet for insects, notably, Coleoptera. Around 400 thousand Coleoptera species have been described to date, including terrestrial and aquatic species (Zhang, 2013; Zhang et al., 2018). True weevils, or Curculionidae, are one of the largest animal families and include major agronomic pests, such as the cereal weevil (Sitophilus spp.), which is responsible for important damage to fields and stored grains (Longstaff, 1981).
As most insects thriving on a nutritionally unbalanced diet, S. oryzae shares a symbiotic relationship with an intracellular Gram-negative bacterium called Sodalis pierantonius (Mansour, 1930; Heddi et al., 1999; Oakeson et al., 2014). S. pierantonius is housed within specific host cells, the bacteriocytes, forming a unique bacteriome organ attached to the gut during all larval stages (Pierantoni, 1927; Mansour, 1930). Throughout S. oryzae’s metamorphosis, the bacteriocytes migrate along the midgut and endosymbionts subsequently infect cells in the apexes of gut caeca (Maire et al., 2020). These coordinated changes result in the formation of multiple bacteriomes along the midgut by the completion of metamorphosis. In these bacteriomes, endosymbionts exponentially increase in number in the emerging adult, before being recycled by an apoptotic-autophagic mechanism following the first week of adult life (Vigneron et al., 2014). Analyzing transcriptomic regulations across weevil’s development is required to fully understand S. oryzae and S. pierantonius relationship, and to uncover key pathways profitable for integrative pest management strategies. We have recently conducted a thorough analysis of the gut (and associated bacteriomes) transcriptome of S. oryzae across its development from larval stages up to bacterial clearance in adults (Figure 1; Ferrarini et al., 2023). In this work, we took advantage of the recent transcriptomic profile of S. oryzae during development (Ferrarini et al., 2023) to select a set of reliable reference genes for qRT-PCR analyses, with high stability and conservation among insects. The identified transcripts will be of use for the comparison of transcript abundance across multiple developmental stages in S. oryzae, gene silencing conditions, and comparisons with artificially obtained aposymbiotic weevils. These genes also have a great potential for being suitable reference transcripts in other insects, as well as in many other eukaryotes.
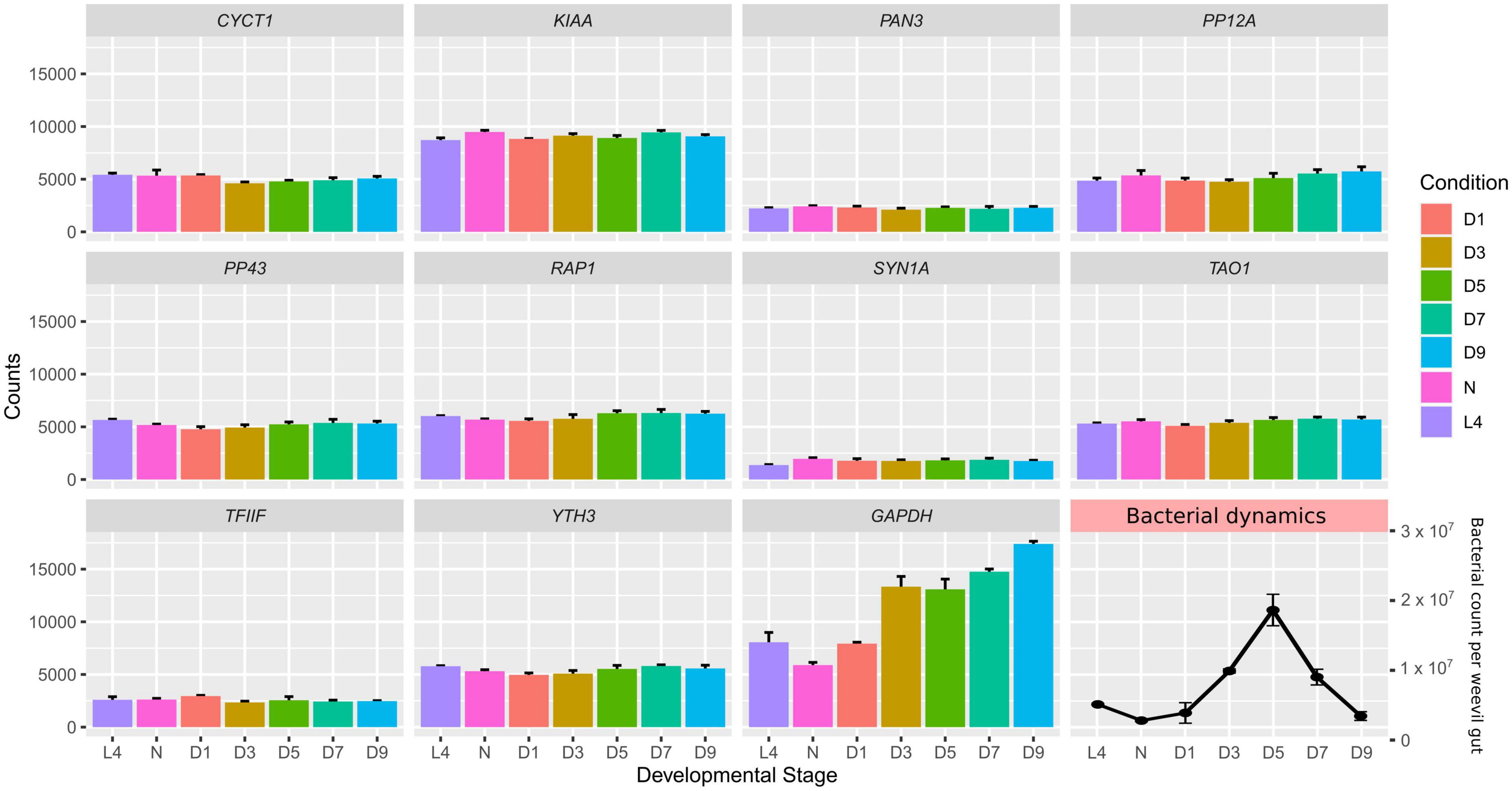
Figure 1. Expression profile of all candidate housekeeping genes in Sitophilus oryzae gut-bacteriome samples analyzed by transcriptomics at various developmental stages. L4: larval stage 4; P: pupae; D1: 1-day-old adults; D3: 3-day-old adults; D5: 5-day-old adults; D7: 7-day-old adults; D9: 9-day-old adults. Data represent normalized counts at each developmental stage (Ferrarini et al., 2023). Error bars represent standard deviation. Bacterial dynamics is represented in the bottom right corner, adapted from Dell’Aglio et al. (2023).
2. Materials and methods
2.1. Insect rearing and sampling
The symbiotic S. oryzae population was sampled in Azergues valley, France in 1984, and has been reared in laboratory conditions ever since. The aposymbiotic strain was obtained by heat treatment in 2010, following a protocol described previously (Nardon, 1973). S. oryzae symbiotic and artificially obtained aposymbiotic weevils were reared in a climate chamber (27.5°C, 70% relative humidity, no light source) on wheat grains as a food source and substrate for egg laying. In these conditions, the timespan between egg laying and the emergence of adults from the grain is 1 month for symbiotic weevils, and 5 weeks for aposymbiotic ones. Adults emerging from the grain are fully formed, although their cuticle is reinforced during the 5 days following emergence from the grain, together with sexual maturation (Maire et al., 2019). The time between the end of metamorphosis and emergence has been calculated to be around 3 days (Vigneron et al., 2014). Since our studies are mainly focused on S. oryzae transcript levels in the gut, from larval stages to the symbiotic clearance (around day 9 of adulthood), we tested candidate reference genes on gut samples (which contains the bacteriomes) harvested from the last larval stage (L4) to day 9 of adulthood (D9), including pupae (P), and intermediate adult stages (D1, D3, D5, and D7).
For sampling, weevils were immersed in buffer TA (35 mM Tris/HCl, 25 mM KCl, 10 mM MgCl2, 250 mM sucrose, pH 7.5) and dissected under a stereomicroscope. Three biological samples per time point were collected, each composed of five guts and their bacteriomes. All samples were collected on ice in 1.5 mL RNAse-free tubes and stored at −80°C until use.
2.2. Transcriptomic analysis
RNAseq datasets were processed in our previous publication (Ferrarini et al., 2023), and raw reads are available at BioProject PRJNA918957. Briefly, quality check was performed with FastQC v0.11.8 (Andrews, 2010), and raw reads were quality trimmed using trim_galore from Cutadapt v0.6.7 (Martin, 2011), then mapped to the S. oryzae’s genome (GCA_002938485.2 Soryzae_2.0) using STAR v2.7.3a (Dobin et al., 2013). Then, uniquely mapping reads were counted with featureCounts v2.0.1 (Liao et al., 2014), and mapping quality was assessed by multiqc v1.13 (Ewels et al., 2016). Differential expression analysis was conducted with SarTOOLS, taking advantage of a global linear model from Deseq2 (Love et al., 2014, 2), and log2 fold changes and adjusted p-values were obtained for sequential pairwise comparisons, i.e., L4 vs. D1, D1 vs. D3 etc., and can be found in the Supplementary material of our previous report (Ferrarini et al., 2023). Most graphic outputs are either performed in R, using ggplot2 (Wickham, 2009) or with GraphPad Prism software.
2.3. Primer design for candidate reference genes
Primers for each candidate reference gene and GAPDH were designed with Primer3 software (Kõressaar et al., 2018, 3), with “best annealing” at 55°C, amplicon length between 70 and 120 nucleotides (Table 2). Forward and reverse primers were always located in two different exons in order to span at least one intron. In the case of multiple predicted isoforms for each transcript, the primers were designed so that they could amplify all of them. Primer sequences were checked by BLAST (Camacho et al., 2009) to ensure their specificity in the target genome.
2.4. RNA extraction and transcript amplification
Total RNA was purified from samples using the RNAqueous Micro kit (Ambion) and its DNAse treatment, following the manufacturer’s instructions. Final RNA concentration was measured with a Nanodrop® spectrophotometer (Thermo Scientific), and RNA quality was checked using agarose gel electrophoresis. Reverse transcription to cDNA was carried out using the iScript cDNA Synthesis Kit (Bio-Rad), which relies on a blend of oligo(dT) and random primers. Reverse transcription was performed starting from 1 μg of total RNA. Transcript amplification by qRT-PCR was performed with a CFX Connect Real-Time detection system (Bio-Rad) using the LightCycler Fast Start DNA Master SYBR Green I kit (Roche Diagnostics) as previously described (Maire et al., 2019). Each reaction contained 5 μl of Master Sybr Green mix, 0.5 μl of each primer (10 μM), 1.5 μl RNAse Free water, and 2.5 μl of 1:5 diluted cDNA. Primers for amplification are listed in Table 2. After 5 min at 95°C, the cycling conditions were: 45 cycles at 95°C for 10 s, 56°C for 20 s, and 72°C for 30 s. A melting curve was obtained at the end of each PCR by heating for 30 s at 66°C, then increasing the temperature up to 95°C (increment rates of 0.11°C/s). For every candidate gene, three biological and two technical replicates were tested for each developmental stage, together with a standard curve made with PCR products as templates, at concentrations spanning from 0.2 fg/μl to 2 pg/μl of amplicon, to calculate the reaction efficiency and ensure linearity of the amplification. The data from each transcript were collected within the same plate.
2.5. Candidate reference gene comparisons
To evaluate the reliability of each candidate reference gene, we compared the average cycle threshold (Ct) of each developmental stage and the percentage covariance (CV%) value for each gene (Boda et al., 2009; Sundaram et al., 2019). We then compared the rankings of various web-based algorithms: GeNorm (Vandesompele et al., 2002), the ΔCt method (Silver et al., 2006), BestKeeper (Pfaffl et al., 2004), and Reffinder (Xie et al., 2012). Each candidate reference gene was compared, ranked and evaluated through the tools described.
3. Results and discussion
3.1. Choice of candidate reference genes based on transcriptome data
To find S. oryzae reference genes to be used across gut and bacteriomes development (from larval stage 4 to 9-day-old adults), we scanned a comprehensive transcriptomic dataset spanning S. oryzae life cycle (Ferrarini et al., 2023). Candidate reference genes should be stable between conditions, highly expressed, and present transcript isoforms suitable for primer design. There are 17 957 genes in S. oryzae (Parisot et al., 2021) and our previous analysis showed that 11 908 were differentially expressed between sequential conditions (Ferrarini et al., 2023). In order to find stable genes across development, we removed genes presenting an adjusted p-value < 0.05 between time points (∼1 500 remaining genes), along with a significant log2 fold change lower than −1 or higher than 1 between two consecutive stages, yielding 100 candidate genes. A selection was made by prioritizing highly expressed genes (mean expression level higher than 1000), in order to improve qRT-PCR detection, decreasing the candidate list to only 19 genes. Among the resulting pre-selected 19 genes, we visually selected those with the most steady expression along the development. We eliminated genes encoding regulators, which are more likely to change in expression according to the experimental conditions. Among the remaining genes, we avoided uncharacterized genes, abundant in the S. oryzae genome. These filtering steps resulted in selecting nine potential reference genes for experimental validation (Table 1 and Figure 1). The selected genes spanned various predicted biological functions, to reduce the risk of co-regulation among them and to facilitate the future use of the same genes in other organisms.
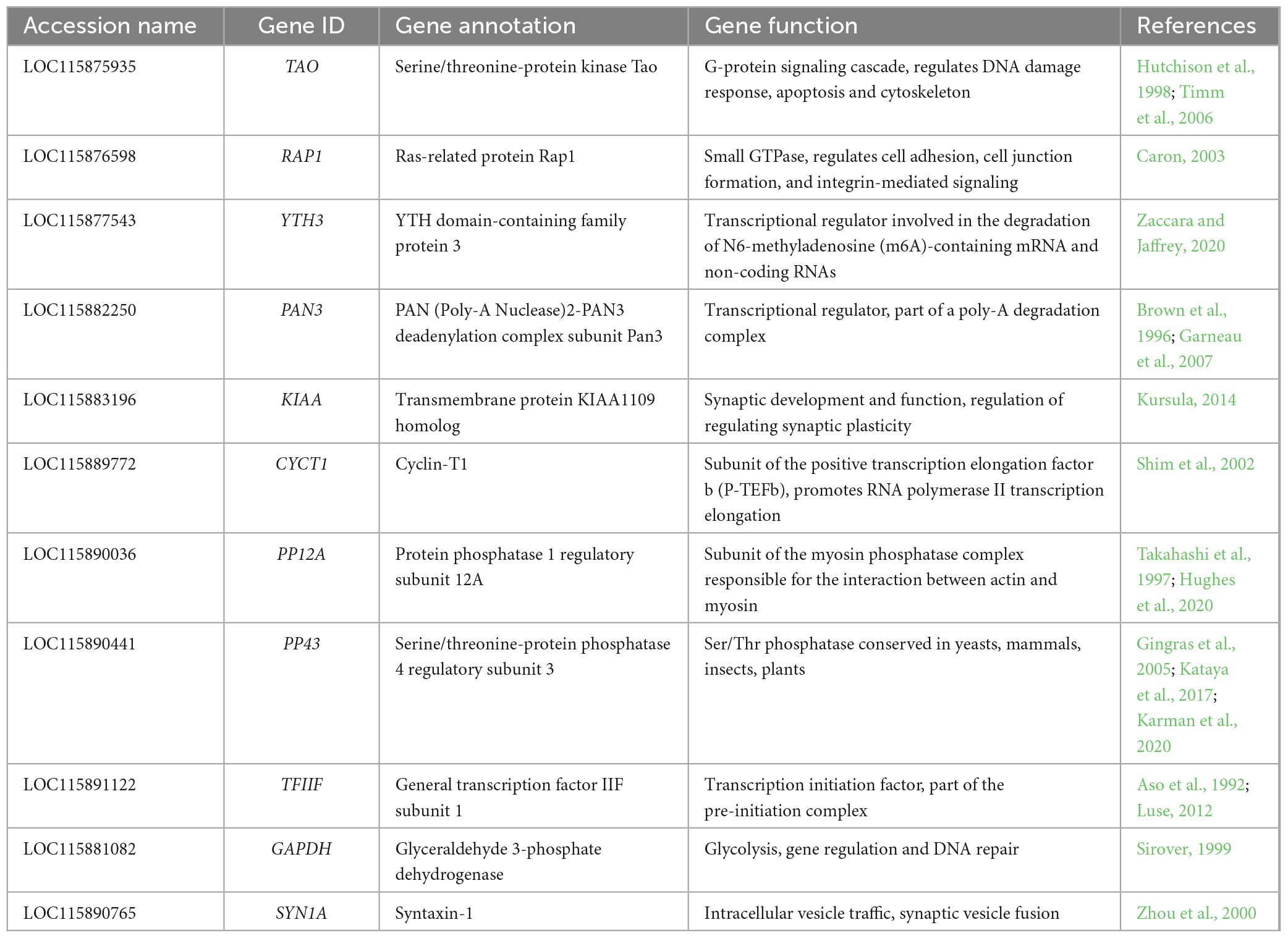
Table 1. candidate reference genes.
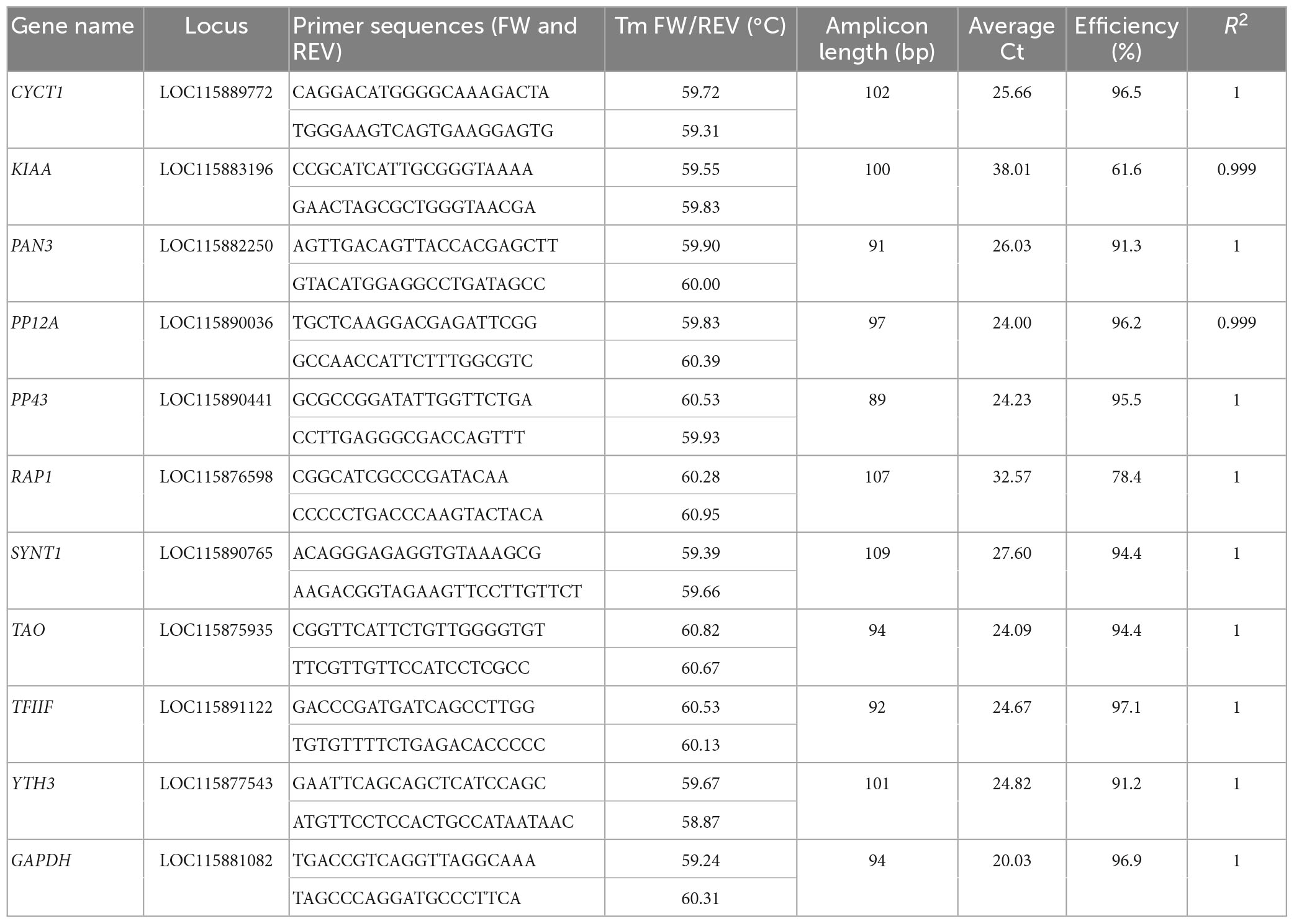
Table 2. Primers and amplification parameters used for each candidate reference gene selected for qRT-PCR normalization.
Finally, we also included GAPDH, a very common reference gene in our model and similar model organisms, and the homolog of syntaxin-1 (SYN1A), a protein involved in intracellular vesicle traffic, especially in neurons (Zhou et al., 2000), previously selected as a suitable reference gene by Lord et al. (2010) for Tribolium castaneum, the most widely studied member of Coleoptera. These genes were included in the study as a point of comparison for the newly selected genes. Both genes showed little variation between time points (Figure 1). Altogether, a set of 11 candidate genes were then submitted for further analysis.
3.2. Expression profiles of candidate reference genes
While the transcriptomic analysis suggests the candidate genes are stable across S. oryzae’s gut development, it is important to verify that it is indeed possible to amplify such transcripts in an accurate manner by qRT-PCR. A preliminary PCR test allowed amplification of all predicted transcripts from gut cDNA pools, obtaining fragments compatible with the predicted amplicon sizes. Subsequent qRT-PCR quantification from gut samples (larval stage L4, pupal stage P, and adult stages D1, D3, D5, D7, and D9) was successful for nine out of eleven genes, leading to acceptable efficiency and linearity (Table 2). Two exceptions were the primers for KIAA and RAP1, which did not lead to acceptable fragment amplification under standard conditions (efficiency values of 61.6 and 78.4, respectively; average Ct values of 38.01 and 32.58, respectively); and were therefore excluded from the list of potential candidates (Table 2). Moreover, although successful, YTH3 expression levels appear lower than expected based on the transcriptomic analysis. Discrepancies between amplification in qRT-PCR and normalized read counts could be due to low primer efficiency, a tendency to primer dimerization, or suboptimal amplification of transcripts in our standardized qRT-PCR protocol, as well as differences in normalization strategies between the two techniques. One should also note that for qRT-PCR, larval samples consisted of bacteriomes and whole guts, while in the previous transcriptomic dataset on which the candidate reference gene selection was based, only larval bacteriomes were dissected (Ferrarini et al., 2023).
The mean Ct values of amplicons for each candidate reference gene at each developmental stage are reported in Figure 2. The gene with the lowest average Ct was GAPDH (mean for all stages: 20.0), followed by PP12A (mean for all stages: 23.99) and PP43 (mean for all stages: 24.66). All average Ct values are considered low (GAPDH) or medium (all other genes) and are therefore suitable for estimating gene expression of various target genes with different expression profiles. Statistical analysis of Ct values revealed that the mean expression level for the majority of the genes is subjected to mild variations between developmental stages (see asterisks in Figure 2), except for GAPDH and SYN1A.
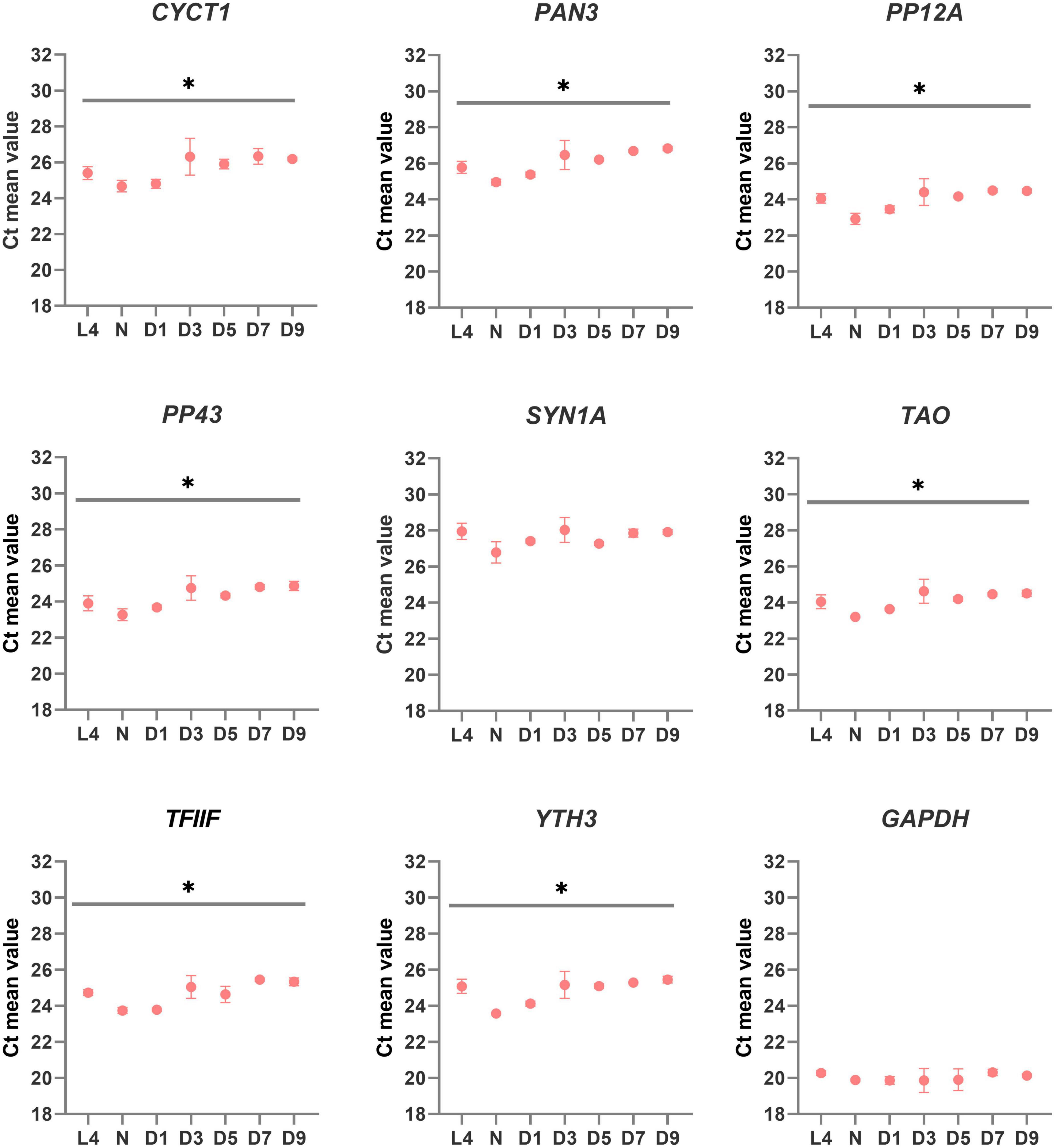
Figure 2. Quantitative real-time PCR (qRT-PCR) expression profile of candidate reference genes in Sitophilus oryzae gut-bacteriome samples. Graphs represent the mean Ct value for each developmental stage. One-way ANOVA was performed to assess differences between the means of all developmental stages. Asterisks denote statistical significance (ANOVA with Kruskal-Wallis test, *p ≤ 0.05). Error bars represent standard deviation.
3.3. Evaluation of expression stability and ranking of candidate reference genes
To identify the most suitable reference gene to compare transcript expression across S. oryzae development, we relied on the comparison of statistical methods recently conducted by Sundaram et al. (2019). After describing the merits and limitations of various comparison algorithms, the authors suggested the following as the best pipeline for the identification of reliable reference genes: (i) visual estimation of expression variation across stages, (ii) analysis of CV% values [(Boda et al., 2009); a cut-off of 50 is recommended]; and (iii) ranking of the remaining candidate reference genes by the NormFinder algorithm (Andersen et al., 2004).
Since the visual estimation of expression variation across stages was already performed and showed mild variation for most genes (Figure 2), we calculated the CV% value for each candidate reference gene. The CV% values of four out of nine genes (YTH3, CYCT1, PAN3, and TFIIF) across all developmental stages are indeed higher than 50 (Figure 3A), reflecting variations observed in particular at the pupal stage (Figure 2). When only adult stages were considered (Figure 3B), only two genes still displayed CV% values higher than 50 (CYCT1 and TFIIF). After excluding genes with CV% higher than 50, we ran the NormFinder algorithm together with other common reference gene ranking algorithms: GeNorm (Vandesompele et al., 2002), the ΔCt method (Silver et al., 2006), BestKeeper (Pfaffl et al., 2004), and Reffinder (Xie et al., 2012). To note, all ranking systems used here are relative to the set of genes that have been chosen and input in the system. Hence, the important information here is the rank that the genes obtain (i.e., their classification), relative to the others. Including the GAPDH and SYN1A genes that are routinely used as “standard” reference genes ensure a point of comparison with the candidate genes we found in our process. Rankings according to all prediction tools are reported in Table 3 for all stages, and in Table 4 for adult stages only. Overall, each of these methods provided good stability values for all the preselected candidate genes, with a consensus for TAO and SYN1A at all stages and for TAO, PP12A, and YTH3 in adult stages only. GAPDH was often ranked last among these set of candidate reference genes.
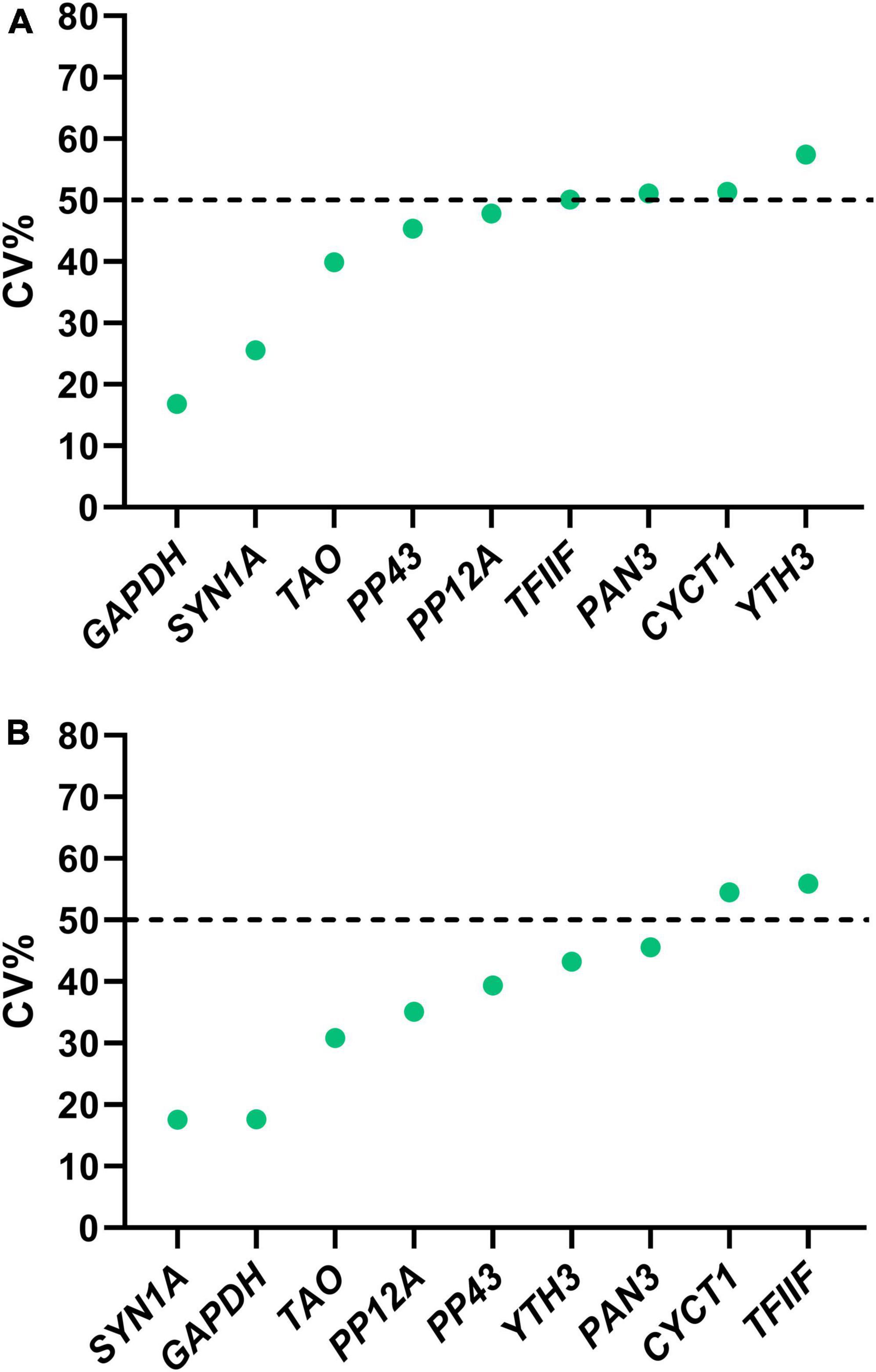
Figure 3. CV% values for the selected candidate reference genes, calculated on the linearized 2-Ct converted Ct values at all Sitophilus oryzae developmental stages considered in Figure 2. (A) CV% measured considering all developmental stages. (B) CV% calculated considering only adult stages. Only genes with a CV% lower than 50 were selected for further analysis.
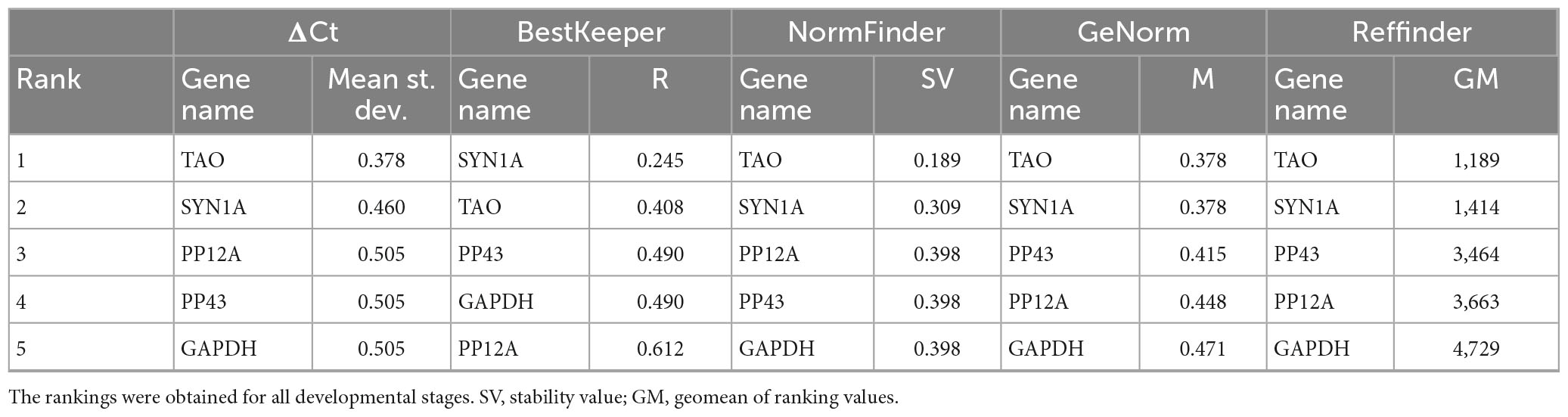
Table 3. Ranking of candidate reference genes for various Sitophilus oryzae developmental stages, according to the ΔCt method, BestKeeper, NormFinder, GeNorm, and Reffinder.
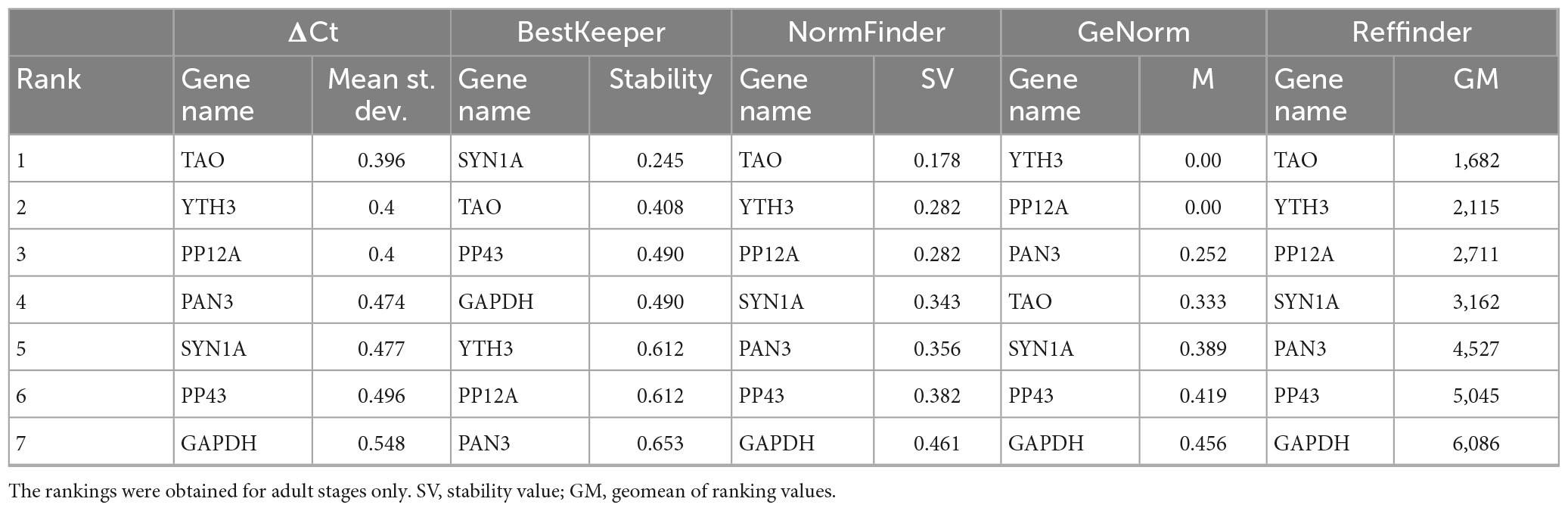
Table 4. Ranking of candidate reference genes for various Sitophilus oryzae developmental stages, according to the ΔCt method, BestKeeper, NormFinder, GeNorm, and Reffinder.
3.4. Determination of the minimum number of reference genes for qPCR normalization
The pairwise variation value (V) between the ranked genes was calculated with the geNorm algorithm and used to estimate the stability of the normalization factor with the addition of one normalization gene at a time. The cut-off value for pairwise variation used was 0.15 (Vandesompele et al., 2002), meaning that as soon as the value of V drops below 0.15 the number of reference genes was considered suitable for correct standardization of gene expression. The V value for the set of candidate reference genes was always lower than 0.15 (Figure 4). This indicates that two reference genes, which is the minimal number of reference genes accepted by the MIQE guidelines (Bustin et al., 2009), are sufficient for standardization of gene expression across S. oryzae development in the gut, from the L4 larval stages to D9 adults. It also shows that any combination of the proposed nine reference genes is suitable for normalization.
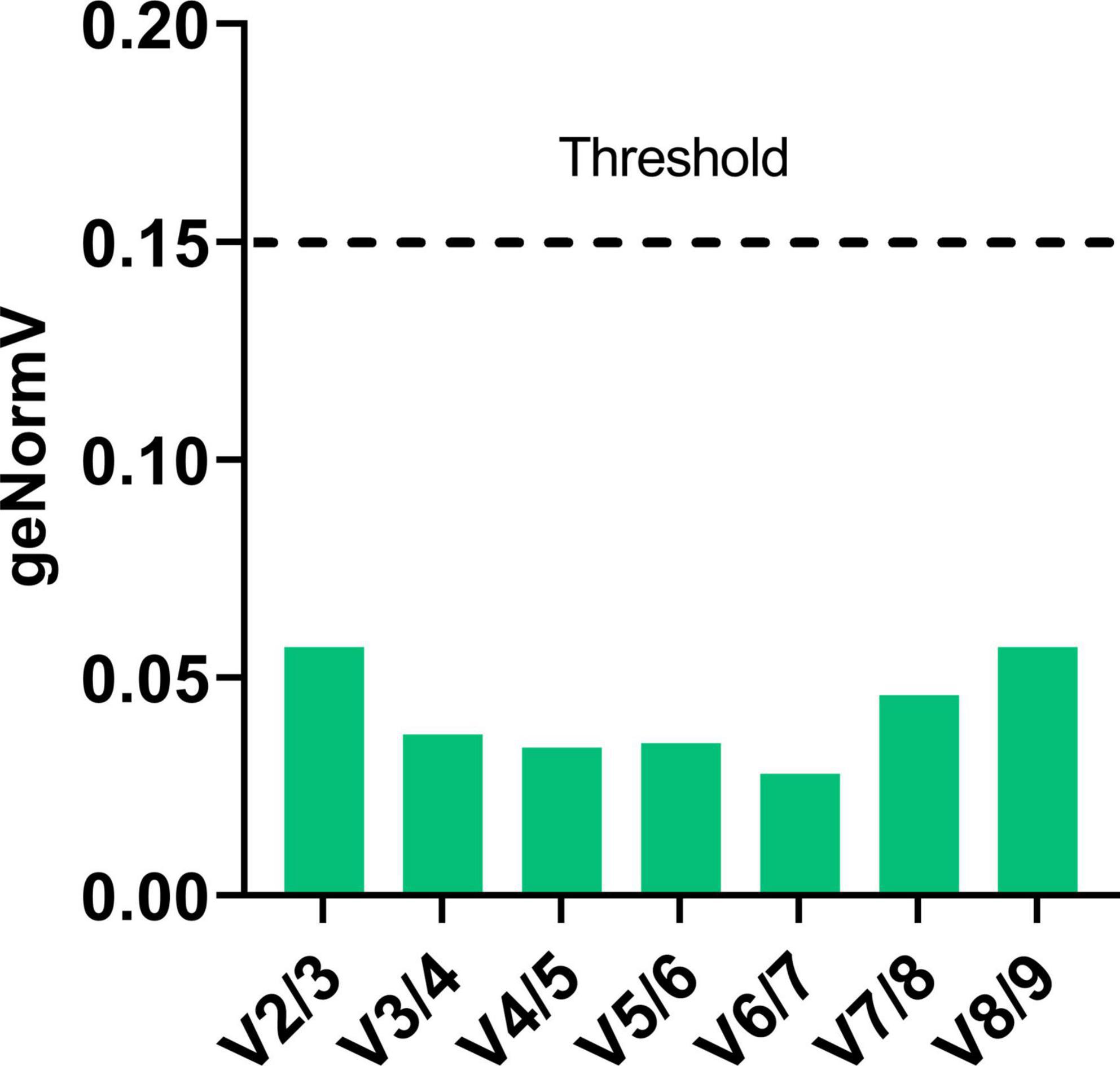
Figure 4. Optimal number of reference genes for normalization in S. oryzae at various developmental stages. The pairwise variation values obtained from GeNorm analyses were used to determine the minimum number of reference genes for normalization. The value of Vn/Vn + 1 < 0.15 indicates that the minimum number of reference genes to be used for qPCR data normalization is n.
3.5. Assessment of candidate reference genes for transcript comparisons between symbiotic and aposymbiotic insects
Weevils artificially deprived of symbionts by heat treatment (aposymbiotic) do survive under laboratory standard conditions, although their fitness is partially compromised, e.g., the larval development takes longer, fertility is lower (Nardon, 1973), and aposymbiotic adults are unable to fly (Nardon et al., 1998). In aposymbiotic weevils, the insect cuticle, which is derived from aromatic amino acids mainly supplied by the bacteria, does not fully develop but remains thinner and more delicate (Vigneron et al., 2014). In order to pinpoint symbiosis-specific transcriptional changes, the transcriptomic profiles obtained for symbiotic weevils (Ferrarini et al., 2023) should be compared with the expression profiles obtained at the same stage in aposymbiotic weevils. We took advantage of existing transcriptomic datasets produced from adult symbiotic and aposymbiotic weevils at different adult stages, to compare the expression of all candidate genes between symbiotic stages (Figure 5; Ferrarini et al., 2023). Differential expression analysis of all nine candidate genes between symbiotic and aposymbiotic weevils shows only three genes are equally expressed between symbiotic conditions: PAN3, TAO, and YTH3. Two of these genes were chosen as suitable candidates for symbiotic analysis, TAO and YTH3 and should constitute a good set for symbiotic/aposymbiotic comparisons.
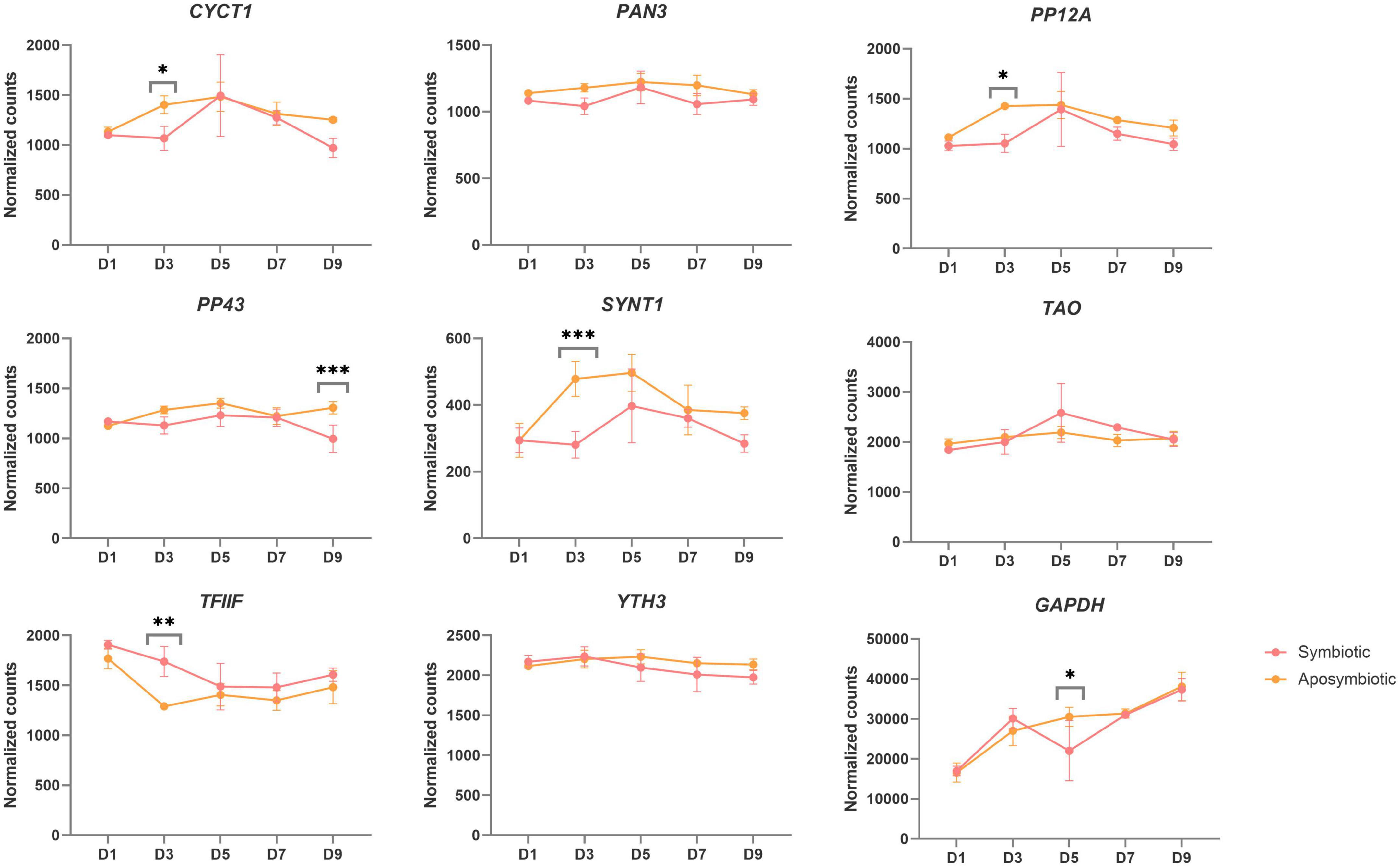
Figure 5. Normalized counts of selected candidate reference genes from previous RNAseq data obtained from symbiotic and aposymbiotic weevils (Ferrarini et al., 2023). Stars denote the adjusted p-value between symbiotic and aposymbiotic weevils at a given time point, upon DESEQ2 analysis (see section “2. Materials and methods”). *Adjpvalue < 0.05, ** < 0.005, *** < 0.0005. Three genes show no differences between symbiotic and aposymbiotic weevils at any given time point and constitute good candidates for symbiotic/aposymbiotic qRT-PCR comparisons: pan3, tao, and yth3.
4. Conclusion
In this work, we relied on transcriptomic data to select a set of nine new candidate reference genes for S. oryzae, which we compared with GAPDH, a very common reference gene (Bustin, 2000), and SYN1A, which has been previously used as a reference gene for T. castaneum (Lord et al., 2010). By combining differential expression data filtering, visual inspection, CV% analysis and a variety of gene stability ranking algorithms, we were able to select stable gut transcripts in S. oryzae adult stages, but also for stages spanning from larvae to adult, including metamorphosis. In particular, all the preselected candidate genes scored well in all ranking systems, with a general preference for TAO and SYN1A when all developmental stages (from larval stage 4 to 9-day-old adults) were considered, and for TAO, PP12A, and YTH3 in adult stages only. Interestingly, for all the various algorithms used to rank the reliability of reference genes, the newly selected genes were always more suitable than the highly popular GAPDH, although the latter ranked better in terms of CV% values.
Our selection approach was guided by the stability of expression levels of various transcripts and their abundance in transcriptomic data (Ferrarini et al., 2023), and on the presumed housekeeping function of each gene on the basis of the available literature and sequence alignments with homologs from other species. Indeed, all the candidates that were successfully quantified by qPCR, and in particular TAO and YTH3, which were chosen for comparing symbiotic and aposymbiotic weevils, have been previously associated with important cell functions, such as DNA repair and RNA transcription, suggesting they are likely good candidates as reference genes for many other tissues and organisms (Table 1).
In conclusion, we identified stable reference genes across S. oryzae development with potential application in various other animal species. This work also demonstrates the potential of transcriptomic data as a guide for reference gene selection to obtain comparable or better performances in comparison to traditional ones–a strategy still neglected in insect studies.
Data availability statement
The original contributions presented in this study are included in the article, further inquiries can be directed to the corresponding authors.
Author contributions
AZ-R and RR conceived the study with the help from AH. MF performed the gene selection from high throughput transcriptomic data, with inputs from CV-M, AV, ED, AH, and AZ-R. AV, ED, and OH produced and treated the gene expression data. AV, ED, and RR performed the statistical analysis. AZ-R, RR, AV, AH, and ED wrote the manuscript. All authors read and approved the manuscript.
Funding
This work was funded by the ANR UNLEASH (ANR UNLEASH-CE20-0015-01–RR) and ANR GREEN (ANR-17-CE20-0031-01–AH).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Andersen, C. L., Jensen, J. L., and Ørntoft, T. F. (2004). Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 64, 5245–5250. doi: 10.1158/0008-5472.CAN-04-0496
Andrews, S. (2010). FastQC A Quality Control tool for High Throughput Sequence Data. Available online at: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed December 23, 2022).
Aso, T., Vasavada, H. A., Kawaguchi, T., Germino, F. J., Ganguly, S., Kitajima, S., et al. (1992). Characterization of cDNA for the large subunit of the transcription initiation factor TFIIF. Nature 355, 461–464. doi: 10.1038/355461a0
Boda, E., Pini, A., Hoxha, E., Parolisi, R., and Tempia, F. (2009). Selection of reference genes for quantitative real-time RT-PCR studies in mouse brain. J. Mol. Neurosci. MN 37, 238–253. doi: 10.1007/s12031-008-9128-9
Brown, C. E., Tarun, S. Z., Boeck, R., and Sachs, A. B. (1996). PAN3 encodes a subunit of the Pab1p-dependent poly(A) nuclease in Saccharomyces cerevisiae. Mol. Cell. Biol. 16, 5744–5753. doi: 10.1128/MCB.16.10.5744
Bustin, S. A. (2000). Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J. Mol. Endocrinol. 25, 169–193. doi: 10.1677/jme.0.0250169
Bustin, S. A., Benes, V., Garson, J. A., Hellemans, J., Huggett, J., Kubista, M., et al. (2009). The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55, 611–622. doi: 10.1373/clinchem.2008.112797
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Caracausi, M., Piovesan, A., Antonaros, F., Strippoli, P., Vitale, L., and Pelleri, M. C. (2017). Systematic identification of human housekeeping genes possibly useful as references in gene expression studies. Mol. Med. Rep. 16, 2397–2410. doi: 10.3892/mmr.2017.6944
Caron, E. (2003). Cellular functions of the Rap1 GTP-binding protein: a pattern emerges. J Cell Sci. 116(Pt 21), 435–440. doi: 10.1242/jcs.00707
Dell’Aglio, E., Lacotte, V., Peignier, S., Rahioui, I., Benzaoui, F., Vallier, A., et al. (2023). Weevil carbohydrate intake triggers endosymbiont proliferation: a trade-off between host benefit and endosymbiont burden. mBio 1–14. doi: 10.1128/mbio.03333-22
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. doi: 10.1093/bioinformatics/bts635
Dveksler, G. S., Basile, A. A., and Dieffenbach, C. W. (1992). Analysis of gene expression: use of oligonucleotide primers for glyceraldehyde-3-phosphate dehydrogenase. PCR Methods Appl. 1, 283–285. doi: 10.1101/gr.1.4.283
Ewels, P., Magnusson, M., Lundin, S., and Käller, M. (2016). MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048. doi: 10.1093/bioinformatics/btw354
Ferrarini, M. G., Vallier, A., Vincent-Monégat, C., Dell’Aglio, E., Gillet, B., Hughes, S., et al. (2023). Coordinated host and endosymbiont gene expression governs endosymbiont growth and elimination in the cereal weevil Sitophilus spp. bioRxiv [Preprint]. doi: 10.1101/2023.04.03.535335
Fu, W., Xie, W., Zhang, Z., Wang, S., Wu, Q., Liu, Y., et al. (2013). Exploring valid reference genes for quantitative real-time PCR analysis in Plutella xylostella (Lepidoptera: Plutellidae). Int. J. Biol. Sci. 9, 792–802. doi: 10.7150/ijbs.5862
Garneau, N. L., Wilusz, J., and Wilusz, C. J. (2007). The highways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol. 8, 113–126. doi: 10.1038/nrm2104
Gingras, A.-C., Caballero, M., Zarske, M., Sanchez, A., Hazbun, T. R., Fields, S., et al. (2005). A novel, evolutionarily conserved protein phosphatase complex involved in cisplatin sensitivity. Mol. Cell. Proteomics MCP 4, 1725–1740. doi: 10.1074/mcp.M500231-MCP200
Heddi, A., Grenier, A.-M., Khatchadourian, C., Charles, H., and Nardon, P. (1999). Four intracellular genomes direct weevil biology: nuclear, mitochondrial, principal endosymbiont, and Wolbachia. Proc. Natl. Acad. Sci. U.S.A. 96, 6814–6819. doi: 10.1073/pnas.96.12.6814
Higuchi, R., Dollinger, G., Walsh, P. S., and Griffith, R. (1992). Simultaneous amplification and detection of specific DNA sequences. Biotechnol. Nat. Publ. Co. 10, 413–417. doi: 10.1038/nbt0492-413
Hughes, J. J., Alkhunaizi, E., Kruszka, P., Pyle, L. C., Grange, D. K., Berger, S. I., et al. (2020). Loss-of-function variants in PPP1R12A: from isolated sex reversal to Holoprosencephaly spectrum and urogenital malformations. Am. J. Hum. Genet. 106, 121–128. doi: 10.1016/j.ajhg.2019.12.004
Hutchison, M., Berman, K. S., and Cobb, M. H. (1998). Isolation of TAO1, a protein kinase that activates MEKs in stress-activated protein kinase cascades. J. Biol. Chem. 273, 28625–28632. doi: 10.1074/jbc.273.44.28625
Karman, Z., Rethi-Nagy, Z., Abraham, E., Fabri-Ordogh, L., Csonka, A., Vilmos, P., et al. (2020). Novel perspectives of target-binding by the evolutionarily conserved PP4 phosphatase. Open Biol. 10, 200343. doi: 10.1098/rsob.200343
Kataya, A. R. A., Creighton, M. T., Napitupulu, T. P., Saetre, C., Heidari, B., Ruoff, P., et al. (2017). PLATINUM SENSITIVE 2 LIKE impacts growth, root morphology, seed set, and stress responses. PLoS One 12:e0180478. doi: 10.1371/journal.pone.0180478
Kõressaar, T., Lepamets, M., Kaplinski, L., Raime, K., Andreson, R., and Remm, M. (2018). Primer3_masker: integrating masking of template sequence with primer design software. Bioinforma. Oxf. Engl. 34, 1937–1938. doi: 10.1093/bioinformatics/bty036
Kubista, M., Andrade, J. M., Bengtsson, M., Forootan, A., Jonák, J., Lind, K., et al. (2006). The real-time polymerase chain reaction. Mol. Aspects Med. 27, 95–125. doi: 10.1016/j.mam.2005.12.007
Kursula, P. (2014). Structural basis of synaptic adhesion and signaling by Calsyntenin-1. Proc. Natl. Acad. Sci. U.S.A. 111, 3956–3957. doi: 10.1073/pnas.1401919111
Liang, L., He, Z., Yu, H., Wang, E., Zhang, X., Zhang, B., et al. (2020). Selection and validation of reference genes for gene expression studies in Codonopsis pilosula based on Transcriptome sequence data. Sci. Rep. 10:1362. doi: 10.1038/s41598-020-58328-5
Liao, Y., Smyth, G. K., and Shi, W. (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinforma. Oxf. Engl. 30, 923–930. doi: 10.1093/bioinformatics/btt656
Longstaff, B. C. (1981). Biology of the grain pest species of the genus Sitophilus (Coleoptera: Curculionidae): a critical review. Prot. Ecol. 3, 83–130.
Lopes, J. M. L., de Matos, E. M., de Queiroz Nascimento, L. S., and Viccini, L. F. (2021). Validation of reference genes for quantitative gene expression in the Lippia alba polyploid complex (Verbenaceae). Mol. Biol. Rep. 48, 1037–1044. doi: 10.1007/s11033-021-06183-6
Lord, J. C., Hartzer, K., Toutges, M., and Oppert, B. (2010). Evaluation of quantitative PCR reference genes for gene expression studies in Tribolium castaneum after fungal challenge. J. Microbiol. Methods 80, 219–221. doi: 10.1016/j.mimet.2009.12.007
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Lü, J., Yang, C., Zhang, Y., and Pan, H. (2018). Selection of reference genes for the normalization of RT-qPCR data in gene expression studies in insects: a systematic review. Front. Physiol. 9:1560. doi: 10.3389/fphys.2018.01560
Luse, D. S. (2012). Rethinking the role of TFIIF in transcript initiation by RNA polymerase II. Transcription 3, 156–159. doi: 10.4161/trns.20725
Maire, J., Parisot, N., Galvao Ferrarini, M., Vallier, A., Gillet, B., Hughes, S., et al. (2020). Spatial and morphological reorganization of endosymbiosis during metamorphosis accommodates adult metabolic requirements in a weevil. Proc. Natl. Acad. Sci. U.S.A. 117, 19347–19358. doi: 10.1073/pnas.2007151117
Maire, J., Vincent-Monégat, C., Balmand, S., Vallier, A., Hervé, M., Masson, F., et al. (2019). Weevil pgrp-lb prevents endosymbiont TCT dissemination and chronic host systemic immune activation. Proc. Natl. Acad. Sci. U.S.A. 116, 5623–5632. doi: 10.1073/pnas.1821806116
Mansour, K. (1930). Memoirs: preliminary Studies on the Bacterial Cell-mass (Accessory Cell-mass) of Calandra Oryzae (Linn.): the Rice Weevil. J. Cell Sci. s2- 73, 421–435. doi: 10.1242/jcs.s2-73.291.421
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12. doi: 10.14806/ej.17.1.200
Nardon, P. (1973). Obtention d’une souche asymbiotique chez le charançon Sitophilus sasakii Tak: différentes méthodes d’obtention et comparaison avec la souche symbiotique d’origine. CR Acad Sci Paris D 277, 981–984.
Nardon, P., Grenier, A., and Heddi, A. (1998). Endocytobiote control by the host in the weevil Sitophilus oryzae, Coleoptera, Curculionidae. Symbiosis 25:237.
Oakeson, K. F., Gil, R., Clayton, A. L., Dunn, D. M., von Niederhausern, A. C., Hamil, C., et al. (2014). Genome degeneration and adaptation in a nascent stage of symbiosis. Genome Biol. Evol. 6, 76–93. doi: 10.1093/gbe/evt210
Pan, H., Yang, X., Siegfried, B. D., and Zhou, X. (2015). A comprehensive selection of reference genes for RT-qPCR analysis in a predatory lady beetle, Hippodamia convergens (Coleoptera: Coccinellidae). PLoS One 10:e0125868. doi: 10.1371/journal.pone.0125868
Parisot, N., Vargas-Chávez, C., Goubert, C., Baa-Puyoulet, P., Balmand, S., Beranger, L., et al. (2021). The transposable element-rich genome of the cereal pest Sitophilus oryzae. BMC Biol. 19:241. doi: 10.1186/s12915-021-01158-2
Pfaffl, M. W., Tichopad, A., Prgomet, C., and Neuvians, T. P. (2004). Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper–Excel-based tool using pair-wise correlations. Biotechnol. Lett. 26, 509–515. doi: 10.1023/b:bile.0000019559.84305.47
Pierantoni, U. (1927). L’organo simbiotico nello svillupo di Calandra oryzae. Rend R. Acad Sci Fis Mat Napoli 27, 21–33. doi: 10.1080/11250006009438308
Shim, E. Y., Walker, A. K., Shi, Y., and Blackwell, T. K. (2002). CDK-9/cyclin T (P-TEFb) is required in two postinitiation pathways for transcription in the C. elegans embryo. Genes Dev. 16, 2135–2146. doi: 10.1101/gad.999002
Silver, N., Best, S., Jiang, J., and Thein, S. L. (2006). Selection of housekeeping genes for gene expression studies in human reticulocytes using real-time PCR. BMC Mol. Biol. 7:33. doi: 10.1186/1471-2199-7-33
Sirover, M. (1999). New insights into an old protein: the functional diversity of mammalian glyceraldehyde-3-phosphate dehydrogenase. Biochim Biophys Acta. 1432, 159–184. doi: 10.1016/s0167-4838(99)00063-0
Sundaram, V. K., Sampathkumar, N. K., Massaad, C., and Grenier, J. (2019). Optimal use of statistical methods to validate reference gene stability in longitudinal studies. PLoS One 14:e0219440. doi: 10.1371/journal.pone.0219440
Takahashi, N., Ito, M., Tanaka, J., Nakano, T., Kaibuchi, K., Odai, H., et al. (1997). Localization of the gene coding for myosin phosphatase, target subunit 1 (MYPT1) to human chromosome 12q15-q21. Genomics 44, 150–152. doi: 10.1006/geno.1997.4859
Timm, T., Matenia, D., Li, X.-Y., Griesshaber, B., and Mandelkow, E.-M. (2006). Signaling from MARK to tau: regulation, cytoskeletal crosstalk, and pathological phosphorylation. Neurodegener. Dis. 3, 207–217. doi: 10.1159/000095258
Vandesompele, J., De Preter, K., Pattyn, F., Poppe, B., Van Roy, N., De Paepe, A., et al. (2002). Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3:RESEARCH0034. doi: 10.1186/gb-2002-3-7-research0034
Vigneron, A., Masson, F., Vallier, A., Balmand, S., Rey, M., Vincent-Monégat, C., et al. (2014). insects recycle endosymbionts when the benefit is over. Curr. Biol. 24, 2267–2273. doi: 10.1016/j.cub.2014.07.065
Xie, F., Xiao, P., Chen, D., Xu, L., and Zhang, B. (2012). miRDeepFinder: a miRNA analysis tool for deep sequencing of plant small RNAs. Plant Mol. Biol doi: 10.1007/s11103-012-9885-2 [Epub ahead of print].
Yim, A. K.-Y., Wong, J. W.-H., Ku, Y.-S., Qin, H., Chan, T.-F., and Lam, H.-M. (2015). Using RNA-Seq data to evaluate reference genes suitable for gene expression studies in soybean. PLoS One 10:e0136343. doi: 10.1371/journal.pone.0136343
Zaccara, S., and Jaffrey, S. R. (2020). A unified model for the function of YTHDF proteins in regulating m6A-modified mRNA. Cell 181, 1582.e18–1595.e18. doi: 10.1016/j.cell.2020.05.012
Zhang, J., Deng, C., Li, J., and Zhao, Y. (2020). Transcriptome-based selection and validation of optimal house-keeping genes for skin research in goats (Capra hircus). BMC Genomics 21:493. doi: 10.1186/s12864-020-06912-4
Zhang, S.-Q., Che, L.-H., Li, Y., Liang, D., Pang, H., Ślipiński, A., et al. (2018). Evolutionary history of Coleoptera revealed by extensive sampling of genes and species. Nat. Commun. 9:205. doi: 10.1038/s41467-017-02644-4
Zhang, Z.-Q. (2013). Animal biodiversity: an outline of higher-level classification and survey of taxonomic richness (Addenda 2013). Zootaxa 3703, 1–82. doi: 10.11646/zootaxa.3703.1.1
Keywords: real-time PCR, insect, symbiosis, transcriptomic, normalization
Citation: Vallier A, Dell’Aglio E, Ferrarini MG, Hurtado O, Vincent-Monégat C, Heddi A, Rebollo R and Zaidman-Rémy A (2023) Transcriptomic-based selection of reference genes for quantitative real-time PCR in an insect endosymbiotic model. Front. Ecol. Evol. 11:1152183. doi: 10.3389/fevo.2023.1152183
Received: 27 January 2023; Accepted: 20 March 2023;
Published: 20 April 2023.
Edited by:
Jennifer L. Matthews, University of Technology Sydney, AustraliaReviewed by:
Daniel Powell, University of the Sunshine Coast, AustraliaJarosław Bryk, University of Huddersfield, United Kingdom
Copyright © 2023 Vallier, Dell’Aglio, Ferrarini, Hurtado, Vincent-Monégat, Heddi, Rebollo and Zaidman-Rémy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rita Rebollo, rita.rebollo@inrae.fr; Anna Zaidman-Rémy, anna.zaidman@insa-lyon.fr
†These authors have contributed equally to this work