
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol. , 19 May 2023
Sec. Behavioral and Evolutionary Ecology
Volume 11 - 2023 | https://doi.org/10.3389/fevo.2023.1076756
This article is part of the Research Topic Adaptations to Subterranean Environments View all 14 articles
 Dahiana Arcila1,2,3*
Dahiana Arcila1,2,3* Melissa Rincon-Sandoval2
Melissa Rincon-Sandoval2 William Hanson2,3
William Hanson2,3 Pamela B. Hart2,3,4
Pamela B. Hart2,3,4 Vanessa L. González5
Vanessa L. González5 Ricardo Betancur-R1,2
Ricardo Betancur-R1,2 Maria E. Bichuette6
Maria E. Bichuette6Molecular studies have shown that Neotropical fishes of the order Characiformes have undergone two independent events of cave colonization. Among these fishes are the Mexican blind cavefish (Astyanax mexicanus), a well-studied model system for cave adaptation, and the lesser-known Brazilian blind characid (Stygichthys typhlops). Although various genomic and transcriptomic approaches have been used to identify genes responsible for cave adaptation in A. mexicanus, these genetic factors have not been explored in an evolutionary comparative framework in cave-adapted characiforms. To address this gap, we assembled a de novo transcriptome for the Brazilian blind characid, identifying 27,845 assembled unigenes, of which 22,580 were assigned as putative one-to-one orthologs to the Mexican cavefish. We then used the package RELAX to analyze 789 genes in cavefishes, identifying 311 genes under intensified or relaxed selection. Our analysis revealed 26 genes with signatures of convergent, relaxed selection linked to vision, circadian cycles, pigmentation, and hematopoiesis processes. Additionally, we conducted differential gene expression analyzes between the snout region and a control tissue sample (muscle), identifying 96 differentially expressed genes associated with cell-surface-bound and calcium-binding proteins. Our study offers insights into the genetic mechanisms underlying cave adaptation in characiform fishes, particularly the Brazilian blind characid. Moreover, our transcriptome dataset and list of genes under convergent, relaxed, and intensified selection serve as a valuable resource for future functional studies of genes involved in cave adaptation. Our work highlights the importance of examining genetic adaptations in multiple independent lineages to better understand the evolutionary processes underlying cave adaptation.
Fishes are among the few evolutionary groups that have undergone multiple independent origins of adaptation to caves, one of the most adverse environments on earth. Together, cavefishes include over 298 species, demonstrating high evolutionary success in their adaption to life in caves (Proudlove, 2004; Protas et al., 2006; Soares and Niemiller, 2013; Proudlove, 2022). Habitat shifts from surface to cave systems are often accompanied by extreme morphological phenotypes, including loss of eyes, lack of pigmentation, and enhanced non-visual sensory structures, providing an excellent system to investigate the role of morphological and genomic convergence associated with this ecological transition. The Mexican blind cave tetra, Astyanax mexicanus, is the most popular model organism for studying cave adaptive evolution (Borowsky, 2008b), with several studies deploying transcriptomic and genomic data in an effort to understand the evolutionary and genetic mechanisms linked to the morphological, physiological, and genetic evolution of cave-obligate forms (Protas et al., 2006; Borowsky, 2008b; Bernardi et al., 2012; Beale et al., 2013; Yoshizawa, 2015; Krishnan and Rohner, 2017; Gross, 2018).
In the last decade, researchers in Southeastern Brazil collected over 20 wild specimens of the enigmatic subterranean characid, Stygichthys typhlops, from the phreatic zone of the Middle Sao Francisco basin (Moreira et al., 2010). This characid is associated with small subterranean drainages and is restricted to the Jaiba region in Minas Gerais (Bichuette and Gallão, 2021). Due to the rarity of its characteristic habitat, the conservation of S. typhlops is of significant concern, and it is currently listed as an endangered species (Romero and McLeran, 2000; Bichuette and Gallão, 2021).
While both Stygichthys typhlops and Astyanax mexicanus belong to the family Characidae (order Characiformes), a diverse group of Neotropical fishes that also includes the popular aquarium “tetras,” phylogenetic evidence indicates that cave adaptation occurred independently in these two species (Figure 1; Oliveira et al., 2011; Arcila et al., 2017; Betancur-R et al., 2019). Stygichthys typhlops has no known surface relatives while A. mexicanus has both cave and surface populations or morphs (Borowsky, 2008a; Moreira et al., 2010; Gross et al., 2013). Like the cave morph of the Mexican blind cave tetra, the Brazilian blind cave characid is cave obligate; as such, the fish exemplifies troglomorphism with no external eye characters (lost circumorbital bones vs. partially fragmented in A. mexicanus) and pigment loss (Moreira et al., 2010). Furthermore, studies on the circadian rhythms of S. typhlops have demonstrated that the species does not exhibit any significant circadian or ultradian rhythms (Moreira et al., 2010; Sampaio et al., 2012).
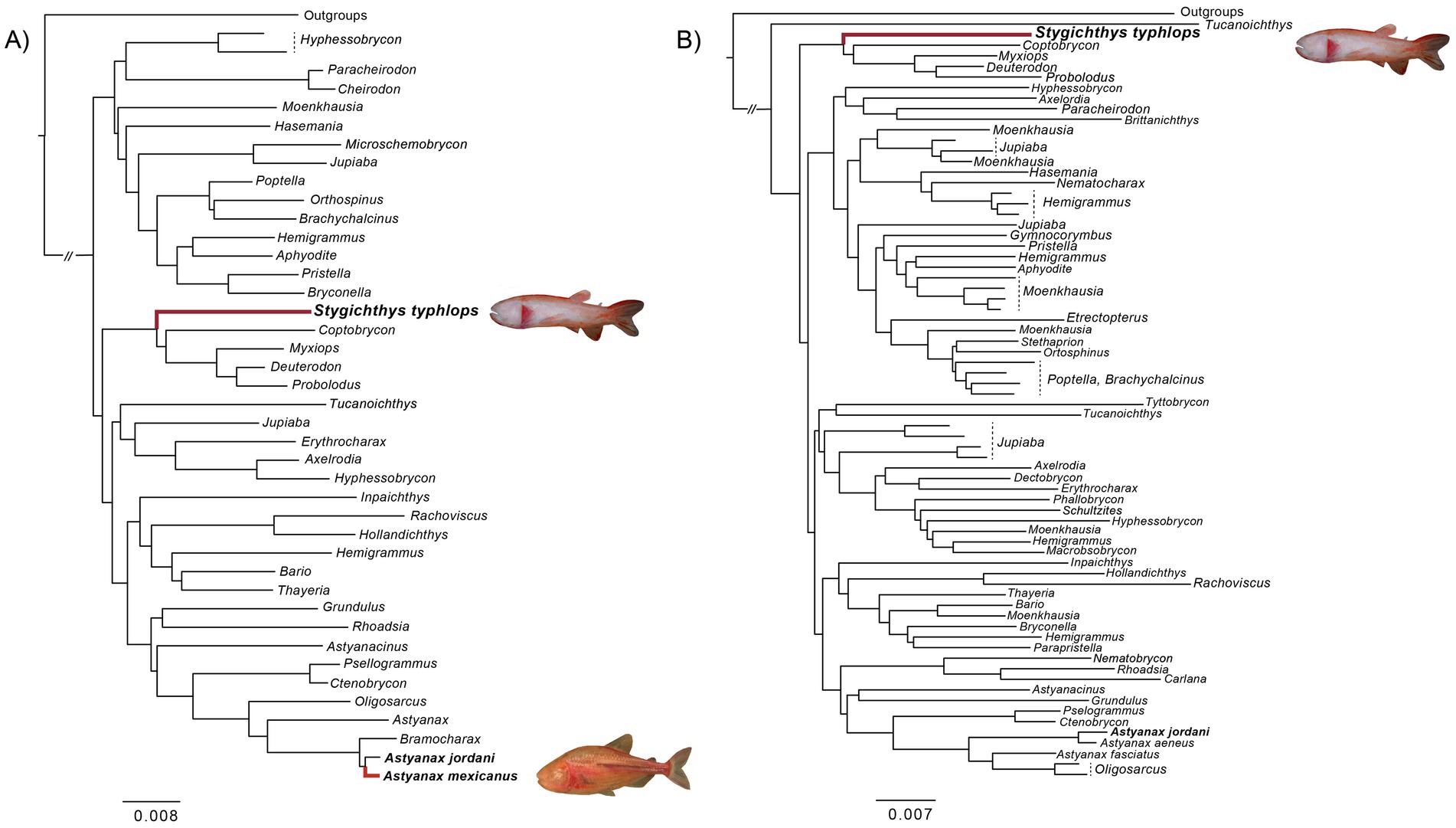
Figure 1. Summary of phylogenomic hypotheses indicating the evolutionary relationships of Stygichthys typhlops and the origins of cave adaptation in the family Characidae. Previous hypotheses based on (A) 1,051 exons markers sequenced for 93 characid species (Betancur-R et al., 2019), and (B) 1,141 ultraconserved markers (UCE) sequenced for 162 characid species (Melo et al., 2021).
The close but non-sister relationship between Stygichthys typhlops and Astyanax mexicanus provides a remarkable system to investigate the genetic bases of convergent adaptation to life in caves at the molecular level. Here, we assemble the first transcriptome of the Brazilian blind cave characid, S. typhlops, and compare it to both surface and blind morphs of A. mexicanus. In addition, we analyzed a set of 789 genes to investigate whether there are signatures of relaxed or intensified selection related to cave phenotypic convergence across other closely related ostariophysian lineages. We also compared tissue-specificity between the snout region (olfactory organs) and muscle by performing differential gene expression analysis for S. typhlops.
The samples used in this study were collected in November 2010 from phreatic water outcrops at “Poço do Mandioqueu,” Jaíba municipality, Minas Gerais state, Brazil (collecting permits ICMBio/SISBIO 28992; financial support for collections: FAPESP). The fish were kept in captivity for 30 days before RNA extraction. Three individuals were sacrificed using an MS-222 overdose and subsequently preserved in RNAlater (Ambion). The samples were frozen at-80C until RNA was extracted. Four different types of tissue samples (muscle, mouth, snout, and eye region) were collected from the right side of each individual. The main purpose was to capture tissue regions that might reveal differential gene expression. However, due to potential concerns with tissue sampling overlapping (e.g., snout vs. mouth or muscle vs. eye region), only the muscle and snout tissues were used for downstream differential gene expression analyzes. All procedures followed the UFAW Handbook on the care and management of laboratory animals.1 Total RNA was extracted using the timing and centrifugation steps specified in the TRIzol (ThermoFisher) protocol. After mRNA purification, samples were treated with Ambion turbo DNA-free DNase to remove residual genomic and rRNA contaminants. Quantity and quality (purity and integrity) of mRNA were assessed using the Agilent 2,200 TapeStation (Agilent Technologies, Santa Clara, CA).
The libraries were prepared using the Illumina TruSeq RNA Sample Preparation kit. The mRNA was purified using poly-T oligo-attached magnetic beads to pull down the poly-A mRNA. After purification, the mRNA was fragmented and copied into first strand cDNA using reverse transcriptase. This was followed by second strand cDNA synthesis using DNA Polymerase I and RNase. The concentration of the cDNA libraries was measured with the qubit dsDNA high-sensitivity (HS) assay kit using the qubit fluorometer (Invitrogen). Concentrations of sequencing runs were normalized based on final concentrations of the fragmented cDNA. Next-generation sequencing (NGS) was carried out using the Illumina HiSeq 2000 platform (Illumina Inc., San Diego, CA) at the University of Chicago Genomics Facility. The 150 bp paired-end raw reads were submitted to the NCBI’s Sequence Read Archive (SRA) database: BioProject PRJNA891276.
The raw read quality was visualized with FastQC v 0.11.5 (Andrews, 2010). Initial removal of low-quality reads and TruSeq multiplex index adaptor sequences (Illumina) was performed using Trimmomatic v 0.32 (Bolger et al., 2014) with the following parameters: ILLUMINACLIP:2:30:7, HEADCROP:13, SLIDINGWINDOW:7:15, MINLEN:25. De novo assembly for the three specimens of S. typhlops was performed using Trinity v 2.8.3 (Haas et al., 2013) with the strand-specific parameter option on. The resulting transcriptome was filtered for ribosomal RNA and microbial contaminants using SortMeRNA v 2.1 (Kopylova and Touzet, 2012) against a non-redundant database compiled from coding-protein genes. TransDecoder v 3.0.0 was used to identify all likely coding regions within our assembled transcripts and filtered by selecting the single best open reading frame (ORFs) per transcript. Only contigs longer than 100 bp were kept for downstream analyzes. We reduced the number of redundant transcripts in the final assembly by clustering highly similar sequences using an amino acid sequence identity threshold of 1.0 with CD-Hit v 4.6.6 (Fu et al., 2012). Quantification for the abundance of transcripts was performed using the pseudo-aligner Kallisto (Bray et al., 2016) in no strand-specific paired-end mode.
After clustering was complete, annotation and mapping were performed within OmicsBox using the BLAST2GO functional annotation pipeline (Conesa et al., 2005). The ORFs were BLASTed against the Ostariophysi nr v5 protein sequences database with a hit filter of 1.0E-6. A reference-based transcriptome was also assembled using A. mexicanus (PRJNA89115) model. This transcriptome was annotated in OmicsBox following the same procedure for de novo annotation. BUSCO v 5.4.3 (Benchmarking Universal Single Copy Orthologs) was used to assess the completeness of both assemblies using the Eukaryota databases as reference (Simao et al., 2015). The de novo assembly was found to be significantly more complete than the A. mexicanus-based assembly for both databases (see Results), and thus the former was used for all downstream analyzes.
For the identification of differentially expressed genes (DEGs) between the nostril and muscle regions, we used the TMM (Robinson and Oshlack, 2010) normalization counts at gene level with EdgeR in Trinity v 2.8.3. For this method, genes with an FDR (false discovery rate) <0.1 were considered to be differentially expressed. Using the Fisher’s exact test implemented in software BLAST2GO with an FDR threshold of 0.05, a GO term enrichment analysis was performed to identify functional categories enriched in DEGs between the nostril and control group. We conducted further Gene Ontology enrichment analyzes using differentially upregulated and downregulated genes from both treatments using ShinyGO v 0.76.2 (Ge et al., 2020).
We retrieved genomes or transcriptomes of 14 Ostariophysian species, including both surface and Pachón morphs of Astyanax mexicanus, from the NCBI database (Supplementary Table S1). We initially mined a set of 1,051 single copy nuclear exons previously identified and optimized for phylogenetic analyzes of ostariophysan fishes (Arcila et al., 2017). Sequences for each exon were aligned with MACSE v 2.03 (Ranwez et al., 2011) after cleaning out potentially non-homologous fragments with the -cleanNonHomologousSequences option. Next, we concatenated all genes alignment using the python package AMAS (Borowiec, 2016). Maximum Likelihood (ML) analyzes were performed in RAxML v 8.2.9 (Stamatakis, 2014) using a by-codon-position partitioning scheme and 30 independent search replicates. The best scoring tree across searches was selected using the GTRGAMMA model. Branch support was assessed using the rapid bootstrap algorithm with 1,000 replicates; the collection of bootstrapped trees was used to draw bipartition frequencies onto the optimal tree.
We identified an extra set of 458 candidate genes from previous studies related to vision, pigmentation, and circadian cycles in cavefishes and subterranean mammals (Emerling and Springer, 2014; McGaugh et al., 2014; Aardema et al., 2021; Policarpo et al., 2021; Appendix 1). From the cavefishes dataset, we first retrieved 404 (of 458) candidate genes using reference alignments for seven ray-finned fishes, which represented species from the orders Cypriniformes, Ophidiiformes, Cichliformes and Percopsiformes (Policarpo et al., 2021), and 14 additional genes using reference sequences from Danio, Astyanax, Sinocyclocheilus, and Lamprologus (Pottin et al., 2011; Meng et al., 2013; McGaugh et al., 2014; Yang et al., 2016; Aardema et al., 2021). From the subterranean mammal dataset, we identified three to five ortholog sequences for ray-finned fishes for 40 additional candidates (Emerling and Springer, 2014) using the NCBI Orthologs database.
We also examined an additional set of 481 genes that were extracted using BUSCO and the Actinopterygii Single-Copy Orthologs library (with 3,640 genes), for a total of 939 genes (458 candidates plus 481 BUSCOs). As the species in the genus Sinocyclocheilus are tetraploids while the other species are diploids, BUSCO analyzes also allowed us to accurately identify orthologous sequences from all species. We then used the hidden Markov model (HMM) approach available in HMMER v 3.2.1 (Wheeler and Eddy, 2013) to extract the 918 orthologous sequences from the 13 ostariophysian genomes and the S. typhlops transcriptome.
The candidate gene sequences extracted were aligned using MACSE v2.03 03 (Ranwez et al., 2011), which takes into account the codon structure of protein-coding nucleotide sequences. The alignments were visually verified using Geneious Prime® v2022.1. We excluded 125 gene alignments that contained four or fewer ostariophysian species. Additionally, we merged four genes that were common to both the Emerling and Springer (2014) and Policarpo et al. (2021) studies, resulting in a total of 789 candidate genes retained for downstream analysis.
We used the RELAX model (Wertheim et al., 2015) implemented in the package HyPhy (Pond et al., 2005) to investigate whether the genes associated with vision, pigmentation, circadian cycles, hematopoiesis, and others, show any signatures of intensified or relaxed selection in foreground branches (test and reference) of cavefishes relative to background (non-cavefish) branches, based on estimates of the selection strength parameter K (using a p value = 0.05). A value of K < 1 indicates that test branch was under significant relaxed selection, whereas a value of K > 1 suggests intensified selection on the test branches. Two alternative schemes were used, each employing test, reference, and background branches. The first scheme included test branches comprising only obligate cavefish species (A. mexicanus cave, Sinocyclocheilus anshuiensis, and S. typhlops), reference branches consisting of facultative cavefish species (Sinocyclocheilus rhinocerous and A. mexicanus surface), and background branches representing the remaining non-cave species in the tree. The second scheme involved S. typhlops as the test branch, the remaining obligate and facultative cavefish species represented as reference branches, and non-cavefish species as background branches. Multiple testing was accounted for with false-discovery rate (FDR) correction (Benjamini and Hochberg, 1995) in R.
Illumina HiSeq 2000 pair-end reads obtained ranged from 13 to 32 million pairs per tissue sample. All biological replicates yielded a total of 190 million paired end reads that were combined to generate a single reference transcriptome assembly. Preliminary analyzes mapping the S. typhlops raw reads to the closest available reference genome, Astyanax mexicanus, resulted in a low percentage of accurately mapped reads (<40%). Thus, we utilized a de novo approach to assemble the transcriptome of S. typhlops. Raw reads were first quality filtered and assembled in Trinity, resulting in 442,897 transcripts, with an N50 length of 1,416 bp (vs. N50 length of 1,572 bp in A. mexicanus Pachón cavefish and 1,521 bp in A. mexicanus surface morpho; Gross et al., 2013). To reduce the number of potentially spurious contigs, a filter was applied based on the abundance and quality of the assembly. For abundance, the isoform expression level and redundance was used as estimated using the Kallisto method, with only highly expressed isoforms being retained. After filtering the nonredundant transcripts using cd-hit-est, the total number of transcripts was reduced to 52,151 contigs ranging from 297 to 6,981 bases and identified as putatively homologs based on BLAST alignment against the NR, InterPro, and GO databases. To further refine the assembly, we considered transcript expression and identified 20,652 transcripts that represented 90% of the total expression data (Ex90), producing an N50 length of 1,027 bp (Ex90N50). Detailed statistics are shown in Figure 2.

Figure 2. Summary of Stygichthys typhlops transcriptome. (A) Summary statistics of sequencing data and the combined de novo transcriptome assembly. (B) Histogram of transcript classifications of biological processes. The BUSCO library of Eukaryota and Actinopterygii orthologs indicated that the transcriptome assembly is 97.6 and 76% complete, respectively.
When comparing the transcripts with the data available for the A. mexicanus genome, a total of 22,528 predicted proteins were aligned to one of S. typhlops transcripts. In addition to the N50 and ExN50 values, transcriptome completeness was assessed in terms of gene content, by searching the transcriptome for the presence or absence of conserved orthologous genes in the Eukaryota and Actinopterygii BUSCO libraries. The S. typhlops assembly was 97.6% complete with only 2.0% fragmented reads, when compared to the Eukaryota ortholog library which includes a total of 255 BUSCO groups. Furthermore, when compared to the Actinopterygii ortholog library, which includes 3,640 BUSCO groups, the assembly was 76% complete with 5.3% fragmented reads.
Significantly DEGs between the snout and muscle regions were detected for all analyzed replicates using edgeR. All three replicate libraries for each treatment showed a high correlation (Pearson r = 0.89–0.99; Supplementary Figure S1) and were used for expression analysis. A total of 96 unigenes were found to be differentially expressed among the snout (olfactory) and muscle tissue samples. Using the snout region as a reference, 65 and 31 unigenes were upregulated and downregulated, respectively. The majority of the DEGs were expressed with fold change values > − 2.0 and < 6.0 (Figures 3A,B). Over 25% of the genes were uncharacterized or incompletely annotated, matching only genes associated with unknown functions. A total of 20 GO terms were associated with the DEG. The most significant GO terms for each set of DEGs across the treatments are shown in Figure 3C. Interestingly, genes related to the Ephrin (cell-surface-bound proteins) and Parvalbumin (calcium binding proteins) were upregulated in the snout tissue samples. Genes related to myosin tail and cardiac muscle contraction were downregulated in the muscle tissue.
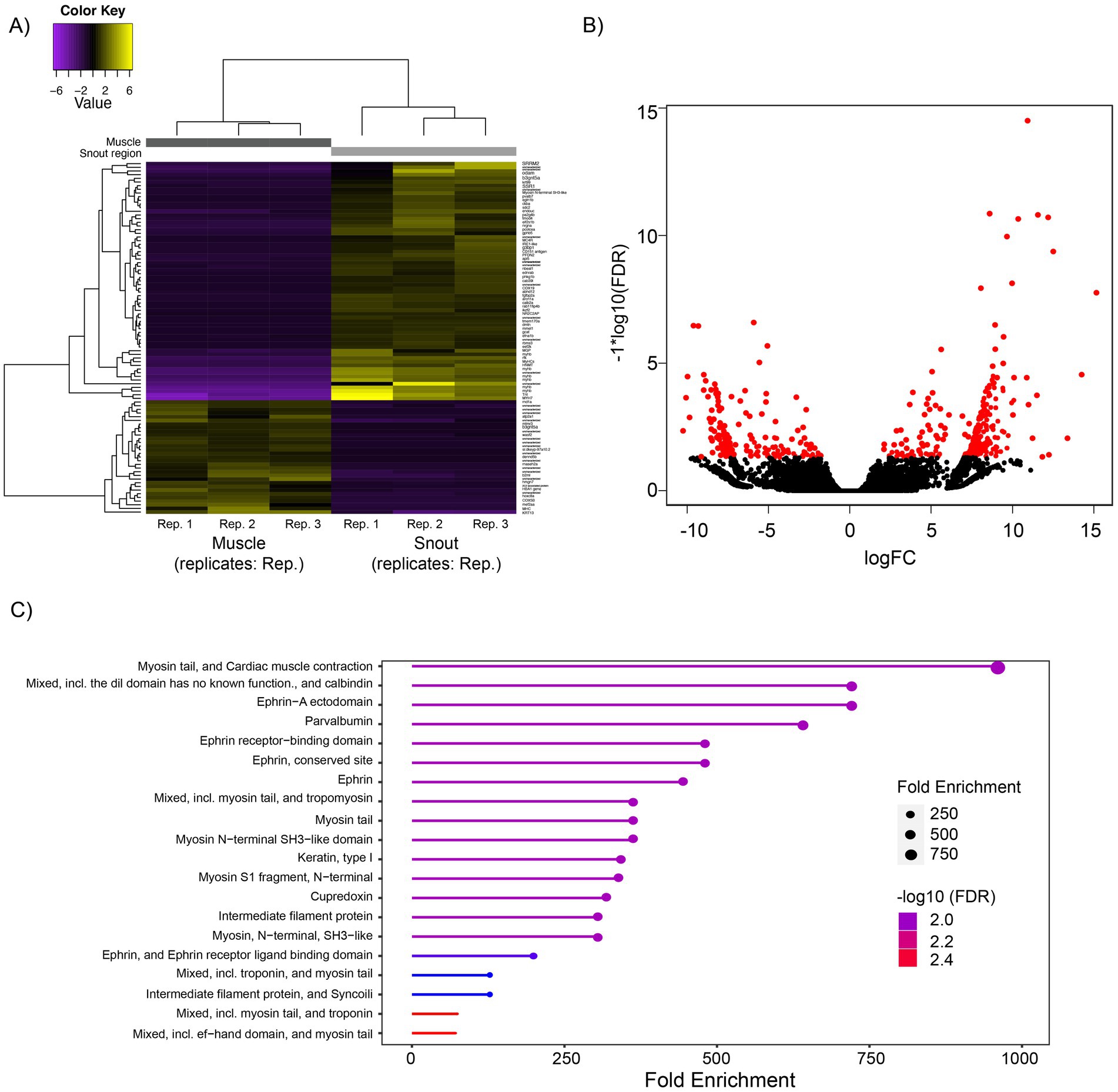
Figure 3. Summary of differentially expressed genes (DEGs). (A) Heatmap of DEGs in Stygichthys typhlops. Both upregulated (yellow) and downregulated (purple) genes with logFC values of 0.3 were clustered. Rows represent single transcripts, and columns represent the replicates for each treatment. Genes were clustered based on expression similarity (Euclidean distance). A complete list of genes DEG is included in Supplementary Figure S1. (B) Volcano plot visualization of DEGs from the two main treatments (muscle vs. snout) using the edgeR method. (C) Gene ontology enrichment analysis for set of DEGs, including the top of 20 GO terms with the highest enrichment values for each pairwise comparison.
Using RELAX with two alternative schemes, we analyzed 789 genes and identified 311 genes under intensified or relaxed selection after assessing their statistical significance using the FDR correction. In the first scheme, we used the obligate cavefish species as the test branches, and the facultative cavefish species as the reference branches. The second scheme employed S. typhlops as the test branch, with other obligate and facultative cavefish species as the reference branches (Figure 4). Our results revealed a consistent pattern across both schemes, with a higher number of genes under relaxed selection in test branches compared to the reference branches (107 vs. 101 for scheme 1, 79 vs. 49 for scheme 2, and 38 vs. 35 for genes shared between schemes), and a lower number of genes under intensified selection in the test branches compared to the reference branches (74 vs. 67 for scheme 1, 88 vs. 62 for scheme 2, and 22 vs. 19 for genes shared between schemes).
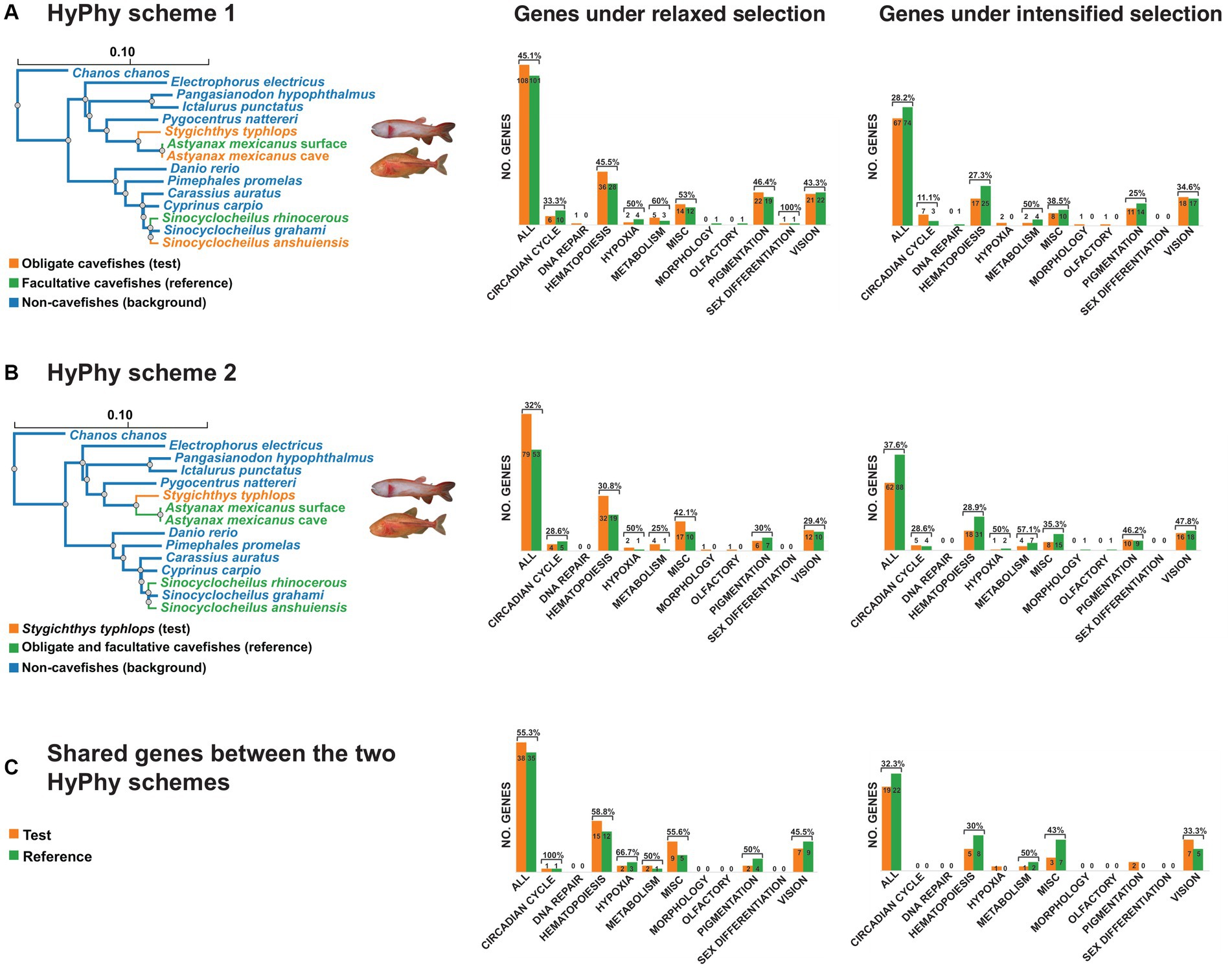
Figure 4. Selection dynamics using RELAX and two alternative schemes in HyPhy for relaxed (K < 1) and intensified (K > 1) selection (p = 0.05). (A) Scheme 1 represents obligate cavefishes as test branches (orange), facultative cavefishes as reference branches (green), and non-cavefishes (blue) as background branches. (B) Scheme 2 shows Stygichthys typhlops as the test branch (orange), other obligate and facultative cavefishes as reference branches (green), and non-cave species as background branches (blue). (C) Genes shared among schemes 1 and 2.
We examined signatures of selection on the 311 genes, of which 174 had a significant p value (p < 0.05) for scheme 1, including 107 genes under relaxed selection and 67 under intensified selection. For scheme 2, 137 genes had a significant p value, with 79 being under relaxed selection and 58 under intensified selection. Of these, 73 genes were identified in both schemes, with 38 genes being under relaxed selection and 19 under intensified selection on the test branches for both schemes (Figure 4). However, we found that 16 out of the 73 shared genes exhibited different signatures of selection. Specifically, seven genes were under relaxed selection and nine genes were under intensified selection for scheme 1. When treating S. typhlops as the sole test branch, nine genes were under relaxed selection and seven genes were under intensified selection.
We annotated the 311 genes examined and found that 103 (32.5%) of them were associated with hematopoiesis processes. Among these, 68 (21.5%) showed evidence of relaxed selection (36 for scheme 1; 32 for scheme 2) and 35 (11.0%) showed intensified selection (17 for scheme 1; 18 for scheme 2). We also identified 67 (21%) genes related to vision, with 33 (10.4%) under relaxed selection (21 for scheme 1; 12 for scheme 2) and 34 (10.7%) under intensified selection (18 for scheme 1; 16 for scheme 2). Pigmentation-related genes accounted for 49 (15.5%) of the total genes, with 22 and 6 under relaxed selection (e.g., gpc3 and atp6p1), and 11 and 6 under intensified selection (e.g., bnc2 and zic2b) for schemes 1 and 2, respectively. Genes associated with the circadian cycle constituted 22 (6.9%) of the total, with 6 and 4 under relaxed selection (e.g., nr1d1 and rorcb), and 7 and 5 under intensified selection (e.g., bmrl1b and cry2b), for schemes 1 and 2, respectively. We also identified a smaller number of genes related to DNA repair, hypoxia, metabolism, morphology, and the olfactory system that exhibited signatures of selection. Furthermore, out of the 311 genes under selection, 47 genes were classified as unknown due to a lack of available annotations (Appendix 2). Interestingly, we identified only two gene families (B3GNT and Rab GTPases) that overlapped between the 96 genes showing differential expression and the 311 genes under selection.
The generation of de novo transcriptome data for non-model organisms has established itself as a powerful approach to identifying novel transcripts and differentially expressed genes. However, among vertebrates, transcriptomic resources for fishes remain relatively underrepresented in public databases and mainly restricted to model species (Sun et al., 2016; Yang et al., 2020). Additionally, transcriptomes of cave-obligate fishes continue to surprise researchers as the molecular pathways related to troglomorphism show unique molecular signatures in each species (Gross et al., 2013; Meng et al., 2013; Casane and Retaux, 2016). We conducted a de novo assembly transcriptome of the Brazilian cavefish, S. typhlops, to provide additional insights into the extreme phenotypic adaptations to cave systems. The lack of a reference genome or transcriptome resulted in the generation of partial and fragmented transcripts. However, the fraction of annotated transcripts in relation to the Mexican cavefish, A. mexicanus, was remarkably high (27,845 transcripts for the cave morpho and 22,090 transcripts for the surface morpho).
A total of 96 genes were differentially expressed between the snout (olfactory) and muscle regions in Stygichthys typhlops. We observed that for the GO analysis, upregulated genes were enriched with more functional categories, including the presence of Ephrin A receptors considered to play a critical role for establishing topographic projections in the olfactory, auditory and somatosensory systems (Prakash et al., 2000; Serizawa et al., 2006; Cramer and Gabriele, 2014). Additional coding protein genes found included the calcium-binding calbindin, parvalbumin, and myosin usually involved in sensory organs of the ear, nose, and throat, which have shown to play an important role in spatial navigation, memory processes, and social interactions in guinea pigs and rats (Yamagishi et al., 1993; Bjerke et al., 2021). Olfaction by way of self-odor has also been shown to be an important part of spatial awareness in the blind cave cyprinid Phreatichthys adruzzii (Annalisa et al., 2006).
We also found the hoxc8a gene upregulated in the muscle region, a candidate gene previously associated with the evolution of rib axial variability in A. mexicanus, resulting in fewer ribs in the cave versus the surface morphs (Gross et al., 2008). The decrease in the number bones and bone density is considered a regressive (reductive) trait in troglomorphic organisms (Wilkens, 1988; Protas and Jeffery, 2012). The upregulation of hoxc8a in S. typhlops could indicate changes in bone structure.
Another differentially expressed gene is cox5b, which encodes for a hypoxia-inducible isoform of COX subunit recently identified in the pike icefish Champsocephalus esox (Rivera-Colón et al., 2022). The absence of primary productivity decreases the oxygen content in caves and cave streams creating hypoxic conditions (Malard and Hervant, 1999; Boggs and Gross, 2021). The heterogeneity of dissolved oxygen content in groundwater has even been proposed as a selective pressure on low metabolic rates in cave-obligate organisms, in lieu of low food supply (Malard and Hervant, 1999). The increased expression of cox5b could indicate an adaptation in S. typhlops to hypoxic conditions.
Additionally, downregulation of myosin tail and cardiac muscle contraction in Stygichthys typhlops muscle tissue may make sense in light of hypoxic conditions. Many fishes in hypoxic conditions have adapted the unique cardiac reaction of a slower heartbeat (i.e., hypoxic bradycardia; Farrell, 2007). Hypoxic bradycardia may have significant benefits to fishes in these conditions, including a slower beating heart generating more force, maintaining cardiac stroke volume (Shiels et al., 2002; Farrell, 2007). Thus, downregulation of genes relating to cardiac muscle contraction may indicate S. typhlops’ cardiac adaptations to low-oxygen groundwater systems.
While all tissues carry out common processes, tissues are distinguished by gene expression dynamics, implying that distinct regulatory programs control tissue specificity. Although we found a higher number of genes differentially expressed in the muscle compared to the snout region in Stygichthys, this result may indicate that an undetermined number of genes can remain undetected due to low expression levels (García-Ortega and Martínez, 2015). Sensory organs vary significantly among cave-adapted species suggesting adaptive responses to the microenvironment. Our results provide new insights to understand the role of differential gene expression among cave organisms as well as additional genomic resources for further transcriptome-wide association studies to test the association between traits and genetically predicted gene expression levels (Hinaux et al., 2016).
Our study adds to the growing evidence that many genes associated with vision, circadian cycles, hematopoiesis, and pigmentation have undergone genome-wide relaxation in cavefish evolution (Hinaux et al., 2013; Calderoni et al., 2016; Mack et al., 2021; Policarpo et al., 2021; Van der Weele and Jeffery, 2022; Zhao et al., 2022). Consistent with previous studies, we identified 26 genes that exhibited signatures of convergent, relaxed selection and 10 genes with convergent, intensified selection on all obligate cavefishes (S. typhlops, A. mexicanus, and S. anshuiensis, scheme 1), as well as in S. typhlops alone (scheme 2). These results provide further evidence that relaxed selection have played a significant role in the adaptation of multiple cavefish lineages to cave environments.
Seven genes associated with vision exhibited convergent, relaxed selection, including TELO2, which has been implicated in cortical visual impairment and cataracts (Albokhari et al., 2023), and PCIF1, which has been associated with binocular phenotypes in vertebrates (Bosten et al., 2015). Additionally, in fishes and other vertebrates, the circadian system plays a crucial role in regulating important genes in metabolic pathways, creating a two-way relationship between circadian clocks and metabolic signals (Froland and Whitmore, 2019). One of the genes that displayed convergent, relaxed selection was Sirtuin 6 (SIRT6), which regulates circadian chromatin recruitment physiology and cellular metabolism in mammals (Masri et al., 2014). However, additional research is needed to understand the role of SIRT6 in modulating the brain and peripheral clocks in cavefish (Delgado et al., 2017). Additionally, we identified under relaxed selection, two previously identified clock genes in zebrafish, nr1d (scheme 1) and rorcb (scheme 2) genes (Policarpo et al., 2021).
In recent transcriptomic studies in A. mexicanus, Sears et al. (2020) identified at least eight genes with reduced level of differential expression associated with blood physiology. In our study, we identified 17 additional genes linked to hematopoiesis processes that were under relaxed selection in both schemes used. Among these genes, we found EGF1, which plays a significant role in blood coagulation in vertebrates (Davidson et al., 2003), and ASXL2, which is a critical regulator in the formation of blood cellular components (Micol et al., 2017). Furthermore, we identified two genes associated with pigmentation in vertebrates that exhibited convergent, relaxed selection in obligated cavefishes. VAMP7 is required for the production of melanin pigments and has been linked to hypopigmentation in mice (Jani et al., 2015; Dennis et al., 2016), while LRP11 is involved in carotenoid metabolism and has been significantly linked to the skin coloration in fishes (Wu et al., 2023).
The genes that displayed signatures of convergent, intensified selection in the test and reference branches analyzed (schemes 1 and 2) were primarily associated with hematopoietic and metabolic processes. Among these genes were MVD, which is necessary for cell survival (Wong et al., 2021); SCARA5, previously identified to exclusively express on epithelial cells in vertebrates (Yap et al., 2015); SLC4A1AP, an ion transporter gene identified in crabs (Shih et al., 2023); STARD7, which is thought to play an essential role in morphogenesis and respiration in mammals (Horibata et al., 2020); and PDK4, previously found to have increased levels of expression in fasting in chickens and mice (Connaughton et al., 2009). The identification of intensified selection on genes associated with hematopoietic and metabolic processes in our dataset may suggest that selection exerts ongoing pressures on a limited number of genes that are crucial for ensuring the survival of fishes in cave environments characterized by limited food availability (Poulson and Lavoie, 2000). This finding underscores the importance of physiological adaptation, such as slow metabolism and fasting, as strategies for coping with food scarcity (Langecker and Longley, 1993). In future studies, it will be interesting to determine the role of genes involved in hematopoiesis processes in the evolution of “constructive” adaptive traits in cavefishes.
Our study identified an intriguing result regarding the PIK3CG gene, which maintains epithelial integrity. We observed that this gene was under relaxed selection in scheme 1, including all obligate cavefishes, but under intensified selection in S. typhlops as the sole test foreground branch in scheme 2. This finding is particularly interesting because recent research has identified PIK3CG as a regulator and responder to multiple physiological processes and suggested that it may contribute to the ability of deep-sea fishes to resist pressure (Bo et al., 2022). The karst aquifer where S. typhlops is found typically has an average depth of 20 to 30 meters, but the thickness of the aquifer can easily exceed 200 meters or more (P. Pessoa personal comm.). It is possible that PIK3CG might have undergone adaptive evolution in S. typhlops, enabling it to overcome physiological challenges common to the deep sea and cave environments of the Middle Sao Francisco basin. In addition, we also identified genes related to the B3GNT and Rab GTPases gene families under intensified selection and upregulated in S. typhlops. The role of these genes families in fishes remain unknown.
The investigation of genes responsible for phenotypic adaptations in obligate cavefishes (A. mexicanus, S. typhlops, and S. anshuiensis) has revealed that different genetic mechanisms can lead to “many ways to build a cavefish,” as suggested in previous studies (Sears et al., 2020; Policarpo et al., 2021; Van der Weele and Jeffery, 2022). However, the identification of 73 shared genes between S. typhlops and other cavefishes suggests that molecular convergence also plays a significant role in cave adaptation, as previously observed in the Oca2 gene associated with the albinism phenotype in vertebrates (Protas et al., 2006); note that we were unable to retrieve Oca2 from S. typhlops.
Previous molecular studies investigating the evolutionary relationships of the diverse family Characidae consistently identified Stygichthys as most closely related to the genera Coptobrycon, Myxiops, Deuterodon, and Probolodus, and as part of the so called “Clade C” (Figure 1). Two alternative placements for Stygichthys have been proposed, either as the sister of the monotypic genus Coptobrycon, based on two mitochondrial and three nuclear markers (Oliveira et al., 2011), or as sister to a clade including Coptobrycon, Myxiops, Deuterodon, and Probolodus, based on exonic and ultraconserved markers (Betancur-R et al., 2019; Melo et al., 2021).
A recent total evidence analysis, based on 520 morphological characters and nine molecular markers for Astyanax and allies, resolved Stygichthys as most closely related to Phycocharax rasbora, Cyanogaster noctivaga, “Hyphessobrycon” elachys and Coptobrycon (Terán et al., 2020). While the sister-group relationships of Stygichthys remain contentious, it is clear that the Mexican and the Brazilian cavefish belong in different clades within “Clade C” of Characidae. The lack of phylogenetic resolution among the early branching lineages of “Clade C,” which encompasses over 500 species and includes some of the most taxonomically challenging genera in the family, suggest that future phylogenetic analyzes aimed at elucidating the sister-group relationships of Stygichthys will require dense taxonomic and genetic sampling to provide new insights into its evolutionary history.
The datasets presented in this study can be found in online repositories. The name of the repository and accession number can be found below: NCBI; PRJNA891276.
The animal study was reviewed and approved by ICMBio/SISBIO 28992.
DA, RB-R, and MB conceived the original idea. WH, MR-S, RB-R, VG, and DA analyzed the data. MB collected samples. VG and DA performed lab work. DA prepared the manuscript. All authors discussed the results, implications, and commented on the manuscript.
This project was supported by the National Science Foundation (NSF) grants to DA (DEB-2144325 and DEB-2015404), RB-R (DEB-1541491) and PH (PRFB-2109469). Fish drawings by E. Santaquiteria.
The computing for this project was performed at the OU Supercomputing Center for Education and Research (OSCER) at the University of Oklahoma. OSCER Director H. Neeman and OSCER Senior System Administrators J. Speckman and H. Severini provided valuable technical expertise. We also thank Jose Carlos Bonilla and Humberto Ortiz for providing support with bioinformatic analyses and access to the High-Performance Computing facility of UPR-RP (funded by INBRE Grant Number P20GM103475 from the National Institute of General Medical Sciences and the National Institutes of Health). We thank members of the FishEvoLab for providing valuable comments on earlier versions of the paper.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2023.1076756/full#supplementary-material
Aardema, M. L., Stiassny, M. L., and Alter, E. S. (2021). Genomic analysis of the only blind cichlid reveals extensive inactivation in eye and pigment formation genes. Genome Biol. Evol. 12, 1392–1406. doi: 10.1093/gbe/evaa144
Albokhari, D., Pritchard, A. B., Beil, A., Muss, C., Bupp, C., Grange, D. K., et al. (2023). TELO2-related syndrome (You-Hoover-Fong syndrome): description of 14 new affected individuals and review of the literature. Am. J. Med. Genet. A 191, 1261–1272. doi: 10.1002/ajmg.a.63142
Andrews, S. (2010). Fast QC: A quality control tool for high throughput sequence data. BibSonomy the blue social bookmark and publication sharing system.
Annalisa, P., Giuseppe, M., Alessandro, C. I., and Roberto, B. I. (2006). Is the perception of their own odour effective in orienting the exploratory activity of cave fishes? Can. J. Zool. 84, 871–876. doi: 10.1139/z06-072
Arcila, D., Ortí, G., Vari, R., Armbruster, J. W., Stiassny, M. L. J., Ko, K. D., et al. (2017). Genome-wide interrogation advances resolution of recalcitrant groups in the tree of life. Nat. Ecol. Evol. 1:20. doi: 10.1038/s41559-016-0020
Beale, A., Guibal, C., Tamai, T. K., Klotz, L., Cowen, S., Peyric, E., et al. (2013). Circadian rhythms in Mexican blind cavefish Astyanax mexicanus in the lab and in the field. Nat. Commun. 4:2769. doi: 10.1038/ncomms3769
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat Methodol. 57, 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
Bernardi, G., Wiley, E. O., Mansour, H., Miller, M. R., Orti, G., Haussler, D., et al. (2012). The fishes of genome 10K. Mar. Genomics 7, 3–6. doi: 10.1016/j.margen.2012.02.002
Betancur-R, R., Arcila, D., Vari, R. P., Hughes, L. C., Oliveira, C., Sabaj, M. H., et al. (2019). Phylogenomic incongruence, hypothesis testing, and taxonomic sampling: the monophyly of characiform fishes. Evolution 73, 329–345. doi: 10.1111/evo.13649
Bichuette, M. E., and Gallão, J. E. (2021). Under the surface: what we know about the threats to subterranean fishes in Brazil. Neotrop. Ichthyol. 19:3. doi: 10.1590/1982-0224-2021-0089
Bjerke, I. E., Yates, S. C., Laja, A., Witter, M. P., Puchades, M. A., Bjaalie, J. G., et al. (2021). Densities and numbers of calbindin and parvalbumin positive neurons across the rat and mouse brain. iScience 24:101906. doi: 10.1016/j.isci.2020.101906
Bo, J., Xu, H., Lv, W., Wang, C., He, S., and Yang, L. (2022). Molecular mechanisms of the convergent adaptation of bathypelagic and abyssopelagic fishes. Genome Biol. Evol. 14:evac109. doi: 10.1093/gbe/evac109
Boggs, T., and Gross, J. B. (2021). Reduced oxygen as an environmental pressure in the evolution of the blind Mexican cavefish. Diversity 13:26. doi: 10.3390/d13010026
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Borowiec, M. L. (2016). AMAS: a fast tool for alignment manipulation and computing of summary statistics. PeerJ. 4:e1660.
Borowsky, R. (2008a). Astyanax mexicanus, the blind Mexican cave fish: a model for studies in development and morphology. CSH Protoc. 2008:emo107. doi: 10.1101/pdb.emo107
Borowsky, R. (2008b). Restoring sight in blind cavefish. Curr. Biol. 18, R23–R24. doi: 10.1016/j.cub.2007.11.023
Bosten, J. M., Goodbourn, P. T., Lawrance-Owen, A. J., Bargary, G., Hogg, R. E., and Mollon, J. D. (2015). A population study of binocular function. Vis. Res. 110, 34–50. doi: 10.1016/j.visres.2015.02.017
Bray, N. L., Pimentel, H., Melsted, P., and Pachter, L. (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527. doi: 10.1038/nbt.3519
Calderoni, L., Rota-Stabelli, O., Frigato, E., Panziera, A., Kirchner, S., Foulkes, N. S., et al. (2016). Relaxed selective constraints drove functional modifications in peripheral photoreception of the cavefish P. andruzzii and provide insight into the time of cave colonization. Heredity (Edinb) 117, 383–392. doi: 10.1038/hdy.2016.59
Casane, D., and Retaux, S. (2016). Evolutionary genetics of the cavefish Astyanax mexicanus. Adv. Genet. 95, 117–159. doi: 10.1016/bs.adgen.2016.03.001
Conesa, A., Götz, S., García-Gómez, J. M., Terol, J., Talón, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Connaughton, S., Chowdhury, F., Attia, R., Song, S., Zhang, Y., Elam, M., et al. (2009). Regulation of pyruvate dehydrogenase kinase isoform 4 (PDK4) gene expression by glucocorticoids and insulin. Mol. Cell. Endocrinol. 315, 159–167. doi: 10.1016/j.mce.2009.08.011
Cramer, K. S., and Gabriele, M. L. (2014). Axon guidance in the auditory system: multiple functions of Eph receptors. Neuroscience 277, 152–162. doi: 10.1016/j.neuroscience.2014.06.068
Davidson, C. J., Hirt, R. P., Lal, K., Snell, P., Elgar, G., Tuddenham, E. G., et al. (2003). Molecular evolution of the vertebrate blood coagulation network. Thromb. Haemost. 89, 420–428. doi: 10.1055/s-0037-1613369
Delgado, M. J., Cerda-Reverter, J. M., and Soengas, J. L. (2017). Hypothalamic integration of metabolic, endocrine, and circadian signals in fish: involvement in the control of food intake. Front. Neurosci. 11:354. doi: 10.3389/fnins.2017.00354
Dennis, M. K., Delevoye, C., Acosta-Ruiz, A., Hurbain, I., Romao, M., Hesketh, G. G., et al. (2016). BLOC-1 and BLOC-3 regulate VAMP7 cycling to and from melanosomes via distinct tubular transport carriers. J. Cell Biol. 214, 293–308. doi: 10.1083/jcb.201605090
Emerling, C. A., and Springer, M. S. (2014). Eyes underground: regression of visual protein networks in subterranean mammals. Mol. Phylogenet. Evol. 78, 260–270. doi: 10.1016/j.ympev.2014.05.016
Farrell, A. P. (2007). Tribute to P. L. Lutz: a message from the heart--why hypoxic bradycardia in fishes? J. Exp. Biol. 210, 1715–1725. doi: 10.1242/jeb.02781
Froland, I. A., and Whitmore, D. (2019). Circadian clocks in fish-what have we learned so far? Biology (Basel) 8:17. doi: 10.3390/biology8010017
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
García-Ortega, L. F., and Martínez, O. (2015). How many genes are expressed in a Transcriptome? Estimation and results for RNA-Seq. PLoS One 10:e0130262. doi: 10.1371/journal.pone.0130262
Ge, S. X., Jung, D., and Yao, R. (2020). ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics 36, 2628–2629. doi: 10.1093/bioinformatics/btz931
Gross, M. (2018). The genome sequence of everything. Curr. Biol. 28, R719–R721. doi: 10.1016/j.cub.2018.06.049
Gross, J. B., Furterer, A., Carlson, B. M., and Stahl, B. A. (2013). An integrated transcriptome-wide analysis of cave and surface dwelling Astyanax mexicanus. PLoS One 8:e55659. doi: 10.1371/journal.pone.0055659
Gross, J. B., Protas, M. E., Conrad, M., Scheid, P. E., Vidal, O., Jeffery, W. R., et al. (2008). Synteny and candidate gene prediction using an anchored linkage map of Astyanax mexicanus. Proc. Natl. Acad. Sci. 105, 20106–20111. doi: 10.1073/pnas.0806238105
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Hinaux, H., Devos, L., Blin, M., Elipot, Y., Bibliowicz, J., Alie, A., et al. (2016). Sensory evolution in blind cavefish is driven by early embryonic events during gastrulation and neurulation. Development 143, 4521–4532. doi: 10.1242/dev.141291
Hinaux, H., Poulain, J., Da Silva, C., Noirot, C., Jeffery, W. R., Casane, D., et al. (2013). De novo sequencing of Astyanax mexicanus surface fish and Pachon cavefish transcriptomes reveals enrichment of mutations in cavefish putative eye genes. PLoS One 8:e53553. doi: 10.1371/journal.pone.0053553
Horibata, Y., Mitsuhashi, S., Shimizu, H., Maejima, S., Sakamoto, H., Aoyama, C., et al. (2020). The phosphatidylcholine transfer protein StarD7 is important for myogenic differentiation in mouse myoblast C2C12 cells and human primary skeletal myoblasts. Sci. Rep. 10:2845. doi: 10.1038/s41598-020-59444-y
Jani, R. A., Purushothaman, L. K., Rani, S., Bergam, P., and Setty, S. R. (2015). STX13 regulates cargo delivery from recycling endosomes during melanosome biogenesis. J. Cell Sci. 128, 3263–3276. doi: 10.1242/jcs.171165
Kopylova, N. L., and Touzet, H. (2012). SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28, 3211–3217. doi: 10.1093/bioinformatics/bts611
Krishnan, J., and Rohner, N. (2017). Cavefish and the basis for eye loss. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 372:20150487. doi: 10.1098/rstb.2015.0487
Langecker, T. G., and Longley, G. (1993). Morphological adaptations of the Texas blind catfishes Trogloglanis pattersoni and Satan eurystomus (Siluriformes: Ictaluridae) to their underground environment. Copeia 4, 976–986. doi: 10.2307/1447075
Mack, K. L., Jaggard, J. B., Persons, J. L., Roback, E. Y., Passow, C. N., Stanhope, B. A., et al. (2021). Repeated evolution of circadian clock dysregulation in cavefish populations. PLoS Genet. 17:e1009642. doi: 10.1371/journal.pgen.1009642
Malard, F., and Hervant, F. (1999). Oxygen supply and the adaptations of animals in groundwater. Freshw. Biol. 41, 1–30. doi: 10.1046/j.1365-2427.1999.00379.x
Masri, S., Rigor, P., Cervantes, M., Ceglia, N., Sebastian, C., Xiao, C., et al. (2014). Partitioning circadian transcription by SIRT6 leads to segregated control of cellular metabolism. Cells 158, 659–672. doi: 10.1016/j.cell.2014.06.050
McGaugh, S. E., Gross, J. B., Aken, B., Blin, M., Borowsky, R., Chalopin, D., et al. (2014). The cavefish genome reveals candidate genes for eye loss. Nat. Commun. 5:5307. doi: 10.1038/ncomms6307
Melo, B. F., Sidlauskas, B. L., Near, T. J., Roxo, F. F., Ghezelayagh, A., Ochoa, L. E., et al. (2021). Accelerated diversification explains the exceptional species richness of tropical Characoid fishes. Syst. Biol. 71, 78–92. doi: 10.1093/sysbio/syab040
Meng, F., Braasch, I., Phillips, J. B., Lin, X., Titus, T., Zhang, C., et al. (2013). Evolution of the eye transcriptome under constant darkness in Sinocyclocheilus cavefish. Mol. Biol. Evol. 30, 1527–1543. doi: 10.1093/molbev/mst079
Micol, J. B., Pastore, A., Inoue, D., Duployez, N., Kim, E., Lee, S. C., et al. (2017). ASXL2 is essential for haematopoiesis and acts as a haploinsufficient tumour suppressor in leukemia. Nat. Commun. 8:15429. doi: 10.1038/ncomms15429
Moreira, C. R., Bichuette, M. E., Oyakawa, O. T., de Pinna, M. C., and Trajano, E. (2010). Rediscovery and redescription of the unusual subterranean characiform Stygichthys typhlops, with notes on its life history. J. Fish Biol. 76, 1815–1824. doi: 10.1111/j.1095-8649.2010.02625.x
Oliveira, C., Avelino, G. S., Abe, K. T., Mariguela, T. C., Benine, R. C., Ortí, G., et al. (2011). Phylogenetic relationships within the speciose family Characidae (Teleostei: Ostariophysi: Characiformes) based on multilocus analysis and extensive ingroup sampling. BMC Evol. Biol. 11, 1–25. doi: 10.1186/1471-2148-11-275
Policarpo, M., Fumey, J., Lafargeas, P., Naquin, D., Thermes, C., Naville, M., et al. (2021). Contrasting gene decay in subterranean vertebrates: insights from cavefishes and Fossorial mammals. Mol. Biol. Evol. 38, 589–605. doi: 10.1093/molbev/msaa249
Pond, S. L., Frost, S. D., and Muse, S. V. (2005). HyPhy: hypothesis testing using phylogenies. Bioinformatics 21, 676–679. doi: 10.1093/bioinformatics/bti079
Pottin, K., Hinaux, H., and Rétaux, S. (2011). Restoring eye size in Astyanax mexicanus blind cavefish embryos through modulation of the Shh and Fgf8 forebrain organising centres. Development 138, 2467–2476. doi: 10.1242/dev.054106
Poulson, T. L., and Lavoie, K. H. (2000). “The trophic basis of subsurface ecosystems” in Ecosystems of the world. eds. H. Wilkens, D. C. Culver, and W. F. Humphreys (Amsterdam: Elsevier), 231–249.
Prakash, N., Vanderhaeghen, P., Cohen-Cory, S., Frisen, J., Flanagan, J. G., and Frostig, R. D. (2000). Malformation of the functional organization of somatosensory cortex in adult ephrin-A5 knock-out mice revealed by in vivo functional imaging. J. Neurosci. 20, 5841–5847. doi: 10.1523/JNEUROSCI.20-15-05841.2000
Protas, M. E., Hersey, C., Kochanek, D., Zhou, Y., Wilkens, H., Jeffery, W. R., et al. (2006). Genetic analysis of cavefish reveals molecular convergence in the evolution of albinism. Nat. Genet. 38, 107–111. doi: 10.1038/ng1700
Protas, M. E., and Jeffery, W. R. (2012). Evolution and development in cave animals: from fish to crustaceans. WIREs Dev. Biol. 1, 823–845. doi: 10.1002/wdev.61
Proudlove, G. S. (2004). Subterranean fishes of the world. An account of the subterranean (hypogean) fishes described up to 2003 with a bibliography 1541–2004. Moulis, France: International Society for Subterranean Biology.
Proudlove, G. (2022). Subterranean fishes of the world. Moulis: International Society for Subterranean Biology.
Ranwez, V., Harispe, S., Delsuc, F., and Douzery, E. J. (2011). MACSE: multiple alignment of coding SEquences accounting for frameshifts and stop codons. PLoS One 6:e22594. doi: 10.1371/journal.pone.0022594
Rivera-Colón, A. G., Rayamajhi, N., Minhas, B. F., Madrigal, G., Bilyk, K. T., Yoon, V., et al. (2022). Genomics of secondarily temperate adaptation in the only non-artarctic icefish. bioRxiv.
Robinson, M. D., and Oshlack, A. A. (2010). Scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11:R25. doi: 10.1186/gb-2010-11-3-r25
Romero, A., and McLeran, A. (2000). Threatened fishes of the world: Stygichthys typhlops Brittan & Böhlke, 1965 (Characidae). Environ. Biol. Fish 57:270. doi: 10.1023/a:1007691926260
Sampaio, F. A. C., Pompeu, P. S., Santos, H., and Ferreira, R. L. (2012). Swimming performance of epigeal and hypogeal species of Characidae, with an emphasis on the troglobiotic Stygichthys typhlops Brittan and Böhlke, 1965. Int. J. Speleol. 41, 9–16. doi: 10.5038/1827-806X.41.1.2
Sears, C. R., Boggs, T. E., and Gross, J. B. (2020). Dark-rearing uncovers novel gene expression patterns in an obligate cave-dwelling fish. J. Exp. Zool. B Mol. Dev. Evol. 334, 518–529. doi: 10.1002/jez.b.22947
Serizawa, S., Miyamichi, K., Takeuchi, H., Yamagishi, Y., Suzuki, M., and Sakano, H. (2006). A neuronal identity code for the odorant receptor-specific and activity-dependent axon sorting. Cells 127, 1057–1069. doi: 10.1016/j.cell.2006.10.031
Shiels, H., Vornanen, M., and Farrell, A. (2002). The force-frequency relationship in fish hearts - a review. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 132, 811–826. doi: 10.1016/S1095-6433(02)00050-8
Shih, Y. J., Yang, Y. M., Luo, S. T., and Liu, J. Y. (2023). Revealing the gene diversity and candidate gene family for adaption to environment depth in Leucosiid crabs comparing the Transcriptome assembly. Water 15:1246. doi: 10.3390/w15061246
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Soares, D., and Niemiller, M. L. (2013). Sensory adaptations of fishes to subterranean environments. Bioscience 63, 274–283. doi: 10.1525/bio.2013.63.4.7
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Sun, Y., Huang, Y., Li, X., Baldwin, C. C., Zhou, Z., Yan, Z., et al. (2016). Fish-T1K (Transcriptomes of 1,000 fishes) project: large-scale transcriptome data for fish evolution studies. Gigascience 5:18. doi: 10.1186/s13742-016-0124-7
Terán, G. E., Benitez, M. F., and Mirande, J. M. (2020). Opening the Trojan horse: phylogeny of Astyanax, two new genera and resurrection of Psalidodon (Teleostei: Characidae). Zool. J. Linnean Soc. 190, 1217–1234. doi: 10.1093/zoolinnean/zlaa019
Van der Weele, C. M., and Jeffery, W. R. (2022). Cavefish cope with environmental hypoxia by developing more erythrocytes and overexpression of hypoxia-inducible genes. Elife 11:69109. doi: 10.7554/eLife.69109
Wertheim, J. O., Murrell, B., Smith, M. D., Kosakovsky Pond, S. L., and Scheffler, K. (2015). RELAX: detecting relaxed selection in a phylogenetic framework. Mol. Biol. Evol. 32, 820–832. doi: 10.1093/molbev/msu400
Wheeler, T. J., and Eddy, S. R. (2013). Nhmmer: DNA homology search with profile HMMs. Bioinformatics 29, 2487–2489. doi: 10.1093/bioinformatics/btt403
Wilkens, H. (1988). Evolution and genetics of Epigean and cave Astyanax fasciatus (Characidae, Pisces). Evol. Biol. 23:271.
Wong, W., Huang, Y., Wu, Z., Kong, Y., Luan, J., Zhang, Q., et al. (2021). Mvda is required for zebrafish early development. Biol. Res. 54:17. doi: 10.1186/s40659-021-00341-7
Wu, H. Y., Chen, K. S., Huang, Y. S., Hsieh, H. Y., and Tsai, H. (2023). Comparative transcriptome analysis of skin color-associated genes in leopard coral grouper (Plectropomus leopardus). BMC Genomics 24:5. doi: 10.1186/s12864-022-09091-6
Yamagishi, M., Ishizuka, Y., Fujiwara, M., Nakamura, H., Igarashi, S., Nakano, Y., et al. (1993). Distribution of calcium binding proteins in sensory organs of the ear, nose and throat. Acta Otolaryngol. Suppl. 506, 85–89. doi: 10.3109/00016489309130248
Yang, J., Chen, X., Bai, J., Fang, D., Qiu, Y., Jiang, W., et al. (2016). The Sinocyclocheilus cavefish genome provides insights into cave adaptation. BMC Biol. 14:1. doi: 10.1186/s12915-015-0223-4
Yang, L., Xu, Z., Zeng, H., Sun, N., Wu, B., Wang, C., et al. (2020). FishDB: an integrated functional genomics database for fishes. BMC Genomics 21:801. doi: 10.1186/s12864-020-07159-9
Yap, N. V. L., Whelan, F. J., Bowdish, D. M. E., and Golding, G. B. (2015). The evolution of the scavenger receptor cysteine-rich domain of the class A scavenger receptors. Front. Immunol. 6:342. doi: 10.3389/fimmu.2015.00342
Yoshizawa, M. (2015). Behaviors of cavefish offer insight into developmental evolution. Mol. Reprod. Dev. 82, 268–280. doi: 10.1002/mrd.22471
Keywords: Teleostei, Characidae, cavefishes, RNA-seq, differential gene expression
Citation: Arcila D, Rincon-Sandoval M, Hanson W, Hart PB, González VL, Betancur-R R and Bichuette ME (2023) Transcriptomic analysis of the Brazilian blind characid, Stygichthys typhlops, reveals convergent selection with Astyanax mexicanus and other cavefishes. Front. Ecol. Evol. 11:1076756. doi: 10.3389/fevo.2023.1076756
Edited by:
Enrico Lunghi, University of L’Aquila, ItalyReviewed by:
Josh Gross, University of Cincinnati, United StatesCopyright © 2023 Arcila, Rincon-Sandoval, Hanson, Hart, González, Betancur-R and Bichuette. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dahiana Arcila, ZGthcmNpbGFAdWNzZC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.