 Melissa T. R. Hawkins
Melissa T. R. Hawkins Mary Faith C. Flores
Mary Faith C. Flores Michael McGowen1
Michael McGowen1 Arlo Hinckley
Arlo Hinckley
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Ecol. Evol., 17 August 2022
Sec. Phylogenetics, Phylogenomics, and Systematics
Volume 10 - 2022 | https://doi.org/10.3389/fevo.2022.984056
This article is part of the Research TopicRecent Advances in Museomics: Revolutionizing Biodiversity ResearchView all 16 articles
The extraction of nucleic acids is one of the most routine procedures used in molecular biology laboratories, yet kit performance may influence the downstream processing of samples, particularly for samples which are degraded, and in low concentrations. Here we tested several commercial kits for specific use on commonly sampled mammalian museum specimens to evaluate the yield, size distribution, and endogenous content. Samples were weighed and had approximately equal input material for each extraction. These sample types are typical of natural history repositories ranged from 53 to 130 years old. The tested protocols spanned spin-column based extractions, magnetic bead purification, phenol/chloroform isolation, and specific modifications for ancient DNA. Diverse types of mammalian specimens were tested including adherent osteological material, bone and teeth, skin, and baleen. The concentration of DNA was quantified via fluorometry, and the size distributions of extracts visualized on an Agilent TapeStation. Overall, when DNA isolation was successful, all methods had quantifiable concentrations, albeit with variation across extracts. The length distributions varied based on the extraction protocol used. Shotgun sequencing was performed to evaluate if the extraction methods influenced the amount of endogenous versus exogenous content. The DNA content was similar across extraction methods indicating no obvious biases for DNA derived from different sources. Qiagen kits and phenol/chloroform isolation outperformed the Zymo magnetic bead isolations in these types of samples. Statistical analyses revealed that extraction method only explained 5% of the observed variation, and that specimen age explained variation (29%) more effectively.
High throughput sequencing (HTS) has revolutionized the ability to recover genomic DNA from many unconventional sources. Most ancient DNA (aDNA) studies have been published since the first high throughput sequencer was available in 2008 (Knapp and Hofreiter, 2010). As such, it became more tangible to obtain nucleic acid sequences from samples which had historically performed poorly with standard Sanger sequencing and polymerase chain reaction (PCR) (Paabo et al., 2004). Degraded samples which did not yield high molecular weight DNA (fragments <1,000 bp), particularly benefited from this technology various starting sources can be considered degraded DNA (feces, eDNA, etc.); however, the focus of this work is dry mammalian museum collections.
Since the exponential decrease in sequencing cost, museum collections have become invaluable sources of degraded samples for genetic and genomic analyses. Natural history repositories house millions of specimens around the world and contain both temporally and geographically wide-ranging specimens for inclusion in genetic studies (Rowe et al., 2011; Bi et al., 2013; Holmes et al., 2016; Lopez et al., 2020; Buckner et al., 2021; Card et al., 2021; Colella et al., 2021). Museum specimens also allow for endangered, extinct, or elusive species to be represented when fresh tissues are not available (Ho and Gilbert, 2010; Fabre et al., 2014; Brüniche-Olsen et al., 2018; White et al., 2018). Additionally, by optimizing methodology for museum specimens, genomic signatures can be generated from type specimens, the individual specimen (or series) from which species descriptions are generated. This is important for the study of taxonomy as well as conservation and biodiversity (Guschanski et al., 2013; Chomicki and Renner, 2015; Zedane et al., 2016; Raxworthy and Smith, 2021).
Despite being beneficial for using such material to recover genetic signatures, the resulting DNA molecules are in low copy number and concentration as well as highly fragmentary (Burrell et al., 2015). Here we test several types of DNA extraction including phenol/chloroform, silica membrane, and magnetic bead isolation to determine if one method is superior for recovering DNA from degraded mammalian museum specimens. In addition to standard quality metrices (DNA concentration, size distribution, etc.) shotgun sequencing was performed to evaluate if any extraction methods appeared to bias the amount of endogenous versus exogenous DNA in each sample as assessed by metagenomic analyses.
A set of mammalian museum specimens were selected to represent common sources of nucleic acids from non-tissue-based museum holdings. A total of 17 samples were included in various comparisons of extraction protocols. First, 12 samples were extracted across three different extraction kits/protocols with approximately the same input mass per sample per extraction (see Table 1). In order to make extractions as comparable as possible all samples were weighed on the same scale, digested overnight, and manually processed in the same way across all three treatments. Depending on the type of sample (e.g., bone, dried tissue/osteocrusts, skin, baleen, and teeth) the amount of physical processing varied. For example, the baleen was shaved via a Dremel tool from a 2″ × 2″ square of baleen from the growth plate, and the fine powder was collected and divided into three replicates. Teeth were ground into a fine powder using a mortar and pestle. In contrast, the adherent muscle tissue and bone fragments required less manipulation and were weighed and divided in thirds for each replicate. This included cutting skin with scissors, or breaking osteocrusts and bone with a blade to allow each replicate as similar sample as possible. None of our sample types were subjected to a prewash as most (osteocrusts and bone fragments) are very fragile and the risk of losing sample outweighed the potential benefits of a prewash. Once weighed and placed in a 2.0 ml tube, the samples were broken down with forceps against the wall of the tube. After overnight digestion the samples were vortexed and evaluated for complete lysis of tissue. If large pieces remained, additional Proteinase K was added, and more physical manipulation of the tissues was performed with sterile instruments (particularly cutting up the skin into smaller fragments). After adding more Proteinase K the samples were vortexed and placed back into the shaker/incubator for another 1–2 h. Specific details for each kit are provided below.
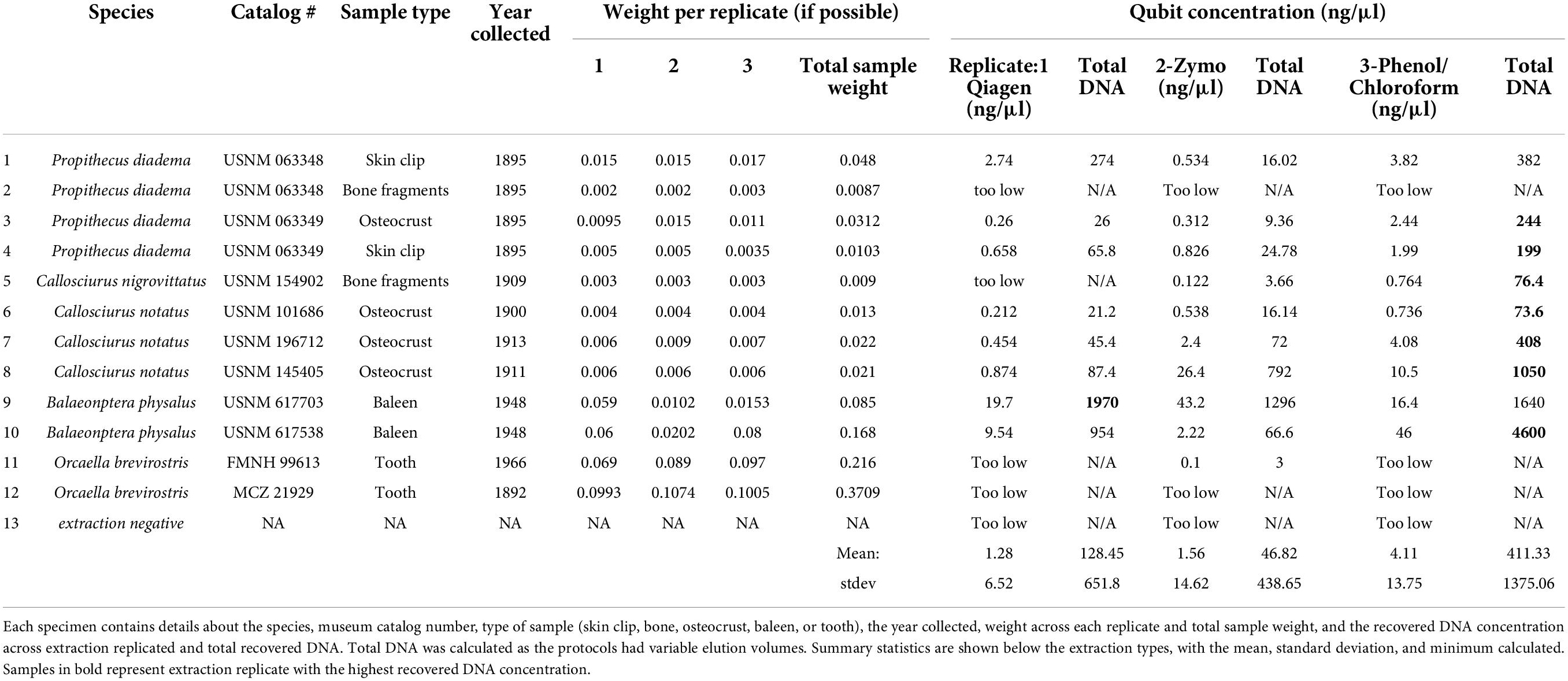
Table 1. A summary of the 12 samples extracted across the QIAamp®, Zymo®, and phenol/chloroform extractions.
The first protocol included using minor modifications (detailed below) to a Qiagen QIAamp DNA extraction kit. First, a Qiagen QIAamp DNA Mini Kit (#51306) was used to extract DNA following the manufacturer’s protocol (180 μl ATL plus 20 μl Proteinase K) with an overnight digestion at 56°C in a shaking incubator. The final elution step was done twice, with 50 μl of AE buffer added to the membrane, incubated, then centrifuged for a total elution volume of 100 μl.
The second kit used in this comparison was the Zymo DNA/RNA Viral MagBead Kit (#R2140). This kit was selected due to the increased recovery of low concentration, short insert size nucleic acids. The digestion was modified to have 10 μl of 10 mg/ml Proteinase K added (optional for viral studies of extracellular molecules, but part of the manufacturer’s protocol for tissue usage) with an overnight digestion at 56°C. The standard elution volume of 30 μl was retained. While performing this extraction it became clear that undigested tissues could potentially interfere with the magnetic bead steps, so upon magnetic separation any remaining undigested particles were removed via pipette tips. It is noteworthy that some museum specimen sample types (osteocrusts and bone) are difficult to fully lyse, but in spin column and phenol/chloroform extractions the particles do not interfere with subsequent steps.
The third extraction protocol followed a standard phenol/chloroform isolation as described in Hawkins et al. (2016), and originally detailed in Leonard et al. (2000). Briefly, an extraction buffer was prepared containing Tris + EDTA (100×), EDTA (0.5 M), NaCl (5 M), and water plus 10% SDS, DTT (400 mg/ml), and 20 μl of 10 mg/ml Proteinase K. The extraction buffer composition can be found in the Supplementary material. After samples were placed in the 1× extraction buffer they were incubated in a shaker/incubator overnight at 56°C. The next day two washes of phenol were used to separate proteins from nucleic acids followed by a chloroform wash. Top aqueous layers were removed and placed in clean tubes at each step and the final product was washed with 2 ml of water (1 ml washes performed twice) via an Amicon Ultra-4 centrifugal column and centrifuged at 3,300 RPM for 9 min. After the final spin, the volume was evaluated in each Amicon filter and an additional 8–12 min of centrifugation was performed to yield approximately 100 μl of purified DNA.
Ancient DNA laboratories have published various modifications to Qiagen spin column-based DNA extractions (Dabney and Meyer, 2019; Hagan et al., 2020; Xavier et al., 2021; Dehasque et al., 2022). Unfortunately we were not able to compare the 12 samples across four extraction protocols without resulting in extremely limited input for each replicate. However, as the aDNA protocol uses similar chemistry, reagents, and procedures as the QIAamp DNA extraction kit, we extracted five additional samples with both the aDNA protocol and the standard QIAamp protocol described above, with one minor modification (the addition of 20 μl of DTT 400 mg/ml to the extraction buffer) since the aDNA protocol also includes the usage of DTT. Samples spanned skin, adherent muscle tissue and nasal turbinates to determine if DNA concentration or size distribution of recovered molecules varied between protocols. The samples used for this comparison are provided in Table 2.
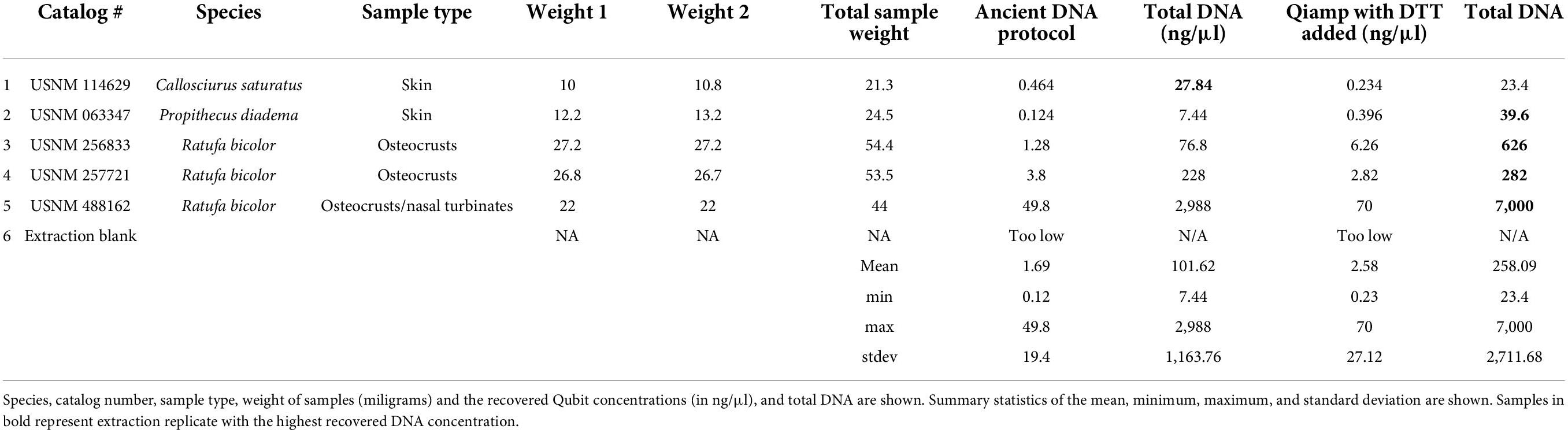
Table 2. Sample details for the ancient DNA extraction method (Hagan et al., 2020) and the modified QIAamp protocol which included DTT.
The aDNA protocol used here adhered closely to that described in Hagan et al. (2020), specifically “Method B,” which uses a Zymo reservoir attached to a MinElute Spin Column to allow for a larger volume of Qiagen Buffer PB (binding buffer) to be mixed with sample lysate following overnight digestion. From the published protocol we made a few modifications for better comparison to our QIAamp extractions. We added 1 ml of 0.5 M EDTA to the weighed sample, then added 100 μl proteinase K, and placed in a shaker incubator overnight at 37°C (recommended for aDNA). After the overnight digest samples were checked, 50 μl of additional proteinase K was added, as well as 20 μl of DTT (Sigma). Samples were vortexed and placed back in the shaker/incubator for two additional hours. We did not add a second milliliter of EDTA, and as such we did not use the full 13 ml of Buffer PB detailed in Hagan et al. (2020), and instead used 7 ml Buffer PB. The 50 ml conical tubes had the Zymo reservoir added, with the MinElute spin column snugly attached. Then a 5 ml tube had the lid removed and was placed inside the Falcon tube to help hold the spin column in place. Finally, the 5 ml tube had a hole drilled in the side with a Dremel to allow the buffer to flow out during centrifugation and prevent contaminating the MinElute column with flowthrough. The remaining spin and wash steps were the same as detailed in Hagan et al. (2020). Two washes of Buffer EB were done with 30 μl each wash for a final elution volume of 60 μl.
After extractions were completed, each sample was quantified via a Qubit (Invitrogen) dsDNA High Sensitivity Kit (# Q33230). From the Qubit concentrations the total DNA yield from each extraction was calculated. This was necessary as the different methods resulted in varying volumes of DNA. We also generated electropherograms of each extract to visualize if the protocols recovered varying size distributions based on the protocol for DNA isolation. An Agilent TapeStation 4200 was used with a high-sensitivity kit to evaluate the size distribution of the extracted DNA.
Dual indexed sequencing libraries were generated for all replicates and all extracts in this study with an Illumina Library Preparation—Kapa Biosystems Kit (Catalog # KK8232). Qubit values were used to pool samples in equimolar ratios with all replicates across this study. Once this was completed qPCR was performed on this pool of all samples with replication and dilution and the average size fragments of 250 bp (from a final TapeStation electropherogram) on an ABI ViiA7 using KAPA library quantification kit (#KK4824).
To evaluate recovered DNA content, shotgun sequencing was performed on an Illumina HiSeq X with an insert length of 150 bp PE. Sequencing was performed at Admera Health Biopharma Services, NJ, United States. Reads were demultiplexed via the BaseSpace Hub. Standard sequence quality filtering was performed with BBDUK (Bushnell, 2014) version 38.84 via Geneious Prime (Biomatters). All Illumina adapters were removed, and low-quality reads were removed from both ends (quality score minimum 20), and reads under 10 bp in length were discarded.
Metagenomic analysis of sample contents was performed to evaluate any biases between the extraction methods. Using the program MEGAN (Huson et al., 2016), we evaluated the major composition of identifiable taxa from each extract and used these results to identify any patterns across samples. DIAMOND (Baǧcı et al., 2021) BLAST was performed prior to importing results to MEGAN, and the DIAMOND “*.daa” were “meganized” and transformed into “*.RMA6” files to make comparisons across the replicates for each individual. Samples were compared using “MeganMap” from February 2022, and trees were made at the Phyla level. Comparisons were made across each sample by the different extraction method.
We used paired t-tests (two tailed p = 0.05) to determine if the quantified differences were statistically significant across extract method. We also performed linear regressions across the sample type, extraction type, age of sample, against recovered DNA concentration to determine which relationships explained our results better.
After comparing the three major kit types [silica-membrane (Qiagen), phenol/chloroform and magnetic beads (Zymo)] we found that most extractions recovered quantifiable DNA across the museum specimens. Samples that recovered detectable concentrations via Qubit recovered quantifications across all three extractions, and the samples which were too low to measure via Qubit were generally not quantified in any extraction method. When quantifiable, the minimum DNA concentrations ranged from 3 ng (Zymo) to 76.3 ng (phenol/chloroform) across replicates. The maximum across methods ranged from 1,296 to 4,600 ng, again with Zymo recovering the least and phenol/chloroform the most. The average yields were: 128.45 ng (Qiagen), 46.82 ng (Zymo), and 411.33 ng (phenol/chloroform) with the standard deviation high across all extraction methods. All quantification results are shown in Table 1. Despite the wide range of DNA concentrations, the yields were not statistically significant in any t-tests, details are provided in the Supplementary material. Linear regressions were performed on total DNA yield versus extraction method, as well as starting sample type. DNA extraction method only explained about 5% of the observed variation in DNA concentrations (Supplementary Table 1, p = 0.25). Alternatively, the starting template explained about 16% of the variation (Supplementary Table 2, p = 0.04). When a regression was performed using sample age and recovered DNA concentration, a total of 29.4% of the variation was explained by age (Supplementary Table 3, p = 0.003).
The recovered DNA concentrations between the aDNA and the QIAamp extraction kits were similar. For these samples recovery did not increase with use of the aDNA protocol. In fact, all concentrations were higher with the modified Qiagen kit. The average yield from the aDNA protocol was 101.62 ng versus 258.09 ng with the modified Qiagen extraction. The minimum for each was 7.44 ng (aDNA) and 23.4 ng (Qiagen), and maximum yield was 2,988 and 7,000 ng for aDNA and Qiagen kits respectively. The highest yield for both kits in this comparison was from the same sample (USNM 488162). Neither a one nor two tailed t-test was significant for this comparison. Details of all these comparisons can be found in Table 2. Regressions showed that extraction method was not significant, and explained 5% of the variation (Supplementary Table 4, p = 0.4), sample type was also not significant, and explained 18% (Supplementary Table 5, p = 0.22). Sample age explained 61% of the variation and was the only significant comparison for these samples (Supplementary Table 6, p = 0.007).
Despite samples being pooled in equimolar ratios, the number of reads varied across replicates. Quality filtering results recovered a general trend in which each sample had the same percentage of reads removed. However, the Zymo extracts appeared to have more reads removed than either Qiagen or phenol/chloroform. The MEGAN analysis showed that the proportion of endogenous and exogenous sequences was fairly consistent across replicates (Figure 1). Due to the variable components in each extract some amount of stochasticity between replicates was expected. Plots of all samples can be found in the Supplementary material.
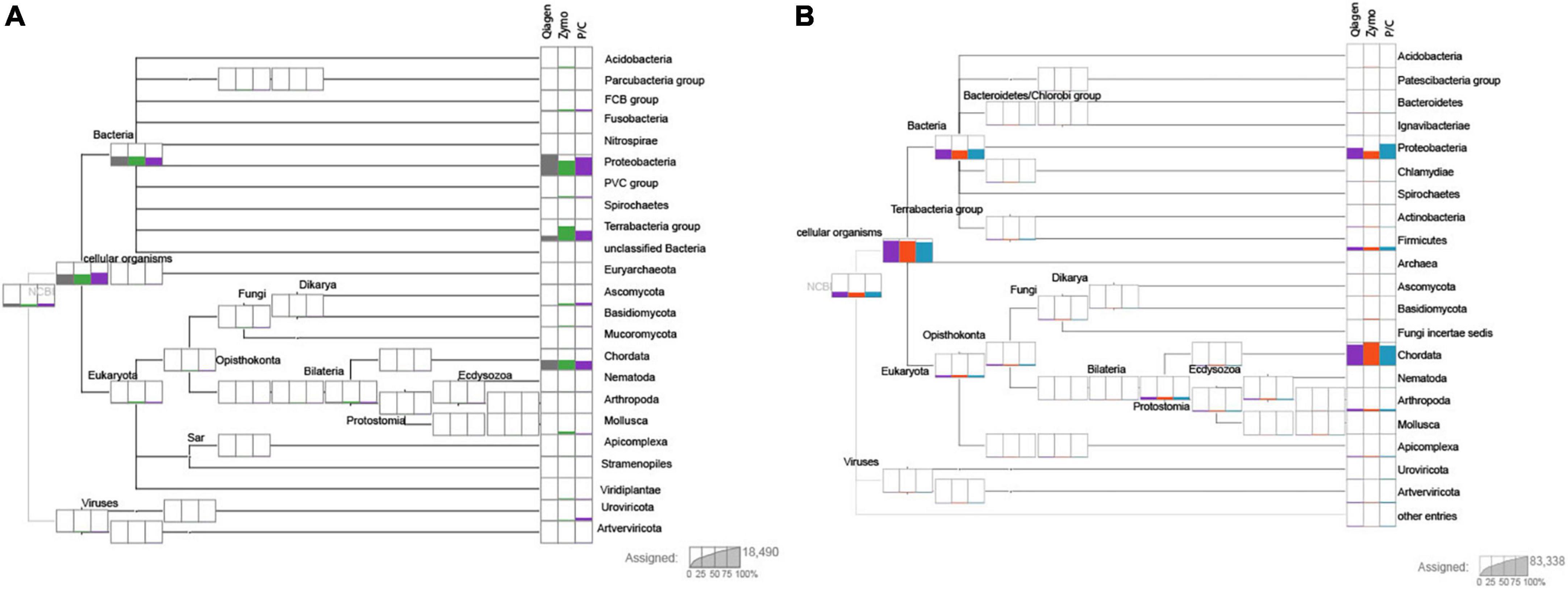
Figure 1. Two individuals after performing DIAMOND (Baǧcı et al., 2021) BLAST and importing through MEGAN (Huson et al., 2016). Sample 1 (USNM 063348, Propithecus diadema, skin clip) and sample 3 (USNM 063349, P. diadema, osteocrusts) are shown in panels (A,B), respectively. The columns at each terminal represent the extraction method, with Qiagen shown first, then Zymo and finally P/C for phenol/chloroform. The number of reads of each group are proportionally represented on each plot. Resolution is at the level of the phylum. Note the stochasticity between extraction method, but generally similar proportions of each phylum are represented across methods. Most samples recovered a high amount of Chordata and Proteobacteria with other phyla having more variable proportions. Individual plots for each sample can be found in the Supplementary material.
The second set of extraction comparisons was between an aDNA protocol (Hagan et al., 2020) and modifications to a Qiagen QIAamp extraction protocol. These samples recovered quite different size distributions as evaluated on the TapeStation (summarized in Supplementary Table 7). The aDNA protocol recovered TapeStation traces for all five samples, ranging in size from 50 to >850 bp. The modified Qiagen extraction only recovered traces for three samples, one of which was too large to determine the average size (obscured the upper marker). The sequencing results were also different between extraction methods, with the aDNA protocol retaining a much higher percentage of starting reads (average of 79%) than the modified Qiagen protocol (55%). Details of quality filtering are shown in Table 3. Subsequent research evaluating biases in length recovery across kits is warranted. It is also worth noting that despite many replicates lacking a visible peak, all samples yielded usable sequence.
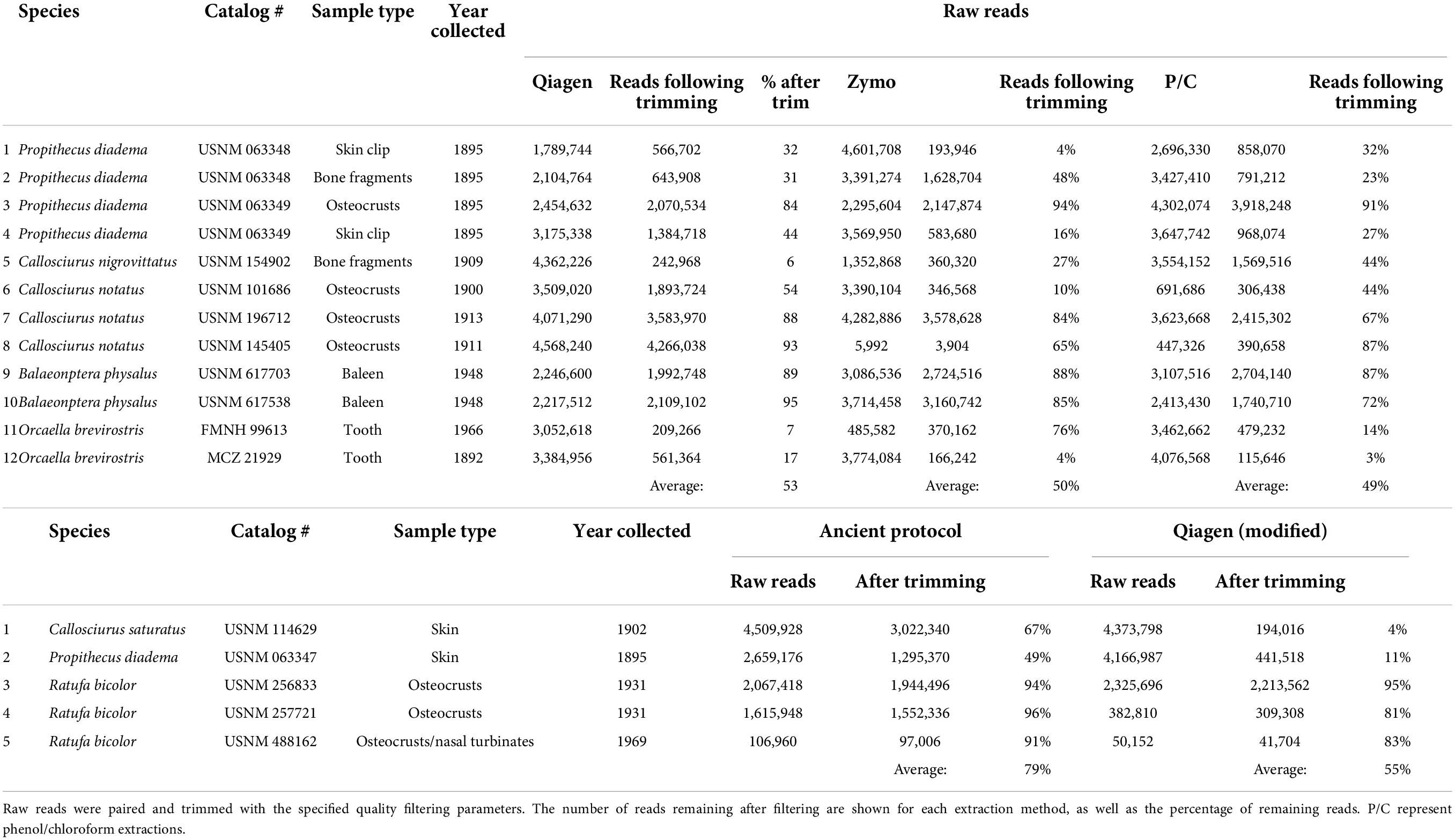
Table 3. Quality filtering results following BBDUK (Dehasque et al., 2022).
The Qiagen and phenol/chloroform protocols performed better than the Zymo kit in all metrics evaluated here. The Zymo kit was the most cost effective but is also marketed toward intracellular viruses and may not be geared for optimization of degraded vertebrate DNA. We modified the protocol as detailed by the manufacturer to lyse tissues but it appears to do so at the cost of losing the smallest fragments. The magnetic bead-based protocol is a quick and less toxic method for nucleic acid isolation, and is scalable for large tissue samples; however, it is not an effective protocol for the museum specimens tested here. Our results mirror those of McDonough (McDonough et al., 2018) supporting a similar DNA yield, fragment size and percentage of starting reads for Qiagen and phenol/chloroform.
One caveat of this study is that it is impossible to know if subsamples are truly representative across replicates. For example, one weighed subsample may contain bone of different density, and thus provide a better template for extraction than another. Similarly, the ratio of endogenous/contaminant DNA may vary among different osteocrust or skin clip subsamples of the same specimen. To our knowledge there is no way to use real-world specimens and account for this variable.
Concentration does not necessarily imply target DNA (Straube et al., 2021). Sample USNM 488162 had the highest DNA yield but also the lowest percentage of raw reads. Previous studies found that the highest concentration was associated with the highest amount of contamination in some samples (Campana et al., 2012; McDonough et al., 2018). The two included baleen samples also had a large amount of exogenous DNA (particularly bacteria). Specimen age has the largest impact on DNA yield. This is in contrast with other studies which showed no correlation between these variables (McDonough et al., 2018; Straube et al., 2021), but in corroboration with other studies (Yuan et al., 2021).
The average DNA yield and size from the ancient protocol was smaller than the modified Qiagen extraction, but the average percentage of reads after trimming was higher for the former than the latter, particularly for the two oldest samples (USNM 114629 and USNM 063347) which had a shorter fragment size (50–60 bp). The aDNA protocol does appear to do exactly as intended by retaining the smallest fragments; however, depending on the quantity and value of individual samples, the modified Qiagen kit may perform nearly as well while removing the smallest fragments. The aDNA protocol is more time consuming and expensive making the Qiagen kit a nearly equivalent option. Individual projects should perform their own cost-benefit analyses to determine which methods to employ.
Shotgun sequencing of samples provides valuable data to understand DNA content within a degraded sample. Studies of aDNA have found that the percent of endogenous DNA varies tremendously based on the preservation, age, and handling conditions following excavation from substrate (Dabney and Meyer, 2019). The most common phyla represented across taxa were Proteobacteria and Chordata. However, our results indicate that extraction methods had less of an effect on DNA recovery than either pre-extraction preservation or contamination. Other considerations for selection of an extraction protocol may be more important than sample contents as it does not appear that a bias was recovered across the extraction methods tested here. From this study we show that the Qiagen extractions (both standard and modified) recovered nearly the same profile of endogenous DNA as the more expensive phenol/chloroform, and more labor intensive aDNA protocol. Samples of the same specimen derived from different input (Figure 1; skin versus osteocrust) also show variation in the amount of endogenous DNA, with osteocrust out performing skin in this sample.
A comparison of kit and reagent costs is an important factor when budgeting for a grant and planning a project. Cost is important, as is efficiency, especially when using limited starting material which can be difficult or impossible to replace or resample. The cost of the kits tested here varied substantially and are detailed in the Supplementary material. Extractions ranged from $3.05 per sample to $11.54 per sample. Overall, the cheapest per sample cost was the Zymo kit, which ultimately had the poorest recovery in terms of concentration and appeared to lose more short fragments based on the TapeStation electropherograms.
Phenol/chloroform extractions were the most expensive ($8.39–11.54), especially when the Amicon Ultra-4 spin columns were used to wash the sample. It is possible to use different more cost-effective centrifugal columns (such as a Qiagen MinElute column), which reduces the cost to approximately $8.40 per sample. In any case, future studies should assess the efficiency of this and other modified phenol/chloroform protocols with alternative cost-effective washing steps following chloroform precipitation.
Based on the cost and endogenous DNA recovery, we show here that QIAamp kits perform well on mammalian museum specimens. With a price point at $3.76 per sample it is difficult to beat the savings, and the kits are well vetted and can be processed in a higher throughput on a QIAcube robot if throughput is a concern (although historical materials should always be performed in small batches with negative controls).
Single-tube and single-strand library preparation methods have been shown to yield better results than other approaches when working with highly degraded DNA (Gansauge et al., 2017; Carøe et al., 2018). Future research should evaluate the performance of combinations of DNA extractions and library preparation methods. The most expensive yet efficient phenol/chloroform extraction might yield better results in combination with a single-tube library preparation, since it has less bead cleaning steps than the KAPA library preparation protocol and will potentially lose fewer short fragments that are retained by the Amicon column during the extraction. However, if funding is limited and savings on DNA extraction are desirable, the QIAamp DNA extraction was fairly comparable to phenol/chloroform, at a much lower price point (under $4 an extraction) versus ∼$11.50 when using the Amicon filters. The phenol/chloroform protocol with a Qiagen spin column clean up saves approximately $3 per sample. Finally, the aDNA protocol did retain the smallest fragments, but it does not appear overly important for samples derived from museum specimens, as the fragmentary sequences are much more difficult to reconstruct, and with a price point of over $8.50/sample. The aDNA protocol did recover higher proportions of Chordata sequences in four of the five tested samples, so individual decisions should be made when determining the best methods to use for each project weighing the extraction cost, and availability of samples.
The datasets presented in this study can be found in online repositories. The data can be found at: https://figshare.com/s/260c150dcfa53a6f405b.
Ethical review and approval was not required for the animal study because all samples from this study involved long preserved museum specimens.
MH conceived the study and analyzed the data. MF, MH, and AH performed the laboratory work. All authors contributed to the article and approved the submitted version.
MH funded the study with discretionary startup funds provided by the Smithsonian Institution. AH was supported by a “Margarita Salas” postdoctoral fellowship funded by the Ministerio de Universidades de España and the European Union “NextGenerationEU.”
We thank Darrin Lunde, Ingrid Rochon from USNM, Adam Ferguson from FMNH, and Mark Omura from MCZ. Jeff Hunt, Katie Murphy, and Carrie Craig are thanked for assistance with laboratory logistics from the Laboratory of Analytical Biology. Some of the computations in this manuscript were conducted on the Smithsonian High Performance Cluster (SI/HPC), Smithsonian Institution (https://doi.org/10.25572/SIHPC).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2022.984056/full#supplementary-material
Baǧcı, C., Patz, S., and Huson, D. H. (2021). DIAMOND+MEGAN: Fast and easy taxonomic and functional analysis of short and long Microbiome sequences. Curr. Protoc. 1:e59. doi: 10.1002/cpz1.59
Bi, K., Linderoth, T., Vanderpool, D., Good, J. M., Nielsen, R., and Moritz, C. (2013). Unlocking the vault: Next-generation museum population genomics. Mol. Ecol. 22, 6018–6032. doi: 10.1111/mec.12516
Brüniche-Olsen, A., Jones, M. E., Burridge, C. P., Murchison, E. P., Holland, B. R., and Austin, J. J. (2018). Ancient DNA tracks the mainland extinction and island survival of the Tasmanian devil. J. Biogeogr. 45, 963–976. doi: 10.1111/jbi.13214
Buckner, J. C., Sanders, R. C., Faircloth, B. C., and Chakrabarty, P. (2021). The critical importance of vouchers in genomics. eLife 10:e68264. doi: 10.7554/eLife.68264
Burrell, A. S., Disotell, T. R., and Bergey, C. M. (2015). The use of museum specimens with high-throughput DNA sequencers. J. Hum. Evol. 79, 35–44. doi: 10.1016/j.jhevol.2014.10.015
Bushnell, B. (2014). BBMap: A fast, accurate, splice-aware aligner. Available online at: https://www.osti.gov/biblio/1241166 (accessed June 27, 2022).
Campana, M. G., Lister, D. L., Whitten, C. M., Edwards, C. J., Stock, F., Barker, G., et al. (2012). Complex relationships between mitochondrial and nuclear DNA preservation in historical DNA extracts. Archaeometry 54, 193–202. doi: 10.1111/j.1475-4754.2011.00606.x
Card, D. C., Shapiro, B., Giribet, G., Moritz, C., and Edwards, S. V. (2021). Museum genomics. Annu. Rev. Genet. 55, 633–659. doi: 10.1146/annurev-genet-071719-020506
Carøe, C., Gopalakrishnan, S., Vinner, L., Mak, S. S. T., Sinding, M.-H. S., Samaniego, J. A., et al. (2018). Single-tube library preparation for degraded DNA. Methods Ecol. Evol. 9, 410–419. doi: 10.1111/2041-210X.12871
Chomicki, G., and Renner, S. S. (2015). Watermelon origin solved with molecular phylogenetics including Linnaean material: Another example of museomics. New Phytol. 205, 526–532. doi: 10.1111/nph.13163
Colella, J. P., Bates, J., Burneo, S. F., Camacho, M. A., Carrion Bonilla, C., Constable, I., et al. (2021). Leveraging natural history biorepositories as a global, decentralized, pathogen surveillance network. PLoS Pathog. 17:e1009583. doi: 10.1371/journal.ppat.1009583
Dabney, J., and Meyer, M. (2019). Extraction of Highly Degraded DNA from Ancient Bones and Teeth. Methods Protoc. 1963, 25–29. doi: 10.1007/978-1-4939-9176-1_4
Dehasque, M., Pečnerová, P., Kempe Lagerholm, V., Ersmark, E., Danilov, G. K., Mortensen, P., et al. (2022). Development and Optimization of a Silica Column-Based Extraction Protocol for Ancient DNA. Genes 13:687. doi: 10.3390/genes13040687
Fabre, P.-H., Vilstrup, J. T., Raghavan, M., Sarkissian, C. D., Willerslev, E., Douzery, E. J., et al. (2014). Rodents of the Caribbean: Origin and diversification of hutias unravelled by next-generation museomics. Biol. Lett. 10:20140266. doi: 10.1098/rsbl.2014.0266
Gansauge, M.-T., Gerber, T., Glocke, I., Korlevic, P., Lippik, L., Nagel, S., et al. (2017). Single-stranded DNA library preparation from highly degraded DNA using T4 DNA ligase. Nucleic Acids Res. 45:e79. doi: 10.1093/nar/gkx033
Guschanski, K., Krause, J., Sawyer, S., Valente, L. M., Bailey, S., Finstermeier, K., et al. (2013). Next-generation museomics disentangles one of the largest primate radiations. Syst. Biol. 62, 539–554. doi: 10.1093/sysbio/syt018
Hagan, R. W., Hofman, C. A., Hübner, A., Reinhard, K., Schnorr, S., Lewis, C. M., et al. (2020). Comparison of extraction methods for recovering ancient microbial DNA from paleofeces. Am. J. Phys. Anthropol. 171, 275–284. doi: 10.1002/ajpa.23978
Hawkins, M. T. R., Leonard, J. A., Helgen, K. M., McDonough, M. M., Rockwood, L. L., and Maldonado, J. E. (2016). Evolutionary history of endemic Sulawesi squirrels constructed from UCEs and mitogenomes sequenced from museum specimens. BMC Evol. Biol. 16:80. doi: 10.1186/s12862-016-0650-z
Ho, S. Y., and Gilbert, M. T. P. (2010). Ancient mitogenomics. Mitochondrion 10, 1–11. doi: 10.1016/j.mito.2009.09.005
Holmes, M. W., Hammond, T. T., Wogan, G. O., Walsh, R. E., LaBarbera, K., Wommack, E. A., et al. (2016). Natural history collections as windows on evolutionary processes. Mol. Ecol. 25, 864–881. doi: 10.1111/mec.13529
Huson, D. H., Beier, S., Flade, I., Górska, A., El-Hadidi, M., Mitra, S., et al. (2016). MEGAN Community Edition - Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLoS Comput. Biol. 12:e1004957. doi: 10.1371/journal.pcbi.1004957
Knapp, M., and Hofreiter, M. (2010). Next Generation Sequencing of Ancient DNA: Requirements, strategies and perspectives. Genes 1, 227–243. doi: 10.3390/genes1020227
Leonard, J. A., Wayne, R. K., and Cooper, A. (2000). Population genetics of ice age brown bears. Proc. Natl. Acad. Sci. U.S.A. 97, 1651–1654. doi: 10.1073/pnas.040453097
Lopez, L., Turner, K. G., Bellis, E. S., and Lasky, J. R. (2020). Genomics of natural history collections for understanding evolution in the wild. Mol. Ecol. Resour. 20, 1153–1160. doi: 10.1111/1755-0998.13245
McDonough, M. M., Parker, L. D., Rotzel McInerney, N., Campana, M. G., and Maldonado, J. E. (2018). Performance of commonly requested destructive museum samples for mammalian genomic studies. J. Mammal. 99, 789–802. doi: 10.1093/jmammal/gyy080
Paabo, S., Poinar, H., Serre, D., Jaenicke-Despres, V., Hebler, J., Rohland, N., et al. (2004). Genetic Analyses from Ancient DNA. Annu. Rev. Genet. 38, 645–679. doi: 10.1146/annurev.genet.37.110801.143214
Raxworthy, C. J., and Smith, B. T. (2021). Mining museums for historical DNA: Advances and challenges in museomics. Trends Ecol. Evol. 36, 1049–1060. doi: 10.1016/j.tree.2021.07.009
Rowe, K. C., Singhal, S., Macmanes, M. D., Ayroles, J. F., Morelli, T. L., Rubidge, E. M., et al. (2011). Museum genomics: Low-cost and high-accuracy genetic data from historical specimens. Mol. Ecol. Resour. 11, 1082–1092. doi: 10.1111/j.1755-0998.2011.03052.x
Straube, N., Lyra, M. L., Paijmans, J. L. A., Preick, M., Basler, N., Penner, J., et al. (2021). Successful application of ancient DNA extraction and library construction protocols to museum wet collection specimens. Mol. Ecol. Resour. 21, 2299–2315. doi: 10.1111/1755-0998.13433
White, L. C., Mitchell, K. J., and Austin, J. J. (2018). Ancient mitochondrial genomes reveal the demographic history and phylogeography of the extinct, enigmatic thylacine (Thylacinus cynocephalus). J. Biogeogr. 45, 1–13. doi: 10.1111/jbi.13101
Xavier, C., Eduardoff, M., Bertoglio, B., Amory, C., Berger, C., Casas-Vargas, A., et al. (2021). Evaluation of DNA extraction methods developed for forensic and ancient dna applications using bone samples of different age. Genes 12:146. doi: 10.3390/genes12020146
Yuan, S. C., Malekos, E., and Hawkins, M. T. R. (2021). Assessing genotyping errors in mammalian museum study skins using high-throughput genotyping-by-sequencing. Conserv. Genet. Resour. 13, 303–317. doi: 10.1007/s12686-021-01213-8
Zedane, L., Hong-Wa, C., Murienne, J., Jeziorski, C., Baldwin, B. G., and Besnard, G. (2016). Museomics illuminate the history of an extinct, paleoendemic plant lineage (Hesperelaea, Oleaceae) known from an 1875 collection from Guadalupe Island, Mexico. Biol. J. Linn. Soc. 117, 44–57. doi: 10.1111/bij.12509
Keywords: museomics, degraded DNA, high throughput sequencing, bone, skin, baleen, osteological tissue
Citation: Hawkins MTR, Flores MFC, McGowen M and Hinckley A (2022) A comparative analysis of extraction protocol performance on degraded mammalian museum specimens. Front. Ecol. Evol. 10:984056. doi: 10.3389/fevo.2022.984056
Received: 01 July 2022; Accepted: 01 August 2022;
Published: 17 August 2022.
Edited by:
Jonathan J. Fong, Lingnan University, ChinaReviewed by:
Paúl M. Velazco, American Museum of Natural History, United StatesCopyright © 2022 Hawkins, Flores, McGowen and Hinckley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Melissa T. R. Hawkins, aGF3a2luc210QHNpLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.