 Ying Wang
Ying Wang Jinjun Cao
Jinjun Cao Xuan Guo1
Xuan Guo1
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol. , 15 August 2022
Sec. Phylogenetics, Phylogenomics, and Systematics
Volume 10 - 2022 | https://doi.org/10.3389/fevo.2022.979847
Nowadays, the position of Peltoperlidae in Systellognatha has been resolved based on morphological analyses. However, there are different opinions based on molecular data. To date, only three peltoperlid mitogenomes are available, and more sampling is needed to obtain precise phylogenetic relationships. In this study, we obtained the complete mitogenomes of Cryptoperla kawasawai (15,832 bp) and Peltoperlopsis sagittata (15,756 bp). Our results show that gene content, gene order, DmTTF binding site, nucleotide composition, codon usage, ribonucleic acid (RNA) structure, and structural elements in the control region are highly conserved in peltoperlids. Heatmap analysis of codon usage shows that the AT-rich codons UUA, AUU, UUU, and AUA were commonly used codons in the Peltoperlidae. Evolutionary rate analyses of protein-coding genes reveal that different genes have been subject to different rates of molecular evolution correlated with the GC content. All tRNA genes in peltoperlid mitogenomes have a canonical cloverleaf secondary structure except for trnS1, whose dihydrouridine arm simply forms a loop. The control region of the family has several distinct structural characteristics and has the potential to serve as effective phylogenetic markers. Phylogenetic analyses support the monophyly of Perloidea, but the monophyly of Pteronarcyoidea is still not supported. The Peltoperlidae is placed as the earliest branch within the Systellognatha, and the estimated phylogenetic relationship is: Peltoperlidae + {(Styloperlidae + Pteronarcyidae) + [Perlidae + (Chloroperlidae + Perlodidae)]}. Our results provide new insight into the phylogeny of this group.
The Plecoptera, also called stoneflies, are a small order of hemimetabolous insects of about 4,000 species worldwide (DeWalt et al., 2022). Because their nymphs dwell in aquatic habitats and are very sensitive to water quality, stoneflies can be used as monitors of healthy streams and rivers (Fochetti and Tierno de Figueroa, 2008). Stoneflies have a low level of vagility, making them ideal for biogeographic and phylogeographic research (Fochetti and Tierno de Figueroa, 2008; Pessino et al., 2014; McCulloch et al., 2016; Stevens et al., 2018). But the phylogenetic position of Plecoptera has long been under debate. Previously phylogenetic studies have indicated that Plecoptera is closely related to different insect taxa (Kukalová-Peck and Brauckmann, 1992). However, more conflicting hypotheses have been proposed by morphological evidence and the sister group of Plecoptera is still inconclusive (Terry and Whiting, 2005; Lin et al., 2010; Simon et al., 2012; Letsch and Simon, 2013; Misof et al., 2014; Song et al., 2016).
Several phylogenetic clades have been defined for Plecoptera, one of which is the infraorder Systellognatha. According to the morphological study of Zwick (2000), six extant families (Chloroperlidae, Perlidae, Perlodidae, Peltoperlidae, Pteronarcyidae, and Styloperlidae) in two superfamilies (Pteronarcyoidea and Perloidea) are included in Systellognatha. Recently, one new family (Kathroperlidae) was added to the superfamily Perloidea (South et al., 2021a). Finally, the infraorder includes seven families. Based on morphological evidence, Zwick (2000) reconstructed the broadly accepted phylogenetic relationship within Plecoptera. However, the phylogeny of Systellognatha remains debatable to date. As summarized in Figure 1, most studies placed Perloidea as a monophyletic group, but the monophyly of Pteronarcyoidea was not well supported. Meanwhile, the relationship among Perloidea was undetermined based on morphological data (Zwick, 2000), while it was recovered as Perlidae + (Chloroperlidae + Perlodidae) by most studies based on the mitochondrial genome (mitogenome) and transcriptome data (Chen et al., 2018; Shen and Du, 2019, 2020; Veale et al., 2019; Wang et al., 2019; South et al., 2021a,b; Mo et al., 2022). These results generated by mitogenome and transcriptome data are inconsistent with those by single or multiple genes and other transcriptome data (Thomas et al., 2000; Terry and Whiting, 2003; Davis, 2013). Therefore, the phylogeny of Systellognatha remains controversial, and more phylogenetic studies are needed.
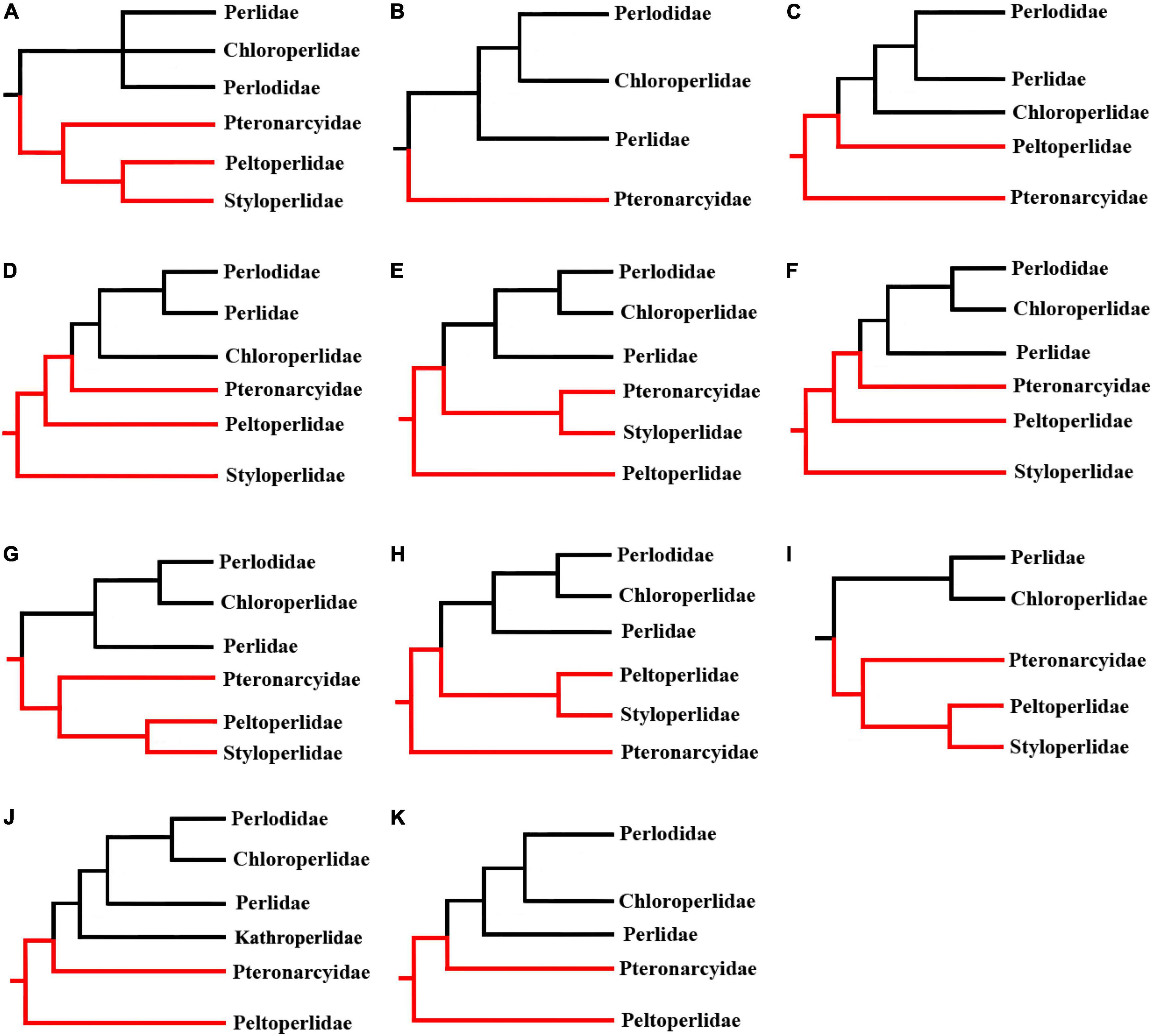
Figure 1. Representations of previous hypotheses for the family-level relationships within Systellognatha. (A) Zwick (2000), morphological data; (B) Davis (2013), transcriptome data; (C) Thomas et al. (2000), 18S gene; (D) Terry and Whiting (2003), six molecular markers; (E) Chen et al. (2018) and Mo et al. (2022), mitogenome data; (F) Shen and Du (2019) and Veale et al. (2019), mitogenome data; (G) Shen and Du (2020), mitogenome data; (H) Wang et al. (2019), mitogenome data; (I) Wang et al. (2018) and Ding et al. (2019), mitogenome data; (J) South et al. (2021a), transcriptome data; (K) South et al. (2021b), transcriptome data (concatenated complete nucleotide data set). Clades with black and red color belong to the superfamily Perloidea and Pteronarcyoidea, respectively.
Peltoperlidae is one of the smallest families of Plecoptera with approximately 50 species distributed in the Nearctic, Palearctic, and Oriental regions (Zwick, 2000; DeWalt et al., 2022). The nymphs are most common in very shallow running water, like seeps on rock faces, but they also occur in a variety of small streams and rivers (DeWalt et al., 2022). The nymphs are easily identified by their “roach-like” appearance, and the adults can be distinguished from other systellognathan stoneflies in the field by their small heads (Zwick, 2000). Peltoperlidae belongs to the superfamily Pteronarcyoidea, and the monophyly of Pteronarcyoidea was supported by Zwick’s (2000) morphological phylogeny. However, phylogenetic relationships among three families in Pteronarcyoidea are very controversial all the time, as evidenced by molecular studies (Figure 1). Taxon sampling in these molecular studies has been limited, and not enough to obtain precise phylogenetic relationships within Systellognatha.
A well-constructed molecular phylogeny could substantially benefit the understanding of evolutionary relationships of major lineages and morphological character evolution, as in this case, resolving superfamily monophyly and the phylogenetic relationships within the Systellognatha. Complete mitogenomes contain more useful evolutionary information than single or multiple genes and have been widely used to investigate insect relationships at different taxonomic scales (Fenn et al., 2008; Dowton et al., 2009; Cameron, 2014), due to their small size, conserved gene components, maternal inheritance, rare recombination, and relatively high evolutionary rate (Boore, 1999; Curole and Kocher, 1999; Barr et al., 2005).
So far, there are approximately forty complete or near complete systellognathan mitogenomes available in GenBank, of which only nine species belong to Pteronarcyoidea. In this study, two complete mitogenomes from the family Peltoperlidae [Cryptoperla kawasawai Maruyama, 2002 and Peltoperlopsis sagittata (Cao et al., 2019)]. were sequenced. We conducted a comparative analysis of those newly sequenced mitogenomes and four published peltoperlid mitogenomes. Finally, we investigated the phylogenetic relationships within Systellognatha.
Adult male specimens of C. kawasawai were collected from Kumakogen town (Ehime Prefecture, Japan; May, 2016) and of P. sagittata from Gaoligong Mountain (Yunnan Provence, China; July, 2016). Before this study, all samples were stored in 100% alcohol and maintained at −20°C. Total genomic deoxyribonucleic acid (DNA) was extracted from the thoracic muscle using the DNeasy tissue kit (Qiagen, Hilden, Germany). NanoDrop One (Thermo Scientific, Waltham, MA, United States) was used to measure the DNA concentration for each sample. DNA samples with qualified concentration (>10 μg) were sent to Berry Genomics Co., Ltd. (Beijing, China) for further detecting. From the genomic DNA, an Illumina TruSeq library with an insert size of 480 bp was generated. The de novo genome sequencing was conducted on an Illumina Hiseq 2500 platform with 500 cycles of paired-end sequencing (250 bp reads).
Trimmomatic v0.30 (Lohse et al., 2012) was used for de novo assembly of high-quality data. A total of 6 Gb clean data were obtained and used in the de novo assembly using IDBA-UD (Peng et al., 2012) with minimum and maximum k values of 45 and 145 bp. COI and srRNA fragments were amplified as bait sequences using PCR (Li et al., 2012). Then, the mitogenome sequence was searched with the bait sequences using BLAST with at least 98% similarity. Two newly sequenced mitogenomes have been deposited in GenBank (Table 1). MITOS web server was used to identify transfer ribonucleic acid genes (tRNAs) (Bernt et al., 2013). Protein-coding genes (PCGs) and ribosomal RNA genes (rRNA) were identified by alignment with homologous genes of previously sequenced stonefly mitogenomes. The graphical maps of two mitogenomes were depicted with CGView Server (Grant and Stothard, 2008). Nucleotide composition and codon usage was obtained using MEGA 6.0 (Tamura et al., 2013). The codon usage was visualized by a heatmap using the online tool CIMminer.1 AT and GC skew were calculated via the following formula: AT-skew = (A − T)/(A + T), GC-skew = (G − C)/(G + C) (Perna and Kocher, 1995). The rates of Ka (the non-synonymous substitution rate) and Ks (the synonymous substitution rate) for each PCG were determined with DnaSP 5.0 (Librado and Rozas, 2009). The tandem repeats in the control region were predicted using the Tandem Repeat Finder server (Benson, 1999).
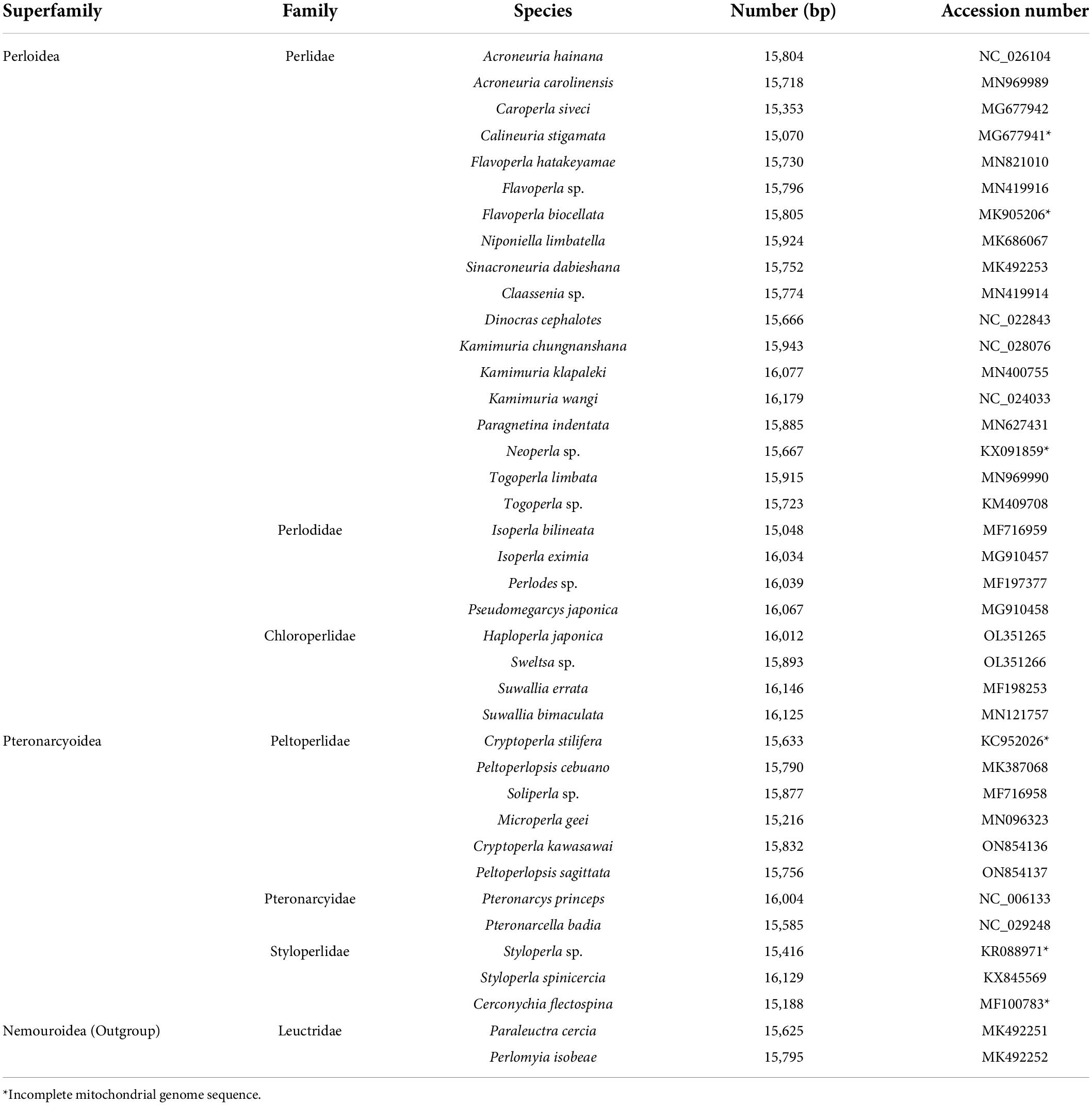
Table 1. List of taxa used in this research.
To analyze the phylogenetic relationships among Systellognatha, thirty-seven systellognathan mitogenomes were involved in our phylogenetic analysis. Two leuctrid mitogenomes (Paraleuctra cercia and Perlomyia isobeae) from infraorder Euholognatha were used as outgroups (Table 1). Two datasets were assembled for phylogenetic analyses: (1) the “PCG matrix” (10,971 bp), including 13 PCGs; (2) the “13 PCGs and two rRNAs (PCGRNA) matrix” (12,750 bp), including 13 PCGs and two rRNAs. PCGs were aligned independently using the MAFFT algorithm within the TranslatorX online platform (Abascal et al., 2010). Before the protein alignment was back-translated to nucleotides, GBlocks (in TranslatorX) with default settings were used to remove ambiguously aligned areas. The G–INS–I alignment strategy in MAFFT 7.0 online was used for rRNA alignment (Katoh and Standley, 2013), and ambiguously aligned sites masked with Gblocks (Castresana, 2000).
Phylogenetic analyses were conducted using maximum likelihood (ML) and Bayesian inference (BI). The best-fit model for each dataset was determined using ModelFinder applying the Akaike Information Criterion (AIC) (Nguyen et al., 2015). ML analyses were inferred using IQ–TREE (Nguyen et al., 2015), and an ultrafast bootstrap approximation with 1,000 replicates. Bayesian analyses were carried out using MrBayes v3.2.6 (Ronquist et al., 2012) with the selected model (GTR + I + G). For MrBayes, runs were as follows: 10 million generations with four chains, sampling every 100 generations, and the first 25% discarded as burn-in.
We successfully obtained two complete mitogenomes of the family Peltoperlidae and submitted the sequences to GenBank (accession numbers: ON854136-ON854137). The genome sizes were 15,832 bp (C. kawasawai) and 15,756 bp (P. sagittata), respectively (Supplementary Tables 1, 2 and Figure 2). The gene order and content of two mitogenomes were typical of Plecoptera (Chen et al., 2018; Cao et al., 2019; Wang et al., 2019) and highly conserved, including 37 genes (22 tRNAs, 13 PCGs, and two rRNAs) and a non-coding control region (CR) (Figure 2). Compared with other systellognathan species, the sizes of two newly sequenced peltoperlid mitogenomes were close to average (15,832 bp, only counting the complete mitogenomes) (Table 1). In the peltoperlid mitogenomes, variation in the length of PCGs, tRNAs, lrRNA (large subunit ribosome gene), and srRNA (small subunit ribosome gene) was inconspicuous. But the length variation was very different in the control region (Table 2), which differed in both the replicates and length of various short repeat sequences within it (Dotson and Beard, 2001).
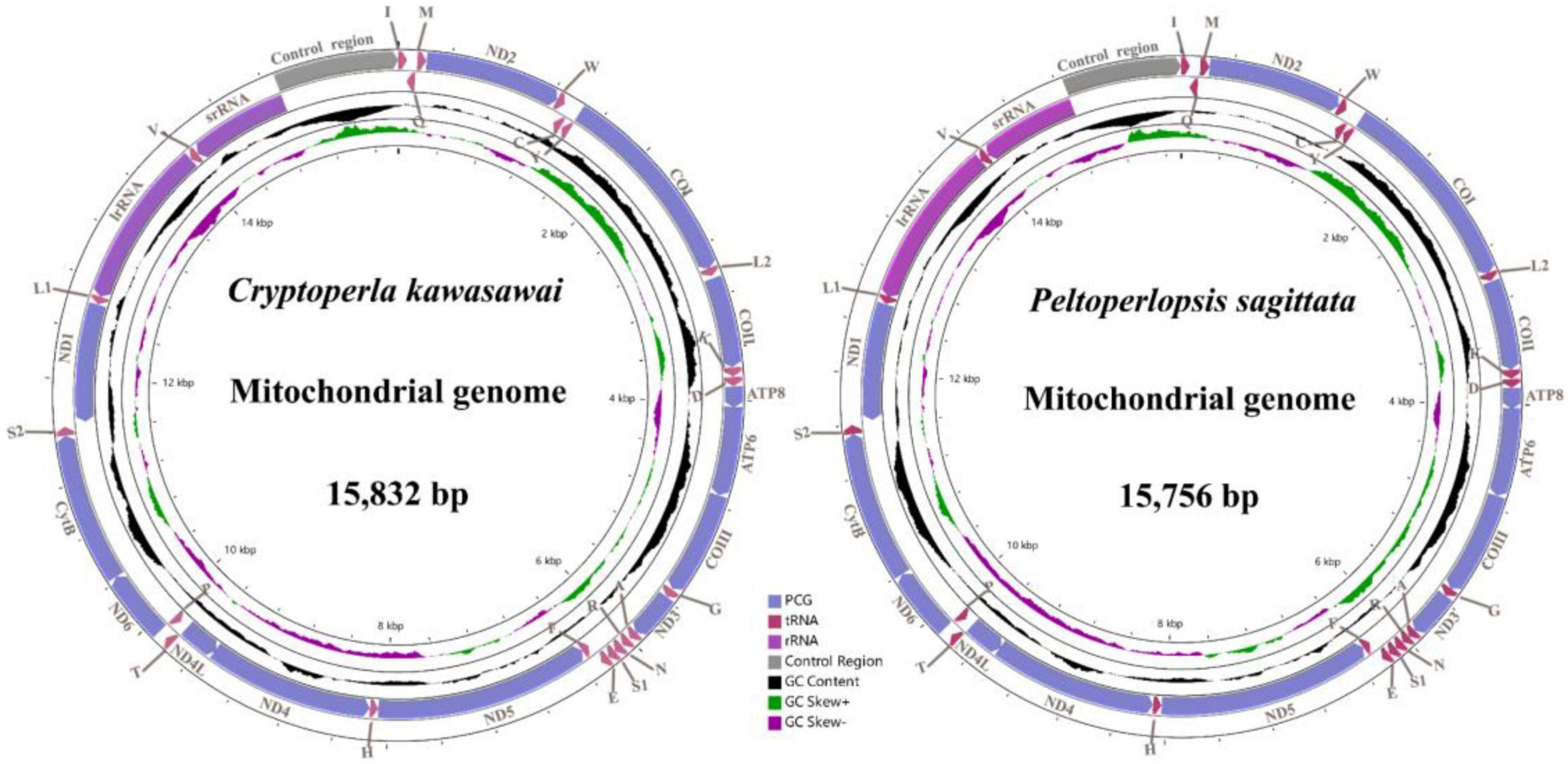
Figure 2. Mitochondrial maps of Cryptoperla kawasawai and Peltoperlopsis sagittata. The direction of gene transcription is indicated by the arrows. tRNA genes are labeled according to single-letter International Union of Pure and Applied Chemistry-International Union of Biochemistry (IUPAC-IUB) abbreviations (L1, UUR; L2, CUN; S1, AGN; S2, UCN).
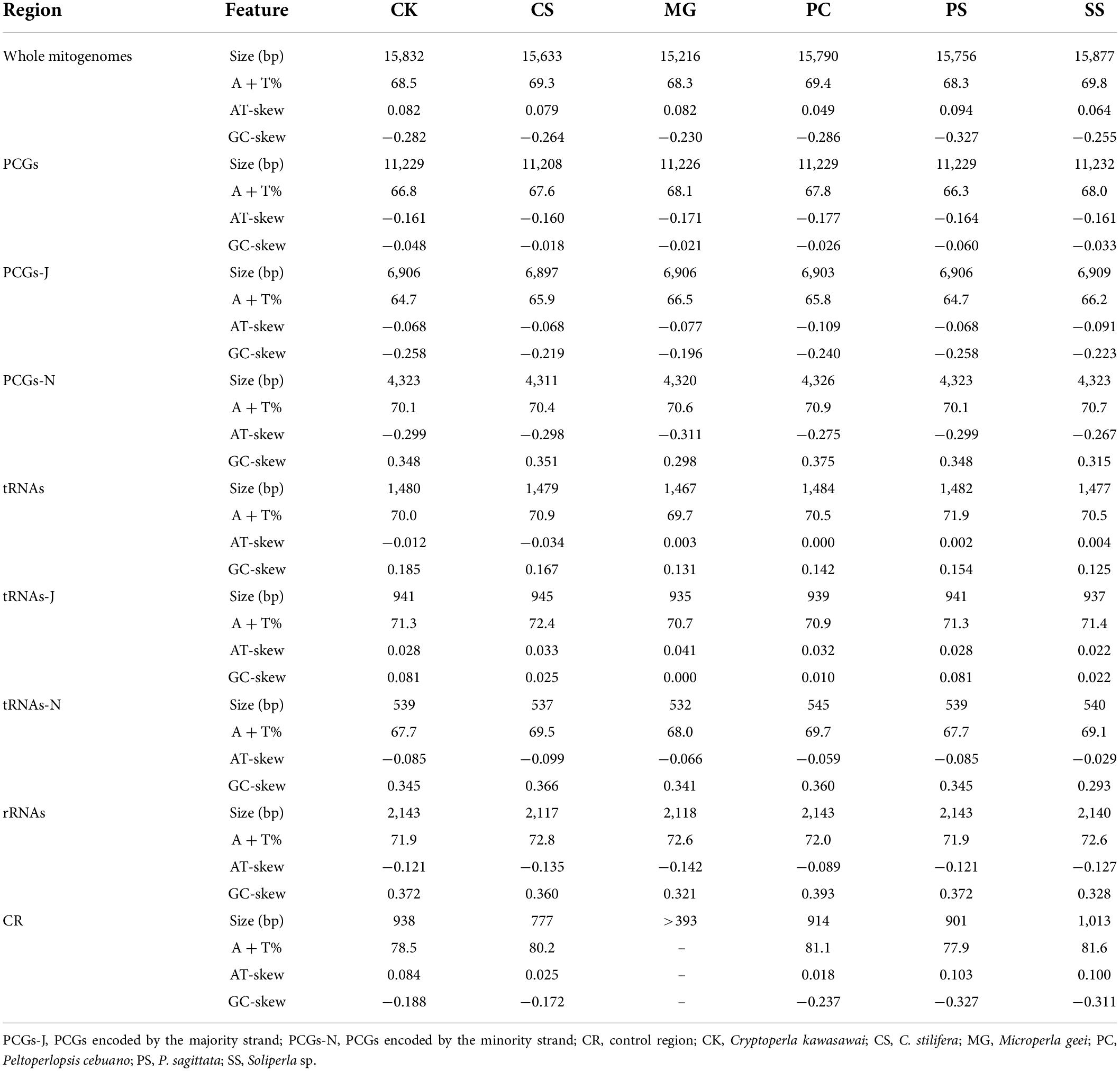
Table 2. Structural features of the mitochondrial genomes across six species of family Peltoperlidae.
Gene overlaps and spacers were presented in several conserved positions in the peltoperlid mitogenomes, such as trnI-trnQ (3 bp), trnW-trnC (−8 bp), COI-trnL2 (−5 bp), ATP8-ATP6 (−7 bp), ND4-ND4L (−7 bp), ND4L-trnT (2 bp), etc (Supplementary Table 3). In Drosophila melanogaster, two conserved non-coding intergenic regions (trnE-trnF and trnS2-ND1) have been considered to be bidirectional transcription termination factor (DmTTF) binding sites (Beckenbach, 2012). In this study, we aligned the sequences of these two regions in six peltoperlid species and D. melanogaster (Supplementary Figure 1). Like other stonefly and insect mitogenomes (Beckenbach and Stewart, 2009; Beckenbach, 2012; Wang et al., 2018; Cao et al., 2019), the DmTTF binding site of trnE-trnF was absent in six peltoperlid mitogenomes (Supplementary Figure 1). However, the DmTTF binding site of trnS2-ND1 (the longest spacer sequence except for the control region) was found in the six species and is highly conserved across insects (Cameron and Whiting, 2008; Beckenbach, 2012; Wang et al., 2018; Cao et al., 2019). In addition, the 7 bp gene pairs (ATP8/ATP6 and ND4/ND4L) are often been found across the Metazoa (Stewart and Beckenbach, 2005; Carapelli et al., 2006), and thought to be translated as a bicstron (Stewart and Beckenbach, 2005).
The nucleotide compositional behavior of mitogenomes can be analyzed by A + T content, AT skew, and GC skew (Hassanin et al., 2005; Wei et al., 2010). The nucleotide composition of the C. kawasawai mitogenome (A = 37.1%, T = 31.4%, G = 11.3%, C = 20.2%) was similar to that of P. sagittata (A = 37.4%, T = 30.9%, G = 10.6%, C = 21.0%). Similar to other published stoneflies, the nucleotide compositions of the six peltoperlid species revealed a strong A and T base bias in all six mitogenomes (Chen et al., 2018; Wang et al., 2018, 2019; Ding et al., 2019; Shen and Du, 2019, 2020; Mo et al., 2022). By comparison, the control region of all six peltoperlid mitogenomes showed a higher A + T content than other major partitions (e.g., PCGs, tRNAs, rRNAs, etc.) (Table 2).
The nucleotide compositions were all strongly skewed away from T in favor of A (the AT-skews were from 0.049 to 0.094) and from G in favor of C (the GC-skews were from −0.230 to −0.327) (Table 2). In most metazoan mitogenomes, the strand skew biases are found to be weakly positive AT-skew and strongly negative GC-skew for the J-strand (Hassanin et al., 2005). Our results show that all the peltoperlid PCGs have a negative AT-skew and negative GC-skew for the J-strand, while all the peltoperlid tRNAs have a positive AT-skew and positive GC-skew for the J-strand (Table 2). This pattern is inconsistent with most insect mitogenomes, but it is also found in many stonefly mitogenomes (Chen et al., 2018; Wang et al., 2018, 2019; Cao et al., 2019). This phenomenon may result from the gene direction, replication, and codon positions (Lindahl, 1993; Wei et al., 2010).
We detected 13 protein-coding genes in two newly sequenced mitogenomes. Similar to other peltoperlid mitogenomes, nine PCGs were encoded on the majority strand (J-strand), and the remaining four PCGs were encoded on the minority strand (N-strand) (Figure 2). Six out of thirteen PCGs (COI, ND1, ND2, ND4, ND5, and ND6) differed in size among the six peltoperlid species (Supplementary Table 4). But in general, the length variation in those genes was limited.
Most PCGs in six peltoperlid species initiated with a typical ATN codon, while ND2 in Peltoperlopsis cebuano initiated with GTG, ND5 in five peltoperlids (one exception: Soliperla sp.) started with GTG, and ND1 in all peltoperlids initiated with TTG (Supplementary Table 4). Most PCGs terminated with the canonical TAA/TAG stop codon in six peltoperlid species. The incomplete stop codon T was found in COII and ND5 genes in most of the six peltoperlid mitogenomes (Supplementary Table 4). These incomplete codons may be the product of the selective pressure to economize the mitogenome size and are presumed to be completed via post-transcriptional polyadenylation (Ojala et al., 1981). In addition, each of the five genes (ATP6, COII, COIII, ND4L, and ND6) used the same start and stop codons, indicating these genes were highly conserved among all species (Supplementary Table 4).
In our study, a heatmap was used to visualize codon usage for the 13 PCGs available in the Peltoperlidae, with the color representing the frequency of codon usage (Figure 3). Within peltoperlids, similar but slightly different patterns were observed. Heatmap analysis showed that the AT-rich codons UUA, AUU, UUU, and AUA were commonly used codons in the Peltoperlidae. Similarly, the biased use of A + T nucleotides was reflected in the codon frequencies. The dendrogram based on codon usage showed a close relationship between C. kawasawai and the clade of Soliperla sp. plus Cryptoperla stilifera, and P. sagittata is the earliest branch within Peltoperlidae. The monophyly of Cryptoperla and Peltoperlopsis was not supported.
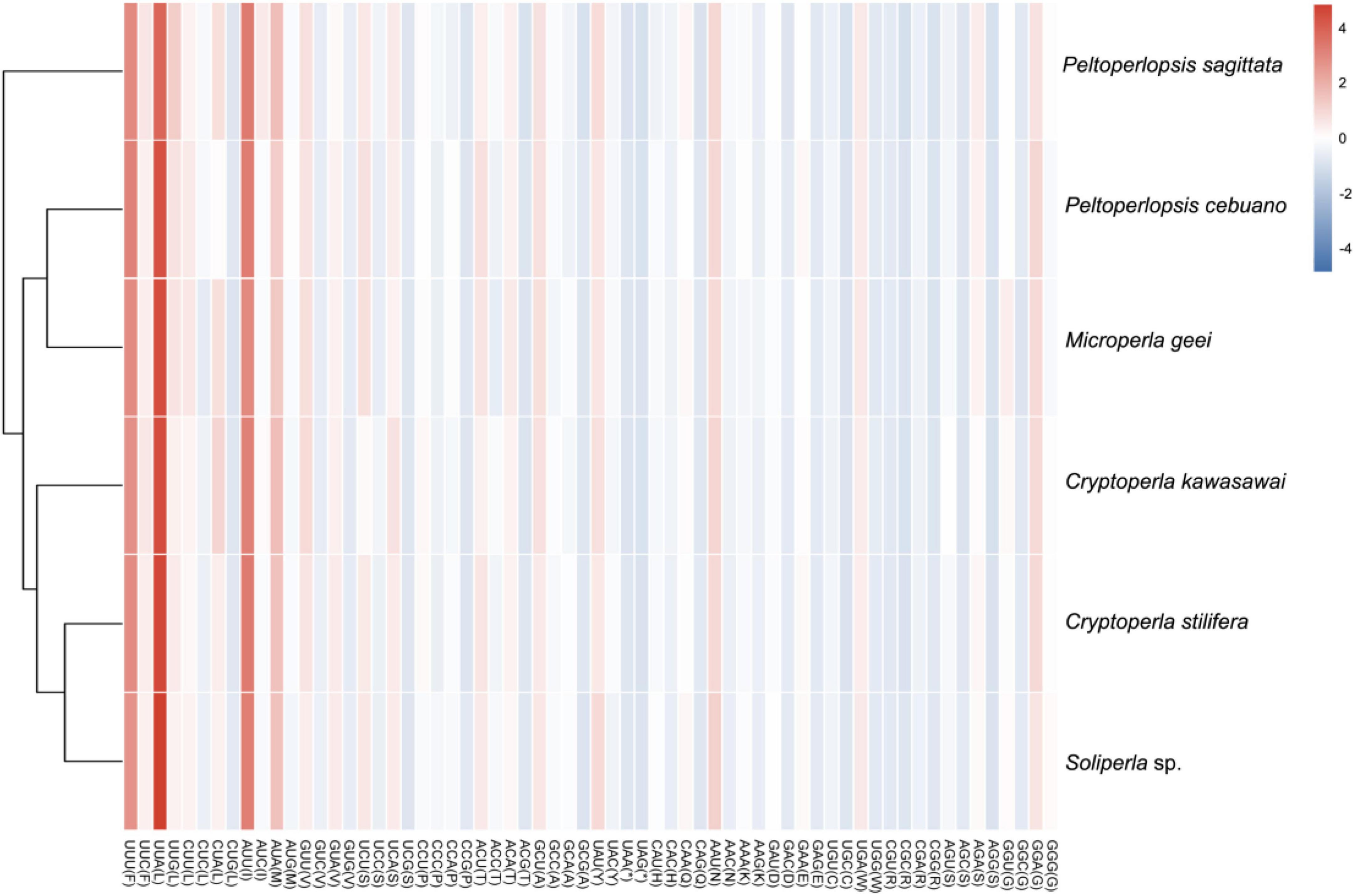
Figure 3. Heatmap of codon usage for protein-coding genes in Peltoperlidae. Red and blue in the heatmap indicate high and low absolute correlation, respectively. The species in the rows of the heat map are sorted by the corresponding cluster tree based on codon usage. The columns indicate 64 codons of the invertebrate mitochondrion.
To better investigate the evolutionary patterns across the 13 PCGs in peltoperlid species, the values of Ka (rates of non-synonymous mutations), Ks (rates of synonymous mutations), and the ratio of Ka/Ks (ω) were calculated for each PCG, respectively (Figure 4). In all PCGs, COIII had the highest Ks, whereas ND6 had the highest Ka and ω values. The ω values for 13 PCGs were far lower than 1 (<0.40), indicating the existence of purifying selection in these genes (Roques et al., 2006). Therefore, all mitochondrial PCGs could be used to analyze phylogenetic relationships within Peltoperlidae. In addition, we found a negative correlation between ω and the G + C content of each PCG in peltoperlid species (y = −24.37x + 36.38, R2 = 0.89, P < 0.05). This result is similar to previous studies and indicates that G + C content may be one important element in determining the evolutionary patterns of PCGs (Li et al., 2012; Yuan et al., 2015; Zhang et al., 2016).

Figure 4. Evolutionary rates of 13 protein-coding genes in the mitochondrial genomes of six peltoperlid species. The left Y-axis provides the substitution rate of the mitochondrial gene, while the right Y-axis provides the G + C content.
All tRNA genes in six peltoperlid species showed classical cloverleaf structures except for trnS1, whose dihydrouridine (DHU) arm simply formed a loop (Figure 5). The lack of a DHU arm in trnS1 was also found in sequenced stonefly mitogenomes, and this phenomenon has been considered a typical feature of metazoan mitochondrial DNA (Lavrov et al., 2004). Although it is not clear if the aberrant tRNAs lose function in this case, some studies proposed a post-transcriptional RNA editing mechanism to keep these tRNA genes functional (Tomita et al., 2001).
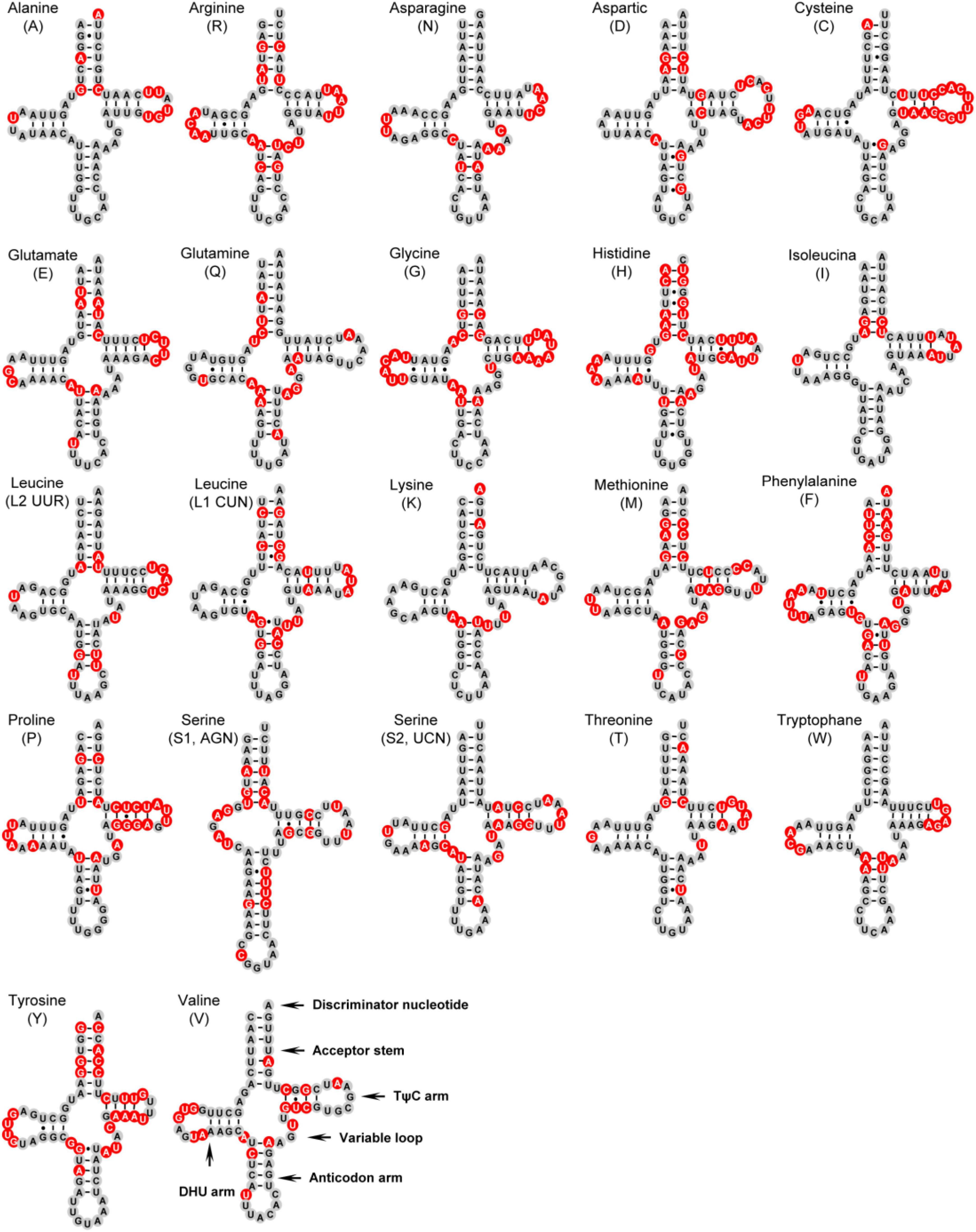
Figure 5. Putative secondary structures of tRNAs found in the Cryptoperla kawasawai mitogenome. Completely conserved sites within the six peltoperlid species were marked by black nucleotides within gray spheres. Inferred Watson–Crick bonds are illustrated by lines, whereas guanine–uracil (GU) bonds are illustrated by dots.
We calculated the percentage of identical nucleotides (%INUC) for each tRNA family of the six peltoperlid mitogenomes (Supplementary Table 5). The%INUC ranged from 54.2% in trnH to 88.7% in trnK, with an average of 72.8%. Eleven tRNAs displayed high levels of conservation (%INUC ≥ 75.0%). Nucleotides in the stems and loops of the tRNAs were relatively conserved (>70%). The most conserved site was the anticodon (AC) loop with an average of 96.1%, and the most variable region was the TψC loop (with an average of 33.2%). In addition, the conservation of each stem, with the exception of the AC loop, was always higher than that of its corresponding loop.
As in the inferred ancestral insect mitogenome pattern, the two rRNA genes were usually separated by a single trnV gene. The lengths of lrRNA in the two newly sequenced mitogenomes were 1,334 and 1,344 bp, and the lengths of srRNA were 809 and 796 bp, respectively (Supplementary Tables 1, 2). The multiple alignments of peltoperlid lrRNAs had 1382 positions and contained 837 conserved positions (60.6%), 518 nucleotide substitutions (37.5%), and 27 indels (1.9%), respectively. The multiple alignments of peltoperlid srRNAs possessed 822 positions and contained 491 conserved positions (59.7%), 319 nucleotide substitutions (38.8%), and 12 indels (1.5%), respectively (Supplementary Figure 2).
The control region is located between srRNA and trnI, including the origin of replication and promoters for transcription initiation (Zhang et al., 1995). A comparison of the control region sequences of six peltoperlid species revealed a few structural elements: (1) a leading sequence adjacent to srRNA with high AT content; (2) one or two tandem repeated sequence blocks consisting of repeat units; (3) the remainder of the control region (Figure 6).
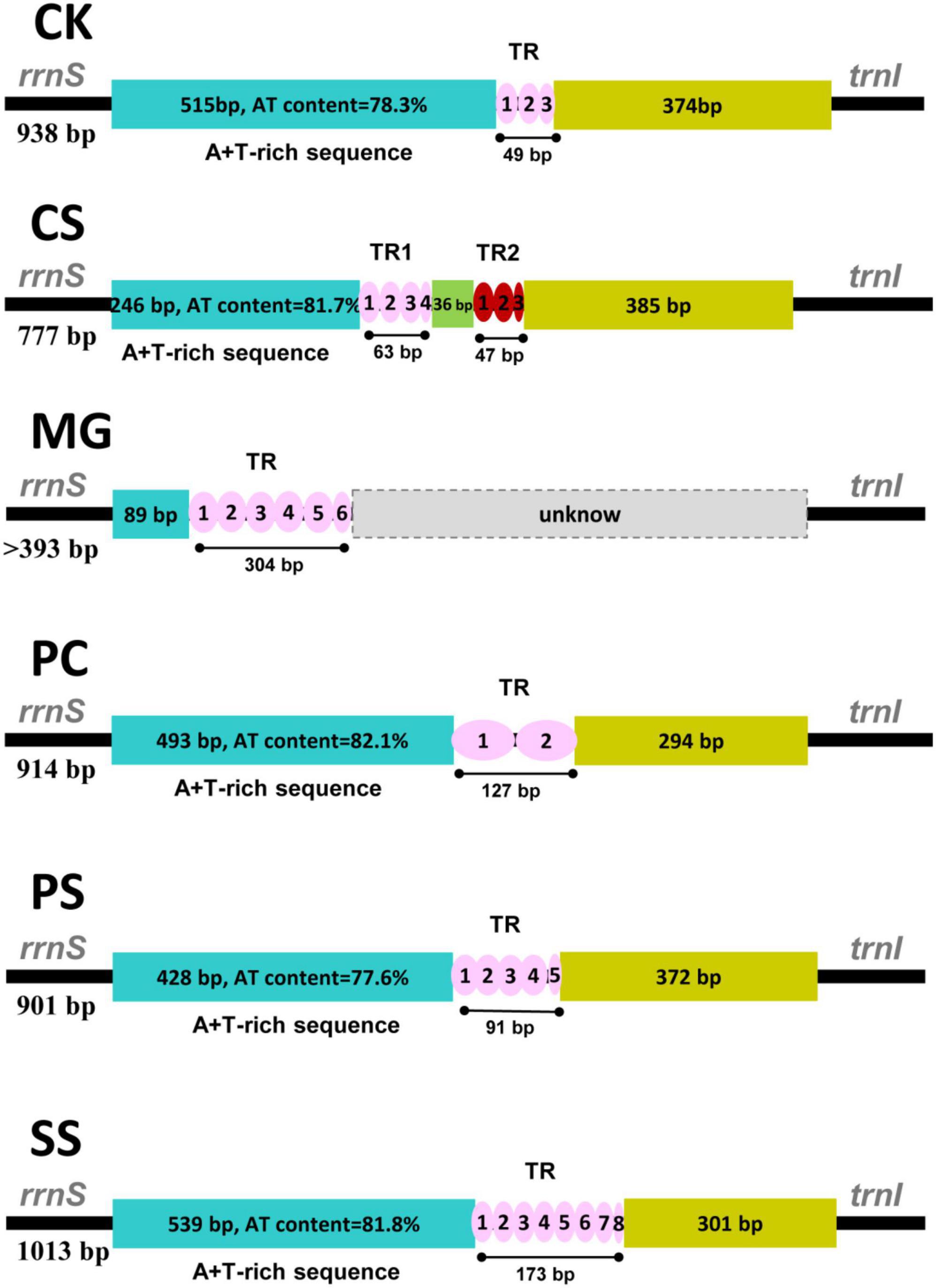
Figure 6. Control region structure of six peltoperlid species. TR, tandem repeat.
Large tandem repeats with two or more copies were detected in all control region sequences examined here. The size and copy number of the repeat unit are different in six peltoperlids, and the size variation of the control region is largely caused by this discrepancy. Overall, the control region of the family exhibited a number of distinctive structural and evolutionary characteristics, such as variable size, conserved structural elements, and abundant tandem repetitions. These properties made this region an effective phylogenetic marker for evolutionary and population genetic studies.
Two datasets (PCG and PCGRNA) were used in the present analyses. The phylogenetic trees generated from BI and ML inferences had identical topologies based on different datasets (Figure 7). Our results showed support values were higher in the BI tree than in the ML tree using the same dataset. All phylogenetic analyses supported the monophyly of each family, although some nodes have lower bootstrap values (BP).

Figure 7. Phylogenetic tree of the thirty-nine sequenced stoneflies. Bayesian inference and Maximum Likelihood analysis inferred from Protein-coding gene (PCG) and 13 PCGs and two rRNAs (PCGRNA) datasets supported the same topological structure. Values at nodes are ML bootstrap values and Bayesian posterior probabilities using the PCG (up) and PCGRNA (down) datasets. The tree was rooted with two outgroups (Paraleuctra cercia and Perlomyia isobeae).
The monophyly of two superfamilies, namely, Perloidea and Pteronarcyoidea are widely accepted and supported by morphological data (Zwick, 2000). However, this has never been well–supported by molecular evidence, especially the monophyly of Pteronarcyoidea (Thomas et al., 2000; Terry and Whiting, 2003; Davis, 2013; Chen et al., 2018; Ding et al., 2019; Wang et al., 2019; Cao et al., 2021; South et al., 2021a,b; Mo et al., 2022). In the current study, the monophyly of the superfamily Perloidea was recovered. The relationships of three families in Perloidea were recovered as: [Perlidae + (Chloroperlidae + Perlodidae)]. Our results are consistent with previous morphological studies (Illies, 1965), transcriptome (Davis, 2013; South et al., 2021a), and mitogenome analyses (Wang et al., 2019; Cao et al., 2021; Mo et al., 2022). In Zwick’s (2000) study, the monophyly of Perloidea was supported based on morphological characteristics, but the relationships within Perloidea were not fully resolved. Our results confirm Chloroperlidae as a sister to Perlodidae [supported in all analyses, BP = 100, Bayesian posterior probability values (PP) = 1.0] and also provide an increasingly clearer view of relationships within Perloidea.
The phylogenetic position of Peltoperlidae has long been under debate. Based on synapomorphic reduction of gills and abdominal ganglia, Zwick (1973) placed Peltoperlidae as sister to Perloidea. Stark and Stewart (1981) suggested Peltoperlidae to be more closely related to Pteronarcyidae than Perloidea based on some characters, such as the synapomorphies of present tridentate lacinia and an apical spine-like process of the tenth tergite in the nymphs and flattened egg shape. Uchida and Isobe (1989) elevated the subfamily Styloperlinae of the Peltoperlidae to familial rank as Styloperlidae and placed Peltoperlidae as sister group to Styloperlidae. In this study, a sister group relationship between Peltoperlidae and remaining Systellognatha was highly supported by all analyses (Figure 7), which was consistent with the hypotheses of Ricker (1952); Terry and Whiting (2003), and South et al. (2021a,b).
Unfortunately, the monophyly of Pteronarcyoidea was still not supported in this study. The Peltoperlidae was placed as the earliest branch within the Systellognatha, and all analyses generated the same relationships within Pteronarcyoidea [the relationship is Peltoperlidae + ((Styloperlidae + Pteronarcyidae) + Perloidea)]. These relationships are well-supported by BI analyses (PP ≥ 0.98). However, the posterior probabilities on some nodes are very low (PP = 0.37–0.52), and relationships within Pteronarcyoidea are still not exactly solved. Although this result differs from the generally accepted hypothesis that the Pteronarcyoidea are monophyletic (Zwick, 2000), no molecular study has proposed this relationship until now. Our results provide new insight into the phylogeny of this group, and analyses with more systellognathan taxa in future studies are needed to test the conclusion from the present study.
In this study, two complete mitogenomes from the family Peltoperlidae (C. kawasawai and P. sagittata) were sequenced. We present the comparative analysis of six peltoperlid mitogenomes and our results show that gene content, gene order, DmTTF binding site, nucleotide composition, codon usage, RNA structure, and structural elements in the control region are highly conserved in peltoperlids. Phylogenetic relationships within Systellognatha support the monophyly of Perloidea, but the monophyly of Pteronarcyoidea is still not supported. Sequencing more mitogenomes representing various taxonomic levels will greatly improve our understanding of phylogenetic relationships in Systellognatha.
The datasets presented in this study can be found in online repositories and the study is deposited in the NCBI repository with accession numbers: ON854136 and ON854137.
YW, JC, and WL conceived and designed the study and critically revised the manuscript. YW, JC, XG, and CG performed the experiments. YW and JC analyzed the data. YW and WL drafted the manuscript. WL and DM helped in the study design. All authors contributed to the article and approved the submitted version.
This study was supported by the Program for Science and Technology Innovation Talents in Universities of Henan (No. 21HASTIT042), the Key Scientific Research Project of Henan Province (Nos. 21A210009 and 22A210004), and the National Natural Science Foundation of China (Nos. 31801999 and 31970402).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2022.979847/full#supplementary-material
Abascal, F., Zardoya, R., and Telford, M. J. (2010). TranslatorX: Multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 38, 7–13. doi: 10.1093/nar/gkq291
Barr, C. M., Neiman, M., and Taylor, D. R. (2005). Inheritance and recombination of mitochondrial genomes in plants, fungi and animals. New Phytol. 168, 39–50. doi: 10.1111/j.1469-8137.2005.01492.x
Beckenbach, A. T. (2012). Mitochondrial genome sequences of Nematocera (lower Diptera): Evidence of rearrangement following a complete genome duplication in a winter crane fly genome. Genome Biol. Evol. 4, 89–101. doi: 10.1093/gbe/evr131
Beckenbach, A. T., and Stewart, J. B. (2009). Insect mitochondrial genomics 3: The complete mitochondrial genome sequences of representatives from two Neuropteroid orders: A dobsonfly (order Megaloptera) and a giant lacewing and an owlfly (order Neuroptera). Genome 52, 31–38. doi: 10.1139/G08-098
Benson, G. (1999). Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. doi: 10.1093/nar/27.2.573
Bernt, M., Donath, A., Jühling, F., Externbrink, F., Florentz, C., Fritzsch, G., et al. (2013). MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319. doi: 10.1016/j.ympev.2012.08.023
Boore, J. L. (1999). Animal mitochondrial genomes. Nucleic Acids Res. 27, 1767–1780. doi: 10.1093/nar/27.8.1767
Cameron, S. L. (2014). Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 59, 95–117. doi: 10.1146/annurev-ento-011613-162007
Cameron, S. L., and Whiting, M. F. (2008). The complete mitochondrial genome of the tobacco hornworm, Manduca sexta, (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene 408, 112–123. doi: 10.1016/j.gene.2007.10.023
Cao, J. J., Wang, Y., and Li, W. H. (2019). Comparative mitogenomic analysis of species in the subfamily Amphinemurinae (Plecoptera: Nemouridae) reveal conserved mitochondrial genome organization. Int. J. Biol. Macromol. 138, 292–301. doi: 10.1016/j.ijbiomac.2019.07.087
Cao, J. J., Wang, Y., Guo, X., Wang, G. Q., Li, W. H., and Murányi, D. (2021). Two complete mitochondrial genomes from Leuctridae (Plecoptera: Nemouroidea): Implications for the phylogenetic relationships among stoneflies. J. Insect Sci. 21, 1–6. doi: 10.1093/jisesa/ieab009
Carapelli, A., Vannini, L., Nardi, F., Boore, J. L., Beani, L., Dallai, R., et al. (2006). The mitochondrial genome of the entomophagous endoparasite Xenos vesparum (Insecta: Strepsiptera). Gene 376, 248–259. doi: 10.1016/j.gene.2006.04.005
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Chen, Z. T., Zhao, M. Y., Xu, C., and Du, Y. Z. (2018). Molecular phylogeny of Systellognatha (Plecoptera: Arctoperlaria) inferred from mitochondrial genome sequences. Int. J. Biol. Macromol. 111, 542–547. doi: 10.1016/j.ijbiomac.2018.01.065
Curole, J. P., and Kocher, T. D. (1999). Mitogenomics: Digging deeper with complete mitochondrial genomes. Trends Ecol. Evol. 14, 394–398. doi: 10.1016/S0169-5347(99)01660-2
Davis, N. G. (2013). Application Of Next-Generation Transcriptomic Tools For Non-Model Organisms: Gene Discovery And Marker Development Within Plecoptera (Insecta). Ph.D. thesis, Provo: Brigham Young University, 4265.
DeWalt, R. E., Maehr, M. D., Hopkins, H., Neu-Becker, U., and Stueber, G. (2022). Plecoptera species file online. Version 5.0/5.0. Available Online at: http://Plecoptera.SpeciesFile.org (accessed July 5, 2022).
Ding, S. M., Li, W. H., Wang, Y., Cameron, S. L., Murányi, D., and Yang, D. (2019). The phylogeny and evolutionary timescale of stoneflies (Insecta: Plecoptera) inferred from mitochondrial genomes. Mol. Phylogenet. Evol. 135, 123–135. doi: 10.1016/j.ympev.2019.03.005
Dotson, E. M., and Beard, C. B. (2001). Sequence and organization of the mitochondrial genome of the Chagas disease vector, Triatoma dimidiate. Insect Mol. Biol. 10, 205–215. doi: 10.1046/j.1365-2583.2001.00258.x
Dowton, M., Cameron, S. L., Austin, A. D., and Whiting, M. F. (2009). Phylogenetic approaches for the analysis of mitochondrial genome sequences data in the Hymenoptera–A lineage with both rapidly and slowly evolving mitochondrial genomes. Mol. Phylogenet. Evol. 52, 512–519. doi: 10.1016/j.ympev.2009.04.001
Fenn, J. D., Song, H., Cameron, S. L., and Whiting, M. F. (2008). A preliminary mitochondrial genome phylogeny of Orthoptera (Insecta) and approaches to maximizing phylogenetic signal found within mitochondrial genome data. Mol. Phylogenet. Evol. 49, 59–68. doi: 10.1016/j.ympev.2008.07.004
Fochetti, R., and Tierno de Figueroa, J. M. (2008). Global diversity of stoneflies (Plecoptera: Insecta) in freshwater. Hydrobiologia 595, 365–377. doi: 10.1007/s10750-007-9031-3
Grant, J. R., and Stothard, P. (2008). The CGView server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 36, W181–W184. doi: 10.1093/nar/gkn179
Hassanin, A., Léger, N., and Deutsch, J. (2005). Evidence for multiple reversals of asymmetric mutational constraints during the evolution of the mitochondrial genome of Metazoa, and consequences for phylogenetic inferences. Syst. Biol. 54, 277–298. doi: 10.1080/10635150590947843
Illies, J. (1965). Phylogeny and zoogeography of the Plecoptera. Annu. Rev. Entomol. 10, 117–140. doi: 10.1146/annurev.en.10.010165.001001
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kukalová-Peck, J., and Brauckmann, C. (1992). Most paleozoic Protorthoptera are ancestral hemipteroids: Major wing braces as clues to a new phylogeny of Neoptera (Insecta). Can. J. Zool. 70, 2452–2473. doi: 10.1139/z92-330
Lavrov, D. V., Brown, W. M., and Boore, J. L. (2004). Phylogenetic position of the Pentastomida and (pan) crustacean relationships. Proc. Biol. Sci. 271, 537–544. doi: 10.1098/rspb.2003.2631
Letsch, H., and Simon, S. (2013). Insect phylogenomics: New insights on the relationships of lower neopteran orders (Polyneoptera). Syst. Entomol. 38, 783–793. doi: 10.1111/syen.12028
Li, H., Liu, H. Y., Song, F., Shi, A. M., Zhou, X. G., and Cai, W. Z. (2012). Comparative Mitogenomic analysis of damsel bugs representing three tribes in the family Nabidae (Insecta: Hemiptera). PLoS One 7:e45925. doi: 10.1371/journal.pone.0045925
Librado, P., and Rozas, J. (2009). DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Lin, C. P., Chen, M. Y., and Huang, J. P. (2010). The compete mitochondrial genome and phylogenomics of a damselfly Euphaea formosa support a basal Odonata within the Pterygota. Gene 468, 20–29. doi: 10.1016/j.gene.2010.08.001
Lindahl, T. (1993). Instability and decay of the primary structure of DNA. Nature 362, 709–715. doi: 10.1038/362709a0
Lohse, M., Bolger, A. M., Nagel, A., Fernie, A. R., Lunn, J. E., Stitt, M., et al. (2012). RobiNA: A user-friendly, integrated software solution for RNA-Seq based transcriptomics. Nucleic Acids Res. 40, W622–W627. doi: 10.1093/nar/gks540
McCulloch, G. A., Wallis, G. P., and Waters, J. M. (2016). A time-calibrated phylogeny of southern hemisphere stoneflies: Testing for Gondwanan origins. Mol. Phylogenet. Evol. 96, 150–160. doi: 10.1016/j.ympev.2015.10.028
Misof, B., Liu, S., Meusemann, K., Peters, R. S., Donath, A., Mayer, C., et al. (2014). Phylogenomics resolves the timing and pattern of insect evolution. Science 346, 763–767. doi: 10.1126/science.1257570
Mo, R. R., Wang, Y., Cao, J. J., Wang, G. Q., Li, W. H., and Murányi, D. (2022). Two complete mitochondrial genomes of the subfamily Chloroperlinae (Plecoptera: Chloroperlidae) and their phylogenetic implications. Arthropod Syst. Phylo. 80, 155–168. doi: 10.3897/asp.80.e78173
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Ojala, D., Montoya, J., and Attardi, G. (1981). tRNA punctuation model of RNA processing in human mitochondria. Nature 290, 470–474. doi: 10.1038/290470a0
Peng, Y., Leung, H. C. M., Yiu, S. M., and Chin, F. Y. L. (2012). IBDA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428. doi: 10.1093/bioinformatics/bts174
Perna, N. T., and Kocher, T. D. (1995). Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 41, 353–358. doi: 10.1007/BF00186547
Pessino, M., Chabot, E. T., Giordano, R., and DeWalt, R. E. (2014). Refugia and postglacial expansion of Acroneuria frisoni Stark & Brown (Plecoptera: Perlidae) in North America. Freshw. Sci. 33, 232–249. doi: 10.1086/675306
Ricker, W. E. (1952). Systematic Studies in Plecoptera, Indiana University Publications Science Series 18. Bloomington: Indiana University Press.
Roques, S., Fox, C. J., Villasana, M, I., and Rico, C. (2006). The complete mitochondrial genome of the whiting, Merlangius merlangus and the haddock, Melanogrammus aeglefinus: a detailed genomic comparison among closely related species of the Gadidae family. Gene 383, 12–23.
Ronquist, F., Teslenko, M., van der Mark, P. V. D., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Shen, Y., and Du, Y. Z. (2019). The mitochondrial genome of Leuctra sp. (Plecoptera: Leuctridae) and its performance in phylogenetic analyses. Zootaxa 4671, 571–580. doi: 10.11646/zootaxa.4671.4.8
Shen, Y., and Du, Y. Z. (2020). The complete mitochondrial genome of Flavoperla biocellata Chu, 1929 (Plecoptera: Perlidae) and the phylogenetic analyses of Plecoptera. PeerJ 8:e8762. doi: 10.7717/peerj.8762
Simon, S., Narechania, A., DeSalle, R., and Hadrys, H. (2012). Insect phylogenomics: Exploring the source of incongruence using new transcriptomic data. Genome Biol. Evol. 4, 1295–1309. doi: 10.1093/gbe/evs104
Song, N., Li, H., Song, F., and Cai, W. Z. (2016). Molecular phylogeny of Polyneoptera (Insecta) inferred from expanded mitogenomic data. Sci. Rep. 6:36175. doi: 10.1038/srep36175
South, E. J., Skinner, R. K., Dewalt, R. E., Davis, M. A., and Myers, L. W. (2021a). A new family of stoneflies (Insecta: Plecoptera), Kathroperlidae, fam. n. with a phylogenomic analysis of the Paraperlinae (Plecoptera: Chloroperlidae). Insect Syst. Diver. 5, 1–27. doi: 10.1093/isd/ixab014
South, E. J., Skinner, R. K., DeWalt, R. E., Kondratieff, B. C., Johnson, K. P., Davis, M. A., et al. (2021b). Phylogenomics of the North American Plecoptera. Syst. Entomol. 46, 287–305. doi: 10.1111/syen.12462
Stark, B. P., and Stewart, K. W. (1981). The Nearctic genera of Peltoperlidae (Plecoptera). J. Kansas Entomol. Soc. 54, 285–311. doi: 10.2307/25084161
Stevens, D. M., Bishop, J., and Picker, M. D. (2018). Phylogenetic analysis reveals high local endemism and clear biogeographic breaks in southern African stoneflies (Notonemouridae, Plecoptera). Zootaxa 4483, 428–454. doi: 10.11646/zootaxa.4483.3.2
Stewart, J. B., and Beckenbach, A. T. (2005). Insect mitochondrial genomics: The complete mitogenome sequence of the meadow spittlebug Philaenus spumarius (Hemiptera: Auchenorrhyncha: Cercopoidae). Genome 48, 46–54.
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Terry, M. D., and Whiting, M. F. (2003). Phylogeny of Plecoptera: Molecular evidence and evolutionary trends. Entomol. Abh. 61, 130–131.
Terry, M. D., and Whiting, M. F. (2005). Mantophasmatodea and phylogeny of the lower neopterous insects. Cladistics 21, 240–257. doi: 10.1111/j.1096-0031.2005.00062.x
Thomas, M. A., Walsh, K. A., Wolf, M. R., McPheron, B. A., and Marden, J. H. (2000). Molecular phylogenetic analysis of evolutionary trends in stonefly wing structure and locomotor behavior. Proc. Natl. Acad. Sci. U.S.A. 97, 13178–13183. doi: 10.1073/pnas.230296997
Tomita, K., Yokobori, S., Oshima, T., Ueda, T., and Watanabe, K. (2001). The cephalopod Loligo bleekeri mitochondrial genome: Multiplied noncoding regions and transposition of tRNA genes. J. Mol. Evol. 54, 486–500. doi: 10.1007/s00239-001-0039-4
Uchida, S., and Isobe, Y. (1989). Styloperlidae, stat. Nov. and Microperlinae, subfam. Nov. with a revised system of the family group Systellognatha (Plecoptera). Spixiana 12, 145–182.
Veale, A. J., Dearden, P. K., and Waters, J. M. (2019). First complete mitochondrial genome of a Gripopterygid stonefly from the suborder Antarctoperlaria: Zelandoperla fenestrata. Mitochondrial DNA Part B 4, 886–888. doi: 10.1080/23802359.2018.1546130
Wang, Y., Cao, J. J., and Li, W. H. (2018). Complete mitochondrial genome of Suwallia teleckojensis (Plecoptera: Chloroperlidae) and implications for the higher phylogeny of stoneflies. Int. J. Mol. Sci. 19:680. doi: 10.3390/ijms19030680
Wang, Y., Cao, J. J., Li, N., Ma, G. Y., and Li, W. H. (2019). The first mitochondrial genome from Scopuridae (Insecta: Plecoptera) reveals structural features and phylogenetic implications. Int. J. Biol. Macromol. 122, 893–902. doi: 10.1016/j.ijbiomac.2018.11.019
Wei, S. J., Shi, M., Chen, X. X., Sharkey, M. J., van Achterberg, C., Ye, G. Y., et al. (2010). New views on strand asymmetry in insect mitochondrial genomes. PLoS One 5:e12708. doi: 10.1371/journal.pone.0012708
Yuan, M. L., Zhang, Q. L., Guo, Z. L., Wang, J., and Shen, Y. Y. (2015). Comparative mitogenomic analysis of the superfamily Pentatomoidea (Insecta: Hemiptera: Heteroptera) and phylogenetic implications. BMC Genomics 16:460. doi: 10.1186/s12864-015-1679-x
Zhang, D. X., Szymura, J. M., and Hewitt, G. M. (1995). Evolution and structural conservation of the control region of insect mitochondrial DNA. J. Mol. Evol. 40, 382–391. doi: 10.1007/BF00164024
Zhang, H. L., Liu, B. B., Wang, X. Y., Han, Z. P., Zhang, D. X., and Su, C. N. (2016). Comparative Mitogenomic analysis of species representing six subfamilies in the family Tenebrionidae. Int. J. Mol. Sci. 17:841. doi: 10.3390/ijms17060841
Zwick, P. (1973). Insecta: Plecoptera. Phylogenetisches system und Katalog. Das Tierreich 94, 1–465.
Keywords: Cryptoperla kawasawai, Peltoperlopsis sagittata, mitochondrial genome, phylogeny, Plecoptera
Citation: Wang Y, Cao J, Guo X, Guo C, Li W and Murányi D (2022) Comparative analysis of mitochondrial genomes among the family Peltoperlidae (Plecoptera: Systellognatha) and phylogenetic implications. Front. Ecol. Evol. 10:979847. doi: 10.3389/fevo.2022.979847
Received: 28 June 2022; Accepted: 22 July 2022;
Published: 15 August 2022.
Edited by:
Yuyu Wang, Agricultural University of Hebei, ChinaReviewed by:
Nan Song, Henan Agricultural University, ChinaCopyright © 2022 Wang, Cao, Guo, Guo, Li and Murányi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weihai Li, bHdoNzk2OUAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.