 Michael J. Bradshaw
Michael J. Bradshaw Guan-Xiu Guan
Guan-Xiu Guan Liam Nokes1,3
Liam Nokes1,3 Uwe Braun
Uwe Braun Shu-Yan Liu
Shu-Yan Liu Donald H. Pfister
Donald H. Pfister
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol. , 14 June 2022
Sec. Phylogenetics, Phylogenomics, and Systematics
Volume 10 - 2022 | https://doi.org/10.3389/fevo.2022.918908
Powdery mildews are a group of economically and ecologically important plant pathogens. In the past 25 years the use of ribosomal DNA (rDNA) in the powdery mildews has led to major taxonomic revisions. However, the broad scale use of rDNA has also revealed multiple species complexes that cannot be differentiated based on ITS + LSU data alone. Currently, there are only two powdery mildew taxonomic studies that took a multi-locus approach to resolve a species complex. In the present study, we introduce primers to sequence four additional regions (CAM, GAPDH, GS, and RPB2) that have the potential to improve support values in both broad and fine scale phylogenetic analyses. The primers were applied to a broad set of powdery mildew genera in China and the United States, and phylogenetic analyses included some of the common complexes. In taxa with nearly identical ITS sequences the analyses revealed a great amount of diversity. In total 154 non-rDNA sequences from 11 different powdery mildew genera were deposited in NCBI’s GenBank, laying the foundation for secondary barcode databases for powdery mildews. The combined and single loci phylogenetic trees constructed generally followed the previously defined species/genus concepts for the powdery mildews. Future research can use these primers to conduct in depth phylogenetic, and taxonomic studies to elucidate the evolutionary relationships of species and genera within the powdery mildews.
Approximately 900 species of powdery mildews (Helotiales, Erysiphaceae) have been described in 19 genera infecting over 10,000 plant species worldwide (Amano, 1986; Braun and Cook, 2012; Marmolejo et al., 2018; Johnston et al., 2019; Kiss et al., 2020). The taxonomy and phylogeny of these obligate plant pathogens has undergone radical change in the past 25 years as molecular methods have been widely applied (Saenz and Taylor, 1999; Braun and Takamatsu, 2000; Mori et al., 2000; Braun and Cook, 2012). The taxonomic rank of the powdery mildews (i.e. Erysiphaceae) has been recently resolved (Johnston et al., 2019; Haelewaters et al., 2021) using multiple loci from the full genomes of three different genera. Most of the molecular phylogenetic work conducted on this important group has focused solely on the ITS and adjacent LSU region. The biotrophic nature of powdery mildews has rendered it difficult to evaluate single copy gene regions and thus, at present, there have been only two multi-locus taxonomic studies (Qiu et al., 2020; Liu M. et al., 2021).
There are several instances of powdery mildew species complexes where morphologically distinct species cannot be delineated by ITS + LSU sequences. Examples include the Erysiphe aquilegiae complex (Bradshaw et al., 2021a), the E. berberidis complex (Liu et al., 2022)1, the E. elevata complex (Tymon et al., 2022), the E. trifoliorum complex (Bradshaw et al., 2021b), the Podosphaera aphanis complex (Liu M. et al., 2021), and the P. xanthii complex (Shin et al., 2019). Generally, the morphological differences among species within these complexes can be difficult to discern except by experts. This has led to misidentifications that have been propagated in the literature and on public databases. Other plant-pathogen complexes have been resolved by using non-ribosomal DNA (rDNA) markers, such as, Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), calmodulin (CAL), glutamine synthetase synthase (GS), β-Tubulin (TUB2), and Actin (ACT) (Aspergillus-Samson et al., 2014; Botrytis-Saito et al., 2016; Penicillium-Ouhibi et al., 2018; Colletotrichum-Khodadadi et al., 2020). Ellingham et al. (2019) evaluated ACT, TUB2, CAL, CHS (chitin synthase), EF1- α (elongation factor 1 alpha), MCM7 (minichromosome maintenance protein 7), and TSR1 (20-S rRNA accumulation 1) to enhance identification accuracy of powdery mildews. The authors found success with MCM7 which led to a broad scale phylogenetic study by Shirouzu et al. (2020).
Markers to improve higher level support, including the powdery mildew phylogenetic backbone, have eluded this group. Recently, Shirouzu et al. (2020) evaluated MCM7, LSU, and SSU (small subunit) sequences to improve the powdery mildew phylogeny. Although their analysis improved tree resolution, low support values (less than 80 posterior values in Maximum likelihood analysis) were still present throughout their trees. Markers for higher level phylogenetic analyses in the powdery mildews are needed since support for the morphologically distinct sections, i.e., Erysiphe sect. Microsphaera, Erysiphe sect. Uncinula etc. has been elusive. Additionally, some genera are paraphyletic (Leveillula is nested within the Phyllactinia clade). With the increase of multi-locus sequences, better resolution and backbone support will be achievable.
The influx of full genome sequences of powdery mildews (there are currently 33 genomes from 14 different species on GenBank) allows the generation of specific powdery mildew primers for a range of protein and single copy genomic regions. In this manuscript, we report multiple genetic markers, and their newly designed primers, that have the potential for broad and fine scale phylogenetic evaluations of the powdery mildews.
Samples from the United States were collected at the University of Washington, Seattle, Washington, United States in 2019, the Arnold Arboretum, Boston, MA, United States, in 2021 and the Harvard University main campus, Cambridge, MA, United States in 2021 (Table 1). Samples from China were collected between 2017 and 2021. One herbarium specimen from the Farlow Herbarium (FH), Harvard University, was evaluated to assess the performance of the newly designed primers on an 83 year old specimen. Freshly collected specimens were deposited at the Farlow Herbarium, Harvard University (FH), and the Herbarium of Mycology of Jilin Agricultural University (HMJAU).
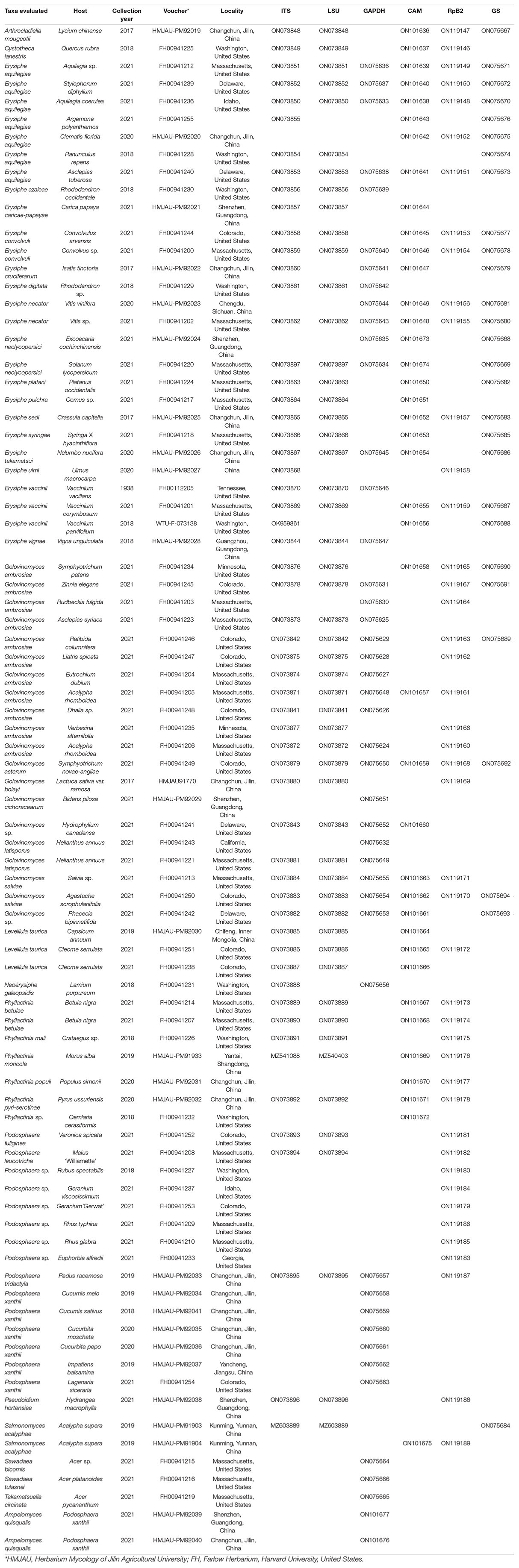
Table 1. Taxa evaluated and their associated barcodes and GenBank numbers.
Loci were chosen based on previous research on plant-fungal pathogen systems. For most of the regions, no powdery mildew sequences were available on GenBank. In cases where no powdery mildew sequences were available, sequences from other closely related fungi were used to blast the powdery mildew full genomes available on GenBank. There were 33 genome assemblies available on GenBank from 14 species. The blast results were downloaded into Geneious version 11.0.22 and aligned. From the alignment, primers were chosen and analyzed in OligoAnalyzer (integrated DNA Technologies). The following parameters were considered: primers ∼20 bps long, G/C content between 40 and 60%, double T’s or double A’s on the 5’ or 3’ end were avoided, primers ended with a GC clamp, hairpins less than ∼45°C, Delta G above ∼−9, and no more than 5°C difference in melting temperature between primer pairs.
DNA extractions were done using the Chelex method (Walsh et al., 1991; Hirata and Takamatsu, 1996). Around 20 chasmothecia or 100 conidia were taken from the leaf surface using a sterile pipette tip for DNA extractions. Polymerase chain reaction (PCR) was carried out for the ITS and LSU region using the primer pair PM10/PM28R (Bradshaw and Tobin, 2020). When PCR was unsuccessful, a nested approach was applied using the primers AITS (Bradshaw and Tobin, 2020) and TW14 (Mori et al., 2000); followed by PM10 and PM28R or AITS and PM11 (Bradshaw and Tobin, 2020); followed by PM10 and PM2 (Cunnington et al., 2003). For the CAL, GAPDH, GS, and RPB2 regions the primer pairs from Table 2 were used.

Table 2. Primers generated in the present study and the genera in which amplicons were generated.
PCR specifications for all the regions were as follows for a 50 μl solution reaction: 35.7 μl molecular grade H2O, 4 μl BSA (20 mg/ml), 1 μl of forward primer and 1 μl reverse primer at a 10 μM concentration, 2 μl of DNA, 5 μl of Buffer, 0.3 μl of TAQ DNA Polymerase (5 units per μl) and 1 μl dNTPs (10 mM). Cycling included initial denaturation at 95°C for 3 min followed by 40 cycles of denaturation at 95°C for 30 s, annealing at 57°C for 1 min, and elongation at 72°C for 2 min and then a final elongation of 72°C for 10 min.
In the United States laboratory crude PCR products were sent to Eurofins (Luxembourg) to be purified and directly sequenced in the forward and reverse directions using the primers above. Samples in China were sent to Sangon Biotech (Shanghai, China) for sequencing in both the forward and reverse direction.
To show the potential of the primers and their ability to anneal to a variety of species in different genera, phylogenetic trees were constructed with a general focus on common powdery mildew complexes. A concatenated, GAPDH-CAM-GS-ITS-LSU-RPB2, tree was generated to show their potential to improve higher level support. In the concatenated tree, sequences from different specimens of Salmonomyces acalyphae were spliced together. Large gaps up to ∼20 bps were manually deleted in the ITS and GAPDH alignments prior to analyses. The regions evaluated were mined from the full genomes of Blumeria graminis (Assembly accession: GCA_905067625.1), Phyllactinia moricola (GCA 019455665), Pleochaeta shiraiana (GCA 019455505), Podosphaera cerasi (GCA 018398735), Podosphaera leucotricha (GCA 013170925), and Podosphaera xanthii (GCA 010015925 and GCA 014884795) to be included in some of the analyses. The full genome of Arachnopeziza araneosa (GCA_00398855) was chosen as an outgroup taxon based on the analyses by Johnston et al. (2019) for the concatenated and single loci trees. An ITS + LSU tree was constructed using all the sequences obtained for comparative purposes. Sequences were aligned and edited using MUSCLE in MEGA11:Molecular Evolutionary Genetics Analysis version 11 (Tamura et al., 2021). A GTR + G + I evolutionary model was used for phylogenetic analyses as it is the most inclusive model of evolution and includes all other evolutionary models (Abadi et al., 2019). A fixed parameter-rich model (such as GTR + G + I) can be used in lieu of running a test to select the most suitable evolutionary model (Abadi et al., 2019).
For all the trees the phylogeny was inferred using Bayesian analysis using a Yule tree prior (Gernhard, 2008) and a strict molecular clock, in the program BEAST version 1.10.4 (Suchard et al., 2018). A single MCMC chain of 106 steps was run, with a burn-in of 10%. Posterior probabilities were calculated from the remaining 9,000 sampled trees. A maximum clade credibility tree was produced using TreeAnnotator version 1.10.4 (part of the BEAST package). Stationarity was confirmed by running the analysis multiple times, which revealed convergence between runs. The resulting tree was visualized using FigTree version 1.3.1 (Rambaut, 2009)3. A maximum likelihood analysis was accomplished using raxmlGUI (Silvestro and Michalak, 2012) under the default settings with a GTR + G + I evolutionary model. Bootstrap analyses were conducted using 1,000 replications (Felsenstein, 1985).
Eight primers were successfully constructed and applied to 11 out of the 19 powdery mildew genera (Table 2). For the GAPDH region we sequenced an herbarium specimen that was 83 years old (1938: FH00112205). We were unable to sequence the specimen from 1938 using the other regions likely due to the size of the amplicons. For the GS, RPB2, and CAM regions multiple specimens were sequenced that were up to 3 years old. In total 55 sequences were generated from the ITS/LSU loci and 310 sequences were generated from non-rDNA loci: 74 from CAM, 134 from the GAPDH, 52 from GS, and 50 from RPB2. Of these 310 sequences, 154 (43 from GAPDH, 38 from CAM, 44 from RPB2, and 29 from GS) were used for phylogenetic analysis and deposited in GenBank. In the course of the study, the GAPDH primers were found to anneal to other fungi. In particular, multiple Ampelomyces sequences were generated that aligned with both GCA 018398575 and GCA 010094095. We deposited 3 GAPDH sequences from Ampelomyces spp. in GenBank that could potentially assist future researchers evaluating GAPDH of Ampelomyces spp.
Amplicons for the specimens obtained were deposited in GenBank (Table 1). Sequences from the GAPDH, GS, CAM, and RPB2 regions were evaluated individually and in a concatenated tree that included ITS + LSU sequences. A separate ITS + LSU tree from the sequences obtained in this study is presented in Supplementary Figure 1. Six phylogenetic trees were constructed and presented (Figures 1–6). For the majority of these regions, no additional sequences were available on GenBank and could not be evaluated for phylogenetic purposes. A maximum clade credibility tree was constructed using Bayesian analyses from the single loci and combined sequences. Posterior probabilities > 90 are displayed followed by bootstrap values greater than 70% for the maximum likelihood (ML) analyses conducted. The representative maximum clade credibility tree is illustrated in Figure 1.
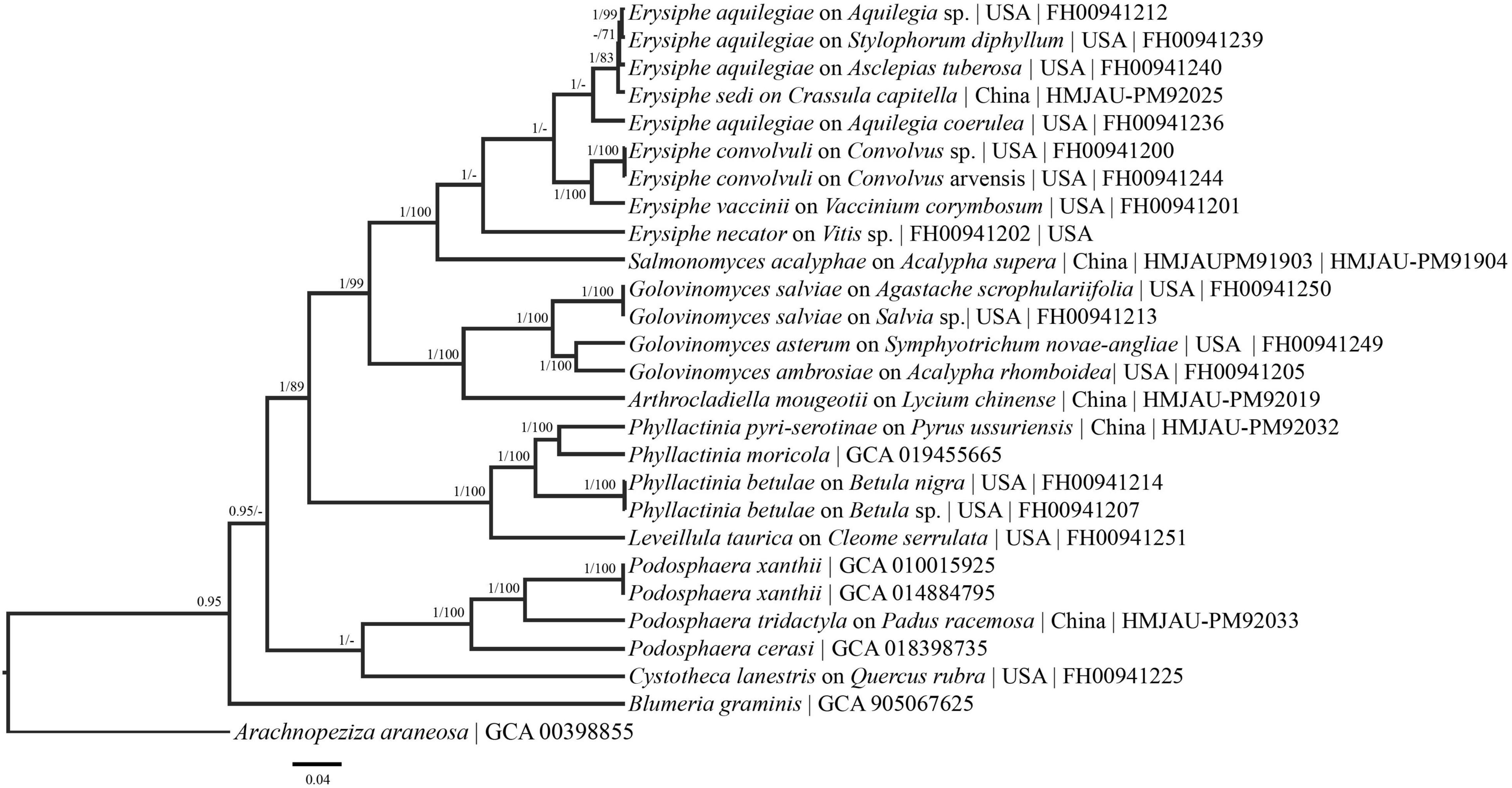
Figure 1. Bayesian maximum clade credibility tree of sequences from the combined GAPDH-CAM-GS-ITS-LSU-RPB2 regions. Posterior probabilities > 90 are displayed followed by bootstrap values greater than 70% for the maximum likelihood (ML) analyses conducted. High support values are present throughout the tree. Taxa names are followed by host taxa, collection locality, and voucher/GenBank numbers.
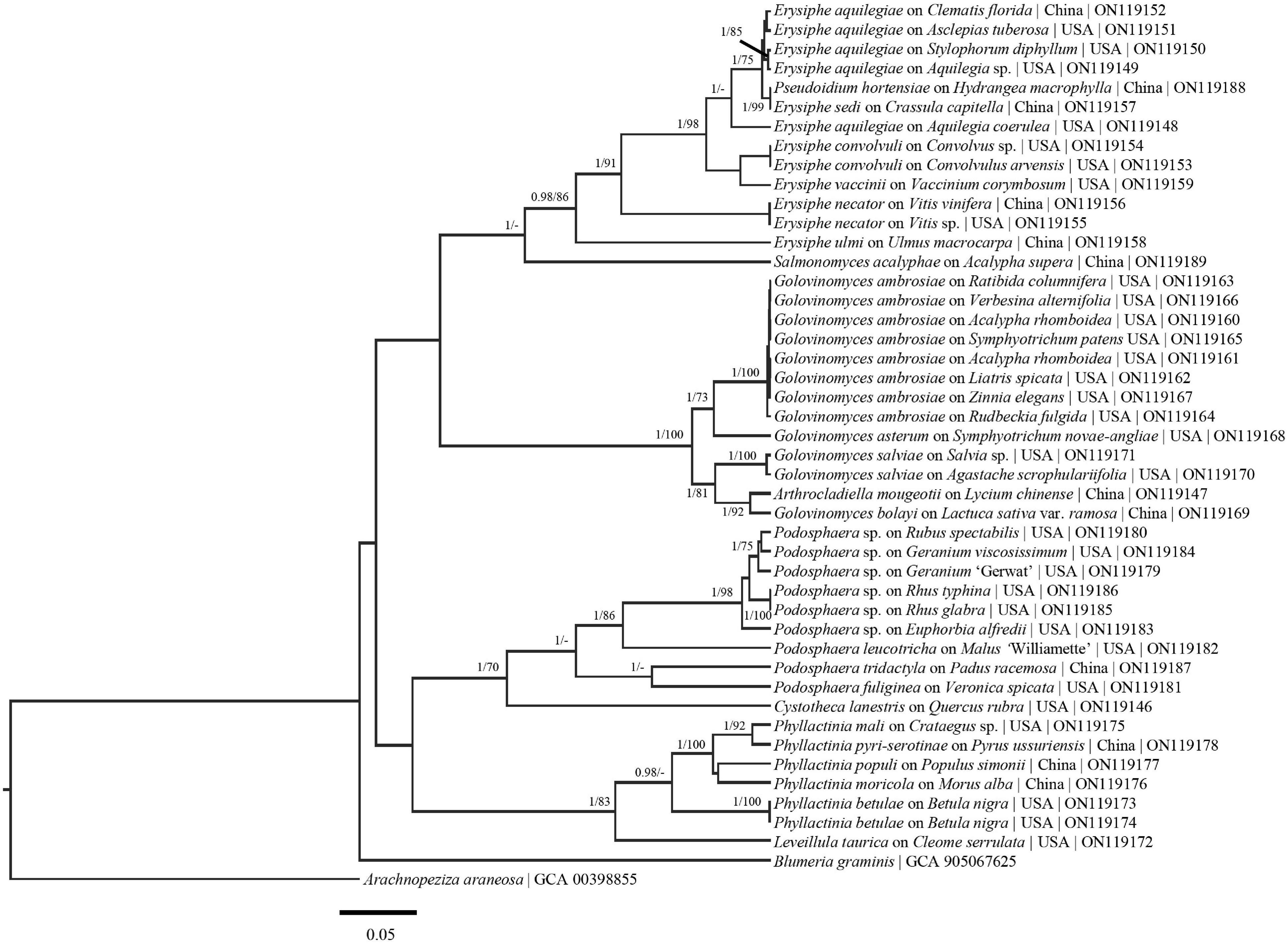
Figure 2. Bayesian maximum clade credibility tree of sequences from the RPB2 region. Posterior probabilities > 90 are displayed followed by bootstrap values greater than 70% for the maximum likelihood (ML) analyses conducted. Generally, previously defined genus/species concepts are seen as well as some broad and fine scale support (E. aquilegiae complex). Taxa names are followed by host taxa, collection locality and GenBank numbers.
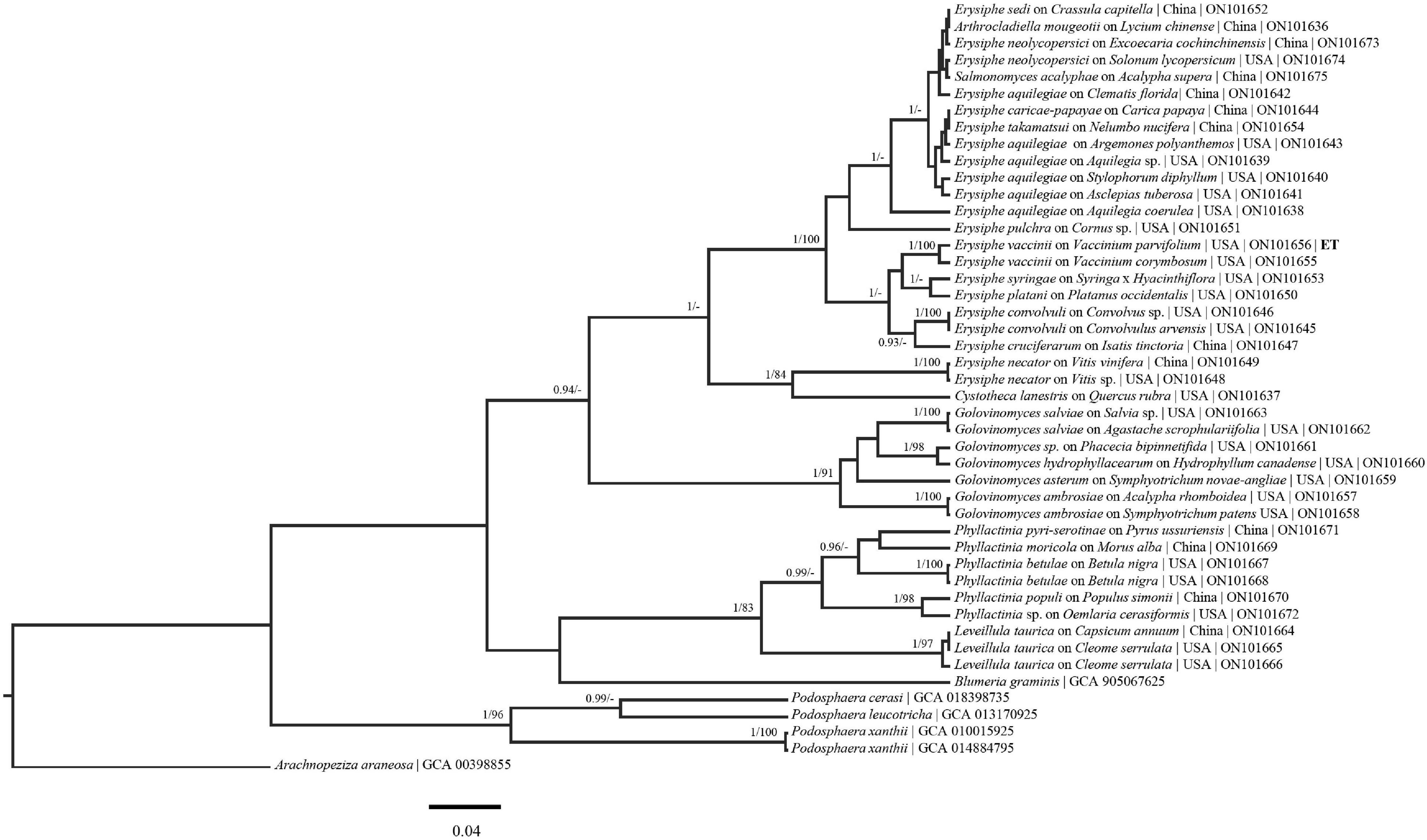
Figure 3. Bayesian maximum clade credibility tree of sequences from the CAM region. Posterior probabilities > 90 are displayed followed by bootstrap values greater than 70% for the maximum likelihood (ML) analyses conducted. Generally, previously defined genus/species concepts are seen as well as some broad scale support. ET, ex epitype. Taxa names are followed by host taxa, collection locality, and GenBank numbers.

Figure 4. Bayesian maximum clade credibility tree of sequences from the GS region. Posterior probabilities > 90 are displayed followed by bootstrap values greater than 70% for the maximum likelihood (ML) analyses conducted. Generally, previously defined genus/species concepts are seen as well as some broad scale support. ET, ex epitype. Taxa names are followed by host taxa, collection locality and GenBank numbers.
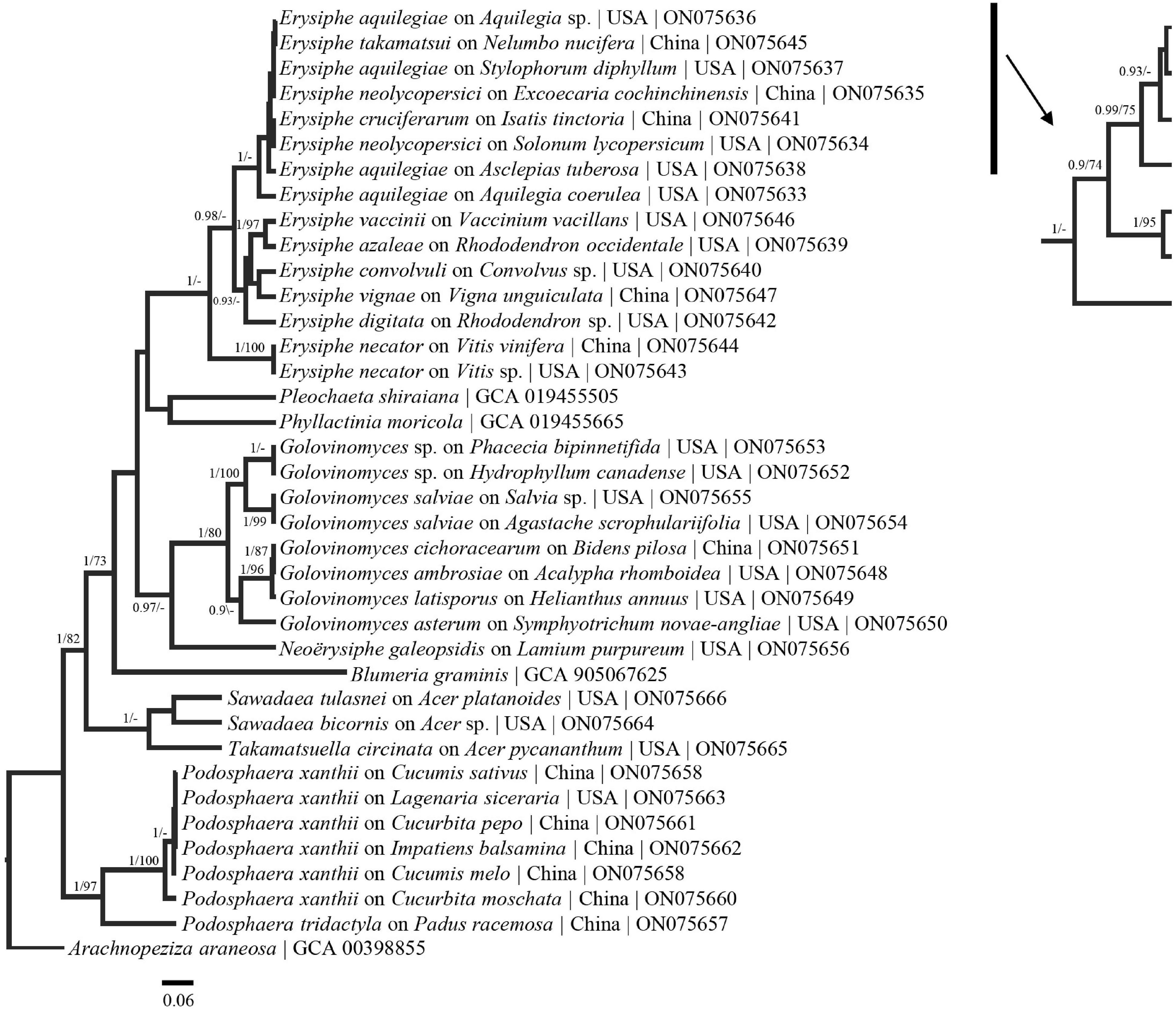
Figure 5. Bayesian maximum clade credibility tree of sequences from the GAPDH region. Posterior probabilities > 90 are displayed followed by bootstrap values greater than 70% for the maximum likelihood (ML) analyses conducted. Generally, previously defined genus/species concepts are seen as well as some broad and fine scale support (E. aquilegiae and P. xanthii complexes). Taxa names are followed by host taxa, collection locality and GenBank numbers.

Figure 6. Bayesian maximum clade credibility tree of GAPDH sequences from species in the Golovinomyces ambrosiae complex. Posterior probabilities > 90 are displayed followed by bootstrap values greater than 70% for the maximum likelihood (ML) analyses conducted. Support exists in the tree for the separation of G. ambrosiae and G. latisporus as defined by Qiu et al. (2020). Taxa names are followed by host taxa, collection locality and GenBank numbers.
The phylogenetic analyses revealed that the different regions have great potential for splitting up the ITS + LSU complexes and increasing the backbone support of the powdery mildews. In the ITS + LSU tree (Supplementary Figure 1) there is no support seen within the E. aquilegiae or G. ambrosiae clade whereas support is seen within the E. aquilegiae clade in the concatenated (Figure 1) and RPB2 tree (Figure 2) and the G. ambrosiae clade in the GAPDH tree (Figure 6). There is also much better higher level support throughout the concatenated tree than in the ITS + LSU tree alone (Supplementary Figure 1). For example, in the concatenated tree (Figure 1), there is high support that E. convolvuli and E. vaccinii form a clade yet no support for this grouping in the ITS + LSU tree. Additionally, in the ITS + LSU, unlike the concatenated tree, there is no support for the placement of Blumeria and the Phyllactinia-Leveillula clade.
The evaluation of protein-coding genes for phylogenetic analyses has largely been understudied in the powdery mildews. Relying solely on rDNA in analyses has led to the recognition of species complexes that group morphologically dissimilar taxa that are genetically similar in ITS and LSU profiles. In the present study, we designed primers for four protein coding genes. We have shown that these genes have potential for refining taxonomic/phylogenetic studies of the powdery mildews. Additionally, the divergent nature of these genes shows their potential in phylogenetic/taxonomic studies.
In recent years the ITS region has come under scrutiny due to its potential for intragenomic variation and its inability to differentiate cryptic species (Seifert et al., 1995; Simon and Weiss, 2008; Kovács et al., 2011; Kiss, 2012). Studies are finding that the ITS and adjacent LSU regions are unable to identify a large percentage of species, emphasizing the importance of secondary markers for certain fungal lineages (Badotti et al., 2017; Vu et al., 2019). Secondary barcodes can be important for understanding the species and evolutionary relationships in the Kingdom Fungi and in particular, the Ascomycota (Tanabe et al., 2005; Stielow et al., 2015; Ellingham et al., 2019; Johnston et al., 2019; Meyer et al., 2019). Recently, secondary markers including TEF1α, TOP1, and PGK have been established for species identification in fungi (Stielow et al., 2015; Meyer et al., 2019).
There have been two powdery mildew phylogenetic/taxonomic publications that evaluated secondary barcodes to resolve complexes. Qiu et al. (2020) used five genes (ITS, LSU, IGS, TUB2, and CHS1) to increase resolution in the Golovinomyces ambrosiae complex. Using solely GAPDH (Figure 6) we were able to delimit taxa of this complex, consistent with Qiu et al. (2020). Liu S. Y. et al. (2021) used four genes (ITS, CHS1, and fragments of two unnamed orthologous genes) to split the Blumeria graminis complex into eight separate species. One limitation of Liu S. Y. et al. (2021) is that using unnamed orthologous genes and their associated primers can be difficult to apply broadly to other powdery mildew taxa.
In the present research we have shown that the genes evaluated have the potential to resolve multiple powdery mildew complexes. For example, the E. aquilegiae complex forms well supported groups in the concatenated tree, as well as in some of the single loci (RpB2, RPB2, and GAPDH) trees (Figures 1, 2, 4, 5). Additionally, groups, such as the Podosphaera xanthii complex, will likely be able to be clarified by taking a multi-locus approach on a range of hosts from throughout the world. GAPDH is especially promising for the P. xanthii complex where in phylogenetic analysis two separate groups formed (Figure 5). The divergent nature of some of these sequences of species with close ITS affinity, provides evidence that the ITS could not be accounting for evolutionary relationships within the powdery mildews. GAPDH is phylogenetically informative for cryptic species (Matsuda et al., 2015; Vélez et al., 2021) in other fungal systems and should be applied broadly to the powdery mildews. We suggest that GAPDH be used in conjunction with ITS for species identification due to the high variation between GAPDH sequences and the GAPDH primers ability to anneal to herbarium specimens.
Higher level support, not observed in previous powdery mildew studies (Bradshaw and Tobin, 2020; Shirouzu et al., 2020), can be seen in the concatenated tree (Figure 1). Additionally, each region evaluated generally follows the species/genus concepts established for the powdery mildews (Figures 1– 5). The phylogenetic analyses presented using these underexplored loci demonstrate their potential to resolve relationships in major clades and to determine the powdery mildew sister group. The exploration of additional loci will also likely lead to major genus level taxonomic changes. For example, in the RPB2 tree (Figure 2) Arthrocladiella is nested in the Golovinomyces clade. This situation is also seen in the CAM and GS tree where Salmonomyces is nested within the Erysiphe clade (Figures 3, 4). When evaluating “6” gene phylogenies, limitations, including single genes having a comparatively large evolutionary pull, need to be considered before any conclusions are made. For example, in the present manuscript the placement of Blumeria (Figure 2) does not align with previous studies (Bradshaw and Tobin, 2020; Shirouzu et al., 2020) that placed Blumeria sister to Podosphaera. It is likely that the RPB2 and GS loci are driving the evolution of Blumeria. Additional taxa, genomic regions and above all, full genome sequences need to be acquired and analyzed to solidify the taxonomy and phylogeny of this group.
The presented sequences now located in GenBank can serve as reference sequences to help future researchers determine the genus and species present in their collections. The primers evaluated were able to anneal to a broad range of powdery mildew species in multiple genera (Table 2). Future research can employ these primers to assist in phylogenetic and taxonomic studies of species complexes to elucidate the evolutionary relationships of the species in question. Furthermore, the sequences evaluated revealed limited contamination showing the primers specificity to powdery mildews except for the GAPDH primers which readily annealed to Ampelomyces quisqualis s. lat. (the mycopathogen of powdery mildew). The secondary barcode sequences provided can be further mined to generate species/genus specific primers and to improve success with herbarium specimens. Interspecies variation in virulence and fungicide resistance as well as the host specific nature of powdery mildews emphasizes the importance of species identification for these economically important plant pathogens.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
MB designed the experiment, wrote the manuscript, conducted the phylogenetic analysis, designed some of the primers, and sequenced the specimens from the United States. G-XG sequenced the specimens from China and submitted all the sequences to GenBank. LN designed some of the primers. UB helped design the experiment. S-YL helped fund the manuscript and assisted with sequencing the specimens from China. DP helped design the experiment and obtain funding for the analyses. All authors assisted with editing the manuscript.
We would like to thank the National Natural Science Foundation of China (31970019 and U21A20177), the Daniel E. Stuntz Memorial Foundation, the Puget Sound Mycological Society, and the Sonoma County Mycological Association for funding this research.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank the curatorial team at FH for helping with the specimens and the staff of the Arnold Arboretum for allowing access to collect fresh specimens. We would also like to thank Daniel Murphy at the Idaho Botanical Garden, Tom G. Potterfield and Elan Alford at the Mt. Cuba Center, Cindy Newlander at the Denver Botanical Garden, Erin Buchholz at the Minnesota Landscape Arboretum, Anna Bower at Lotus land, and Wade Enos at the Atlanta Botanica Garden for helping with specimen collections.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2022.918908/full#supplementary-material
Supplementary Figure 1 | Bayesian maximum clade credibility tree of sequences generated for the current study for the ITS + LSU region.
Abadi, S., Azouri, D., Mayrose, I., and Pupko, T. (2019). Model selection may not be a mandatory step for phylogeny reconstruction. Nat. Commun. 10:934. doi: 10.1038/s41467-019-08822-w
Amano, K. (1986). Host Range and Geographical Distribution of the Powdery Mildew Fungi, 2nd Edn. Tokyo: Scientific Societies Press.
Badotti, F., de Oliveira, F. S., Garcia, C. F., Vaz, A. B. M., Fonseca, P. L. C., Nahum, L. A., et al. (2017). Effectiveness of ITS and sub-regions as DNA barcode markers for the identification of Basidiomycota (Fungi). BMC Microbiol. 17:42. doi: 10.1186/s12866-017-0958-x
Bradshaw, M., Braun, U., Götz, M., Takamatsu, S., Brand, T., Cabrera, M. G., et al. (2021a). Contributions to the phylogeny and taxonomy of the Erysiphaceae (powdery mildews)—part 1. Sydowia 73, 89–112.
Bradshaw, M., Braun, U., Götz, M., and Jurick, W. II (2021b). Phylogeny and taxonomy of powdery mildew caused by Erysiphe species on Lupinus hosts. Mycologia 114, 76–88. doi: 10.1080/00275514.2021.1973287
Bradshaw, M., and Tobin, P. (2020). Sequencing herbarium specimens of a common detrimental plant pathogen (powdery mildew). Phytopathology 110, 1248–1254. doi: 10.1094/PHYTO-04-20-0139-PER
Braun, U., and Cook, R. T. A. (2012). Taxonomic Manual of the Erysiphales (Powdery Mildews). CBS Biodiversity Series No. 11. Utrecht: CBS-KNAW Fungal Biodiversity Centre.
Braun, U., and Takamatsu, S. (2000). Phylogeny of Erysiphe, Microsphaera, Uncinula (Erysipheae) and Cystotheca, Podosphaera, Sphaerotheca (Cystotheceae) inferred from rDNA ITS sequences–Some taxonomic consequences. Schlechtendalia 4, 1–33.
Cunnington, J., Takamatsu, H., Lawrie, S., and Pascoe, A. (2003). Molecular identification of anamorphic powdery mildews (Erysiphales). Australas. Plant Pathol. 32, 421–428.
Ellingham, O., David, J., and Culham, A. (2019). Enhancing identification accuracy for powdery mildews using previously underexploited DNA loci. Mycologia 111, 798–812. doi: 10.1080/00275514.2019.1643644
Felsenstein, J. (1985). Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783e–791e. doi: 10.1111/j.1558-5646.1985.tb00420.x
Gernhard, T. (2008). The conditioned reconstructed process. J. Theor. Biol. 253, 769–778. doi: 10.1016/j.jtbi.2008.04.005
Haelewaters, D., Park, D., and Johnston, P. R. (2021). Multilocus phylogenetic analysis reveals that Cyttariales is a synonym of Helotiales. Mycol. Prog. 20, 1323–1330. doi: 10.1007/s11557-021-01736-2
Hirata, T., and Takamatsu, S. (1996). Nucleotide sequence diversity of rDNA internal transcribed spacers extracted from conidia and cleistothecia of several powdery mildew fungi. Mycoscience 37, 283–288.
Johnston, P. R., Quijada, L., Smith, C., Baral, H. O., Hosoyo, T., Baschien, C., et al. (2019). A multigene phylogeny toward a new phylogenetic classification of Leotiomycetes. IMA fungus 10:1. doi: 10.1186/s43008-019-0002-x
Khodadadi, F., Gonzalez, J. B., Martin, P. L., Giroux, E., Bilodeau, G. J., Peter, K. A., et al. (2020). Identification and characterization of Colletotrichum species causing apple bitter rot in New York and description of C. noveboracense sp. nov. Sci. Rep. 10:11043. doi: 10.1038/s41598-020-66761-9
Kiss, L. (2012). Limits of nuclear ribosomal DNA internal transcribed spacer (ITS) sequences as species barcodes for Fungi. Proc. Natl. Acad. Sci. U.S.A. 109:E1811. doi: 10.1073/pnas.1207143109
Kiss, L., Vaghefi, N., Bransgrove, K., Dearnaley, J. D. W., Takamatsu, S., Tan, Y. P., et al. (2020). Australia: A Continent Without Native Powdery Mildews? The First Comprehensive Catalog Indicates Recent Introductions and Multiple Host Range Expansion Events, and Leads to the Re-discovery of Salmonomyces as a New Lineage of the Erysiphales. Front. Microbiol. 11:1571. doi: 10.3389/fmicb.2020.01571
Kovács, G. M., Jankovics, T., and Kiss, L. (2011). Variation in the nrDNA ITS sequences of some powdery mildew species: do routine molecular identification procedures hide valuable information? Eur. J. Plant Pathol. 131, 135–141.
Liu, M., Braun, U., Takamatsu, S., Hambleton, S., Shoukouhi, P., Bisson, K. R., et al. (2021). Taxonomic revision of Blumeria based on multi-gene DNA sequences, host preferences and morphology. Mycoscience 62, 143–165. doi: 10.47371/mycosci.2020.12.003
Liu, S. Y., Jin, D., Götz, M., Bradshaw, M., Liu, M., Takamatsu, S., et al. (2021). Phylogeny and taxonomy of Podosphaera filipendulae (Erysiphaceae) revisited. Mycoscience 62, 390–394. doi: 10.47371/mycosci.2021.07.001
Marmolejo, J., Siahaan, S. A. S., Takamatsu, S., and Braun, U. (2018). Three new records of powdery mildews found in Mexico with one genus and one new species proposed. Mycoscience 59, 1–7. doi: 10.1016/j.myc.2017.06.010
Matsuda, Y., Takeuchi, K., Obase, K., and Ito, S. (2015). Spatial distribution and genetic structure of Cenococcum geophilum in coastal pine forests in Japan. FEMS Microbiol. Ecol. 91:fiv108. doi: 10.1093/femsec/fiv108
Meyer, W., Irinyi, L., Hoang, M. T. V., Robert, V., Garcia-Hermoso, D., Desnos-Ollivier, M., et al. (2019). Database establishment for the secondary fungal DNA barcode translational elongation factor 1α (TEF1α)1. Genome 62, 160–169. doi: 10.1139/gen-2018-0083
Mori, Y., Sato, Y., and Takamatsu, S. (2000). Evolutionary Analysis of the Powdery Mildew Fungi Using Nucleotide Sequences of the Nuclear Ribosomal DNA. Mycologia 92:74. doi: 10.2307/3761452
Ouhibi, S., Santos, C., Ghali, R., Soares, C., Hedhili, A., Paterson, R., et al. (2018). (2018) Penicillium tunisiense sp. nov., a novel species of Penicillium section Ramosa discovered from Tunisian orchard apples. Int. J. Syst. Evol. Microbiol. 68, 3217–3225. doi: 10.1099/ijsem.0.002962
Qiu, P.-L., Liu, S.-Y., Bradshaw, M., Rooney-Latham, S., Takamatsu, S., Bulgakov, T. S., et al. (2020). Multi-locus phylogeny and taxonomy of an unresolved, heterogeneous species complex within the genus Golovinomyces (Ascomycota, Erysiphales), including G. ambrosiae, G. circumfusus and G. spadiceus. BMC Microbiol. 20:51. doi: 10.1186/s12866-020-01731-9
Saenz, G. S., and Taylor, J. W. (1999). Phylogeny of the Erysiphales (powdery mildews) inferred from internal transcribed spacer ribosomal DNA sequences. Can. J. Bot. 77, 150–168. doi: 10.1139/b98-235
Saito, S., Margosan, D., Michailides, T. J., and Xiao, C. L. (2016). Botrytis californica, a new cryptic species in the B.cinerea species complex causing gray mold in blueberries and table grapes. Mycologia 108, 330–343. doi: 10.3852/15-165
Samson, R. A., Visagie, C., Houbraken, J., Hong, S.-B., Hubka, V., Klaassen, C. H., et al. (2014). Phylogeny, identification and nomenclature of the genus Aspergillus. Stud. Mycol. 78, 141–173. doi: 10.1016/j.simyco.2014.07.004
Seifert, K. A., Wingfield, B. D., and Wingfield, M. J. (1995). A critique of DNA sequence analysis in the taxonomy of filamentous Ascomycetes and ascomycetous anamorphs. Can. J. Bot. 73, 760–767.
Shin, H. D., Meeboon, J., Takamatsu, S., Adhikari, M. K., and Braun, U. (2019). Phylogeny and taxonomy of Pseudoidium pedaliacearum. Mycol. Prog. 18, 237–246. doi: 10.1007/s11557-018-1429-y
Shirouzu, T., Takamatsu, S., Hashimoto, A., Meeboon, J., and Ohkuma, M. (2020). Phylogenetic overview of Erysiphaceae based on nrDNA and MCM7 sequences. Mycoscience 61, 249–258. doi: 10.1016/j.myc.2020.03.006
Silvestro, D., and Michalak, I. (2012). RaxmlGUI: A graphical front-end for RAxML. Organ. Divers. Evol. 12, 335–337. doi: 10.1007/s13127-011-0056-0
Simon, U. K., and Weiss, M. (2008). Intragenomic variation of fungal ribosomal genes is higher than previously thought. Mol. Biol. Evol. 25, 2251–2254. doi: 10.1093/molbev/msn188
Stielow, J. B., Lévesque, C. A., Seifert, K. A., Meyer, W., Iriny, L., Smits, D., et al. (2015). One fungus, which genes? Development and assessment of universal primers for potential secondary fungal DNA barcodes. Persoonia 35, 242–263. doi: 10.3767/003158515X689135
Suchard, M. A., Lemey, P., Baele, G., Ayres, D. L., Drummond, A. J., and Rambaut, A. (2018). Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 4:Vey016. doi: 10.1093/ve/vey016
Tamura, K., Stecher, G., and Kumar, S. (2021). MEGA11: molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 38, 3022–3027. doi: 10.1093/molbev/msab120
Tanabe, Y., Watanabe, M. M., and Sugiyama, J. (2005). Evolutionary relationships among basal fungi (Chytridiomycota and Zygomycota): insights from molecular phylogenetics. J. Gen. Appl. Microbiol. 51, 267–276. doi: 10.2323/jgam.51.267
Tymon, L., Bradshaw, M., Gotz, M., Braun, U., Peever, T. L., and Edmonds, R. L. (2022). Phylogeny and Taxonomy of Erysiphe spp. on Rhododendron with a special emphasis on North American species. Mycologia
Vélez, J. M., Morris, R. M., Vilgalys, R., Labbé, J., and Schadt, C. W. (2021). Phylogenetic diversity of 200+ isolates of the ectomycorrhizal fungus Cenococcum geophilum associated with Populus trichocarpa soils in the Pacific Northwest, USA and comparison to globally distributed representatives. PLoS One 16:e0231367. doi: 10.1371/journal.pone.0231367
Vu, D., Groenewald, M., de Vries, M., Gehrmann, T., Stielow, B., Eberhardt, U., et al. (2019). Large-scale generation and analysis of filamentous fungal DNA barcodes boosts coverage for kingdom fungi and reveals thresholds for fungal species and higher taxon delimitation. Stud. Mycol. 92, 135–154. doi: 10.1016/j.simyco.2018.05.001
Keywords: erysiphaceae, molecular phylogeny, multi-locus, powdery mildews, species complexes
Citation: Bradshaw MJ, Guan G-X, Nokes L, Braun U, Liu S-Y and Pfister DH (2022) Secondary DNA Barcodes (CAM, GAPDH, GS, and RpB2) to Characterize Species Complexes and Strengthen the Powdery Mildew Phylogeny. Front. Ecol. Evol. 10:918908. doi: 10.3389/fevo.2022.918908
Received: 21 April 2022; Accepted: 09 May 2022;
Published: 14 June 2022.
Edited by:
Danny Haelewaters, Ghent University, BelgiumReviewed by:
Arthur Grupe, University of Florida, United StatesCopyright © 2022 Bradshaw, Guan, Nokes, Braun, Liu and Pfister. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael J. Bradshaw, bWJyYWRzaGF3QGZhcy5oYXJ2YXJkLmVkdQ==; Shu-Yan Liu, bGl1c3N5YW5AMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.