 Junyan Chen
Junyan Chen Christopher R. Gissendanner2
Christopher R. Gissendanner2 Chinmay V. Tikhe
Chinmay V. Tikhe Qian Sun
Qian Sun Claudia Husseneder
Claudia Husseneder
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol., 27 May 2022
Sec. Coevolution
Volume 10 - 2022 | https://doi.org/10.3389/fevo.2022.881538
This article is part of the Research TopicCoevolution of Insect-Gut MicrobiomeView all 5 articles
Subterranean termites depend nutritionally on their gut microbiota, which includes protozoa as well as taxonomically and functionally diverse bacteria. Our previous metavirome study revealed a high diversity and novel families of bacteriophages in the guts of Coptotermes formosanus workers from New Orleans, Louisiana, United States. Two assembled bacteriophage genomes (Phages TG-crAlp-04 and 06, family Podoviridae) existed in all colonies and showed similarity to a prophage (ProJPt-Bp1) previously sequenced from a bacterial endosymbiont (Candidatus Azobacteroides pseudotrichonymphae, CAP) of protozoa in the gut of a termite species of the genus Prorhinotermes from Taiwan. In this study the genomes of Phage TG-crAlp-04 and 06 were subjected to detailed functional annotation. Both phage genomes contained conserved genes for DNA packaging, head and tail morphogenesis, and phage replication. Approximately 30% of the amino acid sequences derived from genes in both genomes matched to those of ProJPt-Bp1 phage or other phages from the crAss-like phage group. No integrase was identified; the lack of a lysogeny module is a characteristic of crAss-like phages. Primers were designed to sequence conserved genes of the two phages and their putative host bacterium (CAP) to detect their presence in different termite species from native and introduced distribution ranges. Related strains of the host bacterium were found across different termite genera and geographic regions. Different termite species had separate CAP strains, but intraspecific geographical variation was low. These results together with the fact that CAP is an important intracellular symbiont of obligate cellulose-digesting protozoa, suggest that CAP is a core gut bacterium and co-evolved across several subterranean termite species. Variants of both crAss-like phages were detected in different Coptotermes species from the native and introduced range, but they did not differentiate by species or geographic region. Since similar phages were detected in different termite species, we propose the existence of a core virome associated with core bacterial endosymbionts of protozoa in the guts of subterranean termites. This work provides a strong basis for further study of the quadripartite relationship of termites, protozoa, bacteria, and bacteriophages.
Termites are social insects (Thorne, 1997; Harrison et al., 2018) and play an important role in the environment as the main decomposers of plant material (Noble et al., 2009; Brune, 2014). A typical mature termite colony contains three castes: reproductives consisting of seasonal winged alates and permanent egg-laying queens and kings as well as soldiers and workers. Mature colonies develop alates during swarming season, which disperse to establish new colonies. Soldiers defend the colony and workers are responsible for nest maintenance, brood care, foraging for food and water and feeding the other colony members (Boomsma, 2009). Termite workers produce endogenous enzymes in their guts, including cellulases (Inoue et al., 1997), but lignocellulose digestion by the termites’ enzymes alone is incomplete (Slaytor, 1992; Lo and Eggleton, 2010). Therefore, all termite lineages developed a relationship with specialized symbionts that aid in lignocellulose digestion.
The Formosan subterranean termite Coptotermes formosanus Shiraki and the Asian subterranean termite, Coptotermes gestroi Wasmann, both native to Southeast Asia, are two notorious invasive species that have spread around the globe including to the United States (Austin et al., 2006; Li et al., 2009; Husseneder et al., 2012; Evans et al., 2019; Blumenfeld and Vargo, 2020). Subterranean termites harbor a complex community of symbionts consisting of protozoa, bacteria, and archaea in the guts of their workers to ensure adequate nutrition (Cleveland, 1923; Brune and Dietrich, 2015; Benjamino and Graf, 2016). Protozoa are responsible not only for the hydrolysis of cellulose, but also for the generation of the bulk of the fermentation products that are eventually resorbed by the host (Hungate, 1979). Studies showed that colonies of subterranean termites starved to death if the cellulolytic protozoa were removed (Ohkuma, 2003; Sethi et al., 2014; Peterson et al., 2015). Subterranean termite species have different numbers of protozoa species with recent genetic sequencing studies suggesting a higher diversity than previously described (Koizumi, 1921; Su et al., 2016; Jasso-Selles et al., 2020).
In addition to protozoa, subterranean termite worker guts contain a diverse bacteria community (Husseneder et al., 2010; Brune, 2014). In previous studies based on cloning of 16S rRNA gene amplicons, 213 different bacterial operational taxonomic units (OTUs), i.e., species defined by DNA similarity, were reported from the gut of workers of C. formosanus (Shinzato et al., 2005; Husseneder et al., 2010). This was most likely an underestimation since next-generation sequencing of the 16S rRNA gene uncovered 1,460 different OTUs in the closely related species, C. gestroi (Do et al., 2014). The dominant bacteria taxa, richness and diversity were similar when comparing C. formosanus workers from the native (China) and introduced (Japan, United States) range suggesting a tight co-evolutionary relationship of termites with their core bacteria and only minor effects of location and environment (Husseneder et al., 2010). Bacteria can be free-living in the gut lumen, attached to the gut wall, or associated with the protozoa as endo- or ectosymbionts (Brune, 2014). Bacteria play important roles for termite nutrition, such as nitrogen-fixation, uric acid recycling, sulfate-reduction, acetogenesis, and sustaining an anaerobic environment for protozoa, among others (Brune and Friedrich, 2000; Inoue et al., 2000; Ohkuma, 2003).
The bacteria diversity in the termite gut makes termites good models to investigate the roles of gut bacteriophages (Ottesen et al., 2006; Tikhe and Husseneder, 2018; Schmidlin et al., 2019). Bacteriophages, or phages, are viruses that infect only bacteria and are the most abundant organism in the biosphere (Wommack and Colwell, 2000; Rodriguez-Brito et al., 2010). Typical phages follow a lytic (virulent) cycle characterized by immediate lysis of the host after infection or a lysogenic (temperate) cycle, when the phage integrates into the bacterial chromosome as prophage and replicates with the host bacterium until the lytic cycle is induced (Hobbs and Abedon, 2016; Howard-Varona et al., 2017). Other more unusual phages exist as extrachromosomal elements and behave like temperate phages but without integrating into the host genome (Kirsch et al., 2021).
Previous studies of microbiota of subterranean termites showed a high taxonomical and functional diversity of bacteria and bacteriophages (Husseneder et al., 2010; Benjamino and Graf, 2016; Tikhe and Husseneder, 2018). However, detailed knowledge about the existence, composition, and influence of core microbiota on termite biology is lacking. Our lab was the first to sequence full genomes of bacteriophages from termite guts. We previously isolated and sequenced a lytic phage (Tikhe et al., 2015), a temperate phage (Tikhe et al., 2018a) and a phage with a defective integrase pseudogene (Tikhe et al., 2018b) from hindguts of C. formosanus workers. Metagenomic profiling of C. formosanus workers’ gut bacteriophage community identified a taxonomically diverse phage community in workers from three colonies of C. formosanus collected in New Orleans, Louisiana, United States (Tikhe and Husseneder, 2018). Two of the assembled phage genomes (NCBI Genbank number: LSPY01000004) and LSPY01000006 existed in all collected colonies and showed similarity to a phage previously sequenced from a bacterium (Candidatus Azobacteroides pseudotrichonymphae, CAP) associated with cellulase digesting protozoa (Pseudotrichonympha sp.) in the gut of a subterranean termite species Prorhinotermes japonicus from Lanyu island (Taiwan) (Pramono et al., 2017). Phylogenetic analysis by Yutin et al. (2018) characterized both phages as crAss-like phage (Caudovirales, Podoviridae). Therefore, LSPY01000004 was named “Termite Gut CrAss-like phage 04” abbreviated as “Phage TG-crAlp-04” and LSPY01000006 was named “Termite Gut CrAss-like phage 06” abbreviated as “Phage TG-crAlp-06”). CrAssphages were first identified in 2014 as the most abundant virus in the human gut microbiome (Dutilh et al., 2014; Yutin et al., 2018, 2021; Koonin and Yutin, 2020). Although hundreds of crAss-like phage genomes have been identified in silico, few crAssphages have been isolated in pure culture (Shkoporov et al., 2018, 2021; Guerin et al., 2021).
Research into the core microbiota, which is likely crucial in termite gut functionality, and its role in invasive termite pest species, such as C. formosanus and C. gestroi is expected to shed light on an important part of the symbiotic biology of subterranean termites and might lead in the future to novel pest control methods by disrupting the core symbiosis and thereby possibly impacting nutrition and reproduction of termite colonies (Sethi et al., 2014; Husseneder et al., 2016). In this study we fully annotated the genomes of the two crAss-like phages. We also screened for the presence of these phages and their putative host bacteria species (CAP) in C. formosanus and C. gestroi specimen across their native and introduced range to test whether these phages and their host belong to the core microbiota.
Workers and soldiers from 18 Coptotermes formosanus colonies were collected from their native range (Taiwan) and from their introduced range in the United States (3 colonies from Florida and 4 colonies from New Orleans and Baton Rouge, Louisiana). In addition, 17 Coptotermes gestroi colonies were collected from Taiwan and 6 from Saipan. Eighteen C. formosanus alates, i.e., winged reproductive, were collected in Baton Rouge with light traps consisting of buckets with black lights as described previously (Husseneder and Simms, 2008). All samples from outside Louisiana were preserved in 95% EtOH or DNAlater solution (Thermo Fisher Scientific, Inc., Waltham, MA, United States) immediately after collection from the field and sent to our lab at Louisiana State University Agricultural Center. Termite samples from Louisiana were transported to the lab alive and processed within 24 hours. Termite species were identified based on morphological characteristics of soldiers (Scheffrahn and Su, 2000) by using a stereomicroscope (LEICA MZ16, Meyers Instruments, Houston, TX, United States). Sample details were listed in Supplementary Table 1.
Individual alates and workers were rinsed with 70% EtOH to clean the surface of the body. Then, the gut was removed using sterile forceps and homogenized in phosphate buffered saline with a sterile pestle (Husseneder et al., 2010). Total DNA was extracted from five pooled worker guts per colony and from individual alate guts by using the DNeasy Blood and Tissue kit (Qiagen, Valencia, CA, United States). High molecular weight DNA was confirmed visually on 1% agarose gels and concentration was measured with Qubit™ assays (protocol provided by Invitrogen™, life technologies™).
The genomes of two crAss-like bacteriophages (Phage TG-crAlp-04 and TG-crAlp-06) from our previous study (Tikhe and Husseneder, 2018) were retrieved from NCBI (Genbank Accession numbers: LSPY01000004.1, and LSPY01000006.1). Putative genes were predicted by performing auto-annotation with GeneMarkS and Glimmer gene prediction programs (Besemer et al., 2001; Delcher et al., 2007) and the PHANOTATE program downloaded from GitHub (McNair et al., 2019). The Phage Evidence Collection and Annotation Network (PECAAN) server (Rinehart et al., 2016) was utilized to evaluate genes predicted by GeneMarkS and Glimmer. Both PECAAN and PHANOTATE produced a list of predicted protein-encoding genes for both genomes. For each genome the two gene prediction lists were compared, and all start positions identified in PHANOTATE and PECAAN were manually checked. PHANOTATE and PECAAN gene calls were mostly identical. Multiple criteria were utilized for finalizing called genes and gene starts. These included agreement between the various gene calling programs, coding potential data (generated by host-trained GeneMarkS), sequence similarity matches, gene overlap, and length. In cases where two genes on the forward and reverse strands occupied the same genomic position, one gene was deleted from the final list. In this situation, the genes associated with the best coding potential, and/or those that had the strongest functional call or homology to other phage genes were kept for the final gene list.
To assign putative functions to the predicted genes, the protein sequence encoded by the genes was subjected to BLASTP analysis using default settings. For the majority of genes from both genomes, there was no significant protein sequence similarity detected by BLASTP. In cases where similarity with other proteins in the database was detected we used a minimum cutoff of 1.0E-7 or less when BLASTP data was used as supporting evidence for a putative functional call. Protein sequences were also evaluated using HHpred to identify the probability of structural homology to three-dimensional protein structures found in the Protein Data Bank (wwPDB1) (Sussman et al., 1998). We used a 50% probability when HHpred data was used as supporting evidence for a functional call. In cases where BLASTP and HHpred did not identify the same predicted function, the call with the strongest support was used as the predicted function. In cases where the HHpred output was the only supporting evidence, a functional call was not made if the alignment coverage was < 10%, even if probability values were > 50%. We avoided making functional calls from HHpred alone if the function was only relevant to eukaryotes. However, in the case of the latter, a functional call was made if a conserved protein domain, or general protein function (e.g., hydrolase) could be identified in the HHpred output. Membrane proteins were identified using TMHMM2. All TMHMM calls were confirmed by using the SOSUI transmembrane prediction program3. Aragorn4 and tRNAscan-SE5 were utilized to search for tRNA genes (Laslett and Canback, 2004; Chan et al., 2021).
Detailed genome annotation information was provided in the Supplementary Table 4 for Phage TG-crAlp-04 and Supplementary Table 5 for Phage TG-crAlp-06. Genome maps based on all combined results were drawn using CGView server (Grant and Stothard, 2008).
The genomes of Phage TG-crAlp-04 and TG-crAlp-06 were aligned with other crAss-like phage genomes in the NCBI database, including the prophage discovered in CAP (Pramono et al., 2017) to identify conserved regions. Multiple primer pairs were designed from Geneious software to target signature genes, such as the Large Terminase and Portal Protein gene. In a similar approach, the 16S rRNA gene region of the host bacterium CAP was aligned with closely related sequences (Supplementary Table 4) from NCBI GenBank to design species specific primers for partial amplification of the 16S gene V3 region. Primer sets that resulted in successful amplification of the phage and CAP target region are shown in Table 1. Polymerase chain reactions (PCR) for phage and bacteria DNA templates were performed using One Taq 2X Master Mix with standard buffer (New England BioLabs Inc., Ipswich, MA, United States) on a thermal cycler (Bio-Rad, Hercules, CA, United States). For each reaction, 15 μl of 2X Master Mix, 1–4 μl of DNA (10–15 ng) and 1 μl of each primer (0.5 μM) were combined. Nuclease free water was added to reach a final volume of 30 μl. The amplification protocol was 5 min of activation at 94°C, 25 cycles of 1 min at 94°C, 1 min of annealing at 52–55°C depending on the primer set (Table 1), 1 min of extension at 68°C, and 5 min for final elongation at 72°C. Each PCR product’s size and concentration were estimated by quantitative marker ladder (1 kb DNA ladder, NEB, MA, United States) in 1% agarose gels and concentrations were confirmed by QubitTM 4 fluorometer (ThermoFisher, Waltham, MA, United States). Samples with negative results (referring to the wrong size of PCR product and amplification failures) were repeated with higher DNA concentration and lower annealing temperatures. Negative results (absence of phages) were not included in further analyses. Successfully amplified products were subjected to bidirectional Sanger sequencing (Applied Biosystems™ 3130xl, LSU Genome facility). The forward and reverse sequences were assembled into single contigs. All contigs of each gene region were aligned and truncated to the same length. MUSCLE (v.3.8.425) was used for alignment with the Geneious Prime software’s (version 2020.2) default settings: kmer4_6 (iteration 1) and pctid_kimura (subsequent) for distance measure; UPGMB for clustering method; pseudo for tree rooting; CLUSTALW for sequence weighting scheme (Kearse et al., 2012). The alignment files were then exported in PHYLIP format for phylogenetic tree analysis. Maximum likelihood phylogenetic trees were constructed by using IQtree web server6 with auto substitution model and ultrafast bootstrap analysis (Trifinopoulos et al., 2016; Minh et al., 2020) with 1,000 bootstrap alignments. Bacteria and phage reference sequences of Coptotermes species and closely related genera were downloaded from NCBI GenBank in December 2021 to analyze evolutionary diversification of bacteriophages and host bacteria across geographical regions and closely related species (Supplementary Table 2). For the tanglegram tree shown in Figure 3, cytochrome oxidase II (COII) gene sequences of subterranean termites from similar geographic regions (Supplementary Table 3) as our samples were selected from NCBI GenBank. The COII tree was generated using the same method as described above and aligned to the CAP bacteria phylogenetic tree for comparison.
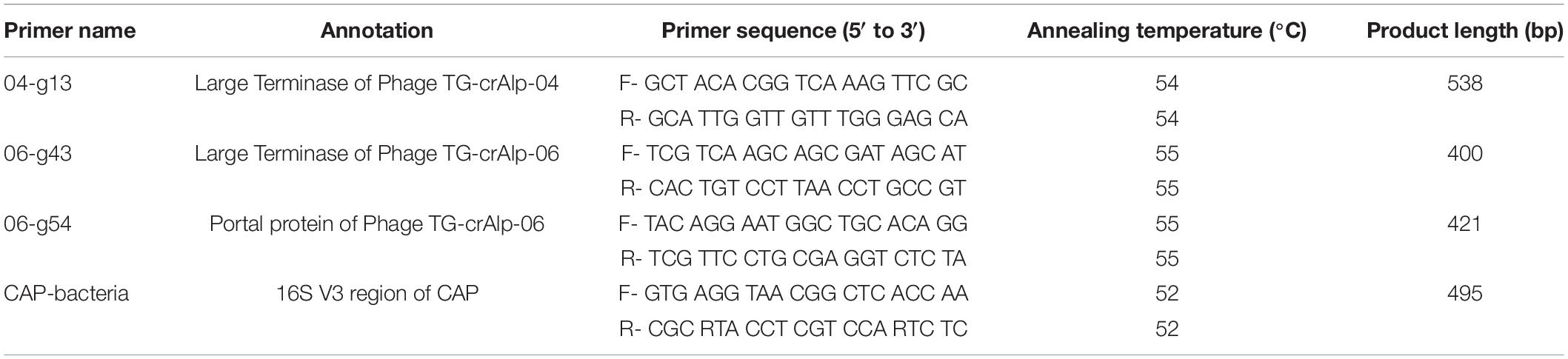
Table 1. Primer pairs designed for phage signature genes and 16S rRNA gene region of host bacteria Candidatus Azobacteroides pseudotrichonymphae (CAP).
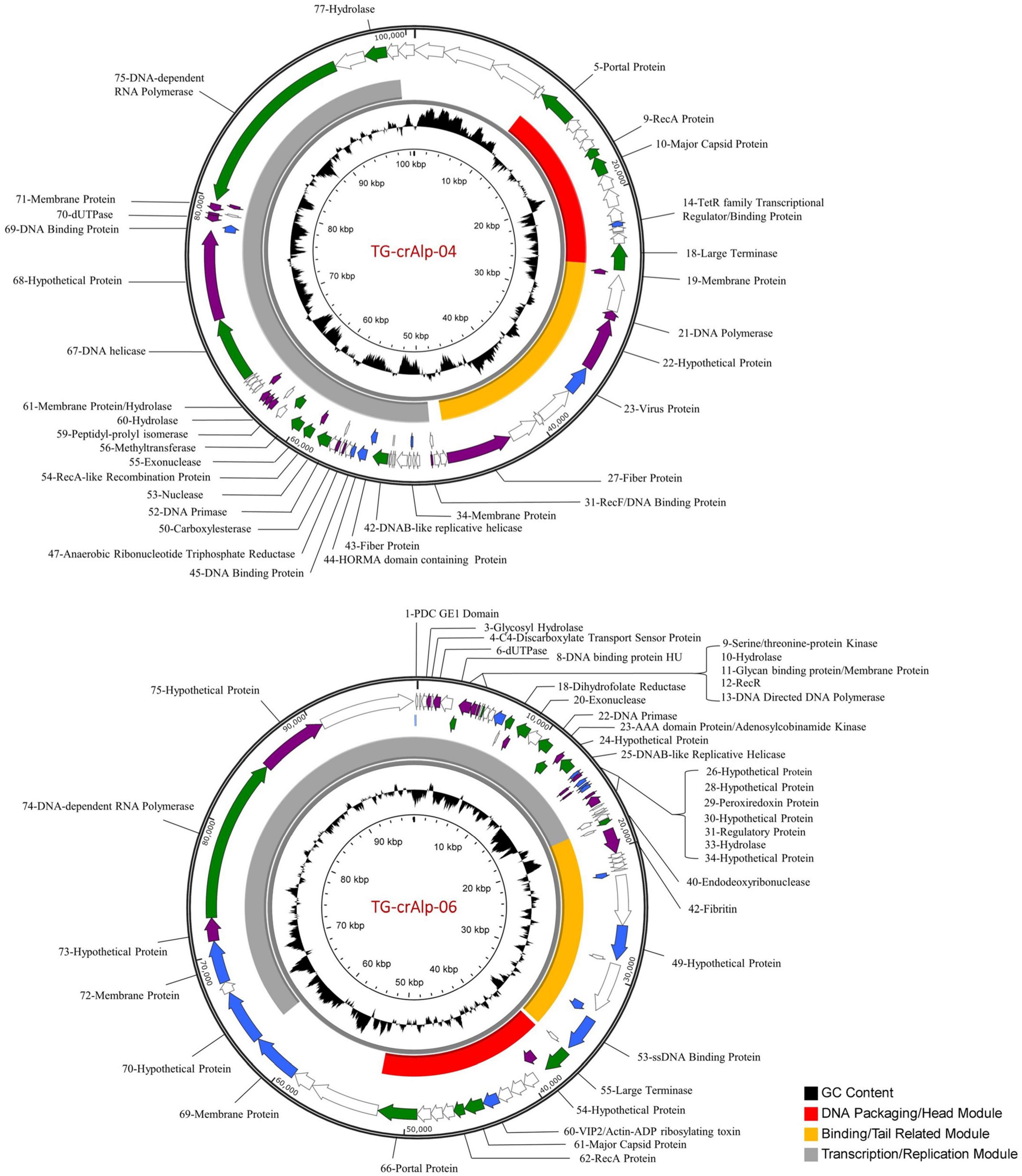
Figure 1. Genome maps of Termite gut crAss-like Phages (TG-crAlp 04 and 06). The outer ring shows orientation of individual genes with color-coded probability of homology: green ≥ 95%, purple 70–95%, blue 50–70%, white < 50%. The middle ring represents phage functional modules (Keary et al., 2014; Koonin and Yutin, 2020). The inner ring displays the GC content. Detailed annotation information is provided in Supplementary Table 4 (for Phage TG-crAlp-04) and S5 (for Phage TG-crAlp-06).

Figure 2. Maximum likelihood tree based on the V3 region of the 16S rRNA gene of Bacteria Candidatus Azobacteroides pseudotrichonymphae (CAP). Reference sequences downloaded from NCBI GenBank are presented with their accession numbers; samples collected for this study are in bold letters. A code for sample location [JP (Japan), TW (Taiwan), BZ (Brazil), LAO (Laos), MA (Malaysia), CN (China Mainland), FL (Florida, United States), United States (New Orleans, LA, United States), BR (Baton Rouge, LA, United States) and LAB (lab reared termites)], termite species [Cf (Coptotermes formosanus), Cg (Coptotermes gestroi), Cc (Coptotermes curvignathus), Ct (Coptotermes testaceus), Ht (Heterotermes tenuis), Hl (Heterotermes longiceps), Pj (Prorhinotermes japonicus), Rs (Reticulitermes speratus), Csp (an unidentified Coptotermes sp.), Cs (Cryptotermes secundus), Cb (Cryptotermes brevis) and Cd (Cryptotermes dudleyi)], and caste [W (worker) and A (alate)] was added to the sequence description if known. In addition, CAP close related bacteria Candidatus Symbiothrix dinenymphae identified in R. speratus termite and Candidatus Armantifilum devescovinae were included as outgroup. Node support values were obtained from 1,000 bootstrap replications. A detailed reference sequence list was submitted in Supplementary Table 2.
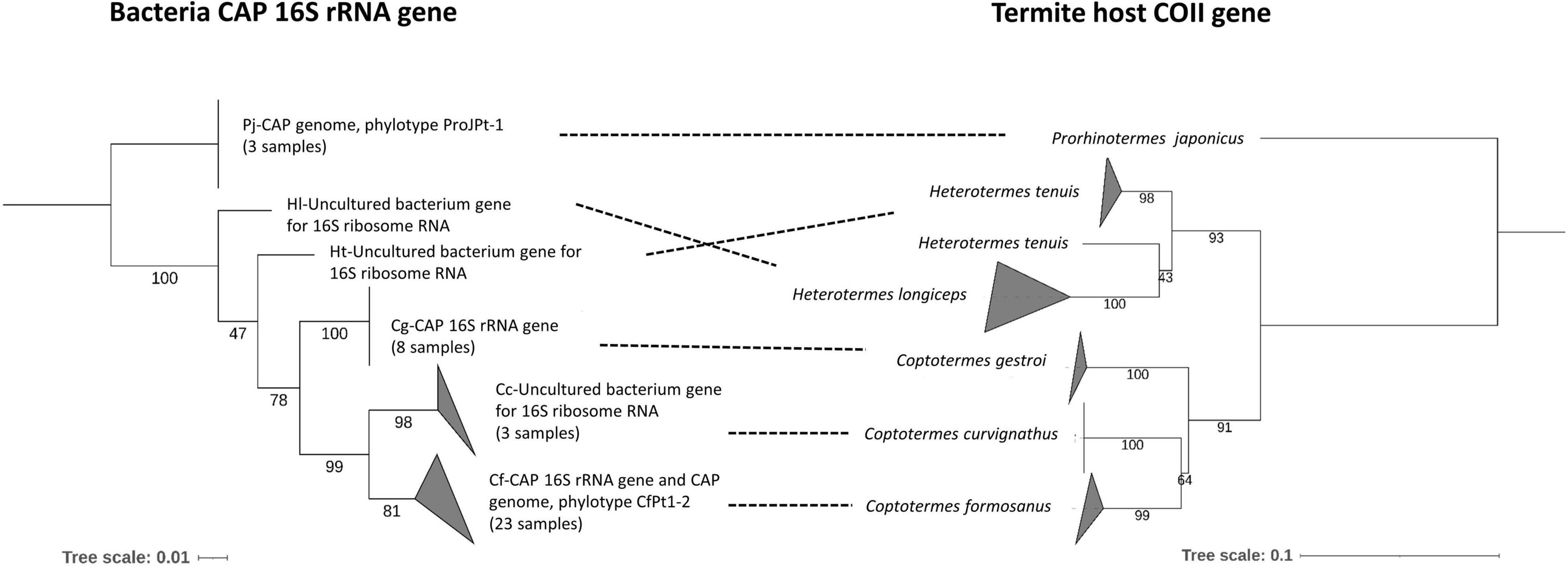
Figure 3. Phylogenetic tanglegram of CAP related bacteria strain and termite hosts. The bacteria tree is a condensed version of Figure 2. The topology of the termite tree was estimated by maximum-likelihood analysis of the cytochrome oxidase II (COII) genes. Dashed lines connect the same termite host species. Node support values were obtained from 1,000 bootstrap replications. A detailed reference sequence list was submitted in Supplementary Table 3.
The genome of Phage TG-crAlp-04 consisted of 100,626 bp (GC content of 39.4%) with a total of 85 protein coding genes predicted by PHANOTATE and 74 genes predicted by GeneMarkS and/or Glimmer (Supplementary Table 4). Seventy-two genes were called by both sets of analyses with complete or partial agreement in genomic position. Phage TG-crAlp-06 had a similar genome size with 98,173 bp (GC content of 39.8%) and 82 protein coding genes predicted by PHANOTATE and 68 genes predicted by GeneMarkS and/or Glimmer (Supplementary Table 5). Sixty-eight genes were called by both analyses with complete or partial agreement in genomic position. While most gene predictions were identical or similar with the different gene prediction tools PHANOTATE predicted more genes. All genes and gene starts from PHANOTATE, GeneMark, and Glimmer were manually evaluated to generate a final gene list of 79 genes for Phage TG-crAlp-04 and 77 genes for Phage TG-crAlp-06. No tRNA genes and integrase genes were identified in both genomes. The final gene set was used for functional annotation and drawing of genome maps (Figure 1).
Predicted gene functions were determined by sequence homology from BLASTP analysis and/or structural homology using HHPred. Predicted functions were assigned for 37 Phage TG-crAlp-04 genes and 34 Phage TG-crAlp-06 genes. Phage TG-crAlp-04 and Phage TG-crAlp-06 shared 12 common predicted gene functions, such as large terminase, portal protein, major capsid protein, DNA-dependent RNA polymerase and DNA binding protein, etc. (Table 2). Additionally, 25 genes encoded in Phage TG-crAlp- 04 and 22 encoded in Phage TG-crAlp-06 had similarity to the CAP phage ProJPt-Bp1 or other crAassphages. Both genomes had similar genome structure and contained the three discernible blocks typically found in all bacteriophages: “DNA packaging/head module”, “Binding/tail related module” and “Transcription/replication module”.
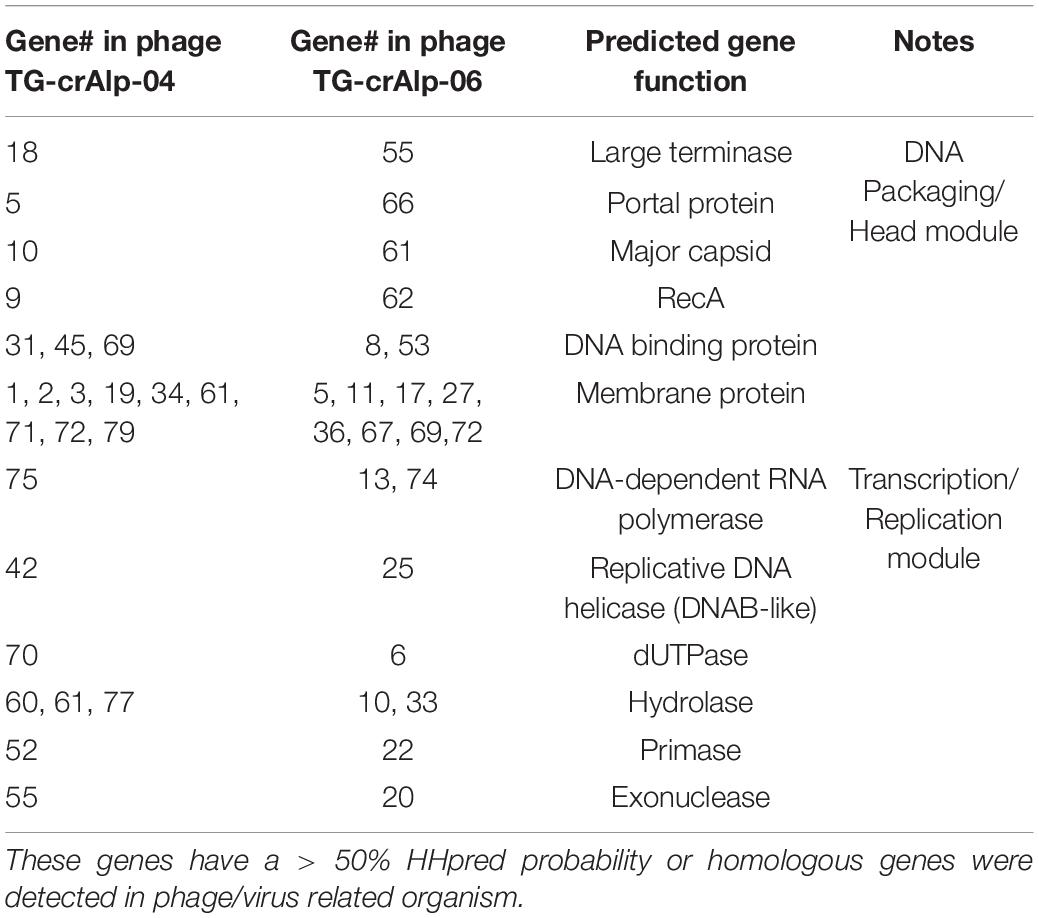
Table 2. List of genes with similar function in both phage genomes.
The “DNA packaging/head module” was overall the most conserved region and consisted of genes encoding for large terminase, portal protein, major capsid, recombination protein A (RecA), along with genes that could not be functionally annotated. All 12 protein encoding genes in the “DNA packaging/head module” of Phage TG-crAlp-06 and 9 out of 13 genes in Phage TG-crAlp-04 showed homology to hypothetical proteins from Azobacteroides phage ProJPt-Bp1 or other crAssphages through BLASTP analysis. Of the three modules, the “DNA packaging/head module” had the highest gene content similarity across the whole genome of both phages when compared to all other crAssphages (69% for Phage TG-crAlp-04; 100% for Phage TG-crAlp-06).
The “Binding/tail related module” was located between the “DNA packaging/head module” and the “Transcription/replication module”. The majority of the proteins encoded by genes in this module for both Phage TG-crAlp-04 and TG-crAlp-06 could not be assigned predicted functions by similarity searching or structural homology. However, based on structural homology, Phage TG-crAlp-04 Gene_27 was predicted to encode a Fiber protein that could be associated with the tail and Phage TG-crAlp-06 Gene_42 was predicted to encode a fibritin protein that functions in connecting the tail and capsid.
In the “Transcription/replication module” of both genomes, we identified several putative enzymes involved in nucleotide metabolism, such as DNAB-like replicative helicase (Gene_42 in Phage TG-crAlp-04, Gene_25 in Phage TG-crAlp-06), DNA primase (Gene_52 in Phage TG-crAlp-04, Gene_22 in Phage TG-crAlp-06), exonuclease (Gene_55 in Phage TG-crAlp-04, Gene_20 in Phage TG-crAlp-06), hydrolase (Gene_60 and 77 in Phage TG-crAlp-04, Gene_10 and 33 in Phage TG-crAlp-06), dUTPase (Gene_70 in Phage TG-crAlp-04, Gene_6 in Phage TG-crAlp-06), and DNA-dependent RNA polymerase (Gene_75 in Phage TG-crAlp-04, Gene_74 in Phage TG-crAlp-06). Furthermore, in Phage TG-crAlp-04, 14 of 39 (35.9%) genes in this module showed homology to hypothetical proteins from Azobacteroides phage ProJPt-Bp1 or other crAssphages. In comparison, only 9 of 48 (18.75%) genes in this this module for Phage TG-crAlp-06 genome were homologs to crAssphages.
Of the genes with predicted functions, some were found in the Phage TG-crAlp-04 genome but not in Phage TG-crAlp-06, and vice versa. For example, the Phage TG-crAlp-04 genome encoded a predicted anaerobic ribonucleotide triphosphate reductase (Gene_47), a carboxylesterase (Gene_50), a methyltransferase (Gene_56), and a peptidyl-prolyl isomerase (Gene_59). Unique genes encoded in the Phage TG-crAlp-06 genome, were a C4-discarboxylate transport sensor protein (Gene_4), a dihydrofolate reductase (Gene_18). While AAA-domain proteins were identified in both genomes, they were different types with an AAA-domain/RecA-like recombination protein (Gene_54) in Phage TG-crAlp-04, and an AAA-domain/Adenosylcobinamide kinase (Gene_23) in Phage TG-crAlp-06.
The putative host bacterium of the phages, Ca. Azobacteroides pseudotrichonymphae (CAP) was successfully amplified in 26 out of 66 samples of C. formosanus and C. gestroi. All amplified sequences aligned with CAP strain references of different Coptotermes species found in the NCBI database (Supplementary Table 2) indicating that the primer design was specific for CAP. Samples that repeatedly failed to amplify either did not contain the target bacteria species in detectable concentrations and/or the primers designed for CAP were not able to detect all strain variants due to mutations in primer binding sites.
A phylogenetic tree was constructed with sequences from our samples consisting of C. formosanus workers and alates and C. gestroi workers from different geographic regions. Related CAP strains from other Coptotermes spp. and termite genera retrieved from the NCBI database were included for reference (Figure 2 and Supplementary Table 2). The majority of CAP sequences of C. formosanus workers and alates from various locations grouped with the originally described CAP reference strain of C. formosanus with less than 8 bps (98.25–100% similarity) nucleotide difference. The adjacent clade formed by C. curvignathus and C. speratus samples from Malaysia, showed 94–97% similarity to the C. formosanus group. Sequences of CAP from C. formosanus, C. curvignathus, and C. testaceus formed a large clade separate from C. gestroi CAP sequences. The sequences of the C. gestroi samples from Taiwan showed a 6–7% difference compared to the C. formosanus samples from various geographic locations, including Taiwan. The original CAP reference strains (phylotype ProJPt-1) described from a Pseudotrichonympha sp. in the gut of Prorhinotermes japonicus formed a clade with a similar CAP strain isolated from symbiotic protozoa, Pseudotrichonympha sp., in the rhinotermitids Heterotermes tenuis and Heterotermes longiceps from Brazil. The interspecific distance of the P. japonicus CAP strains to those from our C. gestroi and C. formosanus samples was 10–12%.
In general, CAP sequences from the same termite host species grouped together and the intraspecific similarity (C. formosanus: 98–100%, C. gestroi: 99–100%, C. curvignathus 97–99%) was greater than the interspecific similarity of CAP strains of other termite species (C. gestroi vs. C. formosanus CAP sequences: 93–94%; C. gestroi vs. C. curvignathus CAP sequences: 92–94%, C. formosanus vs. C. curvignathus CAP sequences: 94–96%).
Intraspecific sequence variation of CAP in C. formosanus specimens was low and there were only very few base pair differences among CAP sequences across geographical regions. Furthermore, we noticed that the amplified bacteria sequence from a C. gestroi sample from Saipan was considerably different from CAP sequences of other Coptotermes spp. (only 78–81% similarity). However, it showed high similarity (98–100%) to the 16S gene sequence of bacteria Candidatus Armantifilum devescovinae (Bacteroidales) (NCBI accession number: FN377755, FN377756 and FN377757). The bacteria Ca. A. devescovinae is an obligate ectosymbiotic bacterium in devescovinid protozoa (Devescovina spp.) in dry-wood termites (Kalotermitidae). Both CAP and bacteria Ca. Armantifilum devescovinae are associated with protozoa and are involved in dinitrogen-fixing processes (Desai and Brune, 2012).
The phylogenetic tree of CAP 16S rRNA gene sequences was compared to a phylogenetic tree of termite hosts constructed from COII sequences retrieved from NCBI GenBank (Figure 3 and Supplementary Table 3). There was high phylogenetic congruence between the phylogenies of the termite host species and CAP. Both trees showed that Coptotermes species evolved as a monophyletic clade with C. formosanus closer to C. curvignathus than C. gestroi.
The primers for Phage TG-crAlp-04 large terminase gene amplified 15 sequences, while primers for Phage TG-crAlp-06 amplified 23 large terminase and 18 portal protein gene sequences from all 66 samples (Supplementary Table 1). No amplification was achieved with the primers designed for Phage TG-crAlp-04 portal protein gene. Despite limited amplification success, sequences were obtained from both Coptotermes species, castes and all collected geographical regions (Figure 4).
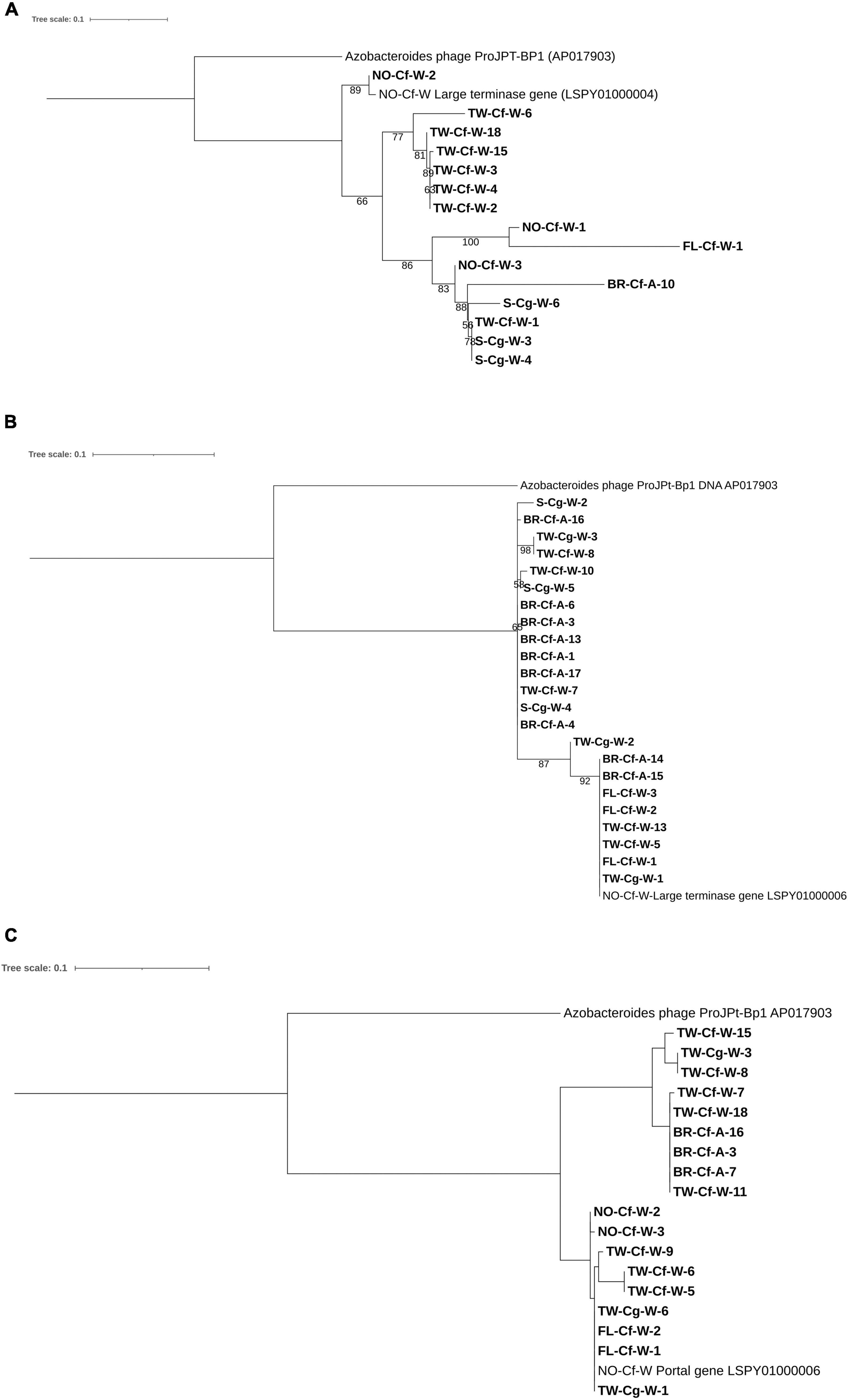
Figure 4. Maximum likelihood trees of (A) Phage TG-crAlp-04 Terminase gene, (B) Phage TG-crAlp-06 Terminase gene and (C) Phage TG-crAlp-06 Portal Protein gene sequences. Support values were obtained from 1,000 bootstrap replications; values greater than 50% are shown. Sample names in bold indicate samples collected in this study and consist of location [TW (Taiwan), S (Saipan), FL (Florida, United States), NO (New Orleans, LA, United States), BR (Baton Rouge, LA, United States)], termite species [Cf (Coptotermes formosanus), Cg (Coptotermes gestroi)], and caste [W (worker) and A (alate)]. Sequences for the Azobacteroides phage from P. japonicus (Pramono et al., 2017) and Phage TG-crAlp-04 and TG-crAlp-06 from the C. formosanus metavirome (Tikhe and Husseneder, 2018) were retrieved from NCBI database and included as references.
The Phage TG-crAlp-04 and Phage TG-crAlp-06 large terminase genes and the portal protein gene of Phage TG-crAlp-06 from the C. formosanus metavirome (Tikhe and Husseneder, 2018) matched closely (67–72%) to Azobacteroides phage ProJPt-BP1 (NCBI accession number: AP017903) (Pramono et al., 2017). Sequences for the Phage TG-crAlp-04 large terminase genes from C. formosanus and C. gestroi samples showed 67–99% similarity to the large terminase gene of the Phage TG-crAlp-04 reference genome assembled from the metavirome, which confirmed target specific amplification. Similarly, sequences of Phage TG-crAlp-06 large terminase and portal protein genes from both Coptotermes species mapped with 92–100% (large terminase gene) and 87–100% (portal protein gene) similarity to the Phage TG-crAlp-06 genome from the metavirome.
Phage TG-crAlp-04 large terminase sequences as well as Phage TG-crAlp-06 large terminase and portal protein gene sequences showed variation among our Coptotermes samples (21, 8, and 11%, respectively) and separate clades in the phylogenetic trees confirmed the existence of gene variants; however, variations in neither gene were strictly associated with termite host species, caste, or geographical region of origin (Figure 4).
In this study, we comprehensively annotated two crAss-like phages from the gut of Coptotermes formosanus workers and described the diversification of these phages and their putative host bacteria across geographical regions and subterranean termite species.
Both phages investigated in this study belong to the crAss-like phages. Their genome sizes (Phage TG-crAlp-04 – 100,626 bp, Phage TG-crAlp-06 – 98,173 bp) fell in the general size range of crAss-like phages which are typically around 100 kb in size (Guerin et al., 2021). Detailed annotation of the genomes of Phage TG-crAlp-04 and TG-crAlp-06 confirmed signature genes from all major phage modules typically found in phages in general and in crAss-like phages (Keary et al., 2014; Yutin et al., 2018; Koonin and Yutin, 2020). Protein sequences encoded by the signature genes in both phages matched to crAss-like phages in the NCBI database, thus, confirming the phylogenetic placement of the termite gut phages in the crAss-like phage group (Tikhe and Husseneder, 2018) similar to crAss-like phages from human gut and environmental metagenomes (Yutin et al., 2018, 2021).
CrAss-like phages are dsDNA phages of the order Caudovirales with Podovirus-like morphology (Yutin et al., 2018). Although crAss-like phages are the most abundant phages in the human gut (Yutin et al., 2018), the effects and roles of crAss-like phages in the human gut or other environments are unclear (Guerin et al., 2021). The detection of crAss-like phages as components of the termite gut microbiota indicates that they may play roles in diverse ecological niches and need to be investigated further.
There are no phages in the current NCBI database (Dec. 2021) that show close similarity to our termite phages at genome level. Thus, an in-depth phage genome annotation was undertaken to decipher the biology of the two phages. The in-depth annotation in the current study provided evidence for the functional prediction for 49% of genes in Phage TG-crAlp-04, and 57% of genes in Phage TG-crAlp-06. These genes include phage signature genes, which are considered conserved due to their important function in the phage life cycle (Mitchell et al., 2002; Hou et al., 2018; Frantzen and Holo, 2019). For example, packaging of the phage concatemeric DNA is initiated by phage terminase enzymes (encoded by Gene_18 in Phage TG-crAlp-04 and Gene_55 in Phage TG-crAlp-06) (Black, 1995; Rao and Feiss, 2015) with the packaging force moving the DNA through a ring made by the portal protein (encoded by Gene_5 in Phage TG-crAlp-04 and Gene_66 in Phage TG-crAlp-06) into the phage capsid (capsid protein encoded by Gene_10 in Phage TG-crAlp-04 and Gene_61 in Phage TG-crAlp-06). RecA gene found in both phages in the “DNA packaging/head module” is considered important for the homologous recombination process (Le et al., 2017). Other genes like dUTPase, hydrolase, nuclease, DNA primase, DNAB-like replicative helicase and DNA-dependent RNA polymerase are considered to be involved in the phage replication process (Häuser et al., 2012; Kerepesi et al., 2016; Afek et al., 2018; Drobysheva et al., 2021; Halgasova et al., 2021). Proteins encoded by all the above-mentioned genes showed homology to Azobacteroides phage or crAss-like phage proteins.
A previously conducted preliminary annotation of phages TG-crAlp-04 and TG-crAlp-06 did not detect typically conserved genes, like terminase, portal protein, and major capsid (Tikhe and Husseneder, 2018). However, in the present study, terminase, portal protein, and major capsid were confirmed with strong support in both phages (Yutin et al., 2018). The discrepancy between former and recent annotation results was likely due to the previous scarcity of references for well annotated crAss-like phages. The study of crAss-like phages has been hampered by the fact that most remain unculturable in lab condition and the hosts for the majority of crAss-like phages are unknown. Recent interest in crAss-like phages resulted in increased availability of references, which helped in the annotation of phages TG-crAlp-04 and TG-crAlp-06 (Yutin et al., 2021; Gulyaeva et al., 2022). No tRNA genes were identified in Phage TG-crAlp-04 and 06. The absence of tRNA genes might indicate a non-virulent life cycle of Phage TG-crAlp-04 and 06 since the presence of tRNA in phage genomes genes has been shown to largely coincide with virulent phages (Bailly-Bechet et al., 2007; Delesalle et al., 2016).
It is believed that bacteria-phage interactions drive co-evolution that shapes microbial ecology, (Blazanin and Turner, 2021). In general, all phages must adsorb to the bacterial surface and introduce their genome into the host. Non-temperate phages replicate and assemble new virions while temperate phages may integrate into the bacterial chromosome as a prophage or be maintained as an episome (Kirsch et al., 2021). Most phages in the termite metavirome study had predicted genes that matched to bacterial prophages and were, thus temperate (Tikhe and Husseneder, 2018). However, neither Phage TG-crAlp-04 nor TG-crAlp-06 contained verifiable genes typically associated with a classical temperate phage. Once a bacteriophage integrated itself into the host chromosome, the prophage will produce a repressor, which self-regulates its own promoter and inhibits all other phage gene expression in the lysogen to maintain a stable status (Fogg et al., 2011; Yoshua et al., 2021). No repressor genes and no phage integrase site-specific recombination sequence were identified in our phage genomes (Fogg et al., 2014). Moreover, integration as prophage into the bacteria chromosome requires functional integrase. Although integrase-like genes were identified in both Phage TG-crAlp-04 and TG-crAlp-06 genomes, the HHpred probability value was very low (less than 50%), and the genes did not have phage/virus related orthologs. Besides, the two major families of integrases, tyrosine and serine integrases (Groth et al., 2000; Fogg et al., 2014), have not been identified in any crAss-like phage genome so far (Shkoporov et al., 2018; Guerin et al., 2021; Yoshua et al., 2021).
The most closely related phage to Phage TG-crAlp-04 and TG-crAlp-06, the ProJPt-Bp1 phage described from the genome sequencing project of Ca. A. pseudotrichonympha, was not integrated into the chromosome or the plasmids of Ca. A. pseudotrichonymphae (Pramono et al., 2017). The phage showed much higher GC content and little sequence similarity when compared to the chromosome or plasmid sequences, was circular or circular permuted and, thus, existed as a separate extrachromosomal element (episome). The lack of related prophage sequences in bacteria chromosomes in NCBI GenBank and the lack of typical genes related to a temperate life cycle suggests that the crAss-like phages of CAP do not necessarily integrate into the host chromosome but exist as episomes. Since the termite gut microbiota comprises a delicate functional ecosystem essential for the survival of the termite host (Brune, 2014; Zeng et al., 2020), we expected a prevalence of temperate over lytic phages in our metavirome study (Tikhe and Husseneder, 2018). The temperate phages would prevent rapid changes in bacteria composition and metabolism, and, thus, maintain the vital balance in the termite gut. In addition, they would replicate along with their bacterial hosts, be horizontally transferred among termite colony members via trophallaxis and vertically to new colonies via alate colony founders. Nevertheless, the phages infecting the dominant bacteria (CAP) of an important protozoan symbiont are not following the classic lysogenic cycle.
Shkoporov et al. (2019) showed that crAss-like phages stably persist alongside their host bacteria in the human gut to form a “personal persistent virome” and the phages can be transferred horizontally via fecal transplants and vertically from mother to infant (Manrique et al., 2016). In addition, persistence was confirmed by co-culture experiments of isolated crAss phages with their Bacteroides host, which showed that phages can stably coexist with their host over time at high concentrations without impacting host abundance (Guerin et al., 2021). The mechanisms by which coexistence is achieved are not entirely clear, but there are several possible ecological models. As previously suggested for phages in the termite gut (Tikhe and Husseneder, 2018), virulent phages that infect well-adapted, dominant and obligate bacteria might switch from lytic to a lysogenic or other benign infection mechanism to benefit from the success of the thriving bacteria host, i.e., they employ the “piggyback-the-winner” strategy; this strategy results in a low phage-to-bacteria ratio, which is less likely to cause abrupt population collapse (Knowles et al., 2016; Guerin et al., 2021; Shkoporov et al., 2021).
While the crAss-like phages might not be equipped to become lysogenic via integration due to the lack of integrase genes, they may employ alternative less aggressive infection methods that allow them to coexist with the host by sustaining replication without disrupting the host population. For example, host strain variations regarding phage receptors would create a mix of phage susceptible and resistant strain allowing phages to replicate while keeping the viral load in check to protect susceptible population. Bacteria have been shown to create herd immunity using phase variation of receptors to switch between phage-susceptible and phage-resistant phenotypes (Turkington et al., 2019). Spatial separation of microbial habitats has been shown to create refuges for phage susceptible host subpopulations (Lourenço et al., 2020). Such spatial heterogeneity is provided by the fact that the phages’ host bacteria (CAP) are endosymbionts of protozoa (Noda et al., 2005). In addition, the existence of Phage TG-crAlp-04 and TG-crAlp-06 as episomes in the bacteria cell might be a mechanism of pseudolysogeny to ensure delayed lysis of infected cells (Cenens et al., 2013; Shkoporov et al., 2021). Such stable and relatively benign interaction suggests that bacteria and phages associated with the protozoan endosymbionts in termite hind guts are in a long-term partnership, maintaining high host population abundance, which is up to 105 CAP bacteria cells per protozoa (Noda et al., 2005).
Although lysis is likely delayed, both phages exist outside the cell in form of virion particles at some point in their phage cycle as evidenced by the fact that their DNA was collected after a filtration process including DNase treatment (Tikhe and Husseneder, 2018). Further studies are needed to determine how crAss-like phages are able to penetrate both protozoa and bacteria cells during the infection process.
The putative host bacterium of both crass-like phages, CAP, was present in C. formosanus and C. gestroi specimens, and matched to related CAP strains from three other Coptotermes spp and rhinotermitid genera retrieved from the NCBI database. This suggests that CAP is a core bacterium in Coptotermes and related genera of subterranean termites with the exception of Reticulitermes. In a pilot study we used the primers developed in this study to test workers of Reticulitermes flavipes for CAP-related bacteria; however, amplification consistently failed. A previous core microbiota study in R. flavipes detected bacteria related to Azobacteroides with 3.44% abundance (Benjamino and Graf, 2016). However, when we compared these sequences to CAP genomes in GenBank the highest similarity was only 88% indicating a rather distant relationship. To date (Jan. 2022) there are no CAP sequences for R. flavipes in NCBI GenBank. This is likely due to the lack of Pseudotrichonympha spp in termites of the genus Reticulitermes (Song et al., 2021) and, in particular, in R. flavipes termites (Lewis and Forschler, 2004).
Previous studies showed the Bacteroidales endosymbiont (later identified as CAP, Ohkuma, 2003) of Pseudotrichonympha cospeciated with protozoa and rhinotermitid species as part of a triplex symbiotic system (Noda et al., 2007). Here we confirmed the phylogenetic congruence between CAP and Coptotermes species as well as closely related genera of subterranean termites. A tight association has been shown to exist for the majority of intracellular endosymbioses in insects (Bandi et al., 1995; Moran and Telang, 1998; Noda et al., 2007; Ikeda-Ohtsubo and Brune, 2009; Strassert et al., 2012; Zheng et al., 2015) and is expected due to the crucial role the endosymbionts play for their respective hosts. The vital functions of CAP as nitrogen fixating and recycling endosymbiont that also imports glucose and xylose as energy and carbon sources for its protozoan host (Hongoh, 2010) and the obligate relationship with Pseudotrichonympha protozoa, which are vital for efficient cellulose digestion of their termite host, strengthen the triple coevolutionary congruence. Furthermore, due to the strict anaerobic nature of the protozoa environmental transfer of symbionts is unlikely; the social lifestyle of termites promotes horizontal transfer within colonies via proctodeal feeding and long-term vertical transfer from the colony founders to offspring, i.e., transfer occurs within a relatively closed system. The high abundance of CAP bacteria in Pseudotrichonympha grassii accounting for over 70% of the bacteria in C. formosanus workers (Noda et al., 2005), facilitates stable transmission during protozoa cell division. Overall, this important mutualistically adapted symbiotic partnership supports cospeciation of symbionts and termites at least in recent coevolutionary history of closely related species (Bourguignon et al., 2018).
Intraspecific sequence analyses showed variation of only a few base pairs in CAP sequences of C. formosanus populations across geographical regions from the native and introduced range. The lack of differentiation is unexpected, since both historical and genetic studies showed that native and introduced C. formosanus populations have been separated for decades if not centuries (Evans et al., 2013, 2019). Colonies of C. formosanus were thought to have been transported from the native range of mainland China and Taiwan to Japan prior to the 17th century (Su, 1987; Vargo et al., 2003). The first introductions to the United States (Haverty et al., 1990) likely involved at least two different sources with Hawaii serving as the bridgehead for invasion of the United States mainland starting in the 1930s (Blumenfeld et al., 2021). Colonies of C. formosanus were first described in Louisiana in 1966 (Spink, 1967), but were likely introduced from the Pacific theater after World War II, which is consistent with their genetic similarity to Hawaiian populations (Husseneder et al., 2012; Blumenfeld et al., 2021); however, a separate introduction occurred into Florida (ca. 1940) likely from southcentral China, which later became mixed with populations from Louisiana and Texas (Blumenfeld et al., 2021). Although the genotypes of the termite host populations have clearly changed across their invasion history (Husseneder et al., 2012; Blumenfeld et al., 2021) there is little distinction between CAP strains, which indicates that CAP is a core bacterium of C. formosanus. As a core bacterium, CAP was also found to be part of the gut symbiont community carried by swarming alates that become the founders of new colonies. Most likely, CAP gets transferred to incipient colonies together with its protozoan host and becomes the starter culture for populating the guts of the worker offspring.
Unlike the results for the host bacterium, phage diversification based on the signature genes encoding terminase and portal protein was not associated with termite speciation. This may indicate that phages could be evolving independently of their termite hosts. However, the taxonomic range in the dataset was limited since crAss-like phages have only been investigated in two Coptotermes species and a single specimen of Prorhinotermes. Future studies will determine whether co-cladogenesis of phage variants and termite speciation occurs when a wider range of taxa are considered and how evolution of these phages is linked to their host bacteria. Nevertheless, the wide-spread presence of phages similar to the crAss-like phages TG-crAlp-04 and TG-crAlp-06 originally described from C. formosanus colonies from New Orleans, LA, in both C. formosanus and C. gestroi specimen across their native and introduced range suggests that these phages are an important part of the gut microbiota of Coptotermes spp. Their broad geographical association with a host bacterium (CAP), which this study identified as a core bacterium in Coptotermes and related termites, suggests that the Phage TG-crAlp-04 and TG-crAlp-06 and closely related variants belong to a core virome. Furthermore, our study showed that swarming alates carry the phages in their guts, ensuring propagation and inoculation of the new colony with these phages.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, MZ008310-MZ008336; https://www.ncbi.nlm.nih.gov/genbank/, OL979637-OL979692. NCBI nucleotide database: https://www.ncbi.nlm.nih.gov/nuccore/.
JC, CG, and CH wrote the manuscript and designed the study. JC conducted the experiments. JC and CG performed the annotations. H-FL and QS provided the termite material and expertise. All co-authors edited the manuscript. All authors contributed to the article and approved the submitted version.
This research was supported by NIH’s IDeA Networks of Biomedical Research Excellence (INBRE) Grants to CG and CH, and by the Jeffrey P. LaFage Memorial Assistantship of the Department of Entomology at the Louisiana State University Agcenter to JC.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank Robert Schlub from University of Guam and Aaron Mullins from University of Florida for providing termite samples. We also would like to thank graduate students Paula Castillo and Joe McCarthy from Sun’s lab for their help with collecting termite samples and species identification. Scott Herke from Louisiana State University provided sequencing services and expertise. In addition, we thank Jin Tao, and Lane Foil from the Louisiana State University Agcenter for valuable comments on the manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2022.881538/full#supplementary-material
Supplementary Table 1 | Information of all collected termite samples used in this project. Information includes “termite species,” “caste,” “gender” (only applicable to alate samples), “collection date,” GPS locations, “labels,” and “collectors.”
Supplementary Table 2 | List of 16S rRNA gene reference sequences with > 97% sequence similarity to Candidatus Azobacteroides pseudotrichonymphae (CAP, Acc. No. AP010656) retrieved from NCBI GenBank in December 2021. Furthermore, two additional CAP genomes (AP017913 and AP0017914) identified from the termite Prorhinotermes japonicus were included. Two bacteria closely related to CAP, i.e., Candidatus Symbiothrix dinenymphae identified in Reticulitermes speratus and Candidatus Armantifilum devescovinae were included as outgroup. Additional information includes sample source, termite species, and collection locations.
Supplementary Table 3 | List of termite COII gene reference sequences used for termite host phylogenetic study. All sequences were retrieved from NCBI GenBank in December 2021.
Supplementary Table 4 | Detailed gene lists and functional annotation data for Phage TG-crAlp-04. Gene lists were generated from manual annotation of Genemark, Glimmer, and PHANOTATE gene predictions (see section “Materials and Methods”). BLASTP results without e-values indicate that no significant similarity was found. Structural homology from HHpred with > 50% probabilities are listed, along with % coverage of the alignment for the query protein. Final functional predictions were made based on assessment of BLASTP and/or HHpred supporting data. The basis for the functional calls is described under “Supporting Evidence/Notes.” Criteria for functional calls can be found in Materials and Methods.
Supplementary Table 5 | Detailed gene lists and functional annotation data for Phage TG-crAlp-06. Gene lists were generated from manual annotation of Genemark, Glimmer, and PHANOTATE gene predictions (see section “Materials and Methods”). BLASTP results without e-values indicate that no significant similarity was found. Structural homology from HHpred with > 50% probabilities are listed, along with % coverage of the alignment for the query protein. Final functional predictions were made based on assessment of BLASTP and/or HHpred supporting data. The basis for the functional calls is described under “Supporting Evidence/Notes”. Criteria for functional calls can be found in Materials and Methods.
Afek, A., Ilic, S., Horton, J., Lukatsky, D. B., Gordan, R., and Akabayov, B. (2018). DNA sequence context controls the binding and processivity of the T7 DNA primase. Iscience 2, 141–147. doi: 10.1016/j.isci.2018.03.019
Austin, J. W., Szalanski, A. L., Scheffrahn, R. H., Messenger, M. T., McKern, J. A., and Gold, R. E. (2006). Genetic evidence for two introductions of the Formosan subterranean termite, Coptotermes formosanus (Isoptera: Rhinotermitidae), to the United States. Fl. Entomol. 89, 183–193. doi: 10.1653/0015-4040200689[183:GEFTIO]2.0.CO;2
Bailly-Bechet, M., Vergassola, M., and Rocha, E. (2007). Causes for the intriguing presence of tRNAs in phages. Gen. Res. 17, 1486–1495. doi: 10.1101/gr.6649807
Bandi, C., Sironi, M., Damiani, G., Magrassi, L., Nalepa, C. A., Laudani, U., et al. (1995). The establishment of intracellular symbiosis in an ancestor of cockroaches and termites. Proc. R. Soc. Lon. 259, 293–299. doi: 10.1098/rspb.1995.0043
Benjamino, J., and Graf, J. (2016). Characterization of the core and caste-specific microbiota in the termite. Reticulitermes flavipes. Front. Microb. 7:171. doi: 10.3389/fmicb.2016.00171
Besemer, J., Lomsadze, A., and Borodovsky, M. (2001). GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 29, 2607–2618. doi: 10.1093/nar/29.12.2607
Black, L. W. (1995). DNA packaging and cutting by phage terminases: control in phage T4 by a synaptic mechanism. Bioessays 17, 1025–1030. doi: 10.1002/bies.950171206
Blazanin, M., and Turner, P. E. (2021). Community context matters for bacteria-phage ecology and evolution. ISME J. 2021, 1–10. doi: 10.1038/s41396-021-01012-x
Blumenfeld, A. J., Eyer, P. A., Husseneder, C., Mo, J., Johnson, L. N., Wang, C., et al. (2021). Bridgehead effect and multiple introductions shape the global invasion history of a termite. Comm. Biol. 4, 1–12. doi: 10.1038/s42003-021-01725-x
Blumenfeld, A. J., and Vargo, E. L. (2020). Geography, opportunity, and bridgeheads facilitate termite invasions to the United States. Biol. Inv. 22, 3269–3282. doi: 10.1007/s10530-020-02322-5
Boomsma, J. J. (2009). Lifetime monogamy and the evolution of eusociality. Philosoph. Trans. R. Soc. B 364, 3191–3207. doi: 10.1098/rstb.2009.0101
Bourguignon, T., Lo, N., Dietrich, C., Šobotník, J., Sidek, S., Roisin, Y., et al. (2018). Rampant host switching shaped the termite gut microbiome. Curr. Biol. 28, 649–654. doi: 10.1016/j.cub.2018.01.035
Brune, A. (2014). Symbiotic digestion of lignocellulose in termite guts. Nat. Rev. Microb. 12:168. doi: 10.1038/nrmicro3182
Brune, A., and Dietrich, C. (2015). The gut microbiota of termites: digesting the diversity in the light of ecology and evolution. Annu. Rev. Microb. 69, 145–166. doi: 10.1146/annurev-micro-092412-155715
Brune, A., and Friedrich, M. (2000). Microecology of the termite gut: structure and function on a microscale. Curr. Opin. Microb. 3, 263–269. doi: 10.1016/S1369-5274(00)00087-4
Cenens, W., Mebrhatu, M. T., Makumi, A., Ceyssens, P. J., Lavigne, R., Van Houdt, R., et al. (2013). Expression of a novel P22 ORFan gene reveals the phage carrier state in Salmonella typhimurium. PLoS Genet. 9:2. doi: 10.1371/journal.pgen.1003269
Chan, P. P., Lin, B. Y., Mak, A. J., and Lowe, T. M. (2021). tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49, 9077–9096. doi: 10.1093/nar/gkab688
Cleveland, L. R. (1923). Symbiosis between termites and their intestinal protozoa. Proc. Natl. Acad. Sci. U S A 9, 424–428. doi: 10.1073/pnas.9.12.424
Delcher, A. L., Bratke, K. A., Powers, E. C., and Salzberg, S. L. (2007). Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23, 673–679. doi: 10.1093/bioinformatics/btm009
Delesalle, V. A., Tanke, N. T., Vill, A. C., and Krukonis, G. P. (2016). Testing hypotheses for the presence of tRNA genes in mycobacteriophage genomes. Bacteriophage 6:e1219441. doi: 10.1080/21597081.2016.1219441
Desai, M. S., and Brune, A. (2012). Bacteroidales ectosymbionts of gut flagellates shape the nitrogen-fixing community in dry-wood termites. ISME J. 6, 1302–1313. doi: 10.1038/ismej.2011.194
Do, T. H., Nguyen, T. T., Nguyen, T. N., Le, Q. G., Nguyen, C., Kimura, K., et al. (2014). Mining biomass-degrading genes through Illumina-based de novo sequencing and metagenomic analysis of free-living bacteria in the gut of the lower termite Coptotermes gestroi harvested in Vietnam. J. Biosci. Bioeng. 118, 665–671. doi: 10.1016/j.jbiosc.2014.05.010
Drobysheva, A. V., Panafidina, S. A., Kolesnik, M. V., Klimuk, E. I., Minakhin, L., Yakunina, M. V., et al. (2021). Structure and function of virion RNA polymerase of a crAss-like phage. Nature 589, 306–309. doi: 10.1038/s41586-020-2921-5
Dutilh, B. E., Cassman, N., McNair, K., Sanchez, S. E., Silva, G. G., Boling, L., et al. (2014). A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Comm. 5, 1–11. doi: 10.1038/ncomms5498
Evans, T. A., Forschler, B. T., and Grace, J. K. (2013). Biology of invasive termites: a worldwide review. Annu. Rev. Entomol. 58, 455–474. doi: 10.1146/annurev-ento-120811-153554
Evans, T. A., Forschler, B. T., and Trettin, C. C. (2019). Not just urban: The Formosan subterranean termite, Coptotermes formosanus, is invading forests in the Southeastern USA. Biolog. Inv. 21, 1283–1294. doi: 10.1007/s10530-018-1899-5
Fogg, P. C., Colloms, S., Rosser, S., Stark, M., and Smith, M. C. (2014). New applications for phage integrases. J. Mole. Biol. 426, 2703–2716. doi: 10.1016/j.jmb.2014.05.014
Fogg, P. C. M., Rigden, D. J., Saunders, J. R., McCarthy, A. J., and Allison, H. E. (2011). Characterization of the relationship between integrase, excisionase and antirepressor activities associated with a superinfecting Shiga toxin encoding bacteriophage. Nucleic Acids Res. 39, 2116–2129. doi: 10.1093/nar/gkq923
Frantzen, C. A., and Holo, H. (2019). Unprecedented diversity of lactococcal group 936 bacteriophages revealed by amplicon sequencing of the portal protein gene. Viruses 11:443. doi: 10.3390/v11050443
Grant, J. R., and Stothard, P. (2008). The CGView Server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 36(Suppl._2), W181–W184. doi: 10.1093/nar/gkn179
Groth, A. C., Olivares, E. C., Thyagarajan, B., and Calos, M. P. (2000). A phage integrase directs efficient site-specific integration in human cells. Proc. Natl. Acad. Sci. 97, 5995–6000. doi: 10.1073/pnas.090527097
Guerin, E., Shkoporov, A. N., Stockdale, S. R., Comas, J. C., Khokhlova, E. V., Clooney, A. G., et al. (2021). Isolation and characterisation of ΦcrAss002, a crAss-like phage from the human gut that infects Bacteroides xylanisolvens. Microbiome 9, 1–21. doi: 10.1186/s40168-021-01036-7
Gulyaeva, A., Garmaeva, S., Ruigrok, R. A., Wang, D., Riksen, N. P., Netea, M. G., et al. (2022). Discovery, diversity, and functional associations of crAss-like phages in human gut metagenomes from four Dutch cohorts. Cell Rep. 38:110204. doi: 10.1016/j.celrep.2021.110204
Halgasova, N., Krajcikova, D., Kraus, D., and Bukovska, G. (2021). The helicase core accessory regions of the phage BFK20 DnaB-like helicase gp43 significantly affect its activity, oligomeric state and DNA binding properties. Virology 558, 96–109. doi: 10.1016/j.virol.2021.02.016
Harrison, M. C., Jongepier, E., Robertson, H. M., Arning, N., Bitard-Feildel, T., Chao, H., et al. (2018). Hemimetabolous genomes reveal molecular basis of termite eusociality. Nat. Ecol. Evol. 2, 557–566. doi: 10.1038/s41559-017-0459-1
Häuser, R., Blasche, S., Dokland, T., Haggård-Ljungquist, E., von Brunn, A., Salas, M., et al. (2012). Bacteriophage protein–protein interactions. Adv. Virus Res. 83, 219–298. doi: 10.1016/B978-0-12-394438-2.00006-2
Haverty, M. I., Nelson, L. J., and Page, M. (1990). Cuticular hydrocarbons of four populations of Coptotermes formosanus Shiraki in the United States. J. Chem. Ecol. 16, 1635–1647. doi: 10.1007/BF01014096
Hobbs, Z., and Abedon, S. T. (2016). Diversity of phage infection types and associated terminology: the problem with ‘Lytic or lysogenic’. FEMS Microb. Lett. 363:7. doi: 10.1093/femsle/fnw047
Hongoh, Y. (2010). Diversity and genomes of uncultured microbial symbionts in the termite gut. Biosci. Biotechnol. Biochem. 74, 1145–1151. doi: 10.1271/bbb.100094
Hou, W., Wang, S., Briggs, B. R., Li, G., Xie, W., and Dong, H. (2018). High diversity of myocyanophage in various aquatic environments revealed by high-throughput sequencing of major capsid protein gene with a new set of primers. Front. Microb. 9:887. doi: 10.3389/fmicb.2018.00887
Howard-Varona, C., Hargreaves, K. R., Abedon, S. T., and Sullivan, M. B. (2017). Lysogeny in nature: mechanisms, impact and ecology of temperate phages. ISME J. 11, 1511–1520. doi: 10.1038/ismej.2017.16
Hungate, R. E. (1979). Evolution of a microbial ecologist. Annu. Rev. Microb. 33, 1–21. doi: 10.1146/annurev.mi.33.100179.000245
Husseneder, C., Donaldson, J. R., and Foil, L. D. (2016). Genetically Engineered Yeast Expressing a Lytic Peptide from Bee Venom (Melittin) Kills Symbiotic Protozoa in the Gut of Formosan Subterranean Termites. PLoS One 11:e0151675. doi: 10.1371/journal.pone.0151675
Husseneder, C., Ho, H. Y., and Blackwell, M. (2010). Comparison of the bacterial symbiont composition of the Formosan subterranean termite from its native and introduced range. Open Microb. J. 4:53. doi: 10.2174/1874285801004010053
Husseneder, C., and Simms, D. M. (2008). Size and heterozygosity influence partner selection in the Formosan subterranean termite. Behav. Ecol. 19, 764–773. doi: 10.1093/beheco/arn041
Husseneder, C., Simms, D. M., Delatte, J. R., Wang, C., Grace, J. K., and Vargo, E. L. (2012). Genetic diversity and colony breeding structure in native and introduced ranges of the Formosan subterranean termite. Coptotermes formosanus. Biol. Invas. 14, 419–437. doi: 10.1007/s10530-011-0087-7
Ikeda-Ohtsubo, W., and Brune, A. (2009). Cospeciation of termite gut flagellates and their bacterial endosymbionts: Trichonympha species and ‘Candidatus Endomicrobium trichonymphae’. Mol. Ecol. 18, 332–342. doi: 10.1111/j.1365-294X.2008.04029.x
Inoue, T., Kitade, O., Yoshimura, T., and Yamaoka, I. (2000). “Symbiotic Associations with Protists,” in Termites: Evolution, Sociality, Symbioses, Ecology, eds T. Abe, D. E. Bignell, and M. Higashi (Dordrecht: Springer), doi: 10.1007/978-94-017-3223-9_13
Inoue, T., Murashima, K., Azuma, J. I., Sugimoto, A., and Slaytor, M. (1997). Cellulose and xylan utilisation in the lower termite Reticulitermes speratus. J. Insect Physiol. 43, 235–242. doi: 10.1016/S0022-1910(96)00097-2
Jasso-Selles, D. E., De Martini, F., Velenovsky, J. F. I. V., Mee, E. D., Montoya, S. J., et al. (2020). The Complete Protist Symbiont Communities of Coptotermes formosanus and Coptotermes gestroi: morphological and Molecular Characterization of Five New Species. J. Eukary. Microb. 67, 626–641. doi: 10.1111/jeu.12815
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Keary, R., McAuliffe, O., Ross, R. P., Hill, C., O’Mahony, J., and Coffey, A. (2014). Genome analysis of the staphylococcal temperate phage DW2 and functional studies on the endolysin and tail hydrolase. Bacteriophage 4:e28451. doi: 10.4161/bact.28451
Kerepesi, C., Szabó, J. E., Papp-Kádár, V., Dobay, O., Szabó, D., Grolmusz, V., et al. (2016). Life without dUTPase. Front. Microb. 7:1768. doi: 10.3389/fmicb.2016.01768
Kirsch, J. M., Brzozowski, R. S., Faith, D., Round, J. L., Secor, P. R., and Duerkop, B. A. (2021). Bacteriophage-Bacteria Interactions in the Gut: from Invertebrates to Mammals. Annu. Rev. Virol. 8, 95–113. doi: 10.1146/annurev-virology-091919-101238
Knowles, B., Silveira, C. B., Bailey, B. A., Barott, K., Cantu, V. A., Cobián-Güemes, A. G., et al. (2016). Lytic to temperate switching of viral communities. Nature 531, 466–470. doi: 10.1038/nature17193
Koizumi, M. (1921). Studies on the intestinal protozoa found in the termites of Japan. Parasitology 13, 235–309. doi: 10.1017/S0031182000012506
Koonin, E. V., and Yutin, N. (2020). The crAss-like phage group: how metagenomics reshaped the human virome. Trends Microb. 28, 349–359. doi: 10.1016/j.tim.2020.01.010
Laslett, D., and Canback, B. (2004). ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32, 11–16. doi: 10.1093/nar/gkh152
Le, S., Serrano, E., Kawamura, R., Carrasco, B., Yan, J., and Alonso, J. C. (2017). Bacillus subtilis RecA with DprA–SsbA antagonizes RecX function during natural transformation. Nucleic Acids Res. 45, 8873–8885. doi: 10.1093/nar/gkx583
Lewis, J. L., and Forschler, B. T. (2004). Protist communities from four castes and three species of Reticulitermes (Isoptera: Rhinotermitidae). Ann. Entomol. Soc. Am. 97, 1242–1251. doi: 10.1603/0013-87462004097[1242:PCFFCA]2.0.CO;2
Li, H. F., Ye, W., Su, N. Y., and Kanzaki, N. (2009). Phylogeography of Coptotermes gestroi and Coptotermes formosanus (Isoptera: Rhinotermitidae) in Taiwan. Ann. Entomol. Soc. Am. 102, 684–693. doi: 10.1603/008.102.0413
Lo, N., and Eggleton, P. (2010). “Termite Phylogenetics and Co-cladogenesis with Symbionts,” in Biology of Termites: a Modern Synthesis, eds D. Bignell, Y. Roisin, and N. Lo (Dordrecht: Springer), 27–50. doi: 10.1007/978-90-481-3977-4_2
Lourenço, M., Chaffringeon, L., Lamy-Besnier, Q., Pedron, T., Campagne, P., et al. (2020). The spatial heterogeneity of the gut limits predation and fosters coexistence of bacteria and bacteriophages. Cell Host Microbe 28, 390–401. doi: 10.1016/j.chom.2020.06.002
Manrique, P., Bolduc, B., Walk, S. T., van der Oost, J., de Vos, W. M., and Young, M. J. (2016). Healthy human gut phageome. Proc. Natl. Acad. Sci. 113, 10400–10405. doi: 10.1073/pnas.1601060113
McNair, K., Zhou, C., Dinsdale, E. A., Souza, B., and Edwards, R. A. (2019). PHANOTATE: a novel approach to gene identification in phage genomes. Bioinformatics 35, 4537–4542. doi: 10.1093/bioinformatics/btz265
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., Von Haeseler, A., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mole. Biol. Evol. 37, 1530–1534. doi: 10.1093/molbev/msaa015
Mitchell, M. S., Matsuzaki, S., Imai, S., and Rao, V. B. (2002). Sequence analysis of bacteriophage T4 DNA packaging/terminase genes 16 and 17 reveals a common ATPase center in the large subunit of viral terminases. Nucleic Acids Res. 30, 4009–4021. doi: 10.1093/nar/gkf524
Moran, N. A., and Telang, A. (1998). Bacteriocyte-associated symbionts of insects. Bioscience 48, 295–304.
Noble, J. C., Müller, W. J., Whitford, W. G., and Pfitzner, G. H. (2009). The significance of termites as decomposers in contrasting grassland communities of semi-arid eastern Australia. J. Arid Environ. 73, 113–119. doi: 10.1016/j.jaridenv.2008.08.004
Noda, S., Iida, T., Kitade, O., Nakajima, H., Kudo, T., and Ohkuma, M. (2005). Endosymbiotic Bacteroidales bacteria of the flagellated protist Pseudotrichonympha grassii in the gut of the termite Coptotermes formosanus. Appl. Env. Microb. 71, 8811–8817. doi: 10.1128/AEM.71.12.8811-8817.2005
Noda, S., Kitade, O., Inoue, T., Kawai, M., Kanuka, M., Hiroshima, K., et al. (2007). Cospeciation in the triplex symbiosis of termite gut protists (Pseudotrichonympha spp.), their hosts, and their bacterial endosymbionts. Mole. Ecol. 16, 1257–1266. doi: 10.1111/j.1365-294X.2006.03219.x
Ohkuma, M. (2003). Termite symbiotic systems: efficient bio-recycling of lignocellulose. Appl. Microbiol. Biotechnol. 61, 1–9. doi: 10.1007/s00253-002-1189-z
Ottesen, E. A., Hong, J. W., Quake, S. R., and Leadbetter, J. R. (2006). Microfluidic digital PCR enables multigene analysis of individual environmental bacteria. Science 314, 1464–1467. doi: 10.1126/science.1131370
Peterson, B. F., Stewart, H. L., and Scharf, M. E. (2015). Quantification of symbiotic contributions to lower termite lignocellulose digestion using antimicrobial treatments. Insect Biochem. Mole. Biol. 59, 80–88. doi: 10.1016/j.ibmb.2015.02.009
Pramono, A. K., Kuwahara, H., Itoh, T., Toyoda, A., Yamada, A., and Hongoh, Y. (2017). Discovery and complete genome sequence of a bacteriophage from an obligate intracellular symbiont of a cellulolytic protist in the termite gut. Microb. Env. 2017:ME16175. doi: 10.1264/jsme2.ME16175
Rao, V. B., and Feiss, M. (2015). Mechanisms of DNA packaging by large double-stranded DNA viruses. Annu. Rev. Virol. 2, 351–378. doi: 10.1146/annurev-virology-100114-055212
Rinehart, C. A., Gaffney, B. L., Smith, J. R., and Wood, J. D. (2016). “PECAAN: Phage Evidence Collection and Annotation Network user guide,” in Western Kentucky University Bioinformatics and Information Science Center (Bowling Green, KY). https://seaphages.org/media/docs/PECAAN_User_Guide_Dec7_2016.pdf (accessed date 7 December-2016).
Rodriguez-Brito, B., Li, L., Wegley, L., Furlan, M., Angly, F., Breitbart, M., et al. (2010). Viral and microbial community dynamics in four aquatic environments. ISME J. 4, 739–751. doi: 10.1038/ismej.2010.1
Scheffrahn, R. H., and Su, N. Y. (2000). “Asian subterranean termite, Coptotermes gestroi (= havilandi) (Wasmann) (Insecta: Isoptera: Rhinotermitidae),” in Extension document EENY-128. Institute of Food and Agricultural Sciences (Gainesville, FL: University of Florida).
Schmidlin, K., Kraberger, S., Fontenele, R. S., De Martini, F., Chouvenc, T., Gile, G. H., et al. (2019). Genome sequences of microviruses associated with Coptotermes formosanus. Microbiol. Res. Announc. 8:16. doi: 10.1128/MRA.00185-19
Sethi, A., Delatte, J., Foil, L., and Husseneder, C. (2014). Protozoacidal Trojan-Horse: use of a ligand-lytic peptide for selective destruction of symbiotic protozoa within termite guts. PLoS One 9:9. doi: 10.1371/journal.pone.0106199
Shinzato, N., Muramatsu, M., Matsui, T., and Watanabe, Y. (2005). Molecular phylogenetic diversity of the bacterial community in the gut of the termite Coptotermes formosanus. Biosci. Biotechnol. Biochem. 69, 1145–1155. doi: 10.1271/bbb.69.1145
Shkoporov, A. N., Clooney, A. G., Sutton, T. D., Ryan, F. J., Daly, K. M., et al. (2019). The human gut virome is highly diverse, stable, and individual specific. Cell Host Microbe 26, 527–541. doi: 10.1016/j.chom.2019.09.009
Shkoporov, A. N., Khokhlova, E. V., Fitzgerald, C. B., Stockdale, S. R., Draper, L. A., Ross, R. P., et al. (2018). ΦCrAss001 represents the most abundant bacteriophage family in the human gut and infects Bacteroides intestinalis. Nat. Comm. 9, 1–8. doi: 10.1038/s41467-018-07225-7
Shkoporov, A. N., Khokhlova, E. V., Stephens, N., Hueston, C., Seymour, S., et al. (2021). Long-term persistence of crAss-like phage crAss001 is associated with phase variation in Bacteroides intestinalis. BMC Biol. 19, 1–16. doi: 10.1186/s12915-021-01084-3
Slaytor, M. (1992). Cellulose digestion in termites and cockroaches: what role do symbionts play? Comp. Biochem. Phys. Part B 103, 775–784. doi: 10.1016/0305-0491(92)90194-V
Song, Y. Q., Zhang, D., Chen, W., Dang, X. X., and Yang, H. (2021). Phylogenetic Identification of Symbiotic Protists of Five Chinese Reticulitermes Species Indicates a Cospeciation of Gut Microfauna with Host Termites. J. Euk. Microb. 2021:e12862. doi: 10.1111/jeu.12862
Spink, W. T. (1967). The Formosan subterranean termite in Louisiana. Circul. Louis. State Univ. 89, 1–12.
Strassert, J. F., Kohler, T., Wienemann, T. H., Ikeda-Ohtsubo, W., Faivre, N., et al. (2012). ‘Candidatus Ancillula trichonymphae’, a novel lineage of endosymbiotic Actinobacteria in termite gut flagellates of the genus Trichonympha. Environ. Microb. 14, 3259–3270. doi: 10.1111/1462-2920.12012
Su, L., Yang, L., Huang, S., Su, X., Li, Y., Wang, F., et al. (2016). Comparative gut microbiomes of four species representing the higher and the lower termites. J. Insect Sci. 16, doi: 10.1093/jisesa/iew081
Su, N. Y. (1987). An overview of the Formosan subterranean termite (Isoptera: Rhinotermitidae) in the world. Res. Exten. Series, Hawaii Instit. Trop. Agricult. Hum. Resourc. 83, 3–15.
Sussman, J. L., Lin, D., Jiang, J., Manning, N. O., Prilusky, J., Ritter, O., et al. (1998). Protein Data Bank (PDB): database of three-dimensional structural information of biological macromolecules. Biolog. Crystallogr. 54, 1078–1084. doi: 10.1107/S0907444998009378
Thorne, B. L. (1997). Evolution of eusociality in termites. Annu. Rev. Ecol. Syst. 28, 27–54. doi: 10.1146/annurev.ecolsys.28.1.27
Tikhe, C. V., Gissendanner, C. R., and Husseneder, C. (2018a). Whole-genome sequence of the novel temperate Enterobacter bacteriophage Tyrion, isolated from the gut of the Formosan subterranean termite. Gen. Announ. 6:1. doi: 10.1128/genomeA.00839-17
Tikhe, C. V., Gissendanner, C. R., and Husseneder, C. (2018b). Whole-Genome Sequence of the Novel Enterobacter Bacteriophage Arya with an Integrase Pseudogene, Isolated from the Gut of the Formosan Subterranean Termite. Gen. Announ. 6:17. doi: 10.1128/genomeA.00838-17
Tikhe, C. V., and Husseneder, C. (2018). Metavirome sequencing of the termite gut reveals the presence of an unexplored bacteriophage community. Frontiers in Microbiology 8:2548. doi: 10.3389/fmicb.2017.02548
Tikhe, C. V., Martin, T. M., Gissendanner, C. R., and Husseneder, C. (2015). Complete genome sequence of Citrobacter phage CVT22 isolated from the gut of the Formosan subterranean termite, Coptotermes formosanus Shiraki. Gen. Announ. 3:4. doi: 10.1128/genomeA.00408-15
Trifinopoulos, J., Nguyen, L. T., von Haeseler, A., and Minh, B. Q. (2016). W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44, W232–W235. doi: 10.1093/nar/gkw256
Turkington, C. J., Morozov, A., Clokie, M. R., and Bayliss, C. D. (2019). Phage-resistant phase-variant sub-populations mediate herd immunity against bacteriophage invasion of bacterial meta-populations. Front. Microb. 10:1473. doi: 10.3389/fmicb.2019.01473
Vargo, E. L., Husseneder, C., and Grace, J. K. (2003). Colony and population genetic structure of the Formosan subterranean termite, Coptotermes formosanus, in Japan. Mole. Ecol. 12, 2599–2608. doi: 10.1046/j.1365-294X.2003.01938.x
Wommack, K. E., and Colwell, R. R. (2000). Virioplankton: viruses in aquatic ecosystems. Microbiol. Mole. Biol. Rev. 64, 69–114. doi: 10.1128/MMBR.64.1.69-114.2000
Yoshua, S. B., Watson, G. D., Howard, J. A., Velasco-Berrelleza, V., Leake, M. C., and Noy, A. (2021). Integration host factor bends and bridges DNA in a multiplicity of binding modes with varying specificity. Nucleic Acids Res. 49, 8684–8698. doi: 10.1093/nar/gkab641
Yutin, N., Benler, S., Shmakov, S. A., Wolf, Y. I., Tolstoy, I., Rayko, M., et al. (2021). Analysis of metagenome-assembled viral genomes from the human gut reveals diverse putative CrAss-like phages with unique genomic features. Nat. Comm. 12, 1–11. doi: 10.1038/s41467-021-21350-w
Yutin, N., Makarova, K. S., Gussow, A. B., Krupovic, M., Segall, A., et al. (2018). Discovery of an expansive bacteriophage family that includes the most abundant viruses from the human gut. Nat. Microb. 3, 38–46. doi: 10.1038/s41564-017-0053-y
Zeng, W., Liu, B., Zhong, J., Li, Q., and Li, Z. (2020). A natural high-sugar diet has different effects on the prokaryotic community structures of lower and higher termites (Blattaria). Environ. Ent. 49, 21–32. doi: 10.1093/ee/nvz130
Keywords: subterranean termite, gut bacteria, crAss-like phage, phylogeny, Candidatus Azobacteroides pseudotrichonymphae
Citation: Chen J, Gissendanner CR, Tikhe CV, Li H-F, Sun Q and Husseneder C (2022) Genomics and Geographic Diversity of Bacteriophages Associated With Endosymbionts in the Guts of Workers and Alates of Coptotermes Species (Blattodea: Rhinotermitidae). Front. Ecol. Evol. 10:881538. doi: 10.3389/fevo.2022.881538
Received: 22 February 2022; Accepted: 10 May 2022;
Published: 27 May 2022.
Edited by:
Carol Lauzon, California State University, East Bay, United StatesReviewed by:
Ikhsan Guswenrivo, National Research and Innovation Agency, IndonesiaCopyright © 2022 Chen, Gissendanner, Tikhe, Li, Sun and Husseneder. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claudia Husseneder, Y2h1c3NlbmVkZXJAYWdjZW50ZXIubHN1LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.