 Omid Jafari1Mehrshad Zeinalabedini2*Diego Robledo3Jorge M. O. Fernandes4*Ali-Akbar Hedayati5Babak Arefnezhad6
Omid Jafari1Mehrshad Zeinalabedini2*Diego Robledo3Jorge M. O. Fernandes4*Ali-Akbar Hedayati5Babak Arefnezhad6
- 1International Sturgeon Research Institute, Iranian Fisheries Science Research Institute, Agricultural Research, Education and Extension Organization, Rasht, Iran
- 2Department of Genomics, Agricultural Biotechnology Research Institute of Iran (ABRII), Karaj, Iran
- 3The Roslin Institute and Royal (Dick) School of Veterinary Studies, University of Edinburgh, Midlothian, United Kingdom
- 4Faculty of Biosciences and Aquaculture, Nord University, Bodø, Norway
- 5Department of Fisheries, Faculty of Fisheries and Environmental Sciences, Gorgan University of Agricultural Sciences and Natural Resources, Gorgan, Iran
- 6OMICS Research Group (Media Teb Gene Co. Ltd.), Tehran, Iran
Understanding the population structure and level of genetic diversity of wild populations is fundamental for appropriate stock management and species conservation. The common carp (Cyprinus carpio) is one of the most important bony fish throughout the Southern coastline of the Caspian Sea, but captures of this species have seen a dramatic reduction during the last decade. As a consequence, a restocking program has been put in place to maintain C. carpio populations, but its impact is not clear. In the present study, the population structure and genetic diversity of C. carpio in the Southern Caspian basin was determined using 17,828 single-nucleotide polymorphism (SNP) markers. A total of 117 individuals collected from four different locations in the southern Caspian basin and a farm were genotyped by genotyping-by-sequencing. The overall Fst obtained was 0.04, indicating a low level of differentiation between populations, and most genetic diversity was attributed to within population variation (97%). The low Fst values suggest that frequent migration events between different locations occur, and three migration events were inferred in the present study. However, each population still showed a distinct genetic profile, which allowed distinguishing the origin of the fish. This indicates that the ongoing restocking program is maintaining the differences between populations to some extent. Nonetheless, high inbreeding and low heterozygosity were detected in all populations, suggesting that additional conservation efforts are required to protect C. carpio populations in the Southern coast of the Caspian Sea.
Introduction
Common carp (Cyprinus carpio) is one of the most widely distributed fish species across the world, and is farmed in Asia, Europe, Africa, and America. This species has been domesticated for thousands of years (Nakajima et al., 2019) and currently represents almost 10% of the world’s freshwater aquaculture production (Xu et al., 2014). Common carp is considered an endemic species across the Caspian and Aral Seas. Carp fisheries along the southern coast of the Caspian Sea have been traditionally important for the region, but the captures of this species have sharply decreased over the past decade, indicating a substantial reduction of its population. Several factors have contributed to this decline, such as overfishing, pollution, and loss of nursery grounds due to the construction of dams. Furthermore, global warming and the associated decrease of water levels may also be playing an important role. In addition to its commercial importance, C. carpio also plays a critical ecological and environmental role by affecting aerobic decomposition of organic matter and nutrient accessibility in the water column, specifically through benthic sediment bioturbation when feeding on benthic organisms (Rahman, 2015). Consequently, the reduction in wild populations of C. carpio in the southern Caspian basin led the Iranian Fisheries Organization (IFRO) to establish a restocking program in 1999. Mature animals coming into the rivers to breed were captured and their reproduction occurred under controlled conditions in captivity. Then both fingerlings and parents are in theory released to the rivers of origin, although field observations suggest that this release may not always happen based on their origin. The effects of this restocking program on population size, genetic diversity, and population structure of C. carpio in the Southern Caspian basin have not been studied, even if understanding these aspects is fundamental to evaluate the success and impact of the restocking program. It is worth to mention that despite the annually high number of released offspring to the rivers (∼50 million larvae in 2020), the capture rate of wild C. carpio in the southern basin of the Caspian Sea is still low.
Population identification plays a key role in the genetic management of natural stocks (Utter, 2004; Waples et al., 2008). Previous studies on the genetic structure of C. carpio in the Southern Caspian basin suggested a low level of genetic diversity between populations (Dorafshan et al., 2013; Laloei et al., 2013) based on a low number of genetic markers. Single-nucleotide polymorphisms (SNPs) are not only valuable genetic markers in population studies but can also provide far denser genotypes with higher precision for genomic approaches such as association mapping and genomic selection (Daw et al., 2005; Bradbury et al., 2015). The lower mutation rate of SNP variants compared to simple sequence repeats (SSRs) makes them more appropriate for individual-based genetic diversity deduction (Lemopoulos et al., 2019). Recent advances in sequencing technologies have provided the opportunity to perform high-throughput genomic studies in non-model species devoid of previous genomic knowledge (Allendorf et al., 2010; Stapley et al., 2010; Li et al., 2012), which has resulted in an increased understanding of the demography, evolution, and adaptive mechanisms of natural populations (Rodríguez-Ezpeleta et al., 2016; Xu et al., 2017).
Genotyping-by-sequencing (GBS) refers to a range of genomic technologies generally based on a reduction of genome complexity for cost-effective genotyping. Many of these techniques rely on the use of restrictions enzymes, varying in the type and number of enzymes (Andrews et al., 2016; Robledo et al., 2018b). GBS methods represent a cost-effective genotyping strategy over whole genome sequencing, offering increased depth of coverage per locus for a fraction of the cost, allowing genotyping a larger number of samples (Andrews et al., 2016). Further, this approach frequently produces genotypes for thousands of genetic markers, generally enough for population-level analyses, such as genetic diversity assessment, genome-wide association studies, or genomic selection (Sonah et al., 2013; Barría et al., 2018; Liu et al., 2018; Robledo et al., 2018a; Zhao et al., 2018). GBS is particularly useful to clarify the structure of populations showing low genetic differentiation, such as in Pacific lamprey [Entosphenus tridentatus, Hess et al. (2013)], Atlantic salmon [Salmo salar, Bourret et al. (2013)], and Chinook salmon [Oncorhynchus tshawytscha, Larson et al. (2014)].
The aim of the present study was to apply GBS-acquired SNP information to reveal the influence of overfishing and the Iranian restocking program on genetic diversity of C. carpio populations across the southern coastline of the Caspian Sea.
Materials and Methods
Sample Collection and DNA Extraction
A total of 117 specimens of C. carpio with an average weight of 673.72 ± 51.11 g (mean ± SD) were used, namely 102 wild animals caught along the southern coast of the Caspian Sea near the cities of Gomishan, Miankaleh, Anzali-wetland, and Anzali (Rezvanshahr) during the winter of 2015 (Figure 1), and 15 additional fish from Sijval farm (the farmed group in this study). Fish were humanely sacrificed with an overdose of tricaine methanesulfonate (Pharmaq Ltd., United Kingdom) at 300 mg/L for 5 min. Caudal fin samples were taken from each animal and kept in ethanol until DNA extraction. Total genomic DNA extraction was performed using the DNeasy Blood and Tissue kit (Qiagen, Germany) following the manufacturer’s protocol. DNA concentration was measured using Qubit 3.0 fluorometer (ThermoFisher Scientific, United States) and a 1% (w/v) agarose gel was used to assess the integrity of the DNA.
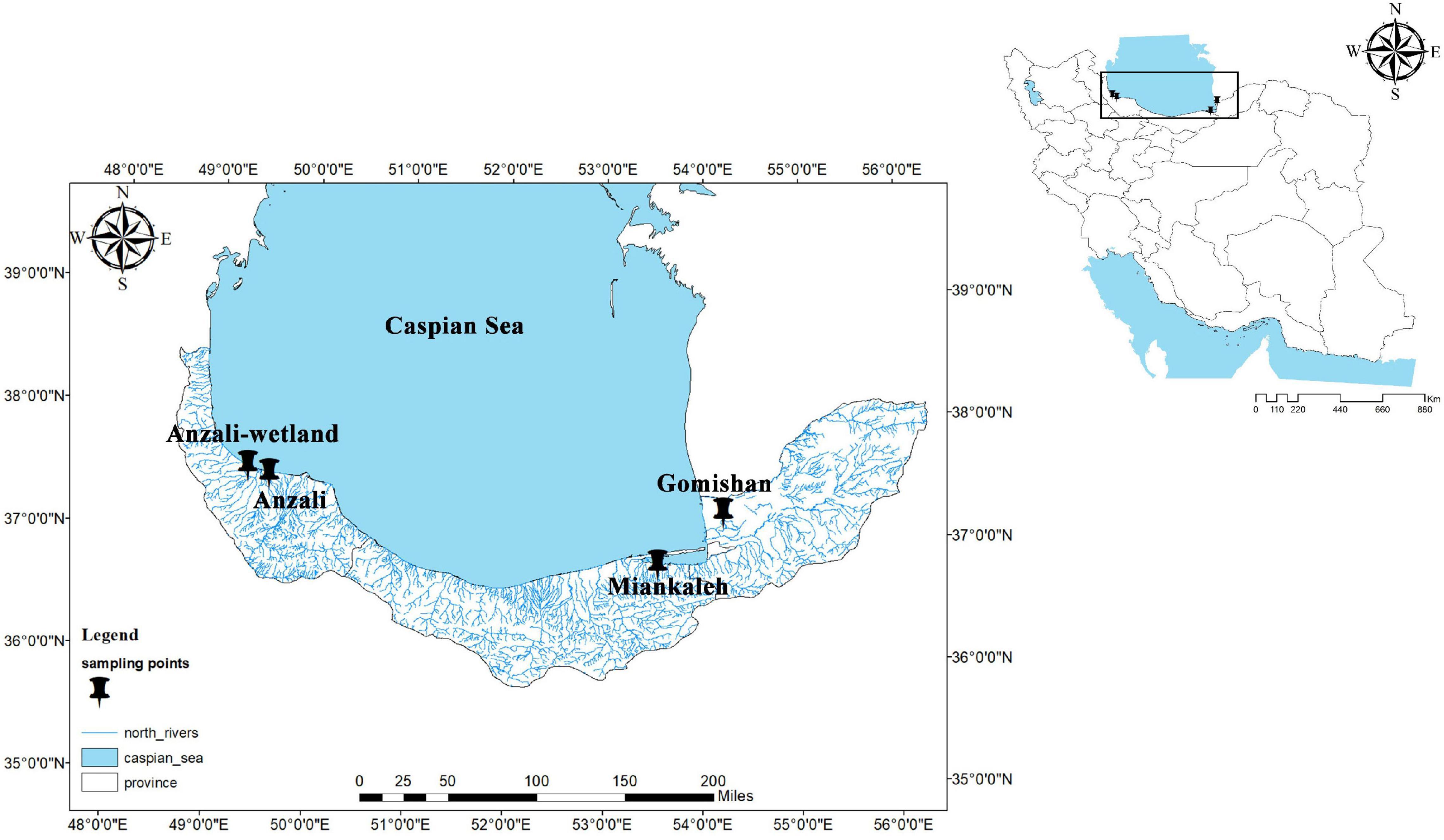
Figure 1. Sampling locations of wild C. carpio specimens across the southern Caspian basin.
Library Construction and Sequencing
Preparation and sequencing of GBS DNA libraries were performed by BGI Genomics (China). Libraries were prepared following the protocol of Elshire et al. (2011) with minor modifications. Briefly, 100 ng of genomic DNA was digested with the restriction enzyme ApeKI (NEB, United States), and afterward common and barcode adaptors were ligated to the digested DNA fragments of each sample and incubated at 22°C for 1 h. After adapter ligation, equal volumes of the DNA products from each sample were pooled. The pool was purified using the QIAquick PCR Purification kit (Qiagen). The adapter-ligated DNA fragments were enriched by PCR amplification with PCR Primer Cocktail and PCR Master Mix. The PCR products were then size selected by agarose gel electrophoresis and the QIAquick Gel Extraction kit (Qiagen) was used for gel purification, retaining fragments between 180 and 480 bp. Quality control of the libraries was performed using the Agilent Technologies 2100 Bioanalyzer and the ABI StepOnePlus Real-Time PCR System. The final GBS libraries were sequenced on a lane of Illumina HiSeq 2000 at the facilities of BGI as 150 bp paired-end reads.
Quality Filtering and Single-Nucleotide Polymorphism Calling
Raw sequencing data (Jafari et al., 2021a) were demultiplexed using the Illumina Experiment Manager ver. 1.16.0. The quality of the individual fastq files was assessed using FASTQC ver. 0.11.5 (Babraham Bioinformatics, Babraham Institute1). SNP calling was performed using the stacks pipeline version 2 (Catchen et al., 2013). Briefly, raw reads were filtered using the process rad tag script to remove low quality reads using a sliding window of 15% of the length of the reads, so that if the average score of the window drops below 90% of base-call accuracy (a raw phred score of 10) the read is discarded. The clean reads were mapped to the most recent reference genome assembly of C. carpio (NCBI; GCF_000951615.1) using Bowtie2 ver. 2.3.2 in –very-sensitive mode (Langmead and Salzberg, 2012). The obtained Sequence Alignment Map files were converted to binary format and sorted with samtools 1.6. Using the gstacks module, loci were built from the paired end data and SNP calling was performed after removing PCR duplicates. The Populations module of stacks 2.3b was used to generate a variant call format (VCF) file with the loci found in at least 65% of the animals (-r 0.65) of a minimum of three (out of five) populations (-p 3), filtering possible sequencing errors, paralogous sequence variants (PSVs), and uninformative polymorphisms. PGDSpider 2.1.1.5 (Lischer and Excoffier, 2012) was used to convert the VCF file to other file formats for downstream applications. The Hardy–Weinberg exact test with genepop ver 4.0.0 was used to identify loci significantly out of equilibrium, which were removed from the data set. Finally, only one loci of those in linkage disequilibrium (r2 > 0.5) was retained in the final data set using GBS_SNP_Filter (Alexander, 2018).
Population Structure and Genetic Diversity
A principal component analyses (PCA) was conducted based on the SNP data using the ape package (Paradis and Schliep, 2019) in R ver. 3.6 (Team, 2013) and the first two principal components were used to evaluate and visualize the distribution of the individuals using ggplot2 package (Wickham, 2009). Population clustering and connectivity were estimated in R using the packages vcfR (Knaus and Grünwald, 2017), ape, and adegenet (Jombart, 2008), estimating the best K parameter based on the lowest Bayesian information criterion (BIC = 54.9). Nei genetic distances (GDs) between individuals and between populations were calculated in R using the poppr package (Kamvar et al., 2014) and visualized using a heatmap. The Fst index between sampling locations was determined using Genepop 4.0.9 (Rousset, 2008). Pairwise population differentiation based on Weir’s and Cockerham’s Fst estimator was obtained using Genodive 2.0b23 (Meirmans and Van Tienderen, 2004). Observed (Ho) and expected heterozygosity (He) were obtained with the package GBS_SNP_Filter. To assess the proportion of between and within population genetic variances, an AMOVA was run in Genodive 2.0b23 pre-replacing missing values by randomly selecting values based on overall allele frequencies. The AMOVA was run with 1000 permutations with an infinite allele model.
The package fineRADstructure, combined with chromopainter and finestructure, was used to understand the structure of the C. carpio populations examined in the present study. This package works based on the nearest neighbor haplotype (coancestry) among sampled individuals by deriving a coancestry matrix, which is a summary of pairs of individuals that had the most similar haplotypes (Malinsky et al., 2018). The TREEMIX package was used to investigate the pattern of migration between different C. carpio sampling locations (Pickrell and Pritchard, 2012).
Population Assignment Test and Effective Population Size
In order to assess the probability of the individuals’ dependency to their sampling site, the Population Assignment test was implemented using the Genodive 2.0b23 Home likelihood statistic method, replacing frequencies of 0 with 0.005. This analysis is based on allele frequencies, and to avoid bias due to the calculation of allele frequencies from the same individuals that are going to be assigned, the program uses the leave one out validation procedure so that a marked individual is removed from its source population before allele frequency calculation. A total of 1000 permutations and a significance threshold of 0.01 were used.
Demographic estimates of the effective population size (Ne) were conducted with the SNeP software V1.11 using default parameters and considering the Sved and Feldman approximation as the recombination model (Barbato et al., 2015).
Outlier Detection and Number of Private Alleles in Each Population
Loci under selection were detected using BAYESCAN v.2.1 (Foll, 2012) with the Fst parameter, considering significant outliers those with a false discovery rate (FDR) p-value < 0.05. Loci with Log10 (PO) > 2 were considered under selection. The outliers were then mapped to the genome reference annotation of C. carpio with the genome data viewer to identify the related genes. Discriminant analysis of principal components (DAPCs) was applied for a new population structure classification based on all identified outliers using adegenet package in R. Furthermore, the number of private alleles in each population was estimated from the VCF file using the Population program of Stacks2.3b.
Results
Sequencing and Single-Nucleotide Polymorphism Calling
After filtering, a total of 388.82 million clean reads were obtained from the 117 C. carpio specimens, with an average of 6.53 million reads per sample. The average data size and Q30 of the reads were 484.49 Mb and 92.5%, respectively. After mapping the clean reads to the C. carpio reference genome, a total of 1,539,147 loci were obtained, and 252,288 of them passed the sample/population quality control. Of the 36,219 SNPs identified in these loci, 17,828 remained after filtering and were used in the subsequent analyses.
Genetic Variation and Heterozygosity Studies
The first (4.27%) and second (2.03%) principal components of the PCA explained 6.3% of the total genetic variation, clustering the farmed and Anzali populations separately from the other populations (Figure 2). The overall Fst value was 0.046, and the pairwise values ranged from 0.02 for Anzali-wetland and Gomishan to 0.07 for Miankaleh-farmed populations (Table 1). The overall mean value of the observed heterozygosity (Ho) was 0.19 ± 0.001, with minimum and maximum values of 0.17 and 0.23 for Anzali and farmed populations, respectively. The mean genetic diversity within populations (Hs) was 0.315, with the farmed fish showing the highest value (Hs = 0.334) and Anzali-wetland and Miankaleh the lowest (Hs = 0.302). The Anzali population had the highest Fis value (Fis = 0.48), while the farmed population had the lowest with Fis = 0.32 (Table 2). The overall genetic differentiation based on the exact G test and Fisher’s method for all populations were highly significant, as shown in Supplementary Table 1.
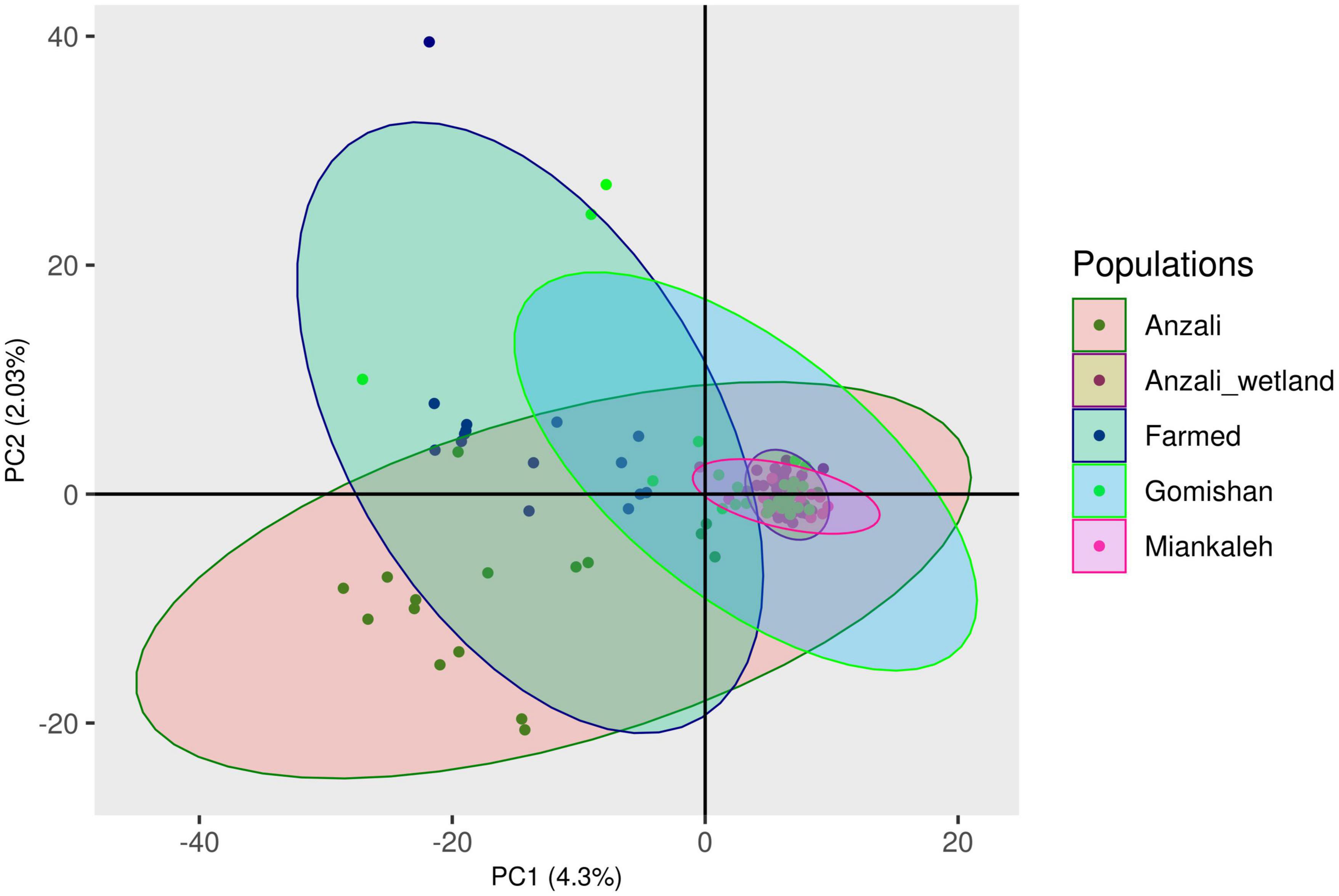
Figure 2. Principal component analysis (PCA) scatter plot based on 17,828 GBS-obtained SNPs on investigated populations of C. carpio.

Table 1. Fst values between different locations of C. carpio based on 17,828 SNP markers.
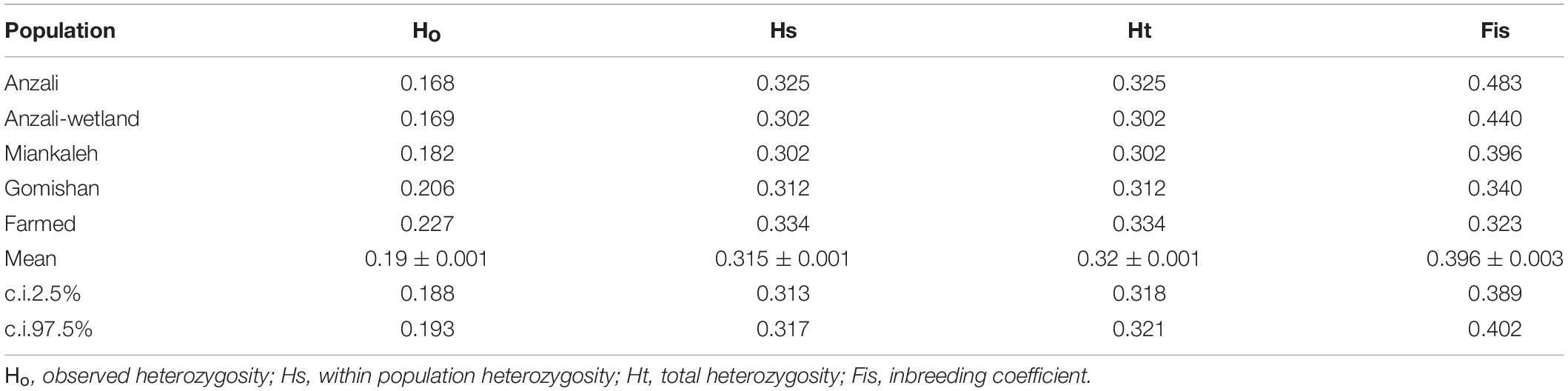
Table 2. Genetic diversity statistics of C. carpio based on 17,828 SNP markers from genotyping by sequencing.
Population Clustering
The Nei based GDs between populations were calculated and visualized (Figure 3). Based on the GD values, the different populations were classified into three groups. The results of AMOVA estimated that within population variation accounted for 97% of the total variation, while variation between populations accounted for only 3% of the total (Table 3).
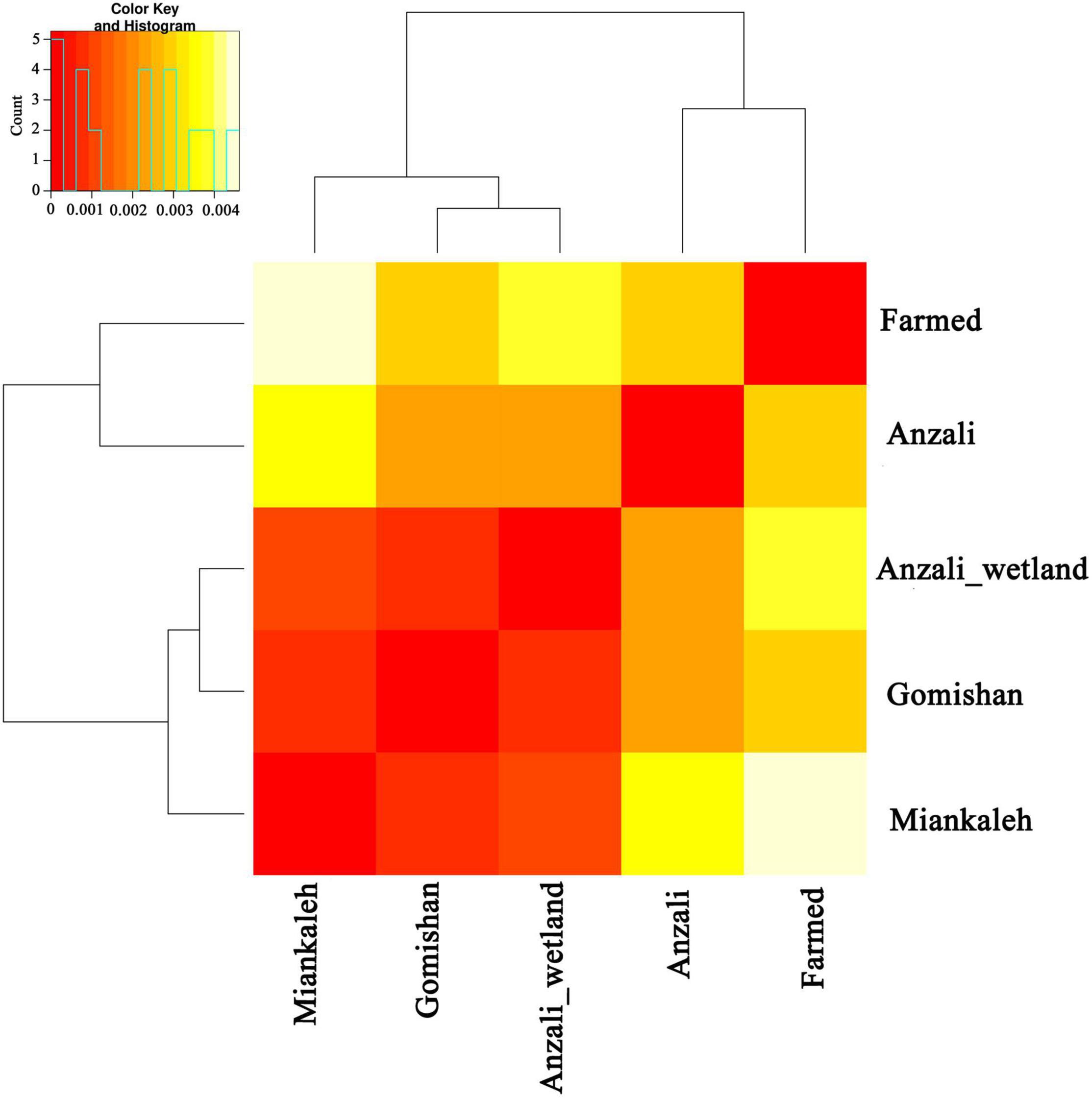
Figure 3. Pairwise genetic distances between C. carpio populations and clustering based on 17,828 SNP markers.

Table 3. Proportion of variation based on AMOVA results from 17,828 SNP markers.
Admixture Analysis and Migrations
The patterns with the highest admixture were observed for Gomishan, Miankaleh, and Anzali-wetland locations, while the Anzali and farmed populations showed the lowest admixture (Figure 4). The admixture results and the BIC parameter classified all studied locations into three separate clusters so that Anzali and Farmed populations (two clusters) showed recognizable patterns, while Anzali-wetland, Gomishan, and Miankaleh (one cluster) had a higher level of admixture structure, consistent with the results of the PCA.
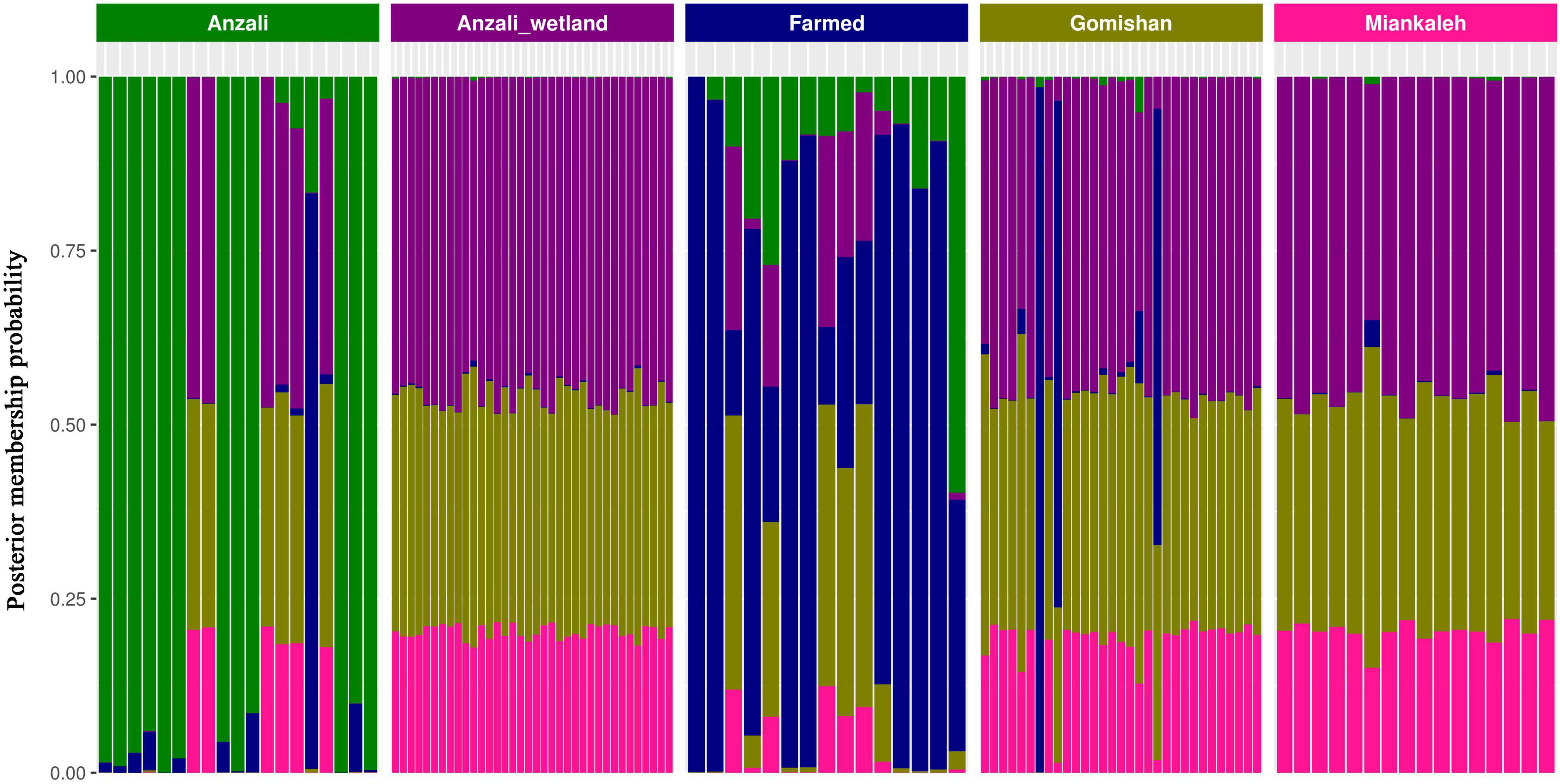
Figure 4. Compoplot showing the assignments of population membership probability against their original populations. The X axis is the sample individuals and the Y axis represents the membership probabilities, fill colors indicate the population of origin.
FineRADstructure analysis based on the nearest neighbor haplotype or coancestry confirmed the admixture pattern and revealed that all animals were related to three main ancestral populations (Figure 5). The population assignment test showed that among all individuals, 25 animals were identified as migrant, having a genetic background indicative of another location (Supplementary Table 2). Furthermore, TREEMIX recognized four migration edges including farmed population to Anzali and Anzali-wetland; Gomishan to Anzali-wetland; and Miankaleh to Gomishan (Figure 6).

Figure 5. Finestructure classification based on the obtained SNP markers from genotyping-by sequencing (GBS) on C. carpio across the southern Caspian basin (different colors in the X and Y axis trees represent different populations; Green: Anzali, Purple: Anzali-wetland, Olive: Gomishan, Pink: Miankaleh, and Blue: Farmed).
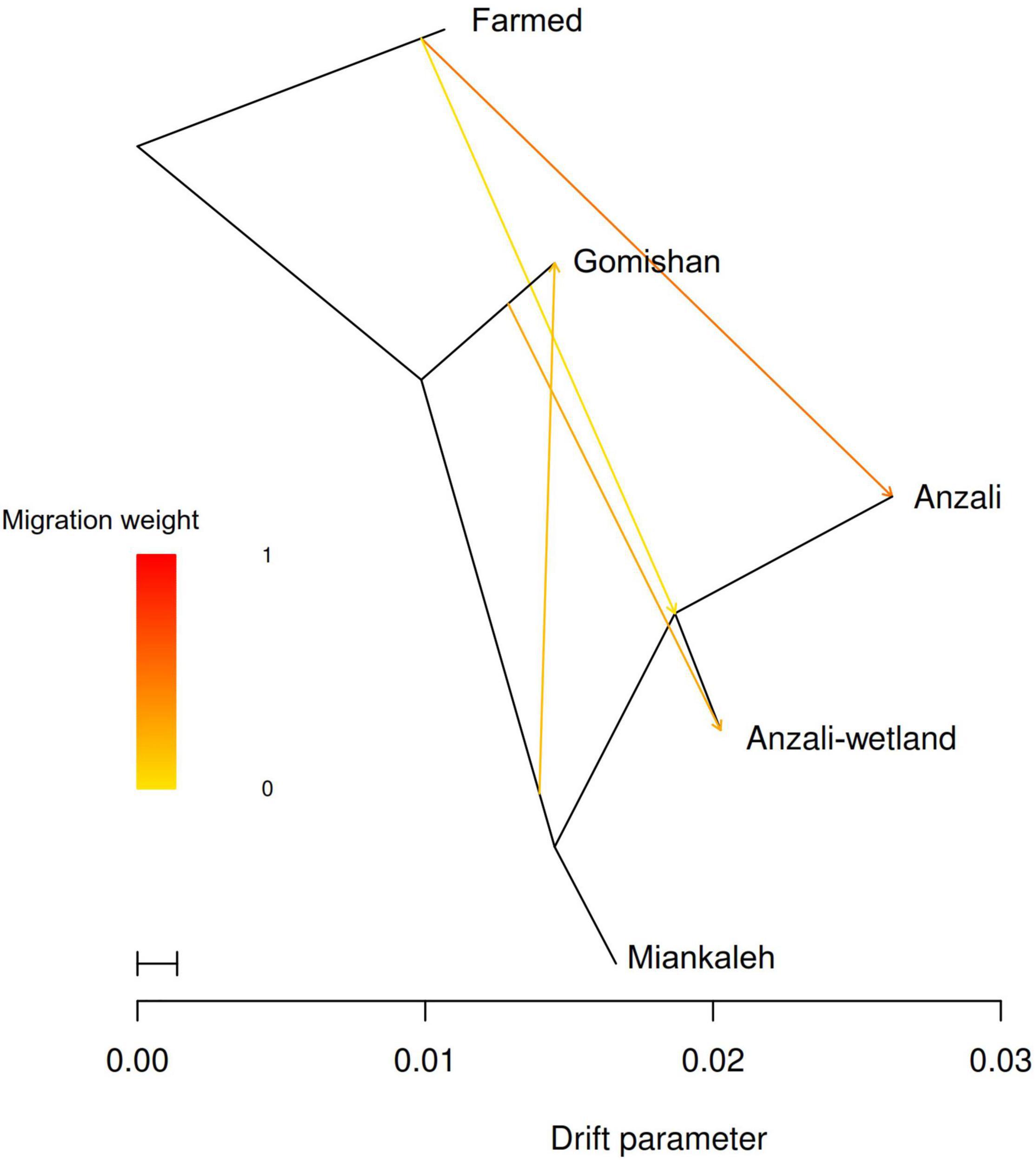
Figure 6. Graph of admixture between 117 individuals of C. carpio fitted using TreeMix (Pickrell and Pritchard, 2012) with three migration edges. The drift parameter is proportional to 2Ne (effective population size) generations and migration weight indicates the proportion of ancestry deriving from the migration edge.
Effective Population Size (Ne)
Demographic estimates of Ne for all individuals obtained up to 578 generations ago, ranged from 41,653 for 578 generations ago to 81 for the latest generation (Table 4). Recent Ne up to 10 generations ago showed a drastic reduction of 88%.
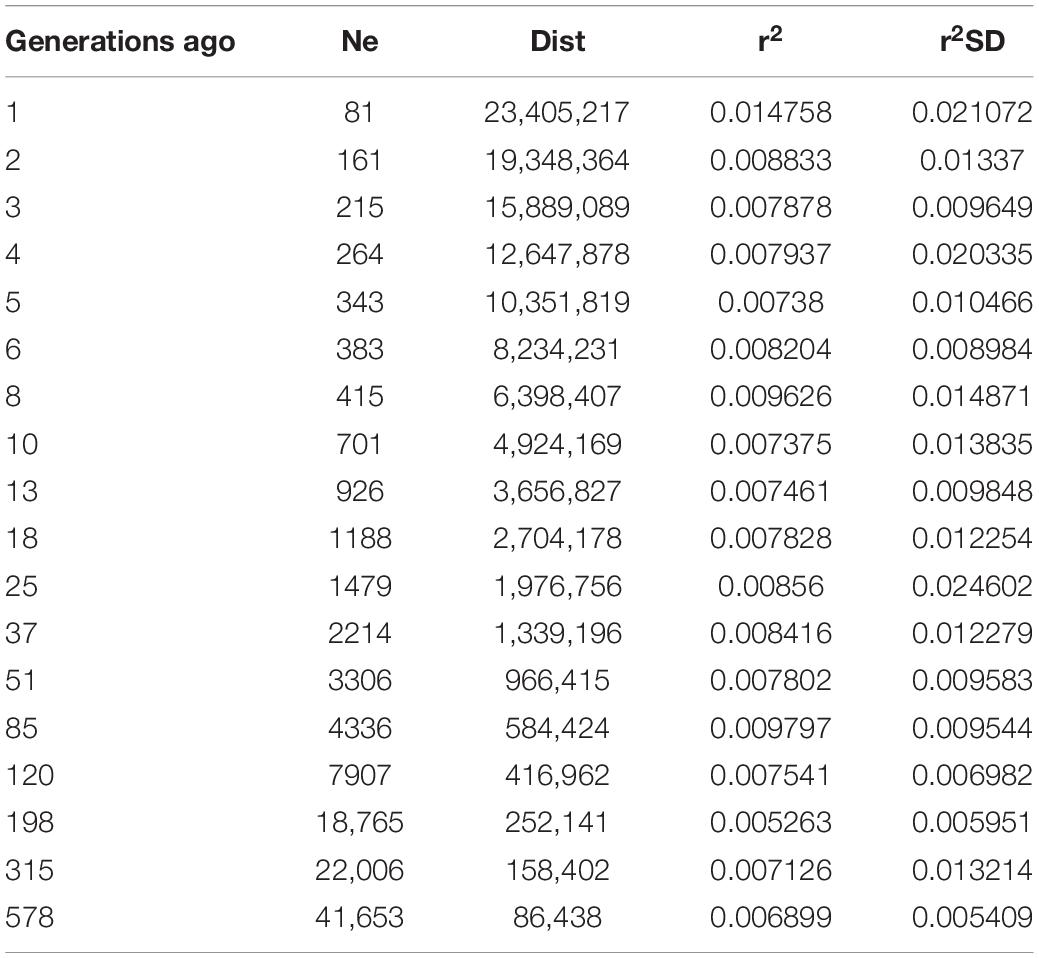
Table 4. Effective population size (Ne) for C. carpio in the southern basin of the Caspian Sea (dist: average distance between loci; r2: average LD; r2SD: LD standard deviation).
Loci Under Putative Selection and Number of Private Alleles
Bayescan identified 240 outlier loci across the genome of C. carpio with positive values for alpha, implying diversifying selection (Supplementary Figure 1A). A total of 109 out of 240 identified outlier loci (45%) were related to 70 unique genes. After filtering based on the Log10 (PO) > 2, 57 loci were identified as strong candidates under selection, connected to 17 unique genes (Supplementary Table 3). Further, the Anzali fish had the largest number of private alleles (663), while the Gomishan population showed the least number of private alleles (79, Supplementary Figure 1B). The DAPC scatter plot based on the limited data set (240 outlier loci identified through BayeScan) classified all the investigated C. carpio individuals into five different groups (Figure 7).
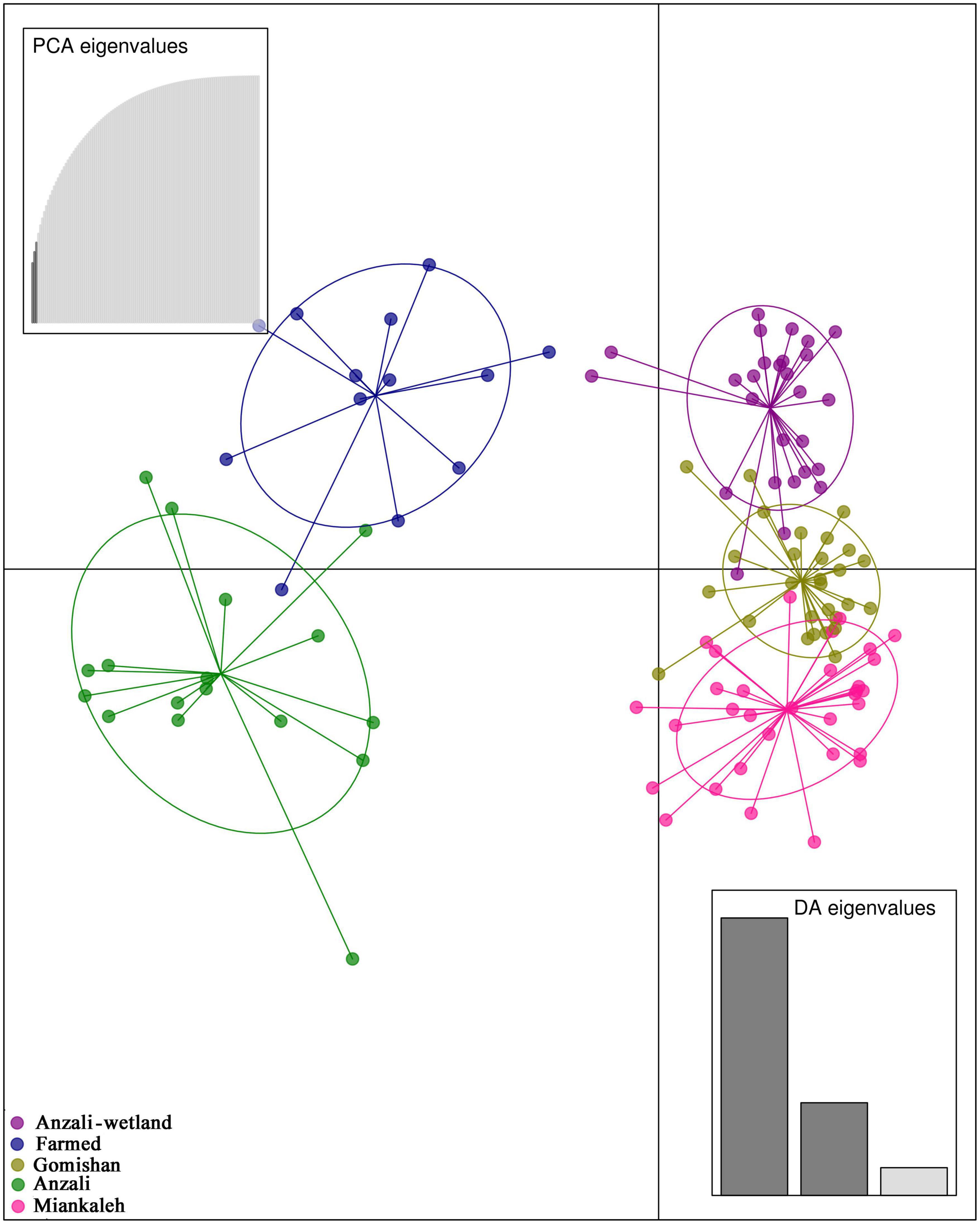
Figure 7. Discriminant analysis of principal components (DAPCs) scatter plot based on the outlier loci (240 SNPs) found through BayeScan.
Discussion
In this study we have obtained 17,828 genetic markers from wild C. carpio captured from four different locations in the Caspian Sea and from one aquaculture farm. This dataset has allowed us to study the genetic diversity of wild common carp populations of the Caspian Sea, their genetic structure and introgression and migration between populations. To the best of our knowledge, this is the first study investigating the population genomics of C. carpio in the southern coastline of the Caspian Sea using a large number of genetic markers, and offers pivotal evidence to assess the Iranian restocking program.
Genetic Diversity and Inbreeding of the Wild and Farmed Populations
All populations studied showed low levels of heterozygosity, in agreement with other studies on Caspian C. carpio (Khalili and Amirkolaie, 2010; Yousefian and Laloei, 2011) and other wild populations of C. carpio, which reported low to medium level of diversity (Li et al., 2007). Mean observed heterozygosity (Ho) estimates suggest that the farmed population has higher genetic diversity (Ho = 0.23) than the wild populations. Amongst the wild populations, Gomishan with Ho = 0.21 showed the highest diversity. Gomishan is considered the main source of animals for the restocking program of C. carpio, as capture rate in this area is higher than other locations in the South of the Caspian Sea. Gomishan is in the southern east of the Caspian Sea, and it is less affected by the industrial pollutants, urban, and agricultural runoff, which are known to have a negative impact on the genetic diversity of fish populations (Mcmillan et al., 2006; Martinez et al., 2018). A small effective population size of the wild animals can be one of the main reasons for the current low level of genetic diversity. However, it should be noted that the farmed C. carpio may have come from different families, resulting in a higher level of genetic diversity compared to the wild animals. Furthermore, the negative genetic and epigenetic effects of stocking programs known as Ryman–Laikre effect on wild fish species such as S. salar have been reported (Hagen et al., 2021). Stocking can lead to a loss of genetic variation, breakdown in genetic structure of wild stocks by increasing the level of inbreeding and reduction in effective population size in spite of the increased census population size (Christie et al., 2012).
All C. carpio populations investigated showed considerable positive values of inbreeding. Anzali-wetland is an isolated area (∼15,000 ha) with a narrow connection to the sea. It is presumed that the C. carpio individuals in the Anzali-wetland have no tendency to migrate outside for breeding, which also could explain the observed low heterozygosity. Freshwater fish species are more prone to experience inbreeding due to the lower connectivity between different habitats, as they are usually geographically isolated from each other. Landínez-García et al. (2020) reported a significant inbreeding and high genetic diversity for the freshwater fish Prochilodus magdalenae using 11 species-specific SSR markers. For example, a high level of inbreeding and genomic relatedness among individuals within the population of northern pike (Esox lucius) in the Irtysh River in China was recognized and the controlled capture was suggested for future conservation of this species (Luan et al., 2021). There are also documented cases of inbreeding amongst marine fishes and, for instance, a high level of relatedness and inbreeding was reported for winter flounder (Pseudopleuronectes americanus) in 6 New York estuaries (O’Leary et al., 2013).
Genetic Differentiation, Population Structure, and Gene Flow
Low to medium level of differentiation was observed among different wild populations of C. carpio and also between wild and farmed animals, respectively. The Fst values for the wild populations were between 0 and 0.051, while wild-farmed comparisons showed Fst values above 0.05, except the Anzali–Farmed comparison (Fst = 0.04). The genetic difference between populations is considered small for Fst below 0.05, whereas Fst values in the range 0.05–0.15 indicate a medium level of differentiation, and Fst above 0.15 corresponds to complete separation (Li et al., 2007). Based on AMOVA, genetic variation between populations accounted for only 3% of the total variation, indicating a high level of introgression among wild populations of C. carpio in the southern Caspian Sea.
Animals from the five sampling sites in the present study were related to three main genetic stocks. The two first clades included the farmed and Anzali populations while the third clade encompassed mainly the individuals from Gomishan, Miankaleh, and Anzali-wetland. C. carpio from different locations were traceable based on their recent coancestry matrix. The Anzali population showed a fair differentiation relative to the other Caspian populations. These results support previous reports based on SSR markers, which genetically discriminated two populations of C. carpio existing in Anzali and Anzali-wetland (Laloei et al., 2013). While both Anzali and Anzali-wetland are located the Guilan province, not only the reproductive behavior of their carp populations is very different, but also their time scale for breeding is slightly different. The sea form of C. carpio migrates to the rivers for spawning between March and May, while the specimens in Anzali-wetland (resident form) have no migratory behavior, completing their life cycle in the wetland and breeding mostly in May (Coad 2020). Further, while we found no sign of isolation by distance, Anzali is geographically relatively far from the other locations, which probably contributes to the larger genetic differences between this site and the other sea form populations of C. carpio.
The decrease of C. carpio wild fisheries in the southern part of the Caspian Sea over the past decade led to the establishment of a restocking program that uses a semi-natural breeding scheme. Migrant adults from the Sea to rivers are collected during the breeding season, bred under controlled conditions and then the progeny is released to the rivers where their parents were collected. Our results suggest that each sampling location maintains a specific genetic signature, which means that this restocking strategy has managed to preserve the genetic differences of wild C. carpio populations. Nonetheless, there is some evidence of admixture between populations.
Four migration edges were identified between different populations of C. carpio, and the latest identified migrations occurred from the farmed population to the Anzali and Anzali-wetland locations. The longest distance migration was observed between Gomishan and Anzali-wetland, which was surprising, while the migrations between Gomishan and Miankaleh or Anzali and Anzali-wetland were more expected because of the closer geographic distances. Besides, the lack of migration form Anzali-wetland C. carpio confirms the assumption that this population is a permanent resident in this lagoon. The observed migrations and the consequent low genetic differentiation could have three possible reasons. The migration events have probably occurred naturally, especially during the breeding season, because the fish can migrate far distances to find suitable nursery grounds. Another factor could be human-induced gene flow during the restocking program. Shen et al. (2019) in their study on population structure and demographic history of grass carp observed a high level of gene flow between geographically isolated river systems and mentioned that the aquaculture activities have probably played a more important role than natural gene flow in the observed low genetic differentiation. The Gorganrod river area close to Gomishan is the region in which restocking for the sea form of Caspian C. carpio happens in practice; however, in Anzali-wetland restocking is done occasionally and the origin of its restocking source is not clear. Accordingly, a mantel test showed no significant relationship between genetic and geographical distances (r = 0.001, p = 0.50, nrepet = 9999). Hence, migration between populations can be considered as the main possible factor for the small genetic differences. As the third alternative explanation, Anzali-wetland in the southwest of the Caspian Sea has a narrow connectivity to the sea and the observed migration between this location and Gomishan in the Southeast may be supported by the historical connectivity between these populations now inhabiting in different locations. This theory was also suggested by Mcglashan and Hughes (2001) to explain the unexpected patterns of low genetic variations among freshwater populations of Hypseleotris compressa.
The Farmed population showed the highest genetic differentiation. The Farmed stocks of C. carpio in Iran have been imported from European domesticated strains, which probably originated from the Caspian Sea (Balon, 1995). Historically, the Caspian Sea and Black Sea were a single mass of water known as Paratethys Sea, which separated 5.5 million years ago. The outputs in the present study support the admixture between farmed and wild C. carpio in the southern basin of the Caspian Sea, probably caused by farmed escapees to the drainages of the Caspian Sea and/or by crossing between wild parents and farmed animals in restocking centers.
Effective Population Size
The results of demographic estimates (Ne) based on SNP data from C. carpio showed a dramatic reduction from 578 generations ago to the present. Demographic estimates of Ne is a practical tool in population genetics to infer the history of populations and to determine the level of variability for a given population (Charlesworth, 2009). The low Ne value in recent generations (81) implies a small number of breeders contributing to genetic diversity in the wild populations of C. carpio across the southern coasts of the Caspian Sea.
Signatures of Selection
Amongst 17,828 obtained SNPs in this study, 240 SNPs (1.34%) were identified as outliers under diversifying selection. During the first stage of new habitat colonization, populations experience different evolutionary forces that contribute to population differentiation, namely genetic drift and directional selection (Shen et al., 2019). Diversifying selection can maintain and increase genetic variation (Gavrilets and Vose, 2005). In populations experiencing strong diversifying selection, extreme phenotypes are favored and the population diversifies into new populations. Hence, the level of differentiation between the new populations increases over time, especially when migrations are absent. For example, a study on three European whitefish (Coregonus lavaretus) morphs showed that diversifying selection was the force driving the observed phenotypic specializations (Häkli et al., 2018). The southern coast of the Caspian Sea varies ecologically and environmentally (Jafari et al., 2021a,b) and the outliers reflecting diversifying selection in different populations are likely involved in local adaptation of C. carpio.
Management Implications
Conservation efforts must aim to preserve the genetic diversity of the different populations of a species (Weissing et al., 2011), and inappropriate management can frequently decrease this genetic diversity. Our results highlight a low genetic diversity in the C. carpio populations of the Southern Caspian Sea, albeit genetic differences between these populations still exist. The low level of differentiation in the presence of migration may be an indicator of the small effective population size leading to a shallow population structure for this species in the southern coasts of the Caspian Sea. Given the global vulnerable status of C. carpio based on the IUCN red list (Freyhof and Kottelat, 2008) and the small effective population size of the Southern Caspian Sea populations, it is essential to consider fishing bans and provide action plans to rehabilitate the nursery grounds. In this scenario, it is fundamental that the restocking programs take into account the distinct genetic backgrounds and make efforts to avoid mixing animals from different locations. The present study is the first report of SNP outliers in C. carpio in the southern Caspian Sea basin. We have shown that a reduced number of SNPs can effectively determine the genetic background of Southern Caspian C. carpio populations. A low-density SNP panel with these SNPs capable of distinguishing the different genetic backgrounds would facilitate fisheries management and restocking programs (Smith et al., 2005; Dann et al., 2013). Furthermore, considering the high level of inbreeding, one of the practical solutions to improve the genetic health of the C. carpio populations in the southern parts of the Caspian Sea would be to introduce wild specimens from the Aral or Black Seas.
Conclusion
The present study is the first investigation of C. carpio population structure at the genomic level along the southern coast of the Caspian Sea. Fish from different locations showed specific genetic signatures, which implies the existence of distinct C. carpio populations. The fact that this diversity is preserved to some extent suggests that the ongoing restocking program has been successful in this regard. However, all populations showed a high level of inbreeding, and therefore better fisheries management and habitat protection are recommended to keep and improve the genetic diversity in order to avoid loss of C. carpio stocks in the Caspian Sea.
Data Availability Statement
The datasets presented in this study can be found in online repositories at https://www.ncbi.nlm.nih.gov/ under the accession number PRJNA763073.
Ethics Statement
The animal study was reviewed and approved by the Ethics Committee of Gorgan University of Agricultural Sciences and Natural Resources (Iran).
Author Contributions
OJ, MZ, and A-AH designed the study. OJ carried out the related field work, sampling and lab work, analyzed the GBS data, and wrote the manuscript. BA and MZ participated in data analysis. OJ, MZ, JF, and DR interpreted the obtained data. JF, DR, and MZ reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
We are grateful to Agricultural Research, Education and Extension Organization (AREEO) for providing the required financial and technical resources and to Nord University (Norway) for additional support.
Conflict of Interest
BA was employed by OMICS Research Group (Media Teb Gene Co. Ltd.).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2022.872176/full#supplementary-material
Footnotes
References
Alexander, A. (2018). GBS_SNP_filter v1.x.x. Available online at: https://github.com/laninsky/GBS_SNP_filter (accessed January, 2021).
Allendorf, F. W., Hohenlohe, P. A., and luikart, G. (2010). Genomics and the future of conservation genetics. Nat. Rev. Genet. 11, 697–709. doi: 10.1038/nrg2844
Andrews, K. R., Good, J. M., Miller, M. R., Luikart, G., and Hohenlohe, P. A. (2016). Harnessing the power of RADSEQ for ecological and evolutionary genomics. Nat. Rev. Genet. 17, 81–92. doi: 10.1038/nrg.2015.28
Balon, E. K. (1995). Origin and domestication of the wild carp, Cyprinus carpio: from Roman gourmets to the swimming flowers. Aquaculture 129, 3–48. doi: 10.1016/0044-8486(94)00227-f
Barbato, M., Orozco-Terwengel, P., Tapio, M., and Bruford, M. W. (2015). Snep: a tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Front. Genet. 6:109. doi: 10.3389/fgene.2015.00109
Barría, A., Christensen, K. A., Yoshida, G. M., Correa, K., Jedlicki, A., Lhorente, J. P., et al. (2018). Genomic predictions and genome-wide association study of resistance against Piscirickettsia salmonis in coho salmon (Oncorhynchus kisutch) using Ddrad sequencing. G3: Genes, Genom. Genet. 8, 1183–1194. doi: 10.1534/g3.118.200053
Bourret, V., Kent, M. P., Primmer, C. R., Vasemägi, A., Karlsson, S., Hindar, K., et al. (2013). SNP-array reveals genome-wide patterns of geographical and potential adaptive divergence across the natural range of A tlantic salmon (Salmo salar). Mol. Ecol. 22, 532–551. doi: 10.1111/mec.12003
Bradbury, I. R., Hamilton, L. C., Dempson, B., Robertson, M. J., Bourret, V., Bernatchez, L., et al. (2015). Transatlantic secondary contact in Atlantic Salmon, comparing microsatellites, a single nucleotide polymorphism array and restriction-site associated DNA sequencing for the resolution of complex spatial structure. Mol. Ecol. 24, 5130–5144. doi: 10.1111/mec.13395
Catchen, J., Hohenlohe, P. A., Bassham, S., Amores, A., and Cresko, W. A. (2013). Stacks: an analysis tool set for population genomics. Mol. Ecol. 22, 3124–3140. doi: 10.1111/mec.12354
Charlesworth, B. (2009). Effective population size and patterns of molecular evolution and variation. Nat. Rev. Genet. 10, 195–205. doi: 10.1038/nrg2526
Christie, M. R., Marine, M. L., French, R. A., Waples, R. S., and Blouin, M. (2012). Effective size of a wild salmonid population is greatly reduced by hatchery supplementation. Heredity 109, 254–260. doi: 10.1038/hdy.2012.39
Dann, T. H., Habicht, C., Baker, T. T., and Seeb, J. E. (2013). Exploiting genetic diversity to balance conservation and harvest of migratory salmon. Can. J. Fish. Aquat. Sci. 70, 785–793. doi: 10.1139/cjfas-2012-0449
Daw, E. W., Heath, S. C., and Lu, Y. (2005). Single-nucleotide polymorphism versus microsatellite markers in a combined linkage and segregation analysis of a quantitative trait. BMC Genet. 6:S32. doi: 10.1186/1471-2156-6-S1-S32
Dorafshan, S., Fallahbagheri, F., Pourkazemi, M., Keivany, Y., and Chakmedouz Qasemi, F. (2013). Genetic analysis of wild common carp, Cyprinus carpio L. in the Anzali wetland, the Caspian Sea. Iran. J. Fish. Sci. 12, 1–11.
Elshire, R. J., Glaubitz, J. C., Sun, Q., Poland, J. A., Kawamoto, K., Buckler, E. S., et al. (2011). A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PloS One 6:e19379. doi: 10.1371/journal.pone.0019379
Freyhof, J., and Kottelat, M. (2008). Cyprinus carpio. The IUCN Red List of Threatened Species. Available online at: https://dx.doi.org/10.2305/IUCN.UK.2008.RLTS.T6181A12559362.en (accessed June 27, 2021).
Gavrilets, S., and Vose, A. (2005). Dynamic patterns of adaptive radiation. Proc. Natl. Acad. Sci. 102, 18040–18045. doi: 10.1073/pnas.0506330102
Hagen, I. J., Ugedal, O., Jensen, A. J., Lo, H., Holthe, E., Bjøru, B., et al. (2021). Evaluation of genetic effects on wild salmon populations from stock enhancement. ICES J. Mar. Sci. 78, 900–909. doi: 10.1093/icesjms/fsaa235
Häkli, K., Østbye, K., Kahilainen, K. K., Amundsen, P. A., and Præbel, K. (2018). Diversifying selection drives parallel evolution of gill raker number and body size along the speciation continuum of European whitefish. Ecol. Evol. 8, 2617–2631. doi: 10.1002/ece3.3876
Hess, J. E., Campbell, N. R., Close, D. A., Docker, M. F., and Narum, S. R. (2013). Population genomics of P acific lamprey: adaptive variation in a highly dispersive species. Mol. Ecol. 22, 2898–2916. doi: 10.1111/mec.12150
Jafari, O., Zeinalabedini, M., Robledo, D., Fernandes, J. M. O., Hedayati, A. A., and Arefnezhad, B. (2021a). Common carp (Cyprinus carpio) genotyping-by-sequencing libraries. NCBI BioProject PRJNA 763073, https://dataviewncbinlmnihgovobjectPRJNA763073reviewercg7besoj8i5u0bm1slce9n89lc.
Jafari, O., Hedayati, S., Poorbagher, H., Ghorbani, R., Abdolhay, H. A., and Zeinolabedini, M. (2021b). Phenotype-environment associations in common carp. Cyprinus carpio Linnaeus 1758, in the southern coast of the Caspian Sea. Iran. J. Fish Sci. 20, 1114–1127.
Jombart, T. (2008). Analyses Multivariées de Marqueurs Génétiques: Développements Méthodologiques, Applications et Extensions. Doctoral dissertation. Villeurbanne: UCB Lyon 1.
Kamvar, Z. N., Tabima, J. F., and Grünwald, N. J. (2014). Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ. 2:e281. doi: 10.7717/peerj.281
Khalili, K., and Amirkolaie, A. K. (2010). Comparison of common carp (Cyprinus carpio L.) morphological and electrophoretic characteristics in the southern coast of the Caspian Sea. J. Fish. Aquat. Sci. 5, 200–207. doi: 10.3923/jfas.2010.200.207
Knaus, B. J., and Grünwald, N. J. (2017). vcfr: a package to manipulate and visualize variant call format data in R. Mol. Ecol. Res. 17, 44–53. doi: 10.1111/1755-0998.12549
Laloei, F., Gilkolaei, S., and Taghavi, M. J. (2013). Genetic diversity and differentiation of common carp (Cyprinus carpio L.) in the southern part of caspian sea by using microsatellite markers. Asian Fish. Sci. 26, 115–127. doi: 10.1186/s12862-018-1219-9
Landínez-García, R. M., Narváez, J. C., and Márquez, E. J. (2020). Population genetics of the freshwater fish Prochilodus magdalenae (Characiformes: Prochilodontidae), using species-specific microsatellite loci. Peer.J. 8:e10327. doi: 10.7717/peerj.10327
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Larson, W. A., Seeb, L. W., Everett, M. V., Waples, R. K., Templin, W. D., and Seeb, J. E. (2014). Genotyping by sequencing resolves shallow population structure to inform conservation of Chinook salmon (Oncorhynchus tshawytscha). Evol. Appl. 7, 355–369. doi: 10.1111/eva.12128
Lemopoulos, A., Prokkola, J. M., Uusi-Heikkilä, S., Vasemägi, A., Huusko, A., Hyvärinen, P., et al. (2019). Comparing Radseq and microsatellites for estimating genetic diversity and relatedness—Implications for brown trout conservation. Ecol. Evol. 9, 2106–2120. doi: 10.1002/ece3.4905
Li, D., Kang, D., Yin, Q., Sun, X., and Liang, L. (2007). Microsatellite DNA marker analysis of genetic diversity in wild common carp (Cyprinus carpio L.) populations. J. Genet. Genom. 34, 984–993. doi: 10.1016/S1673-8527(07)60111-8
Li, J., Li, H., Jakobsson, M., Li, S., Sjödin, P., and Lascoux, M. (2012). Joint analysis of demography and selection in population genetics: where do we stand and where could we go? Mol. Ecol. 21, 28–44. doi: 10.1111/j.1365-294X.2011.05308.x
Lischer, H. E., and Excoffier, L. (2012). PGDSPIDER: an automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 28, 298–299. doi: 10.1093/bioinformatics/btr642
Liu, S., Ferchaud, A. L., Grønkjær, P., Nygaard, R., and Hansen, M. M. (2018). Genomic parallelism and lack thereof in contrasting systems of three-spined sticklebacks. Mol. Ecol. 27, 4725–4743. doi: 10.1111/mec.14782
Luan, P., Huo, T., Ma, B., Song, D., Zhang, X., and Hu, G. (2021). Genomic inbreeding and population structure of northern pike (Esox lucius) in Xinjiang. China. Ecol. Evol. 11, 5657–5668. doi: 10.1002/ece3.7469
Malinsky, M., Trucchi, E., Lawson, D. J., and Falush, D. (2018). RADPAINTER and Fineradstructure: population inference from Radseq data. Mol. Biol. Evol. 35, 1284–1290. doi: 10.1093/molbev/msy023
Martinez, A. S., Willoughby, J. R., and Christie, M. R. (2018). Genetic diversity in fishes is influenced by habitat type and life-history variation. Ecol. Evol. 8, 12022–12031. doi: 10.1002/ece3.4661
Mcglashan, D. J., and Hughes, J. M. (2001). Low levels of genetic differentiation among populations of the freshwater fish Hypseleotris compressa (Gobiidae: Eleotridinae): implications for its biology, population connectivity and history. Heredity 86, 222–233. doi: 10.1046/j.1365-2540.2001.00824.x
Mcmillan, A. M., Bagley, M. J., Jackson, S. A., and Nacci, D. E. (2006). Genetic diversity and structure of an estuarine fish (Fundulus heteroclitus) indigenous to sites associated with a highly contaminated urban harbor. Ecotoxicology 15, 539–548. doi: 10.1007/s10646-006-0090-4
Meirmans, P. G., and Van Tienderen, P. H. (2004). Genotype and Genodive: two programs for the analysis of genetic diversity of asexual organisms. Mol. Ecol. Notes 4, 792–794. doi: 10.1111/j.1471-8286.2004.00770.x
Nakajima, T., Hudson, M. J., Uchiyama, J., Makibayashi, K., and Zhang, J. (2019). Common carp aquaculture in Neolithic China dates back 8,000 years. Nat. Ecol. Evol. 3, 1415–1418. doi: 10.1038/s41559-019-0974-3
O’Leary, S. J., Hice, L. A., Feldheim, K. A., Frisk, M. G., Mcelroy, A. E., Fast, M. D., et al. (2013). Severe inbreeding and small effective number of breeders in a formerly abundant marine fish. PLoS One 8:e66126. doi: 10.1371/journal.pone.0066126
Paradis, E., and Schliep, K. (2019). ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35, 526–528. doi: 10.1093/bioinformatics/bty633
Pickrell, J., and Pritchard, J. (2012). Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 8:e10029671. doi: 10.1371/journal.pgen.1002967
Rahman, M. M. (2015). Role of common carp (Cyprinus carpio) in aquaculture production systems. Front. Life Sci. 8, 399–410. doi: 10.1080/21553769.2015.1045629
Robledo, D., Matika, O., Hamilton, A., and Houston, R. D. (2018a). Genome-wide association and genomic selection for resistance to amoebic gill disease in Atlantic salmon. G3: Genes Genom. Genet. 8, 1195–1203. doi: 10.1534/g3.118.200075
Robledo, D., Palaiokostas, C., Bargelloni, L., Martínez, P., and Houston, R. (2018b). Applications of genotyping by sequencing in aquaculture breeding and genetics. Rev. Aquacult. 10, 670–682. doi: 10.1111/raq.12193
Rodríguez-Ezpeleta, N., Bradbury, I. R., Mendibil, I., Álvarez, P., Cotano, U., and Irigoien, X. (2016). Population structure of Atlantic mackerel inferred from RAD-seq-derived SNP markers: Effects of sequence clustering parameters and hierarchical SNP selection. Mol. Ecol. Resour. 16, 991–1001. doi: 10.1111/1755-0998.12518
Rousset, F. (2008). genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Resour 8, 103–106. doi: 10.1111/j.1471-8286.2007.01931.x
Shen, Y., Wang, L., Fu, J., Xu, X., Yue, G. H., and Li, J. (2019). Population structure, demographic history and local adaptation of the grass carp. BMC Genom. 20:1–16. doi: 10.1186/s12864-019-5872-1
Smith, C. T., Templin, W. D., Seeb, J. E., and Seeb, L. W. (2005). Single nucleotide polymorphisms provide rapid and accurate estimates of the proportions of US and Canadian Chinook salmon caught in Yukon River fisheries. North Am. J. Fish. Manage. 25, 944–953. doi: 10.1577/m04-143.1
Sonah, H., Bastien, M., Iquira, E., Tardivel, A., Légaré, G., Boyle, B., et al. (2013). An improved genotyping by sequencing (GBS) approach offering increased versatility and efficiency of Snp discovery and genotyping. PloS One 8:e54603. doi: 10.1371/journal.pone.0054603
Stapley, J., Reger, J., Feulner, P. G., Smadja, C., Galindo, J., Ekblom, R., et al. (2010). Adaptation genomics: the next generation. Trends Ecol. Evol. 25, 705–712.
Team, R. C. (2013). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Utter, F. (2004). Population genetics, conservation and evolution in salmonids and other widely cultured fishes: some perspectives over six decades. J. Fish Biol. 65, 323–324. doi: 10.1111/j.0022-1112.2004.0559x.x
Waples, R. S., Dickhoff, W. W., Hauser, L., and Ryman, N. (2008). Six decades of fishery genetics: taking stock. Fisheries 33, 76–79. doi: 10.1098/rspb.2000.1128
Weissing, F. J., Edelaar, P., and Van Doorn, G. S. (2011). Adaptive speciation theory: a conceptual review. Behav. Ecol. Sociobiol. 65, 461–480. doi: 10.1007/s00265-010-1125-7
Xu, P., Zhang, X., Wang, X., Li, J., Liu, G., Kuang, Y., et al. (2014). Genome sequence and genetic diversity of the common carp. Cyprinus Carpio. Nat. Genet. 46, 1212–1219. doi: 10.1038/ng.3098
Xu, S., Song, N., Zhao, L., Cai, S., Han, Z., and Gao, T. (2017). Genomic evidence for local adaptation in the ovoviviparous marine fish Sebastiscus marmoratus with a background of population homogeneity. Sci. Rep. 7, 1–12. doi: 10.1038/s41598-017-01742-z
Yousefian, M., and Laloei, F. (2011). Genetic variations and structure of common carp (Cyprinus carpio) populations by use of biochemical, mitochondrial and microsatellite markers. Middle East J. Sci. Res. 7, 339–345.
Zhao, P., Zhou, H.-J., Potter, D., Hu, Y.-H., Feng, X.-J., Dang, M., et al. (2018). Population genetics, phylogenomics and hybrid speciation of Juglans in China determined from whole chloroplast genomes, transcriptomes, and genotyping-by-sequencing (GBS). Mol. Phylogenet. Evol. 126, 250–265. doi: 10.1016/j.ympev.2018.04.014
Keywords: Cyprinus carpio, genomic population structure, conservation, SNPs (single-nucleotide polymorphism), genotyping-by-sequencing (GBS)
Citation: Jafari O, Zeinalabedini M, Robledo D, Fernandes JMO, Hedayati A-A and Arefnezhad B (2022) Genotyping-by-Sequencing Reveals the Impact of Restocking on Wild Common Carp Populations of the Southern Caspian Basin. Front. Ecol. Evol. 10:872176. doi: 10.3389/fevo.2022.872176
Received: 09 February 2022; Accepted: 11 March 2022;
Published: 28 April 2022.
Edited by:
You-Yi Kuang, Heilongjiang River Fisheries Research Institute, Chinese Academy of Fishery Sciences (CAFS), ChinaReviewed by:
Hong Wei Liang, Yangtze River Fisheries Research Institute, Chinese Academy of Fishery Sciences (CAFS), ChinaYingying Zhao, Shenyang Agricultural University, China
Copyright © 2022 Jafari, Zeinalabedini, Robledo, Fernandes, Hedayati and Arefnezhad. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mehrshad Zeinalabedini, mzeinolabedini@abrii.ac.ir; Jorge M. O. Fernandes, jorge.m.fernandes@nord.no