
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol. , 04 August 2022
Sec. Evolutionary and Population Genetics
Volume 10 - 2022 | https://doi.org/10.3389/fevo.2022.866587
 Adriana Vallejo-Trujillo1*
Adriana Vallejo-Trujillo1* Adebabay Kebede2,3
Adebabay Kebede2,3 Maria Lozano-Jaramillo4
Maria Lozano-Jaramillo4 Tadelle Dessie2
Tadelle Dessie2 Jacqueline Smith5
Jacqueline Smith5 Olivier Hanotte1,2*
Olivier Hanotte1,2* Almas A. Gheyas5*
Almas A. Gheyas5*In evolutionary ecology, an “ecotype” is a population that is genetically adapted to specific environmental conditions. Environmental and genetic characterisation of livestock ecotypes can play a crucial role in conservation and breeding improvement, particularly to achieve climate resilience. However, livestock ecotypes are often arbitrarily defined without a detailed characterisation of their agro-ecologies. In this study, we employ a novel integrated approach, combining ecological niche modelling (ENM) with genomics, to delineate ecotypes based on environmental characterisation of population habitats and unravel the signatures of adaptive selection in the ecotype genomes. The method was applied on 25 Ethiopian village chicken populations representing diverse agro-climatic conditions. ENM identified six key environmental drivers of adaptation and delineated 12 ecotypes. Within-ecotype selection signature analyses (using Hp and iHS methods) identified 1,056 candidate sweep regions (SRs) associated with diverse biological processes. While most SRs are ecotype-specific, the biological pathways perturbed by overlapping genes are largely shared among ecotypes. A few biological pathways were shared amongst most ecotypes and the genes involved showed functions important for scavenging chickens, e.g., neuronal development/processes, immune response, vision development, and learning. Genotype-environment association using redundancy analysis (RDA) allowed for correlating ∼33% of the SRs with major environmental drivers. Inspection of some strong candidate genes from selection signature analysis and RDA showed highly relevant functions in relation to the major environmental drivers of corresponding ecotypes. This integrated approach offers a powerful tool to gain insight into the complex processes of adaptive evolution including the genotype × environment (G × E) interactions.
Livestock genetic diversity is an essential prerequisite to achieve resilience to the challenges arising from climatic changes, and the ever-changing dynamics of production systems and consumer demands. Indigenous livestock populations surviving in diverse geographic areas exhibit unique genetic adaptations to their local environmental conditions. In evolutionary ecology, such locally adapted populations of a species are referred to as “ecotypes.” With their extant genetic and adaptive diversity, different ecotypes may hold genetic solutions for many present and future challenges facing the global livestock sector.
Delineating livestock ecotypes in practice, however, has been challenging. Arbitrary and loose criteria have often been applied for defining ecotypes, such as the geographic origin of populations, broad agro-climatic conditions of the populations or some major morphological features (Tadelle et al., 2003; Keambou et al., 2014; Sanarana et al., 2016). Such classifications are not only arbitrary but also are over-simplistic in that they neither take into consideration a detailed characterisation of the ecotype agro-ecologies nor do they attempt to decipher the key environmental drivers and their interactions in shaping ecotypes’ genomes. When a livestock species is introduced into a new environment, a wide range of bioclimatic conditions will interact with the standing genetic variations of the introduced population. Local agro-climatic conditions will exert different selection pressures in different environments and contribute to creating new ecotypes. This complex scenario is resonated in the ecotype definition provided by Lowri (2012) who described the term as “a non-static adaptive variation over many traits across the natural landscape with no discernible boundaries.” This emphasises the need for adopting a “systems” approach, such as environmental modelling, for defining ecotypes that would allow considering complex interaction of a large number of environmental predictors. Integrating such an approach with genomic characterisation will have major implications for the characterisation and conservation of livestock adaptive diversity and will help provide genetic solutions for developing “climate resilient” livestock breeds.
Ecological niche modelling (ENM) – also called species distribution modelling (SDM) – has been extensively applied for predicting distribution of wild animal species and crop plants. One of the most widely used ENM/SDM approaches is implemented in the program MaxEnt (Phillips et al., 2006). Using environmental data from known species-presence locations, MaxEnt predicts the potential geographical range of a species across a landscape and estimates the contribution of environmental variables in shaping the species’ habitats. As a consequence, the method has been a powerful tool in the conservation efforts of wild species (Thorn et al., 2009) and has found applications for other diverse purposes such as assessing the risk from invasive species (Jiménez-Valverde et al., 2011), epidemiological studies (Cardoso-Leite et al., 2014), and estimating the effect of climate change on future distribution of a species (Jeschke and Strayer, 2008). In livestock, however, the use of ENM is still in its infancy with only a few available examples (Pitt et al., 2016; Lozano-Jaramillo et al., 2018; Vajana et al., 2018; Gheyas et al., 2021; Kebede et al., 2021). Pitt et al. (2016) used MaxEnt for inferring the actual and historical distributions of early domestic fowl by modelling the environmental conditions of extant Red Jungle Fowl (RJF) with important conservation implications. Lozano-Jaramillo et al. (2018) used MaxEnt to predict the environmental suitability maps for successful introduction of two exotic chicken breeds across the Ethiopian landscape. Vajana et al. (2018) integrated landscape genomics and ENM to investigate local adaptation of indigenous Ugandan cattle to East Coast Fever. In a recent study (Gheyas et al., 2021), we applied MaxEnt for identifying the key environmental drivers in the agro-ecologies of Ethiopian indigenous chickens, followed by genomic analyses in relation to these variables to identify candidate adaptive genes. In another recent study, Kebede et al. (2021) integrated ENM with phenotypic distribution modelling to study phenotypic differentiation among Ethiopian indigenous chicken populations and used this characterisation for delineating potential ecotypes. The study, however, did not consider the genomic basis of adaptation.
Based on the insights from the above studies, here we present a novel application of ENM for delineating livestock ecotypes. Our approach considers “ecology” as the main driving force for adaptive evolution and hence as the focal point for characterising ecotypes. The ENM-based ecotypes are then used as units of analysis to dissect the underlying genetics of adaptation. We exemplify the application of this approach using the same set of Ethiopian village chicken populations used in our previous study (Gheyas et al., 2021). Ethiopian indigenous chicken populations are an excellent model to demonstrate the utility of this novel approach for a number of reasons. In Ethiopia, indigenous chickens account for 78.85% of the total poultry population (Central Statistical Agency [CSA], 2021) and are predominantly reared in scavenging/semi-scavenging rural agro-ecologies, where the birds are directly exposed to various environmental pressures. A great deal of phenotypic diversity (Wilson, 2010) and genetic plasticity are observed in these indigenous chickens (Bettridge et al., 2018), even though their ancestral gene pool is not very diverse (Gheyas et al., 2021). Ethiopia’s agricultural landscape also varies widely due to its altitudinal topography and climatic variations, with one or two rainy seasons separated by dry seasons. As a result, the country displays diverse agro-climatic zones (n = 18) (Ministry of Agriculture [MoA], 1998). Indigenous chicken populations are found in most agro-ecologies where there is human settlement, indicating their adaptive diversity. The novel method introduced here offers an excellent platform for characterising these populations in relation to their environmental adaptation.
The major steps in our analysis include characterising the agro-ecologies of the investigated chicken populations using ENM, followed by clustering the populations based on their niche similarity to define distinct ecotypes. Within-ecotype selection signature analyses are then performed to identify genomic loci at different stages of positive selection – ongoing and/or near fixation – using iHS (Integrated Haplotype Score) and Hp (Pooled heterozygosity) approaches. Variants overlapping the putative selective sweep regions (SRs) are then analysed using redundancy analysis (RDA) approach to find their correlation with key environmental variables.
The Ethiopian landscape is characterised by its altitudinal topography ranging from 126 m below sea level to 4,620 m above sea level and climatic variation with one or two rainy seasons separated by dry seasons. As a result, the country displays diverse agro-ecological zones (AEZ) and subzones (Mengistu, 2006). The data set included in this analysis comprised 245 chicken samples (Gheyas et al., 2021) from across these different environments. Sampling was carried out as part of the African Chicken Genetic Gains project1 to represent diverse agro-climatic conditions in Ethiopia. The samples originated from 25 villages or Kebeles across 12 districts and six national regions (Supplementary Table 1). During sampling of the chicken populations, a single geographic coordinate registry was recorded for each sampled village. As described in Gheyas et al. (2021), for each of these populations, nine additional geographic coordinates in different grids (separated by 1.2 km2) were selected using Google Earth Pro 7.3.1.4507 (2016). Thereby, for each population 10 geographic coordinates were used to allow ENM-based characterisation of population habitats and from 25 populations, a total of 250 geographic coordinates were used as “presence points.”
Thirty-four environmental variables – 21 climatic, 1 elevation, 8 soil, and 4 vegetation and land cover conditions – were initially chosen for ENM as these were deemed important for chicken biology. Geo-referenced environmental data for these variables were extracted from public databases – viz. WorldClim (Fick and Hijmans, 2017), SoilGrids (Hengl et al., 2014), Spatial Data Access Tool of NASA (ORNL DAAC, 2017), Harmonized World Soil Database (Fischer et al., 2008), and Global Food Security-Support Analysis Data (GFSAD30, 2017) – at a spatial resolution of 30 s (∼1 km2) using the “raster” R package. Based on geodetic datum WGS84, the grids’ dimension and extension were corrected and homogenized for 1 km2 using “rgdal,” “maptools,” “rgeos,” and “raster” packages in RStudio v1.1.419 (R Core Team, 2018).
Prior to ENM, several complementary approaches were applied to evaluate the properties and relationship of the environmental variables and their relative contributions in explaining chicken agro-ecologies. This included a normality test, a Spearman’s rank correlation, and Principal Component Analysis (PCA). MaxEnt v3.4.1 (Phillips et al., 2006) was used for executing ENM. Model optimization was performed by removing highly correlated (Spearman r > 0.6) and low contributing (<4%) environmental variables using “MaxentVariableSelection” (Jueterbock et al., 2016) and choosing the best combination of model parameters – Regularization Multiplier (RM) and Feature Classes (FCs) – using ENMeval (Muscarella et al., 2014), considering all populations together (see Gheyas et al., 2021 for details).
Based on the optimization process, six environmental variables were shortlisted, namely, minimum temperature of coldest month (bio6 – minTemp), precipitation seasonality (bio15 – precSeasonality), precipitation of wettest quarter (bio16 – precWQ), precipitation of driest quarter (bio17 – precDQ), soil organic carbon (SoilOrgC), and land use (LandUse). The best combination of model parameters included Hinge (H), Quadratic (Q) and Product (P) FCs and RM = 3.5. A sub-sample of 25% was used as test data while the remaining 75% was used as training data for model execution. Habitat suitability maps were created in the logistic scale.
Using the shortlisted variables and the optimal model parameters, habitat suitability maps were generated for individual populations. Similarity between suitability maps were assessed in ENMtools (Warren et al., 2010) using two methods: (i) pairwise Pearson correlation (r) and (ii) niche overlap statistic “I.” The r co-efficients capture correlation of corresponding grids between two suitability maps, where +1 indicates total positive linear correlation, 0 is no correlation, and −1 indicates complete negative correlation. Contrarily, the “I” values ranges from 0 indicating no overlap to 1 suggesting identical niche models between corresponding grids (Warren, 2018). The populations were grouped based on their habitat similarity by hierarchical clustering using the R package “cluster.” Both values (“I” and “r”) were converted into “Euclidean” distance. Different hierarchical clustering approaches (minimum and maximum linkage, UPGMA, and Ward method) were evaluated to measure the cluster strength based on their agglomerative coefficients. Dendrograms and heatmaps for each similarity dataset were produced using the “gplots” R package. Population clusters generated by these methods were considered as ecotypes when all population pairs within the cluster showed both the I and r values ≥0.6.
Whole-genome sequencing of individual samples was performed on an Illumina HiSeqX platform with an average coverage of ∼45X. Mapping of sequencing reads was performed against the GRCg6a (Galgal6) reference using the BWA-mem algorithm and variant calling was performed following GATK’s best practice protocol for short variant discovery for germline (Broad Institute, 2015). The protocol involved using GATK’s HaplotypeCaller function which is capable of calling short variants simultaneously via local de novo assembly of haplotypes. The GATK function GenotypeGVCF was then applied for joint genotyping across a cohort of samples followed by variant filtration using the VQSR (Variant Calling Score Recalibration) approach. Refer to Gheyas et al. (2021) and Gheyas et al. (2022) for details of sequence data processing, mapping, and variant calling.
Several analyses were performed to summarise the genetic diversity and population structure, as presented in Gheyas et al. (2021). The previous results showed a mean nucleotide diversity between 0.28 and 0.34 per population, very low levels of population differentiation (based on pairwise Fst), a weak genetic sub-structure across populations (based on PCA analysis using SNP data) and the contribution of three different ancestral gene pools (based in Admixture analyses).
Selection signature analyses were carried out on the ENM defined ecotypes by calculating pooled heterozygosity (Hp) following the method described by Rubin et al. (2010) and iHS using the Hapbin package (Maclean et al., 2015). Both metrics were calculated in overlapping sliding windows of 20 kb with a step size of 10 kb.
For iHS calculation, phasing was performed (after removing missing genotypes) using Beagle v5.1 (Browning and Browning, 2007). Chromosome-specific mean recombination rates (cM/Mb) based on Groenen et al. (2009) or Elferink et al. (2010) (for Chr16) were used for calculating genetic map positions of SNPs. Since no information was available for Chr30–33, a mean rate of 5.4 cM/Mb from across other micro-chromosomes (based on Groenen et al., 2009) was used. iHS analyses were first performed for individual SNPs by setting the minor allele frequency (maf) option as 10% and cut-off value for Extended Haplotype Homozygosity (EHH) equal to 0.1. Afterward, mean values were calculated within windows for standardised iHS (iHS_std) and the absolute value of iHS_std.
Empirical P-values were calculated for both standardised Hp (ZHp) and iHS (iHS_std) by ranking the windows in descending order according to their scores and then dividing the ranks by the total number of windows. The criteria for selecting outlier windows (i.e., potential sweeps) for iHS analysis included: windows with at least 10 SNPs, P-value ≤ 0.01, window mean of | iHS_std| ≥ 2.0, and at least 90% of SNPs in a window with | iHS_std| value ≥ 2.0. For Hp analysis, the criteria were: windows with at least 10 SNPs, P-value ≤ 0.01, and ZHp ≤ −4.0.
Genotype-environment association analysis was performed with an LD-pruned set of SNPs overlapping the SRs (from iHS and Hp analyses) using RDA in Vegan v2.5-4 (Oksanen, 2015) following Forester (2019). LD pruning was performed with PLINK v1.9 (Purcell et al., 2007), using the following parameters: –indep-pairwise, window-size = 10 kb, step-size = 10 SNPs, and r2 = 0.5. Genotype was fitted as a response variable and environmental data (six variables) as the explanatory variables with conditioning on latitude and longitude. Ancestry co-efficients from Admixture analysis were included as covariates to correct for any population structure effect.
For each canonical RDA axis, ANOVA analysis was performed. The significance of the partial model and each axis was calculated with 999 and 99 permutations, respectively.
Candidate regions/variants from selection signature and RDA analyses were investigated for their overlap with chicken genes from Ensembl (release 98). SNPs were annotated using Variant Effect Predictor (VEP) (McLaren et al., 2016). Candidate genes were checked for their overlap with chicken-QTLs from AnimalQTLdb (release 45) (Hu et al., 2013). Only QTLs with P-value < 0.05 and size <1 Mb were considered. Molecular functions, biological processes, and metabolic pathways associated with the candidate genes were analysed using the Panther classification system v.14.0 (Huaiyu et al., 2019).
An overview of our framework for ENM-based delineation of livestock ecotypes and their use for dissecting genomic-environmental adaptation is presented in Figure 1 with summary results from its application on Ethiopian chickens.
Figure 1. A framework for defining livestock ecotypes based on ENM approach and exploring genomic adaptation to the environment.
Different classifications of Ethiopian AEZs are available. Since altitudinal topography constitutes a major feature of the Ethiopian landscape, traditional classifications were driven by this and 4–6 different AEZs were defined, e.g., Berha (lowlands, <500 meters above sea level, m.a.s.l.), Kolla (lowlands, 500–1,500 m.a.s.l.), Weyna Dega (midlands, 1,500–2,300 m.a.s.l.), Dega (highlands, 2,300–3,200 m.a.s.l.), Wurch (highlands, 3,200–3,700 m.a.s.l.), and High Wurch (highlands, >3,700 m.a.s.l.) (Hurni, 1998). Many other classifications have combined rainfall and crop pattern information with these traditional elevation-based zonation for the purpose of crop management, but their denominators were often relative and lacked precision (Hurni, 1998). The FAO/IIASA-defined “Global AEZ 16-class” classification (HarvestChoice/IFPRI, 2009) has also been applied on Ethiopia and identifies eight different zones (Figure 2A). While this classification considers temperature, elevation, and rainfall, still it suffers from lack of precision. For example, it provides only two broad classifications based on temperature – either cool or warm.
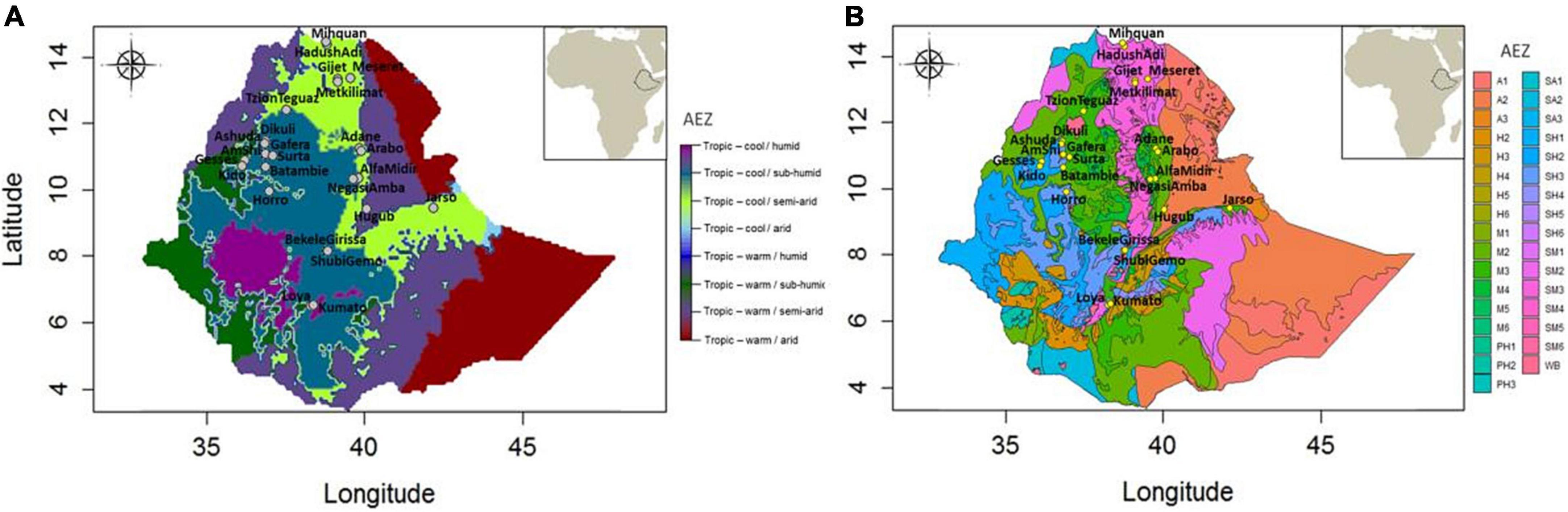
Figure 2. Ethiopian agro-ecological zones and sampling locations. (A) Population locations against Global 16-Class AEZ classification (courtesy of Gheyas et al., 2021), and (B) population locations against the elaborate agro-ecological zonation by MoARD. A1, hot arid lowland plains; A2, warm arid lowland plains; A3, tepid arid mid highlands; SA1, hot semi-arid lowlands; SA2, warm semi-arid lowlands; SA3, tepid semi-arid mid highlands; SM1, hot sub-moist lowlands; SM2, warm sub-moist lowlands; SM3, tepid sub-moist mid highlands; SM4, cool sub-moist mid highlands; SM5, cold sub-moist mid highlands; SM6, very cold sub-moist mid highlands; M1, hot moist lowlands; M2, warm moist lowlands; M3, tepid moist mid highlands; M4, cool moist mid highlands; M5, cold moist sub-afro-alpine to afro-alpine; M6, very cold moist sub-afro-alpine to afro-alpine; SH1, hot sub-humid lowlands; SH2, warm sub-humid lowlands; SH3, tepid sub-humid mid highlands; SH4, cool sub-humid mid highlands; SH5, cold sub-humid sub-afro-alpine to afro-alpine; SH6, very cold sub-humid sub-afro alpine to afro-alpine; H2, warm humid lowlands; H3, tepid humid mid highlands; H4, cool humid mid highlands; H5, cold humid sub-afro-alpine to afro-alpine; H6, very cold humid sub-afro-alpine; PH1, hot per-humid lowlands; PH2, warm per-humid lowlands; PH3, tepid per-humid mid highland; WB, waterbody.
The most elaborate zoning is from Ethiopia’s Ministry of Agriculture and Rural Development (Ministry of Agriculture and Rural Development [MoARD], 2005) with the identification of a staggering 32 different AEZs2 (Figure 2B), taking into account climate, physiography, soil properties, vegetation, and crop patterns. Although this provides a high-resolution classification, the system is developed mainly for agricultural crop management and therefore may not be directly relevant for livestock species.
The MaxEnt based ecological modelling (ENM) for characterising livestock agro-ecologies offers a number of advantages over these established classifications. ENM provides the opportunity to incorporate species-relevant environmental variables and allows examination of a large number of variables. In our study, we initially examined 34 different variables (Figure 3A). Elevation and climatic parameters representing temperature, rainfall patterns, and atmospheric humidity were included, as these may challenge the physiological tolerance of chickens. Soil property variables were included as these could affect food availability for scavenging birds. Vegetation and land use variables were incorporated due to their potential effects on the type of food chickens have access to, as well as their role in providing shelter from predators and inclement weather. Figure 3A shows the relative contribution of these variables in explaining Ethiopian chicken agro-ecologies and marks the most important uncorrelated variables selected by MaxEntVariableSelection for final modelling. ENM’s ability to examine individual variable contribution is extremely important for gaining insight into which variables may play key roles in driving adaptation. Shortlisting of the variables on the other hand is important to avoid “overfitting” of the models and to remove collinearity from the model predictors that can otherwise increase model uncertainty and reduce efficiency (De Marco Júnior and Corrêa Nóbrega, 2018).
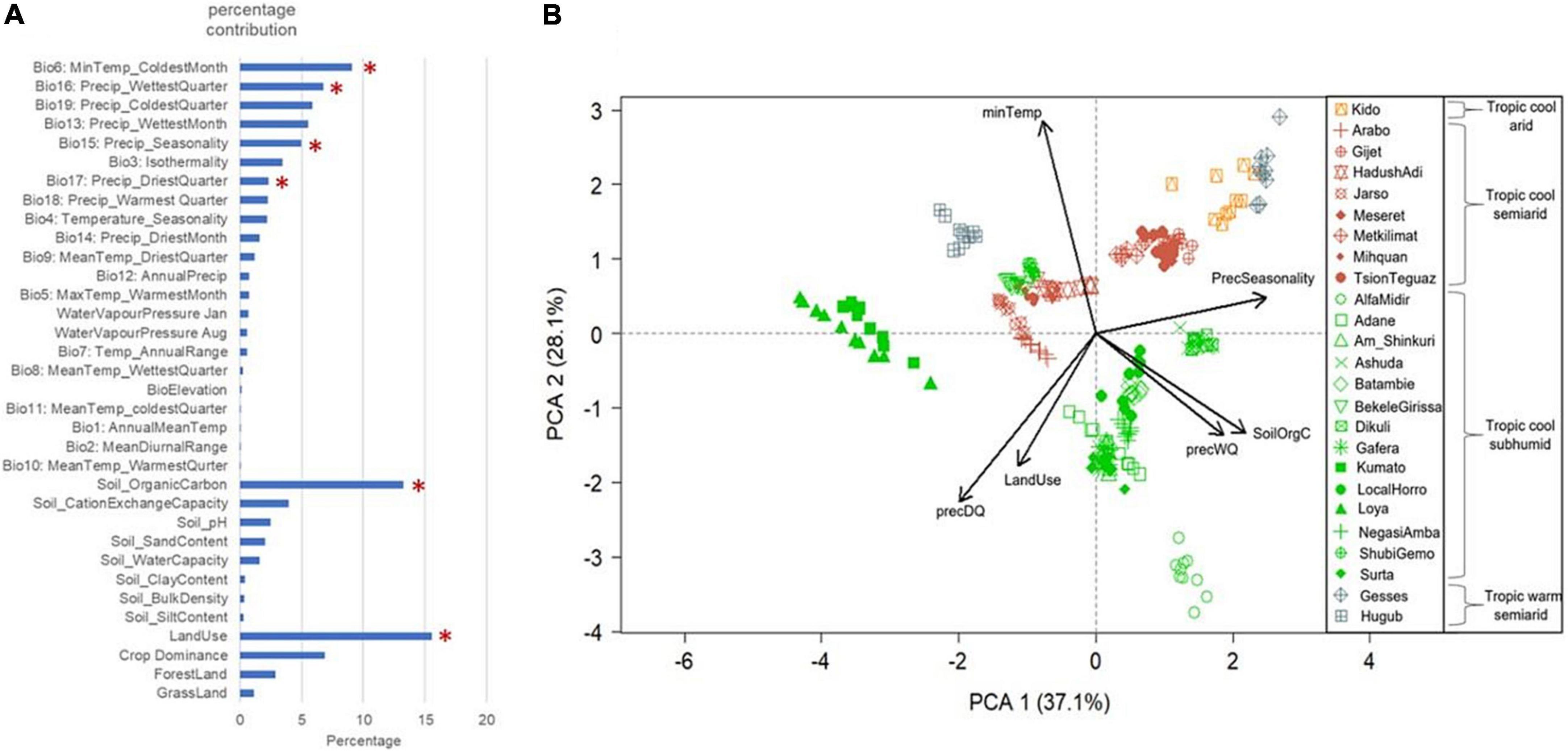
Figure 3. Environmental variables used in ENM for characterising Ethiopian chicken agro-ecologies. (A) Relative contribution of the 34 agro-climatic variables examined in Ethiopian chicken agro-ecologies. Asterisks mark the variables that were shortlisted for final modelling. (B) PCA plot of populations based on six shortlisted variables. The colour coding shows the population classification based on Global AEZ-16. Arrow direction shows the contributing variables in spreading the samples and arrow length indicates relative contribution.
Figure 3B compares the partitioning of the populations based on the six key environmental factors selected by ENM to FAO/IIASA AEZ classification. Some populations clustered together in the PCA plot are from different FAO/IIASA AEZs (e.g., Kido from the cool-arid zone and Gesses from the warm-semiarid zones or Bekele Girissa and Shubi Gemo from the cool-subhumid and Jarso, Hadush Adi and Mihquan from the cool-semiarid zones). Others from the same AEZ are far apart in the PCA plot. For example, 14 of the 25 populations belonging to the broad cool-subhumid AEZ are split into 3–4 major clusters (e.g., Loya, Alfa Midir, and Ashuda, with different environmental parameters playing key roles in their habitat structures). This signifies the importance of ENM in characterising the agro-ecologies and identifying the key environmental drivers in population habitats.
As described in section “Materials and methods,” MaxEnt models were executed first on all populations combined and then on individual populations using the six selected variables and the best parameter settings. The predictive power of all the models, as assessed by AUC (area under ROC curve) values, were high (>0.99) for both training and test data. Similarly, the AUC values for the individual environmental variables for each population were generally higher than 0.6, with several exceptions, like bio6 (minTemp) and bio15 (precSeasonality), which in some populations had values less than 0.6 but greater than 0.5, which still represent moderate potential in predicting suitable conditions (Supplementary Table 2).
The combined analysis of populations allowed assessment of the effect of individual variables on the predictive power of the model (Figure 4). The jackknife result of test and training gains is an important metric for assessing variable contribution and model performance (Figures 4A,B). Jackknife results indicated that, in our model, bio6 (minTemp), bio16 (precWQ), LandUse, and soilOrgC were the variables that had the most useful information when used in isolation of other predictors. On the contrary, bio15 (precSeasonality) decreased the gain most when excluded from the analysis, indicating that this variable contained the most information that was not present in any other predictors.
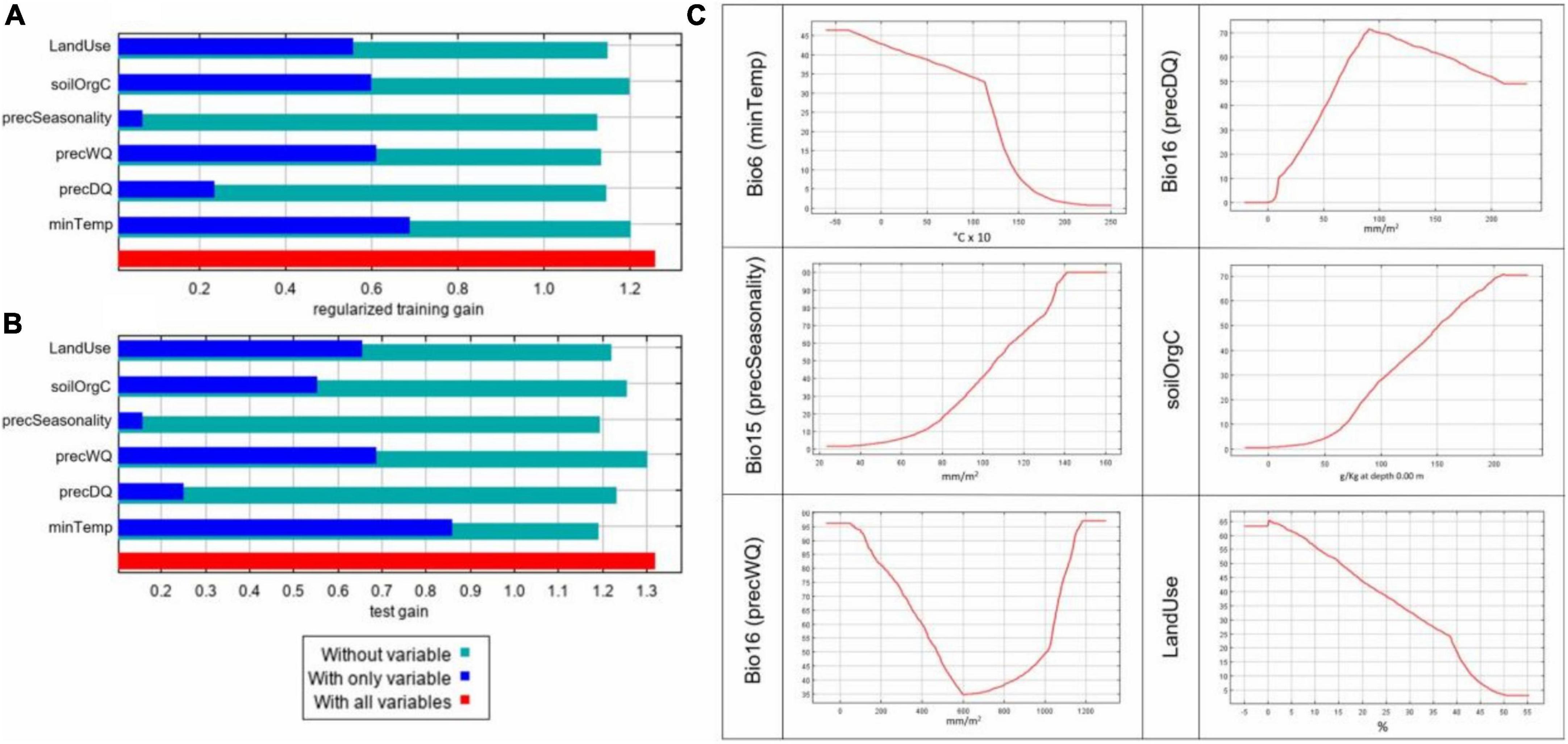
Figure 4. Predictive powers of individual environmental variables in ENM. Results from the combined model based on all 25 populations using 6 selected environmental predictors showing (A) Jacknife of training gains for models with or without a variable, (B) Jacknife of test gain for models with or without a variable, and (C) marginal response curves for individual variables. The X-axis in the response plots shows the environmental values and Y-axis shows the logistic output for probability of presence.
We also inspected the marginal response curves (Figure 4C) from the overall model to examine how the predicted suitability varies as each environmental variable changes while keeping all the other variables at their average sample value. The curves for minTemp and LandUse show that the probability of suitability decreases with rise in these parameters. On the other hand, when precSeasonality and soilOrgC increase, the likelihood of chicken presence increases. For precWQ, the response curve drops sharply with increase in the variable’s value until 600 mm/m2 but then rises sharply again, indicating its potential interaction with other environmental variables. In contrast, the likelihood of chicken presence increases when precipitation in precDQ rises to 90 mm/m2 and drops slowly until 200 mm/m2.
In the combined population model, the contributions of the six variables ranged between 10% for LandUse and 24% for SoilOrgC. The remaining 66% of contributions came from climatic variables: minTemp (21%), precWQ and precSeaonality (both 16%), and precDQ (13%). The relative contributions of the variables in different populations, however, varied widely as shown in Supplementary Figure 1 and not all variables contributed to the characterisation of every population agro-ecology. Supplementary Figure 2 shows the environmental suitability maps for individual populations across the Ethiopian landscape.
Pearson correlation (r) and Niche Overlap (I) statistics were generated by pairwise comparison of the suitability maps for clustering the populations with similar niche for defining ecotypes (Supplementary Tables 3a,b). These two metrics provide complementary information. The Niche Overlap metric is a measure of similarity of occurrences between two populations. In contrast, correlation statistic compares the underlying models (Warren and Seifert, 2011). The correlation metric is generated by comparing all corresponding grid cells between two maps. On the other hand, the Niche Overlap statistic only checks for the proportion of overlap of grid cells that were predicted as suitable (i.e., with logistic suitability score = 1) between two maps. In our study, the results of these two metrics were significantly correlated (Spearman correlation coefficient = 0.6; P < 0.0001).
Among the different hierarchical clustering approaches evaluated, the Ward method was selected as it had the largest agglomerative coefficient (0.746). Clustering of the populations based on the above two similarity metrics showed different overall topography (Figures 5A,B). For delineating ecotypes, we considered the clustering based on one or both approaches as a guide but the definitive criterion was that all pairwise comparisons within an ecotype showed niche overlap (I) ≥ 0.6 and correlation co-efficient (r) ≥ 0.6.
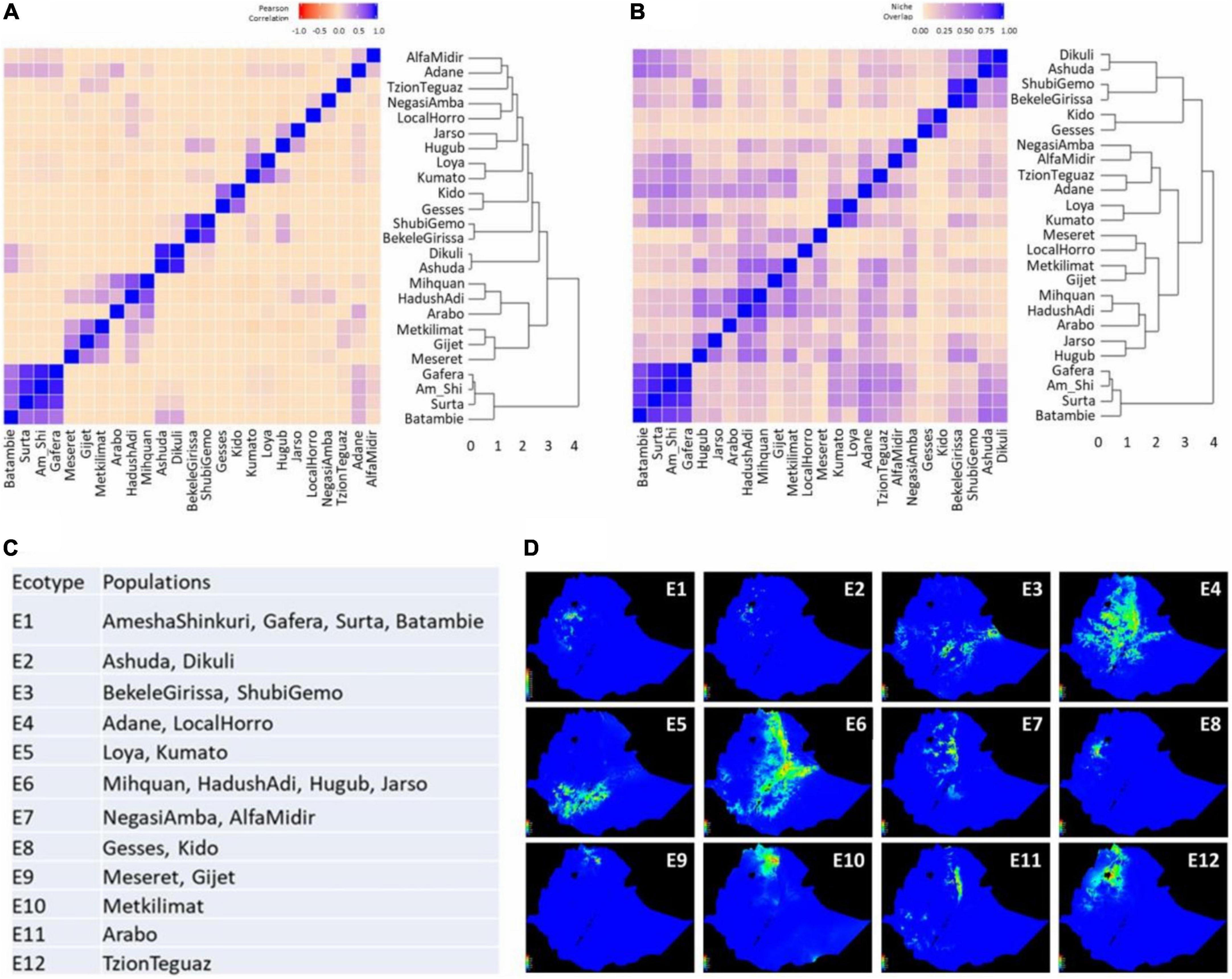
Figure 5. Delineation of ecotypes based on niche similarity among 25 Ethiopian chicken populations examined. (A) Dendrogram and heatmap of pairwise Pearson correlation (I) among 25 populations; (B) dendrogram and heatmap of pairwise niche overlap statistic (r) among 25 populations; (C) 12 delineated ecotypes and their population composition; and (D) suitability maps of 12 delineated ecotypes.
Both approaches grouped identically 16 out of the 25 populations. Consistent clusters were: (1) Amesha Shinkuri, Gafera, Surta, and Batambie; (2) Ashuda and Dikuli; (3) Bekele Girissa and Shubi Gemo; (4) Gesses and Kido; (5) Kumato and Loya; (6) Hugub and Jarso; and (7) Hadush Adi and Mihquan. The first five clusters were used to define individual ecotypes whereas the last two clusters were merged into a single ecotype, as both niche similarity matrics fulfilled the criterion of ≥0.6 for all population pairs after merging.
For the remaining nine populations, an examination of (r) and (I) values was performed to resolve their ecotype membership. Only three populations – Arabo, Metkilmat, and TzionTeguaz – could not be grouped with any other populations and hence were considered as separate ecotypes. Thereby, 12 ecotypes were identified for the 25 populations studied (Figures 5C,D), reflecting the large heterogeneity of the Ethiopian agro-ecological landscape. Notably, even though many populations belong to the same broad AEZ, multiple ecotypes were delineated from them based on ENM, e.g., the cool/sub-humid zone overlaps ecotypes E1–5 and E7 (Supplementary Table 1). Contrarily, populations in E6 originate from two broad AEZs – cool/semi-arid and warm/semi-arid. Although in most cases, geographically close populations were clustered into ecotypes (E1–E3, E5, E7–E9), that did not hold true in all cases. For instance, the E4 populations – Adane and Horro – are far apart geographically (∼300 km). Similarly in E6, Mihquan and Hadush Adi populations are geographically close (∼40 km) but are far from Hugub and Jarso (∼570–660 km). In contrast, Adane and Arabo are geographically close (∼10 km) but were placed in separate ecotypes. These results demonstrate the large heterogeneity in environmental conditions in Ethiopia even within a short geographic distance, and thereby shows the importance of ENM-based characterisation of agro-ecologies. The relative contributions of the 6 environmental variables in the 12 ecotypes are presented in Supplementary Figure 3.
About 14M autosomal SNPs detected in Gheyas et al. (2021) were used for selection signature analyses in the present study. Genetic variant data from all populations constituting each ENM-defined ecotype were combined, resulting in 11M to 13M SNPs per ecotype. Selection signature analyses were then performed within each ecotype using Hp and iHS approaches.
The genome-wide mean values of Hp were similar across all the ecotypes, ranging from 0.29 ± 0.06 for E7 and 0.33 ± 0.05 for E10 (Table 1). However, the number of Hp candidate windows varied widely from only 7 for E7 to 279 for E10. Adjacent or overlapping candidate windows were merged to define the SRs. This resulted in 2 to 77 SRs per ecotype and a total of 365 regions from all ecotypes combined (Table 1 and Supplementary Table 4).
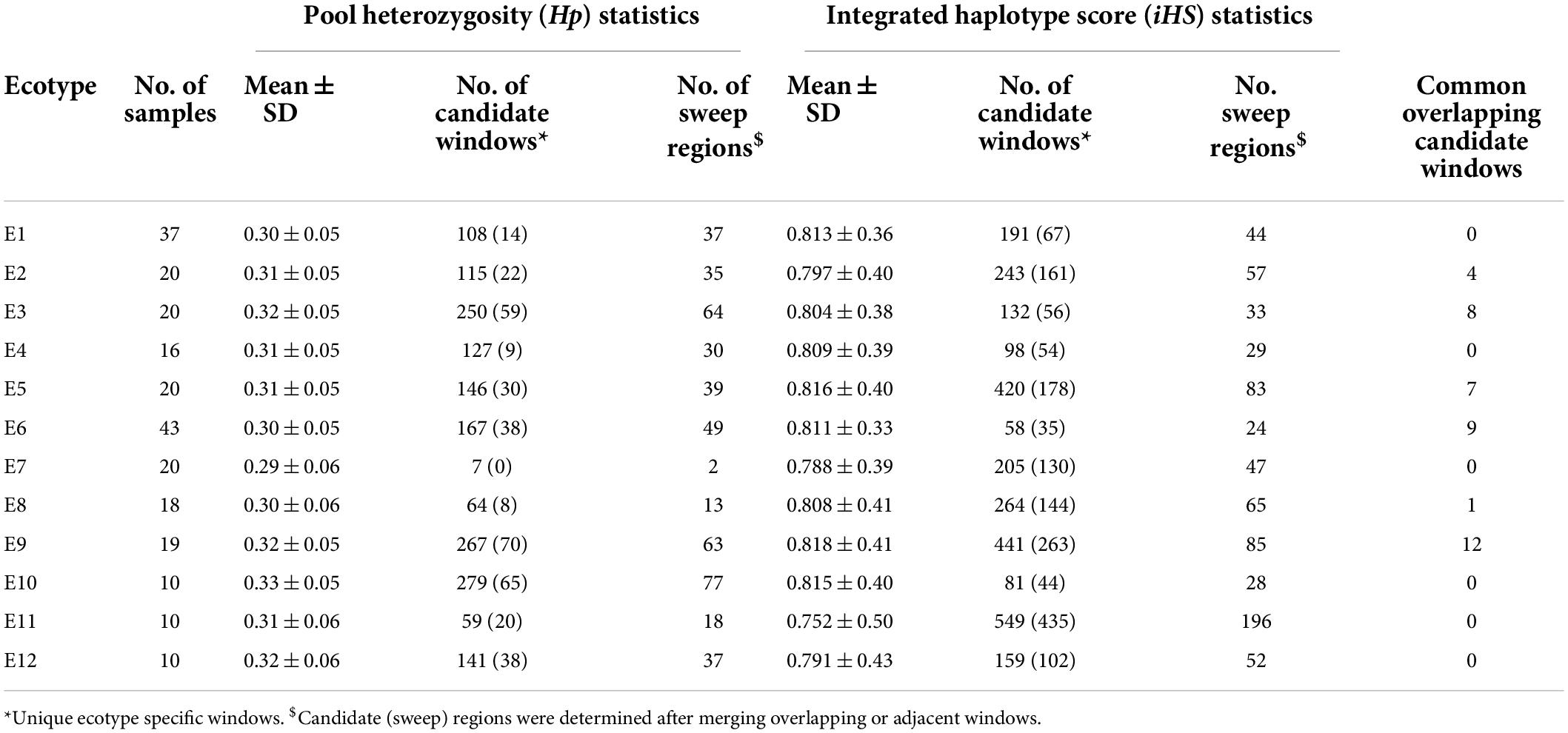
Table 1. Genome-wide pool heterozygosity (Hp) and integrated haplotype score (iHS) for the 12 proposed chicken ecotypes in Ethiopia.
The genome-wide mean values of iHS were also similar among the ecotypes, ranging from 0.75 ± 0.5 (E11) to 0.81 ± 0.4 (E9) but again the number of candidate sweep windows showed large variation, ranging from 58 (E6) to 549 (E11) (Table 1). Interestingly, although E10 produced the largest number of candidate signals with Hp analysis, this ecotype produced one of the lowest number of candidate windows (only 81) with iHS analysis. Merging the overlapping candidate iHS windows resulted in 24–196 SRs in different Ecotypes (Table 1). A complete list of unique SRs (n = 1,056) from both methods can be found in Supplementary Table 4 along with overlapping genes. The SRs vary in size from 20 to 360 kb and together cover roughly 4% of the chicken genome.
We found a weak negative correlation between Hp and iHS (−0.043 ± 0.06, P-value = 0.00), and ZHp and iHS_std (−0.18 ± 0.06, P-value = 0.00) values (Supplementary Figures 4a,b). This shows a complementarity of the methods, which is expected as Hp identifies regions that are fixed or near fixation (Rubin et al., 2010), whereas iHS detects ongoing selection (Voight et al., 2006). Despite this, we found a few SRs (n = 17) which were commonly detected by both methods either within the same ecotype (n = 8) or from different ecotypes (Supplementary Table 5 and Supplementary Figure 5). Regions commonly detected by both methods within the same ecotype would indicate it is close to reaching fixation but not yet fixed. In contrast, the same SR detected by Hp in one ecotype and iHS in another, may pinpoint different levels of selection in different ecotypes.
The percentage of shared sweep windows among ecotypes varied from 1 to 13% (Supplementary Figure 6). E11 generally showed the lowest level of shared sweeps (1–2%) with any other ecotypes whereas E3 generally shared the largest proportion with many other ecotypes (e.g., it shared 10–13% of its sweep windows with five other ecotypes).
We also investigated the shared SR genes among ecotypes. About 22% (n = 125) of the genes overlapping Hp-based SRs were shared by at least two ecotypes (Supplementary Table 6a). The TSHR (Thyroid Stimulating Hormone Receptor) gene was detected ubiquitously in all ecotypes. This gene has previously been detected as a selection signal (Rubin et al., 2007; Gheyas et al., 2015), and therefore represents an old sweep possibly associated with chicken domestication (Rubin et al., 2007; Karlsson et al., 2016). Several other Hp candidate genes were detected in the majority of the ecotypes, e.g., ENSGALG00000047413 (11 ecotypes) and ENSGALG00000052351 (10 ecotypes) – both LncRNAs with possible cis-acting regulation on nearby genes (e.g., TSHR and FUT8, respectively), TSNARE1 (9 ecotypes) and SCN2A (8 ecotypes) – both neurobehavioral genes, and CACNA2D3 (7 ecotypes) with voltage-gated calcium channel activity regulating ion transmembrane transport. In comparison to the Hp analysis, only 10% (122 of 1,180) of the iHS-genes were shared by 2 to 5 ecotypes at most (Supplementary Table 6b).
Genes overlapping the SRs from different ecotypes were checked for their functional classification according to Panther Pathways (Supplementary Figure 7). Although only 1,253 genes (∼5% of all chicken genes) intersected SRs from different ecotypes, they showed a hit for 55% (98 of 177) of the Panther pathways, indicating their involvement in a vast array of physiological processes. About 77% (75 of 98) of these pathways are represented by SR genes from multiple ecotypes but the number of genes contributed by the ecotypes varied. A few pathways are represented by genes from all or most ecotypes. For instance, Alzheimer disease-amyloid secretase pathway (P00003), and Heterotrimeric G-protein signalling pathways (P00026 and P00027) had hits from all 12 ecotypes with representation by 1–6 genes per ecotype. Similarly, Integrin signalling pathway (P00034) and PDGF signalling pathway (P00047) have hits from 10 different ecotypes. Investigation of the individual genes from these ubiquitous pathways shows involvement in a myriad of biological processes, but prominently in processes which are expected to be necessary for the survival of scavenging chickens, irrespective of ecotypes; for instance, roles in nervous system development, neurological processes and/or cognitive functions (APBA2, MAPK6, MAPK13, MAPK14, PCSK2, CACNA1C, BACE2, PKN2, GPSM1, MTNRIB, DRD3, HRH1, GRM5, GRB2, CRKL, MAP2K2, MICALL1, NTN4, RND2, FOS, MTOR, PRKAR2B, and DLC1); eye development, visual perception, and visual learning (MTNRIB, GRK7, DRD3, HRH1, and COL5A1) – important for foraging for food; immune response, inflammatory response, and wound healing (MAPK14, RASGRP1, GRB2, HRH1, CREB3L3, CHUK, CRKL, FOS, and MTOR); and apoptotic process (MAPK14, PKN2, DRD3, ACTN1, VAV2, and DLC1) – an important stress response (Kawamata et al., 1998; Chen et al., 2017; Yan, 2017; Huentelman et al., 2019; Asih et al., 2020; Genecards Database, 2021; Uniprot Consortium, 2021). Other notable involvements include regulation of cardiovascular and renal development and functions (CACNA1C, VAV2, MAPK14, DLC1, PCSK2, COL5A1, CRKL, MTOR, PRKAR2B, DRD3, NTN4, RND2, and ADCY2) (Uniprot Consortium, 2021).
About 12% (n = 128) of the SRs overlapped with known chickens QTLs (Figure 6 and Supplementary Table 7), indicating potential association with different phenotypic traits, e.g., reproductive efficiency (n = 28 hits), egg quality (n = 21), growth (n = 20), feather pecking behaviour (n = 14), feed conversion efficiency or FCR (n = 12), immunity and health (n = 11), body fat (n = 10), body temperature (n = 4), feed intake (n = 4), and feather pigmentation (n = 4).
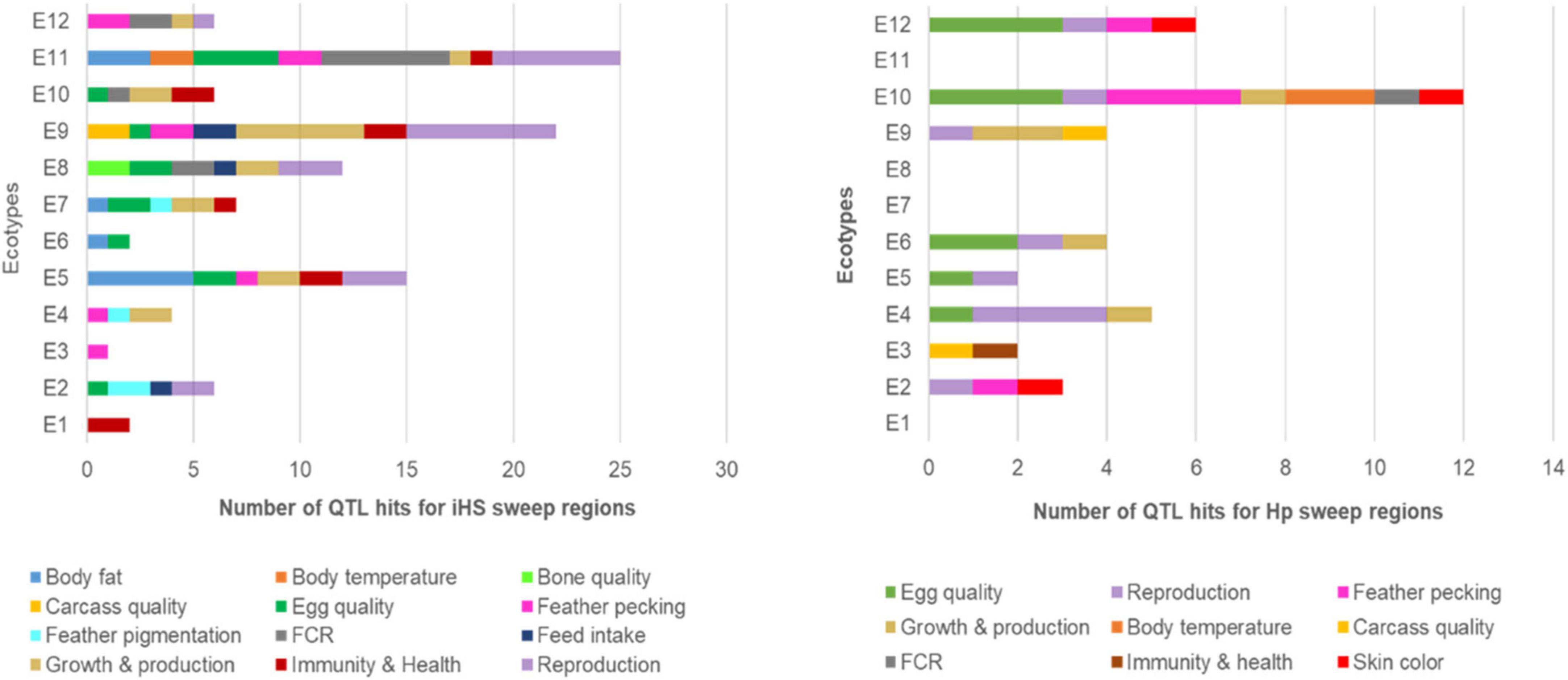
Figure 6. Overlap of the sweep regions from Hp and iHS analyses with chicken QTLs.
Finally we closely inspected the strongest signalling SR from each ecotype to see if a functional relevance can be drawn between the overlapping genes and the ecotype’s environment (Supplementary Tables 8a,b). Strikingly, in most cases a strong relevance is observed through the involvement of the genes in various stress responses, neuronal processes, immune responses and transcriptional regulations. The environmental association of the gene functions is more prominent for ecotypes where only a few environmental variables act as major driving forces. E7 perhaps offers the most striking example where the minTemp contributes ∼99% in characterising the ecotype’s agro-ecology. This ecotype has the lowest temperature environment (as low as near 0°C) due to its very high-altitude location (>3,000 m.a.s.l.). The strongest iHS-sweep from this ecotype overlaps several genes: TEAD3 – involved in organ size control (and therefore may have a crucial role in determining the size of heart and lung for maximising oxygen utility at high altitude); TULP1 – has photoreceptor function (possibly important for adaptation to intense light and UV radiation stress at high altitude); and FKBP5 – an important modulator of stress response (including acute stress) due to its crucial role in the regulation of the hypothalamic-pituitary-adrenal (HPA) axis. Similarly, in E2, with minTemp contribution of 76%, the strongest iHS-sweep overlaps the DDIT3 gene which is a multifunctional transcription factor in endoplasmic reticulum and plays an essential role in the response to a wide variety of cell stresses.
We also find genes AGO4 and CLSPN from E9 and SCARNA7 and TRIM59 from E10 iHS-SRs; both ecotypes with a major environmental contribution from precSeasonality (84 and 80%, respectively). These ecotypes show major variation in rainfall between dry (∼10 mm/m2) and wet seasons (>400 mm/m2). AGO4 is involved in the gene silencing pathway and has been found upregulated during drought conditions in plants, CLSNP is involved in DNA repair, SCARNA7 has role in RNA methylation (and thereby has modulatory effect on expression of genes) and TRIM59 is involved in innate immunity. In ecotypes where multiple environmental variables have large contributions, we find genes involved predominantly in nervous system development/processes and transcriptional regulation, possibly to facilitate a concerted regulation of a wide variety of physiological functions.
While the above examples show genes from iHS analysis, the strongest Hp-based SRs were often detected from many ecotypes, possibly representing old sweeps. For instance, the strongest signals from E1–E3, E5–E7, and E10 overlapped with either the TSHR gene or its linked gene, GTF2A1. For some other ecotypes, we find genes involved in neurological processes (e.g., EFHC2 in E8 and BEGAIN in E9) and immune system development (CACNA2D3 in E12). The strongest Hp-signal from E4 overlaps with several genes: GRK7 – with a role in visual perception, RNF7 – involved in protein ubiquitination pathway and response to redox state, and ATPIB3 which serves as an ion pump across the plasma membrane that is essential for transepithelial transport including nutrient uptake. These genes appear highly relevant for adaptation to the major environmental drivers of the E4 ecotype – LandUse (49%) and SoilOrgC (43%), with both serving as proxies of food availability for scavenging chickens which require the ability to find food and assimilate available nutrients (Gheyas et al., 2021).
As a multivariate linear regression approach, RDA allows simultaneous interrogation of many response variables (SNP genotypes) with many predictors (environmental variables) (Capblancq et al., 2018). RDA was performed using 44,716 LD-pruned SNPs (∼9% of the total number of SNPs) overlapping the SRs.
The RDA model showed highly significant (P-value = 0.001) deviation from the null hypothesis (no linear relationship between the SNP data and the environmental predictor). The model explained 1.9% of the genetic variance indicating that only a small proportion of the SR-SNPs have an association with environmental parameters. Each of the six RDA axes explained 10–30% of the variance captured by the model (Figure 7A). Only the first five axes were significant (P-value ≤ 0.01), and these together accounted for 90% of the total captured variance. The SNP loadings (Figure 7B) from these 5 axes were used to determine outliers, i.e., SNPs showing significant association with environment.
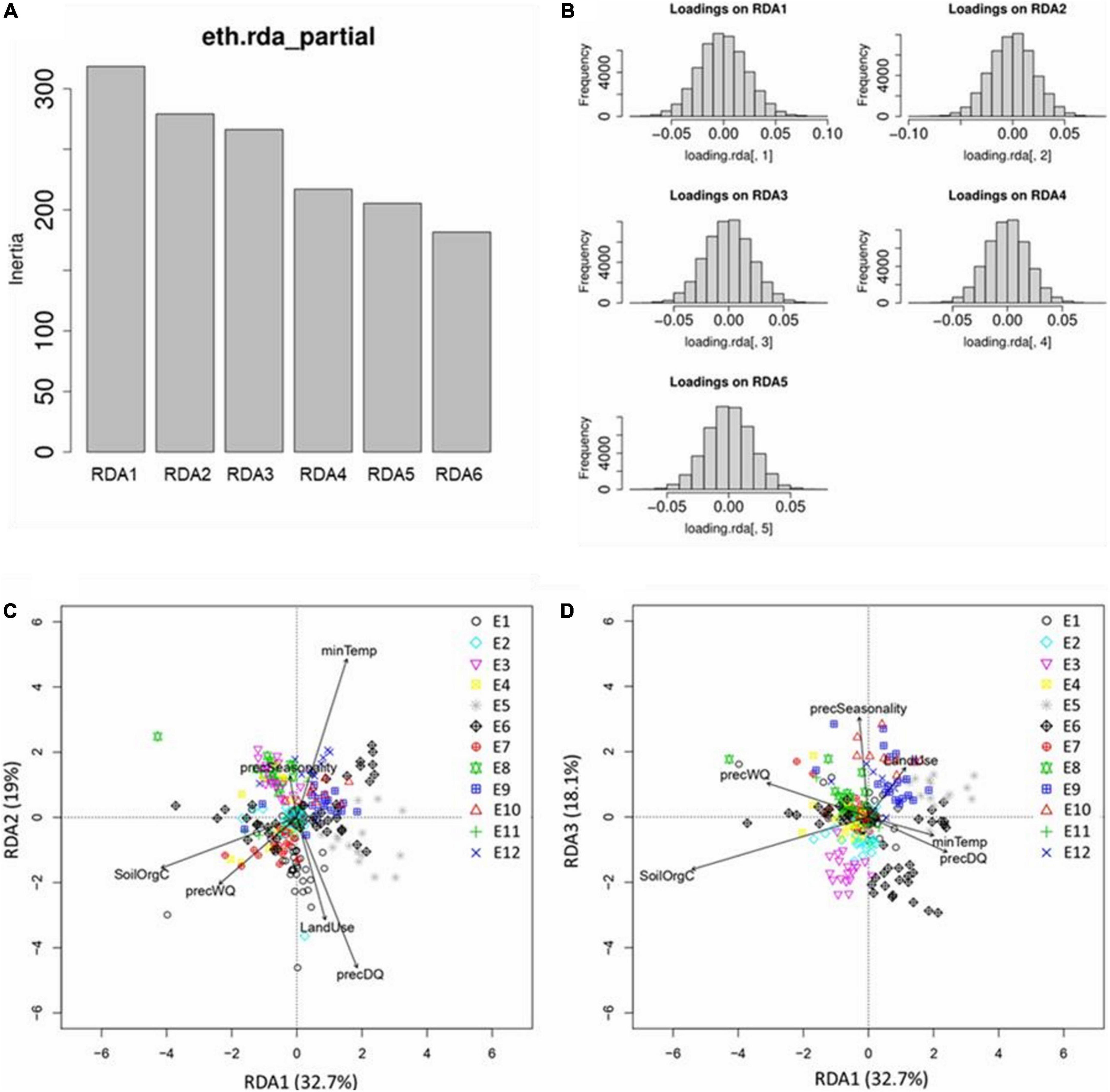
Figure 7. Results from RDA analysis. (A) Projection of SNPs and environmental variables into the RDA space RDA1–RDA2 and (B) RDA1–RDA3; (C) variance (inertia) explained by the six RDA axes; and (D) distribution of SNP loadings for significantly constrained axes (RDA1–RDA5).
Redundancy analysis plots show the distribution of the chicken samples from different ecotypes in relation to the RDA axes which are a linear combination of the predictor variables (Figures 7C,D). Some interesting relationships can be identified. For example, Figure 7C (axis1 vs. axis2) shows that E1 genotypes are positively related to precDQ and LandUse, whereas E3 and E8 are negatively correlated with these two variables. Similarly, E12 genotypes are positively correlated with minTemp and negatively correlated with precWQ, whereas an opposite scenario for E7 is observed. By contrast, E9 individuals are positively associated with LandUse in Figure 7D (axis1 vs. axis3).
Interestingly, individuals from E6 are dispersed in subgroups in both RDA plots. E6 comprises four different populations (Mihquan, HadushAdi, Hugub, and Jarso), and this is indeed one of the few ecotypes where the clustering based on niche overlap and Pearson correlation gave ambiguous results. These populations were included in the same ecotypes as all pairs showed values ≥0.6 for both the similarity metrics. For other ecotypes, no sub-clustering is evident when RDA plots are explored, confirming the overall robustness of our ecotype delineation. Regarding the E6, however, we need to keep in mind the limitation of RDA, which can capture only linear association between genotype and environment. Therefore, in showing the distribution of the samples in the ordination space, RDA did not consider any non-linear association due to G × E interaction. ENM, on the other hand, is blind to any specific genotype-environment association; instead its characterisation is entirely dependent on the environmental conditions of the populations with the assumption that similar environmental pressure will lead to similar selective pressure on the genome. ENM-based ecotype characterisation is therefore expected to capture selective pressure from all sources (linear and non-linear).
Given a normal distribution of the SNP loadings in all axes (Figure 7B), SNPs that exceeded SD >3 (P-value = 0.0027) in both tails were extracted as outliers. With this threshold, 616 SNPs (1.4% of total 44,716 SNPs tested) were found as outliers (Supplementary Table 9). The number of outliers varied in relation to the strongest correlated environmental variable, from 72 for precSeasonality to 130 for SoilOrgC. The strength of the environmental correlation of the outlier SNPs was generally low to moderate (r ≈ 0.1–0.4) (Supplementary Figure 8). The maximum correlation value was 0.42, identified for precWQ.
To gain an understanding of the biological functions of the genes associated with the outlier SNPs in relation to the correlated variables, a closer investigation was made on 30 RDA-outliers – which showed relatively large environmental correlation coefficients (r ≥ 0.3 for most predictors and ≥0.29 and ≥0.28 for PrecDQ and LandUse respectively as no outliers for these passed the first threshold) (Table 2). Notably, all these SNPs came from iHS-detected SRs. Highly relevant gene function or phenotypic associations are observed in most cases. For example, PANK2 gene was detected in association with minTemp (or elevation). This gene is involved in neuronal and vascular development and respiration – important functions for thermo-tolerance and stresses at high-altitude. Two genes, LDLRAD3 and GPX7, detected in association with precipitation variables (precSeasonality and precDQ), were previously reported in association with heat stress in chicken (Del Vesco et al., 2015; Wang et al., 2020). These results are in agreement as the ability to cope with heat stress relies not only on the water availability but also on the environmental humidity – both factors being affected by rainfall patterns. GPX7 has also been found associated with drought resistance in plants (Cruz de Carvalho, 2008). LDLRAD3, in our study, has also been detected in association with SoilOrgC – a proxy of scavenging conditions and food availability for chickens. Interestingly this gene has been found associated with lifespan expansion in a previous mouse study (Tyshkovskiy et al., 2019).
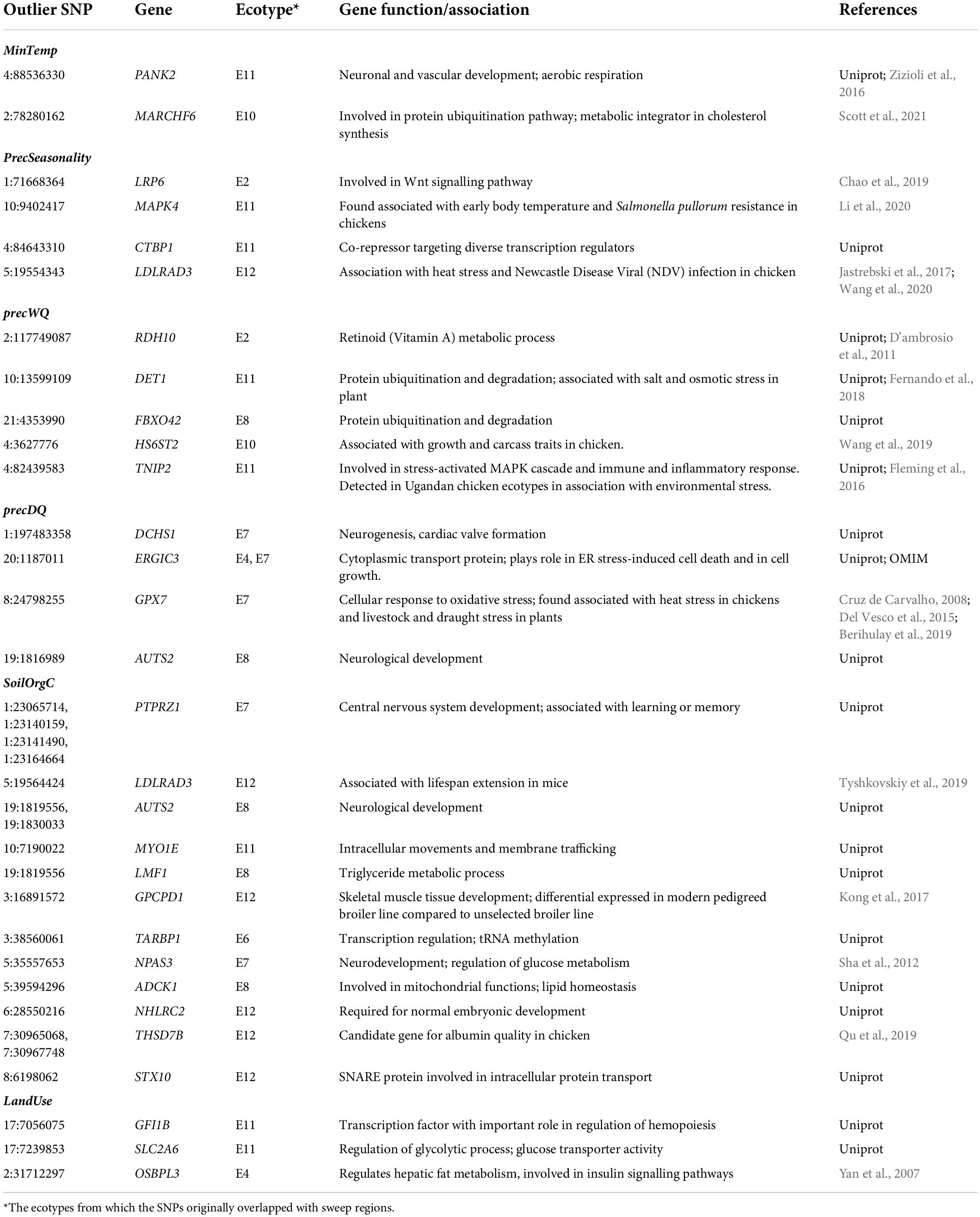
Table 2. Selected RDA SNPs outliers overlapping protein-coding genes.
In association with precipitation variables we also find several genes which are candidates for disease response, e.g., MAPK4 – a candidate for Salmonella resistance in chicken, LDLRAD3 – associated with Newcastle Disease Virus infection in chicken, and TNIP2 – involved in immunity and inflammatory responses (Table 2). Since the rainfall pattern affects the prevalence of various pathogens, these detections appear very relevant. Furthermore, some genes detected in association with climatic factors (i.e., temperature and rainfall variables) are involved in broad regulatory and stress response pathways such as MARCHF6 and DET1 – protein ubiquitination and degradation pathways, LRP6 – Wnt signalling pathway, ERGIC3 – Endoplasmic Reticulum stress induced cell death and cell growth process, CTBP1 – co-repression of diverse transcription regulators, and RDH10 – Retinoid (Vitamin A) metabolic process with implications in many physiological processes including growth, development, immune system, and reproduction. On the contrary, the genes detected in association with SoilOrgC and LandUse (both potentially affect the nature and availability of food for foraging chickens as well as their foraging ability) show involvement in fat and carbohydrate metabolic processes (LMF1, ADCK1, SLC2A6, and OSBPL3), growth, development and reproductive success (GPCPD1, NHLRC2, and THSD7B), and neurological development (PTPRZ1, AUTS2, and NPAS3).
The 616 RDA outliers intersected 349 SRs (33%), thereby providing indication of the major environmental variables exerting selection pressure on these specific SRs. The overlap of only a minority of SRs with the RDA-outliers can be explained by the fact that RDA identified only linear genotype-environment association across all samples. Any SR resulting from a complex G × E interaction – thereby showing non-linear association – will not be detected by RDA. Outlier SNPs associated with all six environmental variables overlapped SRs from most ecotypes, confirming the combined contribution of different variables in each ecotype and thereby justifying the use of ENM for ecotype delineation and adaptation analysis. Also, some of the SRs overlap with several outliers associated with different agro-ecological variables. For instance, the SR on chr1:132930000_132960000 overlaps with RDA-outliers associated with SoilOrgC, minTemp and precDQ. This indicates that we are detecting the interaction of environmental variables that are collectively driving selection pressure on specific genomic regions. Such observation again highlights the importance and utility of using ENM-defined ecotypes for adaptation analysis.
The RDA analysis was performed to directly correlate SNP genotypes from individual samples with environmental parameters, without considering the ecotype effect. We wanted to further investigate how the ecotype allele frequencies of the outlier SNPs fluctuate with the ecotype average of the correlated environmental variables. The aim of this investigation was to gain an insight into whether the allele frequency shows a linear or non-linear trend with environmental variation. The investigation was made on the 30 strongest RDA-outliers (5 per environmental variable) by fitting linear and non-linear trend lines in scatter plots of allele frequency against ecotype average of environmental variables (Supplementary Table 10). Figure 8 shows scatter plots of five example SNPs (one for each variable). These plots as well as Supplementary Table 10 show that, in all cases, non-linear regression generated a larger R2 value, i.e., better fit to the data. This result corroborates our assumption that ENM can capture complex G × E interaction in delineating ecotypes.

Figure 8. Allele frequency fluctuation of example RDA outliers with agro-climatic predictors (mean value) across ecotypes. One outlier (with the strongest RDA r-value) per predictor is shown as an example.
By integrating interdisciplinary approaches – ecological modelling with genomics – this study presents a novel framework for the identification and characterisation of indigenous livestock ecotypes showing genetic adaptation to distinct agro-climatic conditions. Exemplified here with Ethiopian village chickens, the framework is fully transferable to any other livestock species, with important implications for conservation of adaptive biodiversity and breeding improvement towards achieving climate resilience. Unlike traditional adaptation studies, our approach directly models landscape heterogeneity based on a large spectrum of environmental variables, thereby providing the opportunity to identify the key environmental pressures in livestock ecologies as well as capture the complex interplay of major variables in driving adaptive evolution at the genome level.
The benefits of our approach are visible at different levels. First, our method offers an opportunity to classify otherwise non-descript indigenous livestock populations into potential ecotypes based on a detailed characterisation of their agro-ecologies. As demonstrated in our results, classical AEZs are often insufficient in classifying livestock populations into ecotypes as those either lack resolution and precision or do not reflect the most relevant environmental predictors for the species in question. Contrarily, our method, based on ENM, offers not only the resolution and species specificity but also flexibility to incorporate any number of environmental variables in the characterisation process.
Another major advantage of our approach is its ability to capture complex interactions of many environmental variables, including the G × E interaction in shaping the livestock genome. This is possible through the use of different feature classes during the execution of ENM; e.g., we used three FCs: Quadratic (variance), Product (covariance, i.e., captures interactions), and Hinge (linear response) (Phillips and Dudík, 2008; Merow et al., 2013). The consequence of this is reflected in selective sweep detection. We find that even though the same variables may have a major contribution in defining multiple ecotypes (e.g., E9 and E10 have precSeasonality as the major driver with >80% contribution), the same candidate sweeps were not detected (only 10% of the candidate sweep windows are shared between these ecotypes), indicating possible interaction of other drivers in shaping adaptive evolution. Predicting G × E interaction is challenging and failure to take this into consideration has resulted in poorer performance of improved breeds in environmental conditions different from their original performance setting (Wakchaure et al., 2016). ENM-based characterisation of the agro-ecologies offers an interesting opportunity to assess the most suitable condition for any breed or population across a landscape. This has recently been tried for predicting suitable agro-ecologies for the introduction of exotic chicken breeds in Ethiopia (Lozano-Jaramillo et al., 2018).
While most of the currently available methods of environmental association analysis (EAA) in landscape genomics – like RDA – capture only linear correlation between genotype and environment (Rellstab et al., 2015), our approach of integrating ENM with selective sweep analysis allowed capture of both linear and non-linear genetic responses to environmental pressure. That non-linear association plays a major contribution in driving adaptive evolution is reflected by the fact that we only detected a low to moderate level of correlation in the RDA analysis and found only one-third of the detected SR to overlap with RDA outliers. Moreover, a basic inspection of the fluctuation of allele frequency of RDA outliers with environmental variables (Figure 8 and Supplementary Table 10) demonstrated a greater power of non-linear regression in explaining variance compared to the linear approach. Modelling non-linear response is not yet well developed (Rellstab et al., 2015). The few available methods allowing non-linear EAA include SAM (Joost et al., 2007) and SAMβADA (Stucki et al., 2017), which apply logistic regression methods. These methods only allow testing association of the presence/absence of an allele with environmental variables where interpretation of heterozygous genotypes becomes difficult. Moreover, unlike RDA, these are univariate analyses, allowing testing of only one genetic marker at a time, which fails to account for covariation among environmental variables and/or genetic markers (Capblancq and Forester, 2021). Therefore, no attempt was made to apply non-linear EAA in the present study.
In respect of genomic analysis, our approach has some added benefits of reducing false discovery rate (FDR) from the confounding effect of demography (e.g., genetic drift, founder effect, etc.). This is because multiple populations have been clustered together into most ecotypes, thereby increasing heterogeneity across the genome, except around the loci under selection pressure. To minimize FDR, we also employed a stringent criteria for identifying sweep signals. Instead of taking only the top 1% windows (empirical P-value < 0.01), as is frequently applied in selection signature analyses, we employed further filtration based on standardised score (or Z score) with the same threshold applied to all ecotypes. This has resulted in a large variations in detected candidate sweeps from different ecotypes, indicating differential selection pressures.
The application of the proposed framework requires some special considerations regarding the demographic history of the livestock populations involved. In its current form it is applicable on indigenous livestock populations which are raised in extensive farming practices and are not under any systematic selection effort. However, unlike wild species, livestock populations like chicken have undergone selection following domestication, which would be reflected in their genomes. Furthermore chicken in Africa is an imported species. Since its introduction in Africa from Asia around a few thousand years ago (Woldekiros and D’Andrea, 2017), Ethiopian indigenous chickens have evolved to adapt to their local agro-climatic conditions, although this timescale may not be enough for complete fixation of the genomic regions under adaptive selection. Moreover, the environmental conditions are also constantly changing due to climate change and human interventions, creating further dynamism in adaptive selection. With these factors considered, we deployed two different approaches (Hp and iHS) for detecting different types of selection signatures. The Hp method was used to detect genomic regions which have reached fixation or near fixation, potentially due to strong selective pressures (either old, e.g., domestication-related or relatively new), whereas the iHS method was applied to identify recent ongoing selections which are yet to reach fixation. As a proof-of-concept our study highlighted the following: first, the TSHR gene, which is considered an old sweep and possibly associated with the chicken domestication process (Rubin et al., 2007; Karlsson et al., 2016), was ubiquitously detected in all the studied ecotypes by the Hp method. Secondly, the major RDA outlier SNPs came from iHS-detected SR, indicating their linear environmental association across all ecotypes. This is congruent with the effect of ongoing selection under changing environment.
Notably, the candidate genes detected in the present study has little overlap with those detected in our previous study (Gheyas et al., 2021) although both used the same set of populations for environmental adaptation analyses. The major reason behind this is that the two studies differed in their focus and scope. In our previous study, the selection signatures were detected by comparing extreme populations for different environmental parameters (e.g., high temperature vs. low temperature), so that the identified candidates can be targeted in breeding programmes in relation to these specific environmental variables either via marker-assisted selection or gene editing. Multiple populations were combined in each High and Low groups to minimize spurious signals from population effect or from other environmental factors. Contrarily, the main focus of the current study is to consider all key environmental drivers together and assess their combined effect on genome, so that ecotypes can be delineated and conserved/managed effectively. The current analysis can, therefore, capture the complex interplay of many environmental factors in modulating adaptive response at genome level and G × E interactions. Contrary to the previous study, which used Fst and XPEHH to capture differential selection, the present study applied Hp and iHS for detecting within-ecotype signatures.
One of the limitations of our study is that we only used chicken populations that were available to us. Although the sampling was performed to represent major Ethiopian AEZs, no specific environmental-gradation approach was applied to inform the sampling, as has been done in a recent paper by Kebede et al. (2021). Consequently, though fully valid, our study may not have surveyed all possible agro-climatic clines and the ecotypes presented here may not be an exhaustive list from Ethiopia. Another potential shortcoming of our approach may arise from gene flow among ecotypes which may weaken adaptive advantage. For indigenous livestock species – with limited mobility – gene flow would generally be restricted among geographically close populations, which are also likely to be clustered together under the same ecotype due to their environmental similarity; in such cases gene flow is not an interference for local adaptation. However, gene flow among distant populations or ecotypes may still occur due to human interventions, which will weaken the genomic signatures of adaptation and our method will fail to detect them.
Although our study has considered a large array of environmental data, it could not incorporate some other potentially important drivers of adaptation, e.g., pathogenic or parasitic data, due to lack of publicly available data on such prevalence. Our approach, however, offers the exciting opportunity for such inclusion in future analyses. Indeed by integrating ecological concepts with genomics, our method opens up opportunities for interdisciplinary research. This approach can be employed to study the impact of climate change on indigenous livestock populations or to predict disease pre-disposition based on environmental conditions. This methodology will therefore have important implications for sustainable farming through climate resilience and conservation programs oriented to small-holder farmers, relying on local ecosystem production.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ebi.ac.uk/ena, PRJEB39275; https://www.ebi.ac.uk/eva/, PRJEB46494.
The animal study was reviewed and approved by the ILRI Animal Care and Use Committee (IAUC). Written informed consent was obtained from the owners for the participation of their animals in this study.
OH, AAG, AV-T, and JS conceived the research project. AK, TD, and OH led the collection of samples and population metadata. ML-J contributed to the ENM analytical approach. AV-T and AAG performed the analyses and led the writing of the manuscript but all authors contributed critically to the drafts.
This research was funded in part by the Bill & Melinda Gates Foundation (BMGF) and with UK aid from the UK Foreign, Commonwealth and Development Office (Grant Agreement OPP1127286). The study constituted part of AV-T’s Ph.D. research that was funded by Vice-Chancellor Scholarship for Research Excellence International at University of Nottingham and Administrative Department of Science, Technology and Innovation (Colciencias) – Colombian Government (Call 2015 N°728). This study was carried out under the auspices of the Centre for Tropical Livestock Genetics and Health (CTLGH), established jointly by The University of Edinburgh, SRUC (Scotland’s Rural College), and the International Livestock Research Institute. The findings and conclusions contained within were those of the authors and do not necessarily reflect positions or policies of the BMGF nor the United Kingdom Government. This research was conducted as part of the Consultative Group on International Agricultural Research (CGIAR) Research Program on Livestock and was supported by contributors to the CGIAR Trust Fund.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Prof. Nick Sparks (CTLGH, SRUC) for his valuable support in conducting this research. We would also like to thank Edinburgh Genomics (Edinburgh, United Kingdom) for producing the sequence data used in this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2022.866587/full#supplementary-material
Asih, P. R., Prikas, E., Stefanoska, K., Tan, A. R. P., Ahel, H. I., and Ittner, A. (2020). Functions of p38 MAP kinases in the central nervous system. Front. Mol. Neurosci. 13:570586. doi: 10.3389/fnmol.2020.570586
Berihulay, H., Abied, A., He, X., Jiang, L., and Ma, Y. (2019). Adaptation mechanisms of small ruminants to environmental heat stress. Animals 9:75. doi: 10.3390/ani9030075
Bettridge, J., Psifidi, A., Terfa, Z., Desta, T. T., Lozano-Jaramillo, M., Dessie, T., et al. (2018). The role of local adaptation in sustainable village chicken production. Nat. Sustain. 1, 574–582. doi: 10.1038/s41893-018-0150-9
Broad Institute (2015). Best Practices for Variant Calling with the GATK. Available Online at: https://gatk.broadinstitute.org/hc/en-us/articles/360035535932-Germline-short-variant-discovery-SNPs-Indels [accessed June 20, 2022].
Browning, S. R., and Browning, B. L. (2007). Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 81, 1084–1097. doi: 10.1086/521987
Capblancq, T., and Forester, B. R. (2021). Redundancy analysis: a Swiss army knife for landscape genomics. Methods Ecol. Evol. 12, 2298–2309. doi: 10.1111/2041-210X.13722
Capblancq, T., Luu, K., Blum, M., and Bazin, E. (2018). Evaluation of redundancy analysis to identify signatures of local adaptation. Mol. Ecol. Resour. 18, 1223–1233. doi: 10.1111/1755-0998.12906
Cardoso-Leite, R., Vilarinho, A. C., Carneiro, M., Fajar, A., Cestari, G., and Guillermo-Ferreira, R. (2014). Recent and future environmental suitability to dengue fever in Brazil using species distribution model. Trans. R. Soc. Trop. Med. Hyg. 108, 99–104. doi: 10.1093/trstmh/trt115
Central Statistical Agency [CSA] (2021). Report on Livestock and Livestock Characteristics, Vol. II., Addis Ababa: CSA.
Chao, L., Niu, S., Yan, S., Bai, C., Yu, X., Hou, J., et al. (2019). Low-density lipoprotein receptor-related protein 1 regulates muscle fibre development in cooperation with related genes to affect meat quality. Poult. Sci. 98, 3418–3425. doi: 10.3382/ps/pez168
Chen, L.-L., Wang, Y.-B., Song, J.-X., Deng, W.-K., Lu, J.-H., Ma, L.-L., et al. (2017). Phosphoproteome-based kinase activity profiling reveals the critical role of MAP2K2 and PLK1 in neuronal autophagy. Autophagy 13, 1969–1980. doi: 10.1080/15548627.2017.1371393
Cruz de Carvalho, M. H. (2008). Drought stress and reactive oxygen species: production, scavenging and signalling. Plant Signal. Behav. 3, 156–165. doi: 10.4161/psb.3.3.5536
D’ambrosio, D. N., Clugston, R. D., and Blaner, W. S. (2011). Vitamin A metabolism: an update. Nutrients 3, 63–103. doi: 10.3390/nu3010063
De Marco Júnior, P., and Corrêa Nóbrega, C. (2018). Evaluating collinearity effects on species distribution models: an approach based on virtual species simulation. PLoS One 13:e0202403. doi: 10.1371/journal.pone.0202403
Del Vesco, A., Gasparino, E., de Oliveira-Grieser, D., Zancanela, Z., Menck Soares, M. A., and Rodrigues de Oliveira Neto, A. (2015). Effects of methionine supplementation on the expression of oxidative stress-related genes in acute heat stress-exposed broilers. Br. J. Nutr. 113, 549–559. doi: 10.1017/S0007114514003535
Elferink, M. G., van As, P., Veenendaal, T., Crooijmans, R., and Groenen, M. (2010). Regional differences in recombination hotspots between two chicken populations. BMC Genet. 11:11. doi: 10.1186/1471-2156-11-11
Fernando, V. C. D., Al Khateeb, W., Belmonte, M. F., and Schroeder, D. F. (2018). Role of Arabidopsis ABF1/3/4 during det1 germination in salt and osmotic stress conditions. Plant Mol. Biol. 97, 149–163. doi: 10.1007/s11103-018-0729-6
Fick, S. E., and Hijmans, R. J. (2017). Worldclim2: new 1-km spatial resolution climate surfaces for global land areas. Int. J. Climatol. 37, 4302–4315. doi: 10.1002/joc.5086
Fischer, G., Nachtergaele, F., Prieler, S., Van Velthuizen, H. T., Verelst, L., and Wiberg, D. (2008). Global Agro-Ecological Zones Assessment of Agriculture (GAEZ 2008). Luxemburg: IIASA.
Fleming, D., Koltes, J., Markey, A., Schmidt, C. J., Ashwell, C. M., Rothschild, M. F., et al. (2016). Genomic analysis of Ugandan and Rwandan chicken ecotypes using a 600k genotyping array. BMC Genomics 17:407. doi: 10.1186/s12864-016-2711-5
Forester, B. (2019). Detecting Multilocus Adaptation Using Redundancy Analysis (RDA). Available Online at: https://popgen.nescent.org/2018-03-27_RDA_GEA.html [accessed November 24, 2019].
Genecards Database (2021). CACNA1C Gene. Available Online at: https://www.genecards.org/cgi-bin/carddisp.pl?gene=CACNA1C [accessed October 10, 2021].
GFSAD30 (2017). Global Food Security Analysis-Support Data at 30 Meters Project. Available Online at: https://www.usgs.gov/centers/wgsc/science/global-food-security-support-analysis-data-30-m/ [accessed April 11, 2019].
Gheyas, A., Boschiero, C., Eory, L., Ralph, H., Kuo, R., Woolliams, J. A., et al. (2015). Functional classification of 15 million SNPs detected from diverse chicken populations. DNA Res. 22, 205–217. doi: 10.1093/dnares/dsv005
Gheyas, A., Vallejo-Trujillo, A., Kebede, A., Dessie, T., Hanotte, O., and Smith, J. (2022). Whole genome sequences of 234 indigenous African chickens from Ethiopia. Sci. Data 9:53. doi: 10.1038/s41597-022-01129-4
Gheyas, A., Vallejo-Trujillo, A., Kebede, A., Lozano-Jaramillo, M., Dessie, T., Smith, J., et al. (2021). Integrated environmental and genomic analysis reveals the drivers of local adaptation in African indigenous chickens. Mol. Biol. Evol. 38, 4268–4285. doi: 10.1093/molbev/msab156
Groenen, M., Wahlberg, P., Foglio, M., Cheng, H. H., Megens, H.-J., Crooijmans, R., et al. (2009). A high-density SNP-based linkage map of the chicken genome reveals sequence features correlated with recombination rate. Genome Res. 19, 510–519. doi: 10.1101/gr.086538.108
HarvestChoice/IFPRI (2009). AEZ 16-Class. Washington, DC: International Food Policy Research Institute.
Hengl, T., De-Jesus, J. M., MacMillan, R. A., Batjes, N. H., Heuvelink, G. B. M., Samuel-Rosa, A., et al. (2014). SoilGrids1km - global soil information based on automated mapping. PLoS One 9:e105992. doi: 10.1371/journal.pone.0105992
Hu, Z. L., Park, C. A., Wu, X. L., and Reecy, J. M. (2013). Animal QTLdb: an improved database tool for livestock animal QTL/association data dissemination in the post-genome era. Nucleic Acids Res. 41, D871–D879. doi: 10.1093/nar/gks1150
Huaiyu, M., Anushya, M., Dustin, E., Huang, X., and Thomas, P. D. (2019). PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 47, D419–D426. doi: 10.1093/nar/gky1038
Huentelman, M., De Both, M., Jepsen, W., Piras, J. S., Talboom, J. S., Willeman, M., et al. (2019). Common BACE2 polymorphisms are associated with altered risk for Alzheimer’s disease and CSF amyloid biomarkers in APOE ε4 non-carriers. Sci. Rep. 9:9640. doi: 10.1038/s41598-019-45896-4
Hurni, H. (1998). Agroecological Belts of Ethiopia. Explanatory Notes on Three Maps at a Scale of 1:1,000,000. Centre for Development and Environment. Bern: University of Bern.
Jastrebski, S., Lamont, S., and Schmidt, C. (2017). Chicken hepatic response to chronic heat stress using integrated transcriptome and metabolome analysis. PLoS One 12:e0181900. doi: 10.1371/journal.pone.0181900
Jeschke, J., and Strayer, D. (2008). Usefulness of bioclimatic models for studying climate change and invasive species. Ann. N. Y. Acad. Sci. 1134, 1–24. doi: 10.1196/annals.1439.002
Jiménez-Valverde, A., Peterson, A. T., Soberón, J., Aragón, P., and Lobo, J. M. (2011). Use of niche models in invasive species risk assessments. Biol. Invasions 13, 2785–2797. doi: 10.1007/s10530-011-9963-4
Joost, S., Bonin, A., Bruford, M. W., Després, C., Conord, C., Erhardt, G., et al. (2007). A spatial analysis method (SAM) to detect candidate loci for selection: towards a landscape genomics approach to adaptation. Mol. Ecol. 16, 3955–3969. doi: 10.1111/j.1365-294X.2007.03442.x
Jueterbock, A., Smolina, I., Coyer, J. A., and Hoarau, G. (2016). The fate of the Arctic seaweed Fucus distichus under climate change: an ecological niche modelling approach. Ecol. Evol. 6, 1712–1724. doi: 10.1002/ece3.2001
Karlsson, A., Fallahsharoudi, A., Johnsen, H., Hagenblad, J., Wright, D., Andersson, L., et al. (2016). A domestication related mutation in the thyroid stimulating hormone receptor gene (TSHR) modulates photoperiodic response and reproduction in chickens. Gen. Comp. Endocrinol. 228, 69–78. doi: 10.1016/j.ygcen.2016.02.010
Kawamata, T., Taniguchi, T., Mukai, H., Kitagawa, M., Hashimoto, T., Maeda, K., et al. (1998). A protein kinase, PKN, accumulates in Alzheimer neurofibrillary tangles and associated endoplasmic reticulum-derived vesicles and phosphorylates Tau protein. J. Neurosci. 18, 7402–7410. doi: 10.1523/JNEUROSCI.18-18-07402.1998
Keambou, T. C., Hako, B. A., Ommeh, S., Bembide, C., Ngono, E. P., Manuela, Y., et al. (2014). Genetic diversity of the Cameroon Indigenous chicken ecotypes. Int. J. Poult. Sci. 13, 279–291.
Kebede, F. G., Koman, H., Dessie, T., Setegn, W. A., Hanotte, O., and Bastiaansen, J. W. M. (2021). Species and phenotypic distribution models reveal population differentiation in Ethiopian indigenous chickens. Front. Genet. 12:723360. doi: 10.3389/fgene.2021.723360
Kong, B. W., Hudson, N., Seo, D., Lee, S., Khatri, B., Lassiter, K., et al. (2017). RNA sequencing for global gene expression associated with muscle growth in a single male modern broiler line compared to a foundational Barred Plymouth Rock chicken line. BMC Genomics 18:82. doi: 10.1186/s12864-016-3471-y
Li, X., Nie, C., Liu, Y., Chen, Y., Lv, X., Wang, L., et al. (2020). The genetic architecture of early body temperature and its correlation with Salmonella pullorum resistance in three chicken breeds. Front. Genet. 10:1287. doi: 10.3389/fgene.2019.01287
Lowri, D. B. (2012). Ecotype and the controversy over stages in the formation of new species. Biol. J. Linn. Soc. 106, 241–257. doi: 10.1111/j.1095-8312.2012.01867.x
Lozano-Jaramillo, M., Bastiaansen, J. W. M., Dessie, T., and Komen, H. (2018). Use of geographic information system tools to predict animal breed suitability for different agro-ecological zones. Animal 13, 1536–1543. doi: 10.1017/S1751731118003002
Maclean, C. A., Hong, N. P. C., and Prendergast, J. G. D. (2015). Hapbin: an efficient program for performing haplotype-based scans for positive selection in large genomic datasets. Mol. Biol. Evol. 32, 3027–3029. doi: 10.1093/molbev/msv172
McLaren, W., Gil, L., Hunt, S. E., Singh Riat, H., Ritcie, G. R. S., Thormann, A., et al. (2016). The ensembl variant effect predictor. Genome Biol. 17:122. doi: 10.1186/s13059-016-0974-4
Mengistu, A. (2006). ETHIOPIA: Country Pasture/Forage Resource Profiles. Rome: Food and Agriculture Organization of the United Nations (FAO).
Merow, C., Smith, M. J., and Silander, J. A. (2013). A practical guide to MaxEnt for modeling species’ distributions: what it does, and why inputs and settings matter. Ecography 36, 1058–1069. doi: 10.1111/j.1600-0587.2013.07872.x
Ministry of Agriculture and Rural Development [MoARD] (2005). Major Agro-Ecological Zones of Ethiopia. Forestry, Land Use and Soil Conservation Department. Addis Ababa: Ministry of Agriculture and Rural Development.
Ministry of Agriculture [MoA], (1998). Agro-Ecological Zones of Ethiopia. Natural Resources Management and Regulatory Department. With Support of German Agency for Technical Cooperation (GTZ). Addis Ababa: Ministry of Agriculture.
Muscarella, R., Galante, P. J., Soley-Guardia, M., Boria, R. A., Kass, J. M., Uriarte, M., et al. (2014). ENMeval: an R package for conducting spatially independent evaluations and estimating optimal model complexity for MaxEnt ecological niche models. Methods Ecol. Evol. 5, 1198–1205. doi: 10.1111/2041-210X.12261
Oksanen, J. (2015). Multivariate Analysis of Ecological Communities in R: Vegan Tutorial. Available Online at: https://cran.r-project.org/web/packages/vegan/vegan.pdf [accessed November 24, 2019].
Phillips, S. J., Anderson, R. P., and Shapire, R. E. (2006). Maximum entropy modelling of species geographic distributions. Ecol. Model. 190, 231–259. doi: 10.1016/j.ecolmodel.2005.03.026
Phillips, S. J., and Dudík, M. (2008). Modelling of species distributions with MaxEnt: new extensions and a comprehensive evaluation. Ecography 31, 161–175. doi: 10.1111/j.2007.0906-7590.05203.x
Pitt, J., Gillingham, P., Maltby, M., and Stewart, J. R. (2016). New perspectives on the ecology of early domestic fowl: an interdisciplinary approach. J. Archaeol. Sci. 74, 1–10.
Purcell, S., Neale, B., Todd-Brown, K., Tomas, L., Ferreira, M. A., Bender, D., et al. (2007). PLINK: a toolset for whole-genome association and population-based linkage analysis. Am. J. Hum. Genet. 81, 559–575. doi: 10.1086/519795
Qu, L., Shen, M., Guo, J., Wang, X., Dou, T., Hu, Y., et al. (2019). Identification of potential genomic regions and candidate genes for egg albumen quality by a genome-wide association study. Arch. Anim. Breed. 62, 113–123. doi: 10.5194/aab-62-113-2019
R Core Team (2018). R: A Language and Environment for Statistical Computing. Available Online at: https://www.R-project.org/ (accessed February 24, 2021).
Rellstab, C., Gugerli, F., Eckert, A. J., Hancock, A. M., and Holderegger, R. (2015). A practical guide to environmental association analysis in landscape genomics. Mol. Ecol. 24, 4348–4370. doi: 10.1111/mec.13322
Rubin, C. J., Lindberg, J., Fitzsimmons, C., Savolainen, P., Jensen, P., Lundeberg, J., et al. (2007). Differential gene expression in femoral bone from red junglefowl and domestic chicken, differing for bone phenotypic traits. BMC Genomics 8:208. doi: 10.1186/1471-2164-8-208
Rubin, C. J., Zody, M. C., Eriksson, J., Meadow, J. R. S., Sherwood, E., Webster, M. T., et al. (2010). Whole-genome resequencing reveals loci under selection during chicken domestication. Nature 464, 587–591. doi: 10.1038/nature08832
Sanarana, Y., Visser, C., Bosman, L., Nephawe, K., Maiwashe, A., and van Marle-Köster, E. (2016). Genetic diversity in South African Nguni cattle ecotypes based on microsatellite markers. Trop. Anim. Health Prod. 48, 379–385. doi: 10.1007/s11250-015-0962-9
Scott, N., Sharpe, L., and Brown, A. (2021). The E3 ubiquitin ligase MARCHF6 as a metabolic integrator in cholesterol synthesis and beyond. Biochim. Biophys. Acta 1866:158837. doi: 10.1016/j.bbalip.2020.158837
Sha, L., MacIntyre, L., Machell, J. A., Kelly, M. P., Porteous, D. J., Brandon, N. J., et al. (2012). Transcriptional regulation of neurodevelopmental and metabolic pathways by NPAS3. Mol. Psychiatry 17, 267–279. doi: 10.1038/mp.2011.73
Stucki, S., Orozco-Terwengel, P., Forester, B. R., Duruz, S., Colli, L., Masembe, C., et al. (2017). High performance computation of landscape genomic models including local indicators of spatial association. Mol. Ecol. Resour. 17, 1072–1089. doi: 10.1111/1755-0998.12629
Tadelle, D., Kijora, C., and Peter, K. J. (2003). Indigenous chicken ecotypes in Ethiopia: growth and feed utilization potentials. Int. J. Poult. Sci. 2, 144–152.
Thorn, J. S., Nijman, V., Smith, D., and Negaris, K. A. I. (2009). Ecological niche modelling as a technique for assessing threats and setting conservation priorities for Asian slow lorises (Primates: Nycticebus). Divers. Distrib. 15, 289–298. doi: 10.1111/j.1472-4642.2008.00535.x
Tyshkovskiy, A., Bozaykut, P., Borodinova, A., Gerashchenko, M. V., Ables, G. P., Garrat, M., et al. (2019). Identification and application of gene expression signatures associated with lifespan extension. Cell Metab. 30, 573–593. doi: 10.1016/j.cmet.2019.06.018
Uniprot Consortium (2021). Uniprot: the universal protein knowledgebase in 2021. Nucleic Acids Res. 49, D480–D489. doi: 10.1093/nar/gkaa1100
Vajana, E., Barbato, M., Colli, L., Milanesi, M., Rochat, E., Fabrizi, E., et al. (2018). Combining landscape genomics and ecological modelling to investigate local adaptation of indigenous Ugandan cattle to east coast fever. Front. Genet. 9:385. doi: 10.3389/fgene.2018.00385
Voight, B. F., Kudaravalli, S., Wen, X., and Pritchard, J. K. (2006). A map of recent positive selection in the human genome. PLoS Biol. 4:e72. doi: 10.1371/journal.pbio.0040072
Wakchaure, R., Ganguly, S., and Kumar, P. (2016). “Genotype X environment interaction in animal breeding: a review,” in Biodiversity Conservation in Changing Climate, eds M. O. Abdeen and R. K. Maheshwari (Delhi: Lenin Media Private Limited), 60–73.
Wang, X., Li, Z., Guo, Y., Wang, Y., Sun, G., Jiang, R., et al. (2019). Identification of a novel 43 bp insertion in the heparan sulfate 6-O-sulfotransferase 3 (HS6ST3) gene and its associations with growth and carcass traits in chickens. Anim. Biotechnol. 30, 252–259. doi: 10.1080/10495398.2018.1479712
Wang, Y., Saelao, P., Kern, C., Jin, S., Gallardo, R. A., Kelly, T., et al. (2020). Liver transcriptome responses to heat stress and Newcastle disease virus infection in genetically distinct chicken inbred lines. Genes 11:1067. doi: 10.3390/genes11091067
Warren, D. (2018). Why add Correlations for Suitability Scores. Available Online at: http://enmtools.blogspot.com/ [accessed April 12, 2019].
Warren, D. L., Glor, R. E., and Turelli, M. (2010). ENMTools: a toolbox for comparative studies of environmental niche models. Ecography 33, 607–611. doi: 10.1111/J.1600-0587.2009.06142.X
Warren, D. L., and Seifert, S. N. (2011). Ecological niche modelling in MaxEnt: the importance of model complexity and the performance of model selection criteria. Ecol. Appl. 21, 335–342. doi: 10.1890/10-1171.1
Wilson, R. T. (2010). Poultry production and performance in the federal democratic republic of Ethiopia. Worlds Poult. Sci. J. 66, 441–454. doi: 10.1017/S0043933910000528
Woldekiros, H. S., and D’Andrea, A. C. (2017). Early evidence for domestic chickens (Gallus gallus domesticus) in the Horn of Africa. Int. J. Osteoarchaeol. 27, 329–341.
Yan, D., Lehto, M., Rasilainen, L., Metso, J., Ehnolm, C., Ylä-Herttuala, S., et al. (2007). Oxysterol binding protein induces upregulation of SREBP-1c and enhances hepatic lipogenesis. Arterioscler. Thromb. Vasc. Biol. 27, 1108–1114. doi: 10.1161/ATVBAHA.106.138545
Yan, R. (2017). Physiological functions of the β-site amyloid precursor protein cleaving enzyme 1 and 2. Front. Mol. Neurosci. 10:97. doi: 10.3389/fnmol.2017.00097
Zizioli, D., Tiso, N., Guglielmi, A., Saraceno, C., Busolin, G., Giuliani, R., et al. (2016). Knock-down of pantothenate kinase 2 severely affects the development of the nervous and vascular system in zebrafish, providing new insights into PKAN disease. Neurobiol. Dis. 85, 35–48. doi: 10.1016/j.nbd.2015.10.010
Keywords: environmental adaptation, ecological niche modelling, Ethiopian village chicken, redundancy analysis, selection signature analysis
Citation: Vallejo-Trujillo A, Kebede A, Lozano-Jaramillo M, Dessie T, Smith J, Hanotte O and Gheyas AA (2022) Ecological niche modelling for delineating livestock ecotypes and exploring environmental genomic adaptation: The example of Ethiopian village chicken. Front. Ecol. Evol. 10:866587. doi: 10.3389/fevo.2022.866587
Received: 31 January 2022; Accepted: 15 July 2022;
Published: 04 August 2022.
Edited by:
Pablo Orozco-terWengel, Cardiff University, United KingdomReviewed by:
Giri Athrey, Texas A&M University, United StatesCopyright © 2022 Vallejo-Trujillo, Kebede, Lozano-Jaramillo, Dessie, Smith, Hanotte and Gheyas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adriana Vallejo-Trujillo, YWRyaS52YWxsZWpvLnRydWppbGxvQGdtYWlsLmNvbQ==; YXZhbGxlam9AZWQuYWMudWsu; Olivier Hanotte, T2xpdmllci5IYW5vdHRlQG5vdHRpbmdoYW0uYWMudWs=; by5oYW5vdHRlQGNnaWFyLm9yZw==; Almas A. Gheyas, YWxtYXMuZ2hleWFzQHJvc2xpbi5lZC5hYy51aw==; YWxtYXMuZ2hleWFzQHN0aXIuYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.