 Silvia Eckert
Silvia Eckert Jasmin Herden
Jasmin Herden Marc Stift
Marc Stift Walter Durka
Walter Durka Mark van Kleunen
Mark van Kleunen Jasmin Joshi6,7
Jasmin Joshi6,7

94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol. , 29 April 2022
Sec. Evolutionary and Population Genetics
Volume 10 - 2022 | https://doi.org/10.3389/fevo.2022.856453
This article is part of the Research Topic Genomic Basis of Adaptations to New Environments in Expansive and Invasive Species View all 7 articles
Biological invasions may result from multiple introductions, which might compensate for reduced gene pools caused by bottleneck events, but could also dilute adaptive processes. A previous common-garden experiment showed heritable latitudinal clines in fitness-related traits in the invasive goldenrod Solidago canadensis in Central Europe. These latitudinal clines remained stable even in plants chemically treated with zebularine to reduce epigenetic variation. However, despite the heritability of traits investigated, genetic isolation-by-distance was non-significant. Utilizing the same specimens, we applied a molecular analysis of (epi)genetic differentiation with standard and methylation-sensitive (MSAP) AFLPs. We tested whether this variation was spatially structured among populations and whether zebularine had altered epigenetic variation. Additionally, we used genome scans to mine for putative outlier loci susceptible to selection processes in the invaded range. Despite the absence of isolation-by-distance, we found spatial genetic neighborhoods among populations and two AFLP clusters differentiating northern and southern Solidago populations. Genetic and epigenetic diversity were significantly correlated, but not linked to phenotypic variation. Hence, no spatial epigenetic patterns were detected along the latitudinal gradient sampled. Applying genome-scan approaches (BAYESCAN, BAYESCENV, RDA, and LFMM), we found 51 genetic and epigenetic loci putatively responding to selection. One of these genetic loci was significantly more frequent in populations at the northern range. Also, one epigenetic locus was more frequent in populations in the southern range, but this pattern was lost under zebularine treatment. Our results point to some genetic, but not epigenetic adaptation processes along a large-scale latitudinal gradient of S. canadensis in its invasive range.
Heritable variation within a plant species is essential to adapt to new environments in response to natural selection (Darwin, 1859). Invasive plant species often have lower genetic variation in their new habitat due to founder effects or bottleneck events (Dlugosch and Parker, 2008), which makes it a challenge to explain their successful spread (Dlugosch et al., 2015; Estoup et al., 2016). Some clonal plants have become dominant in multiple habitats across a broad geographical range with only a single or a few genotypes, e.g., Reynoutria japonica (Hollingsworth and Bailey, 2000). It is still unclear how such species managed to adapt to a wide range of environmental conditions without genetic diversity to generate heritable variation for selection to act on.
Epigenetic mechanisms in plants are increasingly discussed as a potential explanation because these mechanisms may provide an additional source of phenotypic variation in genetically impoverished invasive species (Banerjee et al., 2019). Cytosine methylation is an adaptive epigenetic mechanism that may cause phenotypic plasticity and transgenerational evolutionary change (Bewick and Schmitz, 2015; Münzbergová et al., 2019). Methylated cytosines frequently occur in regulatory regions of genes, such as CpG islands or as part of gene-body methylations (Platt et al., 2015). Although it has been suggested that cytosine methylation is associated with local adaptation and could counteract possible bottleneck events during introduction processes (Banerjee et al., 2019), the results of the few studies analyzing this have been inconsistent (Sork, 2018; Herden et al., 2019). Therefore, further studies are needed to understand whether and how cytosine methylation is linked to transgenerational and persistent adaptation of plants in new environments.
Several studies have shown that variation in cytosine methylation might be associated with differentiation between local habitat conditions or along climatic gradients: for example in the clonal invasive species Reynoutria japonica (Richards et al., 2012) and the poleward range-expanding apomictic plant species Taraxacum officinale (Preite et al., 2015). Since variation in cytosine methylation was suggested as a response to changing environments (Münzbergová et al., 2019), it could also be involved in the formation of altitudinal (Alexander et al., 2009; Moran et al., 2017) and latitudinal clines across the invasive range (Kollmann and Bañuelos, 2004; Eckert et al., 2021), or related to invasion dynamics due to landscape heterogeneity (Deutschewitz et al., 2003; Eschtruth and Battles, 2009). Invasive plant species are exposed to novel environmental conditions often combined with high disturbance levels in their new range (Theoharides and Dukes, 2007). Therefore, to locally adapt, rapid, and heritable changes are crucial to counter impoverished genetic variation during introduction (Theoharides and Dukes, 2007). Heritable epigenetic mechanisms like changes in cytosine methylation patterns may expand phenotypic variability or the expression of cryptic genotypes in response to changing environmental cues (Marin et al., 2020; Mounger et al., 2021).
Several studies have shown the evolutionary significance of cytosine methylation in inbreeding and clonal non-native plant species (Douhovnikoff and Dodd, 2015; Dong et al., 2019), but studies on non-native outcrossing plants are still underrepresented. As rare examples for studies on native outcrossers using genetically diverse seed material, epigenetic variation was detected, e.g., for Viola cazorlensis (Herrera and Bazaga, 2010), and Plantago lanceolata (Gáspár et al., 2019). Moreover, variation in DNA methylation of genomic regions containing transposable elements has been suggested to mediate phenotypic variation in response to environmental change thus triggering rapid evolutionary processes (Baduel and Colot, 2021). Schmid et al. (2018) even suggest that epigenetic variation may be more involved in adaptive processes among genetically diverse species compared to asexually reproducing ones. Thus, studying invasive outcrossing plant species in the context of cytosine methylation related to climatic variation is necessary to infer the adaptive potential of cytosine methylation, and its contribution to invasion success given the increasing number of non-native plants worldwide (Seebens et al., 2017). To the best of our knowledge, our study is the first to analyze the spatial structuring of non-clonal offspring of an invasive plant species at the epigenetic level quantifying its contribution based on experimental cytosine demethylation at the seedling stage.
For this purpose, we focused on Solidago canadensis s.l., a perennial Asteraceae, which is invasive in Central Europe, as well as in many other parts of the world, and which has formed latitudinal clines that persist even when plants are grown in common-garden environments (Weber and Schmid, 1998; Li et al., 2016; Eckert et al., 2021). Eckert et al. (2021) found that in Central Europe these clines persisted in offspring that have been treated with the cytosine demethylation agent zebularine. Although this suggests that variation in cytosine methylation does not play a role in generating adaptive variation along geographic clines, it might still reflect fine-scale spatial separation between populations as has been found, for example, for Helleborus foetidus (Herrera et al., 2016) and Fragaria vesca (De Kort et al., 2020). Additionally, De Kort et al. (2020) suggested that population history rather than short-term environmental stress might shape epigenetic signatures. For S. canadensis, tracing established populations back to their donor population in the native range is hardly feasible, but this species has been known in Central Europe since the seventeenth century (Aiton, 1813). Therefore, a reasonable period of time might have passed for S. canadensis populations in Central Europe to develop epigenetic and genetic signatures at varying spatial scales that are reflected in phenotypic variation. Because the effect of clinal variation in S. canadensis related to climatic and latitudinal gradients has been analyzed before (Weber and Schmid, 1998; Eckert et al., 2021), we focused on possible associations between (epi)genetic variation and phenotypic variation, and to what extent these associations might have been affected by the zebularine treatment. For this purpose, we incorporated the effect of spatial structure in the invasive range. Genetic structuring may vary at different spatial scales due to unequal gene flow (Ward, 2006). We addressed this by comparing amplified fragment length polymorphisms after digesting genomic DNA with methylation-insensitive (i.e., AFLP; Vos et al., 1995) and methylation-sensitive restriction enzymes (MSAP; Reyna-López et al., 1997) for S. canadensis—an outcrossing species—analyzing plants grown from seeds and sampled along a European latitudinal gradient (Eckert et al., 2021).
We analyzed leaf tissue from offspring that originated from 25 populations (Eckert et al., 2021) and scored AFLP and MSAP patterns for each individual to answer three main questions: (a) To what extent is genetic and epigenetic variation of this species explained by spatial genetic autocorrelation and isolation-by-distance (IBD) patterns along the sampled latitudinal gradient? We hypothesized that S. canadensis populations are epigenetically differentiated despite their high degree of admixture at microsatellite markers (Eckert et al., 2021), and that this reflects spatially autocorrelated genetic and epigenetic variation but not an IBD pattern. Similar to findings in clonal and apomictic species, this may show that outcrossing and admixed invasive plant species exhibit heritable epigenetic variation that is sensitive to spatial heterogeneity in the invasive range and expressed in the phenotype. IBD is characterized by continuously distributed populations where geographically restricted gene flow will lead to distance-based population-level kinship (Wright, 1946; Malécot, 1948). This assumption is, however, violated in the presence of spatial autocorrelation patterns that might have been generated via transport corridors, i.e., highways and railways, in the invasive range (Hansen and Clevenger, 2005). Also, IBD assumes genotype-independent spatial variation in fitness imposed by the environment (Heywood, 1991) and only locally occurring genetic drift (Hardy and Vekemans, 1999). In contrast, spatial population “networks” could have arisen from hybridization of previously isolated native lineages (Hastings et al., 2004) or in interaction with environmental stochasticity resulting in variation of individual fitness and spread rates (O’Reilly-Nugent et al., 2016). We were also interested whether (b) there is an effect of demethylation treatment on putative relationships between epigenetic and phenotypic variation. We expected that zebularine treatment would remove putative associations between epigenetic and phenotypic variation due to the induced loss of methylated loci during cell division. This may indicate that non-random heritable epigenetic changes in this species are at least partly involved in generating phenotypic variation in the invasive range. And finally, we asked (c) whether AFLP and MSAP-based genome scans (BAYESCAN, BAYESCENV, LFMM, and RDA) reveal markers that are potentially associated with climatic variation or spatial genetic autocorrelation patterns across the latitudinal gradient sampled. The former would indicate that epigenetic signatures might be involved in adaptive processes associated with climatic conditions of the sampled S. canadensis populations in the invasive European range. The latter would imply that adaptive epigenetic responses might be unevenly shaped in the outcrossing S. canadensis due to patchy habitat conditions in its invasive range along the latitudinal gradient in Central Europe.
Solidago canadensis s.l. is a perennial Asteraceae native to North America and introduced to Europe in the seventeenth century (Aiton, 1813; Weber, 2000), and to several other continents (van Kleunen et al., 2019). This species reaches heights of over two meters and flowers in Europe from July to October (Schmeil et al., 1993). Multiple shoots re-sprout in the next spring from rhizomes after the aboveground parts have died back in winter (Weber, 2000). Flowers are self-sterile (Melville and Morton, 1982; Schmid and Dolt, 1994), although some degree of autofertility has been found (Razanajatovo and van Kleunen, 2016). This species heavily relies on anemochory with seeds equipped with a feathery pappus (Tackenberg et al., 2003). Solidago canadensis s.l. is represented by several cytotypes in its native range in North America, but so far, only diploid individuals have been found in the invasive range in Central Europe (van Kleunen and Schmid, 2003). In its non-native range, S. canadensis is frequently found in ruderal and disturbed sites (van Kleunen and Schmid, 2003) and, due to its ability to outcompete local flora, has been classified as an invasive species in Central Europe (Freisetzungsverordnung AS 2008 4377, 2008; Bundesamt für Naturschutz, 2013).
Leaf samples were collected from plants grown in a common-garden experiment in the Botanical Garden of the University of Konstanz. The experiment is described in detail in Eckert et al. (2021). In brief, S. canadensis seeds were collected between 2014 and 2015 from 25 wild populations along a latitudinal gradient in Central Europe, with at least five mother plants per population. Information on exact sampling locations, elevation (m a.s.l.), estimated numbers of ramets in the source populations, and the sample sizes of maternal lines are given in Supplementary Table 1. Two batches of seed subsamples were germinated per mother plant, one was treated with an aqueous solution of 25 μM of zebularine during germination and the other was mock-treated with water. Zebularine treatment results in non-specific and dose-dependent genome-wide cytosine demethylation (Zhou et al., 2002), whereby zebularine inhibits DNA methyltransferases leading to hypomethylation during mitosis (Baubec et al., 2009; Champion et al., 2010). Zebularine-derived hypomethylation (hereafter demethylation) has reportedly been shown to be heritable and to modify abiotic and biotic stress responses in different plant species without changing the underlying DNA sequence (Baubec et al., 2009; Verhoeven and van Gurp, 2012).
In total, 400 S. canadensis plants (206 control plants; 194 zebularine−treated plants; 224 maternal lines) were grown in an (incomplete) randomized-block design for 2 years (2015–2016). For each individual, the following phenotypic traits were obtained: initial seedling size, phenology (as days until flowering), plant height, the number of ramets counted in spring of the second year, and total and reproductive-to-total aboveground biomass harvested at the end of the first year. Eckert et al. (2021) found latitudinal clines in all traits (even pronounced in some traits measured on zebularine-treated individuals) except for the number of ramets and the initial seedling size. Latitude of source populations was significantly correlated with climate variables derived from the WorldClim v2.0 database (Fick and Hijmans, 2017). Therefore, we restricted the phenotypic variables in our study to the ones that showed latitudinal variation in Eckert et al. (2021). We refer to Eckert et al. (2021) for further details about the zebularine treatment, the corresponding common-garden experiment and the scoring of plant traits.
For this study, we collected intact leaves from 395 surviving and healthy S. canadensis plants (Eckert et al., 2021) at the onset of flowering in August 2015, and stored them in dry silica gel at room temperature. Genomic DNA was extracted from 10 to 20 mg of leaf tissue using an extraction kit (E.Z.N.A. Plant DNA Kit, omega BIO-TEK Inc., Norcross, Georgia, United States) and about 15% of the samples were randomly chosen to serve as duplicates for later error rate analysis. To assess genetic and epigenetic variation, we performed AFLP and MSAP analysis using protocols detailed in Schulz et al. (2014) and Gáspár et al. (2019) (see also Supplementary Method 1 for changes applied to these protocols). For AFLP analysis, both the restriction and ligation step were combined in one reaction using 500 ng of genomic DNA with the standard enzymes EcoRI and MseI in the AFLP analysis. In each analysis, we used four primer combinations in the selective amplification step (see Supplementary Table 2 for primer sequences) with the selective EcoRI primers labeled with the fluorescent dyes FAM, VIC, NED, and PET. The amplified products were measured on an ABI 3130 Genetic Analyzer (Applied Biosystems, Waltham, Massachusetts, United States) using the GeneScan LIZ 500 sizing standard (Thermo Fisher Scientific, Waltham, Massachusetts, United States).
For MSAP analyses, we followed the same procedures, except that MseI was replaced in two parallel reactions by the isoschizomers HpaII and MspI, which differ in their sensitivity to methylated cytosines (Schulz et al., 2013). The combination of these enzymes with different sensitivity allows the distinction between a maximum of four methylation states for each locus, which is non-methylated if a signal is present in both cases, fully or partially methylated if a signal is present in either of these cases and ambiguous in its status if no signal is present at the specific locus and individual. Selective primer combinations were selected based on pre-testing and quality checking.
After peak sizing of each fragment, each primer combination was analyzed separately and binned using GeneMapper Software v5.1 (Thermo Fisher Scientific, Waltham, Massachusetts, United States). Only peaks with a minimum relative fluorescence unit (RFU) height of 10 within the range of 50–500 base-pairs (bp) were binned as peaks. Binned peaks did not exceed a width of 1.5 bp and peaks were ignored if they overlapped with shoulder peaks or had an odd shape indicating contamination. After peak quality assessment, the final AFLP dataset received an overall error rate of 3.92% on average based on 44 replicates. The final MSAP dataset received an overall error rate of 7.16% on average based on 40 replicates. Details of the genotyping approach are provided in Supplementary Method 2.
To determine the methylation state per peak (hereafter locus), binary tables were analyzed using the MSAP_calc.R script by Schulz et al. (2013) and the scoring option Mix1, which for each locus distinguishes two epigenetic states, methylated (hereafter MSAP-m) and non-methylated loci (hereafter MSAP-n; Schulz et al., 2013). In total, 385 S. canadensis plants (199 control plants and 186 zebularine-treated plants, Supplementary Table 1) were successfully genotyped with an initial number of 856 peaks and, after quality scoring, comprised a final number of 361 polymorphic AFLP loci. Additionally, 336 of these plants (172 control plants and 164 zebularine-treated plants) were successfully epigenotyped with an initial number of 1,396 peaks each and a final number of 187 polymorphic methylated MSAP-m loci and 182 non-methylated MSAP-n loci. Both datasets are openly available in a public repository (Eckert et al., 2022).
To check for population structure along the sampled latitudinal gradient, we used the software STRUCTURE v2.3.4 (Pritchard et al., 2000) on AFLP data of control plants assuming K = 1–11 genetic clusters (Supplementary Figure 1). For each STRUCTURE run, we applied the admixture model and considered the sampling location (LOCPRIOR parameter) with a burn-in of 1,000,000 and 500,000 repetitions, respectively (Porras-Hurtado et al., 2013). Additionally, we tested for isolation-by-distance using the Mantel test with n = 9,999 permutations correlating the geographical log-distance between populations with their corresponding genetic distance as the standardized Wright’s FST (Rousset, 1997; Figure 1).
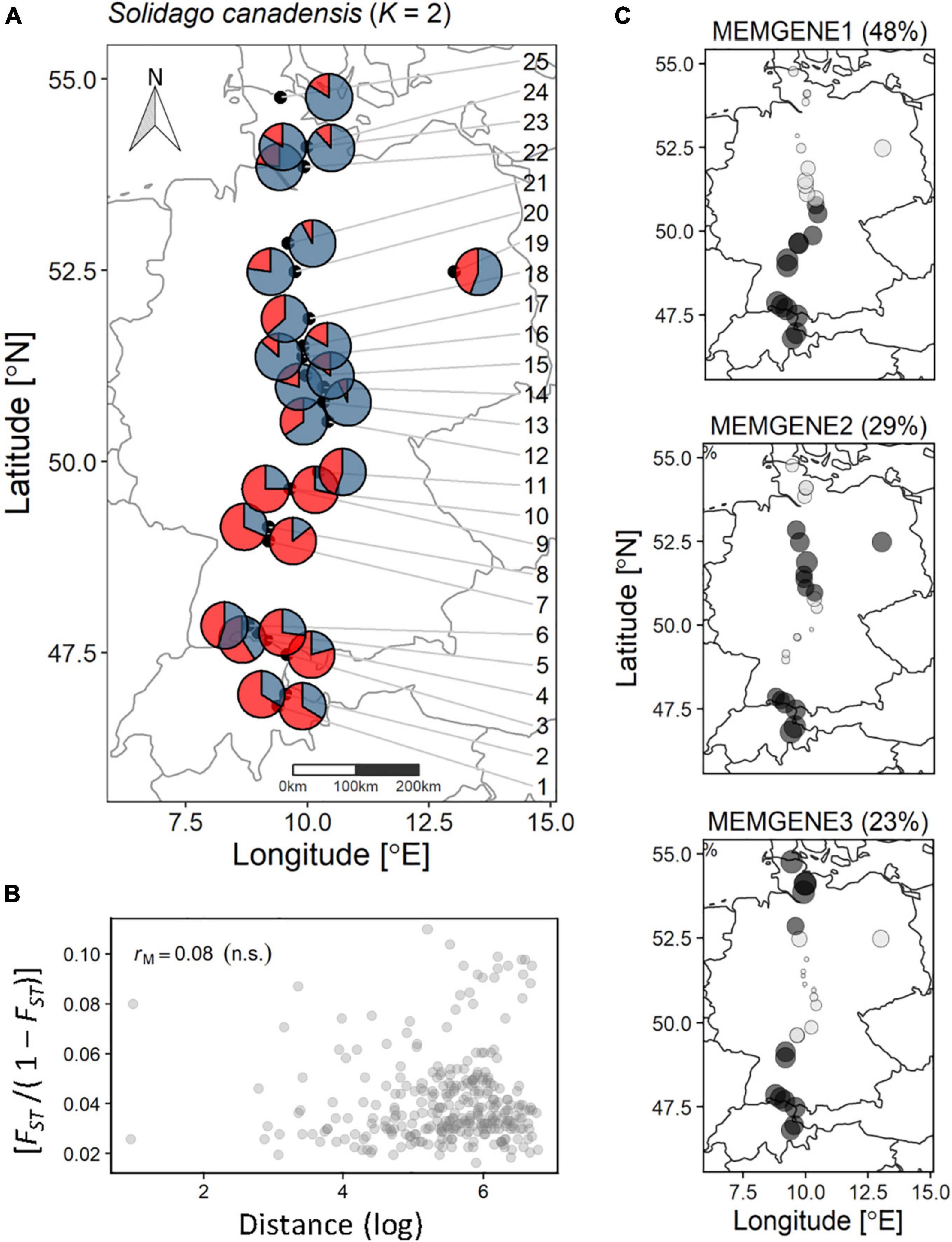
Figure 1. Genetic structuring and spatial genetic neighborhood of the 25 sampled populations of S. canadensis along the latitudinal gradient. (A) Pie charts per population displaying the average probability per cluster (K – 2). The population IDs are connected with arrows pointing to their location. (B) Standardized pairwise genetic diversity [FST/(1 - FST)] vs. the pairwise log-distance between each population analyzed withe the Mantel test for isolation-by-distance. rM, Mantel statistic; n.s., not significant. (C) The percentage of variation in spatial genetic autocorrelation axes (MEMGENE1, MEMGENE2, MEMGENE3) explained from Moran’s eigenvector maps. Dark gray circles denote positive values and light gray circles denote negative values. Circles of similar size and shade denote shared latent genetic neighborhood.
To infer the impact of uneven gene flow in the invasive range, we checked for spatial genetic and epigenetic patterns using Moran’s eigenvector maps (MEM; Dray et al., 2006; Griffith and Peres-Neto, 2006) and the mgQuick-function in the R package MEMGENE v1.0.1 (Galpern et al., 2014; Figure 1). To find the set of eigenvectors that are significantly associated with spatial genetic and epigenetic variation, respectively, we used the geographic coordinates of the source populations and Nei’s unbiased between-population distance (Nei, 1977) calculated with GENALEX v6.5 (Peakall and Smouse, 2012). The mgQuick-function calculates all Moran’s eigenvectors based on the population coordinates and reduces them to a significant set using both forward and backward selection of positive and negative eigenvectors against genetic and epigenetic distance, respectively, via permutation tests. Explained genetic and epigenetic variation of each eigenvector is then assessed using redundancy analysis (RDA). Finally, mgQuick calculates MEMGENE axes, i.e., spatial genetic neighborhoods from eigenvectors based on a principal components analysis (PCA) of the fitted values from RDA representing orthogonal vectors maximizing spatial autocorrelation. We checked significant MEMGENE axes separately for genetic (AFLP) and epigenetic (MSAP-m, MSAP-n) datasets using non-treated control samples. To test the extent to which MEMGENE axes were associated with coordinates of source populations, we calculated a correlation matrix using Pearson’s correlation coefficient. Details of MEMGENE calculations are provided in Supplementary Figures 2, 3.
To investigate whether the zebularine treatment has shifted the presence/absence patterns of AFLP, MSAP-m and MSAP-n loci per sample, we conducted a principal coordinates analysis (PCoA; also known as multi-dimensional scaling) coupled with a multivariate dispersion test (Anderson et al., 2006) based on 9,999 permutations using the betadisper-function from the VEGAN v2.5-6 package (Figure 2). We expected that the zebularine treatment would not affect genetic variation (AFLP and MSAP-n loci) but reduce the number of MSAP-m loci present. To assess between-individual dissimilarity, we used the Sørensen-Dice coefficient (Dice, 1945; Sørensen, 1948) which counts pairwise presence signals as matches. We used the Sørensen-Dice coefficient because we expected that the zebularine treatment would reduce the presence of methylated markers due to its demethylating characteristic. To infer among-population variation against within-population variation, we conducted an analysis of molecular variance (AMOVA; Table 1) according to Excoffier et al. (1992) on control plants based on 9,999 permutations using the poppr.amova-function from the R package POPPR v2.8.6. For both, MSAP-m and MSAP-n, we assessed — at the population level — the total number of loci present, the percentage of polymorphic loci, and the Shannon diversity HS per population and treatment using the MSAP_CALC v1.3 R script (Schulz et al., 2013; see details in Supplementary Figures 4, 5). For epigenetic diagnostics, we used all corresponding plants per population but also confined this analysis using only sibling pairs (maternal lines) present in both control and zebularine-treatment groups. To check whether the frequencies of epigenetic loci (MSAP-m, MSAP-n) changed due to the zebularine-treatment, we compared control plants vs. zebularine-treated individuals for each locus on a population level. Therefore, we calculated the log2-fold change (log2FC) which is common to evaluate the direction of gene expressions (e.g., McCarthy and Smyth, 2009) and visualized log2FCs per locus and population via heatmaps and box-plots (see details in Supplementary Figure 6). For the calculation of log2FC, we used only maternal lines that were present in both control and zebularine-treated subsamples.
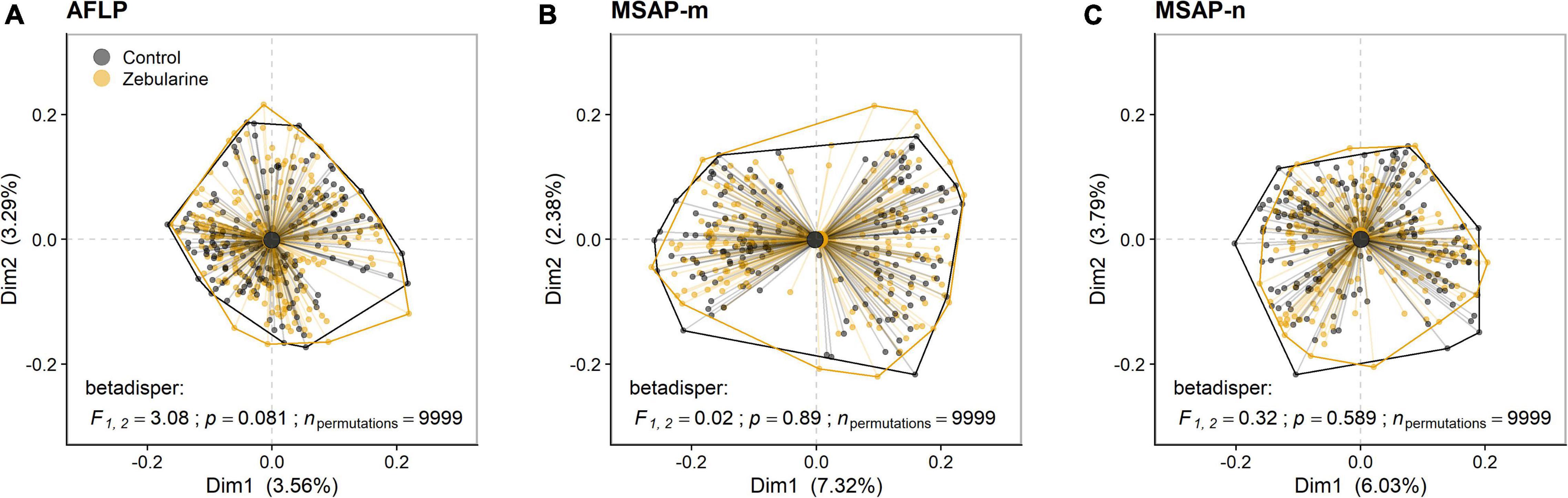
Figure 2. Biplots of principal coordinates analysis (PCoA) based on genetic (AFLP) and epigenetic (MSAP-m, MSAP-n) datasets from 25 S. canadensis populations. The first two PCoA axes (Dim1, Dim2) denote the explained variances for (A) genetic loci (AFLP), (B) methylated epigenetic loci (MSAP-m), and (C) non-methylated epigenetic loci (MSAP-n) for both control (black) and zebularine-treated individuals (orange). Group dispersion is displayed with convex hulls and group centroids. Dissimilarity between samples was determined using the Sørensen-Dice index and multivariate homogeneity of group dispersion (betadisper) was assessed at the significance level of p < 0.05 using permutation tests (npermutations = 9,999). F, empirical F-distribution value with corresponding degrees of freedom.
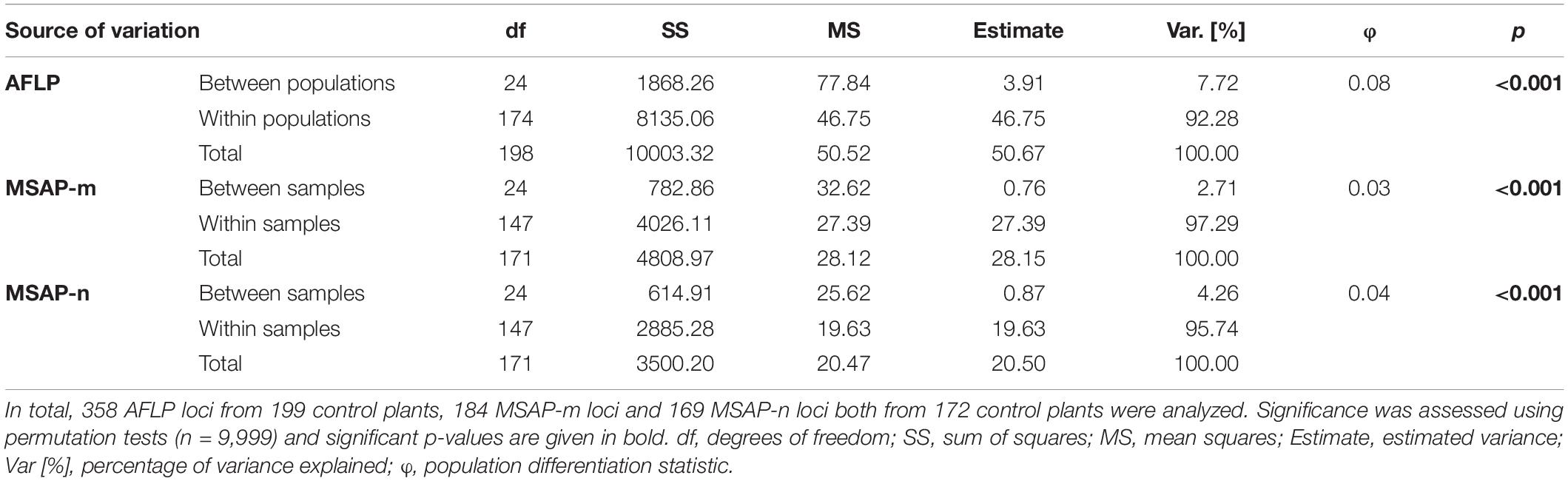
Table 1. Analysis of molecular variance from genetic (AFLP) and epigenetic (MSAP-m, MSAP-n) datasets.
To check whether zebularine treatment affects the relationship between (epi)genetic and phenotypic population-level variation, we conducted (partial) Mantel tests with n = 9,999 permutations. All (partial) Mantel tests were conducted separately for control and zebularine-treated plants using only plants that were both successfully genotyped and epigenotyped (169 control plants; 159 zebularine-treated plants; Table 2). In partial Mantel tests comparing epigenetic (MSAP-m, MSAP-n) and phenotypic variation, we controlled for genetic variation (AFLP) and spatial genetic neighborhoods from MEMGENE analysis. We applied Nei’s fixation index (Nei, 1977) as (epi)genetic population-level distance and population-level Euclidean distance for both trait variation and spatial genetic autocorrelation data. Trait variation was assessed using the population-average empirical quantile dispersion coefficient (Bonett, 2006) based on plant height and flowering phenology in both years, first-year total biomass and first-year reproductive-to-total biomass ratio, respectively. In addition to the unadjusted p-values from (partial) Mantel tests, we also provide adjusted p-values based on the false discovery rate (Benjamini and Hochberg, 1995; Yekutieli and Benjamini, 1999), but only the unadjusted p-values were used for interpretation.
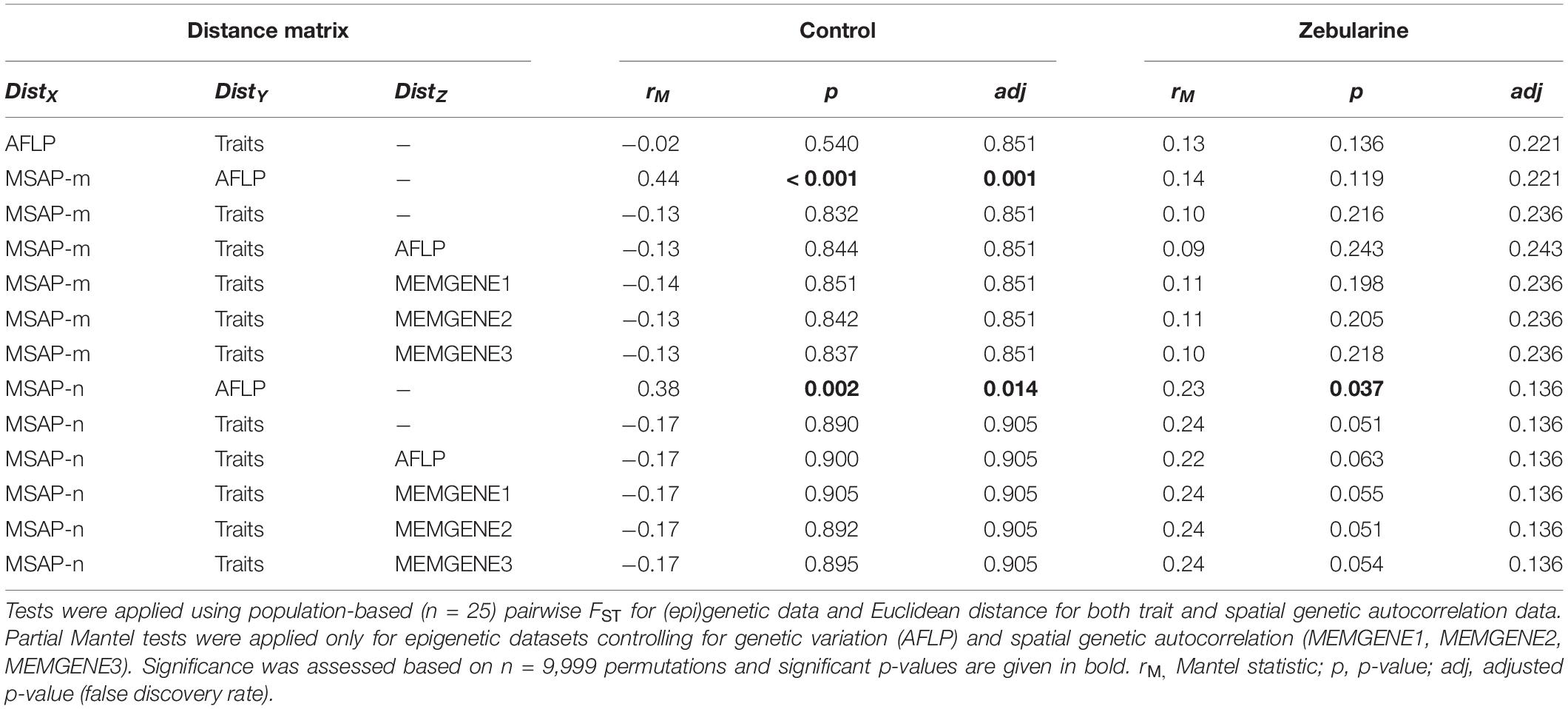
Table 2. (Partial) Mantel tests conducted between phenotypic (Traits), genetic (AFLP) and epigenetic variation (MSAP-m; MSAP-n) separately for control and zebularine-treated plants.
To screen for (epi)genetic loci of potential adaptive significance (hereafter referred to as outlier loci), we ran genome scans. As suggested by Meirmans (2015), we applied four complementary genome-scan methods that are commonly combined to minimize the risk of false positives: BAYESCAN v2.1 (Foll and Gaggiotti, 2008), BAYESCENV v1.1 (de Villemereuil and Gaggiotti, 2015), redundancy analysis (RDA; Capblancq et al., 2018) and latent factor mixed models (LFMM; Frichot et al., 2013). We ran all methods separately on genetic (AFLP; except RDA) and epigenetic (MSAP-m, MSAP-n) data using non-treated control samples. Detected putative outlier loci per dataset were pooled from each genome-scan approach and visualized as Venn diagrams (Venn, 1881; Figure 3). Conservatively, we only retained outlier loci for further statistical analyses that were detected by at least two approaches.
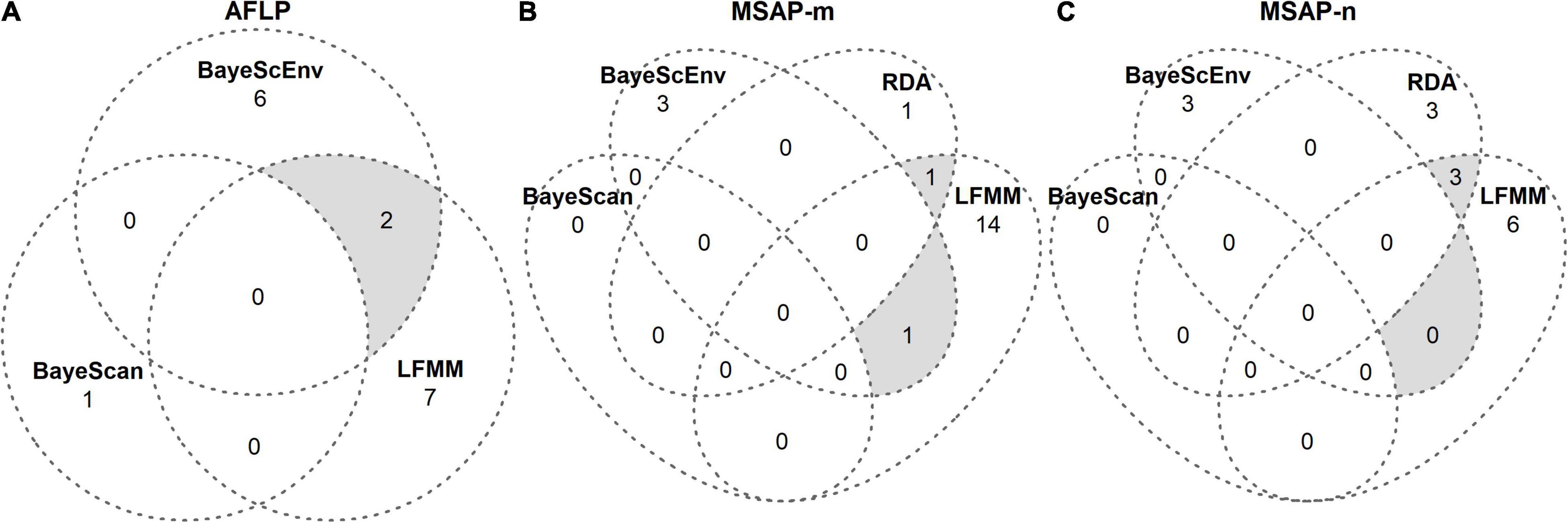
Figure 3. Venn diagrams from genome scans for outlier loci in S. canadensis. Outlier loci were detected using the genome-scan approaches BAYESCAN, BAYESCENV, LFMM, and RDA separately on (A) genetic (AFLP) and (B,C) epigenetic (MSAP-m, MSAP-n) datasets. Details of these approaches are given in Supplementary Method 3. Gray areas denote outlier loci that were jointly detected by different approaches and, to avoid false positives, only these loci were further analyzed in logistic mixed-effects models (Tables 3, 4).
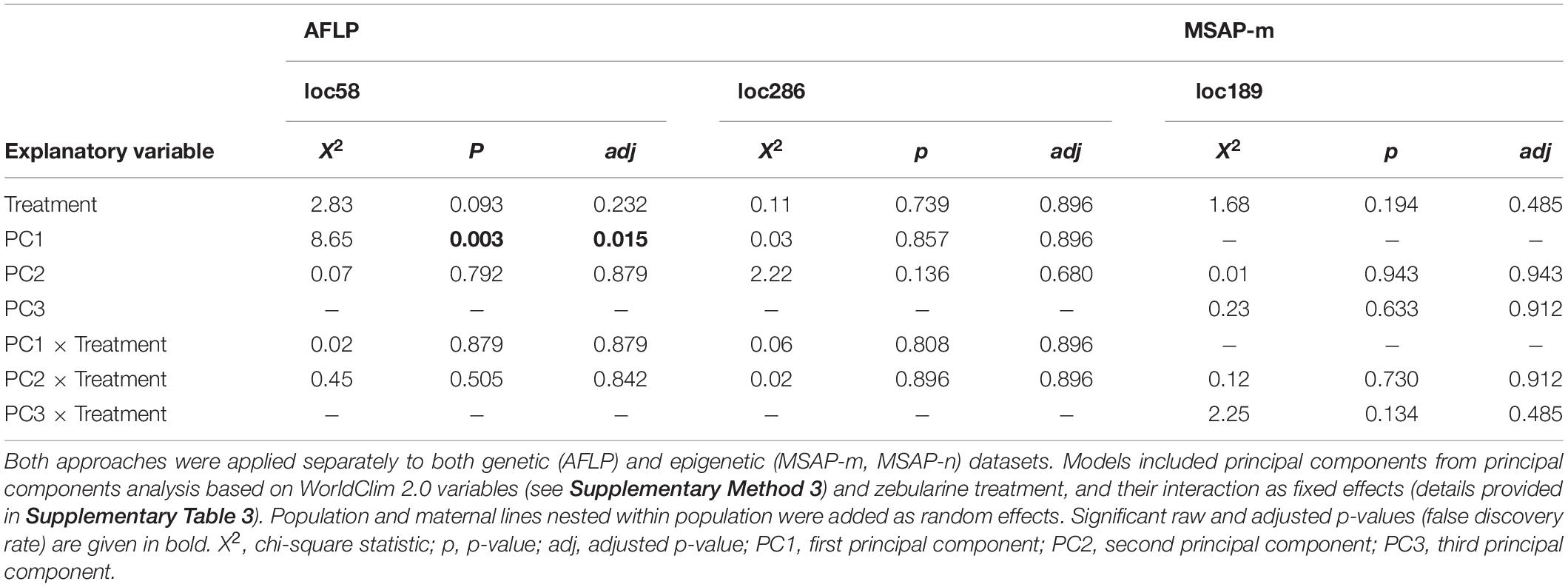
Table 3. Summary of likelihood-ratio tests from logistic mixed-effects models applied to outlier loci detected jointly via the genome-scan approaches BAYESCENV and LFMM.
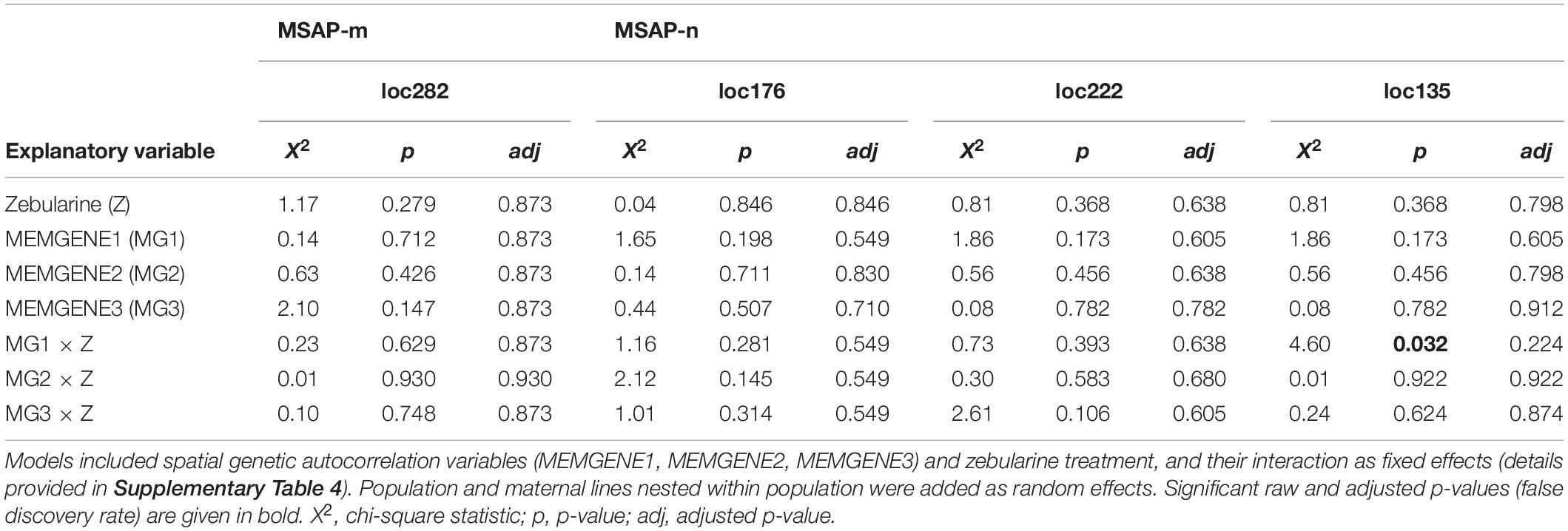
Table 4. Summary of likelihood-ratio tests from logistic mixed-effects models applied to outlier loci detected jointly via the genome-scan approaches LFMM and RDA.
The applied approaches vary in their algorithm but also in whether they are capable of including environmental variables in their calculations. BAYESCAN uses logistic regression to estimate locus-specific and population-specific contributions to the multilocus FST, distinguishing between two selection models, diversifying and balancing selection (Supplementary Figure 7). In contrast to BAYESCAN, both BAYESCENV and LFMM rely on environmental variables as a reference to check for loci of potential adaptive significance (Supplementary Figures 8–13). BAYESCENV implements the same algorithm as BAYESCAN on a modified set of models, a neutral model, a locus-specific model, and the local adaptation model linked to the corresponding environmental variables of interest. LFMM corrects for confounding effects due to underlying population structure by applying admixture coefficient estimation as a first step. We used the same WorldClim 2.0 variables as in Eckert et al. (2021) but accounted for collinearity of these variables via PCA and retained only the first three principal components (PC1, PC2, PC3) for the above-mentioned genome-scan approaches according to the broken-stick criterion (Supplementary Figure 8). To infer outlier loci that might be associated with spatial genetic neighborhoods of populations, we applied RDA on the detected MEMGENE axes as environmental predictors on both epigenetic datasets (MSAP-m, MSAP-n; Supplementary Figure 14) following Forester et al. (2018). Details and settings of the different genome-scan approaches are provided in Supplementary Method 3.
To infer the variation of outlier loci along the latitudinal climatic gradient and across spatial genetic neighborhoods, and whether these were affected by the zebularine treatment, we applied logistic mixed-effects models (LogMM). All models were fitted separately using the glmer-function from the LME4 v1.1-23 package and incorporated the binary (presence/absence) information of each outlier locus as response variable. The sampled source population and maternal lines nested within populations were included as random-intercept factors to account for non-independence of plants from the same maternal lines within populations. For each model, the second-order Akaike information criterion, and the marginal and the conditional R2 were calculated using the AICc-function and the r.squaredGLMM-function, respectively, from the MUMIN v1.43.17 package. We provide details of the model parameters in Supplementary Tables 3, 4.
Fixed factors of each fitted model depended on the environmental variables with which the outlier loci were detected (AFLP: loc58, loc286; MSAP-m: loc189, loc282; MSAP-n: loc135, loc176, loc222; see summary in Supplementary Table 5). Models fitted with loci detected with genome-scan approaches that were based solely on PCs from a principal components analysis (PCA) derived from WorldClim 2.0 variables (WorldClim-PCA) included zebularine treatment, corresponding principal components (PCs) and their interaction as fixed factors. We provide details on dimensionality reduction using PCA based on WorldClim 2.0 variables in Supplementary Figure 8. Models fitted with outlier loci detected with genome-scan approaches based on both PCs from WorldClim-PCA and spatial genetic neighborhoods included zebularine treatment, all three MEMGENE axes and their interaction as fixed factors. Because spatial autocorrelation could already point to variation in local climatic conditions (Herrera et al., 2016), PCs from WorldClim-PCA were not included in these latter models. To facilitate model convergence, fixed factors were z-transformed and shifted to positive space by adding a constant, i.e., the highest negative value plus 0.1. For each model, we checked the assumption that the log-odds of the response and each continuous fixed factor was linear applying the Box-Tidwell procedure (Box and Tidwell, 1962). In rare cases where linearity was violated, fixed factors were squared. Models showed no influential values, which was checked for each model separately using Cook’s distance (Cook, 1986), and only moderate levels of multicollinearity among covariates, which was checked with the variance inflation factor (Dormann et al., 2013).
Fixed effects were tested for significance using likelihood-ratio tests (LRT) by comparing the full model to a model without interaction and the non-interaction model with models where single terms have been iteratively removed (Lewis et al., 2011). In addition to the unadjusted p-values from LRTs, we also provide adjusted p-values using the false discovery rate (Benjamini and Hochberg, 1995; Yekutieli and Benjamini, 1999), but interpretation was based only on the unadjusted p-values. Predicted marginal-effect values were visualized with the GGPLOT2 v3.3.0 package and all results were obtained using R v4.0.2 (R Core Team, 2020).
We found weak but significant spatial genetic structuring in the analyzed S. canadensis populations along the sampled latitudinal gradient. Based on coordinates of S. canadensis source populations, three MEMGENE axes (hereafter MEMGENE1, MEMGENE2, MEMGENE3) significantly explained 13.2% of the genetic variation underlying Moran’s eigenvector maps (MEMs; n = 1,000 permutations; Figure 1C). On each MEMGENE axis, shared genetic neighborhoods among populations are given as similar population-level values and their corresponding signs (negative or positive; Supplementary Figure 2). MEMGENE1 explained 48% of variation underlying MEMs and divided the analyzed populations into a southern (populations 1–13) and a northern genetic subgroup (populations 14–25). This corresponds to results of admixture coefficients estimation via LEA within the genome-scan framework (Supplementary Figure 10 and Supplementary Method 3) as well as to the STRUCTURE analysis where K = 2 was detected as the most probable number of genetic clusters according to Evanno et al. (2005) (see population-averaged cluster probabilities from STRUCTURE analysis in Figure 1A). MEMGENE1 significantly correlated to latitude of the sampled source populations (R2 = −0.94; p < 0.001), whereas MEMGENE2 and MEMGENE3 axes were independent of the latitudinal gradient (Supplementary Figure 3).
MEMGENE2 (29% of variation explained underlying MEMs) and MEMGENE3 (23% of variation explained underlying MEMs) split populations into multiple alternating subgroups where subgroups at the top of the sampled latitudinal gradient as well as in the southern part (MEMGENE2) or only in the southern part (MEMGENE3) split the remaining subgroup (Figure 1C). AMOVA showed that there was significant genetic differentiation among populations explaining around 8% of genetic variation (Table 1), but this was not reflected in IBD along the sampled latitudinal gradient (Figure 1B). Coordinates of source populations did not significantly explain any epigenetic variation in the MEMGENE analysis and no pronounced epigenetic structuring was found with LEA (Supplementary Figures 11, 12). In contrast, AMOVA showed significant epigenetic differentiation between populations explaining 2.7% (MSAP-m) and 4.3% (MSAP-n), respectively, of epigenetic variation (Table 1).
We found that more than one-third (37.9%; 66 loci) of all MSAP-m loci showed a fourfold decrease (log2FC ≤ −2) in occurence across zebularine-treated individuals compared to control plants, but also a fourfold increase (log2FC ≥ 2) was present in 22.9% loci (40 loci) pointing to a bias for decreased methylation from the zebularine treatment. In addition, half of the MSAP-n loci (49.4%; 77 loci) showed a fourfold decrease in occurrence, whereas only 18.5% (29 loci) showed a fourfold increase. On average, up to 20.2 epigenetic loci per population at least halved in frequency whereas up to 15.9 epigenetic loci at least doubled in frequency (Supplementary Figure 6). As expected, zebularine treatment did not affect genetic between-individual distance as revealed by PCoA and betadisper analyses (Figure 2A). Surprisingly, the overall between-individual distance for epigenetic loci was also not significantly reduced by the applied demethylation treatment (Figures 2B,C). Similarly, population-level descriptors of epigenetic diversity, i.e., the number of epigenetic loci present, the percentage of polymorphic epigenetic loci and the Shannon diversity index, were not significantly affected by the zebularine treatment (Supplementary Figures 4, 5). Additionally, epigenetic population-level variation correlated with genetic variation as revealed by Mantel tests (Table 2). This relationship, however, vanished for MSAP-m but not for MSAP-n loci when looking at zebularine-treated plants of the same populations (Table 2). Population-level genetic and epigenetic diversity did not significantly reflect phenotypic population-level variation (Table 2). This was also the case for epigenetic population-level variation when controlling for genetic diversity and spatial genetic neighborhoods (Table 2).
Our genome-scan approaches only revealed few outlier loci in S. canadensis populations that showed signs of adaptive significance. BAYESCAN detected only a single genetic outlier locus (label: loc185; size: 135 bp; marker sequence: ACG-CAT) but no epigenetic outlier loci (Figure 3A). BAYESCENV and LFMM, which relied on WorldClim 2.0 variables as environmental predictors derived from PCA (Supplementary Figure 8), detected up to 15 outlier loci in AFLP (BAYESCENV: 8; LFMM: 9), MSAP-m (BAYESCENV: 4; LFMM: 15), and MSAP-n datasets (BAYESCENV: 3; LFMM: 9). From this pool of putative outlier loci, both approaches jointly detected six genetic and epigenetic outlier loci (AFLP: n = 2; MSAP-m: n = 1; MSAP-n: n = 0; Figures 3A–C). One of these jointly detected genetic loci (label: loc58; size: 58 bp; marker sequence: AAC-CCT) was significantly less probable in zebularine-treated plants as revealed by LogMM, although with an increasing probability along PC1 in both control and zebularine-treated plants (Figure 4A and Table 3). Any other jointly detected outlier locus was not significantly affected by the zebularine treatment and did not significantly vary with climatic variation among source populations (Table 3 and Supplementary Table 3).
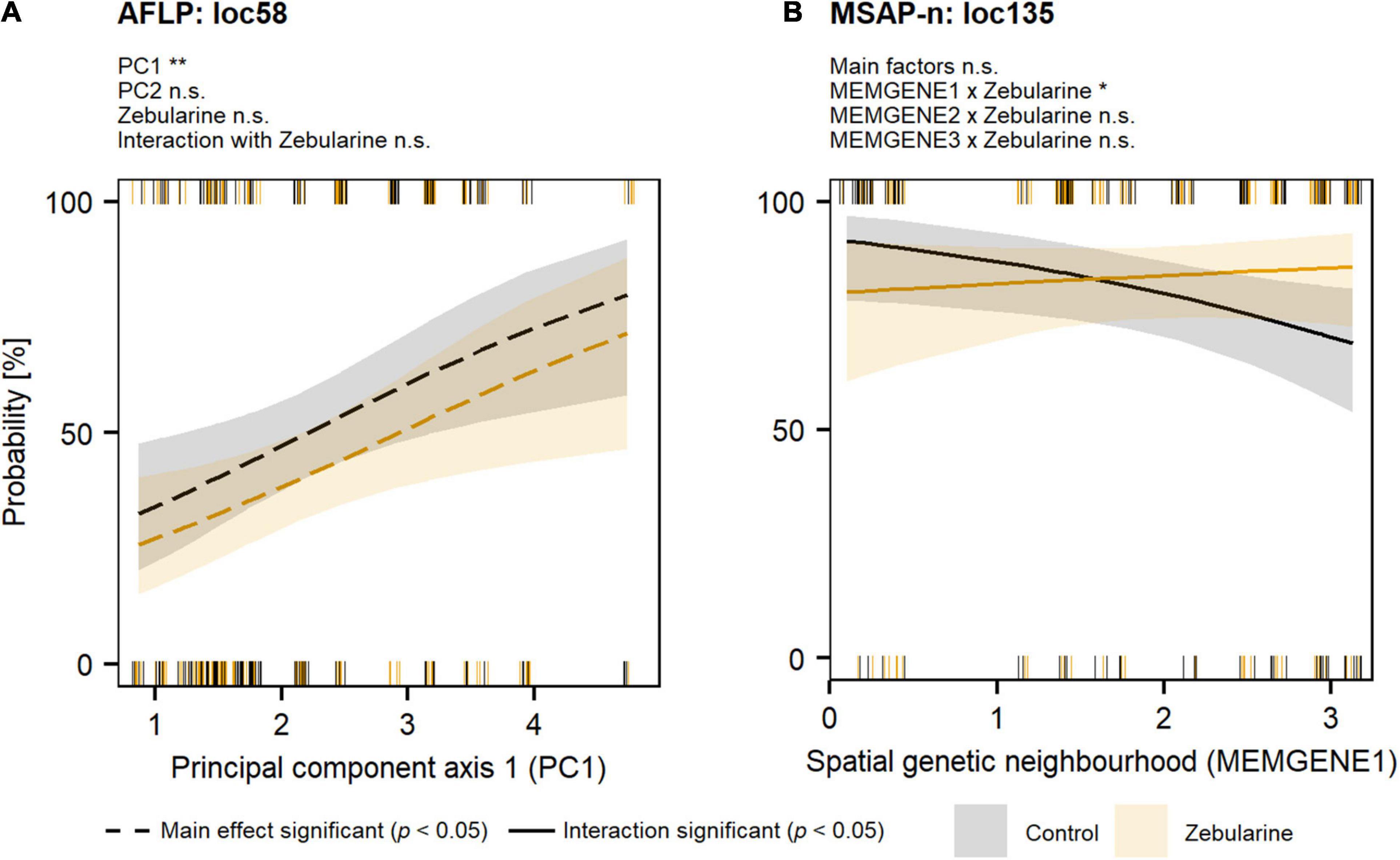
Figure 4. Probability of genetic (AFLP) and epigenetic (MSAP-n) outlier loci for S. canadensis along climatic (WorldClim-PCA) and spatial genetic autocorrelation (MEMGENE) gradients. Outlier loci (see Supplementary Method 3) were analyzed with logistic mixed-effects models followed by likelihood-ratio tests (Tables 3, 4; significance levels: **p < 0.01; *p < 0.05; n.s., not significant). Individual responses per group (control: black; zebularine treatment: orange) are given as rugs at the bottom (loci absent) and at the top (loci present). Predicted marginal-effect values are indicated by different line types (dashed—main effect significant; solid—effect of interaction with the zebularine treatment significant). (A) Genetic outlier locus (loc58; Supplementary Table 3) analyzed with a model including principal components, zebularine treatment, and the interaction with zebularine treatment as fixed factors. (B) Epigenetic outlier locus (loc135; Supplementary Table 4) analyzed with a model including MEMGENE variables, zebularine treatment, and the interaction with zebularine treatment as fixed factors.
The RDA approach, which was applied only on epigenetic datasets (MSAP-m, MSAP-n) and relied on spatial genetic neighborhoods (MEMGENE axes; see Supplementary Method 3) as environmental predictors, detected eight epigenetic outlier loci (Figures 3B,C). From this pool, a subset of four putative outlier loci, that were jointly detected by additional approaches to reduce false positives, was further analyzed (Table 4). One of these epigenetic outlier loci (label: loc135; size: 135 bp; marker sequence: AGG-CGA) was significantly less probable with increasing values of the spatial genetic neighborhoods (MEMGENE1), however, this was evident for control plants only but not for zebularine-treated individuals (Figure 4B). All other jointly detected putative epigenetic outlier loci were not significantly affected by either the zebularine treatment or spatial genetic neighborhoods of source populations (Table 4 and Supplementary Table 4).
Cytosine methylation has received growing attention in recent years as a potential epigenetic driver of adaptation (Banerjee et al., 2019; Richards and Pigliucci, 2021). In our study, we analyzed standard (AFLP) vs. methylation-sensitive (MSAP) amplified fragment length polymorphisms derived from 25 populations of the invasive plant species S. canadensis s.l. in Central Europe. We showed that S. canadensis populations in the invasive range formed spatial genetic neighborhoods, but that no spatial epigenetic patterns were evident. We also showed that among-population genetic variation was not correlated with epigenetic variation under zebularine treatment. Finally, we detected one genetic locus which increased in frequency along a climatic gradient, and another epigenetic locus which decreased in frequency along a spatial genetic autocorrelation gradient for control, but not for zebularine-treated plants. Our results thus point to some genetic adaptation processes in S. canadensis in the invasive range, but also indicate that these processes have not resulted in spatially structured epigenetic variation.
In contrast to clonal, apomictic or selfing species, outcrossing invasive species, such as S. canadensis, should be more affected in their genetic structuring at different spatial scales due to unequal gene flow and dispersal barriers (Ward, 2006). Spatial spread has been found to be often non-uniform, non-linear or to proceed from several locations simultaneously (Hastings et al., 2004). Based on Moran’s eigenvector maps, we found that genetic variation, but not epigenetic variation reflects the spatial distribution of S. canadensis plants. Epigenetic variation has been linked to rapid phenotypic evolution (Zhang et al., 2013), which might be beneficial for outcrossing invasive species, e.g., in ruderal sites which are characterized by spatial heterogeneity and unstable environmental conditions creating potential for epigenetic memory and priming (Turgut-Kara et al., 2020). Inheriting the parental epigenome, however, was also suggested to being less beneficial after long-distance dispersal into a new range and advantageous only in environments where gene expression states were already fine-tuned to cope with local conditions (Verhoeven and Preite, 2014). Linking epigenetic diversity of the outcrossing species S. canadensis to spatial variation in the invasive range in Central Europe, we found that long-term spread within the invasive range did not result in spatial epigenetic structuring. The results of our study indicate that cytosine methylation appears to be less significant in adaptation processes of outcrossing invasive plant species that exhibit high levels of admixture. Our findings support the idea that, in contrast to clonal or apomictic non-native plant species that might exhibit a high degree of epigenetic differentiation (Verhoeven and Preite, 2014), in outcrossing and admixed invasive plant species epigenetic differentiation might rarely exceed genetic variation and thus will likely not result in population structuring.
Spatial heterogeneity has been found to create and to increase time lags during invasion and to act as an environmental filter during the different invasion stages (Theoharides and Dukes, 2007). Rather than environmental selection on genotypic composition, the spatial genetic neighborhoods might also reflect that the analyzed S. canadensis populations vary in their stage of introduction or could be affected by bottleneck as well as by multiple introduction events at different spatial scales (Ward, 2006). Herrera et al. (2016) found that epigenetic between-individual similarity in Helleborus foetidus, a native plant species for which clonal propagation is exceptional, was greater than genetic similarity at the shortest distances. They pointed out that differences in local environmental features might play a similar or even greater role than spatial distance for epigenetic population structure, arguing for isolation-by-environment rather than IBD (Herrera et al., 2016). Our study focused on climatic variation and spatial autocorrelation along a latitudinal gradient, therefore, we did not include additional local site-specific features into our analyses. For example, both local soil properties and functional diversity have been found to affect the performance of S. canadensis in its invasive range in Central Europe (Czortek et al., 2020). Therefore, future studies on S. canadensis in Central Europe might more strongly emphasize local and site-specific conditions and their impact on epigenetic variation.
Ancestry estimation using the LEA framework indicated two epigenetic clusters, however, not structured along the latitudinal gradient under study. This is in line with Lele et al. (2018), who analyzed genetic and epigenetic differentiation in the non-clonal plant species Vitex negundo var. heterophylla in its native range and found that adaptation to heterogeneous habitat conditions was mainly genetically driven. The results of this study, however, also indicated a weak connection of epigenetic diversity and adaptive phenotypes. In our study, we did not find a significant relationship between epigenetic population-level diversity and trait variation, but there was low but significant epigenetic population-level differentiation as revealed by AMOVA. This epigenetic population-level differentiation may be linked to adaptation involving traits that were not measured in this study and that could be correlated to local environmental features. Our results, however, speak against an adaptive role of spatial epigenetics in S. canadensis in the invasive range. The three significant spatial genetic autocorrelation axes found in our study, however, might indicate that adaptive dynamics in S. canadensis, if present, take place at different spatial scales simultaneously and in a non-linear fashion.
Besides evident spatial genetic neighborhoods, S. canadensis populations were divided along the latitudinal gradient by two genetic clusters as revealed by the STRUCTURE and LEA approaches although with no IBD dynamics. This finding is surprising given that Eckert et al. (2021) found an overall lack of genetic population structure along this gradient based on seven microsatellite markers. In contrast to microsatellites, AFLPs are dominant markers with no information about heterozygosity, but have been found to be more efficient in individual-based population-assignment when there is weak population structuring (Campbell et al., 2003). Yet, population differentiation in S. canadensis based on AFLP analysis was weak and only a small amount of genetic variation could be explained by spatial patterns. While our results support the microsatellite analysis in Eckert et al. (2021), weak population differentiation is an unexpected result given the large latitudinal gradient and the long residence time of Solidago canadensis in Central Europe. Future approaches, however, might focus on more sophisticated molecular techniques such as SNPs (Manel et al., 2010) or spatial modeling based on large numbers of loci to map ecological adaptation (Fitzpatrick and Keller, 2015). The importance of including spatial heterogeneity into modeling plant invasion has been demonstrated in several studies (Herrera et al., 2016; Štajerová et al., 2017). Based on their results for Helleborus foetidus, Herrera et al. (2016) even argued that genetic and epigenetic differentiation might develop independently of each other leading to contrasting spatial patterns from spatially divergent selection. More studies are needed to assess whether this is the case for S. canadensis.
To the best of our knowledge, our study is the first to analyze heritable epigenetic spatial structuring in non-clonal offspring of an invasive plant species by quantifying effects of experimental cytosine demethylation. Although the demethylation agent zebularine shows dose-dependent downsides, such as delayed height and root growth (Cho et al., 2011; Eckert et al., 2021), it has been useful to detect stress-related transgenerational epigenetic variation in clonal and apomictic plant species (Richards et al., 2012; Preite et al., 2015). In our study, we found one epigenetic locus that was affected by the zebularine treatment, i.e., the presence of this locus varied depending on spatial variation. Variation in epigenetic patterns using the AFLP/MSAP technique has already been demonstrated in different plant species in their natural habitat. For example, Schulz et al. (2014) have detected one epigenetic locus in Viola elatior whose probability varied with the amount of photosynthetic active radiation present. Additionally, changes in methylation related to temperature have been shown for different plant species, e.g., Arabidopsis thaliana (Finnegan et al., 1998) and Festuca rubra (Münzbergová et al., 2019). The underlying plant material for our study, however, was only sampled once, at the end of the first year of the common-garden experiment. Therefore, we cannot exclude that there were changes in methylation profiles of control plants resulting from the common-garden environment itself, e.g., epigenetic shifts due to microclimatic conditions in the common-garden environment. In general, the majority of the analyzed 369 epigenetic loci (MSAP-m: 187 loci; MSAP-n: 182 loci) remained nearly unchanged, when zebularine effects were compared within populations. When there were changes in frequency, they were bidirectional: Up to 44.1% of epigenetic loci, i.e., MSAP-m and MSAP-n loci combined, showed a fourfold decrease, but up to 21.3% showed a fourfold increase in frequency. This non-directional change might be attributed to an increase in the level of transposable elements due to zebularine treatment, as found for Arabidopsis thaliana ecotypes (Konečná et al., 2021). With our approach, we could link the variation in two of these anonymous loci to climatic and spatial genetic variation, but more specific associations will need further analyses of the underlying mechanistic context, e.g., using next-generation approaches (Schield et al., 2016; Werner et al., 2020).
Although more epigenetic loci decreased than increased in mean frequency in zebularine-treated population subsets, the applied zebularine treatment did not result in a global change in the number of loci per population, percentage of polymorphic loci and Shannon diversity. Unfortunately, we did not sample at the beginning of the growing season, so the long-term stability of the applied zebularine treatment and, thus, persistent detectable changes remains to be tested. While we saw visible changes in plant phenotypes during the germination phase, the concentration applied during the germination phase might have been too low to cause persistent detectable changes. Hence, possible treatment differences, which may have been present at the beginning, may no longer have been visible in methylation profiles of later stages. In fact, plant DNA has been shown to be capable of compensating for experimental demethylation through repair mechanisms (Liu et al., 2015). If this is the case here, then this implies that experimental cytosine demethylation in S. canadensis can be compensated for within one growing season and, that cytosine methylation appears to be short-term only in this species.
Genetic and epigenetic population-level diversity in S. canadensis were strongly correlated. This is not surprising given that epigenetic variation is frequently found to be genetically controlled (Richards et al., 2017). In our study, however, we found that zebularine treatment decoupled genetic from epigenetic (MSAP-m) diversity. Still, there are plant species in which epigenetic variation may also exceed genetic variation or more strongly relate to a distinct environmental variable, e.g., grazing intensity in Plantago lanceolata populations in Germany (Gáspár et al., 2019). Although epigenetic variation might play a role in plastic responses to environmental challenges in the field in S. canadensis, the results of our study suggest that cytosine methylation most likely does not play a crucial role in transgenerational adaptive changes in its invasive range in Central Europe.
Solidago canadensis is often found in anthropogenically disturbed habitats (Weber, 2000). These habitats are often hotspots of continued introduction of non-natives and it seems that introduction rates for S. canadensis have not declined since this species has been introduced in Central Europe in the seventeenth century (Weber, 2000). Thus, variation in (epi)genotypes, i.e., variation in the presence of (epi)genetic loci, could also be due to multiple introductions and secondary spread pointing to unrelated genotypes. A similar scenario, has been assumed for the invasive hexaploid S. canadensis populations in China (Guo et al., 2016). We do not know the donor populations of introduced S. canadensis, so direct comparisons with populations in their native North American range were not possible. Future studies should do intensive sampling in the native range and analyze genome-wide nucleotide variation to infer possible source populations (e.g., Vallejo-Marín et al., 2021). It would also be interesting to expand the latitudinal gradient in the invaded area in future studies to include aspects of land use and urbanization related to genetic and epigenetic effects at different scales (e.g., Gáspár et al., 2019).
Multiple studies have demonstrated elevational and latitudinal phenotypic clines in fitness-related traits for S. canadensis in the invasive range in Central Europe, and some of these clines persisted when offspring were grown in a common-garden environment (Weber and Schmid, 1998; Moran et al., 2017; Eckert et al., 2021). These phenotypic clines should thus be reflected at the genetic level, but possibly likewise at the epigenetic level. Applying three genome-scan approaches and subsequent logistic mixed-effects models, we were able to detect one genetic and one epigenetic locus putatively under selection. One of these genetic loci was significantly associated with climatic variation along the latitudinal gradient and one epigenetic locus was significantly correlated to spatial genetic variation. A comparably low number of two genetic outlier loci with signatures of selection were jointly detected for Viola elatior (Schulz et al., 2014) when applying multiple genome-scan approaches, though compared to a much higher number of 39 detected epigenetic outliers. Conversely, Herrera and Bazaga (2010) found that only 10 out of 23 candidate loci were significantly associated with epigenetic differentiation in Viola cazorlensis pointing to a link between genetic and epigenetic divergence. The low number of only two candidate outlier loci detected in our study might be attributed to ongoing introductions that maintain admixture in S. canadensis and, thus, introduce unrelated (epi)genotypes (Verhoeven et al., 2011; Dlugosch et al., 2015). In this case, the outlier loci would be false positives despite their detection by multiple genome-scan approaches. It could also be a sign of a yet cryptic differentiation, which could be obscured by already pre-existing (epi)genetic variation from the invasion process of this species. Therefore, it remains to be tested whether this might be due to adaptive processes or genetic drift. Likewise, putative non-neutral epigenetic changes might have triggered the Baldwin effect, i.e., plastic responses to environmental cues induced via epigenetic variation might eventually be replaced by genetic change (Simpson, 1953; Bräutigam and Cronk, 2018). This process is yet hypothetical and still needs to be tested in plants (Bräutigam and Cronk, 2018).
Our study of S. canadensis in Europe revealed the presence of one genetic locus associated with climatic variation and one epigenetic locus associated with spatial variation along the studied latitudinal gradient. Interestingly, not all spatial patterns detected with Moran’s eigenvector maps and introduced into the genome screening using RDA resulted in the same, if any, putative loci with signatures of selection. This could suggest that within the analyzed latitudinal gradient, small-scale heterogeneous dynamics and non-linear spatial corridors (Hansen and Clevenger, 2005) between populations might affect allele frequency more strongly than isolation-by-distance processes along the gradient under study. For example, De Kort et al. (2020) used Fragaria vesca offspring collected as seeds from both a steep altitudinal (<2 km) and a wide spatial (>500 km) gradient and have grown individuals in a controlled environment to characterize DNA methylation signatures at different spatial scales. Their study demonstrated that epigenetic differentiation arises at varying spatial scales with CG methylation divergence more pronounced at the fine-scale altitudinal gradient. Since plant material for our analyses was collected at the end of the first growing season in the common-garden, we can only map one point in the lifetime of the plants. Many studies on epigenetic structuring among plant populations collected the samples directly in the field (see e.g., Herrera and Bazaga, 2010; Bewick and Schmitz, 2015; Gáspár et al., 2019; De Kort et al., 2020). Under field conditions, epigenetic structure, if present, could be directly affected by the local environment of the species. In our case, because we sampled in a common garden, the results would be due to transgenerational epigenetic inheritance. Still, it remains to be tested whether variation in the detected loci will be transgenerationally inherited and which underlying mechanistic processes these loci might trigger.
No genetic and epigenetic outlier loci were detected jointly by all three (for epigenetic loci four) genome-scan approaches. This could be because each of these approaches followed different strategies: BAYESCAN is able to detect stabilizing vs. diversifying selection if present (Foll and Gaggiotti, 2008), and BAYESCENV (de Villemereuil and Gaggiotti, 2015) and LFMM (Frichot et al., 2013) were developed to detect loci associated with environmental variables. Although the AFLP/MSAP approach is still useful for non-model organisms in detecting population structuring, it does not provide the regulatory context of the genomic sequence, i.e., linking gene-expression states to complex trait variation. Therefore, future approaches might overcome this limitation by focusing on more advanced methods of genome-wide association, such as QTL analysis (Long et al., 2011), bisulfite sequencing (Lu et al., 2020) or epiGBS (Schield et al., 2016; Werner et al., 2020).
Stochastic epigenetic changes, e.g., epimutations, were found to greatly exceed the frequency of DNA mutations providing a further source for phenotypic variation (van der Graaf et al., 2015). Transgenerational epigenetic variation in cytosine methylation has been related to phenotypic plasticity (Verhoeven and van Gurp, 2012; Zhang et al., 2013) and has been suggested to be a promising mechanism in explaining the invasion success of introduced plant species (Banerjee et al., 2019). Still, only few studies analyzed cytosine methylation in genetically diverse plant species when signals of population structure were weak (but see e.g., Herrera et al., 2019). We showed that genetic variation in the outcrossing invasive plant S. canadensis is non-linearly distributed across the analyzed latitudinal gradient. Moreover, artificial demethylation with zebularine decoupled genetic from epigenetic population-level distance and altered the frequency of one epigenetic locus. It is striking, however, that S. canadensis showed only few signatures of selection and a surprisingly low level of population differentiation in Central Europe. Epigenetic diversity was not linked to phenotypic variation, pointing to either developmental instability (Klingenberg, 2019) or unrelated but robust epigenotypes derived from ongoing introductions. Based on our results, we conclude that S. canadensis exhibits weak-to-absent transgenerational epigenetic variation in the invasive range.
The original contributions presented in the study are publicly available in the Zenodo repository (doi: 10.5281/zenodo.6388135). The R code generated during the statistical analysis process is publicly available in a GitHub repository linked to Zenodo (doi: 10.5281/zenodo.6406884).
MvK, JJ, and MS designed the study. JH collected the samples. WD carried out the laboratory analyses. SE analyzed the data with input from WD. SE wrote the first draft of the manuscript. All authors contributed to the revision of the manuscript and agreed to be accountable for the content of this work.
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under the project number 491466077 and by the following grants: KL 1866/10-1 and JO 777/8-1.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the following people for help and support: The gardeners at the Botanical Garden of Konstanz, especially Otmar Ficht and Claudia Martin, for helping with plant rearing and maintaining; Timo Scheu for helping with seed collection; Katya Stift for extracting DNA from all samples and replicates; Florian Schreckenberg for technical support with the software used. We also thank Sina Konitzer-Glöckner and Beate Rüter. We are deeply indebted to Ina Geier and Martina Herrmann (UFZ Halle) for laboratory measurement of the samples, and for explaining the AFLP and MSAP laboratory procedure. We are grateful for funding by the Deutsche Forschungsgemeinschaft (DFG) as part of the SPP1819 (KL1866/10-1 and JO 777/8-1) and for financial support by the IMPRS for Organismal Biology and the Potsdam Graduate School.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2022.856453/full#supplementary-material
Aiton, W. T. (1813). Hortus Kewensis; Or, a Cataloque of the Plants Cultivated in the Royal Botanic Garden at Kew. 2. Edition Enlarged. London: Longman, Hurst, Rees, Orme, and Brown.
Alexander, J. M., Naylor, B., Poll, M., Edwards, P. J., and Dietz, H. (2009). Plant invasions along mountain roads: the altitudinal amplitude of alien asteraceae forbs in their native and introduced ranges. Ecography 32, 334–344. doi: 10.1111/j.1600-0587.2008.05605.x
Anderson, M. J., Ellingsen, K. E., and McArdle, B. H. (2006). Multivariate dispersion as a measure of beta diversity. Ecol. Lett. 9, 683–693. doi: 10.1111/j.1461-0248.2006.00926.x
Baduel, P., and Colot, V. (2021). The epiallelic potential of transposable elements and its evolutionary significance in plants. Phil. Trans. R. Soc. B. 376:20200123. doi: 10.1098/rstb.2020.0123
Banerjee, A. K., Guo, W., and Huang, Y. (2019). Genetic and epigenetic regulation of phenotypic variation in invasive plants – linking research trends towards a unified framework. NeoBiota 49, 77–103. doi: 10.3897/neobiota.49.33723
Baubec, T., Pecinka, A., Rozhon, W., and Mittelsten Scheid, O. (2009). Effective, homogeneous and transient interference with cytosine methylation in plant genomic DNA by zebularine. Plant J. 57, 542–554. doi: 10.1111/j.1365-313X.2008.03699.x
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. B Met. 57, 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
Bewick, A. J., and Schmitz, R. J. (2015). DNA methylation: epigenetics in the wild. eLife 4:e07808. doi: 10.7554/eLife.07808
Bonett, D. G. (2006). Confidence interval for a coefficient of quartile variation. Comput. Stat. Data Anal. 50, 2953–2957. doi: 10.1016/j.csda.2005.05.007
Box, G. E. P., and Tidwell, P. W. (1962). Transformation of the independent variables. Technometrics 4, 531–550. doi: 10.1080/00401706.1962.10490038
Bräutigam, K., and Cronk, Q. (2018). DNA methylation and the evolution of developmental complexity in plants. Front. Plant Sci. 9:1447. doi: 10.3389/fpls.2018.01447
Bundesamt für Naturschutz (2013). BfN Skripten 352. Available online at: https://neobiota.bfn.de/invasivitaetsbewertung/gefaesspflanzen.html [Accessed January, 9, 2022].
Campbell, D., Duchesne, P., and Bernatchez, L. (2003). AFLP utility for population assignment studies: analytical investigation and empirical comparison with microsatellites. Mol. Ecol. 12, 1979–1991. doi: 10.1046/j.1365-294X.2003.01856.x
Capblancq, T., Luu, K., Blum, M. G. B., and Bazin, E. (2018). Evaluation of redundancy analysis to identify signatures of local adaptation. Mol. Ecol. Resour. 18, 1223–1233. doi: 10.1111/1755-0998.12906
Champion, C., Guianvarc’h, D., Sénamaud-Beaufort, C., Jurkowska, R. Z., Jeltsch, A., Ponger, L., et al. (2010). Mechanistic insights on the inhibition of C5 DNA methyltransferases by zebularine. PLoS One 5:e12388. doi: 10.1371/journal.pone.0012388
Cho, S.-W., Ishii, T., Matsumoto, N., Tanaka, H., Eltayeb, A. E., and Tsujimoto, H. (2011). Effects of the cytidine analogue zebularine on wheat mitotic chromosomes. Chromosome Sci. 14, 23–28. doi: 10.11352/scr.14.23
Cook, R. D. (1986). Assessment of local influence. J. Roy. Stat. Soc. B Met. 48, 133–155. doi: 10.1111/j.2517-6161.1986.tb01398.x
Czortek, P., Królak, E., Borkowska, L., and Bielecka, A. (2020). Impacts of soil properties and functional diversity on the performance of invasive plant species Solidago canadensis L. on post-agricultural wastelands. Sci. Total Environ. 729:139077. doi: 10.1016/j.scitotenv.2020.139077
Darwin, C. (1859). On the Origin of Species by Means of Natural Selection, Or, The Preservation of Favoured Races in the Struggle for Life. London: J. Murray.
De Kort, H., Panis, B., Deforce, D., Van Nieuwerburgh, F., and Honnay, O. (2020). Ecological divergence of wild strawberry DNA methylation patterns at distinct spatial scales. Mol. Ecol. 29, 4871–4881. doi: 10.1111/mec.15689
Deutschewitz, K., Lausch, A., Kühn, I., and Klotz, S. (2003). Native and alien plant species richness in relation to spatial heterogeneity on a regional scale in Germany. Global Ecol. Biogeogr. 12, 299–311. doi: 10.1046/j.1466-822X.2003.00025.x
Dice, L. R. (1945). Measures of the amount of ecologic association between species. Ecology 26, 297–302. doi: 10.2307/1932409
Dlugosch, K. M., and Parker, I. M. (2008). Founding events in species invasions: genetic variation, adaptive evolution, and the role of multiple introductions. Mol. Ecol. 17, 431–449. doi: 10.1111/j.1365-294X.2007.03538.x
Dlugosch, K. M., Anderson, S. R., Braasch, J., Cang, F. A., and Gillette, H. D. (2015). The devil is in the details: genetic variation in introduced populations and its contributions to invasion. Mol. Ecol. 24, 2095–2111. doi: 10.1111/mec.13183
Dong, B.-C., Yu, F.-H., and Roiloa, S. R. (2019). Editorial: ecoepigenetics in clonal and inbreeding plants: transgenerational adaptation and environmental variation. Front. Plant Sci. 10:622. doi: 10.3389/fpls.2019.00622
Dormann, C. F., Elith, J., Bacher, S., Buchmann, C., Carl, G., Carré, G., et al. (2013). Collinearity: a review of methods to deal with it and a simulation study evaluating their performance. Ecography 36, 27–46. doi: 10.1111/j.1600-0587.2012.07348.x
Douhovnikoff, V., and Dodd, R. S. (2015). Epigenetics: a potential mechanism for clonal plant success. Plant Ecol. 216, 227–233. doi: 10.1007/s11258-014-0430-z
Dray, S., Legendre, P., and Peres-Neto, P. R. (2006). Spatial modelling: a comprehensive framework for principal coordinate analysis of neighbour matrices (PCNM). Ecol. Model. 196, 483–493. doi: 10.1016/j.ecolmodel.2006.02.015
Eckert, S., Herden, J., Stift, M., Durka, W. van Kleunen, M., and Joshi, J. (2022). Data from: traces of genetic but not epigenetic adaptation in the invasive goldenrod Solidago canadensis despite the absence of population structure. Zenodo. doi: 10.5281/zenodo.6388135
Eckert, S., Herden, J., Stift, M., Joshi, J., and van Kleunen, M. (2021). Manipulation of cytosine methylation does not remove latitudinal clines in two invasive goldenrod species in Central Europe. Mol. Ecol. 30, 222–236. doi: 10.1111/mec.15722
Eschtruth, A. K., and Battles, J. J. (2009). Assessing the relative importance of disturbance, herbivory, diversity, and propagule pressure in exotic plant invasion. Ecol. Monogr. 79, 265–280. doi: 10.1890/08-0221.1
Estoup, A., Ravigné, V., Hufbauer, R., Vitalis, R., Gautier, M., and Facon, B. (2016). Is there a genetic paradox of biological invasion? Annu. Rev. Ecol. Evol. Syst. 47, 51–72. doi: 10.1146/annurev-ecolsys-121415-032116
Evanno, G., Regnaut, S., and Goudet, J. (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x
Excoffier, L., Smouse, P. E., and Quattro, J. M. (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131, 479–491. doi: 10.1093/genetics/131.2.479
Fick, S. E., and Hijmans, R. J. (2017). WorldClim 2: new 1-km spatial resolution climate surfaces for global land areas. Int. J. Climatol. 37, 4302–4315. doi: 10.1002/joc.5086
Finnegan, E. J., Genger, R. K., Kovac, K., Peacock, W. J., and Dennis, E. S. (1998). DNA methylation and the promotion of flowering by vernalization. Proc. Natl. Acad. Sci. U.S.A. 95, 5824–5829. doi: 10.1073/pnas.95.10.5824
Fitzpatrick, M. C., and Keller, S. R. (2015). Ecological genomics meets community-level modelling of biodiversity: mapping the genomic landscape of current and future environmental adaptation. Ecol. Lett. 18, 1–16. doi: 10.1111/ele.12376
Foll, M., and Gaggiotti, O. (2008). A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a bayesian perspective. Genetics 180, 977–993. doi: 10.1534/genetics.108.092221
Forester, B. R., Lasky, J. R., Wagner, H. H., and Urban, D. L. (2018). Comparing methods for detecting multilocus adaptation with multivariate genotype–environment associations. Mol. Ecol. 27, 2215–2233. doi: 10.1111/mec.14584
Freisetzungsverordnung AS 2008 4377 (2008). 814.911 Verordnung Über Den Umgang Mit Organismen In Der Umwelt, Anhang 2 (Art. 15 Abs. 2). Available online at: https://www.fedlex.admin.ch/eli/cc/2008/614/de [Accessed January 9, 2022].
Frichot, E., Schoville, S. D., Bouchard, G., and François, O. (2013). Testing for associations between loci and environmental gradients using latent factor mixed models. Mol. Biol. Evol. 30, 1687–1699. doi: 10.1093/molbev/mst063
Galpern, P., Peres-Neto, P. R., Polfus, J., and Manseau, M. (2014). MEMGENE: spatial pattern detection in genetic distance data. Methods Ecol. Evol. 5, 1116–1120. doi: 10.1111/2041-210X.12240
Gáspár, B., Bossdorf, O., and Durka, W. (2019). Structure, stability and ecological significance of natural epigenetic variation: a large-scale survey in Plantago lanceolata. New Phytol. 221, 1585–1596. doi: 10.1111/nph.15487
Griffith, D. A., and Peres-Neto, P. R. (2006). Spatial modeling in ecology: the flexibility of eigenfunction spatial analyses. Ecology 87, 2603–2613.
Guo, X., Ren, M., Ding, J., and Stephen, N. (2016). Do the introductions by botanical gardens facilitate the invasion of Solidago canadensis (Asterceae) in China? Weed Res. 56, 442–451. doi: 10.1111/wre.12227
Hansen, M. J., and Clevenger, A. P. (2005). The influence of disturbance and habitat on the presence of non-native plant species along transport corridors. Biol. Conserv. 125, 249–259. doi: 10.1016/j.biocon.2005.03.024
Hardy, O. J., and Vekemans, X. (1999). Isolation by distance in a continuous population: reconciliation between spatial autocorrelation analysis and population genetics models. Heredity 83, 145–154. doi: 10.1046/j.1365-2540.1999.00558.x
Hastings, A., Cuddington, K., Davies, K. F., Dugaw, C. J., Elmendorf, S., Freestone, A., et al. (2004). The spatial spread of invasions: new developments in theory and evidence. Ecol. Lett. 8, 91–101. doi: 10.1111/j.1461-0248.2004.00687.x
Herden, J., Eckert, S., Stift, M., Joshi, J., and van Kleunen, M. (2019). No evidence for local adaptation and an epigenetic underpinning in native and non-native ruderal plant species in Germany. Ecol. Evol. 9, 9412–9426. doi: 10.1002/ece3.5325
Herrera, C. M., and Bazaga, P. (2010). Epigenetic differentiation and relationship to adaptive genetic divergence in discrete populations of the violet Viola cazorlensis. New Phytol. 187, 867–876. doi: 10.1111/j.1469-8137.2010.03298.x
Herrera, C. M., Medrano, M., and Bazaga, P. (2016). Comparative spatial genetics and epigenetics of plant populations: heuristic value and a proof of concept. Mol. Ecol. 25, 1653–1664. doi: 10.1111/mec.13576
Herrera, C. M., Medrano, M., Pérez, R., Bazaga, P., and Alonso, C. (2019). Within-plant heterogeneity in fecundity and herbivory induced by localized DNA hypomethylation in the perennial herb Helleborus foetidus. Am. J. Bot. 106, 798–806. doi: 10.1002/ajb2.1291
Heywood, J. S. (1991). Spatial analysis of genetic variation in plant populations. Annu. Rev. Ecol. Syst. 22, 335–355. doi: 10.1146/annurev.es.22.110191.002003
Hollingsworth, M. L., and Bailey, J. P. (2000). Evidence for massive clonal growth in the invasive weed Fallopia japonica (Japanese Knotweed). Bot. J. Linn. Soc. 133, 463–472. doi: 10.1111/j.1095-8339.2000.tb01589.x
Klingenberg, C. P. (2019). Phenotypic plasticity, developmental instability, and robustness: the concepts and how they are connected. Front. Ecol. Evol. 7:56. doi: 10.3389/fevo.2019.00056
Kollmann, J., and Bañuelos, M. J. (2004). Latitudinal trends in growth and phenology of the invasive alien plant Impatiens glandulifera (Balsaminaceae). Divers. Distrib. 10, 377–385.
Konečná, K., Sováková, P. P., Anteková, K., Fajkus, J., and Fojtová, M. (2021). Distinct responses of arabidopsis telomeres and transposable elements to zebularine exposure. Int. J. Mol. Sci. 22:468. doi: 10.3390/ijms22010468
Lele, L., Ning, D., Cuiping, P., Xiao, G., and Weihua, G. (2018). Genetic and epigenetic variations associated with adaptation to heterogeneous habitat conditions in a deciduous shrub. Ecol. Evol. 8, 2594–2606. doi: 10.1002/ece3.3868
Lewis, F., Butler, A., and Gilbert, L. (2011). A unified approach to model selection using the likelihood ratio test. Methods Ecol. Evol. 2, 155–162. doi: 10.1111/j.2041-210X.2010.00063.x
Li, J., Du, L., Guan, W., Yu, F.-H., and van Kleunen, M. (2016). Latitudinal and longitudinal clines of phenotypic plasticity in the invasive herb Solidago canadensis in China. Oecologia 182, 755–764. doi: 10.1007/s00442-016-3699-x
Liu, C.-H., Finke, A., Díaz, M., Rozhon, W., Poppenberger, B., Baubec, T., et al. (2015). Repair of DNA damage induced by the cytidine analog zebularine requires ATR and ATM in Arabidopsis. Plant Cell 27, 1788–1800. doi: 10.1105/tpc.114.135467
Long, Y., Xia, W., Li, R., Wang, J., Shao, M., Feng, J., et al. (2011). Epigenetic QTL mapping in Brassica napus. Genetics 189, 1093–1102. doi: 10.1534/genetics.111.131615
Lu, H., Xue, L., Cheng, J., Yang, X., Xie, H., Song, X., et al. (2020). Polyploidization-driven differentiation of freezing tolerance in Solidago canadensis. Plant Cell Environ. 43, 1394–1403. doi: 10.1111/pce.13745
Manel, S., Joost, S., Epperson, B. K., Holderegger, R., Storfer, A., Rosenberg, M. S., et al. (2010). Perspectives on the use of landscape genetics to detect genetic adaptive variation in the field. Mol. Ecol. 19, 3760–3772. doi: 10.1111/j.1365-294X.2010.04717.x
Marin, P., Genitoni, J., Barloy, D., Maury, S., Gibert, P., Ghalambor, C. K., et al. (2020). Biological invasion: the influence of the hidden side of the (epi) genome. Funct. Ecol. 34, 385–400. doi: 10.1111/1365-2435.13317
McCarthy, D. J., and Smyth, G. K. (2009). Testing significance relative to a fold-change threshold is a TREAT. Bioinformatics 25, 765–771. doi: 10.1093/bioinformatics/btp053
Meirmans, P. G. (2015). Seven common mistakes in population genetics and how to avoid them. Mol. Ecol. 24, 3223–3231. doi: 10.1111/mec.13243
Melville, M. R., and Morton, J. K. (1982). A biosystematic study of the Solidago canadensis (Compositae) complex. I. The Ontario populations. Can. J. Bot. 60, 976–997. doi: 10.1139/b82-123
Moran, E. V., Reid, A., and Levine, J. M. (2017). Population genetics and adaptation to climate along elevation gradients in invasive Solidago canadensis. PLoS One 12:e0185539. doi: 10.1371/journal.pone.0185539
Mounger, J., Ainouche, M. L., Bossdorf, O., Cavé-Radet, A., Li, B., Parepa, M., et al. (2021). Epigenetics and the success of invasive plants. Philos. Trans. R. Soc. B 376:20200117. doi: 10.1098/rstb.2020.0117
Münzbergová, Z., Latzel, V., Šurinová, M., and Hadincová, V. (2019). DNA methylation as a possible mechanism affecting ability of natural populations to adapt to changing climate. Oikos 128, 124–134. doi: 10.1111/oik.05591
Nei, M. (1977). F-statistics and analysis of gene diversity in subdivided populations. Ann. Hum. Genet. 41, 225–233. doi: 10.1111/j.1469-1809.1977.tb01918.x
O’Reilly-Nugent, A., Palit, R., Lopez-Aldana, A., Medina-Romero, M., Wandrag, E., and Duncan, R. P. (2016). Landscape effects on the spread of invasive species. Curr. Landsc. Ecol. Rep. 1, 107–114. doi: 10.1007/s40823-016-0012-y
Peakall, R., and Smouse, P. E. (2012). GenAlEx 6.5: genetic analysis in excel. population genetic software for teaching and research—an update. Bioinformatics 28, 2537–2539. doi: 10.1093/bioinformatics/bts460
Platt, A., Gugger, P. F., Pellegrini, M., and Sork, V. L. (2015). Genome-wide signature of local adaptation linked to variable CpG methylation in oak populations. Mol. Ecol. 24, 3823–3830. doi: 10.1111/mec.13230
Porras-Hurtado, L., Ruiz, Y., Santos, C., Phillips, C., and Carracedo, Á, and Lareu, M. (2013). An overview of STRUCTURE: applications, parameter settings, and supporting software. Front. Genet. 4:98. doi: 10.3389/fgene.2013.00098
Preite, V., Snoek, L. B., Oplaat, C., Biere, A., van der Putten, W. H., and Verhoeven, K. J. F. (2015). The epigenetic footprint of poleward range-expanding plants in apomictic dandelions. Mol. Ecol. 24, 4406–4418. doi: 10.1111/mec.13329
Pritchard, J. K., Stephens, M., and Donelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959. doi: 10.1093/genetics/155.2.945
R Core Team (2020). The R Project For Statistical Compouting. Available online at: https://www.r-project.org/ [Accessed January 9, 2022].
Razanajatovo, M., and van Kleunen, M. (2016). Non-invasive naturalized alien plants were not more pollen-limited than invasive aliens and natives in a common garden. Funct. Ecol. 30, 1511–1520. doi: 10.1111/1365-2435.12633
Reyna-López, G. E., Simpson, J., and Ruiz-Herrera, J. (1997). Differences in DNA methylation patterns are detectable during the dimorphic transition of fungi by amplification of restriction polymorphisms. Mol. Gen. Genet. 253, 703–710. doi: 10.1007/s004380050374
Richards, C. L., Alonso, C., Becker, C., Bossdorf, O., Bucher, E., Colomé-Tatché, M., et al. (2017). Ecological plant epigenetics: evidence from model and non-model species, and the way forward. Ecol. Lett. 20, 1576–1590. doi: 10.1111/ele.12858
Richards, C. L., and Pigliucci, M. (2021). Epigenetic inheritance. A decade into the extended evolutionary synthesis. Paradigmi 38, 463–494. doi: 10.30460/99624
Richards, C. L., Schrey, A. W., and Pigliucci, M. (2012). Invasion of diverse habitats by few Japanese knotweed genotypes is correlated with epigenetic differentiation. Ecol. Lett. 15, 1016–1025. doi: 10.1111/j.1461-0248.2012.01824.x
Rousset, F. (1997). Genetic differentiation and estimation of gene flow from f-statistics under isolation by distance. Genetics 145, 1219–1228. doi: 10.1093/genetics/145.4.1219
Schield, D. R., Walsh, M. R., Card, D. C., Andrew, A. L., Adams, R. H., and Castoe, T. A. (2016). EpiRADseq: scalable analysis of genomewide patterns of methylation using next-generation sequencing. Methods Ecol. Evol. 7, 60–69. doi: 10.1111/2041-210X.12435
Schmeil, O., Fitschen, J., and Senghas, K. (1993). Flora Von Deutschland Und Angrenzender Länder: Ein Buch Zum Bestimmen Der Wildwachsenden Und Häufig Kultivierten Gefäßpflanzen. 89th Revised And Expanded Edition. Wiebelsheim: Quelle and Meyer.
Schmid, B., and Dolt, C. (1994). Effects of maternal and paternal environment and genotype on offspring phenotype in Solidago altissima L. Evolution 48, 1525–1549. doi: 10.1111/j.1558-5646.1994.tb02194.x
Schmid, M. W., Heichinger, C., Coman Schmid, D., Guthörl, D., Gagliardini, V., Bruggmann, R., et al. (2018). Contribution of epigenetic variation to adaptation in Arabidopsis. Nat. Commun. 9:4446. doi: 10.1038/s41467-018-06932-5
Schulz, B., Eckstein, R. L., and Durka, W. (2013). Scoring and analysis of methylation-sensitive amplification polymorphisms for epigenetic population studies. Mol. Ecol. Resour. 13, 642–653. doi: 10.1111/1755-0998.12100
Schulz, B., Eckstein, R. L., and Durka, W. (2014). Epigenetic variation reflects dynamic habitat conditions in a rare floodplain herb. Mol. Ecol. 23, 3523–3537. doi: 10.1111/mec.12835
Seebens, H., Blackburn, T. M., Dyer, E. E., Genovesi, P., Hulme, P. E., Jeschke, J. M., et al. (2017). No saturation in the accumulation of alien species worldwide. Nat. Commun. 8:14435. doi: 10.1038/ncomms14435
Sørensen, T. (1948). A Method Of Establishing Groups Of Equal Amplitude In Plant Sociology Based On Similarity Of Species Content And Its Application To Analyses Of The Vegetation On Danish Commons: Biologiske Skarifter. V. Kopenhagen: Det Kongelige Danske Videnskabernes Selskabs.
Sork, V. L. (2018). Genomic studies of local adaptation in natural plant populations. J. Hered. 109, 3–15. doi: 10.1093/jhered/esx091
Štajerová, K., Šmilauer, P., Brùna, J., and Pyšek, P. (2017). Distribution of invasive plants in urban environment is strongly spatially structured. Landsc. Ecol. 32, 681–692. doi: 10.1007/s10980-016-0480-9
Tackenberg, O., Poschlod, P., and Bonn, S. (2003). Assessment of wind dispersal potential in plant species. Ecol. Monogr. 73, 191–205.
Theoharides, K. A., and Dukes, J. S. (2007). Plant invasion across space and time: factors affecting nonindigenous species success during four stages of invasion. New Phytol. 176, 256–273. doi: 10.1111/j.1469-8137.2007.02207.x
Turgut-Kara, N., Arikan, B., and Celik, H. (2020). Epigenetic memory and priming in plants. Genetica 148, 47–54. doi: 10.1007/s10709-020-00093-4
Vallejo-Marín, M., Friedman, J., Twyford, A. D., Lepais, O., Ickert-Bond, S. M., Streisfeld, M. A., et al. (2021). Population genomic and historical analysis suggests a global invasion by bridgehead processes in Mimulus guttatus. Commun. Biol. 4:327. doi: 10.1038/s42003-021-01795-x
van der Graaf, A., Wardenaar, R., Neumann, D. A., Taudt, A., Shaw, R. G., Jansen, R. C., et al. (2015). Rate, spectrum, and evolutionary dynamics of spontaneous epimutations. Proc. Natl. Acad. Sci. U.S.A. 112, 6676–6681. doi: 10.1073/pnas.1424254112
van Kleunen, M., and Schmid, B. (2003). No evidence for an evolutionary increased competitive ability in an invasive plant. Ecology 84, 2816–2823. doi: 10.1890/02-0494
van Kleunen, M., Pyšek, P., Dawson, W., Essl, F., Kreft, H., Pergl, J., et al. (2019). The global naturalized alien flora (GloNAF) database. Ecology 100:e02542. doi: 10.1002/ecy.2542
Venn, J. (1881). “Diagrammic representation,” in Symbolic Logic (London: Macmillan and Co.), 100–125.
Verhoeven, K. J. F., and Preite, V. (2014). Epigenetic variation in asexually reproducing organisms. Evolution 68, 644–655. doi: 10.1111/evo.12320
Verhoeven, K. J. F., and van Gurp, T. P. (2012). Transgenerational effects of stress exposure on offspring phenotypes in apomictic dandelion. PLoS One 7:e38605. doi: 10.1371/journal.pone.0038605de
Verhoeven, K. J. F., Macel, M., Wolfe, L. M., and Biere, A. (2011). Population admixture, biological invasions and the balance between local adaptation and inbreeding depression. Proc. Roy. Soc. B 278, 2–8. doi: 10.1098/rspb.2010.1272
Villemereuil, P., and Gaggiotti, O. E. (2015). A new FST-based method to uncover local adaptation using environmental variables. Methods Ecol. Evol. 6, 1248–1258. doi: 10.1111/2041-210X.12418
Vos, P., Hogers, R., Bleeker, M., Reijans, M., van de Lee, T., Hornes, M., et al. (1995). AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res. 23, 4407–4414. doi: 10.1093/nar/23.21.4407
Ward, S. (2006). Genetic analysis of invasive plant populations at different spatial scales. Biol. Invasions 8, 541–552. doi: 10.1007/s10530-005-6443-8
Weber, E. (2000). Biological flora of central europe: Solidago altissima L. Flora 195, 123–134. doi: 10.1016/S0367-2530(17)30960-X
Weber, E., and Schmid, B. (1998). Latitudinal population differentiation in two species of Solidago (Asteraceae) introduced into Europe. Am. J. Bot. 85, 1110–1121. doi: 10.2307/2446344
Werner, O., Prudencio, ÁS., de la Cruz-Martínez, E., Nieto-Lugilde, M., Martínez-Gómez, P., and Ros, R. M. (2020). A cost reduced variant of epi-genotyping by sequencing for studying DNA methylation in non-model organisms. Front. Plant Sci. 11:694. doi: 10.3389/fpls.2020.00694
Yekutieli, D., and Benjamini, Y. (1999). Resampling-based false discovery rate controlling multiple test procedures for correlated test statistics. J. Stat. Plan. Infer. 82, 171–196. doi: 10.1016/S0378-3758(99)00041-5
Zhang, Y.-Y., Fischer, M., Colot, V., and Bossdorf, O. (2013). Epigenetic variation creates potential for evolution of plant phenotypic plasticity. New Phytol. 197, 314–322. doi: 10.1111/nph.12010
Keywords: AFLP, MSAP, cytosine methylation, spatial autocorrelation, genome scan
Citation: Eckert S, Herden J, Stift M, Durka W, van Kleunen M and Joshi J (2022) Traces of Genetic but Not Epigenetic Adaptation in the Invasive Goldenrod Solidago canadensis Despite the Absence of Population Structure. Front. Ecol. Evol. 10:856453. doi: 10.3389/fevo.2022.856453
Received: 17 January 2022; Accepted: 14 March 2022;
Published: 29 April 2022.
Edited by:
Anna Fijarczyk, Laval University, CanadaReviewed by:
Ellen Pagel, University of Regensburg, GermanyCopyright © 2022 Eckert, Herden, Stift, Durka, van Kleunen and Joshi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Silvia Eckert, c2lsdmlhLmVja2VydEB1bmktcG90c2RhbS5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.