
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol., 02 March 2022
Sec. Population, Community, and Ecosystem Dynamics
Volume 10 - 2022 | https://doi.org/10.3389/fevo.2022.813488
 Saloua Biyada1*
Saloua Biyada1* Mohammed Merzouki1
Mohammed Merzouki1 Taisija Dėmčėnko2
Taisija Dėmčėnko2 Dovilė Vasiliauskienė2Eglė Marčiulaitienė3Saulius Vasarevičius3
Dovilė Vasiliauskienė2Eglė Marčiulaitienė3Saulius Vasarevičius3 Jaunius Urbonavičius2
Jaunius Urbonavičius2
In this study, the effect of initial feedstock concentration on the microbial community structure and dynamics during textile waste composting processes was investigated using the next-generation sequencing approach. For this, three mixtures were prepared with different textile waste concentrations mixed with green waste and paper and cardboard waste for composting, to choose the proportion that will provide a mature final compost. A comprehensive characterization of the microbial communities associated with different textile waste concentrations during composting was achieved. It was noted that by increasing the concentration of textile waste, microbial communities (bacterial and fungal) change. Genera and species belonging to Actinobacteria, Firmicutes, Chloroflexia, Rozellomycota, Mortierellomycota, Aphelidiomycota, Ascomycota, and Mucoromycota were the most abundant in the mixtures containing either 40 or 60% of textile waste, whereas some of the species were absent at 80% of textile waste in the mix; this difference was also reflected by their enzymatic activities. Generally, these phyla are associated with composting, and they play a major role in recalcitrant molecular decaying. Ultimately, it can be concluded that the shift most likely occurred in microbial communities during composting probably owing to the interaction between changes in the nutrient concentration and microbial communities. This investigation proves that the concentration of textile waste significantly affects the microbial communities and demonstrates that a high concentration of textile waste is not suitable to grant a good maturity of compost.
Textile waste is considered a serious environmental and health problem worldwide due to its high concentration of chemical products including heavy metals (Talouizte, 2014; Talouizte et al., 2017; Biyada et al., 2019). Otherwise, these wastes are a treasure trove of organic matter, which can be converted into a nutrient-rich organic fertilizer and consequently contribute to the rehabilitation of soils by its soil-improving qualities (Biyada et al., 2020a,2021). To recover these wastes and understate or even eliminate their harmful effect on the environment and public health, various strategies have been proposed and developed to transform them into usable resources (Neher et al., 2013). Wherefore, composting is considered as one of the most sustainable management technologies for organic waste, which is a biological process that degrades organic matter into a useful product aerobically (Soobhany et al., 2015; Temporal-lara et al., 2016).
Successful composting is dependent on microbial community changing, which is affected by physical-chemical conditions and nutritional characteristics of feedstock composted (lignocellulosic compounds, protein, etc.) (Partanen et al., 2010). The correct management of all these factors could be attributed to the generation of a suitable final compost (Partanen et al., 2010). The identity of the microbial communities associated with the composting process, and therefore their metabolic abilities for organic matter biodegradation, depends considerably on the nutritional properties of feedstock composted (Maccready et al., 2013; Song et al., 2014). In fact, many studies devoted the effect of changing the nature of feedstock on microbial communities during composting (Neher et al., 2013; Song et al., 2014). Contrariwise, there is still a narrowed understanding of how microbial communities are affected by changing the concentration of feedstock that occurs during the composting process and their metabolic pathways. In this regard, a deeper understanding of the microbial communities depending on the variation feedstock concentration would significantly contribute to improving their efficiency. This goal could be achieved using the high-throughput sequencing approach; this molecular method has become popular recently due to its high potential to provide a complete genome of all microbial communities (bacterial and fungal) in the compost (Quast et al., 2013; López-gonzález et al., 2015).
No previous studies have been conducted to describe the microbial communities (bacterial and fungal) and their enzymatic activities in the case of industrial waste in general and textile waste especially, using different concentrations of the initial feedstock. Following the above mentioned, this study depicts a comprehensive comparison of both bacterial and fungal communities associated with compost from textile waste mixed with green waste, paper, and cardboard using a high-throughput sequencing approach, and the identification of the different enzymatic activities associated with the microbial communities present in compost mixture. During this investigation, the influence of the feedstock concentration on the change in microbial community structure and their metabolic pathways was highlighted, thus allowing to choose the best combination for improving the composting performance of the textile waste and produce maturity.
The details of composting methods were described previously (Biyada et al., 2020a). Mixtures were set up for composting with shredded wastes (between 20 and 30 cm) labeled “Mix A, B, and C.” Different combinations used are summarized in Table 1, whereas the composition of feedstock used is summarized in Table 2 (Biyada et al., 2020b). An in-silo composter of approximately 200 L was used for composting. Appropriate mixing of the feedstock has been achieved, and the silos were turned at least three times per week for 44 weeks. Samples were collected in triplicate according to the four cardinal positions, later placed in polyethylene bags, and stored at 4°C until further analysis.
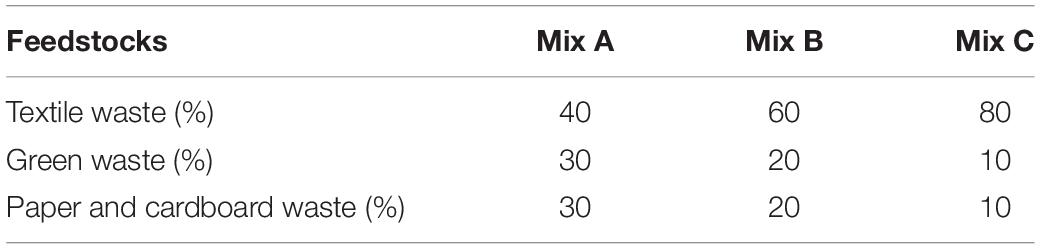
Table 1. Different combinations of feedstock used during the composting of textile waste.
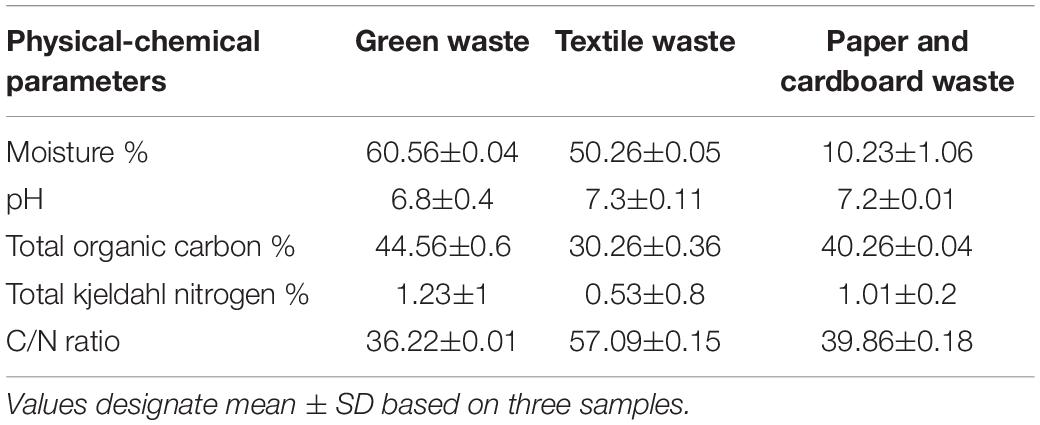
Table 2. Physical-chemical characteristics of the feedstock.
The C/N ratio of compost samples was computed by finding the percentage of total C and N which were analyzed using a TOC analyzer (Shimadzu-V CSN) (Mancinelli et al., 2017). The measurement of heavy metal concentrations was carried out using inductively coupled plasma spectrometry (ICP-AES) according to Alsac (2007). Cellulose activity was measured according to Biyada (Biyada et al., 2020b). Physical-chemical analysis was accomplished at various phases of composting (1 week and 44 weeks), and these data of the same sample were published previously (Biyada et al., 2020b; Table 3). The obtained results were tested statically using ANOVA (normalized ANOVA) using GraphPad Prism.
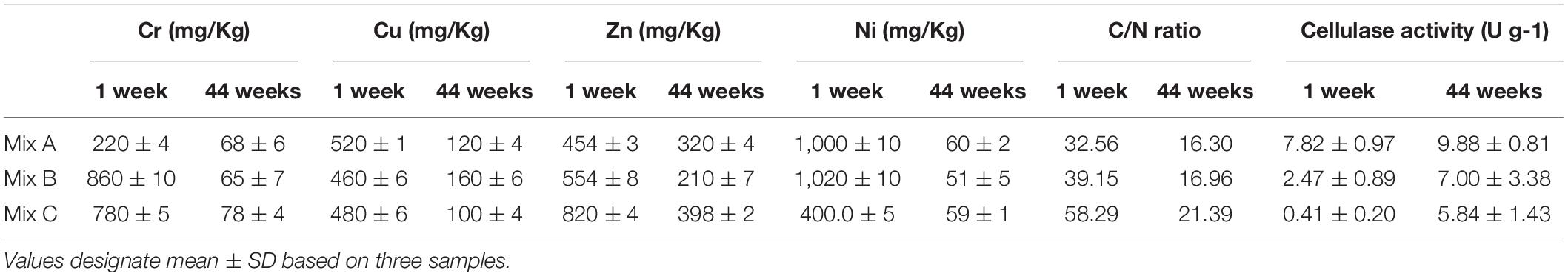
Table 3. Contents of heavy metals (Cr, Cu, Zn, and Ni), C/N ratio, and cellulase activity throughout the textile waste composting process of Mix A, B, and C (Biyada et al., 2020b).
DNA was extracted directly from solid compost samples according to the PureLink Microbiome DNA Purification Kit recommendation. Amplification of 16S rRNA and ITS genes and sequencing was performed at the Genomic Analysis Platform Macrogen (South Korea). For amplification of 16S rRNA gene targeting the V3-V4 region from 16S rRNA primers specific to bacteria was used:
Forward: 5′-TCGTCGGCAGCGTCAGATGTGTATAAGAG ACAGCCTACGGGNGGCWGCAG
Reverse: 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAG ACAGGACTACHVGGGTATCTAATCC.
For the fungal samples, the amplification of the internal transcribed spacer ITS2 region was carried out using the following primers:
Forward: ITS3 (5′-GCATCGATGAAGAACGCAGC-3′).
Reverse ITS4 (5′-TCCTCCGCTTATTGATATGC-3′).
To analyze the 16S rRNA gene fragments for bacteria, Mothur version 1.44.3 was used. To achieve this, raw sequences were filtered and aligned using the SILVA version 132 database (Schloss et al., 2009). For bacterial species identification, Ribosomal Database Project (RDP) 16S database was used. Fungal sequences were first trimmed using CUTADAPT and then analyzed using DADA2 Sequences that were identified using the UNITE version 8.2 database (Callahan et al., 2016). Enzymatic activities were identified using the UniProt (Universal Protein) database. For this, search queries containing identified species (bacterial and fungal) together with enzyme codes of interest were used to find records in the UniProt database.
Twelve FASTQ files were generated by NGS and corresponded to the pair-end sequencing (forward and reverse) of compost samples from mixture A, B, and C. Six FASTQ files corresponded to 16S rRNA and the rest corresponded to ITS regions. Tables 4, 5 illustrate the amount of 16S rRNA and ITS sequences during the analysis by Mothur and DADA2. Bacteria and fungi rDNA libraries were built for each mixture. From the total of bacterial sequences, 6.87, 11.86, and 4.75% of Actinobacteria were obtained, respectively, for mixtures A, B, and C (Table 4).
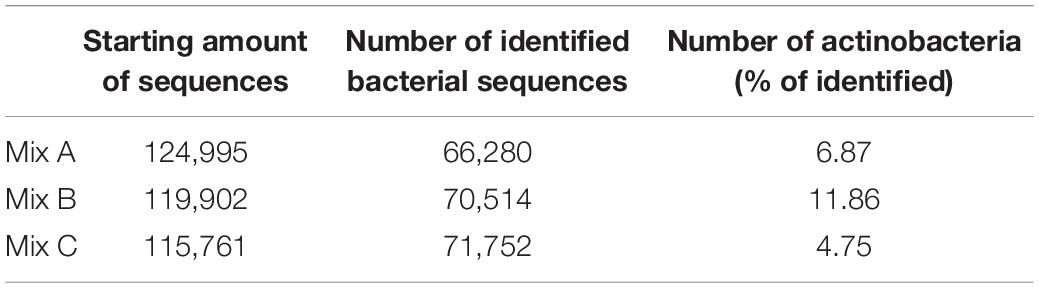
Table 4. Amount of 16S rRNA sequences generated and identified in each mixture A, B, and C using Mothur.

Table 5. Amount of ITS sequences generated in each mixture A, B, and C using DADA2.
The taxa were studied in order to reveal their relative abundance in the compost samples. Figure 1 illustrates the bacterial community in the mixtures analyzed (A, B, and C), thus showing that the bacterial composition in the compost samples varied in taxonomic structure and diversity depending on the concentration of feedstock in each mixture.
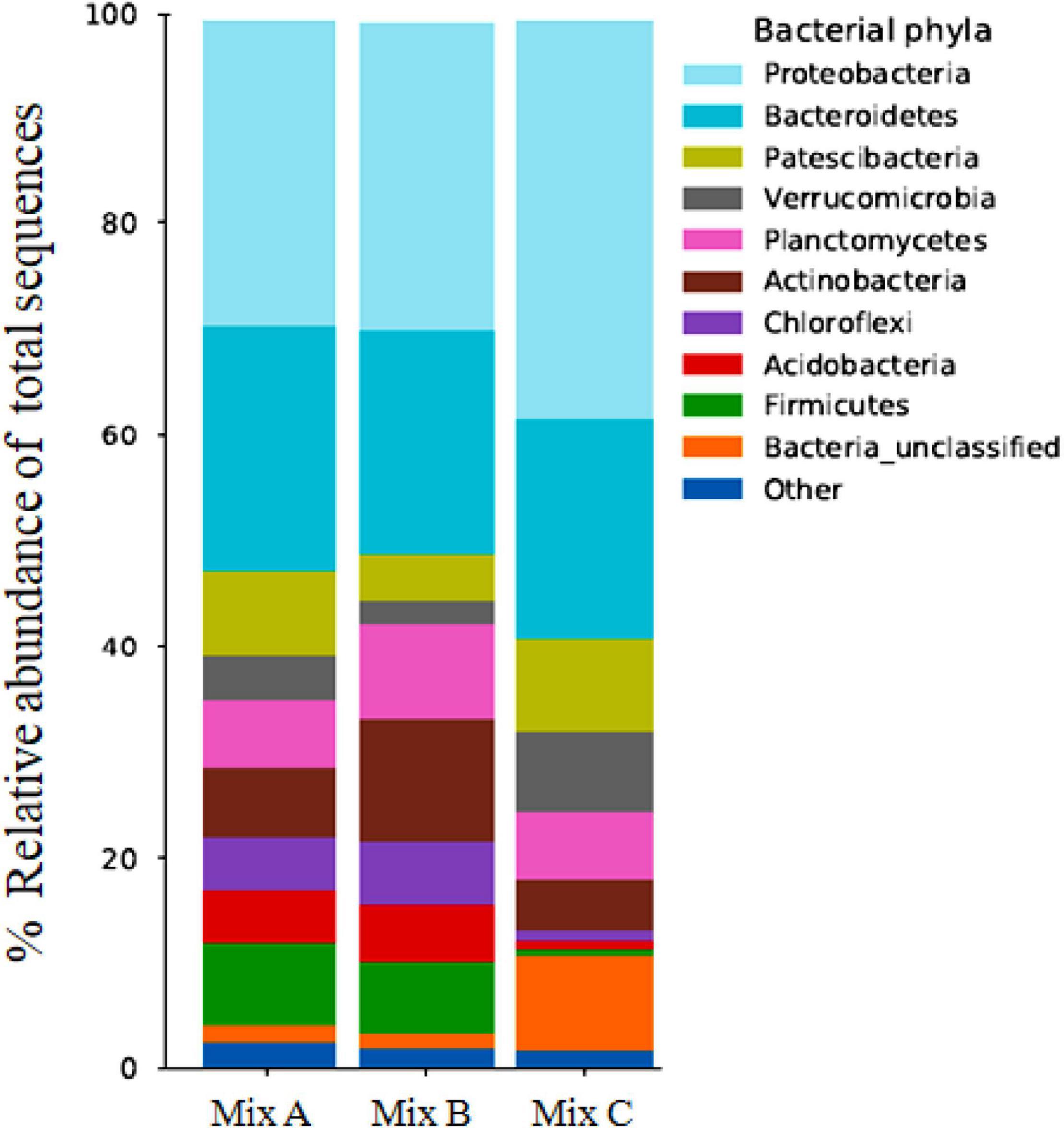
Figure 1. Relative abundance of bacterial phyla in compost samples (Mix A, B, and C). “Uncultured” denotes sequences similar to bacteria that were reported in the database as uncultured bacteria. “Other” denotes bacterial sequences with similarity to classes other than the major bacterial phyla. “Unclassified” denotes bacterial sequences with no close similarity to sequences in the nucleotide database.
Bacterial diversity was represented by four majority phyla, which were selected as the most dominant, with a maximum representation of Proteobacteria (25, 28, and 33%), Bacteroidetes (20, 19, and 19%), Actinobacteria (6, 11, and 5%), and Firmicutes (11, 9, and 0.5%), respectively, for composts A, B, and C (Figure 1), and these taxa have been commonly associated with compost. Within these phyla, the taxonomic distribution comprised mostly α-Proteobacteria (13, 18, and 10%), Bacilli (9, 6, and 0.3%), Actinobacteria (3, 7, and 3%), and γ-Proteobacteria (7, 7, and 20%), respectively, for mixtures A, B, and C, which were identified as the most abundant class.
Several classes such as β-Proteobacteria (0.8, 2, and 1%), Clostridia (1, 3, and 0.06%), and Flavobacteriia (5, 2, and 3%), respectively, for mixtures A, B, and C were present as a minority in compost samples. Even though the same trends were observed for all mixtures (dominance of Proteobacteria and Bacteroidetes), differences were detected from one mixture to another. In this regard, it was observed that the proportion of Actinobacteria, Firmicutes, and Chloroflexia was greater for mixtures A and B than mixture C.
Figure 2 illustrates the bacterial distribution at the genera level for each composting mix, and Table 6 shows the most abundant species in each mixture. It is noteworthy that there is a difference in the diversity within the samples from the three mixtures analyzed. The dominant genera within these mixtures were represented by Cytophage, Steroidobacter, Pseudoxanthomonas, Cytophage, Devosia, Flavobacterium, Streptomyces, Paenibacillus, Pseudomonas, Chelatococcus, Achromobacter, Mycobacterium, Clostridium, Nitrobacter, and Nocardioides. In fact, it should be noted that Mycobacterium, Clostridium, Nitrobacter, and Bifidobacterium are highly present in mixture A. Mixture B was most abundantly colonized by Flavobacterium, Pseudoxanthomonas, Paenibacillus, Pseudomonas, Chelatococcus, Nocardioides, Enterobacter, Chitinophaga, and Nitrosomonas, whereas mixture C is characterized by a relatively higher abundance of Steroidobacter, Devosia, Streptomyces, Achromobacter, and Cytophaga (Figure 2). It should also be noted that many genera and species are present in mixtures A and B and absent in mixture C, and this is the case of Nitrobacter, Chelatococcus (C. asaccharovorans), Chitinophaga (C. cymbidii), Bifidobacterium (B. longum), and Nitrosomonas (N. ureae and N. communis) (Figure 2 and Table 6).
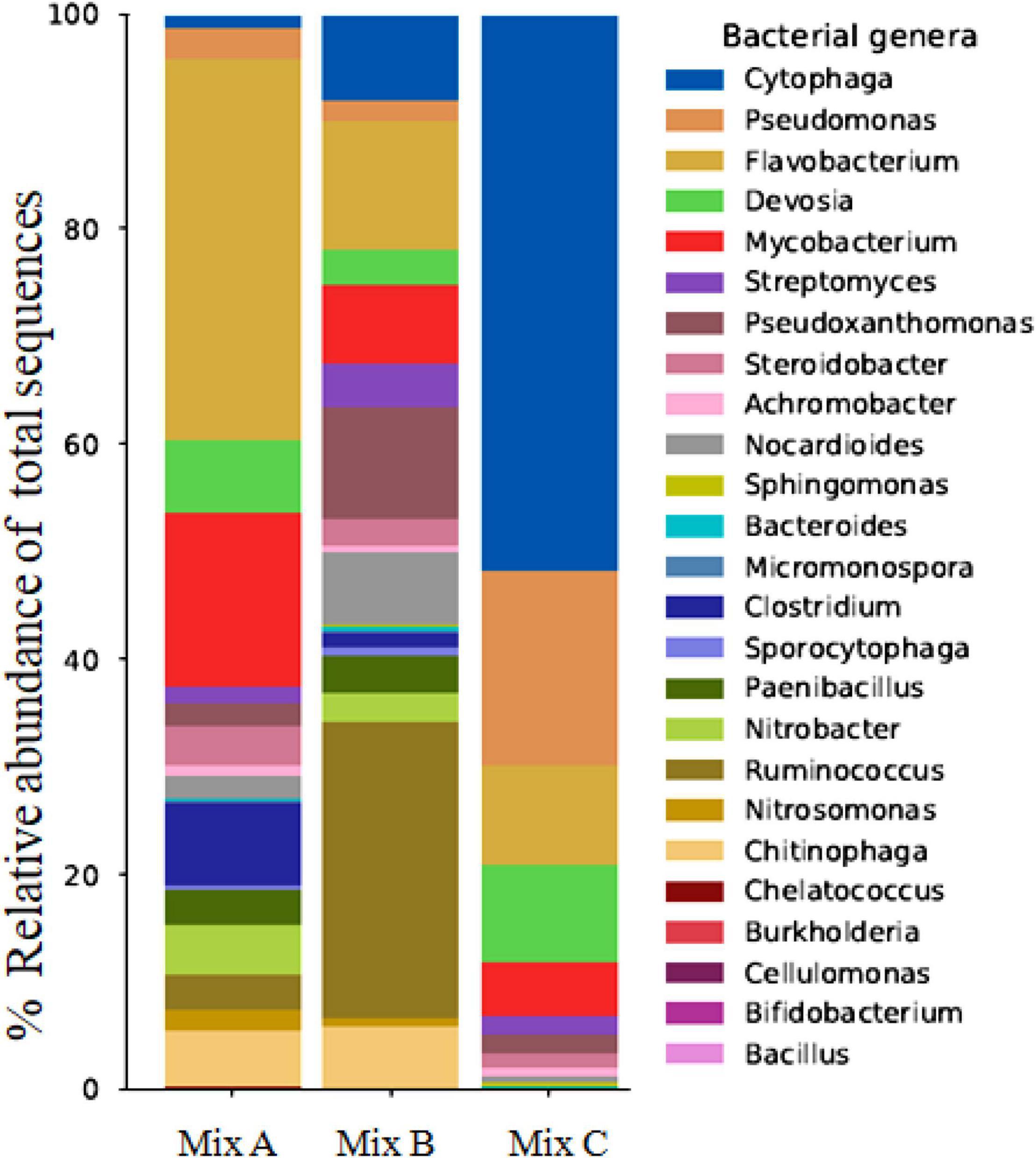
Figure 2. Relative abundance of bacterial genera in compost samples (Mix A, B, and C).
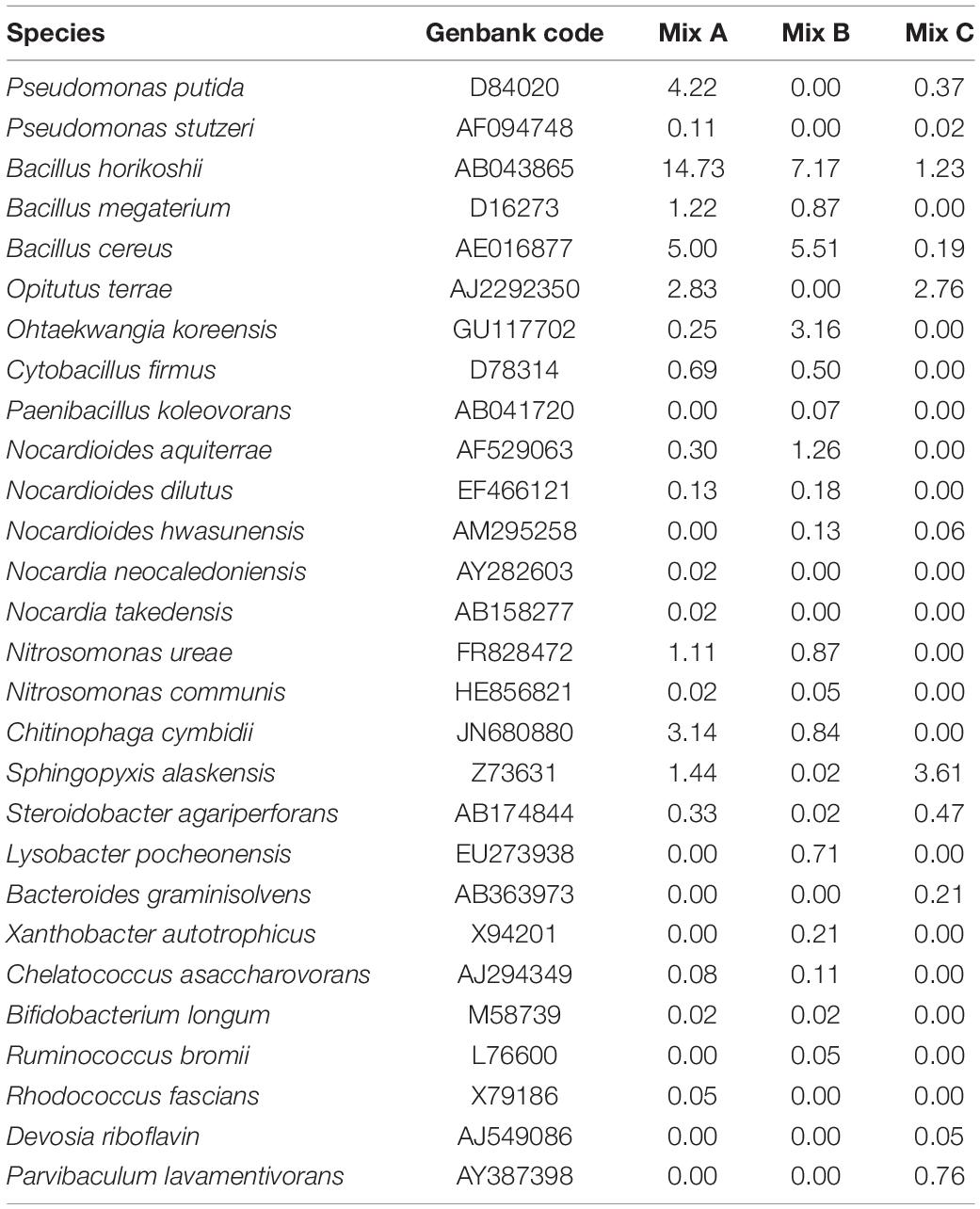
Table 6. The most abundant bacterial species in each mixture (Mix A, B, and C).
The difference between the three mixtures was also detected using the UniProt database. Several enzymatic activities were identified using the UniProt database. It is well known that the absence of some species in mixture C compared with mixtures A and B correlated with the disappearance of some enzymatic activities in this mixture and the appearance of these enzyme activities in mixtures A and B (Table 7).
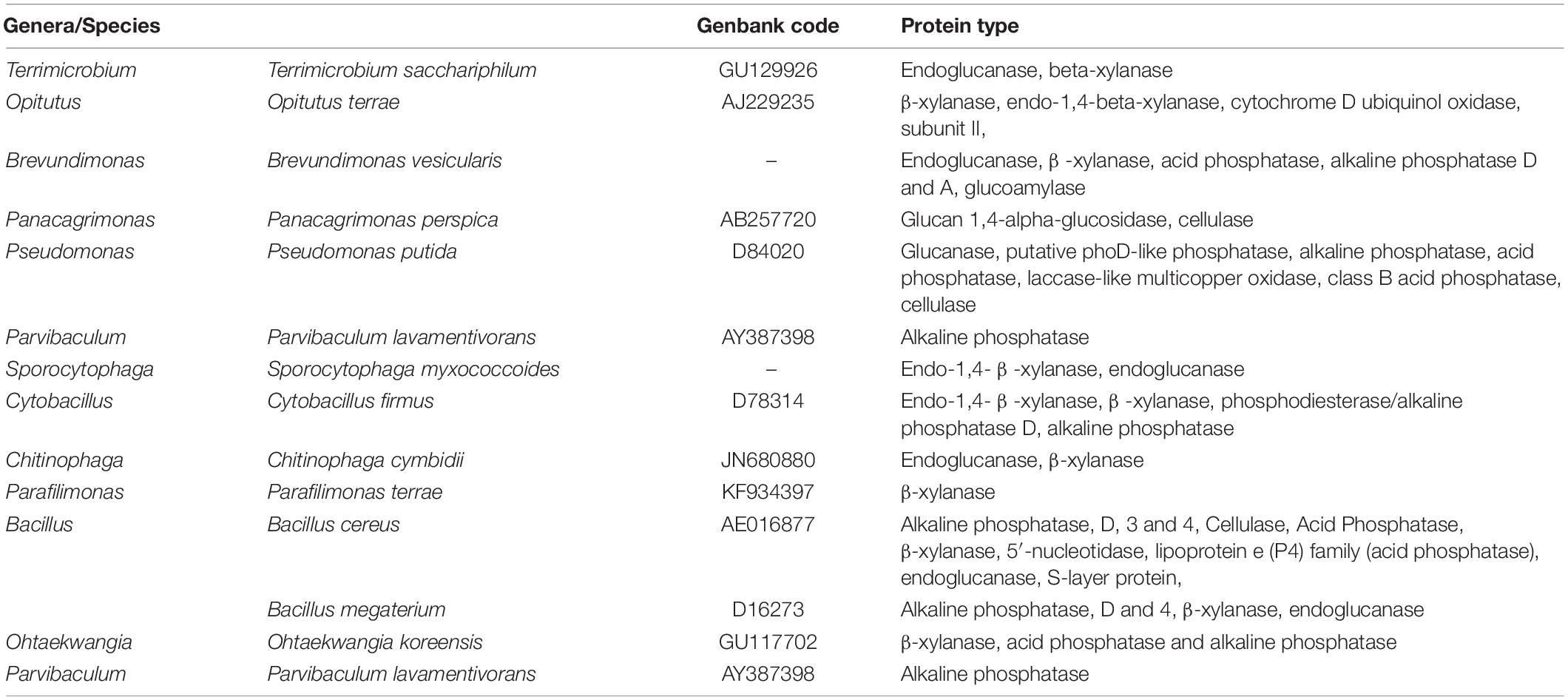
Table 7. Enzymatic profile detected in some bacterial species involved in organic matter degradation throughout the composting using different concentrations of textile waste, according to the UniProt database.
Figure 3 depicts the most dominant fungal phyla detected in the different mixtures and were compared. Samples of different mixtures showed several fungal similarities. The fungal communities in compost samples were largely dominated by Rozellomycota (31, 4, and 0.1%), Basidiomycota (17, 25, and 93%), Ascomycota (19, 37, and 4%), Mucoromycota (11, 29, and 3%), Mortierellomycota (9, 5, and 0%), and Aphelidiomycota (9, 0, and 0%), respectively, for mixtures A, B, and C. In mixture A, the most dominated fungi identified belonged to Rozellomycota, Mortierellomycota, as well as Aphelidiomycota. For mixture B, the highest number of phyla presented in compost samples with species belonging to Ascomycota and Mucoromycota, whereas for mixture C, species belonging to Basidiomycota phylum were identified as the most dominant in compost samples. It is noteworthy that there is disappearance in genera belonging to Mortierellomycota and Aphelidiomycota in mixture C. In the taxonomic distribution at the class level, members belonging to Tremellomycetes (11, 22, and 2%), Sordariomycetes (10, 26, and 2%), Saccharomycetes (1, 4, and 0.6%), Agaricomycetes (2, 2, and 90%), and Eurotiomycetes (4, 2, and 0.09%), respectively, for mixtures A, B, and C were present in majority.
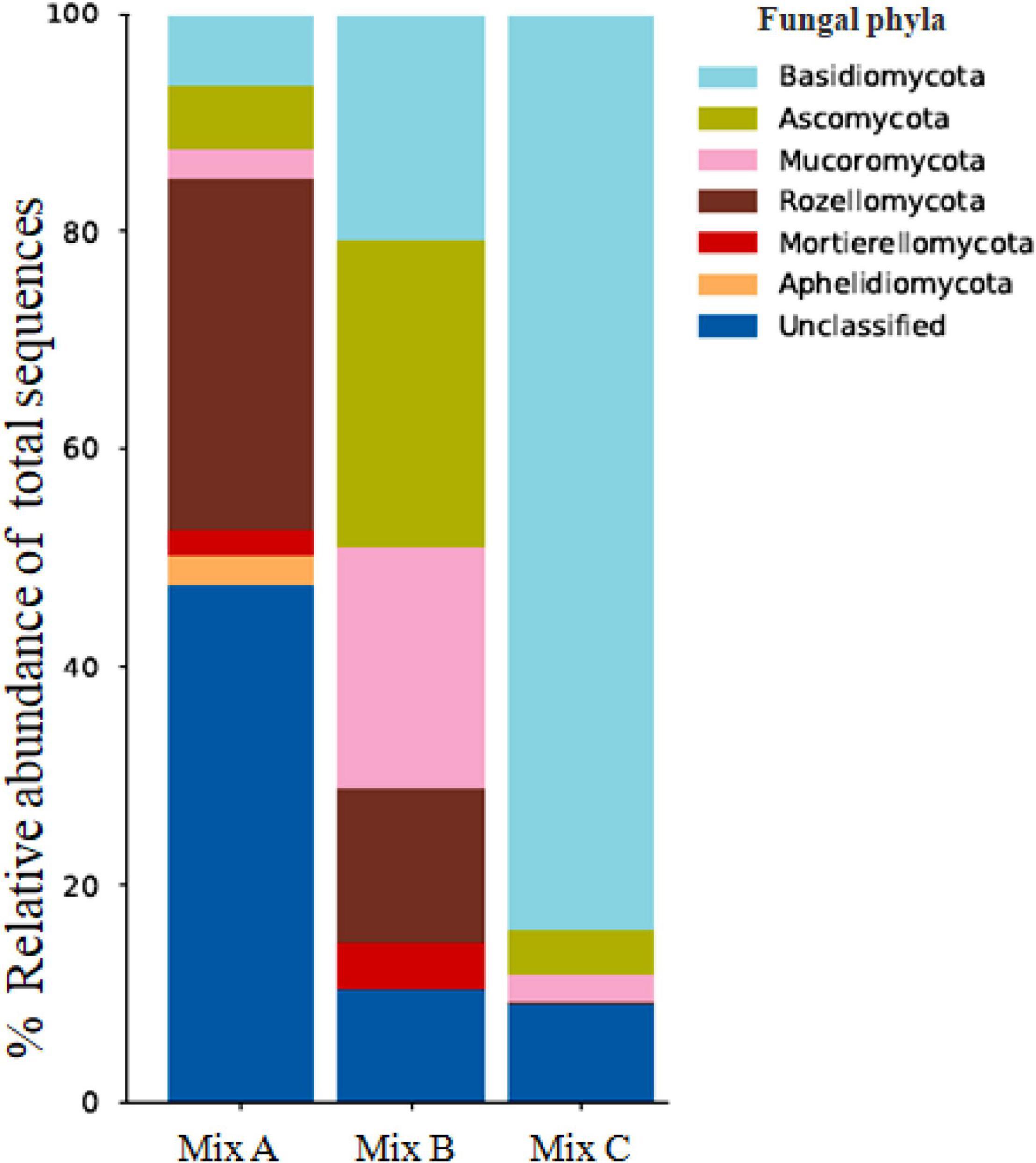
Figure 3. Relative abundance of fungal phyla in compost samples (Mix A, B, and C). “Unclassified” denotes bacterial sequences with no close similarity to sequences in the nucleotide database.
As for bacteria, a difference was observed from one mixture to another. The most dominant fungal were identified as Fusarium, Aspergillus, Penicillium, Rhizopus, and Apiotrichum, which is especially found in mixture B. For mixture A, Acremonium, Cyphellophora, and Phialophora were dominated (Figure 4). In contrast, Saccharomyces was highly dominant in mixture C, with the total disappearance of species belonging to Aspergillus (Table 8).
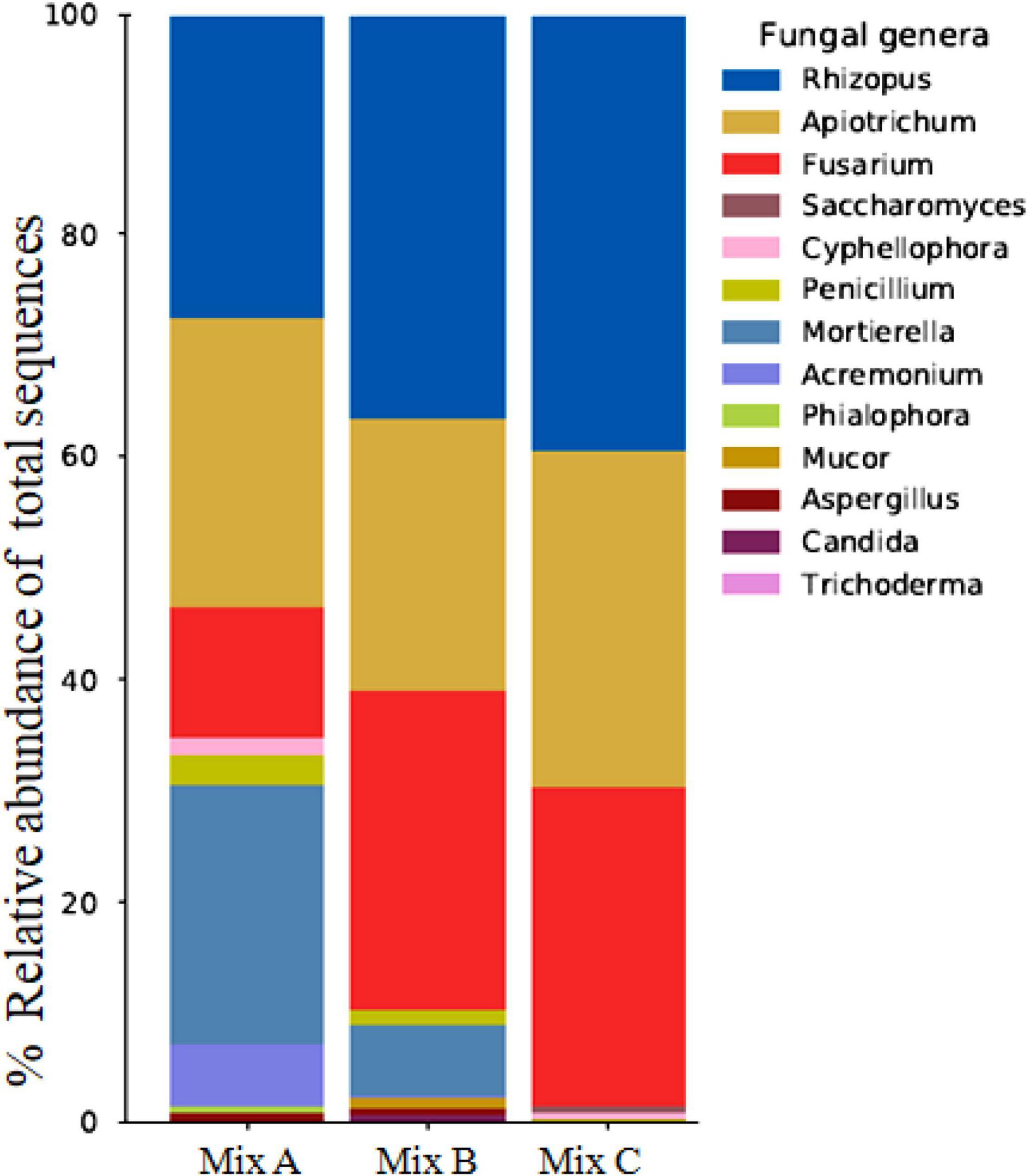
Figure 4. Relative abundance of fungal genera in compost samples (Mix A, B, and C).
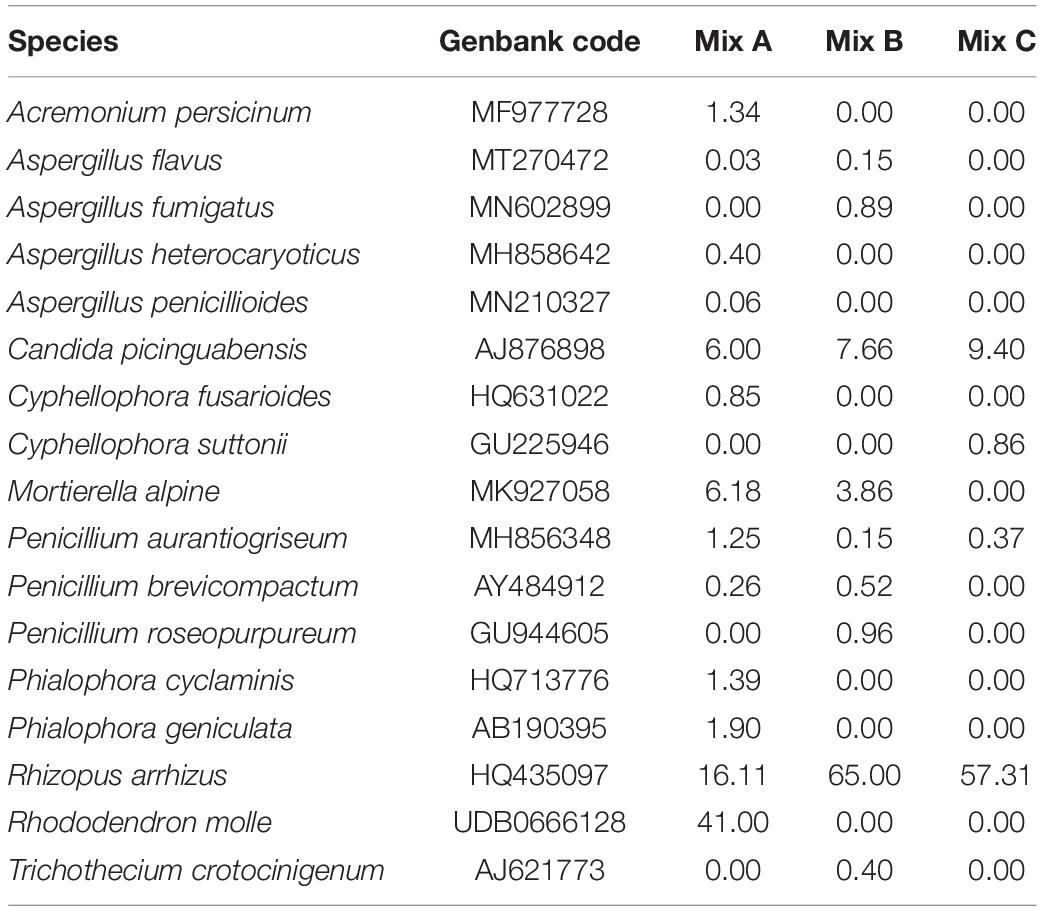
Table 8. The most abundant fungi species in each mixture (Mix A, B, and C).
As for bacterial communities, the UniProt database was used to identify different enzymatic activities; it was noted that many species are present in mixtures A and B but absent in mixture C; leading to the absence of their enzymatic activities (Table 9).
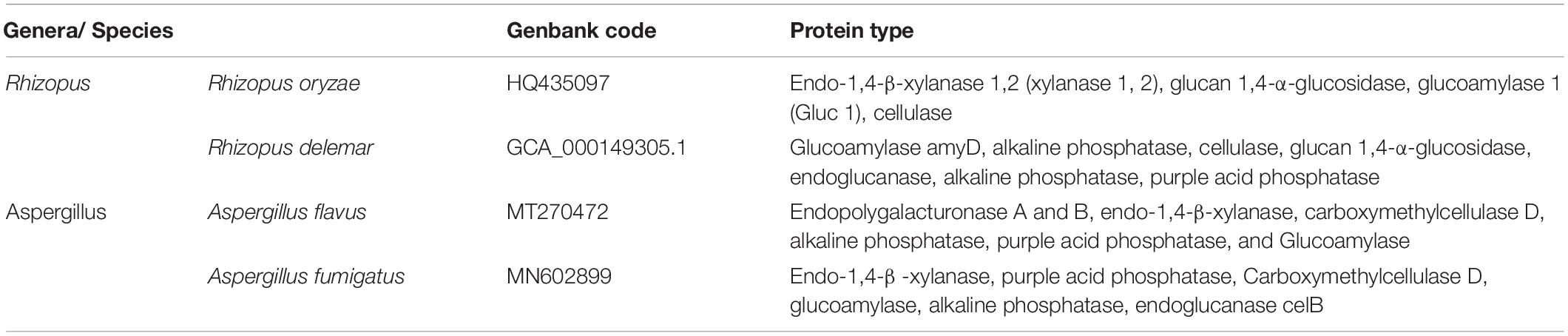
Table 9. Enzymatic profile detected in fungal communities involved in organic matter degradation throughout composting using different concentrations of textile waste, according to the UniProt database.
Composting cannot be understood without studying the microbial communities and their role in organic matter biotransformations. Recently, the microbial community changes during the composting process have attracted the attention of researchers. In this respect, this study provides insight into microbial changes across different textile waste concentrations through composting using the next-generation sequencing approaches. The level of bacteria and fungi diversity was considerably different from one mixture to another, and also the bacterial diversity was higher than the fungal diversity, which could be assigned to their small size and their ability to proliferate on a wide range of substrate even with high concentrations of heavy metals such as Bacillus, Pseudomonas, etc. (Barje et al., 2013; El Fels et al., 2014; Sharma et al., 2015; Łaba et al., 2019; Iravani and Varma, 2020), which is the case in this study. The comparison of the three study mixtures (A, B, and C) showed similarities at the phylum level for bacterial communities with a slight difference in the relative abundance of each phylum for each mixture. In contrast, the major difference was recorded with fungal communities, and the results obtained depicted that the diversity was higher for mixtures A and B, and lower for mixture C. Also, at the genus and species level, big differences in the distribution of the dominant species whether for bacterial or even fungal were noticed from one mixture to another, with a greater abundance in mixtures A and B compared with mixture C. This difference is probably owing to the better adaptation of these phyla to the initial feedstock concentration used in mixtures A and B compared with that in mixture C since the latter contains a high concentration of textile waste, thus explaining the difference in the physical-chemical results (C/N and concentration of heavy metals) and also the difference regarding the enzymatic activities (cellulase) recorded (Biyada et al., 2020b).
Furthermore, it is not wise to assume the presence of a standard composting microbiota in different composting samples since the concentration of feedstock determines the composition of microorganisms inside the compost mixture that nevertheless does not prevent that some of them might be primarily active agents in different kinds of composting processes, thus sharing similar characteristics during organic matter decaying. This is the case of Aspergillus, Fusarium, or even Penicillium, which are usually associated with lignocellulose material as feedstock during composting, and described as prevalent fungi, and Flavobacterium, Streptomyces, Paenibacillus, and Pseudomonas, for bacterial communities, as described in this study. These microorganisms have a tremendous metabolic ability during composting with lignocellulosic material as feedstock, and this was corroborated by the bioinformatic analysis using the UniPro database, thus proving the ability of these genera to produce a wide range of enzymatic activities. These findings are in agreement with those proved in previous studies (Chin et al., 2001; Yoon et al., 2011; Kim et al., 2013; Neher et al., 2013; Antunes et al., 2016; De Corato et al., 2018). At the same time, the predominance of these genera depends on their adaptation to the compost environment and subsequently their capacity to create specific combinations between nutrients and composting conditions, which could be explaining the variation in the abundance of microorganisms during textile waste composting. Additionally, this difference could be also linked to the microorganisms that play a decisive role during composting and which could determine the path of lignocellulosic compound degradation; these findings are in agreement with those elicited by Neher et al. (2013).
Otherwise, it should be noted that the temporal shifts in microbial communities (bacterial and fungal) during the composting were influenced chiefly by environmental conditions (temperature, humidity, pH, and electrical conductivity) and concentration of the feedstock used, which is confirmed previously (Vargas-garcía et al., 2014; Biyada et al., 2020b). It is notoriously shown that the bioavailability of the substrate stimulates microbial growth and consequently the degradation of organic matter. In fact, the presence of recalcitrant molecules with a high concentration which is the case of textile waste could be a limiting factor in microbial growth owing to their non-bioavailability because they are not easily dissipated by microorganisms, which is probably the case of mixture C. Actually, the high level of C/N in mixture C (59.29) at the beginning of composting, which is high compared with mixtures A and B, could be assigned to the poor humification of organic matter and/or low distribution of microbial activity inside of the silos (Biyada et al., 2020a). This is confirmed by microorganism diversity in this study, which is lower for mixture C than that present in mixtures A and B.
Furthermore, the low abundance of microbial communities in mixture C could be referred to the high concentration of heavy metals recorded with mixture C (Biyada et al., 2020b), which mainly results from the high concentration of textile waste in this mixture compared with the other ones (A and B). Despite their richness in lignocellulosic fiber, textile waste is considered as a fountainhead of heavy metals, owing to the use of chemicals and dyes produce in the production chain of textiles, such as sodium carbonate, sulfuric acid, acetic acid, copper sulfate sodium sulfate, ammonium chloride, and the pigments (Talouizte et al., 2017). The use of these products could preclude microorganism proliferation and consequently the degradation of organic matter. It is noticeable that mixture C presents a high concentration of heavy metals (especially of Zn) compared with mixtures A and B, which is considered one of the most toxic metals. The effect of Zn on the microbial enzyme activities (acid phosphatase, alkaline phosphatase, cellulase, b-D-glucosidase, and b-D-fructofuranosidase) was recorded (Kunito et al., 2001). These findings could also explain the variation of microorganism abundance in the three mixtures tested.
By analyzing carefully the results obtained, it can be noticed that the major difference between the three mixtures was observed for the actinomycete and fungal communities, which are less abundant and/or not found at all in mixture C compared with the other mixtures. According to several authors, a wide range of species belonging to actinomycete and fungal phyla have participated in the degradation of organic matter in general and cellulosic and lignocellulosic substrates in particular through their enzymatic systems (Tuomela et al., 2000; Verlag et al., 2000; Lynd et al., 2002; Tiquia, 2005; Raut et al., 2008; Vargas-garcía et al., 2014). Their presence indicates a more efficient and faster composting process, which is the case of mixtures A and B. The low abundance of these phyla could affect the rate of organic matter degradation, which is the case for mixture C in this study.
Eventually, it can be concluded that the nutritional value of initial feedstock and the concentration of heavy metals in different mixtures are probably the major factors affecting the microbial communities during textile waste composting. At the same time, the composition of the microbial communities throughout the composting of lignocellulosic materials is depending on the greater or lesser degree of the bioavailability of the nutrients.
Microorganisms are abundant and diverse in compost. Microbial communities are organized and affected by the feedstock concentration, which is demonstrated during this study. By increasing the concentration of textile waste in the mixtures used and therefore the concentration of heavy metals especially Zn, the abundance of species belonging to fungal and actinomycete communities decreased and consequently the rate of organic matter degradation. By analyzing the composition of microbiota associated with different concentrations of the feedstock used, the most effective control of the composting process can be achieved. Ultimately, it can be concluded that mixtures A and B, containing either 40 or 60% of textile waste, could be the best combination for the valorization of such waste using composting.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
SB, MM, and JU: conceptualization. SB, DV, JU, EM, and SV: methodology. SB and TD: software. MM and JU: validation. SB: formal analysis, investigation, data curation, and writing – original draft. JU, SV, and SB: resources. SB, MM, JU, and SV: writing, review, and editing. SB and JU: visualization. MM, JU, and SV: supervision. MM: project administration. JU: funding acquisition. All authors have read and agreed to the published version of the manuscript.
This research was supported by the National Center for Scientific Research in Rabat (CNRST) and the Research Council of Lithuania (LMT) grant S-MIP-19-61 to JU. The visit of SB to Vilnius Gediminas Technical University was supported by an Erasmus + program. The article processing charge (APC) was funded by the Vilnius Gediminas Technical University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alsac, N. (2007). Dosage des métaux lourds (As, Cd, Cr, Cu, Ni, Pb, Zn et Hg) dans les sols par ICP-MS. Ann. Toxicol. Anal. 19, 37–41. doi: 10.1051/ata:2007006
Antunes, L. P., Martins, L. F., Pereira, R. V., Thomas, A. M., Barbosa, D., Lemos, L. N., et al. (2016). Microbial community structure and dynamics in thermophilic composting viewed through metagenomics and metatranscriptomics. Sci. Rep. 6:38915. doi: 10.1038/srep38915
Barje, F., El Fels, L., El Hajjouji, H., Winterton, P., and Hafidi, M. (2013). Biodegradation of organic compounds during co-composting of olive oil mill waste and municipal solid waste with added rock phosphate. Environ. Technol. 34, 2965–2975. doi: 10.1080/09593330.2013.796009
Biyada, S., Merzouki, M., Elkarrach, K., and Benlemlih, M. (2020a). Spectroscopic characterization of organic matter transformation during composting of textile solid waste using UV – visible spectroscopy, infrared spectroscopy and X-ray diffraction (XRD). Microchem. J. 159:105314. doi: 10.1016/j.microc.2020.105314
Biyada, S., Merzouki, M., Imtara, H., Alajmi, M. F., Elkarrach, K., Mechchate, H., et al. (2021). Advanced characterization of organic matter decaying during composting of industrial waste using spectral methods. Processes 9, 1–13.
Biyada, S., Merzouki, M., Imtara, H., and Benlemlih, M. (2019). Assessment of the maturity of textile waste compost and their capacity of fertilization. Eur. J. Sci. Res. 154, 399–412.
Biyada, S., Merzouki, M., Taisija, D., Vasiliauskienė, D., Urbonavicius, J., Marčiulaitienė, E., et al. (2020b). Evolution of microbial composition and enzymatic activities during the composting of textile waste. Appl. Sci. 10:3758. doi: 10.3390/app10113758
Callahan, B. J., Mcmurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2?: high-resolution sample inference from illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Chin, K., Liesack, W., and Janssen, P. H. (2001). Opitutus terrae gen. nov., sp. nov., to accommodate novel strains of the division ‘Verrucomicrobia’ isolated from rice paddy soil. Int. J. Syst. Evol. Microbiol. 51, 1965–1968. doi: 10.1099/00207713-51-6-1965
De Corato, U., Salimbeni, R., De Pretis, A., Patruno, L., Avella, N., Lacolla, G., et al. (2018). Microbiota from ‘next-generation green compost’ improves suppressiveness of composted municipal-solid-waste to soil-borne plant pathogens. Biol. Control 124, 1–17. doi: 10.1016/j.biocontrol.2018.05.020
El Fels, L., El Ouaqoudi, F. Z., Barje, F., Hafidi, M., and Ouhdouch, Y. (2014). Two culture approaches used to determine the co-composting stages by assess of the total microflora changes during sewage sludge and date palm waste co-composting. J. Environ. Health Sci. Eng. 12:132. doi: 10.1186/s40201-014-0132-4
Iravani, S., and Varma, R. S. (2020). Bacteria in heavy metal remediation and nanoparticle biosynthesis bacteria in heavy metal remediation and nanoparticle biosynthesis. ACS Sustain. Chem. Eng. 14, 5395–5409. doi: 10.1021/acssuschemeng.0c00292
Kim, J., Alkawally, M., Brady, A. L., Dunfield, P. F., Rijpstra, W. I. C., and Damste, J. S. S. (2013). Chryseolinea serpens gen. nov., sp. nov., a member of the phylum Bacteroidetes isolated from soil. Int. J. Syst. Evol. Microbiol. 63, 657–660. doi: 10.1099/ijs.0.039404-0
Kunito, T., Saeki, K., Goto, S., and Hayashi, H. (2001). Copper and zinc fractions affecting microorganisms in long-term sludge-amended soils. Bioresour. Technol. 79, 135–146. doi: 10.1016/s0960-8524(01)00047-5
Łaba, W., Rodziewicz, A., and Choin, A. (2019). Enhancement of pig bristles waste bioconversion by inoculum of keratinolytic bacteria during composting. Waste Manag. 84, 269–276. doi: 10.1016/j.wasman.2018.11.052
López-gonzález, J. A., Vargas-garcía, M. C., López, M. J., Suárez-estrella, F., Jurado, M., and Moreno, J. (2015). Biodiversity and succession of mycobiota associated to agricultural lignocellulosic waste-based composting. Bioresour. Technol. 187, 305–313. doi: 10.1016/j.biortech.2015.03.124
Lynd, L. R., Weimer, P. J., Van Zyl, W. H., and Pretorius, I. S. (2002). Microbial cellulose utilization?: fundamentals and biotechnology. Microbiol. Mol. Biol. Rev. 66, 506–577. doi: 10.1128/MMBR.66.3.506
Maccready, J. S., Elbert, N. J., Quinn, A. B., and Potter, B. A. (2013). An assessment of bacterial populations in a static windrow compost pile. Compost Sci. Util. 21, 110–120. doi: 10.1080/1065657X.2013.837272
Mancinelli, E., Baltrėnaitė, E., Baltrėnas, P., Marčiulaitienė, E., Limane, B., and Bartkevic, V. (2017). Dissolved organic carbon content and leachability of biomass waste biochar for trace metal (Cd, Cu and Pb) speciation modelling. J. Environ. Eng. Landscape Manag. 25, 354–366. doi: 10.3846/16486897.2017.1339047
Neher, D. A., Weicht, T. R., Bates, S. T., Leff, J. W., and Fierer, N. (2013). Changes in bacterial and fungal communities across compost recipes, preparation methods, and composting times. PLoS One 8:e79512. doi: 10.1371/journal.pone.0079512
Partanen, P., Hultman, J., Paulin, L., Auvinen, P., and Romantschuk, M. (2010). Bacterial diversity at different stages of the composting process. BMC Biotechnol. 10:94. doi: 10.1186/1471-2180-10-94
Quast, C., Klindworth, A., Pruesse, E., Schweer, T., Horn, M., and Glo, F. O. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41, 1–11. doi: 10.1093/nar/gks808
Raut, M. P., William, S. P. M. P., Bhattacharyya, J. K., Chakrabarti, T., and Devotta, S. (2008). Microbial dynamics and enzyme activities during rapid composting of municipal solid waste – a compost maturity analysis perspective. Bioresour. Technol. 99, 6512–6519. doi: 10.1016/j.biortech.2007.11.030
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Sharma, D., Kanchi, S., and Bisetty, K. (2015). Biogenic synthesis of nanoparticles?: a review. Arab. J. Chem. 12, 3576–3600. doi: 10.1016/j.arabjc.2015.11.002
Song, C., Li, M., Jia, X., Wei, Z., Zhao, Y., Xi, B., et al. (2014). Comparison of bacterial community structure and dynamics during the thermophilic composting of different types of solid wastes?: anaerobic digestion residue, pig manure and chicken manure. Microb. Biotechnol. 7, 424–433. doi: 10.1111/1751-7915.12131
Soobhany, N., Mohee, R., and Kumar, V. (2015). Comparative assessment of heavy metals content during the composting and vermicomposting of municipal solid waste employing Eudrilus eugeniae. Waste Manag. 39, 130–145. doi: 10.1016/j.wasman.2015.02.003
Talouizte, H. (2014). Traitement Physique Et Biologique Des Effluents De Textile De La Ville De Fčs Et Evaluation De Leurs Effets Génotoxique Et Phytotoxique. Morocco: Sidi Mohamed Ben Abdellah University.
Talouizte, H., Merzouki, M., and Benlemlih, M. (2017). Treatment of real textile wastwater using SBR technology?: effect of sludge age and operational parameters. J. Biotechnol. Lett. 4, 79–83.
Temporal-lara, B., Melendez-pastor, I., Gómez, I., and Navarro-pedreño, J. (2016). Wastewater biosolid composting optimization based on UV-VNIR spectroscopy monitoring. Sensors 16:1919. doi: 10.3390/s16111919
Tiquia, S. M. (2005). Microbiological parameters as indicators of compost maturity. J. Appl. Microbiol. 99, 816–828. doi: 10.1111/j.1365-2672.2005.02673.x
Tuomela, M., Vikman, M., Hatakka, A., and Itävaara, M. (2000). Biodegradation of lignin in a compost environment: a review. Bioresour. Technol. 72, 169–183. doi: 10.1016/S0960-8524(99)00104-2
Vargas-garcía, M. C., Jos, M. L. J. A., Jurado, M., Moreno, J., and Su, F. (2014). Enzymatic characterization of microbial isolates from lignocellulose waste composting?: chronological evolution pez-Gonz a. J. Environ. Manag. 145, 137–146. doi: 10.1016/j.jenvman.2014.06.019
Verlag, F., Lednickal, D., and Margo, J. M. (2000). Isolation and identification of cellulolytic bacteria involved in the degradation of natural cellulosic fibres. Syst. Appl. Microbiol. 23, 292–299. doi: 10.1016/S0723-2020(00)80017-X
Keywords: textile waste, feedstock concentration, microbial communities, next-generation sequencing, enzymatic activities
Citation: Biyada S, Merzouki M, Dėmčėnko T, Vasiliauskienė D, Marčiulaitienė E, Vasarevičius S and Urbonavičius J (2022) The Effect of Feedstock Concentration on the Microbial Community Dynamics During Textile Waste Composting. Front. Ecol. Evol. 10:813488. doi: 10.3389/fevo.2022.813488
Received: 11 November 2021; Accepted: 28 January 2022;
Published: 02 March 2022.
Edited by:
Leonardo Erijman, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), ArgentinaReviewed by:
Suzan Pantaroto De Vasconcellos, Federal University of São Paulo, BrazilCopyright © 2022 Biyada, Merzouki, Dėmčėnko, Vasiliauskienė, Marčiulaitienė, Vasarevičius and Urbonavičius. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saloua Biyada, c2Fsb3VhLmJpeWFkYUB1c21iYS5hYy5tYQ==; c2Fsb3VhYml5YWRhQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.