 Zelin Chen
Zelin Chen Shaobo Ma1,3†
Shaobo Ma1,3† Geng Qin
Geng Qin Qiang Lin
Qiang Lin- 1CAS Key Laboratory of Tropical Marine Bio-Resources and Ecology, South China Sea Institute of Oceanology, Chinese Academy of Sciences, Guangzhou, China
- 2Center for Ocean Mega-Science, Chinese Academy of Sciences, Qingdao, China
- 3University of Chinese Academy of Sciences, Beijing, China
- 4Key Laboratory of South China Sea Fishery Resources Exploitation & Utilization, Ministry of Agriculture, South China Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Guangzhou, China
Diffusing fluid from methane seepage in cold seep field creates zones with physicochemical gradients and divergent ecosystems like the mussel beds and clam beds. Three species of brittle stars (Ophiuroidea) were discovered in the Haima cold seep fields, of which Ophiophthalmus serratus and Histampica haimaensis were found on top of or within mussel beds and clam beds, whereas Amphiura sp. was only collected from muds in the clam bed assemblage. Here, we evaluated the genetic signatures of micro-environmental adaptation of brittle stars to cold seep through the comparison of mitogenomes. This study provided two complete mitogenome sequences of O. serratus and Amphiura sp. and compared with those of H. haimaensis and other non-seep species. We found that the split events of the seep and non-seep species were as ancient as the Cretaceous period (∼148–98 Mya). O. serratus and H. haimaensis display rapid residue mutation and mitogenome rearrangements compared to their shallow or deep-sea relatives, in contrast, Amphiura sp. only show medium, regardless of nucleotide mutation rate or mitogenome rearrangement, which may correlate with their adaptation to one or two micro-ecosystems. Furthermore, we identified 10 positively selected residues in ND4 in the Amphiura sp. lineage, suggesting important roles of the dehydrogenase complex in Amphiura sp. adaptive to the cold seep environment. Our results shed light on the different evolutionary strategies during colonization in different micro-environments.
Introduction
The deep sea represents an extreme and harsh environment of darkness, hypoxia, high hydrostatic pressure, low temperature, and low energy, and is generally characterized by low biomass. However, chemosynthetic habitats including hydrothermal vents and cold seeps scatter the deep seas as biodiversity hot spots (Levin, 2005). Cold seeps are seafloor manifestations where reduced fluid include hydrogen sulfide, methane, petroleum, and other hydrocarbon-rich migration through sediments from the subsurface to the seabed and into the water column. Since the first exploration of the cold seep biological communities at the Florida Escarpment in 1983 (Paull et al., 1984), hundreds of seep areas were exhibited at Gulf of Mexico, Monterey Bay, Eastern Mediterranean, Japan and Kurile trenches, and South China Sea, etc., ranging from 400 to >6,000 m depth (Sibuet and Olu, 1998; Feng et al., 2018).
More than 600 macrofaunal species have been reported in these seep areas (German et al., 2011). Several animals are holobionts, like the tube-dwelling “fan worm” annelids Laminatubus and Bispira, which is hosted and nourished by aerobic methane-oxidizing bacteria Methylococcales (Goffredi et al., 2020). Other animals are epibionts or feeding on bacterial mats, mud, debris, or other animals, many of which may only be visitors. Seep or vent sites create water columns with different composition of methane, hydrogen sulfide, etc. and form different macrofaunal communities. A comparison between vent and seep squat lobster Shinkaia crosnieri identified differentially expressed genes with amino acid substitutions related to oxidation resistance and xenobiotic detoxification, indicating adaption to the vent or seep condition (Xiao et al., 2020). However, the origin and colonization of these seep animals remains unclear. We speculated that species occupying different niches in the same cold-seep area may facilitate different adaptive strategies (Zhu et al., 2020).
Mitochondria (MT) are the center of energy production, through oxidative phosphorylation. The mitogenome contains 13 protein-coding genes (PCGs), encoding essential subunits of OXPHOX complexes, the pivot of oxidative phosphorylation and aerobic respiration. Mutations in the PCGs may facilitate or detriment animals and promote adaption to many special ecosystems or behaviors such as high altitude, dark caves and flying. Coupling a high daily energy consumption for flying, sparrow mitochondria oxidize NADH and succinate more than 1.8 times faster than rat mitochondria (Kuzmiak et al., 2012). The deep-sea and chemosynthetic vent or cold-seep environment also impact the MT genes in inhabiting marine animals and positively selected residues are identified in a number of vent and cold-seep aborigines, one site of ATP6 was identified to be positively selected in the cold-seep gastropod Phymorhynchus buccinoides (Du et al., 2020), and deep-sea vesicomyids were supposed to be more sensitive to non-synonymous substitution in MT genes than the sublittoral bivalves (Liu et al., 2018).
Brittle stars (Ophiuroidea) are common macrofaunal taxa in deep-sea benthic ecosystem. There are more than 2,000 ophiuroid species in the world ocean, of which more than half dwell in the deep sea (>1,000 m), but less than 20 species are recorded endemic to the seep chemosynthesis ecosystem (Rodrigues et al., 2011; Okanishi et al., 2020). Three ophiuroid species were highly abundant in the newly exhibited Haima cold seep in the South China Sea, which belong to three different families: Ophiothamnidae, Ophiacanthidae, and Amphiuridae (Dong et al., 2021; Li et al., 2021; Nethupul et al., 2022). Histampica haimaensis (Family: Ophiothamnidae) is distributed across both mussel beds and clam beds (Li et al., 2021), of which mussel beds dominated by Gigantidas haimaensis feature ecosystems at active seepage in Haima cold seep areas, while living clams are rare and occur with half shells buried in mud (Liang et al., 2017; Feng et al., 2018).
In the present study, we observed that the Histampica and Ophiacanthidae, but not Amphiuridae species, were on top of or within mussels and clams, but only Amphiuridae were collected from the muddy sediments at the remotely operated vehicle (ROV) site with clams. We hypothesize that different micro-ecosystems may shape the mitogenome of the three species in different way. Therefore, we sequenced the complete mitogenomes of two species (Ophiacanthidae: Ophiophthalmus serratus and Amphiuridae: Amphiura sp.) at the Haima cold seep. Combined with the newly sequenced mitochondrial genome of the other Haima dominant species Histampica haimaensis. We conducted a phylogenetic analysis of PCGs and compared gene order of ophiuroids among non-reduced and cold seep ecosystems, to reveal their potential adaptive evolution to chemosynthetic habitats, providing insight into different evolutionary strategies for cold seep micro-ecosystem adaption.
Results
Phylogenetic relationship of cold-seep brittle stars
Three species (Histampica haimaensis, Ophiophthalmus sp., and Amphiura sp.) of brittle stars were collected from three diving sites (ROV1, ROV3, and ROV4, same as R1, R3, and R4 in Ke et al., 2022) at the Haima cold seep in the northwest South China Sea (Ke et al., 2022). ROV1 was dominated by mussel beds (Gigantidas haimaensis), whereas ROV4 was dominated by clam beds (Calyptogena marissinica), and there is no active seepage at ROV3. Ophiophthalmus sp. and H. haimaensis occupied all three stations, include the mussel beds and clam beds, and were observed on top or within mussels and clams. Amphiura sp. was only collected from the muddy sediment at ROV3 and ROV4 (in the assemblage of clam beds with lower biodiversity) (Figures 1A,B). It was rare to observe Amphiura sp. from the video, perhaps because of its smaller body size and burrowing lifestyle as its congeners like A. chiajei and A. filiformis (Skold and Gunnarsson, 1996).
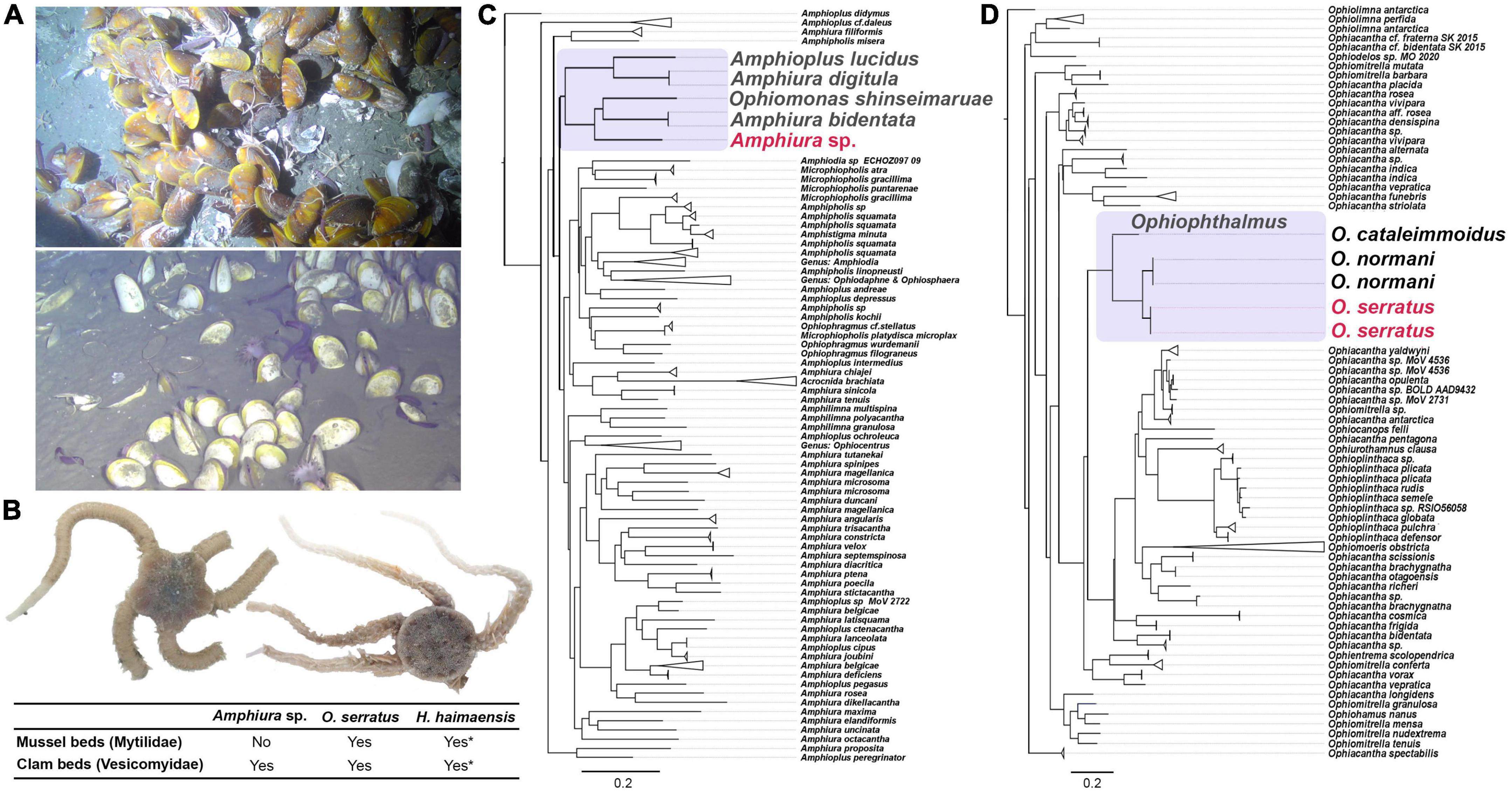
Figure 1. Description of two brittle stars collected from Haima cold seep. (A) Two dominant ecosystems of the Haima cold seep: Gigantidas haimaensis mussel bed (top) and Calyptogena marissinica clam bed (bottom); (B) sample of brittle stars Amphiura sp. (left) and Ophiophthalmus serratus (right) and their occupied ecosystem(s); stars means refer to Li’s study (Li et al., 2021); (C) phylogenetic relationship using maximum likelihood analysis of COX1 sequences of Amphiuridae, our sample was sister of Amphiura bidentata and Ophiomonas shinseimaruae, but the identity is as low as 81.5 and 80.9%, respectively; (D) phylogenetic relationship by ML analysis of Ophiacanthidae COX1 sequences. The nucleotide similarity between O. serratus and O. normani is ∼92%.
The COX1 phylogenetic relationship and NCBI alignment indicated that Amphiura sp. was sister to Amphiura bidentata and recently new deep-sea species Ophiomonas shinseimaruae (Okanishi et al., 2021; Figure 1C) and was not similar to the other Amphiura sp. discovered from the Weijia Guyot seamount (Na et al., 2019). The sequence identity between our Amphiura sp. and A. bidentata (or O. shinseimaruae) was as low as 81.5% (80.9%), indicating it may be a new species or a species without COX1 barcode in the NCBI database. The COX1 sequence of Ophiophthalmus sp. is most similar to Ophiophthalmus normani (Figure 1D), and the COX1 nucleic identity between Ophiophthalmus sp. and the newly reported Ophiophthalmus serratus (Nethupul et al., 2022) is 97.9%. Combining with the morphological evidence (Supplementary Figure 1), we declared it as O. serratus.
Mitochondrial genome characterization
We sequenced and assembled the MT genome of two ophiuroids, Amphiura sp. (Family Amphiuridae) and O. serratus (Family Ophiacanthidae), collected from the active cold seep fields—Haima cold seep. The assembled MT genome sizes are 16,147 and 16,705 bp respectively, longer than those of H. haimaensis (15,759) from the same seep field. The MT genome size of Amphiura sp. was medium in Amphiuridae (15,348–16,907 bp), while mitogenome size of O. serratus was ∼1 kbp long than that of O. linea (15,845) in the same family. The MT genome of O. serratus had an overall higher A + T% (71.42%) than that of Amphiura sp. (62.04%) and H. haimaensis (61.50%), also higher than O. linea (68.58%). O. serratus. also had the highest AT dinucleotide content (12.27%) compared to the other ophiuroids. In addition, the A + T content and AT dinucleotide of deep-sea or Antarctic ophiuroids is higher than those of shallow ophiuroids (Wilcoxon rank sum exact test, p = 0.094 and 0.012, respectively). The higher A + T content of mtDNA was also characterized in deep-sea starfish, supposed to be combined outcome of random genetic drift and deep-sea adaption (Sun et al., 2022). In contrast to the positive GC-skew in H. haimaensis (Li et al., 2021), the GC-skew (i.e., [G-C]/[G + C]) of Amphiura sp. and O. serratus were both negative (−0.28 and −0.14 respectively), similar to all other studied ophiuroids (except H. haimaensis).
Amphiura sp. contains 37 MT genes, including 13 PCGs, two rRNA, and 22 tRNA genes, whereas O. serratus encodes another complete tRNA-Met (anticodon CAT), which may derive from a tandem duplication event. All other reported Ophiuroidea MT genomes contain 37 genes, except O. brevirima, which has a shorter tRNA-Leu. The largest intergenic regions (495 and 654 bp) were located following ND6 in both Amphiura sp. and O. serratus, which were also present in other species of the same family Amphiuridae and Ophiacanthidae respectively. There were windows with low GC contents and high GC skew within this region (Figure 2) and a poly-G stretch, indicating it may be the control region of the mitogenomes in both species (Bronstein et al., 2018).
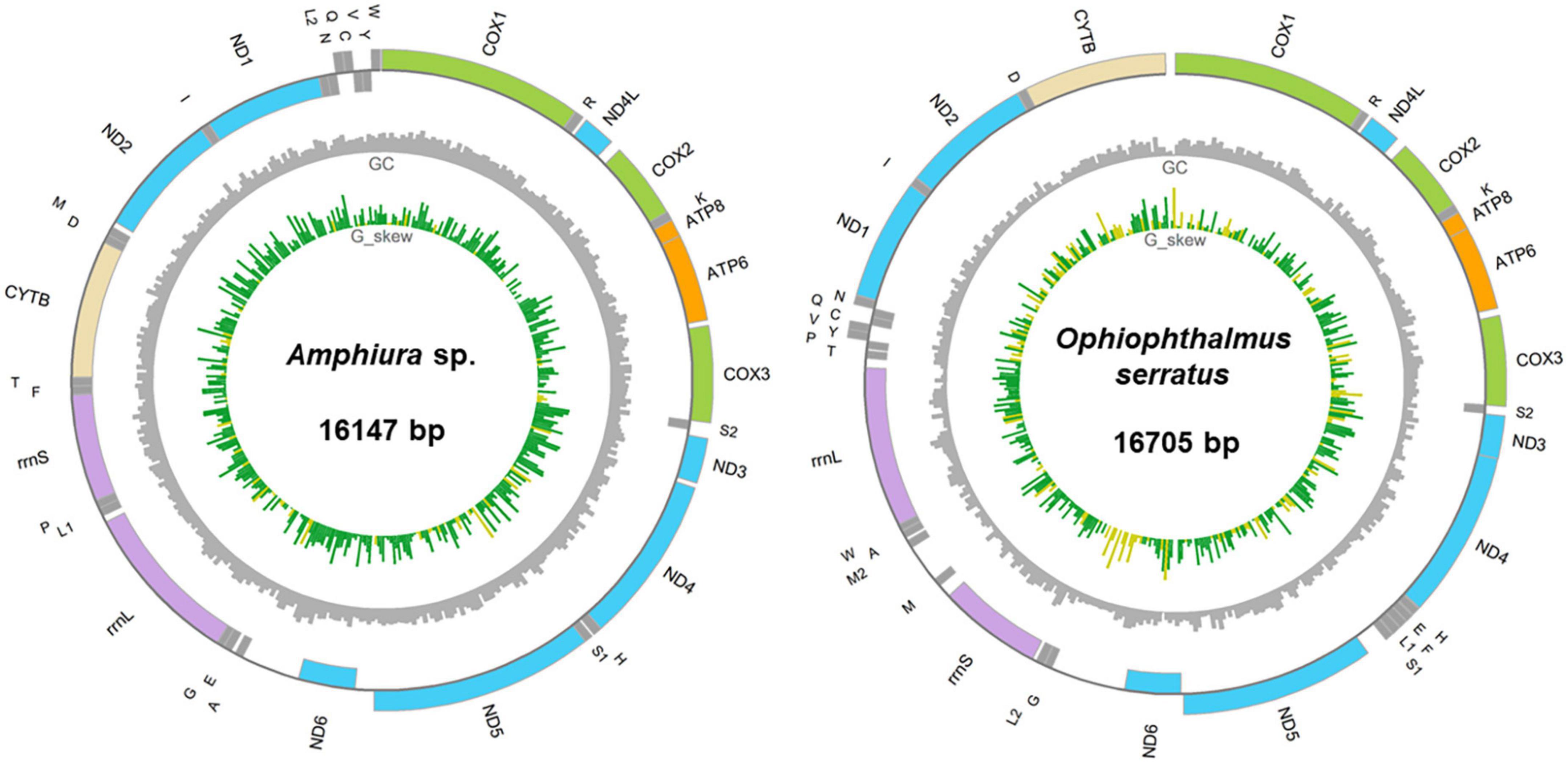
Figure 2. Circular map of two sequenced cold-seep ophiuroids, Amphiura sp. and Ophiophthalmus serratus. Circular tracks from inside to outside is GC skew, GC percentage with window size 40 bp and genes. Yellow bars in the GC skew track indicated positive value and green ones were negative value. Outward (inward) genes are on the heavy (light) strand.
Codon usage and relative synonymous codon usage (RSU) values in the PCGs of Amphiura sp. and O. serratus were calculated. The most and least used amino acid of PCGs in both species are isoleucine (11.17 and 15.65%) and cysteine (1.20 and 1.08%), respectively, similar to other Amphiura and Ophiacantha (Ophiacanthidae). The correlation between Amphiura sp. and deep-sea (1,950 m) Amphiura sp. JN-2020 is 0.987, higher then compared to the two shallow water species A. digitula (0.979) and A. sinicola (0.949). The most commonly used codons are TTT (Phe), ATA (Ile), ATT (Ile), TTA (Leu), CTA (Leu) and GGA (Gly) in Amphiura sp., and TTA (Leu), ATA (Ile), TTT (F), ATT (Ile), AAA (Asn) and GGA (Gly) in O. serratus, showing an A + T bias in both species, especially in O. serratus.
Phylogenetic divergence of Ophiuroidea in South China Sea cold seep fields
Maximum likelihood (ML) phylogenetic tree was constructed for all PCGs from 30 ophiuroids, including the three cold seed species from the South China Sea. These three species were derived from three different clades (Figure 3, colored boxes around species names), and divergent in the early Cretaceous (∼148–98 Mya) from their most common ancestors (Figure 3). The divergence time is comparable to that between Bathymodiolinae and its shallow-water sister group Modiolinae roughly 110 million years ago (Mya) (Zhang et al., 2021), which is consistent with the suggestion that many vent and seep taxa underwent adaptive radiation during the Mesozoic era (Kiel and Little, 2006).
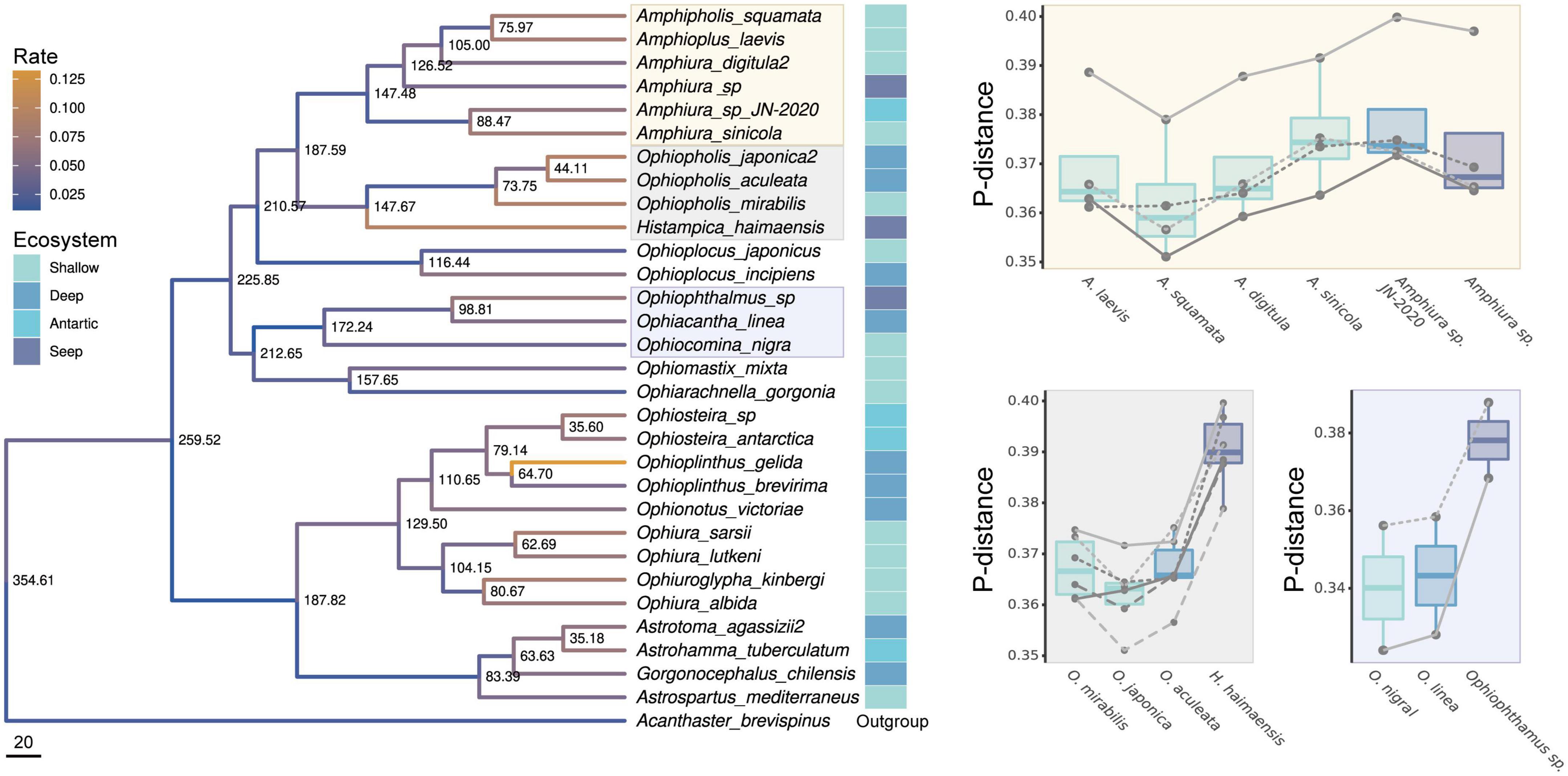
Figure 3. Reconstructed phylogenetic relationship of brittle star mitogenome. Node labels show the dating time of the BEAST2 analysis. Branches were colored by the rate. Three clades consist of shallow, deep-sea and cold-seep species were labeled in colored boxes. For each clade, p-distances between in-clade species to its sister-clade species were illustrated on the right panels of corresponding colors as boxes around the species names, with boxes colored by ecosystem; each line represents a sister-clade species.
O. serratus and H. haimaesis showed a higher nucleic mutation rate than their shallow and deep-sea relatives (0.00662 vs. 0.00506 and 0.00898 vs. 0.00840 respectively), while Amphiura sp. showed a lower nucleic mutation rate (0.00437 vs. 0.00511–0.00767). We also compared the p-distance between the species in each clade reference with their sister clades. The p-distance between O. serratus and clade (O. mixta + O. gorgonia) are larger than between shallow (O. nigra) or deep-sea (O. linea) species and the same clade (mean: 0.378 vs. 0.340 (O. linea, p = 0.33) and 0.343 (O. nigra, p = 0.33), though not significant because of small testing sample sizes (Figure 3, gray panel); same pattern in the case of H. haimaesis [mean: 0.391 vs. 0.367 (O. mirabilis), 0.362 (O. japonica), and 0.367 (O. aculeata), p = 0.002] (Figure 3, bottom right panel), suggesting rapid nucleotide evolution in these two seep species. However, there was no difference (or slightly lower) between Amphiura sp. and its shallow or deep-sea relatives (Figure 3, top right panel).
Divergent gene order for broader niches
MT gene order was thought to be conserved in brittle stars (Galaska et al., 2019). The gene orders of O. mirabilis, O. nigra and O. gorgonia, which belong to three different families, were the same and set as the gene order of their most recent common ancestor (Figure 4, node “B” in box) and Amphilepidida. The gene orders of other nodes were reconstructed using MGRA based on the reversal distance in the ML phylogenetic topology.
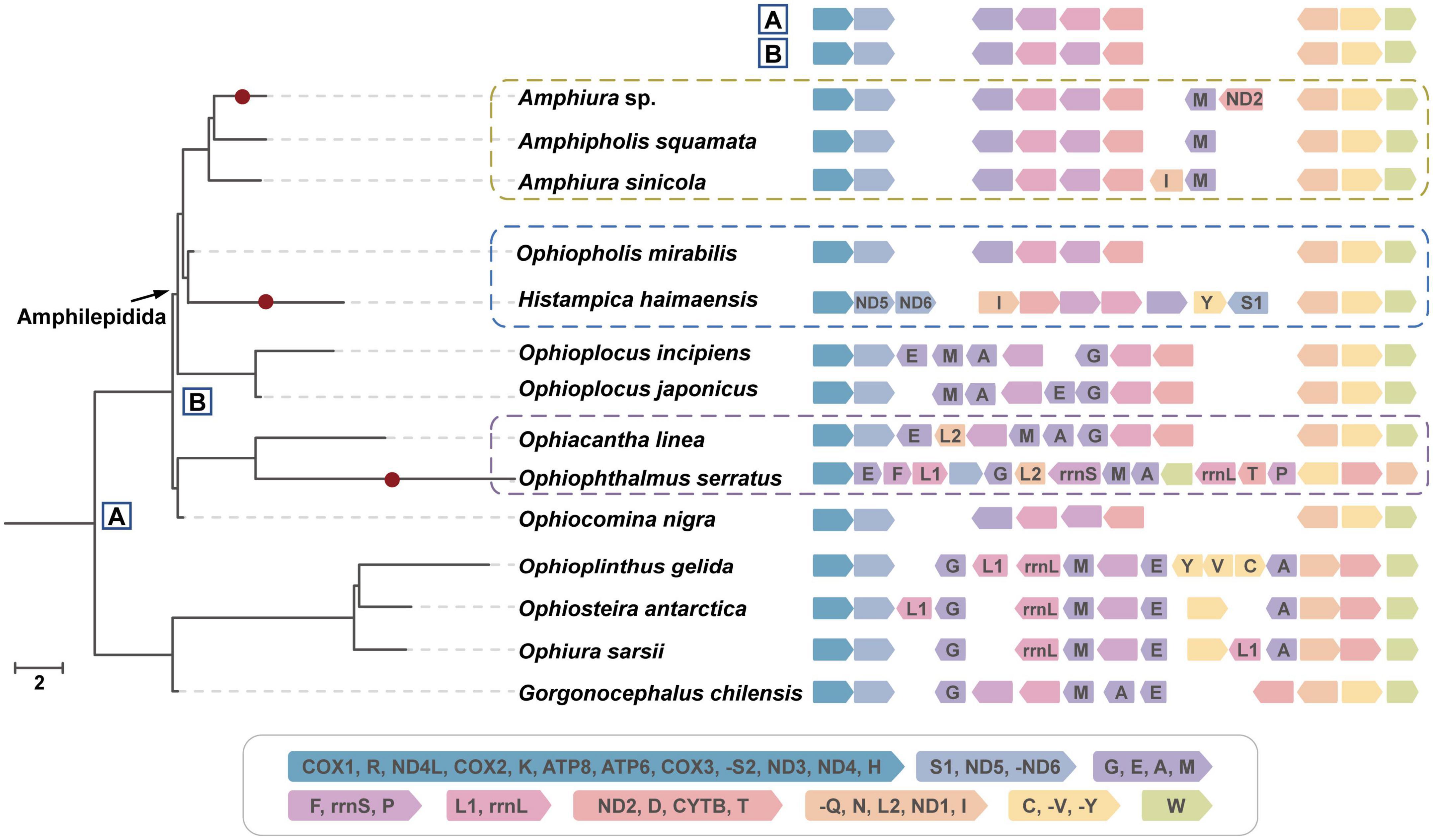
Figure 4. Rearrangement of MT genes. The same phylogenetic topology as the ML tree was showed on left, with branch length represented reversal distance. Gene order of each species were showed on the right, with two ancestors (box A: Ophiuroidea and box B: Ophintegrida) on top. MT genes were colored according to the blocks in the legend panel, where tRNA genes are labeled by one uppercase letter amino acid abbreviations. Blocks with arrow point to right means the same order as those in the legend, otherwise, it is reversed.
Genes of the Ophiuroidea can be classified into eight blocks, of which COX1-R-ND4L-COX2-K-ATP8-ATP6-COX3-S2-ND3-ND4-H is always conserved in all 30 ophiuroids. The gene order of species in different families showed different stability. Family Gorgonocephalidae and Euryalidae show most conserved gene orders, while Ophiothamnidae and Ophiacanthidae display very divergent gene orders. The gene order of O. serratus and Amphiura sp. were unique. The reversal distance between O. serratus and its MRCA was nine, which was larger than that between O. linea and MRCA (five) (Figure 4, violet panel). The reversal distance of the H. haimaensis branch was seven. The other cold-seep brittle Amphiura sp. only showed a slight difference compared with other species among congeners, such as the specimen from the Weijia Guyot seamount (1,995 m depth) in the West Pacific (Na et al., 2019). Taking ancestor “B” as reference, the reversal distance between the cold-seep species and “B” were much larger or at least the same as their non-seep congener, including those of deep-sea, suggesting the cold-seep environment may increase genome instability accelerate the rearrangement of MT genome.
Distinct positive selection signal among cold-seep brittle stars
Low temperature and high concentrations of hydrogen sulfide and methane in cold seep areas may affect the function of MT genes and exert selective stress on them. We used PAML to detect positive selection in two cold seep brittle stars (O. serratus and Amphiura sp.). The ω (dN/dS) value calculated using the one-ratio model (M0) ranged from 0.01663 to 0.06457 for the 13 PCGs, suggesting that these genes were constrained to maintain their functions. The LRT test of the two-ratio branch model compared to the one-ratio model showed that the two-ratio branch model is significantly better than the one-ration model using O. serratus as foreground for ND1, ND5, and ND6 (p < 0.05), but we found no significant difference between the two model when using Amphiura sp. as foreground.
Positive signals were detected in gene ND5 for O. serratus and CYTB, ND4, and ND4L for Amphiura sp. based on the LTR test of branch site model A. Only one site (CYTB:341:S, mutation: E or Q to S) was detected as positive selection (Bayes Empirical Bayes (BEB) posterior probability > 0.95) for foreground lineage O. serratus, whereas 10 sites on genes COX2, CYTB, ND2, ND4, and ND4L were detected as positive selection (BEB posterior probability > 0.95) for foreground lineage Amphiura sp. using BEB analysis (Yang, 2005). Five of the 10 Amphiura sp. positively selected sites lie on the ND4 genes and induce amino acid changes, suggesting that OXPHOS complex I in Amphiura sp. may play an important role in its adaptation to the cold-seep reducing environment. In addition, we identified three amino acid sites (ATP6:212, CYTB:247, and ND5:473) with positive selection in any of the three cold seep species and contained different amino acid from non-seep species.
Discussion
Our observation showed that O. serratus colonized broader niches or ecosystem than Amphiura sp. in Haima cold seep. O. serratus also displayed higher mutation rate in its clade than Amphiura sp. Higher mutation rate may accumulate fixed beneficial mutations allowing the organism to adapt to new environment (Taddei et al., 1997). Rapid sequence mutation was also found in deep-sea hyperthermophilic anaerobic archaeon Thermococcus eurythermalis through mutation accumulation (MA) experiment (Gu et al., 2021) and hypoxia stress in high altitude was supposed to accelerate mutation of aging-associated genes (Li et al., 2017). The rapid nucleic mutation of O. serratus and H. haimaesis may account for their epibenthic lifestyle in broader niches (both mussel and clam beds), compared to Amphiura sp.’s adaption to the mud sediments and only clam beds.
The gene order of species in other Echinoderms such as Asteroidea and Echinoidea is extensively conserved even for some the deep-sea Mariana Trench Freyastera benthophila (Mu et al., 2018), in contrast, our results suggest high gene order diversity in the Ophiuroidea mitogenome, especially in the cold seep species. Genome rearrangement plays a major role in adaptation and speciation (Merot et al., 2020), and different species adaptive to different niches display different mitogenome footprints. The environment parameters between ROV1 and ROV3/4 were quite different, like δ13C of sediment organic matter (SOM) showed low value (−39.25) at ROV1 but high values at ROV3/4; while only smaller difference exists between SOM δ13C and δ13C of particulate organic matter at ROV3 and ROV4 (Ke et al., 2022). The rapid MT genome rearrangement of O. serratus and H. haimaensis, combined with rapid nucleotide evolution may help them adapt to more volatile sediment-seep water interphase environment and all three ROV sites, whereas Amphiura sp. maintained a much more stable MT genome, perhaps because of its supposed burrowing lifestyle and more stable environment at ROV3 and ROV4. Accelerated genome rearrangement is considered to have occurred during the early evolution of vertebrates and could lead to amazing phylogenetic diversification (Hufton et al., 2008). In addition, the instability of the MT genome could be an indicator of nuclear genome instability, which may reshape brittle stars adapted to the special deep-sea cold seep environment. Further whole genome sequencing and functional analysis of adaptive genes are needed to illustrate how brittle stars could inhabit cold seep fields via reorganization of their genomes.
Amphiura sp. may adapt to the cold seep mud condition using a different strategy, through rapid amino acid modification of ND4, coding a subunit of OXPHOS complex I (or NADH dehydrogenase complex). OXPHOS complex I functions as a proton pump, mutations on which was associated with increased activity and the hypoxia-high-altitude human population, shorten lifespan of killifish (Baumgart et al., 2016; Gonçalves, 2019). It is considered criterial in the adaptive evolution of vertebrates (Dawson and Scott, 2022; Verma et al., 2022). The lowland silly chicken embryos showed higher complex I activity in brain mitochondria under hypoxia incubation condition than normoxic incubation condition, whereas the native hypoxia-adaptive Tibet chicken embryos showed no difference (Bao et al., 2007). ND2, ND4, and ND5 were identified as positively selected along the exclusive deep-sea mussels Bathymodiolinae branch (Zhang et al., 2021) and hydrothermal vent alvinocaridid shrimps Shinkaicaris leurokolos (Sun et al., 2018), and ND3 and ND5 revealed positive selection in the deep-sea crab Chaceon granulates (Zhang et al., 2020). Li had identified three residues on ND1, ND2, and ND5 under positive selection in cold-seep brittle star H. haimaensis, suggesting the important function of OXPHOS complex I in adaption to the environment conditions of deep-sea and cold seep (Li et al., 2021).
There is small overlap of positively selected genes and residues among the three cold-seep brittle stars although they occupied the same cold seep. No convergent or parallel amino acid changes were identified among cold-seep brittle stars, although we identified three positively selected amino acids that changed between all cold seep brittle stars and their non-seep relatives. In contrast, five convergent amino acid changes and 29 parallel amino acid changes in 11 genes have been found in deep-sea fishes (Shen et al., 2019). Divergent repositories of positive selection signals suggest that different organisms adopt different mechanisms of the OXPHOS system and modify them to adapt to similar environments, which may be constrained by their genomic materials (Burskaia et al., 2021). Further comparisons between the whole genomes and gene contents of the cold seep species are needed to decode the detailed effects of each cold-seep micro-ecosystem.
Conclusion
In the present study, we provide complete MT genomes of two brittle stars (Amphiura sp. and O. serratus) collected from Haima cold seep fields. O. serratus revealed relatively high A + T and AT-dinucleotide genomic content. we found that the two species display divergent evolutionary signals, although both evolve at least as rapid as their deep-sea or shallow water relatives. O. serratus showed rapid mitogenome evolution via both nucleotide mutation and genome rearrangement, compared to the medium evolutionary rate of Amphiura sp., which may contribute to its broader adaptive potential (in both mussel and clam beds), compared to the more stable burrowing environment for Amphiura sp. We identified one positive selected residue in CYTB in the O. serratus lineage and 10 in ND4 in the Amphiura sp. lineage, indicating that the function of NADH dehydrogenase complex may be important in their adaption to the reducing cold seep environment.
Materials and methods
Sample preparation and DNA extraction
Specimens of the two brittle star species was collected from three sites ROV1, 3, and 4, same as R1, R3 and R4 in Ke’s study (Ke et al., 2022), using the ROV HAIMA, from the Haima active cold seep area on the north slope of the South China Sea, between Hainan and Paracel Islands in 2020. The specimen was not a member of an endangered or protected species, and no specific permits were required. The specimens were preserved in 95% ethanol and stored at 4°C. DNA was extracted using a Genomic DNA Kit (Tiangen Co., Beijing, China) according to the manufacturer’s instructions. Locations of three sites ROV1, 3, and 4 are the same as R1, R3, and R4 in Ke’s study (Ke et al., 2022).
Sequencing assembly and annotation
For each sample, 1 μg extracted DNA was used as the input material and sequencing libraries were generated using the VAHTS Universal DNA Library Prep Kit for MGI (Vazyme, Nanjing, China) following the manufacturer’s recommendations and index codes were added to attribute sequences to each sample. The library quantification and size was measured using Qubit 3.0 Fluorometer (Life Technologies, Carlsbad, CA, USA) and Bioanalyzer 2100 system (Agilent Technologies, CA, USA). Subsequent sequencing was performed on a BGI MGI-SEQ 2000 platform. Paired end reads were assembled using Novoplasty v4.3.1 (Dierckxsens et al., 2017) (Genome Range = 15,000–18,000 and K-mer = 27), taken other complete mitogenome as seed. The assembled mitogenome was re-circularized to move COX1 to head. All mitogenome were annotated on MITOS1 (Donath et al., 2019). Circular maps of mitogenomes were plotted in R (version 4.0.3) using ggplot2 and in-house R scripts.
Sequence alignment and phylogenetic analysis
For each PCG, amino acid sequence from the 31 mitogenomes were aligned using MAFFT v7.475 with setting “–ep 0.2 –maxiterate 1000 –localpair” and transform back to CDS alignment. Conserve regions of the 13 PCGs were kept by GBlock v0.91b (Castresana, 2000) with default setting and concatenated. COX1 sequences from the two sequenced mitogenome was BLAST against the NCBI nt database to decide the most similar species, result in two families Amphiuridae and Ophiacanthidae respectively. CDS Sequences of COX1 of family Amphiuridae and Ophiacanthidae were downloaded from NCBI database and only two sequences were kept for each species for phylogenetic analysis. Maximum likelihood phylogenetic reconstruction was conducted by RAxML v8.2.12 (Stamatakis, 2014) on the CDS sequences, with substitution model GTR + G + I and the rapid bootstrap analysis, for both the COX1 sequences of the two families and the concatenated PCGs of the 31 mitogenomes. BEAST2 (Bouckaert et al., 2019) was performed to date the internal ancestor nodes of the 31 mitogenomes, with calibrated time points: Normal distribution with Offset = 180, Mean = 10, and SD = 10 My between Gorgonocephalus and Ophiura, Normal distribution with Offset = 260, Mean = 10, and SD = 15 for the ancestor of all Ophiuroidea.
Positive selection analysis
All positive selection analyses were performed via “codeml” from the PAML v4.9j (Yang, 2007). The ML tree was used as tree topology. Branch models with one-ratio, two-ratio (background, cold seep foreground), four-ratio (background, three cold seep ratios for the three cold seep species) and free-ratio were used in the mitogenome analysis. The branch-site model was used to determine whether these genes have undergone positive selection in the foreground lineage. Bayes Empirical Bayes (BEB) analysis was used to calculate the Bayesian posterior probability of the positively selected sites (posterior probability > 0.95).
Gene order reconstruction
Gene orders of 37 mito-genes from the 30 ophiuroids and the outgroup starfish Acanthaster brevispinus were converted to MGRA compatible fasta file in GRIMM format. Additional genes beyond the 37 genes were removed before further analysis. MGRA (Alekseyev and Pevzner, 2009) was performed for ancestor gene order reconstruction and rearrangement distance calculation, using the ML phylogenetic tree as reference topology. Though Ophiopholis aculeata have an annotated tRNA-Thr, it is much divergent from other tRNA-Thr and may not be a true tRNA-Thr. However, we kept the tRNA-Thr in O. aculeata to make sure all genomes contain the same gene contents.
Data availability statement
The datasets presented in this study can be found in online Genbank repositories. The links of the repository and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, OP273954 and OP432684.
Author contributions
ZC, GQ, and QL contributed to the conception and design of the study. ZC and GQ performed the analysis and wrote the manuscript. SM collected samples and public data and performed wet-lab experiments. MQ and BZ revised the manuscript and polished figures. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This study was supported by the National Key Research and Development Program of China (2021YFF0502803), the National Natural Science Foundation of China (41890853), Special Fund for Strategic Pilot Technology Chinese Academy of Sciences (XDB42030204), the Key Deployment Project of Center for Ocean Mega-Research of Science, Chinese Academy of Sciences (COMS2020Q14), the Hainan Provincial Joint Project of Sanya Yazhou Bay Science and Technology City (320LH046), and the Development Fund of South China Sea Institute of Oceanology of the Chinese Academy of Sciences (SCSIO202202).
Acknowledgments
We thank all the scientists and crew on the R/V Haiyangdizhi-06 and the operation teams of the ROV HAIMA and ROPOS for their assistance in sampling and providing valuable videos. We would also thank Yu Chen for their help in processing specimens on board.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2022.1027139/full#supplementary-material
Footnotes
References
Alekseyev, M. A., and Pevzner, P. A. (2009). Breakpoint graphs and ancestral genome reconstructions. Genome Res. 19, 943–957. doi: 10.1101/gr.082784.108
Bao, H., Zhao, C., Li, J., Zhang, H., and Wu, C. (2007). A comparison of mitochondrial respiratory function of Tibet chicken and silky chicken embryonic brain. Poult. Sci. 86, 2210–2215. doi: 10.1093/ps/86.10.2210
Baumgart, M., Priebe, S., Groth, M., Hartmann, N., Menzel, U., Pandolfini, L., et al. (2016). Longitudinal RNA-Seq analysis of vertebrate aging identifies mitochondrial complex I as a small-molecule-sensitive modifier of lifespan. Cell Syst. 2, 122–132. doi: 10.1016/j.cels.2016.01.014
Bouckaert, R., Vaughan, T. G., Barido-Sottani, J., Duchene, S., Fourment, M., Gavryushkina, A., et al. (2019). BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 15:e1006650. doi: 10.1371/journal.pcbi.1006650
Bronstein, O., Kroh, A., and Haring, E. (2018). Mind the gap! The mitochondrial control region and its power as a phylogenetic marker in echinoids. BMC Ecol. Evol. 18:80. doi: 10.1186/s12862-018-1198-x
Burskaia, V., Artyushin, I., Potapova, N. A., Konovalov, K., and Bazykin, G. A. (2021). Convergent adaptation in mitochondria of phylogenetically distant birds: Does it exist? Genome Biol. Evol. 13:evab113. doi: 10.1093/gbe/evab113
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Dawson, N. J., and Scott, G. R. (2022). Adaptive increases in respiratory capacity and O2 affinity of subsarcolemmal mitochondria from skeletal muscle of high-altitude deer mice. FASEB J. 36:e22391. doi: 10.1096/fj.202200219R
Dierckxsens, N., Mardulyn, P., and Smits, G. (2017). NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45:e18. doi: 10.1093/nar/gkw955
Donath, A., Juhling, F., Al-Arab, M., Bernhart, S. H., Reinhardt, F., Stadler, P. F., et al. (2019). Improved annotation of protein-coding genes boundaries in metazoan mitochondrial genomes. Nucleic Acids Res. 47, 10543–10552. doi: 10.1093/nar/gkz833
Dong, D., Li, X., Yang, M., Gong, L., Li, Y., Sui, J., et al. (2021). Report of epibenthic macrofauna found from Haima cold seeps and adjacent deep-sea habitats, South China sea. Mar. Life Sci. Technol. 3, 1–12. doi: 10.1007/s42995-020-00073-9
Du, L. P., Cai, S. Y., Liu, J., Liu, R. Y., and Zhang, H. B. (2020). The complete mitochondrial genome of a cold seep gastropod Phymorhynchus buccinoides (Neogastropoda: Conoidea: Raphitomidae). PLoS One 15:e0242541. doi: 10.1371/journal.pone.0242541
Feng, D., Qiu, J.-W., Hu, Y., Peckmann, J., Guan, H., Tong, H., et al. (2018). Cold seep systems in the South China Sea: An overview. J. Asian Earth Sci. 168, 3–16. doi: 10.1016/j.jseaes.2018.09.021
Galaska, M. P., Li, Y., Kocot, K. M., Mahon, A. R., and Halanych, K. M. (2019). Conservation of mitochondrial genome arrangements in brittle stars (Echinodermata, Ophiuroidea). Mol. Phylogenet. Evol. 130, 115–120. doi: 10.1016/j.ympev.2018.10.002
German, C. R., Ramirez-Llodra, E., Baker, M. C., and Tyler, P. A., and ChEss Scientific Steering Committee. (2011). Deep-water chemosynthetic ecosystem research during the census of marine life decade and beyond: A proposed deep-ocean road map. PLoS One 6:e23259. doi: 10.1371/journal.pone.0023259
Goffredi, S. K., Tilic, E., Mullin, S. W., Dawson, K. S., Keller, A., Lee, R. W., et al. (2020). Methanotrophic bacterial symbionts fuel dense populations of deep-sea feather duster worms (Sabellida, Annelida) and extend the spatial influence of methane seepage. Sci. Adv. 6:eaay8562. doi: 10.1126/sciadv.aay8562
Gonçalves, V. F. (2019). “Mitochondrial genetics,” in Mitochondria in health and in sickness, Vol. 1158, eds A. Urbani and M. Babu (Singapore: Springer), 247–255. doi: 10.1007/978-981-13-8367-0_13
Gu, J. H., Wang, X. J., Ma, X. P., Sun, Y., Xiao, X., and Luo, H. W. (2021). Unexpectedly high mutation rate of a deep-sea hyperthermophilic anaerobic archaeon. ISME J. 15, 1862–1869. doi: 10.1038/s41396-020-00888-5
Hufton, A. L., Groth, D., Vingron, M., Lehrach, H., Poustka, A. J., and Panopoulou, G. (2008). Early vertebrate whole genome duplications were predated by a period of intense genome rearrangement. Genome Res. 18, 1582–1591. doi: 10.1101/gr.080119.108
Ke, Z., Li, R., Chen, Y., Chen, D., Chen, Z., Lian, X., et al. (2022). A preliminary study of macrofaunal communities and their carbon and nitrogen stable isotopes in the Haima cold seeps, South China Sea. Deep Sea Res. 1 Oceanogr. Res. Pap. 184:103774. doi: 10.1016/j.dsr.2022.103774
Kiel, S., and Little, C. T. S. (2006). Cold-seep mollusks are older than the general marine mollusk fauna. Science 313, 1429–1431. doi: 10.1126/science.1126286
Kuzmiak, S., Glancy, B., Sweazea, K. L., and Willis, W. T. (2012). Mitochondrial function in sparrow pectoralis muscle. J. Exp. Biol. 215, 2039–2050. doi: 10.1242/jeb.065094
Levin, L. A. (2005). “Ecology of cold seep sediments: Interactions of fauna with flow, chemistry and microbes,” in Oceanography and marine biology – An annual review, Vol. 43, eds R. N. Gibson, R. J. A. Atkinson, and J. D. M. Gordon (Boca Raton, FL: CRC Press-Taylor & Francis Group), 1–46. doi: 10.1201/9781420037449.ch1
Li, Q., Li, Y., Na, J., Han, X., Paterson, G. L. J., Liu, K., et al. (2021). Description of a new species of Histampica (Ophiuroidea: Ophiothamnidae) from cold seeps in the South China Sea and analysis of its mitochondrial genome. Deep Sea Res. 1 Oceanogr. Res. Pap. 178:103658. doi: 10.1016/j.dsr.2021.103658
Li, Y., Wang, M. S., Otecko, N. O., Wang, W., Shi, P., Wu, D. D., et al. (2017). Hypoxia potentially promotes Tibetan longevity. Cell Res. 27, 302–305. doi: 10.1038/cr.2016.105
Liang, Q. Y., Hu, Y., Feng, D., Peckmann, J., Chen, L. Y., Yang, S. X., et al. (2017). Authigenic carbonates from newly discovered active cold seeps on the NorthWestern slope of the South China Sea: Constraints on fluid sources, formation environments, and seepage dynamics. Deep Sea Res. 1 Oceanogr. Res. Pap. 124, 31–41. doi: 10.1016/j.dsr.2017.04.015
Liu, H. L., Cai, S. Y., Liu, J., and Zhang, H. B. (2018). Comparative mitochondrial genomic analyses of three chemosynthetic vesicomyid clams from deep-sea habitats. Ecol. Evol. 8, 7261–7272. doi: 10.1002/ece3.4153
Merot, C., Oomen, R. A., Tigano, A., and Wellenreuther, M. (2020). A roadmap for understanding the evolutionary significance of structural genomic variation. Trends Ecol. Evol. 35, 561–572. doi: 10.1016/j.tree.2020.03.002
Mu, W., Liu, J., and Zhang, H. (2018). The first complete mitochondrial genome of the mariana trench Freyastera benthophila (Asteroidea: Brisingida: Brisingidae) allows insights into the deep-sea adaptive evolution of Brisingida. Ecol. Evol. 8, 10673–10686. doi: 10.1002/ece3.4427
Na, J., Zhang, D., Cheng, H., Yang, J., Zhang, R., Chen, W., et al. (2019). The complete mitochondrial genome of a deep sea ophiuroid of the genus Amphiura (Ophiuroidea: Amphiuridae). Mitochondrial DNA B Resour. 4, 3709–3710. doi: 10.1080/23802359.2019.1679047
Nethupul, H., Stöhr, S., and Zhang, H. (2022). Review of Ophioplinthaca Verrill, 1899 (Echinodermata, Ophiuroidea, Ophiacanthidae), description of new species in Ophioplinthaca and Ophiophthalmus, and new records from the Northwest Pacific and the South China Sea. ZooKeys 1099, 155–202. doi: 10.3897/zookeys.1099.76479
Okanishi, M., Kato, M., Watanabe, H. K., Chen, C., and Fujita, T. (2020). Large populations of two new species of Ophiambix (Echinodermata, Ophiuroidea) discovered on Japanese hot vents and cold seeps. Raffles Bull. Zool. 68, 196–213. doi: 10.26107/RBZ-2020-0017
Okanishi, M., Matsuo, T., and Fujita, T. (2021). A new species of the genus Ophiomonas Djakonov (Echinodermata: Ophiuroidea: Amphilepididae) from the deep-sea of Japan. Zool. Stud. 60:59. doi: 10.6620/ZS.2021.60-59
Paull, C. K., Hecker, B., Commeau, R., Freemanlynde, R. P., Neumann, C., Corso, W. P., et al. (1984). Biological communities at the Florida Escarpment resemble hydrothermal vent taxa. Science 226, 965–967. doi: 10.1126/science.226.4677.965
Rodrigues, C. F., Paterson, G. L. J., Cabrinovic, A., and Cunha, M. R. (2011). Deep-sea ophiuroids (Echinodermata: Ophiuroidea: Ophiurida) from the Gulf of Cadiz (NE Atlantic). Zootaxa 2754, 1–26. doi: 10.11646/zootaxa.2754.1.1
Shen, X., Pu, Z., Chen, X., Murphy, R. W., and Shen, Y. (2019). Convergent evolution of mitochondrial genes in deep-sea fishes. Front. Genet. 10:925. doi: 10.3389/fgene.2019.00925
Sibuet, M., and Olu, K. (1998). Biogeography, biodiversity and fluid dependence of deep-sea cold-seep communities at active and passive margins. Deep Sea Res. 1 Oceanogr. Res. Pap. 45, 517–567. doi: 10.1016/S0967-0645(97)00074-X
Skold, M., and Gunnarsson, J. S. G. (1996). Somatic and germinal growth of the infaunal brittle stars Amphiura filiformis and A. chiajei in response to organic enrichment. Mar. Ecol. Prog. Ser. 142, 203–214. doi: 10.3354/meps142203
Stamatakis, A. (2014). RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Sun, S. E., Xiao, N., and Sha, Z. L. (2022). Mitogenomics provides new insights into the phylogenetic relationships and evolutionary history of deep-sea sea stars (Asteroidea). Sci. Rep. 12:4656. doi: 10.1038/s41598-022-08644-9
Sun, S., Hui, M., Wang, M., and Sha, Z. (2018). The complete mitochondrial genome of the alvinocaridid shrimp Shinkaicaris leurokolos (Decapoda, Caridea): Insight into the mitochondrial genetic basis of deep-sea hydrothermal vent adaptation in the shrimp. Comp. Biochem. Physiol. Part D Genomics Proteomics 25, 42–52. doi: 10.1016/j.cbd.2017.11.002
Taddei, F., Radman, M., MaynardSmith, J., Toupance, B., Gouyon, P. H., and Godelle, B. (1997). Role of mutator alleles in adaptive evolution. Nature 387, 700–702. doi: 10.1038/42696
Verma, R. K., Kalyakulina, A., Mishra, A., Ivanchenko, M., and Jalan, S. (2022). Role of mitochondrial genetic interactions in determining adaptation to high altitude human population. Sci. Rep. 12:2046. doi: 10.1038/s41598-022-05719-5
Xiao, Y., Xu, T., Sun, J., Wang, Y., Wong, W. C., Kwan, Y. H., et al. (2020). Population genetic structure and gene expression plasticity of the deep-sea vent and seep squat lobster Shinkaia crosnieri. Front. Mar. Sci. 7:587686. doi: 10.3389/fmars.2020.587686
Yang, Z. (2005). Bayes empirical bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 22, 1107–1118. doi: 10.1093/molbev/msi097
Yang, Z. (2007). PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Zhang, B., Wu, Y. Y., Wang, X., Jiang, W., Yin, J. P., and Lin, Q. (2020). Comparative analysis of mitochondrial genome of a deep-sea crab Chaceon granulates reveals positive selection and novel genetic features. J. Oceanol. Limnol. 38, 427–437. doi: 10.1007/s00343-019-8364-x
Zhang, K., Sun, J., Xu, T., Qiu, J. W., and Qian, P. Y. (2021). Phylogenetic relationships and adaptation in deep-sea mussels: Insights from mitochondrial genomes. Int. J. Mol. Sci. 22:1900. doi: 10.3390/ijms22041900
Keywords: cold seep adaption, Ophiuroidea, rapid genome rearrangement, micro-environment, brittle star
Citation: Chen Z, Ma S, Qin G, Qu M, Zhang B and Lin Q (2022) Strategy of micro-environmental adaptation to cold seep among different brittle stars’ colonization. Front. Ecol. Evol. 10:1027139. doi: 10.3389/fevo.2022.1027139
Received: 24 August 2022; Accepted: 31 October 2022;
Published: 23 November 2022.
Edited by:
Ferhat Matur, Dokuz Eylül University, TurkeyReviewed by:
Benny K. K. Chan, Academia Sinica, TaiwanOlga Zueva, Carnegie Mellon University, United States
Copyright © 2022 Chen, Ma, Qin, Qu, Zhang and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiang Lin, bGlucWlhbmdAc2NzaW8uYWMuY24=
†These authors have contributed equally to this work