 Xun Xu
Xun Xu Bao-Sheng Wang
Bao-Sheng Wang Hui Yu
Hui Yu- 1Key Laboratory of Plant Resource Conservation and Sustainable Utilization and Guangdong Provincial and Key Laboratory of Digital Botanical Garden, South China Botanical Garden, The Chinese Academy of Sciences, Guangzhou, China
- 2Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou), Guangzhou, China
- 3Centre for Plant Science, The Chinese Academy of Sciences, Core Botanical Gardens, Guangzhou, China
Understanding how intraspecies divergence results in speciation has great importance for our knowledge of evolutionary biology. Here we applied population genomics approaches to a fig wasp species (Valisia javana complex sp 1) to reveal its intraspecies differentiation and the underlying evolutionary dynamics. With re-sequencing data, we prove the Hainan Island population (DA) of sp1 genetically differ from the continental ones, then reveal the differed divergence pattern. DA has reduced SNP diversity but a higher proportion of population-specific structural variations (SVs), implying a restricted gene exchange. Based on SNPs, 32 differentiated islands containing 204 genes were detected, along with 1,532 population-specific SVs of DA overlapping 4,141 genes. The gene ontology (GO) enrichment analysis performed on differentiated islands linked to three significant GO terms on a basic metabolism process, with most of the genes failing to enrich. In contrast, population-specific SVs contributed more to the adaptation than the SNPs by linking to 59 terms that are crucial for wasp speciation, such as host reorganization and development regulation. In addition, the generalized dissimilarity modeling confirms the importance of environment difference on the genetic divergence within sp1. Hence, we assume the genetic divergence between DA and the continent due to not only the strait as a geographic barrier, but also adaptation. We reconstruct the demographic history within sp1. DA shares a similar population history with the nearby continental population, suggesting an incomplete divergence. Summarily, our results reveal how geographic barriers and adaptation both influence the genetic divergence at population-level, thereby increasing our knowledge on the potential speciation of non-model organisms.
Introduction
The origination of new species usually starts from intraspecies diversity and population subdivision. The division of one population into several descendances generally occurs. Several hypotheses were proposed to explain the evolutionary dynamics driving the descendant populations into new species such as adaptation (Martin et al., 2013), chromosomal structure variant (Korunes et al., 2021), and absence of gene flow (Hill et al., 2021). However, the intraspecies divergence, which is the very beginning of speciation, has received much less attention than fully separated species.
Fig trees (Ficus, Moraceae) and fig wasp (Hymenoptera: Chalcidoidea: Agaonidae) form a specialized co-evolution system, containing the most complex speciation processes (Herre et al., 2008; Bronstein, 2009; Cruaud et al., 2012). Female fig wasps exclusively recognize the host fig tree for oviposition. The host tree, on the other hand, provides their ovules as food for the larvae and urn-shaped inflorescences (figs) as a mating environment for the adults. When the new wasp generation finish mating, they are ready to disperse. The stamens mature simultaneously and stain the pollen on the wasps when they leave. Thus, the wasps transfer pollen to other trees when entering figs. For successful oviposition, wasps must specifically recognize the volatile organic compounds (VOCs) of the host, and match the narrow entrance (i.e., ostiole) of enclosed fig (Berg, 1989; Bronstein and McKey, 1989; Borges, 2015; Chen Y. et al., 2016). Hence, morphologically distinguished figs are usually pollinated by distinct genera (Ramirez, 1970; Cruaud et al., 2012). For the fig tree, successful pollination depends on the strictly consistent life cycle with the wasp (Bronstein, 1988; Anstett et al., 1997; Cook and Rasplus, 2003). Therefore, the fig tree and fig wasp had long been considered as a co-evolution system. That is to say, one fig wasp species inhabits in one particular fig species, and one fig species is only pollinated by one particular fig wasp species. Early molecular phylogenetic evidence supports the co-divergence of fig trees and fig wasp (Weiblen, 2002; Ronsted et al., 2005). However, recently studies have proven that the speciation of wasps seems out of step with the host. Multiple pollinator wasps were found co-existing in a single host species (Darwell et al., 2014; Yang et al., 2015; Darwell and Cook, 2017; Yu et al., 2019). Those wasps are genetically and morphologically distinct. The phylogeny among those species is complex, including populations under different phases of speciation and fully separated new species (Liu et al., 2013; Tian et al., 2015; Souto-Vilaros et al., 2019). One explanation is that the insects have much shorter longevity than trees (Petit and Hampe, 2006; Thomas et al., 2010). For instance, when a reduce of gene flow has led to divergence in the wasp after many generations, the host tree population is still probably in one generation without any genetic differentiation. The adaptation to different niches is an alternative hypothesis for the rapid speciation (Darwell and Cook, 2017), accompanied by others such as geographical isolation (Liu et al., 2013), hybridization (Renoult et al., 2009), host switching (Cruaud et al., 2012; Wang et al., 2021), and pollinator sharing (Bernard et al., 2020).
Ficus hirta Vahl. is a wide-spread species with the most varied pollinator wasps, Valisia javana Mayr (Wiebes, 1993; Yu et al., 2019). Recently, analysis based on neutral gene markers and microsatellites proved V. javana to be a complex including nine cryptic species (referred as sp1–sp9) (Yu et al., 2019). The sp1 is also in the process of divergence: the population on Hainan Island, China presents genetic differences from those of southeast Asia (Tian et al., 2015; Yu et al., 2019). However, whole-genome level analysis on fig wasps, especially on V. javana complex, is still rare. The previous studies are based on relatively few genetic markers, insufficient for revealing genome-wide pattern and detecting the evolutionary dynamics within sp1.
Analysis of whole-genome re-sequencing data mainly focused on single-nucleotide polymorphism (SNPs); other types of variants such as structural variations have long been ignored (Feuk et al., 2006; Frazer et al., 2009; Eichler et al., 2010). Structural variation (SV) is a major source of genomic variations affecting large regions (here defined as more than 50 bp), including deletions (DEL), insertions (INS), inversions (INV), duplications (DUP), and translocations (TRA) (Feuk et al., 2006; Wellenreuther et al., 2019; Ho et al., 2020). The evolutionary importance of SVs is proven in multiple species such as Atlantic salmon (Bertolotti et al., 2020), domesticated crop (Gaut et al., 2018), and the Corvus genus (Weissensteiner et al., 2020). Nevertheless, knowledge about their evolutionary contributions remains limited.
Here, we take sp1 of the V. javana complex as an example, and combine both SNPs and SVs to detect its intraspecies genetic divergence. To be specific, we ask: (1) do the Hainan and continental populations of sp1, differ genetically at the whole-genome level and (2) whether the variation patterns differ among populations? Hainan island is divided from the continent by Qiongzhou Strait and is under a tropical maritime climate, while the continental habitats of sp1 are mainly under a subtropical continental monsoon climate. Hence, we have interest in the effect of the physical barrier caused by Qiongzhou Strait and local adaptation caused by environmental difference. Finally, we reconstruct the population-level demographic history within sp1 to realize the divergent process.
Materials and Methods
Sampling, DNA Extraction and Sequencing
From 2006 to 2014, we sampled figs containing mature fig wasps across Southeast Asia. See Figure 1A is the map of sampling locations and Table 1 provides details. Three locations were included. One is from Hainan Island (DA) and two are from the continent (VH and SCBG). The distances between each two sampling locations are 822 km (SCBG-VH), 498 km (SCBG-DA), and 532 km (VH-DA). In each location, figs were collected from trees 3–5 m away from each other. We chose 10–30 figs from each site and sealed them individually in fine-mesh bags under ambient temperature until the mature wasps emerge. The sp1 wasps were identified and collected, then preserved in 95% ethanol under −20°C until DNA extraction. The DNA pool of each sample were created for all mature female wasps in a single fig by mixing extraction of genomic DNA. We finally obtained 26 samples for DNA extraction. Genomic DNA was extracted following the method outlined in a previous study (Tian et al., 2015). The genomic DNA was fragmented with the S220 Ultrasonic Processor (Covaris, United States). After end repairing and phosphorylating, common adapters were ligated to the fragments for denaturing and amplifying. The prepared libraries were sequenced with the Illumina HiSeq2500 (Illumina, United States). The DNA extraction and library preparation were accomplished by NovoGene Company (Beijing, China).
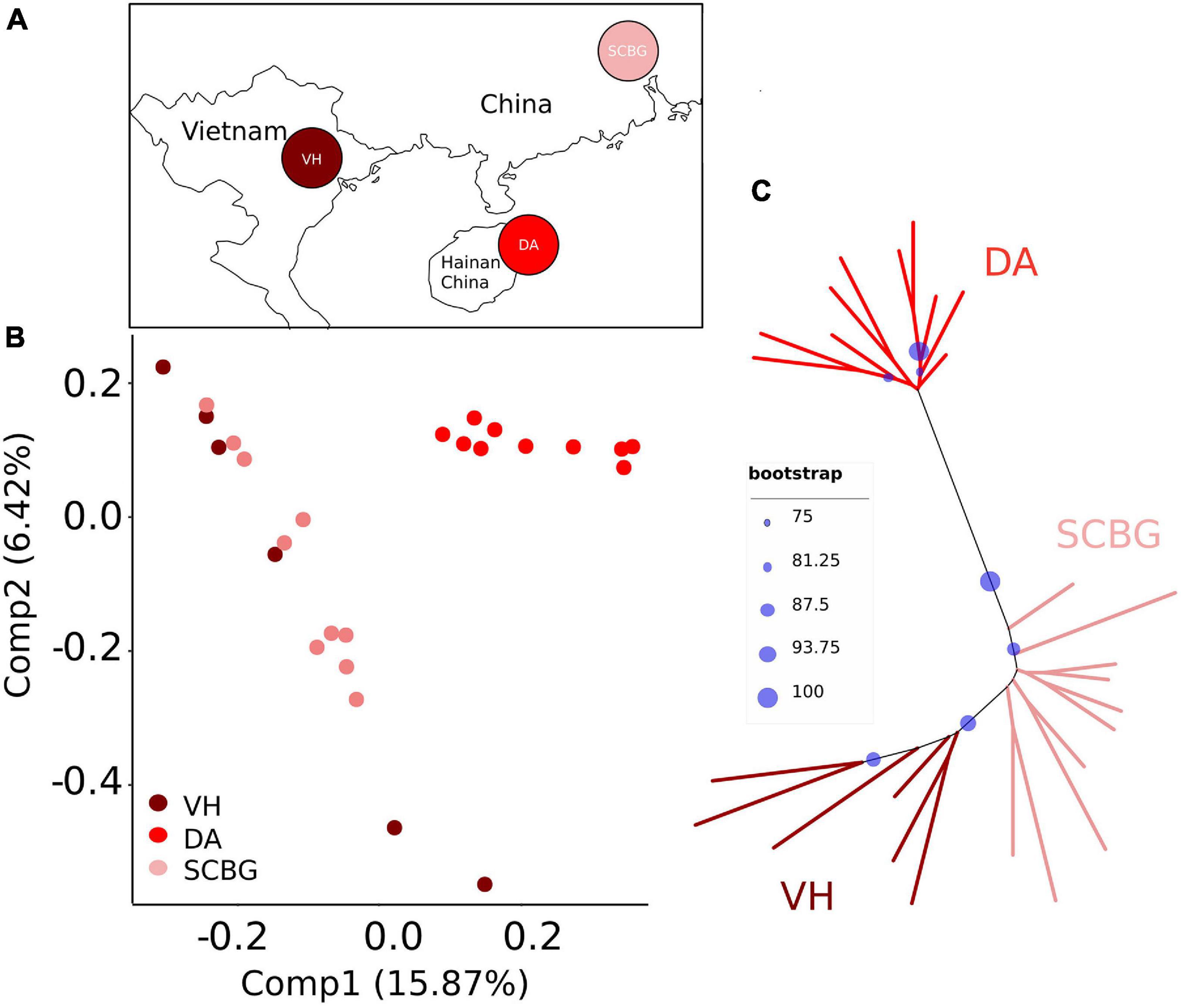
Figure 1. (A) Sampling locations. Sampling sites are Vinh Yen, Vietnam (VH, dark red); Hainan Island, China (DA, red); and Guangzhou, China (SCBG, pink). The map is constructed by R package maps 3.3.0 (Becker et al., 2018). (B) Principal component analyses (PCA) based on SNPs for populations. (C) Maximum likelihood (ML) tree based on SNPs. The bootstraps are indicated with blue spots. Population codes and color scheme for genetic groups is the same in (A–C).
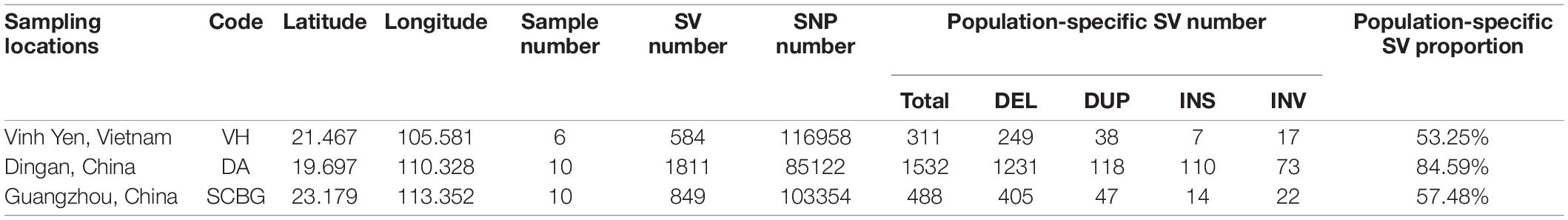
Table 1. Sample list and statistic of variants.
Calling and Filter of Single-Nucleotide Polymorphisms
Reads were evaluated with FastQC 1.0.0 (Andrews, 2010). Adapter sequences in reads were trimmed. Reads containing > 10% N bases or Q no more than 5 were removed. High-quality reads were mapped with BWA-MEM (Li and Durbin, 2011) to the sp1 genome under accession number GWHBDGE00000000, National Genomics Data Center. After being sorted and indexed by SAMtools (Li et al., 2009), the BAM files were then duplicate marked and underwent base quality recalibration with Picard Toolkit 2.17.3 (Broad Institute, 2018). The SNPs of each sample were separately called from resulting BAM files and filtered by GATK 4.1.0.0. (McKenna et al., 2010) with suggested parameters. The samples from the same population were merged into a single file with BCFtools 1.2 (Danecek and McCarthy, 2017) and filtered by VCFtools 0.1.16 (Danecek et al., 2011) with parameters as follows: 10 < read depths < 100, max missing < 0.5, and minor allele frequency > 0.03. All SNPs located within 3 bp of an InDel were removed. Finally, SNPs of each population were merged for the following analysis.
Genetic Structure Within sp1
To detect the intraspecies structure within sp1, we performed principal component analysis (PCA) and maximum likelihood (ML) tree based on SNPs. The PCA was conducted by R package SNPRelate (Zheng et al., 2012). PCA result was visualized with R package ggplot2 (Wickham, 2016). The phylogenetic tree was constructed with 1,000 bootstraps by iQtree (Nguyen et al., 2015). Command “-m MFP” was set for automatic detecting of the optimal model of nucleotide substitution. The bootstrap value was adjusted by command “-bnni.” The final tree was visualized by iTOL version 5 (Letunic and Bork, 2021). Only SNPs with minor allele frequencies more than 0.05 and with max missing less than 0.5 were used for PCA. Only SNPs at fourfold degenerated sites (4DTv) were used for ML tree. The 4DTv were then filtered by minor allele frequencies more than 0.02 and with max missing less than 0.5. To test the effects of filtering threshold setting, we ran multiple filtering with alternative parameters as follows: max missing < 0.5 or 0.7; without Hardy-Weinberg equilibrium (HWE) setting or HWE with p-value > 0.0001, > 0.001, > 0.1, and > 0.5; minor allele frequencies >0.02, 0.03, and 0.05; and minimum distance between any two sites >1000 and >5000. All filtering was performed with VCFtools 0.1.16 (Danecek et al., 2011).
Population Genetics Analysis
We separately calculated nucleotide diversity (π) (Nei and Li, 1979) and Tajima’s D (Tajima, 1989) of each population and relative divergence (Fst) (Weir and Cockerham, 1984) between each population. All calculations were performed with VCFtools 0.1.16 (Danecek et al., 2011) in non-overlap 100-kb windows. We also estimated the linkage disequilibrium (LD) decay with PopLDdecay (Zhang et al., 2019) in the three populations. Only SNPs were included. To compare the diversity pattern among populations, we firstly performed a Shapiro-Wilk test (Shapiro and Wilk, 1965) on the data for normality in frequentist statistics. Based on the results, the unpaired two-samples Wilcoxon rank-sum test (Wilcoxon, 1946) was applicated among π and Tajima’s D of each population, and among the Fst of each combination. Wilcoxon rank-sum test is a method to inspect whether the samples are from two populations with different medians when their distributions are not normal. The tests were performed respectively with “shapiro.test()” and “wilcox.test()” in R 3.6.3 (R Core Team, 2020).
Identification and Gene Ontology Enrichment of Differentiated Islands
To identify the differentiated islands between Hainan (DA) and continental populations (VH and SCBG), the continental ones were merged as a single mainland population. Then we calculated the Fst (Weir and Cockerham, 1984) between DA and mainland in 100-kb non-overlap window with VCFtools (Danecek et al., 2011). The results were Z-transformed with R 3.6.3 (R Core Team, 2020). In the present study, highly differentiated islands were defined as windows with Fst three standard deviations from the mean (i.e., Z-Fst > 3). To assess the signatures of selection, we compared π and Tajima’s D between the islands and background with the same methods in 2.4. Genes of every identified island were assigned gene ontology (GO) enrichment analyses by Ontologize 1.0 (Bauer et al., 2008). The GO terms were inferred from the annotation of sp1, the same reference genome as reads mapping. GO terms with p < 0.05 were considered significant.
Demography
MSMC2 (Schiffels and Durbin, 2014; Malaspinas et al., 2016) was employed to reconstruct the historical change of effective population size (Ne). MSMC2 is capable of estimating the continuous change of Ne over time by a Markovian approximation of the coalescent-with-recombination process. The input SNPs were sorted and aligned by TASSEL (Bradbury et al., 2007). SNPs with minimum frequency below 0.05 were filtered. Then Beagle 5.2 (Browning et al., 2018) was used for genotype phasing. Nasonia (Hymenoptera: Chalcidoidea: Pteromalidae) is a well-studied genus of Chalcidoidea and phylogenetically close to Valisia (King, 1961; Wylie, 1965; Werren et al., 2010). The substitution rate at silent sites of Nasonia species was about 1.4 × 10–8 per site per year (Oliveira et al., 2008). Since mutation includes not only substitutions but more types, the mutation rate is at least twice higher than the substitution rate (Press et al., 2019). Thus, in the present study, the mutation rate for sp1 was estimated as 2.8 × 10–8 per site per year. The generation time was 0.25 per year according to our field observation. The longest seventh contigs were used. MSMC2 estimation was carried out under the default parameters. Four samples from each population were chosen randomly per run. Ten independent runs were performed separately for each population.
Calling, Statistic and Annotation of Structural Variations
Since translocation (TRA) may involve more than one contig, short-read data is not suitable for translocation calling. Hence in the present study, SVs only contained deletions (DEL), insertions (INS), inversions (INV), and tandem duplications (DUP). To reduce noise caused by individual-specific SVs, we separately called SVs of each population used both DELLY 0.8.3 (Rausch et al., 2012) and Manta 1.6.0 (Chen X. et al., 2016) by respective default parameters. The results of each population were then merged by SURVIVOR 1.0.3 (Jeffares et al., 2017) with the parameters “1000 2 1 0 0 30.” Thus, only SVs reported by both software within 1000 bp were retained. The results were sorted by BCFtools 1.10 (Danecek and McCarthy, 2017) and indexed by GATK 4.1.6.0 (McKenna et al., 2010). The number and types of SVs was counted with svprops built in DELLY (Rausch et al., 2012). To reveal the divergence pattern of SVs among populations, we extracted the population-specific SVs by Bedtools 2.25.0 (Quinlan and Hall, 2010), then annotated their distribution regions with snpEff 4.3 (Cingolani et al., 2012). GO enrichment analysis was performed on genes overlapping with every population-specific SV by the same methods as described above.
Contributions of Environment vs. Geographic Distance
For estimating the effects of environment and geographic distance on the genetic distance, we applied the generalized dissimilarity modeling (GDM) (Ferrier et al., 2007) with R package GDM (Manion et al., 2016).
The sp1 was recently identified as a separated species within V. javana complex (Yu et al., 2019). Hence, the habitat records of V. javana is too vague for predicting the habitats of sp1. To expand the sampling area, we inducted 13 reliable sampling sites from previous studies (Tian et al., 2015; Yu et al., 2019; Supplementary Table 1). Wasps of the 13 sites were identified as sp1 by COI gene markers. Because of the low nucleotide diversity within sp1, the pairwise distance is barely distinguished among sites. Hence, we constructed the genetic matrix with Nei’s D (Nei, 1972), which is a standardized genetic distance ranging from 0 to 1. Furthermore, for reducing the effect of the similarity of sequences from the same population, one COI sequence was randomly chosen from each site. Three random samplings of COI sequences were conducted. We then separately calculated Nei’s D based on each random sampling with “nei.dist()” in R package poppr (Kamvar et al., 2014). Then the 19 Bioclim variables at 2.5 m resolution (Hijmans et al., 2005; Fick and Hijmans, 2017) of the 13 sites were obtained. According to Myers et al. (2019), nine of the 19 variables have a low correlation between variables (<0.7). Those are BIO1 (Annual Mean Temperature), BIO2 (Mean Diurnal Range), BIO3 (Isothermality), BIO4 (Temperature Seasonality), BIO8 (Mean Temperature of Wettest Quarter), BIO9 (Mean Temperature of Driest Quarter), BIO12 (Annual Precipitation), BIO14 (Precipitation of Driest Month), and BIO15 (Precipitation Seasonality). We thus only used the nine variables to present environmental differences. In the GDM, we set the genetic distance as the response variable, and respectively set three independent variables: (1) environment differences and geographic distance, (2) environment differences only, and (3) geographic distance only. The most fitted model and the variable importance were quantified with “gdm.varImp()” in the GDM package. The “gdm.varImp()” returns percent deviance were explained and the p-value was fitted for each model. The model with p < 0.01 and highest explained percent deviance was considered as the most suitable for estimating the variable importance and variable significance. We further used MaxEnt 3.4.1 (Phillips et al., 2006) to evaluate the most important environment variables based on all the 19 Bioclim variables. MaxEnt is capable of parsing the contributions of each variable regardless of their correlation (Elith et al., 2011; Myers et al., 2019). The analysis was set with 10 replicates, with 25% of samples as the training data. Receiver operating characteristic curve (ROC) (or area under curve, AUC) and jackknife method were used to measure fitness and variable importance, respectively.
Results
Sequencing, Mapping, and Single-Nucleotide Polymorphisms Calling
A total of 45.8 Gb clean data and 30.3 Gb filtered BAM files were generated from 26 samples. The average sequencing depth is 12.82× and the average alignment to the reference genome is 97.01% (Supplementary Table 2). After filtering, 305,434 SNPs remained for further analysis (Table 1). The SNP number of DA is the least (85,122), much lower than VH (116,958) and SCBG (103,354).
Intraspecies Genetic Structure of Sp 1
We conducted a PCA and an ML tree based on SNPs to explore the genome-wide genetic structure within sp1. See Materials and Methods for parameters of SNP filtering. The PCA proves that DA was clearly separated from the mainland populations (VH and SCBG), even with overlap on Comp 1 (15.87%) and Comp 2 (6.42%) (Figure 1B). On the ML tree (Figure 1C), all samples fell into two distinct clades in which DA forms a monophyletic one. VH and SCBG do not form clear sub-clades separated from each other. Therefore, our results are consistent with previous studies that showed that genetical divergence exists between DA and the continental populations. We noticed that MAF and HWE (>0.0001 or >0.001) barely affected the filtering results let alone the genetic structure results. Different max missing, sites distance, and severe HWE settings presented similar results (Supplementary Figure 1).
Genetic Diversity
To compare the diversity pattern among the three populations, the Tajima’s D and π of each population were calculated on 100-kb non-overlap windows. The LD decay was variable among the three populations (Supplementary Figure 2). DA has the highest decay ratio while the continental populations present strong linkage. The distributions of π and Tajima’s D of each population and Fst of each combination were all non-normal according to the Shapiro-Wilk test (Supplementary Table 3). Therefore, the Wilcoxon rank-sum test was performed for comparing their medians (Supplementary Table 4). Tajima’s D of both continental populations is significantly higher than DA with p < 0.01 (Figure 2A). The π presents a similar pattern with D (Figure 2B). Hence, we assume that the polymorphism in DA is significantly reduced. The median of Fst scores between DA and SCBG is significantly higher (p < 0.05) than that between DA and VH, and both of them are significantly higher (p < 0.01) than the one between VH and SCBG (Figure 2C). The Fst result confirms the clear genetic structure between DA and the continental populations, while the continental populations barely differ. The upper quartiles of Fst between DA and the continental populations are below 0.2 (Figure 2C), indicating the genetic differentiation is still moderate due to incomplete block of gene flow or recent divergence.
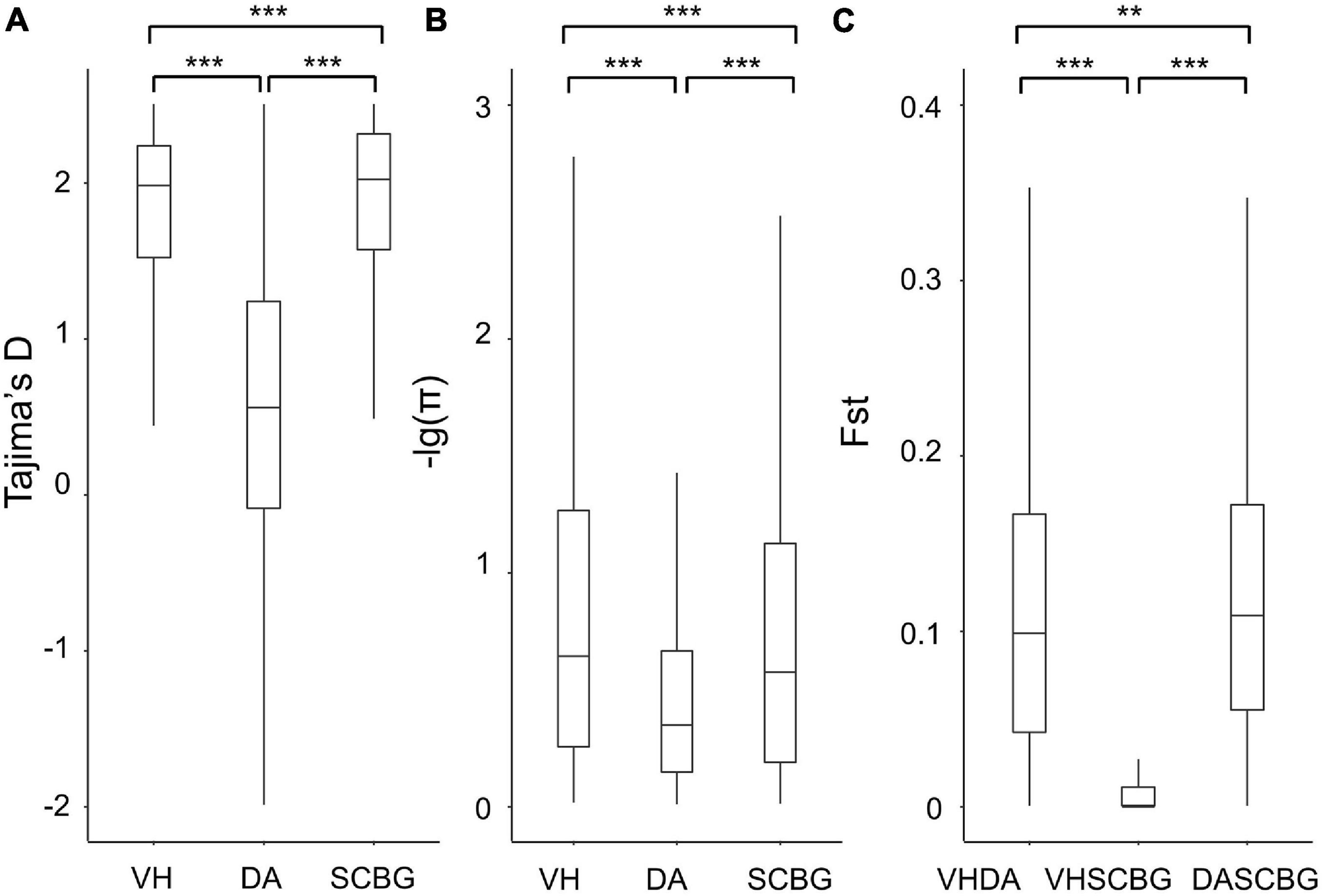
Figure 2. Boxplots of Tajima’s D (A), genetic diversity (π) (B), and relative divergence (Fst) (C) between populations with non-overlap 100-kb windows. VH, DA, and SCBG present populations from Vinh Yen, Vietnam; Hainan Island, China; and Guangzhou, China, respectively. Outliers are not shown. Significance of Wilcoxon rank-sum test are presented on the top (**0.01 < p < 0.05; ***p < 0.01).
Identification and Gene Ontology Enrichment of Differentiated Islands
A total of 32 differentiated islands with Z-Fst > 3 were identified between DA and the mainland (Supplementary Figure 3 and Supplementary Table 5). Continuous islands were merged into a single one. The distribution of π and Tajima’s D within or outside islands are all non-normal, except the Tajima’s D of islands in DA (Supplementary Table 3). Hence, we still applied the Wilcoxon rank-sum test for comparison. The differentiated islands of DA have significantly lower π and Tajima’s D (Figures 3A,B and Supplementary Table 4), with p < 0.05 and < 0.01, respectively. In the mainland, π of differentiated islands is significantly reduced (p < 0.05) but the Tajima’s D shows no significant difference (Figures 3A,B). The GO enrichment analysis of the 32 windows detected 204 genes, but only linked to three significant GO terms with p < 0.05. These 3 GO terms are sodium:potassium-exchanging ATPase complex (GO:0005890), cation-transporting ATPase complex (GO:0090533), and glycerol-3-phosphate catabolic process (GO:0046168). Each of the GO term contains three study terms, which means that most of the 204 genes fail to be enriched by any term.
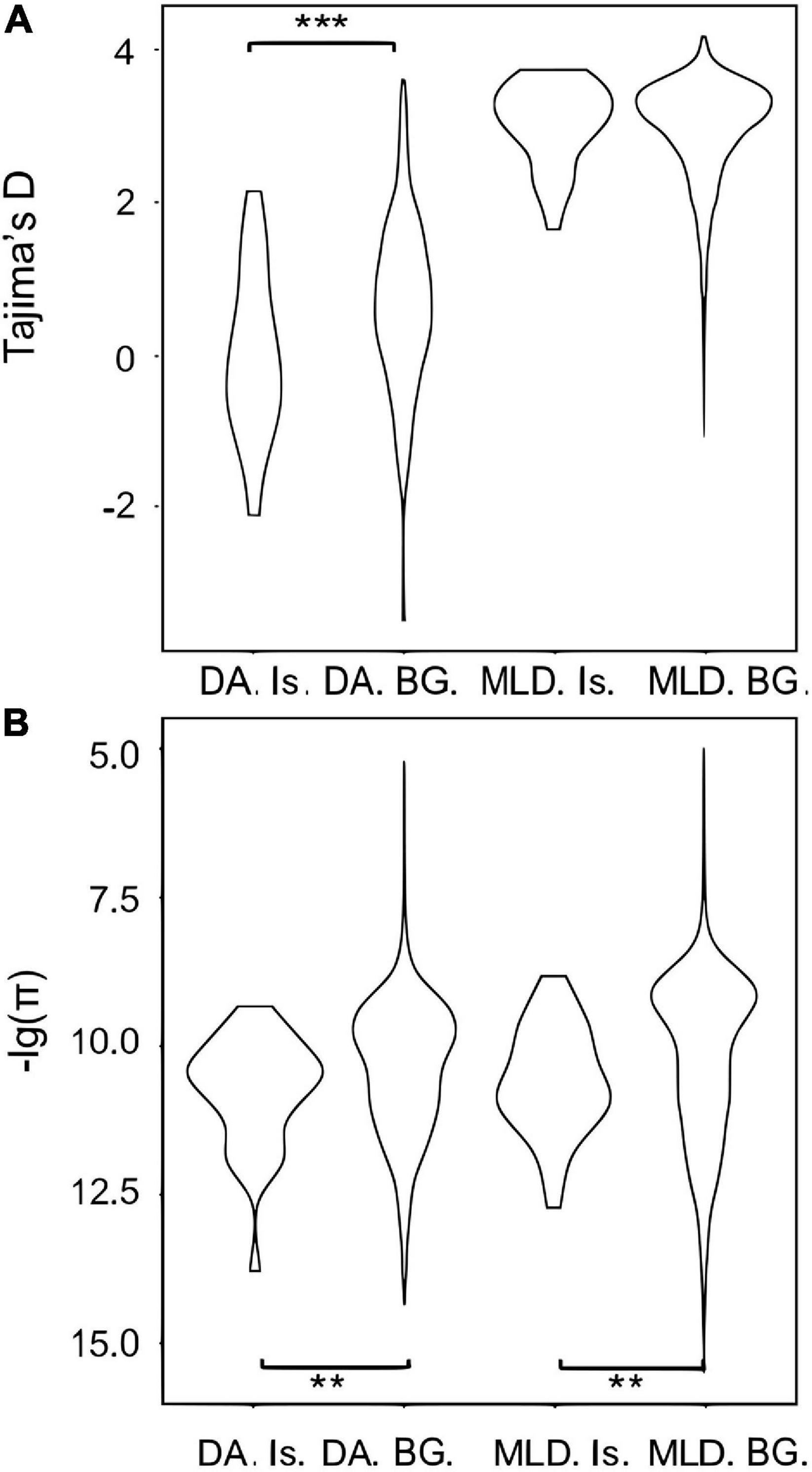
Figure 3. Violin plots for (A) Tajima’s D and (B) genetic diversity (π) in the Hainan and continental populations in differentiated islands (Is.) and the background (BG.). DA and MLD represent Hainan and the merged continental populations, respectively. Significant results of t-test are represented by asterisks (**0.01 < p < 0.05; ***p < 0.01).
Demography
To explore whether the demographic history differs between island and continental populations, we estimated change of the effective population size (Ne) along time. Due to the very short generation time (0.25 year) and rapid mutation rate (2.8 × 10–8 per site per year), the estimation of demographic history can only be traced back 4,000 generations (1,000 years) at most. According to the result, three populations had declined 1,000 years ago (ya) then expanded lately (Figure 4). The Ne of VH has been always higher than the other two populations with a recent explosion. Nevertheless, the demography of SCBG and DA has been similar. Thus, the recent demographic history between the Hainan and continental populations, at least between DA and SCBG, has not separated.
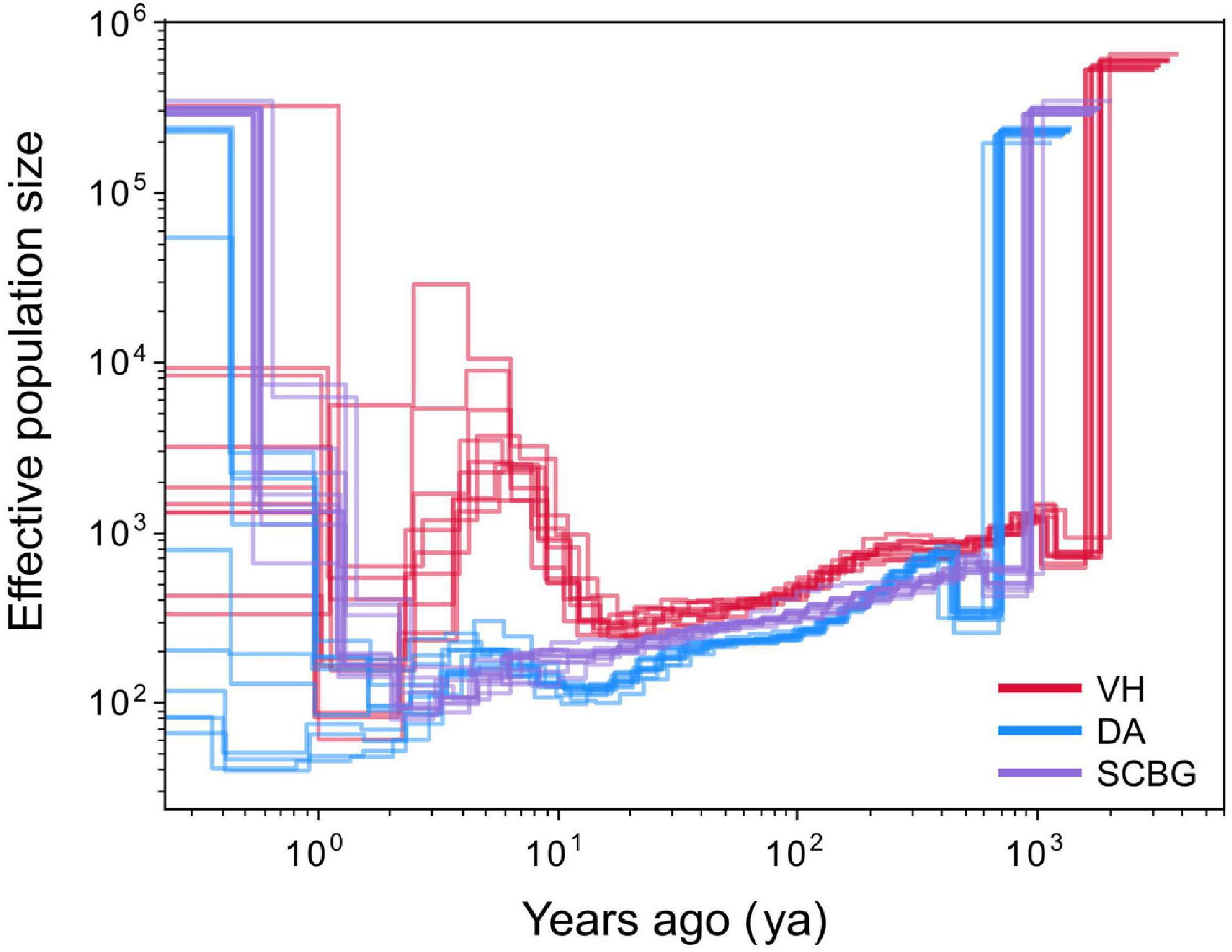
Figure 4. Demographic history of island (DA) and continental (VH and SCBG) populations. The demographic history is reconstructed from the seven longest contigs by multiple sequential Markovian coalescence (MSMC) model. Inferred fluctuations in effective population size (Ne) from 1,000 years ago (ya) to the present based on the 0.25-year generation time and 2.8 × 10–8 per site per generation mutation rate assumptions. Lines represent each one of ten independent estimates, in which four samples from one population were chosen randomly. Red, blue, and purple present populations from Vinh Yen, Vietnam (VH); Hainan island, China (DA); and Guangzhou, China (SCBG), respectively.
Statistics and Annotation of Structural Variations
We obtained a total of 3,244 SVs and 1,881 population-specific SVs (Table 1). DA has the most SV (1,811), exponentially higher than VH (584) and SCBG (849). The number of population-specific SV in DA (1,532) are also multiples higher than the two continental populations (VH: 311; SCBG: 488). As a result, DA has the highest population-specific SV proportion (84.59% vs. VH: 53.25% and SCBG: 57.48%). Hence, in addition to SNPs, SVs contribute to the genetic difference of DA as well. All three populations showed a similar pattern on the type of SVs (Figure 5A). In every population, DEL takes the highest proportion. The population-specific SVs are mostly located at inter-gene regions and introns (Figure 5B). In VH and DA, an unusually high proportion of SV is on splice site donner and transcript, respectively. The results imply that the population-specific SVs may lead to transcriptomic differences in VH and DA. We failed to detect any gene that overlaps the population-specific SVs of VH or SCBG. 4,141 genes were discovered overlapping the population-specific SVs of DA. The GO enrichment analysis of those 4,141 genes enriched to 59 significant GO terms (p < 0.05) (Figure 6). The GO terms, such as signaling pathway, cell communication, or stimulus response, are important for specific host cognization. Besides, GO terms linked to development regulation imply a flexible life cycle.
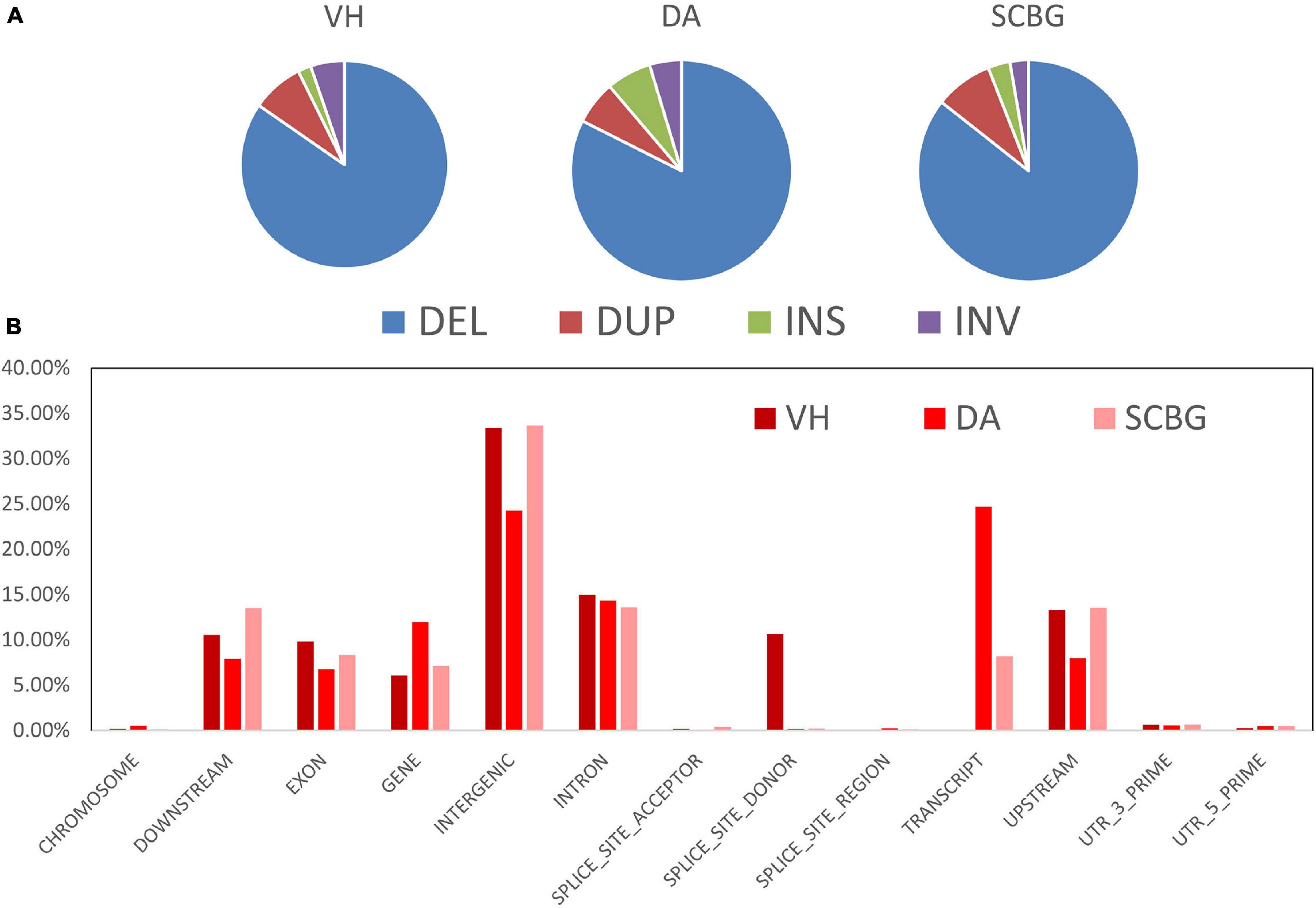
Figure 5. Types and distribution patterns of structural variations (SVs). (A) Pie chart of SV types in each population. DEL, DUP, INS, and INV indicate deletions, duplications, insertions, and inversions, respectively. (B) Bar chart of the population-specific SV proportion in different genome regions.

Figure 6. Gene ontology enrichment results of population specific structural variations.
Contributions of Environment vs. Geographic Distance
To compare the effects of environment and geographic distance, and evaluate the most important environment variables, we applied the GDM to estimate the correlation between genetic distance and environment difference and/or geographic distance. We performed three random samples from the COI sequences. In each run, one sequence was selected from each site. We then separately calculated the Nei’s D as response variable. The GDM results based on the three samples (random 1–3) were presented in Table 2. Three GDM analyses returned a similar conclusion, in which models involving both environment and geographic distance as potential predictor variables explained the most deviance (52.590–57.894%). When only the environment was included, the predictor variables still explained almost half of the deviance (49.778–53.433%). However, when only geographic distance included, the predictor variables explained 11.693–13.791% of the deviance, or even failed to predict response variable. The result confirmed the findings of previous studies (Tian et al., 2015; Yu et al., 2019), which detected the lack of genetic isolation by distance within the mainland populations of sp1. The most important Bioclim variable was either BIO8 (Mean Temperature of Wettest Quarter) or BIO9 (Mean Temperature of Driest Quarter); both present the synchronization of temperature and humidity. Therefore, we assume that the genetic distance in sp1 is due to environment, implying differentiation driven by adaptation. Heat stress accompanied by dryness stress could be a crucial influence. We further estimated the importance of all 19 Bioclim variables. The AUC values of the result are not excellent but acceptable (training AUC: 0.8231; test AUC: 0.5897), due to the strapped number of sampling sites. According to the jackknife method, BIO5 (Max Temperature of Warmest Month, Percent contribution = 50.613%) contributes the most to the distribution of sp1, while the model highly depends on BIO7 (Temperature Annual Range, permutation importance = 54.902%). The result is consistent with GDM, both of which predict the importance of heat stress.
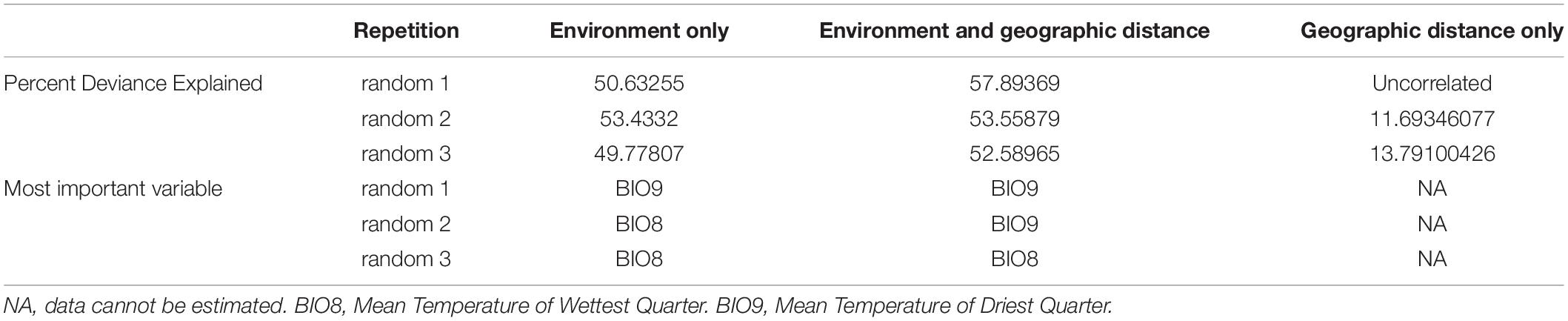
Table 2. Results of the generalized dissimilarity modeling analyses.
Discussion
Genome-Wide Patterns of Divergence Within sp1
The potential divergence between the Hainan and the continental populations of sp1 was hinted by neutral gene markers (Tian et al., 2015; Yu et al., 2019). Our results confirm the genetic structure within sp1 at genome-level, suggesting the Hainan population (DA) has been genetically differing from the continental populations (VH and SCBG). Furthermore, we reveal the divergence pattern across the whole genome.
The PCA (Figure 1B) and ML tree (Figure 1C) based on re-sequencing data both confirm the previous pattern. In addition, the Fst between DA and either VH or SCBG is significantly higher than that between the two continental populations (Figure 2C). All those results indicate a larger genetic distance between DA and continental populations than within the continental ones. We additionally proved that both SNPs and SVs contributed to the genetic difference of DA. The SNP number (Table 1), Tajima’s D (Figure 2A), and π (Figure 2B) of DA are immensely lower than the continental populations. The population-specific SVs of DA, however, are larger in number and higher in proportion than the other two populations (Table 1). Thus, the genetic difference of DA is caused by a decrease on SNP polymorphism and a differentiation on chromosome structure. The LD decayed rapidly in DA but maintained strong linkage in both of the continental populations. Such a phenomenon could be due to the influence of a high proportion of SVs. Moreover, the pattern of variant localization is heterogeneous across the genome but differs among populations. Firstly, we detected 32 differentiated islands with Z-Fst > 3 based on the SNP. Those differentiated islands have significantly reduced diversity than the genomic background (Figure 3 and Supplementary Table 4), and cluster on a few contigs (Supplementary Figure 3 and Supplementary Table 5). Secondly, the proportion of SVs varies among genomic regions (Figure 5B). SVs are significant components of genomic variants and affect large genomic regions (Feuk et al., 2006; Wellenreuther et al., 2019; Ho et al., 2020). The present study discovered a great number of population-specific SVs in DA (1,532), which is times higher than in VH (311) or SCBG (488). DA also presents a higher proportion of SVs in the transcript regions than the others. Therefore, SVs cause divergence not only in genome but also in transcriptome. We detected 4,141 genes overlapping the population-specific SVs of DA, more than the 204 genes detected in the differentiated islands based on SNPs. Furthermore, the functions of those 204 genes are still vague, indicating that they are probably hypothetical genes without thorough study. One SV may overlaps a significant amount of genes and lead to enormous genetic diversity in the process of evolution (Bertolotti et al., 2020; Weissensteiner et al., 2020) and domestication (Liu et al., 2019; Kou et al., 2020). We assume that the divergence between DA and the continental populations is more likely caused by SVs than SNPs.
Our results show some differences from the previous studies. According to mitochondrial gene COI, the Tajima’s Ds of all sp1 populations are significantly negative (Tian et al., 2015). In the present study, however, the whole-genome data reveal an overall positive D (Figure 2A). A probable explanation is that the evolution rate of cytoplasmic genome is much slower than nuclear genome. Therefore, gene markers surely have fewer variants than the nuclear genome.
Geographical Barrier Contributes to the Genetic Divergence
The severe reduced SNP polymorphism and high proportion of population-specific SV in DA represent a signature of gene flow restriction from the continental populations. The Qiongzhou Strait may provide a geographical barrier. Qiongzhou Strait separates Hainan Island from Southeast China, which is about 30 km wide and opened nearly 8,000 years ago (Zhao et al., 2007; Ni et al., 2014). In the pollinator wasp, the lack of genetic structure across a large distance on continent was reported, such as Valisia (Tian et al., 2015) and Wiebesia species (Liu et al., 2015) across Southeast China, or Ceratosolen fusciceps from Southeast China to Thailand (Kobmoo et al., 2010). However, population fragmentation also occurs when their habitats are islands. The signatures of isolation-by-distance was discovered in a Wiebesia species among the islands of Zhoushan Archipelago (Liu et al., 2013), even though the same species are genetically homogeneous over a distance of 1,000 km across Southeast China (Liu et al., 2015). It is thus reasonable to hypothesize that for pollinator wasp, the dispersal capability might be weakened by water bodies, or limited by islands. The dispersal of wasps totally depends on the adult female. After emerging from figs, an adult female wasp only survives for 1–2 days and is vulnerable to abiotic stresses. For them, high temperature and dryness are lethal while moisture is a relief (Ware and Compton, 1994; Dunn et al., 2008; Jevanandam et al., 2013; Gigante et al., 2020). Thus, the dispersal of wasps could be limited by the drastically changing weather above the Qiongzhou Strait, which is open to direct sunlight, and often experiences high temperatures and strong winds. The host of sp1, F. hirta, is a dioecious shrub existing in secondary jungle and the edge of forests. Pollinator wasps of such a host prefer the understory with gentle air (Harrison and Rasplus, 2006; Chen et al., 2011). It is reasonable to assume that the sensitivity and/or preference on habitats delimit the dispersal of wasp, and then restrict the gene flow. Previous studies (Tian et al., 2015; Yu et al., 2019) and our own (Table 2) all confirm the limited influence of the geographic distance. To be clear, the Qiongzhou Strait contributes to the gene flow barrier not because of its width but its unfavorable weather, suggesting that adaptation may also be an underlying drive.
The Role of Adaptation in the Genetic Divergence
The climate of Hainan Island is a tropical maritime climate while the habitats of sp1 continental populations are mainly under subtropical continental monsoon climate. As we have discussed before, in the absence of moisture, heat and dryness lead to high mortality of fig wasp (Dunn et al., 2008; Jevanandam et al., 2013; Gigante et al., 2020). Our own results also prove that the synchronization of temperature and humidity is the most important influence underlying the genetic divergence within sp1 (Table 2). Furthermore, heat and dryness are major restrictions of sp1 distribution (See “Contributions of Environment vs. Geographic Distance”). Therefore, it is reasonable to say that local adaptation contributes to the genetic structure between Hainan and the mainland population of sp1. In addition, different from the densely populated continent, Hainan Island is a biodiversity hotspot, still maintaining anatural tropical rainforest (Li, 1995; Luo et al., 2020). Human activity such as industrial pollution or pesticide may also cause a strong influence, for example, the industrial melanism of moth (Cook et al., 1970; Cook and Saccheri, 2013).
The differentiated islands in DA as well as islands in the mainland show decreased diversity. The results of GO enrichment on the differentiated islands also failed to linked to any genes that maybe related with speciation (See “Identification and Gene Ontology Enrichment of Differentiated Islands”). Hence, we assume that background selection before population dividing is a more likely causation. The GO enrichment analyses performed on the genes overlapping population-specific SVs, however, presented a more informative result. No gene was detected in the population-specific SVs of VH or SCBG. On the contrary, enormously higher number of genes (4,141) overlap the population-specific SVs of DA. We then performed GO enrichment analysis on those 4,141 genes and obtained 59 significant GO terms with p < 0.05 (Figure 6). It is worth noting that most of the 59 GO terms link to regulation of signaling pathway and external stimulus response. Pollinator wasps recognize the host through VOCs produced by figs (Weiblen, 2002; Chen Y. et al., 2016). Geographic divergence of VOCs was reported in F. hirta, the host of sp1 (Deng et al., 2021). The floral odors of F. hirta in Hainan clearly distinguished from those in the continent. The differences that occur in hosts could also drive the adaptation of pollinator wasps. For example, Ficus tikoua presents morphological differences of figs along its habitat (Deng et al., 2016). Its pollinator wasps show a genetic structure that loosely corresponds to the distribution of those morphologically different figs (Deng et al., 2020). Hence, the intra-species divergence of sp1 is probably a consequence of the co-evolution with its host. The 59 GO terms also link to the developmental process, indicating that DA may present difference in developmental regulation or life cycle. On the basis of our field observation, the figs mature much slower in cold periods, and the life cycle of fig wasps still strictly corresponds to the development of figs. Even in the same sampling location, the life cycle of fig wasp changes throughout seasons. Therefore, a flexible life cycle could facilitate adaptation to different climates.
Besides the divergence of genome, the differences on transcription and alternative splicing may result in the local adaptation of each population as well. The distribution of SVs’ localization differs among populations (Figure 5B). VH had an unusually higher proportion of population-specific SVs on the donor of splice sites and DA on the transcript region. Therefore, studies on transcriptome would provide more information. In brief, we confirm that local adaptation is an important force underlying the genetic divergence between DA and the continental populations. For adapting the divergence of its host population and the tropic environment, the Hainan wasp population may evolve special mechanisms for host recognition and life cycle regulation. SVs contribute more than SNPs to the genetic basis of adaptation.
Structural variation is an important source of materials for evolution (Frazer et al., 2009; Weissensteiner et al., 2020). Analyses based on SVs are still rare in insects. Most studies focus on the well-studied model species, Drosophila melanogaster (Dopman and Hartl, 2007; Emerson et al., 2008; Zichner et al., 2013; Long et al., 2018). Those works collected SVs across D. melanogaster genome, and calculated the SV types. In D. melanogaster, deletions also take the largest part of SVs. Long et al. (2018) annotated the distribution of SVs in exons and pointed out that the types and numbers of SVs highly differed among populations as well as in sp1. Hence, SVs contribute to the genetic divergence within D. melanogaster. A recent preprint study on Manduca sexta sheds light on the importance of a single inversion which enhances adaptation of the Z chromosome (Mongue and Kawahara, 2020). Generally speaking, SVs attract much less attention than SNPs. The present study highlights its importance in local adaptation. Nevertheless, it is still challenging to precisely annotate SVs with short-read sequencing data (Tattini et al., 2015). Complex SVs such as TRA are excluded in our study and its effect on the intraspecies divergence of Sp1 unfortunately remains unknown. The high proportion of DEL in SVs seems unusual in sp1 and D. melanogaster. Transposon activity is a possible explanation, but more accurate calling of SVs is required for transposon annotation. Further, the methods for downstream analysis for SV are much less than for SNP. Suitable methods for SV are urgently required.
The Demographic Reconstruction Within Sp 1
The reconstruction of a demographic history for fig wasp is intractable due to their relatively short adult longevity and life cycle. The varied mutation rates along the genome complicate the demographic estimation even more. Thirdly, frequent mutation within the population reduces the robustness of its demography reconstruction when repeated sampling is applied. Most important, local extinction-recolonization usually occurs in insects (Harrison, 2000; Devoto et al., 2005; Liu et al., 2013; Lever et al., 2014). The factors affecting fig production could also affect the survival of wasps. For example, high temperature and dryness are not only lethal for adult wasp, but also cause stress for fig trees. The fig trees in Borneo failed to produce figs during a drought, which then caused local extinction of their pollinator wasp (Harrison, 2001). The demography of fig wasp is therefore remarkably changeable even in a very recent period. The demographic studies of wasps usually based on field observation in a relatively short duration focus on the yearly fluctuations of demographic factors (e.g., number, daily survival rates, and sex ratio) (Forouzan et al., 2013; Asadi et al., 2019). For example, the 3-year field experiments (1997–1999) in a social wasp species (Polistes fuscatus) (Nadeau and Stamp, 2003), and the observation of a primitively eusocial wasp (Ropalidia fasciata), in Japan began in 1983 (Ito, 1996; Ito and Kasuya, 2005). Some other studies estimated a rough demographic history based on a few highly conservative gene markers (Wang et al., 2013; Tian et al., 2015; Yu et al., 2019). To our knowledge, the present study is the first that attempted to reconstruct the demographic history of fig wasp by whole-genome re-sequencing data. The demographic histories of the three populations were traced back 4,000 generations (1,000 ya). The Ne of all populations declined since 1,000 ya with a recent expansion (Figure 5). In one continental population VH, a recent population expansion was detected. However, the demography history does not differ between DA and the other continental population, SCBG. The similar results of DA and SCBG indicate that Hainan and continental populations still share an evolutionary process due to incompletely blocked gene flow, or incomplete lineage sorting. VH is geographically remote from DA and SCBG (Figure 1A) and locates in a landlocked habitat. Hence, we assume that the climatic contrasts between inland and coastal areas is the critical factor. Unfortunately, we failed to provide a more accurate estimation on the divergence time of sp1. Comparing with the whole-genome re-sequencing data and population genomics, we suggest the transcriptome approaches because the transcriptome is more conservative than genome. To be specific, we hope a molecular clock based on single copy genes could provide more reliable evidence for the divergence time estimation of fig wasp. We also performed the demography reconstruction with an approximated mutation rate according to the substitution rate at silent sites of Nasonia (Oliveira et al., 2008). Specific estimation of Valisia species mutation rate, for accuracy, will offer more interesting information.
Data Availability Statement
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive in National Genomics Data Center (https://ngdc.cncb.ac.cn/), China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences, under project number: PRJCA005939, accession number CRA004600 that are publicly accessible at https://ngdc.cncb.ac.cn/gsa.
Author Contributions
HY conducted the sample collection and preparation of wasp materials. XX conducted the bioinformatic analyses and wrote the manuscript. B-SW and HY conceived of the study. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (Grants Nos. 31630008 and 31971568), Key Special Project for Introduced Talents Team of Southern Marine Science and Engineering Guangdong Laboratory (Guangzhou; GML2019ZD0408), and Natural Science Foundation of Guangdong Province (Grants No. c20140500001306).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Vu Quang Nam (Vietnam National Foundation for Science and Technology Development under grant numbers 106.11-2012.82) for sample collection and help in the field. We are grateful to our former colleague Guo Jun-Cheng (South China Botanical Garden, The Chinese Academy of Sciences) for his help in the genome DNA extraction and raw filtering of re-sequencing data. We also thank Ke Fu-Shi (South China Botanical Garden, The Chinese Academy of Sciences), John Nason (Iowa State University), and Jordan Satler (Iowa State University) for advice on data analysis.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2021.764828/full#supplementary-material
Supplementary Figure 1 | Principal component analyses (PCA) and maximum likelihood (ML) tree with alternative filtering thresholds. (A,B) Filter with parameters: max missing <0.5, MAF > 0.03, thin = 1000, hwe = 0.1; (C,D) Filter with parameters: max missing <0.5, MAF > 0.03, thin = 1000; (E,F) Filter with parameters: max missing <0.5, MAF > 0.03, thin = 5000; (G,H) Filter with parameters: max missing <0.5, MAF > 0.03, thin = 1000, hwe = 0.5; (I,J) Filter with parameters: max miss <0.7, MAF > 0.03, thin = 5000. In (A,C,E,G,I), the three populations are presented as follows: Vinh Yen, Vietnam (VH, dark red); Hainan Island, China (DA, red); and Guangzhou, China (SCBG, pink). In (B,D,F,H,J), DA is indicated with red. VH and SCBG are considered as one population (Mainland), indicated with black.
Supplementary Figure 2 | Linkage disequilibrium (LD) decay analysis.
Supplementary Figure 3 | Manhattan plot of Z-normalized relative divergence (Fst) in non-overlap 100-kb windows. The red line represents the regions with Z-normalized Fst > 3.
Supplementary Table 1 | List of sampling sites in the generalized dissimilarity modeling.
Supplementary Table 2 | Statistics of sequencing.
Supplementary Table 3 | Results of Shapiro-Wilk normality test.
Supplementary Table 4 | Results of Wilcoxon rank sum test.
Supplementary Table 5 | List of differentiated islands.
Abbreviations
DEL, deletions; DUP, duplications; GO, gene ontology; INS, insertions; INV, inversions; ML, maximum likelihood; Ne, effective population size; PCA, principal component analysis; SNP, single-nucleotide polymorphisms; SV, structural variation; TRA, translocations; UTR, untranslated region; VOCs, volatile organic compounds; ya, years ago.
References
Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Cambridge: Babraham Bioinformatics, Babraham Institute.
Anstett, M. C., HossaertMcKey, M., and Kjellberg, F. (1997). Figs and fig pollinators: evolutionary conflicts in a coevolved mutualism. Trends Ecol. Evol. 12, 94–99. doi: 10.1016/s0169-5347(96)10064-1
Asadi, M., Rafiee-Dastjerdi, H., Nouri-Ganbalani, G., Naseri, B., and Hassanpour, M. (2019). Lethal and sublethal effects of five insecticides on the demography of a parasitoid wasp. Int. J. Pest Manag. 65, 301–312. doi: 10.1080/09670874.2018.1502899
Bauer, S., Grossmann, S., Vingron, M., and Robinson, P. N. (2008). Ontologizer 2.0-a multifunctional tool for GO term enrichment analysis and data exploration. Bioinformatics 24, 1650–1651. doi: 10.1093/bioinformatics/btn250
Becker, R. A., Wilks, A. R., Brownrigg, R., Minka, T. P., and Deckmyn, A. (2018). Maps: Draw Geographical Maps. Available online at: https://cran.r-project.org/web/packages=maps. (accessed August 18, 2020).
Berg, C. C. (1989). Classification and distribution of Ficus. Experientia 45, 605–611. doi: 10.1007/bf01975677
Bernard, J., Brock, K. C., Tonnell, V., Walsh, S. K., Wenger, J. P., Wolkis, D., et al. (2020). New species assemblages disrupt obligatory mutualisms between figs and their pollinators. Front. Ecol. Evol. 8:564653. doi: 10.3389/fevo.2020.564653
Bertolotti, A. C., Layer, R. M., Gundappa, M. K., Gallagher, M. D., Pehlivanoglu, E., Nome, T., et al. (2020). The structural variation landscape in 492 Atlantic salmon genomes. Nat. Commun. 11:5176. doi: 10.1038/s41467-020-18972-x
Borges, R. M. (2015). How to be a fig wasp parasite on the fig-fig wasp mutualism. Curr. Opin. Insect. Sci. 8, 34–40. doi: 10.1016/j.cois.2015.01.011
Bradbury, P. J., Zhang, Z., Kroon, D. E., Casstevens, T. M., Ramdoss, Y., and Buckler, E. S. (2007). TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635. doi: 10.1093/bioinformatics/btm308
Broad Institute (2018). Picard Toolkit. GitHub Repository: Broad Institute. Available online at: http://broadinstitute.github.io/picard/ (accessed November 18, 2020).
Bronstein, J. L. (1988). Mutualism, antagonism, and the fig-pollinator interaction. Ecology 69, 1298–1302. doi: 10.2307/1941287
Bronstein, J. L. (2009). The evolution of facilitation and mutualism. J. Ecol. 97, 1160–1170. doi: 10.1111/j.1365-2745.2009.01566.x
Bronstein, J. L., and McKey, D. (1989). The fig-pollinator mutualism-a model system for comparative biology. Experientia 45, 601–604. doi: 10.1007/bf01975676
Browning, B. L., Zhou, Y., and Browning, S. R. (2018). A one-penny imputed genome from next-generation reference panels. Am. J. Hum. Genet. 103, 338–348. doi: 10.1016/j.ajhg.2018.07.015
Chen, X., Schulz-Trieglaff, O., Shaw, R., Barnes, B., Schlesinger, F., Källberg, M., et al. (2016). Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222. doi: 10.1093/bioinformatics/btv710
Chen, Y., Huang, M., Wu, W., Wang, A., Bao, T., Zheng, C., et al. (2016). The floral scent of Ficus pumila var. pumila and its effect on the choosing behavior of pollinating wasps of Wiebesia pumilae. Acta Ecol. Sin. 36, 321–326. doi: 10.1016/j.chnaes.2016.06.008
Chen, Y., Jiang, Z. X., Compton, S. G., Liu, M., and Chen, X. Y. (2011). Genetic diversity and differentiation of the extremely dwarf Ficus tikoua in Southwestern China. Biochem. Syst. Ecol. 39, 441–448. doi: 10.1016/j.bse.2011.06.006
Cingolani, P., Platts, A., Wang, L. L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92. doi: 10.4161/fly.19695
Cook, J. M., and Rasplus, J. Y. (2003). Mutualists with attitude: coevolving fig wasps and figs. Trends Ecol. Evol. 18, 241–248. doi: 10.1016/s0169-5347(03)00062-4
Cook, L. M., Askew, R. R., and Bishop, J. A. (1970). Increasing frequency of the typical form of the peppered moth in Manchester. Nature 227, 1155–1155. doi: 10.1038/2271155a0
Cook, L. M., and Saccheri, I. J. (2013). The peppered moth and industrial melanism: evolution of a natural selection case study. Heredity 110, 207–212. doi: 10.1038/hdy.2012.92
Cruaud, A., Ronsted, N., Chantarasuwan, B., Chou, L. S., Clement, W. L., Couloux, A., et al. (2012). An extreme case of plant-insect codiversification: figs and fig-pollinating wasps. Syst. Biol. 61, 1029–1047. doi: 10.1093/sysbio/sys068
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
Danecek, P., and McCarthy, S. A. (2017). BCFtools/csq: haplotype-aware variant consequences. Bioinformatics 33, 2037–2039. doi: 10.1093/bioinformatics/btx100
Darwell, C. T., Al-Beidh, S., and Cook, J. M. (2014). Molecular species delimitation of a symbiotic fig-pollinating wasp species complex reveals extreme deviation from reciprocal partner specificity. BMC Evol. Biol. 14:189. doi: 10.1186/s12862-014-0189-9
Darwell, C. T., and Cook, J. M. (2017). Cryptic diversity in a fig wasp community-morphologically differentiated species are sympatric but cryptic species are parapatric. Mol. Ecol. 26, 937–950. doi: 10.1111/mec.13985
Deng, J. Y., Fu, R. H., Compton, S. G., Hu, D. M., Zhang, L. S., Yang, F., et al. (2016). Extremely high proportions of male flowers and geographic variation in floral ratios within male figs of Ficus tikoua despite pollinators displaying active pollen collection. Ecol. Evol. 6, 607–619. doi: 10.1002/ece3.1926
Deng, J. Y., Fu, R. H., Compton, S. G., Liu, M., Wang, Q., Yuan, C., et al. (2020). Sky islands as foci for divergence of fig trees and their pollinators in southwest China. Mol. Ecol. 29, 762–782. doi: 10.1111/mec.15353
Deng, X. X., Bruno, B., Peng, Y. Q., Yu, H., Cheng, Y. F., Kjellberg, F., et al. (2021). Plants are the drivers of geographic variation of floral scents in a highly specialized pollination mutualism: a study of Ficus hirta in China. BMC Ecol. Evol. doi: 10.21203/rs.3.rs-192226/v1
Devoto, M., Medan, D., and Montaldo, N. H. (2005). Patterns of interaction between plants and pollinators along an environmental gradient. Oikos 109, 461–472. doi: 10.1111/j.0030-1299.2005.13712.x
Dopman, E. B., and Hartl, D. L. (2007). A portrait of copy-number polymorphism in Drosophila melanogaster. Proc. Natl. Acad. Sci. U. S. A. 104, 19920–19925. doi: 10.1073/pnas.0709888104
Dunn, D. W., Yu, D. W., Ridley, J., and Cook, J. M. (2008). Longevity, early emergence and body size in a pollinating fig wasp-implications for stability in a fig-pollinator mutualism. J. Anim. Ecol. 77, 927–935. doi: 10.1111/j.1365-2656.2008.01416.x
Eichler, E. E., Flint, J., Gibson, G., Kong, A., Leal, S. M., Moore, J. H., et al. (2010). VIEWPOINT Missing heritability and strategies for finding the underlying causes of complex disease. Nat. Rev. Genet. 11, 446–450. doi: 10.1038/nrg2809
Elith, J., Phillips, S. J., Hastie, T., Dudik, M., Chee, Y. E., and Yates, C. J. (2011). A statistical explanation of MaxEnt for ecologists. Divers. Distribut. 17, 43–57. doi: 10.1111/j.1472-4642.2010.00725.x
Emerson, J. J., Cardoso-Moreira, M., Borevitz, J. O., and Long, M. (2008). Natural selection shapes genome-wide patterns of copy-number polymorphism in Drosophila melanogaster. Science 320, 1629–1631. doi: 10.1126/science.1158078
Ferrier, S., Manion, G., Elith, J., and Richardson, K. (2007). Using generalized dissimilarity modelling to analyse and predict patterns of beta diversity in regional biodiversity assessment. Divers. Distribut. 13, 252–264. doi: 10.1111/j.1472-4642.2007.00341.x
Feuk, L., Carson, A. R., and Scherer, S. W. (2006). Structural variation in the human genome. Nat. Rev. Genet. 7, 85–97. doi: 10.1038/nrg1767
Fick, S. E., and Hijmans, R. J. (2017). WorldClim 2: new 1-km spatial resolution climate surfaces for global land areas. Int. J. Climatol. 37, 4302–4315. doi: 10.1002/joc.5086
Forouzan, M., Safaralizadeh, M. H., Shirazi, J., Safavi, S. A., and Rezaei, M. (2013). Biology and demography of Trissolcus basalis (Hym.: scelionidae) on eggs of two different hosts. J. Entomol. Soc. Iran. 33, 69–85.
Frazer, K. A., Murray, S. S., Schork, N. J., and Topol, E. J. (2009). Human genetic variation and its contribution to complex traits. Nat. Rev. Genet. 10, 241–251. doi: 10.1038/nrg2554
Gaut, B. S., Seymour, D. K., Liu, Q. P., and Zhou, Y. F. (2018). Demography and its effects on genomic variation in crop domestication. Nat. Plants 4, 512–520. doi: 10.1038/s41477-018-0210-1
Gigante, E. T., Lim, E. J., Crisostomo, K. G., Cornejo, P., and Rodriguez, L. J. (2020). Increase in humidity widens heat tolerance range of tropical Ceratosolen fig wasps. Ecol. Entomol. 46, 573–581. doi: 10.1111/een.13003
Harrison, R. D. (2000). Repercussions of El Nino: drought causes extinction and the breakdown of mutualism in Borneo. Proc. R. Soc. B-Biol. Sci. 267, 911–915. doi: 10.1098/rspb.2000.1089
Harrison, R. D. (2001). Drought and the consequences of El Nino in Borneo: a case study of figs. Population Ecol. 43, 63–75. doi: 10.1007/pl00012017
Harrison, R. D., and Rasplus, J. Y. (2006). Dispersal of fig pollinators in Asian tropical rain forests. J. Trop. Ecol. 22, 631–639. doi: 10.1017/s0266467406003488
Herre, E. A., Jander, K. C., and Machado, C. A. (2008). Evolutionary ecology of figs and their associates: recent progress and outstanding puzzles. Annu. Rev. Ecol. Evol. Syst. 39, 439–458. doi: 10.1146/annurev.ecolsys.37.091305.110232
Hijmans, R. J., Cameron, S. E., Parra, J. L., Jones, P. G., and Jarvis, A. (2005). Very high resolution interpolated climate surfaces for global land areas. Int. J. Climatol. 25, 1965–1978. doi: 10.1002/joc.1276
Hill, P., Wapstra, E., Ezaz, T., and Burridge, C. P. (2021). Pleistocene divergence in the absence of gene flow among populations of a viviparous reptile with intraspecific variation in sex determination. Ecol. Evol. 11, 5575–5583. doi: 10.1002/ece3.7458
Ho, S. S., Urban, A. E., and Mills, R. E. (2020). Structural variation in the sequencing era. Nat. Rev. Genet. 21, 171–189. doi: 10.1038/s41576-019-0180-9
Ito, Y. (1996). Reconstruction of Ropalidia fasciata nests by progeny females after a typhoon and its significance in the social evolution of wasps. Ecol. Res. 11, 79–87. doi: 10.1007/bf02347822
Ito, Y., and Kasuya, E. (2005). Demography of the Okinawan eusocial wasp Ropalidia fasciata (Hymenoptera: vespidae) I. Survival rate of individuals and colonies, and yearly fluctuations in colony density. Entomol. Sci. 8, 41–64. doi: 10.1111/j.1479-8298.2005.00099.x
Jeffares, D. C., Jolly, C., Hoti, M., Speed, D., Shaw, L., Rallis, C., et al. (2017). Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat. Commun. 8:14061. doi: 10.1038/ncomms14061
Jevanandam, N., Goh, A. G. R., and Corlett, R. T. (2013). Climate warming and the potential extinction of fig wasps, the obligate pollinators of figs. Biol. Lett. 9:4. doi: 10.1098/rsbl.2013.0041
Kamvar, Z. N., Tabima, J. F., and Grunwald, N. J. (2014). Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. Peerj 2:e281. doi: 10.7717/peerj.281
King, P. E. (1961). A possible method of sex ratio determination in parasitic hymenopteran Nasonia vitripennis. Nature 189, 330–331. doi: 10.1038/189330a0
Kobmoo, N., Hossaert-McKey, M., Rasplus, J. Y., and Kjellberg, F. (2010). Ficus racemosa is pollinated by a single population of a single agaonid wasp species in continental South-East Asia. Mol. Ecol. 19, 2700–2712. doi: 10.1111/j.1365-294X.2010.04654.x
Korunes, K. L., Machado, C. A., and Noor, M. A. F. (2021). Inversions shape the divergence of Drosophila pseudoobscura and Drosophila persimilis on multiple timescales. Evolution 75, 1820–1834. doi: 10.1111/evo.14278
Kou, Y., Liao, Y., Toivainen, T., Lv, Y., Tian, X., Emerson, J. J., et al. (2020). Evolutionary genomics of structural variation in Asian rice (Oryza sativa) domestication. Mol. Biol. Evol. 37, 3507–3524. doi: 10.1093/molbev/msaa185
Letunic, I., and Bork, P. (2021). Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Lever, J. J., van Nes, E. H., Scheffer, M., and Bascompte, J. (2014). The sudden collapse of pollinator communities. Ecol. Lett. 17, 350–359. doi: 10.1111/ele.12236
Li, H., and Durbin, R. (2011). Inference of human population history from individual whole-genome sequences. Nature 475, 493–496. doi: 10.1038/nature10231
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, Y. (1995). Biodiversity of tropical forest and its protection strategies in Hainan Island, China. Lin Ye Ke Xue Yan Jiu 8, 455–461.
Liu, C., Ran, X. Q., Yu, C. Y., Xu, Q., Niv, X., Zhao, P. J., et al. (2019). Whole-genome analysis of structural variations between Xiang pigs with larger litter sizes and those with smaller litter sizes. Genomics 111, 310–319. doi: 10.1016/j.ygeno.2018.02.005
Liu, M., Compton, S. G., Peng, F. E., Zhang, J., and Chen, X. Y. (2015). Movements of genes between populations: are pollinators more effective at transferring their own or plant genetic markers? Proc. R. Soc. B-Biol. Sci. 282:9. doi: 10.1098/rspb.2015.0290
Liu, M., Zhang, J., Chen, Y., Compton, S. G., and Chen, X. Y. (2013). Contrasting genetic responses to population fragmentation in a coevolving fig and fig wasp across a mainland-island archipelago. Mol. Ecol. 22, 4384–4396. doi: 10.1111/mec.12406
Long, E., Evans, C., Chaston, J., and Udall, J. A. (2018). Genomic structural variations within five continental populations of Drosophila melanogaster G3-GENES. Genom. Genet. 8, 3247–3253. doi: 10.1534/g3.118.200631
Luo, H., Dai, S., Li, M., Li, Y., Zheng, Q., and Hu, Y. (2020). Relative roles of climate changes and human activities in vegetation variables in Hainan Island Remote Sens. Environment 32, 154–161.
Malaspinas, A. S., Westaway, M. C., Muller, C., Sousa, V. C., Lao, O., Alves, I., et al. (2016). A genomic history of Aboriginal Australia. Nature 538, 207–214. doi: 10.1038/nature18299
Manion, G., Lisk, M., Ferrier, S., Nieto-Lugilde, D., and Fitzpatrick, M. C. (2016). gdm: Functions for Generalized Dissimilarity Modeling. R Package.
Martin, S. H., Dasmahapatra, K. K., Nadeau, N. J., Salazar, C., Walters, J. R., Simpson, F., et al. (2013). Genome-wide evidence for speciation with gene flow in Heliconius butterflies. Genome Res. 23, 1817–1828. doi: 10.1101/gr.159426.113
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303.
Mongue, A. J., and Kawahara, A. Y. (2020). Population differentiation and structural variation in the genome of Manduca sexta across the United States. bioRxiv [preprint] 2020.11.01.364000. doi: 10.1101/2020.11.01.364000
Myers, E. A., Xue, A. T., Gehara, M., Cox, C., Rabosky, A. R. D., Lemos-Espinal, J., et al. (2019). Environmental heterogeneity and not vicariant biogeographic barriers generate community-wide population structure in desert-adapted snakes. Mol. Ecol. 28, 4535–4548. doi: 10.1111/mec.15182
Nadeau, H., and Stamp, N. (2003). Effect of prey quantity and temperature on nest demography of social wasps. Ecol. Entomol. 28, 328–339. doi: 10.1046/j.1365-2311.2003.00514.x
Nei, M., and Li, W. H. (1979). Mathematical-model for studying genetic-variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. U. S. A. 76, 5269–5273. doi: 10.1073/pnas.76.10.5269
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Ni, Y., Xia, Z., and Ma, S. (2014). The opening of Qiongzhou Strait: evidence from sub-bottom profiles. Hai Yang Di Zhi Yu Di Si Ji 34, 79–82.
Oliveira, D., Raychoudhury, R., Lavrov, D. V., and Werren, J. H. (2008). Rapidly evolving mitochondrial genome and directional selection in mitochondrial genes in the parasitic wasp Nasonia (Hymenoptera : pteromalidae). Mol. Biol. Evol. 25, 2167–2180. doi: 10.1093/molbev/msn159
Petit, R. J., and Hampe, A. (2006). Some evolutionary consequences of being a tree. Annu. Rev. Ecol. Evol. Syst. 37, 187–214. doi: 10.1146/annurev.ecolsys.37.091305.110215
Phillips, S. J., Anderson, R. P., and Schapire, R. E. (2006). Maximum entropy modeling of species geographic distributions. Ecol. Modell. 190, 231–259. doi: 10.1016/j.ecolmodel.2005.03.026
Press, M. O., Hall, A. N., Morton, E. A., and Queitsch, C. (2019). Substitutions are boring: some arguments about parallel mutations and high mutation rates. Trends Genet. 35, 253–264. doi: 10.1016/j.tig.2019.01.002
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033
R Core Team. (2020). R: A Language and Environment for Statistical Computing. R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Ramirez, W. B. (1970). Host specificity of fig wasps (Agaonidae). Evolution 24, 680–691. doi: 10.1111/j.1558-5646.1970.tb01804.x
Rausch, T., Zichner, T., Schlattl, A., Stütz, A. M., Benes, V., and Korbel, J. O. (2012). DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28, i333–i339. doi: 10.1093/bioinformatics/bts378
Renoult, J. P., Kjellberg, F., Grout, C., Santoni, S., and Khadari, B. (2009). Cyto-nuclear discordance in the phylogeny of Ficus section Galoglychia and host shifts in plant-pollinator associations. BMC Evol. Biol. 9:18. doi: 10.1186/1471-2148-9-248
Ronsted, N., Weiblen, G. D., Cook, J. M., Salamin, N., Machado, C. A., and Savolainen, V. (2005). 60 million years of co-divergence in the fig-wasp symbiosis. Proc. R. Soc. B-Biol. Sci. 272, 2593–2599. doi: 10.1098/rspb.2005.3249
Schiffels, S., and Durbin, R. (2014). Inferring human population size and separation history from multiple genome sequences. Nat. Genet. 46, 919–925. doi: 10.1038/ng.3015
Shapiro, S. S., and Wilk, M. B. (1965). An analysis of variance test for normality (complete samples). Biometrika 52, 591–611. doi: 10.2307/2333709
Souto-Vilaros, D., Machac, A., Michalek, J., Darwell, C. T., Sisol, M., Kuyaiva, T., et al. (2019). Faster speciation of fig-wasps than their host figs leads to decoupled speciation dynamics: snapshots across the speciation continuum. Mol. Ecol. 28, 3958–3976. doi: 10.1111/mec.15190
Tajima, F. (1989). Statistical-method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595.
Tattini, L., D’Aurizio, R., and Magi, A. (2015). Detection of genomic structural variants from next-generation sequencing data. Front. Bioeng. Biotechnol. 3:92. doi: 10.3389/fbioe.2015.00092
Thomas, J. A., Welch, J. J., Lanfear, R., and Bromham, L. (2010). A generation time effect on the rate of molecular evolution in invertebrates. Mol. Biol. Evol. 27, 1173–1180. doi: 10.1093/molbev/msq009
Tian, E. W., Nason, J. D., Machado, C. A., Zheng, L. N., Yu, H., and Kjellberg, F. (2015). Lack of genetic isolation by distance, similar genetic structuring but different demographic histories in a fig-pollinating wasp mutualism. Mol. Ecol. 24, 5976–5991. doi: 10.1111/mec.13438
Wang, G., Zhang, X. T., Herre, E. A., McKey, D., Machado, C. A., Yu, W. B., et al. (2021). Genomic evidence of prevalent hybridization throughout the evolutionary history of the fig-wasp pollination mutualism. Nat. Commun. 12:14. doi: 10.1038/s41467-021-20957-3
Wang, H., Hsieh, C., Huang, C., Kong, S., Chang, H., Lee, H., et al. (2013). Genetic and physiological data suggest demographic and adaptive responses in complex interactions between populations of figs (Ficus pumila) and their pollinating wasps (Wiebesia pumilae). Mol. Ecol. 22, 3814–3832. doi: 10.1111/mec.12336
Ware, A. B., and Compton, S. G. (1994). Dispersal of adult female fig wasps: 2. Movements between trees. Entomol. Exp. Appl. 73, 231–238. doi: 10.1111/j.1570-7458.1994.tb01860.x
Weiblen, G. D. (2002). How to be a fig wasp. Annu. Rev. Entomol. 47, 299–330. doi: 10.1146/annurev.ento.47.091201.145213
Weir, B. S., and Cockerham, C. C. (1984). Estimating F-statistics for the analysis of population-structure. Evolution 38, 1358–1370. doi: 10.2307/2408641
Weissensteiner, M. H., Bunikis, I., Catalan, A., Francoijs, K. J., Knief, U., Heim, W., et al. (2020). Discovery and population genomics of structural variation in a songbird genus. Nat. Commun. 11:11. doi: 10.1038/s41467-020-17195-4
Wellenreuther, M., Merot, C., Berdan, E., and Bernatchez, L. (2019). Going beyond SNPs: the role of structural genomic variants in adaptive evolution and species diversification. Mol. Ecol. 28, 1203–1209. doi: 10.1111/mec.15066
Werren, J. H., Richards, S., Desjardins, C. A., Niehuis, O., Gadau, J., Colbourne, J. K., et al. (2010). Functional and evolutionary insights from the genomes of three parasitoid Nasonia species. Science 327, 343–348. doi: 10.1126/science.1178028
Wiebes, J. T. (1993). Agaonidae (Hymenoptera Chalcidoidea) and Ficus (Moraceae): fig wasps and their figs, xi (Blastophaga) s.l. Proc. Koninklijke Nederlandse Akademie Wetenschappen-Biol. Chem. Geol. Phys. Med. Sci. 96, 347–367.
Wilcoxon, F. (1946). Individual comparisons of grouped data by ranking methods. J. Econ. Entomol. 39, 269–270. doi: 10.2307/3001968
Wylie, H. G. (1965). Some factors that reduce reproductive rate of Nasonia vitripennis (walk) at high adult population densities. Can. Entomol. 97, 970–977. doi: 10.4039/Ent97970-9
Yang, L., Machado, C. A., Dang, X., Peng, Y., Yang, D., Zhang, D., et al. (2015). The incidence and pattern of copollinator diversification in dioecious and monoecious figs. Evolution 69, 294–304. doi: 10.1111/evo.12584
Yu, H., Tian, E. W., Zheng, L. N., Deng, X. X., Cheng, Y. F., Chen, L. F., et al. (2019). Multiple parapatric pollinators have radiated across a continental fig tree displaying clinal genetic variation. Mol. Ecol. 28, 2391–2405. doi: 10.1111/mec.15046
Zhang, C., Dong, S. S., Xu, J. Y., He, W. M., and Yang, T. L. (2019). PopLDdecay: a fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 35, 1786–1788. doi: 10.1093/bioinformatics/bty875
Zhao, H. T., Wang, L., and Yuan, J. Y. (2007). Origin and time of Qiongzhou Strait. Hai Yang Di Zhi Yu Di Si Ji 27, 33–40.
Zheng, X. W., Levine, D., Shen, J., Gogarten, S. M., Laurie, C., and Weir, B. S. (2012). A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 28, 3326–3328. doi: 10.1093/bioinformatics/bts606
Keywords: genetic divergence, adaptation, geographic barrier, population genetics, Ficus hirta, Valisia javana
Citation: Xu X, Wang B-S and Yu H (2021) Intraspecies Genomic Divergence of a Fig Wasp Species Is Due to Geographical Barrier and Adaptation. Front. Ecol. Evol. 9:764828. doi: 10.3389/fevo.2021.764828
Received: 26 August 2021; Accepted: 04 October 2021;
Published: 10 November 2021.
Edited by:
Alison G. Nazareno, Federal University of Minas Gerais, BrazilReviewed by:
Rodrigo Pereira, University of São Paulo, BrazilFernando Farache, Goiano Federal Institute (IFGOIANO), Brazil
Copyright © 2021 Xu, Wang and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Yu, eXVodWlAc2NpYi5hYy5jbg==