
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol., 30 August 2021
Sec. Phylogenetics, Phylogenomics, and Systematics
Volume 9 - 2021 | https://doi.org/10.3389/fevo.2021.734462
 Roderick B. Gagne1,2†
Roderick B. Gagne1,2† Simona Kraberger3†Rebekah McMinn1Daryl R. Trumbo4
Simona Kraberger3†Rebekah McMinn1Daryl R. Trumbo4 Charles R. Anderson Jr.5Ken A. Logan6
Charles R. Anderson Jr.5Ken A. Logan6 Mathew W. Alldredge5Karen Griffin5
Mathew W. Alldredge5Karen Griffin5 Sue Vandewoude1*
Sue Vandewoude1*Monitoring pathogens in wildlife populations is imperative for effective management, and for identifying locations for pathogen spillover among wildlife, domestic species and humans. Wildlife pathogen surveillance is challenging, however, as sampling often requires the capture of a significant proportion of the population to understand host pathogen dynamics. To address this challenge, we assessed the ability to use hunter-collected teeth from puma across Colorado to recover genetic data of two feline retroviruses, feline foamy virus (FFV) and feline immunodeficiency virus (FIVpco) and show they can be utilized for this purpose. Comparative phylogenetic analyses of FIVpco and FFV from tooth and blood samples to previous analyses conducted with blood samples collected over a nine-year period from two distinct areas was undertaken highlighting the value of tooth derived samples. We found less FIVpco phylogeographic structuring than observed from sampling only two regions and that FFV data confirmed previous findings of endemic infection, minimal geographic structuring, and supported frequent cross-species transmission from domestic cats to pumas. Viral analysis conducted using intentionally collected blood samples required extensive financial, capture and sampling efforts. This analysis illustrates that viral genomic data can be cost effectively obtained using tooth samples incidentally-collected from hunter harvested pumas, taking advantage of samples collected for morphological age identification. This technique should be considered as an opportunistic method to provide broad geographic sampling to define viral dynamics more accurately in wildlife.
Diseases of wildlife are increasingly threatening animal populations resulting in negative economic impacts, the loss of biodiversity, and risk to livestock and human health (Artois et al., 2009; Heard et al., 2013; Watsa, 2020). In some cases, disease has led to species extinctions (Skerratt et al., 2007; Heard et al., 2013); for example, the fungal parasite chytridiomycosis is a major contributor to global amphibian declines where over one third of amphibian species are now threatened or have already gone extinct (Cohen, 2003; Skerratt et al., 2007; Wake and Vredenburg, 2008). In other cases disease results in a major die off of already endangered populations, such as with Hemorrhagic septicemia, caused by a bacterial infection, in the endangered saiga antelope that resulted in the loss of half the remaining population in only a few weeks (Nicholls, 2015; Kock et al., 2018). Wildlife diseases have major economic impacts both from the spillover to domestic animals, such as Brucella from multiple wildlife species to domestic cattle, or the loss of game species; for example, chronic wasting disease in ungulates (Bengis et al., 2002; Wandeler et al., 2003; Bishop, 2004; Gomo et al., 2012; Zimmer et al., 2012; Kamath et al., 2016). Additionally, the majority of emerging infectious diseases in humans are a result of spillover from other animal species (Taylor et al., 2001; Belay et al., 2017). As a result we have seen a widespread increase in the need for identifying pathogens present in animal populations, defining disease dynamics among populations, determining spillover events, and the development of control strategies to mitigate disease spread (Artois et al., 2009; Ryser-Degiorgis, 2013; Watsa, 2020).
Wildlife pathogens surveillance is hampered by the difficulty of sampling a wide variety of species, many of which are elusive and challenging to capture. Overcoming this roadblock presents expensive and logistical challenges (Stallknecht, 2007; Watsa, 2020). Active disease surveillance, or the practice of direct sample collection for pathogen analysis, provides the greatest likelihood of detecting diseases in the population and limiting biases of data (Stallknecht, 2007; Artois et al., 2009). This approach is often costly and requires a significant amount of effort, thus limiting the number of species, number of animals, and the geographic area in which information can be collected (Stallknecht, 2007; Watsa, 2020). Opportunistic sample collection, termed “passive surveillance” can take advantage of sampling roadkill or other deceased animals, and presents a cost effective approach that can help to identify disease outbreaks (Hawkins et al., 2006; Lawson et al., 2015; Barron et al., 2018), this does however have its own caveats (Stallknecht, 2007; Santos et al., 2011). Carcasses can be quickly removed from the environment, via scavengers or other processes, undergo degradation when carcasses are not sampled in a timely manner or many carcasses are simply not detected and reported (Wobeser and Wobeser, 1992; Santos et al., 2011). In addition, samples often are biased towards animals that have been killed or are closer to human development (Ryser-Degiorgis, 2013). Hunter-collected sampling offers a promising approach to disease surveillance, as samples represent a large geographic representation of animals that can be sampled in a timely manner post-death (Brook et al., 2009; Lawson et al., 2015). Though this approach does still harbor some sample biases, for example favoring larger males (Belsare et al., 2020), disease data obtained from hunter samples can limit biases, logistical effort and costs.
Tooth samples are often required to be collected and submitted by hunters or directly collected by the permitting agency since morphological analyses can reveal age of the animal which is helpful for population management. Teeth can be relatively easily collected by trained technicians and sufficient amounts of DNA can be extracted from them for nucleic acid based diagnostic testing (Wandeler et al., 2003). The potential to recover pathogens from animal teeth remains largely unexplored, though in humans, pathogen data has been recovered from dental pulp of corpses (Barbieri et al., 2017). A benefit of using wildlife tooth samples is that potential to recover genomic information of pathogens, allowing characterization of pathogen strains and variants (Fitak et al., 2019). Genomic data from pathogens can inform their origin, geographic spread, cross-species transmissions, and host connectivity (Biek et al., 2006; Baele et al., 2018; Kraberger et al., 2020; Fountain-Jones et al., 2021). Increasingly, genomic data is used to determine spread in real time and identify environmental features associated with pathogen gene-flow (Holmes and Grenfell, 2009; Baele et al., 2017; Kozakiewicz et al., 2018), further highlighting the utility of obtaining pathogen genomic data routinely and with minimal cost from wildlife samples.
Pumas are a widespread large North American felid with varying management and population statuses that in the United States run a gamut, from endangered species status of the Florida panther and special protection in California, to game animal status in most western states, and complete lack of protection in Texas (“Non-game, Exotic, Endangered, Threatened and Protected Species” 2020, “Florida Panther – Florida Panther – U.S. Fish and Wildlife Service” n.d.). Pumas are capable of long distance movement (e.g., >1,000 Km over a lifetime (Hawley et al., 2016) and thus viral genomic signatures in specific areas may not translate to larger geographic regions. Infectious diseases have resulted in mortality of threatened puma populations, including viral spillover from domestic cats (Brown et al., 2008; Carver et al., 2016; Chiu et al., 2019; Kraberger et al., 2020). In particular, pumas are infected with several retroviruses that have been shown to reach high prevalence in populations, including a puma specific feline immunodeficiency virus (FIVpco) and feline foamy virus (FFV) (Franklin et al., 2007a, b; Lee et al., 2017; Kechejian et al., 2019). Assessment of viral infections of pumas has revealed cross-species transmission events, landscape features that influence viral structure, and disease-related die-offs in some populations (Lee et al., 2017; Chiu et al., 2019; Kraberger et al., 2020; Fountain-Jones et al., 2021). Genomic variation in FIVpco has been found to be structured, in part, by geographic location, and specific landscape features are associated with viral spread (Fountain-Jones et al., 2021) in contrast, genetic data of FFV has shown a lack of phylogenetic structure related to geography and instead show patterns of multiple spillover events from domestic cats to pumas (Kraberger et al., 2020). The difficulties in obtaining puma samples suitable for recovering diagnostic and genomic data from viruses has constrained these studies to a limited number of distinct geographic areas.
We analyzed DNA from teeth collected from 200 hunter-harvested pumas across Colorado to assess the capacity to use this tissue opportunistically for wildlife disease surveillance. We asked: (1) can viruses be detected in tooth samples of pumas? (2) is genomic material recovered of optimal quality for sequence analysis? and (3) how do viral sequences recovered from puma teeth inform geographical structure of viruses across the state compared to prior analyses conducted with active surveillance? This work illustrates that hunter-harvested samples utilized for disease surveillance provide pathogen data that is temporally linked, well-characterized with respect to individual animal demography, and spatially distinct.
In the state of Colorado, hunter-harvested pumas are required to be reported to Colorado Parks and Wildlife (CPW) in order to record demographic information. This includes the removal of a premolar tooth from each individual primarily to estimate the age for harvest data analyses. DNA was extracted by CPW personnel as described below promptly after receiving the samples using the Qiagen DNeasy Blood and Tissue Kit, with a few modifications; including increased incubation time of 40 min and increased vortexing to remove tissue from the tooth. Teeth from 200 individual puma harvested between 2013 and 2014 were utilized in this proof of concept study. Blood samples from puma live captured between 2005 and 2014 from the Uncompahgre Plateau, and Front Range, regions of Colorado, United States were used for comparison (Carver et al., 2016; Kraberger et al., 2020; Fountain-Jones et al., 2021). Blood was also collected from two puma from Nevada and one animal from New Mexico. The Qiagen DNeasy Blood and Tissue Kits were also utilized to recover nucleic acid from blood samples as per manufacturer’s instructions and as described in Kraberger et al. (2020) and Fountain-Jones et al. (2021).
DNA samples were screened by highly sensitive polymerase chain reaction (PCR) assays for Feline immunodeficiency Virus – Pco (FIVpco, the puma specific FIV) and feline foamy virus (FFV). For FIVpco, primers in a conserved region of the vif gene were used for initial screening (Supplementary Table 1). PCR products were purified with 5 μL ExoSAP-It (USB, Affymetrix, Cleveland, OH, United States) and sent to a commercial lab for Sanger sequencing (GENEWIZ, United States). Samples that were positive were subjected to PCR to FIVpco env and pol genes matching the phylogenetic group of recovered vif gene (Supplementary Table 1). Nested PCR protocols were performed using Kapa HiFi Hotstart DNA polymerase (Kapa Biosystems, United States) as per the product protocol and an annealing temperature based on the primer melting temperature. Given the low volume availability of DNA and high level of genetic conservation seen for FFV in felids, we screened all samples directly for FFV pol and env genes using protocols and primers previously described. Sequences were recovered, cloned and sequenced as described below.
FIVpco and FFV sequence data for the pol and env gene were collated from blood samples taken from live puma captured in the Uncompahgre Plateau region, and Front Range, Colorado, United States. Nucleic acid from blood samples were screened for the presence of FIVpco as described in Fountain-Jones et al. (2021). Specific env and pol primers were utilized (Supplementary Table 2). Nested PCR protocols for all FIVpco screens were performed using Kapa HiFi Hotstart DNA polymerase (Kapa Biosystems, United States) as per the product protocol and an annealing temperature based on the primer melting temperature. Sequences were recovered, cloned and sequenced as outline below. Sequence data for pol and/or env genes of FIVpco isolated from blood of three pumas collected in Nevada and New Mexico were also analyzed here (Supplementary Table 1). Env gene of 63 FIVpco-positive puma samples and 5 pol sequences recovered from these animals, but not previously described, are included in this analysis (Supplementary Table 1). FFV pol and env gene sequences used for phylogenetic analysis were obtained previously (Kraberger et al., 2020; Supplementary Table 1). CSU and CPW Institutional Animal Care and Use Committees reviewed and approved previous live capture work from which samples were included here (CSU IACUC protocol 05-061A).
Polymerase chain reaction products were run on 0.7% agarose gels, amplicons were excised, purified, and cloned using pJET 1.2 vector (Thermo Fisher Scientific, United States) and XL blue Escherichia coli competent cells. Plasmids with viral sequences were purified and Sanger sequenced at GENEWIZ (USA). Forward and reverse reads were assembled, trimmed, and checked using Geneious V7.0.6 (“Geneious” 2019).
Datasets of sequences for the env and pol of FIVpco and FFV were compiled from samples recovered in this study from teeth, in previous studies using blood (Kraberger et al., 2020; Fountain-Jones et al., 2021), and an additional representative subset of those available in GenBank. These datasets were aligned using MUSCLE (Edgar, 2004), and recombination analyses were performed using RDP (v5.5) (Martin et al., 2015). Automask was implemented for similar sequences and events were considered credible if they were detected by three or more methods with p-values < 0.05, coupled with phylogenetic support. Recombination-free datasets were used to construct maximum likelihood phylogenies for each of the datasets with RaxML using RDP (Martin et al., 2015).
Samples were grouped into four identifier geographic clusters based on the major potential barriers to movement. The continental divide represented north to south barrier, and Interstate 70 highway represented east to west barrier. Sections were designated northwest, northeast, southwest and southeast.
A total of 200 samples were screened for two endemic feline retroviruses previously described in Colorado pumas, FIVpco and FFV (Kraberger et al., 2020; Fountain-Jones et al., 2021). Given the limited amount of DNA available for analysis, we undertook an initial screen using a highly conserved region in the FIVpco vif gene. Forty-six of the 200 samples (23%) were PCR positive and were confirmed by sanger sequencing (Table 1). Based on phylogenetically determined genetic groupings of the vif gene from each of the positive samples, we screened for the pol and env genes. We successfully cloned and sequence the pol gene regions from 7/46 (15%) samples and the env gene regions from 17/46 (37%) samples.
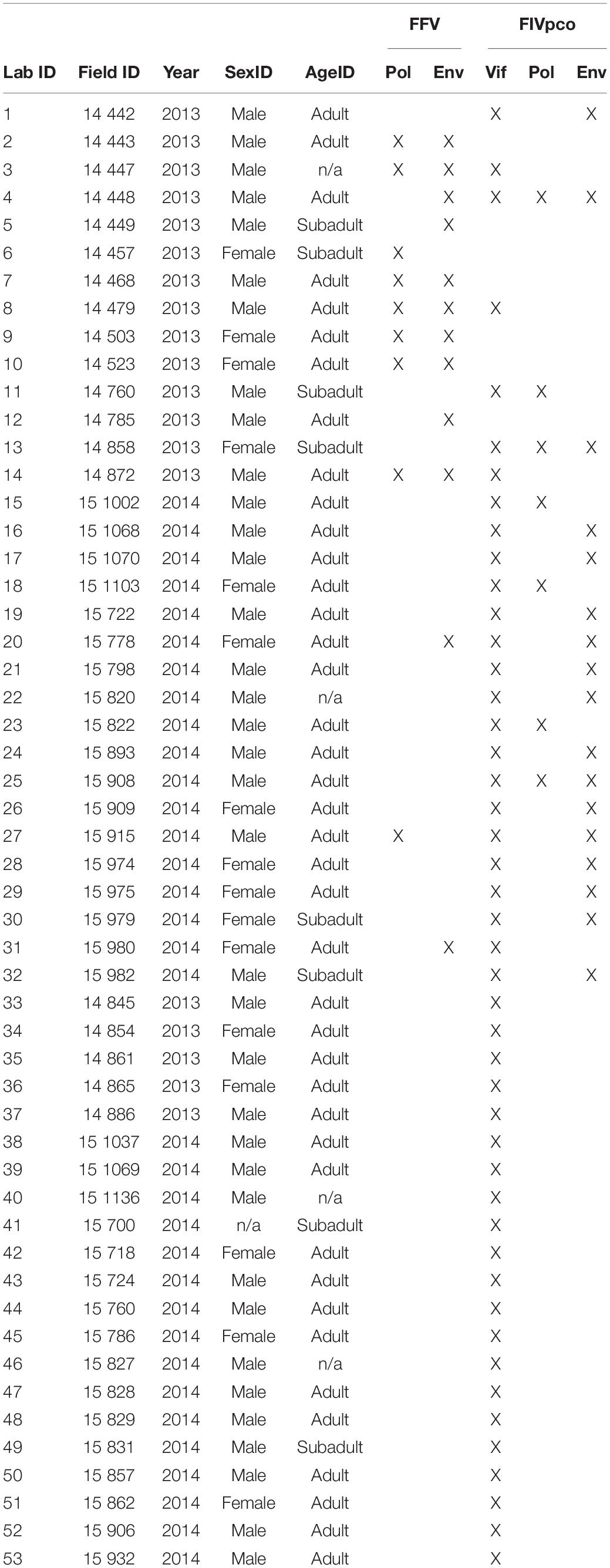
Table 1. Details of tooth derived FIVpco and FFV sequences from Colorado.
The FFV genome is highly conserved and considering the low DNA volume available for recovery of viruses, we screened for the presence of FFV using pol and env PCR. This resulted in 14 out of 200 samples (7%) detected as positive for FFV. Pol sequences were recovered from 9/14 (64%) and env gene sequences were recovered from 12/14 (85%). Both gene sequences were isolated from 7/14 (50%) (Table 1). For seven of these samples, we were able to isolate both genes. Of these seven samples, we were able to detect and sequence at least one gene region from FIVpco and FFV each virus showing they were coinfected.
We detected FFV in seven percent of the tooth samples, in comparison previous evaluation of other pumas from Colorado detected FFV in 61% (qPCR, detection of viral genome) or 77% (ELISA, detection of antibodies) (Kechejian et al., 2019; Dannemiller et al., 2020). We detected FIVpco in 23% of the tooth samples, which was closer to the 48% detected via qPCR from other Colorado puma blood samples (Fountain-Jones et al., 2021). This indicates that while this technique was invaluable for evaluation of viral phylogeny across the landscape, it may underestimate disease prevalence, however, screening with a highly sensitive qPCR approach, which targets small genetic regions, may help to mitigate this. Our goal in this study was to identify viral genomes, which required us to utilize PCR to achieve longer amplicons than qPCR, so these analyses are not head-to-head comparisons. The tooth samples likely had viral degradation, given the nature of the sample. Some degradation of these samples is supported by our ability to detect a larger number of FIVpco positives from the vif screen targeting a shorter portion of the sequence (∼400 bp) than from longer regions from the env (∼2,500 bp) or pol (∼3,500 bp) regions.
Genetic signatures of viruses help us to understand transmission dynamics, allow inferences about host connectivity on the landscape and assist in identifying variants. Previous analyses of blood-derived FIVpco genetic sequences from pumas captured in the Uncompahgre Plateau and Front Range region of Colorado are phylogenetic structured by location (Lee et al., 2014; Fountain-Jones et al., 2021). Conversely, FFV, due to its lower mutation rate, higher prevalence in pumas, and ability to infect domestic cats and pumas, does not show differ between these two sites (Kraberger et al., 2020). Previous analyses offered a limited view of Colorado puma viral phylogeny since it evaluated puma “populations” in geographically distinct (∼300 km apart) regions separated by the continental divide and a four-lane interstate highway (I-70). Viral tooth samples from hunter-collected pumas provide an opportunity to examine a broadly representative viral population distributed more evenly across the state across a larger geographic range. Additionally, assessment of tooth samples afforded the ability to analyze samples collected at a similar time. Recombination-free phylogenies of all FIVpco (puma env and pol sequences from Colorado and surrounding states) and FFV (env and pol sequences from pumas captured in Colorado and domestic cats from Colorado, as well as the broader US and globally) were generated and mapped to show sampling location of tooth and blood derived samples (Figures 1, 2). Four identifier geographic clusters were designated north west, north east, south west and south east based upon impermeable landscape features (Figures 1, 2). Neither the continental divide nor Interstate 70 influenced viral phylogenies. FFV phylogenies showed little geographic structuring for either pol and env genes. FFV clustering occurred across several clades with puma and domestic cat isolates. The FIVpco phylogeny, however, does still show geographic structuring with clades composed of sequences predominantly from one region (Figure 1). FIVpco sequences from pumas captured in Nevada and New Mexico clustered most closely with those from southern Colorado and Wyoming, forming distinct clades.
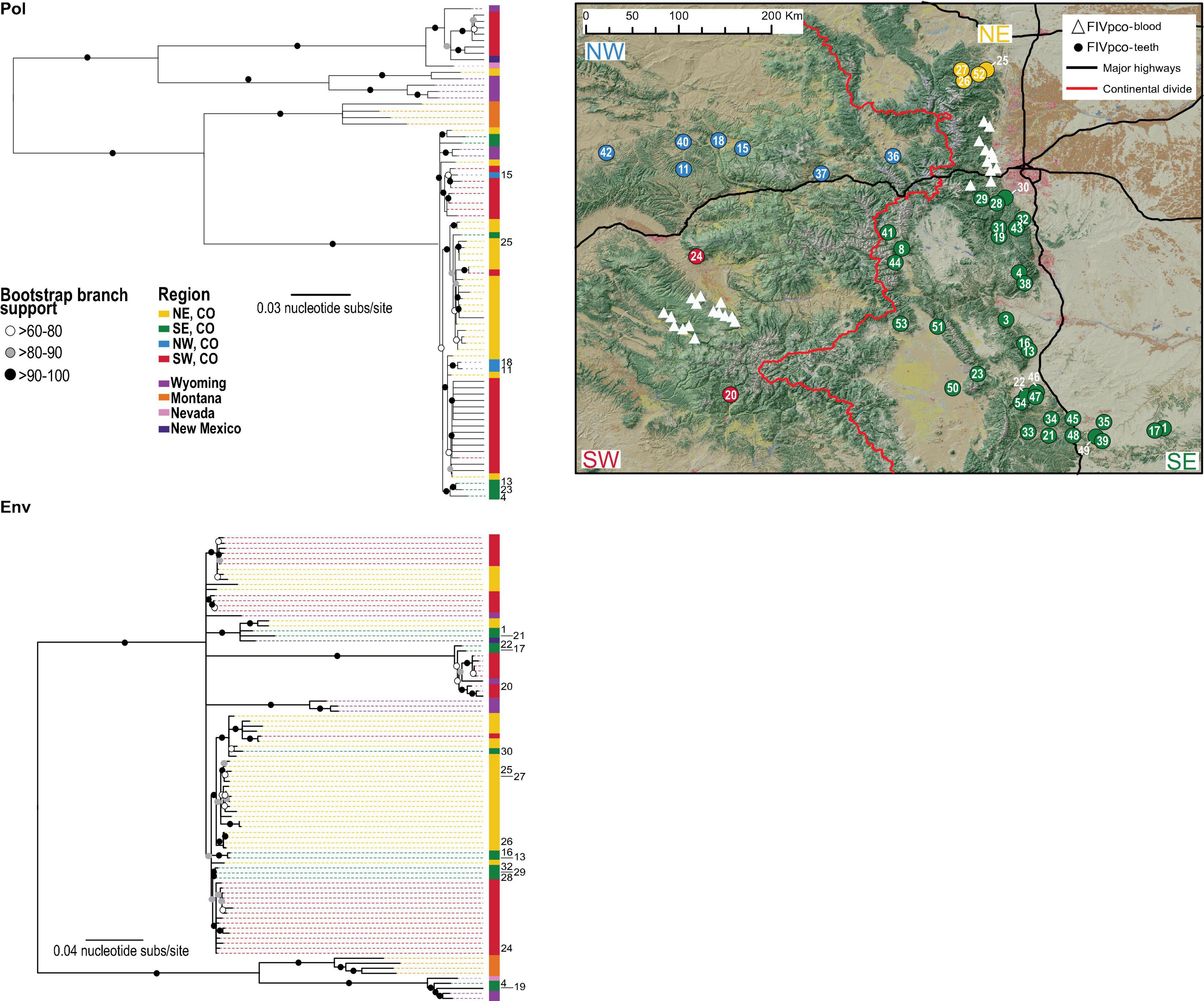
Figure 1. FIVpco from statewide tooth samples support host connectivity across the state. Recombination-free maximum likelihood phylogenies of the pol and env gene regions from FIVpco recovered from statewide puma tooth samples (denoted by the host ID number) and blood samples from Colorado and neighboring states. Key and color bar next to the phylogeny shows the region or state from which the sequences were collected. Map of Colorado indicates location of samples for which genetic sequence(s) of FIVpco have been recovered in this or previous studies (Lee et al., 2014; Fountain-Jones et al., 2021). Blood sample analysis is illustrated by white triangles and viral sequences extracted from teeth are designated by circles. Regions designated correspond to state by the Interstate 70 and continental divide.
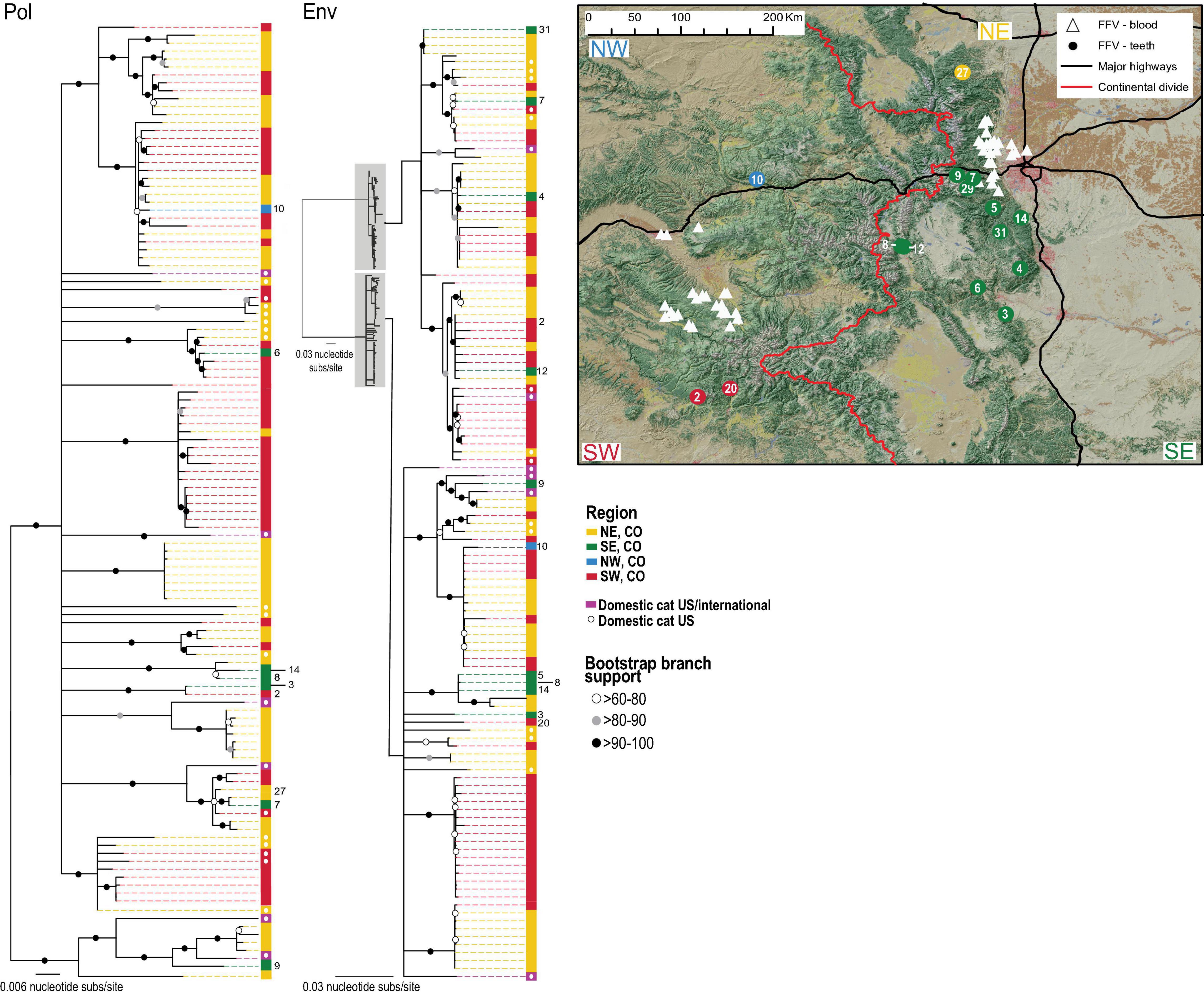
Figure 2. Phylogeny supports FFV is endemic in the Colorado puma population. Recombination-free maximum likelihood phylogenies of FFV pol and env gene regions isolated from statewide puma tooth samples (denoted by the host ID number), and blood samples from Colorado puma and domestic cats. FFV from Puma and cats from US states and domestic cats globally were additionally sourced. Key and color bar next to the phylogeny shows the region or state, and host, from which the sequences were collected. Map of Colorado highlights location of samples for which genetic sequence(s) of FFV have been recovered in this or previous studies (Kraberger et al., 2020). Blood sample analysis is illustrated by white triangles and viral sequences extracted from teeth are designated by circles. Regions designated correspond to state by the Interstate 70 and continental divide.
Mitigation and control of wildlife disease is an increasingly important component of wildlife management. Hunter-harvested samples can be an important component of surveillance programs, and we demonstrate that hunter-collected teeth can not only determine infection status but permit the recovery of viral genetic information. Our results indicate that DNA extracted from teeth of wildlife is suitable for amplification of pathogen genomic sequences. This approach can be used to augment other sampling efforts to better inform the geographic structuring of viruses, and potentially other pathogens, at large spatial scales. The recovery of viral sequencing data from hunter-collected puma teeth demonstrate the utility of these samples to inform pathogen spread in harvested animals.
Few studies have evaluated presence of viruses from tooth samples, though both FIV and FFV infections of domestic cats are thought to be transmitted by bite wounds and thus can be detected in saliva (Miller et al., 2017; Cavalcante et al., 2018). Because viral infection may not be readily detected in the oral cavity, this methodology may not be suitable for evaluation of all pathogens (Bedarida et al., 2011; Siravenha et al., 2016). The ability to use tooth collected samples for diagnostics could be enhanced by targeting smaller regions of amplification. A comparison of tooth DNA and paired blood or lymphoid samples is required to truly compare sensitivity and specificity of both methods.
Feline immunodeficiency virus and feline foamy virus sequences isolated from tooth samples supported limited geographic structuring of retroviral infections of Colorado pumas across the state. FIVpco had more indication of geographic structuring than FFV, likely a result of the frequent cross-species transmission of FFV from domestic cats into pumas and low mutation rate (Kraberger et al., 2020). Phylogenies of FIVpco revealed individuals from the same geographic region tended to cluster together, consistent with previous results showing that in certain regions, spatial proximity was associated with spread of FIVpco (Fountain-Jones et al., 2021). The absence of clear geographic clusters indicates signals of phylogeographic clustering may dissipate with finer scale sampling efforts. These findings illustrate that Interstate 70, which bisects the state into northern and southern regions, and the continental divide, which bisects the state into eastern and western regions, were not impermeable to viral transmission. This is consistent with previous reports that high elevation ridgelines did not limit viral spread (Fountain-Jones et al., 2021) but in contrast to reduced geneflow noted across the continental divide, attributed to the fact that pumas avoid high ridgelines and alpine environments (Sweanor et al., 2000; Gustafson et al., 2019; Trumbo et al., 2019). Additionally, Interstate 70 did not have an apparent effect on viral structuring.
These results support the potential for genomes from rapidly evolving viral genomics to inform host connectivity more so than the genetics of the animals itself (Biek et al., 2006; Gagne et al., 2021). The results mirror studies evaluating interstate barriers to bobcat gene flow compared to FIVlru phylogeny, that illustrated Southern Californian interstates are more permeable to viral transmission than host gene flow (Lee et al., 2012; Ernest et al., 2014; Kozakiewicz et al., 2019, 2020). Because host genetic identities reflect dispersal related to reproductive events, it is less temporally sensitive than viral phylogeography, given the high evolutionary rates of viruses (Richardson et al., 2016; Gagne et al., 2021).
The addition of viral sequences from tooth samples demonstrated that potential geographic barriers (i.e., continental divide, major highways) did not appear to limit the spread of viruses in Colorado pumas. Our findings have important implications for puma population health should more virulent viruses, such as FeLV, arise in Colorado puma populations (Chiu et al., 2019). Further, this work illustrates a novel method for informing apex predator movement and may inform management of predator and prey populations. While additional validation is needed to determine the potential of tooth samples for diagnostic work that demonstrates pathogen prevalence, this work illustrates the benefits of incidentally-collected tooth samples as a tool for wildlife disease surveillance.
All sequence data has been uploaded to Genbank, NCBI (accession: MZ398368-MZ398467).
We handled all pumas in accordance with approved CPW Animal Care and Use Committee (ACUC) capture and handling protocols (ACUC file #08-2004, ACUC protocol #03-2007 ACUC 16-2008). Samples were provided to Colorado State University for diagnostic evaluation. Colorado State University and Colorado Parks and Wildlife (CPW) Institutional Animal Care and Use Committees reviewed and approved this work prior to initiation (CSU IACUC protocol 05-061A).
SK, SV, MA, and CA conceived the project. MA, KL, and CA conducted the field sampling and collections. SK, RG, RM, and KG performed the laboratory work. SK, RG, and DT analyzed the data. SK, RG, and SV wrote the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by NSF-EEID award 1413925.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank wildlife managers, veterinarians, and support staff at Colorado Parks and Wildlife Service. We also thank the members of the SV Research Group for valuable feedback on this work.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2021.734462/full#supplementary-material
Supplementary Table 1 | Details of blood derived FIVpco and FFV sequences from Colorado, New Mexico and Nevada. Those highlighted in red were recovered as part of this study.
Supplementary Table 2 | Primers utilized to recover FIVpco and FFV sequences.
Artois, M., Bengis, R., Delahay, R. J., Duchêne, M.-J., Duff, J. P., Ferroglio, E., et al. (2009). Wildlife disease surveillance and monitoring. Manag. Dis. Wild Mamm. 2009, 187–213.
Baele, G., Dellicour, S., Suchard, M. A., Lemey, P., and Vrancken, B. (2018). Recent advances in computational phylodynamics. Curr. Opin. Virol. 31, 24–32. doi: 10.1016/j.coviro.2018.08.009
Baele, G., Suchard, M. A., Rambaut, A., and Lemey, P. (2017). Emerging concepts of data integration in pathogen phylodynamics. Syst. Biol. 66, e47–e65.
Barbieri, R., Mekni, R., Levasseur, A., Chabrière, E., Signoli, M., Tzortzis, S., et al. (2017). Paleoproteomics of the Dental Pulp: The plague paradigm. PLoS One 12:e0180552. doi: 10.1371/journal.pone.0180552
Barron, E. S., Swift, B., Chantrey, J., Christley, R., Gardner, R., Jewell, C., et al. (2018). A study of tuberculosis in road traffic-killed badgers on the edge of the British bovine TB epidemic area. Sci. Rep. 8, 1–8.
Bedarida, S., Dutour, O., Buzhilova, A. P., de Micco, P., and Biagini, P. (2011). Identification of viral DNA (Anelloviridae) in a 200-year-old dental pulp sample (Napoleon’s Great Army. Kaliningrad, 1812). Infect. Genet. Evol. 11, 358–362. doi: 10.1016/j.meegid.2010.11.007
Belay, E. D., Kile, J. C., Hall, A. J., Barton-Behravesh, C., Parsons, M. B., Salyer, S., et al. (2017). Zoonotic disease programs for enhancing global health security. Emerg. Infect. Dis. 23:S65.
Belsare, A. V., Gompper, M. E., Keller, B., Sumners, J., Hansen, L., and Millspaugh, J. J. (2020). An agent-based framework for improving wildlife disease surveillance: A case study of chronic wasting disease in Missouri white-tailed deer. Ecol. Model. 417:108919. doi: 10.1016/j.ecolmodel.2019.108919
Bengis, R., Kock, R., and Fischer, J. (2002). Infectious animal diseases: the wildlife/livestock interface. Rev. Sci. Tech. Int. Off. Epizoot. 21, 53–65. doi: 10.20506/rst.21.1.1322
Biek, R., Drummond, A. J., and Poss, M. (2006). A virus reveals population structure and recent demographic history of its carnivore host. Science 311, 538–541. doi: 10.1126/science.1121360
Bishop, R. C. (2004). The economic impacts of chronic wasting disease (CWD) in Wisconsin. Hum. Dimens. Wildl. 9, 181–192. doi: 10.1080/10871200490479963
Brook, R. K., Kutz, S. J., Veitch, A. M., Popko, R. A., Elkin, B. T., and Guthrie, G. (2009). Fostering community-based wildlife health monitoring and research in the Canadian North. EcoHealth 6, 266–278. doi: 10.1007/s10393-009-0256-7
Brown, M. A., Cunningham, M. W., Roca, A. L., Troyer, J. L., Johnson, W. E., and O’Brien, S. J. (2008). Genetic characterization of feline leukemia virus from Florida panthers. Emerg. Infect. Dis. 14:252. doi: 10.3201/eid1402.070981
Carver, S., Bevins, S. N., Lappin, M. R., Boydston, E. E., Lyren, L. M., Alldredge, M., et al. (2016). Pathogen exposure varies widely among sympatric populations of wild and domestic felids across the United States. Ecol. Appl. 26, 367–381. doi: 10.1890/15-0445
Cavalcante, L. T., Muniz, C. P., Jia, H., Augusto, A. M., Troccoli, F., Medeiros, S., et al. (2018). Clinical and molecular features of feline foamy virus and feline leukemia virus co-infection in naturally-infected cats. Viruses 10:702. doi: 10.3390/v10120702
Chiu, E. S., Kraberger, S., Cunningham, M., Cusack, L., Roelke, M., and Vandewoude, S. (2019). Multiple Introductions of Domestic Cat Feline Leukemia Virus in Endangered Florida Panthers. Emerg. Infect. Dis. 25:92. doi: 10.3201/eid2501.181347
Cohen, J. E. (2003). Human population: the next half century. Science 302, 1172–1175. doi: 10.1126/science.1088665
Dannemiller, N. G., Kechejian, S., Kraberger, S., Logan, K., Alldredge, M., Crooks, K. R., et al. (2020). Diagnostic Uncertainty and the epidemiology of feline foamy Virus in pumas (Puma concolor). Sci. Rep. 10, 1–7.
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Ernest, H. B., Vickers, T. W., Morrison, S. A., Buchalski, M. R., and Boyce, W. M. (2014). Fractured genetic connectivity threatens a southern California puma (Puma concolor) population. PLoS One 9:e107985. doi: 10.1371/journal.pone.0107985
Fitak, R. R., Antonides, J. D., Baitchman, E. J., Bonaccorso, E., Braun, J., Kubiski, S., et al. (2019). The expectations and challenges of wildlife disease research in the era of genomics: forecasting with a horizon scan-like exercise. J. Hered. 110, 261–274. doi: 10.1093/jhered/esz001
Fountain-Jones, N. M., Kraberger, S., Gagne, R. B., Trumbo, D. R., Salerno, P. E., Chris Funk, W., et al. (2021). Host relatedness and landscape connectivity shape pathogen spread in the puma, a large secretive carnivore. Commun. Biol. 4:12. doi: 10.1038/s42003-020-01548-2
Franklin, S. P., Troyer, J. L., Terwee, J. A., Lyren, L. M., Boyce, W. M., Riley, S., et al. (2007a). Frequent transmission of immunodeficiency viruses among bobcats and pumas. J. Virol. 81, 10961–10969. doi: 10.1128/jvi.00997-07
Franklin, S. P., Troyer, J. L., TerWee, J. A., Lyren, L. M., Kays, R. W., Riley, S. P., et al. (2007b). Variability in assays used for detection of lentiviral infection in bobcats (Lynx rufus), pumas (Puma concolor), and ocelots (Leopardus pardalis). J. Wildl. Dis. 43, 700–710. doi: 10.7589/0090-3558-43.4.700
Gagne, R. B., Crooks, K., Craft, M. E., Chiu, E. S., Fountain-Jones, N. M., Malmberg, J. L., et al. (2021). Parasites as conservation tools. Conserv. Biol. 2021:13719.
Gomo, C., de Garine-Wichatitsky, M., Caron, A., and Pfukenyi, D. M. (2012). Survey of brucellosis at the wildlife–livestock interface on the Zimbabwean side of the Great Limpopo Transfrontier Conservation Area. Trop. Anim. Health Prod. 44, 77–85. doi: 10.1007/s11250-011-9890-5
Gustafson, K. D., Gagne, R. B., Vickers, T. W., Riley, S. P. D., Wilmers, C. C., Bleich, V. C., et al. (2019). Genetic source-sink dynamics among naturally structured and anthropogenically fragmented puma populations. Conserv. Genet. 20, 215–227. doi: 10.1007/s10592-018-1125-0
Hawkins, C. E., Baars, C., Hesterman, H., Hocking, G. J., Jones, M. E., Lazenby, B., et al. (2006). Emerging disease and population decline of an island endemic, the Tasmanian devil Sarcophilus harrisii. Biol. Conserv. 131, 307–324. doi: 10.1016/j.biocon.2006.04.010
Hawley, J. E., Rego, P. W., Wydeven, A. P., Schwartz, M. K., Viner, T. C., Kays, R., et al. (2016). Long-distance dispersal of a subadult male cougar from South Dakota to Connecticut documented with DNA evidence. J. Mammal. 97, 1435–1440. doi: 10.1093/jmammal/gyw088
Heard, M. J., Smith, K. F., Ripp, K., Berger, M., Chen, J., Dittmeier, J., et al. (2013). Increased threat of disease as species move towards extinction. Conserv. Biol. J. Soc. Conserv. Biol. 27:1378. doi: 10.1111/cobi.12143
Holmes, E. C., and Grenfell, B. T. (2009). Discovering the phylodynamics of RNA viruses. PLoS Comput. Biol. 5:e1000505. doi: 10.1371/journal.pcbi.1000505
Kamath, P. L., Foster, J. T., Drees, K. P., Luikart, G., Quance, C., Anderson, N. J., et al. (2016). Genomics reveals historic and contemporary transmission dynamics of a bacterial disease among wildlife and livestock. Nat. Commun. 7, 1–10. doi: 10.1038/ncomms11448
Kechejian, S. R., Dannemiller, N., Kraberger, S., Ledesma-Feliciano, C., Malmberg, J., Roelke Parker, M., et al. (2019). Feline Foamy Virus is Highly Prevalent in Free-Ranging Puma concolor from Colorado. Florida and Southern California. Viruses 11:359. doi: 10.3390/v11040359
Kock, R. A., Orynbayev, M., Robinson, S., Zuther, S., Singh, N. J., Beauvais, W., et al. (2018). Saigas on the brink: Multidisciplinary analysis of the factors influencing mass mortality events. Sci. Adv. 4:eaao2314. doi: 10.1126/sciadv.aao2314
Kozakiewicz, C. P., Burridge, C. P., Funk, W. C., Craft, M. E., Crooks, K. R., Fisher, R. N., et al. (2020). Does the virus cross the road? Viral phylogeographic patterns among bobcat populations reflect a history of urban development. Evol. Appl. 2020:12927.
Kozakiewicz, C. P., Burridge, C. P., Funk, W. C., Salerno, P. E., Trumbo, D. R., Gagne, R. B., et al. (2019). Urbanization reduces genetic connectivity in bobcats (Lynx rufus) at both intra−and inter−population spatial scales. Mol. Ecol. 28:15274.
Kozakiewicz, C. P., Burridge, C. P., Funk, W. C., VandeWoude, S., Craft, M. E., Crooks, K. R., et al. (2018). Pathogens in space: advancing understanding of pathogen dynamics and disease ecology through landscape genetics. Evol. Appl. 11, 1763–1778. doi: 10.1111/eva.12678
Kraberger, S., Fountain-Jones, N. M., Gagne, R. B., Malmberg, J., Dannemiller, N. G., Logan, K., et al. (2020). Frequent cross-species transmissions of foamy virus between domestic and wild felids. Virus Evol. 6:vez058.
Lawson, B., Petrovan, S. O., and Cunningham, A. A. (2015). Citizen science and wildlife disease surveillance. EcoHealth 12, 693–702. doi: 10.1007/s10393-015-1054-z
Lee, J. S., Bevins, S. N., Serieys, L. E., Vickers, W., Logan, K. A., Aldredge, M., et al. (2014). Evolution of puma lentivirus in bobcats (Lynx rufus) and mountain lions (Puma concolor) in North America. J. Virol. 88, 7727–7737. doi: 10.1128/jvi.00473-14
Lee, J. S., Ruell, E. W., Boydston, E. E., Lyren, L. M., Alonso, R. S., Troyer, J. L., et al. (2012). Gene flow and pathogen transmission among bobcats (Lynx rufus) in a fragmented urban landscape. Mol. Ecol. 21, 1617–1631. doi: 10.1111/j.1365-294x.2012.05493.x
Lee, J., Malmberg, J. L., Wood, B. A., Hladky, S., Troyer, R., Roelke, M., et al. (2017). Feline immunodeficiency virus cross-species transmission: implications for emergence of new lentiviral infections. J. Virol. 91, e2116–e2134.
Martin, D. P., Murrell, B., Golden, M., Khoosal, A., and Muhire, B. (2015). RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015:1.
Miller, C., Boegler, K., Carver, S., MacMillan, M., Bielefeldt-Ohmann, H., and VandeWoude, S. (2017). Pathogenesis of oral FIV infection. PLoS One 12:e0185138. doi: 10.1371/journal.pone.0185138
Richardson, J. L., Brady, S. P., Wang, I. J., and Spear, S. F. (2016). Navigating the pitfalls and promise of landscape genetics. Mol. Ecol. 25, 849–863. doi: 10.1111/mec.13527
Ryser-Degiorgis, M.-P. (2013). Wildlife health investigations: needs, challenges and recommendations. BMC Vet. Res. 9, 1–17. doi: 10.1186/1746-6148-9-223
Santos, S. M., Carvalho, F., and Mira, A. (2011). How long do the dead survive on the road? Carcass persistence probability and implications for road-kill monitoring surveys. PLoS One 6:e25383. doi: 10.1371/journal.pone.0025383
Siravenha, L. G., Siravenha, L. Q., Madeira, L. D. P., Oliveira-Filho, A. B., Machado, L. F. A., Martins Feitosa, R. N., et al. (2016). Detection of HCV persistent infections in the dental pulp: a novel approach for the detection of past and ancient infections. PLoS One 11:e0165272. doi: 10.1371/journal.pone.0165272
Skerratt, L. F., Berger, L., Speare, R., Cashins, S., McDonald, K. R., Phillott, A. D., et al. (2007). Spread of chytridiomycosis has caused the rapid global decline and extinction of frogs. EcoHealth 4, 125–134.
Stallknecht, D. (2007). Impediments to wildlife disease surveillance, research, and diagnostics. Wildl. Emerg. Zoonotic Dis. Biol. Circumst. 2007, 445–461. doi: 10.1007/978-3-540-70962-6_17
Sweanor, L. L., Logan, K. A., and Hornocker, M. G. (2000). Cougar dispersal patterns, metapopulation dynamics, and conservation. Conserv. Biol. 14, 798–808. doi: 10.1046/j.1523-1739.2000.99079.x
Taylor, L. H., Latham, S. M., and Woolhouse, M. E. (2001). Risk factors for human disease emergence. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 356, 983–989.
Trumbo, D. R., Salerno, P. E., Logan, K. A., Alldredge, M. W., Gagne, R. B., Kozakiewicz, C. P., et al. (2019). Urbanization impacts apex predator gene flow but not genetic diversity across an urban-rural divide. Mol. Ecol. 28, 4926–4940. doi: 10.1111/mec.15261
Wake, D. B., and Vredenburg, V. T. (2008). Are we in the midst of the sixth mass extinction? A view from the world of amphibians. Proc. Natl. Acad. Sci. U.S.A. 105, 11466–11473. doi: 10.1073/pnas.0801921105
Wandeler, P., Smith, S., Morin, P. A., Pettifor, R., and Funk, S. (2003). Patterns of nuclear DNA degeneration over time—a case study in historic teeth samples. Mol. Ecol. 12, 1087–1093. doi: 10.1046/j.1365-294x.2003.01807.x
Watsa, M. (2020). Rigorous wildlife disease surveillance. Science 369, 145–147. doi: 10.1126/science.abc0017
Wobeser, G., and Wobeser, A. G. (1992). Carcass disappearance and estimation of mortality in a simulated die-off of small birds. J. Wildl. Dis. 28, 548–554. doi: 10.7589/0090-3558-28.4.548
Keywords: feline foamy virus, feline immunodeficiency virus, mountain lion, cougar, wildlife disease
Citation: Gagne RB, Kraberger S, McMinn R, Trumbo DR, Anderson CR Jr, Logan KA, Alldredge MW, Griffin K and Vandewoude S (2021) Viral Sequences Recovered From Puma Tooth DNA Reconstruct Statewide Viral Phylogenies. Front. Ecol. Evol. 9:734462. doi: 10.3389/fevo.2021.734462
Received: 01 July 2021; Accepted: 02 August 2021;
Published: 30 August 2021.
Edited by:
Jonathan J. Fong, Lingnan University, ChinaReviewed by:
Colin Parrish, Cornell University, United StatesCopyright © 2021 Gagne, Kraberger, McMinn, Trumbo, Anderson, Logan, Alldredge, Griffin and Vandewoude. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sue Vandewoude, U3VlLlZhbmRld291ZGVAQ29sb1N0YXRlLkVEVQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.