 Oriane Loiseau1,2*
Oriane Loiseau1,2* Talita Mota Machado3
Talita Mota Machado3 Margot Paris4
Margot Paris4 Darina Koubínová1
Darina Koubínová1 Kyle G. Dexter2Leonardo M. Versieux5
Kyle G. Dexter2Leonardo M. Versieux5 Christian Lexer6†
Christian Lexer6† Nicolas Salamin1
Nicolas Salamin1- 1Department of Computational Biology, University of Lausanne, Lausanne, Switzerland
- 2School of Geosciences, University of Edinburgh, Edinburgh, United Kingdom
- 3Programa de Pós Graduação em Biologia Vegetal, Universidade Federal de Minas Gerais, Belo Horizonte, Brazil
- 4Department of Biology, Unit Ecology and Evolution, University of Fribourg, Fribourg, Switzerland
- 5Departamento de Botânica e Zoologia, Centro de Biociências, Universidade Federal do Rio Grande do Norte, Natal, Brazil
- 6Department of Botany and Biodiversity Research, University of Vienna, Vienna, Austria
The tropics hold at least an order of magnitude greater plant diversity than the temperate zone, yet the reasons for this difference are still subject to debate. Much of tropical plant diversity is in highly speciose genera and understanding the drivers of such high species richness will help solve the tropical diversity enigma. Hybridization has recently been shown to underlie many adaptive radiations, but its role in the evolution of speciose tropical plant genera has received little attention. Here, we address this topic in the hyperdiverse Bromeliaceae genus Vriesea using genome skimming data covering the three genomic compartments. We find evidence for hybridization in ca. 11% of the species in our dataset, both within the genus and between Vriesea and other genera, which is commensurate with hybridization underlying the hyperdiversity of Vriesea, and potentially other genera in Tillandsioideae. While additional genomic research will be needed to further clarify the contribution of hybridization to the rapid diversification of Vriesea, our study provides an important first data point suggesting its importance to the evolution of tropical plant diversity.
Introduction
Hybridization, the exchange of genetic material between lineages, is a widespread evolutionary phenomenon of crucial importance in the evolution of diversity (Marques et al., 2019). Gene flow between species may have deleterious effects on species cohesion by breaking species boundaries and causing genetic homogenization. There is however increasing evidence that hybridization can promote genetic diversity, adaptation and speciation, although the exchange of genetic material per se might not be sufficient to create new diversity without a key role of natural selection acting on the relevant parts of the genome to maintain them (Schumer et al., 2018). In particular, hybridization has been shown to be involved in many adaptive radiations, such as Darwin’s finches (Grant and Grant, 2019), African rift lakes cichlid fishes (Meier et al., 2017; Irisarri et al., 2018; Svardal et al., 2020), Heliconius butterflies (Kozak et al., 2021), and Hawaiian silver swords (Barrier et al., 1999). In comparison with the low rate of mutation, hybridization can immediately elevate the standing genetic variation upon which selection can act (Seehausen, 2013). This has the potential to accelerate the adaptive divergence of populations in face of environmental variability, novel habitats or previously underutilized niches. Elevated rates of speciation following hybridization can create a positive feedback as the evolution of many closely related species in one area may in turn provide new opportunities for hybridization (Seehausen, 2004).
Interspecific gene flow can promote diversification through the direct generation of a new species via hybrid speciation or through adaptive introgression, the latter being the transfer of only parts of the genome which may confer a selective advantage (Harrison and Larson, 2014). The identification of these mechanisms has been helped by two important advances in the field. First, the emergence of the genome scale datasets that are becoming standard in evolutionary studies provided the raw data to tackle these questions. Second, the development of statistical methods that can formally test the presence of introgression against a null model that involves only incomplete lineage sorting (ILS; e.g., Joly et al., 2009; Green et al., 2010; Blanco-Pastor et al., 2012; Blischak et al., 2018). A remaining open question is the prevalence of hybridization or adaptive introgression in the context of diversifying lineages (Pfennig, 2021).
Although hybridization has been detected across a wide range of organisms, it is particularly frequent in plants, and is acknowledged as one of the main drivers of the evolution of angiosperms (Soltis and Soltis, 2009). However, little is known about the contribution of hybridization in the evolution of diverse tropical clades which make up a large part of plant diversity. In this study, we focus on the Bromeliaceae, which is, with ca. 3,500 species, one of the most diverse and iconic plant families of the Neotropical flora, particularly abundant in Andean and Atlantic rainforests (Smith and Downs, 1974; Benzing, 2000; Gouda et al., 2018). The family harbors several exceptionally diverse young clades, such as the core tillandsioids and the tank-forming epiphytic bromelioids (Givnish et al., 2014). The increased rates of diversification which underlie these rapid radiations may have been spurred by several key innovations such as the tank habit, CAM photosynthesis or hummingbird pollination (Givnish et al., 2014; Silvestro et al., 2014). Hybridization is also thought to have promoted bromeliad diversity (Palma-Silva et al., 2016) but although both artificial (Vervaeke et al., 2004; Wagner et al., 2015; de Souza et al., 2017) and natural hybrids have been reported (e.g., Palma-Silva et al., 2011), the frequency and exact contribution of this phenomenon to bromeliad diversity are still to be determined. While some studies suggest that hybridization generates intraspecific genetic diversity in isolated populations (Lexer et al., 2016; Neri et al., 2018; Mota et al., 2019), it may also prevent genetic differentiation and blur species boundaries (Goetze et al., 2017). To date, most of the research on hybridization in bromeliads focuses on the population level and studies with a macroevolutionary perspective are lacking.
Here, we take a phylogenomic approach to the study of hybridization in the Tillandsioideae, the largest of the three subfamilies of bromeliads, with a focus on the diverse genus Vriesea (231 species, Gouda et al., 2018). Using genome skimming we provide an unprecedented molecular dataset for 108 species of Vriesea and 39 species from related genera. We use phylogenetic inference and coalescent-based approaches to test for the presence of hybridization during the evolutionary history of the group. Our results provide evidence for clear cyto-nuclear discordance in the phylogenetic history of Tillandisoideae. We further detect signatures of ancient hybridization at both the inter- and intrageneric level. Our study suggests that hybridization has occurred repeatedly throughout the evolutionary history of bromeliads and may have played an important role in triggering the rapid evolution of this group.
Materials and Methods
Genomic Dataset
We used an extended version of the chloroplast genomic dataset generated with genome skimming by Machado et al. (2020) containing nuclear, chloroplast and mitochondrial data. Genome skimming, also referred as low-coverage whole genome sequencing, allows easy and cost-effective obtention of DNA sequences for phylogenomic analyses across a wide range of evolutionary divergences (Dodsworth, 2015). This technique is particularly effective for the high-depth sequencing of the high-copy fraction of the genome such as plastids and ribosomal nuclear DNA (Coissac et al., 2016). It also offers the possibility to obtain shallow coverage of single copy nuclear DNA, and thousands of low-depth nuclear markers can be obtained with a sufficient sequencing effort. Sampling included 147 species of 11 genera in the subfamily Tillandsioideae, plus Ananas comosus as an outgroup. In total, there were 108 species of Vriesea s.l., representing c. 46% of the total diversity of the genus. For the other genera the number and proportion of species were as follows (species numbers according to Gouda et al., 2018): Alcantarea (11 species; 26% of the diversity), Goudaea (2; 100%), Guzmania (4; 1%), Lutheria (2; 50%), Mezobromelia (1; 20%), Racinaea (1; 78%), Stigmatodon (4; 22%), Tillandsia (9; 1%), Werauhia (3; 1%), Zizkaea (1; 100%). Protocols of DNA extraction, library preparation and sequencing, as well as the bioinformatic pipeline for quality checking of reads, and SNP calling are described in Machado et al. (2020). Briefly, library preparation was performed using KAPA LTP library preparation kits (Roche, Basel, Switzerland) and unique barcodes for each library were chosen from a set of 60 dual-index primers designed in Loiseau et al. (2019) to allow multiplexing. The use of a high number of Illumina dual-indexed primers, designed with a minimum edit distance of 4 between each of the 7-bp indexes, reduces the probability of conversion by sequencing, amplification errors and index hopping (Kircher et al., 2012; Costello et al., 2018). After sequencing in an Illumina HiSeq 3000 Genome Analyzer (Illumina, San Diego, California, United States), quality controlling and read trimming, pair-end reads (2 × 150 bp) were mapped onto the Tillandsia adpressiflora Mez pseudo-reference genome built in de la Harpe et al. (2020) using BOWTIE2 v.2.2.5 (Langmead and Salzberg, 2012). After identification of PCR duplicates, realignment of reads around indels and base calibration, SNPs were called using UnifiedGenotyper of GATK v.3.6 (McKenna et al., 2010) and the EMIT_ALL_SITES option to recover both variant and invariant sites. We used Vcftools v.0.1.13 (Danecek et al., 2011) to filter only high-quality sites called with Q values higher than 20 and less than 50% missing data. For phylogenomic inferences invariant sites were filtered out and separate alignments of SNPs were produced for the nuclear, chloroplast and mitochondrial genomes.
Phylogenetic Inferences
We performed phylogenetic reconstruction using both maximum-likelihood and coalescent-based methods. Since using only variable sites for phylogenetic inference can lead to incorrect estimation of branch lengths and topology (Leaché et al., 2015), we employed methods designed for SNP datasets. First, we used RAxML v.8.2.10 (Stamatakis, 2014) to infer separate phylogenetic trees for the alignments of chloroplast, mitochondrial and nuclear SNPs. For each analysis we applied a GTR + G model of substitution and an ascertainment bias correction to account for the absence of constant sites in the alignments. Bootstrap replicates were performed using the -autoMRE option which executes a maximum of 1,000 bootstrap replicate searches but stops if support values reach convergence earlier. Secondly, to account for any potential effects of ILS in phylogeny estimation, we used SVDquartet (Chifman and Kubatko, 2014, 2015), a coalescent-based method designed for SNP data implemented in PAUP∗ (Swofford, 2001) to infer the nuclear phylogenetic tree. SVDquartet is a coalescent method for inferring phylogenetic relationships based on phylogenetic invariants which are mathematical functions representing the expected frequencies of site patterns in an alignment. It first infers the quartet topology of all subsets of four taxon in the dataset and then agglomerate them in order to obtain the species tree with all taxa. Although SVDquartet can also estimate phylogenetic relationships for non-recombining loci, we did not run it on our chloroplast or mitochondrial datasets because they contained < 10,000 SNPs and the performance of this method is highly dependent on the number of SNPs.
Divergence Time Estimates
We temporally calibrated the nuclear phylogenetic tree using penalized-likelihood implemented in the program treePL v1.0 (Smith and O’meara, 2012). We chose this approach because joint estimation of the topology and divergence times from SNP data (e.g., SNAPP, Bryant et al., 2012) was computationally too intensive to be applied to our large dataset. Given the absence of fossil data for bromeliads, we used a secondary calibration to perform the divergence time estimation. We implemented the age estimate of the crown node of core Tillandsioideae from Bouchenak-Khelladi et al. (2014) as a calibration for the root node of our tree, which encompasses the core Tillandsioideae, using the boundaries of the 95% HPD as minimum and maximum constraints (7.2417–12.984 My). Although their study is a monocotyledon-wide analysis, their sampling of Tillandsioideae is as good as other studies focusing on Bromeliaceae, and their dating analysis is based on nine fossil calibration points. Their sampling did not include any member of the tribe Vrieseeae, and we were unable to calibrate any shallower node in our tree. We performed cross-validation to estimate the value of the best-fitting smoothing parameter whose value can vary between 0 (each branch has its own substitution rate) and 1 (strict clock model).
Hybridization Detection
In order to estimate levels of hybridization across our dataset, while accounting for ILS, we used the program HyDe v0.4.3 (Blischak et al., 2018), a coalescent method which tests for hybridization in the presence of ILS using phylogenetic invariants (Kubatko and Chifman, 2019), similarly to the D statistics (Patterson et al., 2012). HyDe can detect both recent and ancient hybridization without the need for an a priori hypothesis, considers ambiguous sites, and can be applied to large genomic datasets. For all possible quartets in a dataset comprising one outgroup population, two parental populations (P2 and P3) and one hybrid population (P1), HyDe estimates the amount of admixture using a gamma statistic. Gamma values are comprised between 0 and 1, where γ = 0.5 means 50:50 hybrid and values close to 0 or 1 indicate a low level of asymmetrical admixture by introgression. We tested all four-taxon combinations comprising one outgroup and a triplet of ingroup taxa. For all tests Ananas comosus was set as the outgroup taxon using the python script run_hyde.py1. The program applies a conservative Bonferronni correction and outputs the combinations with a significant signal for hybridization at a level of < 0.05. Unless it applied to population data, this approach does not determine whether the significantly introgressed individuals represent the results of a hybrid speciation event, but suggests at a minimum that these individuals have genetic material from at least two different lineages in the phylogeny. We summarized the result of HyDe by recording the genera of the two parental species for each significant triplet, and counted for each hybrid the frequency of the different generic combinations. This allowed us to evaluate whether the detected hybrids most likely reflected intra- or intergeneric hybridization. We then constructed a network diagram using the fonction qgraph from the R package Qgraph v.1.5 (Epskamp et al., 2012) in order to display the frequency at which each genus was involved as a parental species (P2 or P3) for hybrids in other genera across all significant combinations.
Results
Sequencing
After base calling and filtering, 496,594 high-quality sites were obtained for the nuclear genome with a coverage depth of 22.2 reads per position and 19.3% missing data. For the chloroplast and mitochondrial genomes, 68,854 high-quality bases at 53.9X (5.8% missing data) and 19,403 high quality bases at 11.2X (18.2% of missing data) were recovered, respectively. From the SNP calling 116,478 SNPs were obtained for the nuclear, 5,171 SNPs for the chloroplast and 1,069 SNPs for the mitochondrial genomes. Sites presenting a pattern of a single nucleotide and an ambiguity code are considered by RaxML as invariant sites and we excluded them from the alignments, which led to a total dataset of 103,244 nuclear SNPs, 3,913 chloroplast SNPs, and 970 mitochondrial SNPs.
Phylogenetic Relationships
Intergeneric phylogenetic relationships were well-supported in all inferred trees. The chloroplast topology is similar to the one recovered in Machado et al. (2020), with subtribe Cipuropsidinae (composed of the genera Goudaea, Zizkaea, Lutheria, and Werauhia) sister to subtribe Vrieseinae (composed of Vriesea s.str., Stigmatodon, and Alcantarea, Figure 1). Our study adds new results for the mitochondrial phylogeny, which has a similar topology to the plastid tree. However, the nuclear topology resolves the subtribe Cipuropsidinae as sister to the tribe Tillandsieae (Figure 1). Furthermore, within the Cipuropsidinae, Lutheria is recovered as sister to Werauhia in the chloroplast and mitochondrial trees (with bootstrap support of 100 and 98, respectively, Supplementary Figures 1, 2), whereas in the nuclear tree, it forms a clade together with Goudea, Zizkaea and the Cipuropsis-Mezobromelia complex (with bootstrap support of 98; Figure 1 and Supplementary Figure 3). The genus Goudea, represented by the two species G. ospinae and G. chrysostachys, is recovered as monophyletic in the nuclear phylogeny but not in the chloroplast and mitochondrial trees (Figure 1 and Supplementary Figures 1–3). The coalescent-based phylogenetic tree obtained with SVDquartet from the nuclear data has a similar topology to the phylogenetic tree inferred with RAxML but an overall lower bootstrap support (Supplementary Figure 4). It differs, however, in the position of Werauhia, which is found in a sister position to the tribe Tillandsieae, and Guzmania, which is recovered nested within Tillandsia (with low support, Supplementary Figure 4). The monophyletic Vriesea s. str is characterized by small branch lengths and low internal support in all phylogenetic trees, yet most of the 12 clades described in Machado et al. (2020), which correspond to morphological or geographic groups, are recovered in the chloroplast and nuclear phylogenies, but not in the mitochondrial phylogeny.
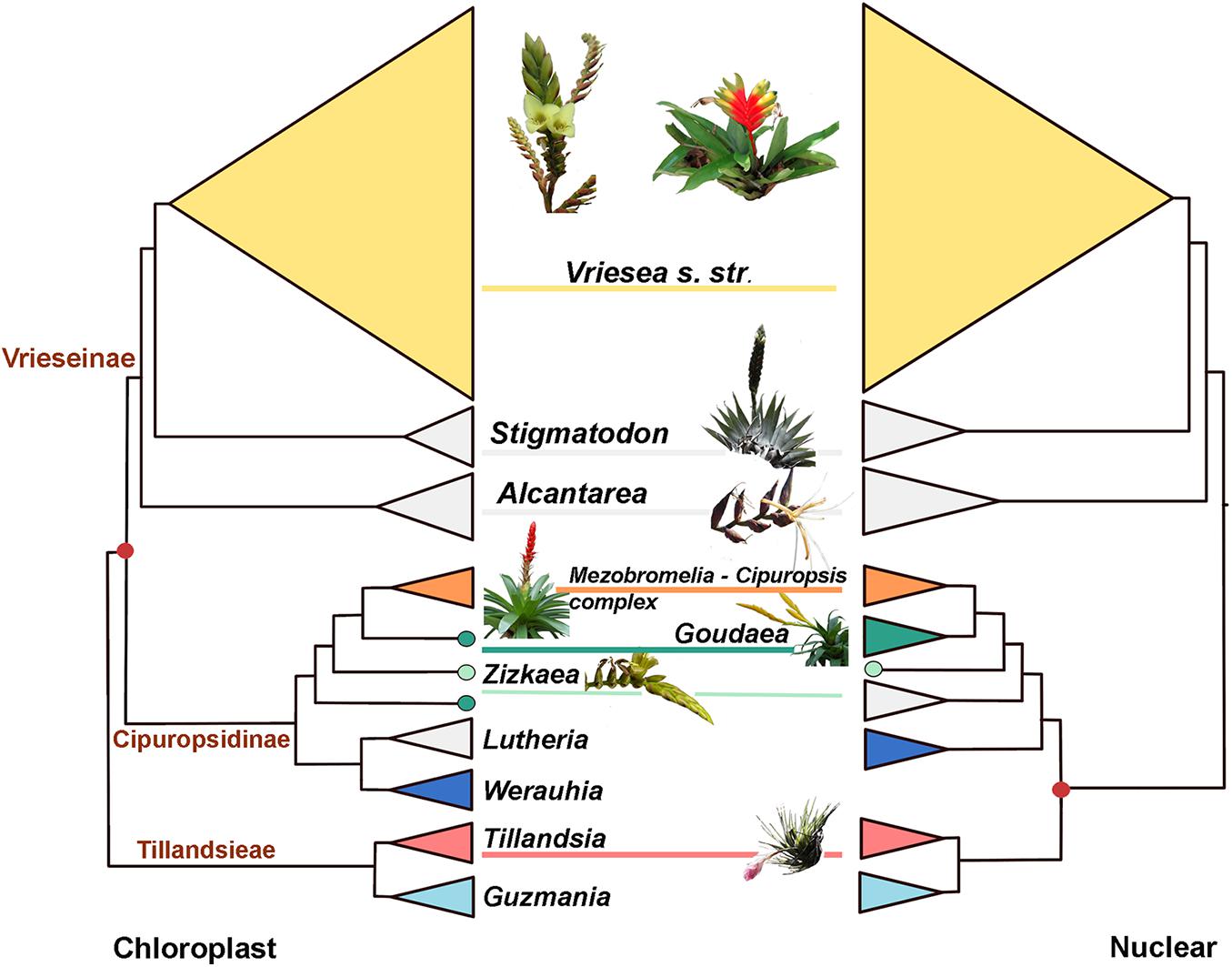
Figure 1. Phylogeneticincongruence in core Tillandsioideae. Summary cladograms of the chloroplast (left) and nuclear (right) phylogenetic trees. The red dot indicates the incongruent node associated with the placement of subtribe Cipuropsidinae. Genera indicated as follow (from up to down): V. s. str., Vriesea sensu stricto; Sti, Stigmatodon; Alc, Alcantarea; Cip- Mez, Vriesea—Cipuropsis/Mezobromelia complex; Gou, Goudeae; Ziz, Zizkaea; Lut, Lutheria; Wer, Werauhia; Till, Tillandsia; Guz, Guzmania. Colors indicate genera where hybrids were detected (see Figure 2). Photos by Eric Gouda (Goudaea, Mezobromelia and Zizkaea) and Talita Mota Machado (Alcantarea, Stigmatodon, Tillandsia and Vriesea s. str.).
Divergence Times
The best smoothing parameter found by treePL was 0.01, which allows for substantial substitution rate variation among lineages. The estimated crown age of the core Tillandsioideae was 12.98 My. The two main clades, the tribe Tillandsieae + the Cipuropsidinae and subtribe Vrieseinae have crown ages of 11.24 and 7.66 My, respectively (Figure 2). The genus Vriesea s. str has a crown age of 6.18 My and following the initial divergence a V. drepanocarpa, the rest of the group began to diversify 3.91 My ago and most of its diversification occurred during the Pleistocene.
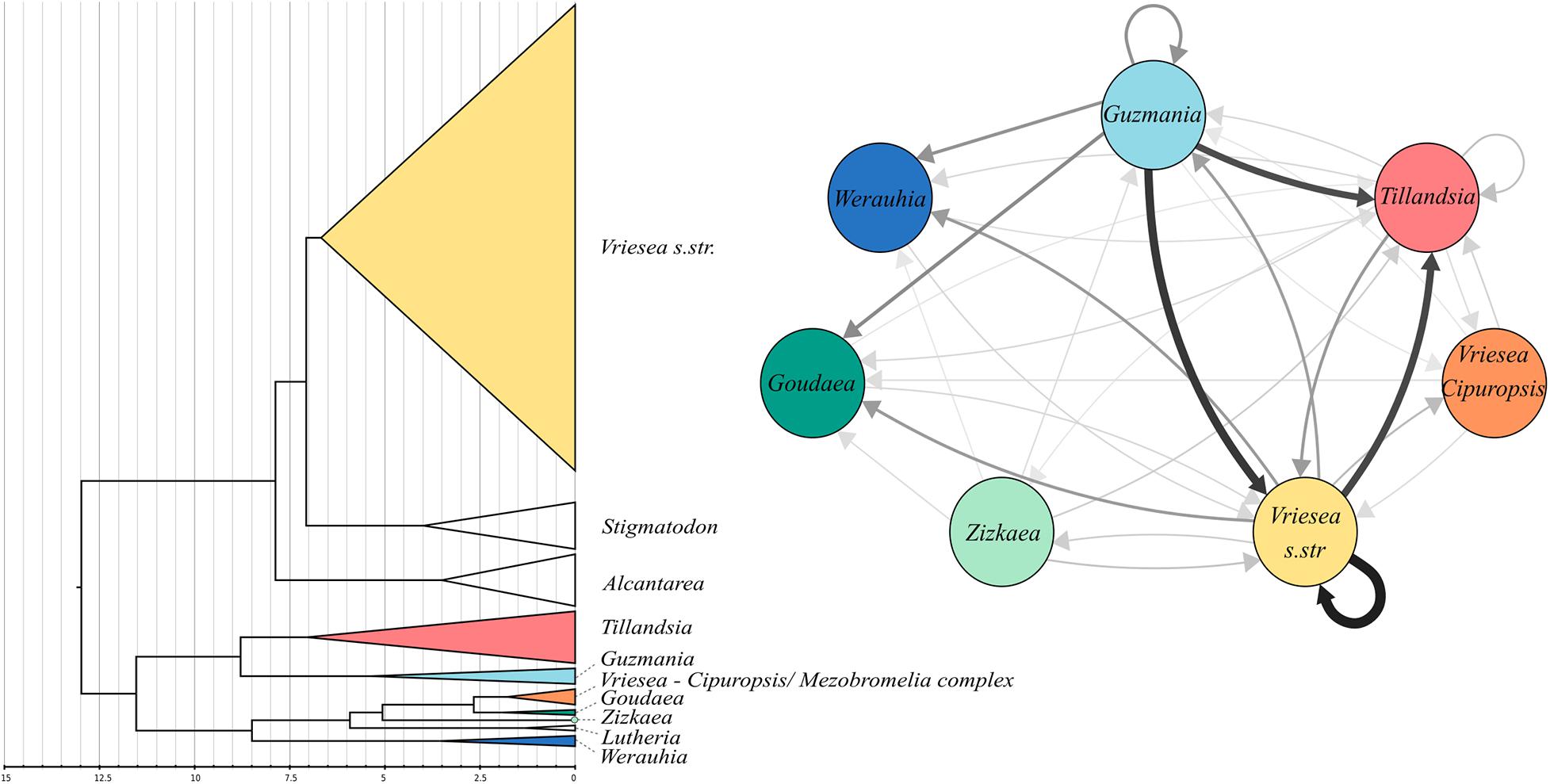
Figure 2. Left: Dated nuclear phylogeny of coreTillandsioids (intrageneric branches not shown). Colors indicate clades were hybrids have been detected. Right: network diagram of the putative origin of the detected hybrids across all significant triplets identified by HyDe. The width and color of the arrows are proportional to the frequency at which one genus is detected as a parental lineageaccross all hybrids.
Introgression and Hybridization
The HyDe analyses detected significant introgression in 287 out of the 1,687,425 possible triplets, after the conservative Bonferroni correction (Supplementary Table 1). For simplicity, we will call these cases “hybrids,” although we fully recognize that it is difficult to distinguish between hybrids and introgressive hybridization from the analyses performed. These 287 combinations contain 17 species identified as potentially of hybrid origin, which included nine species of Vriesea s.str., the species V. dubia that belongs to the Vriesea-Cipuropsis clade, three species of Tillandsia as well as Goudaea chrysostachys, Guzmania berteroniana, Werauhia gladioliflora, and Zizkaea tuerckheimii (Table 1). Nine of them represented products of intergeneric hybridization. The boxplots of gamma values from all significant tests for each of the 17 hybrids are shown in Figure 3. Nine of these (representing V. medusae plus the intergeneric hybrids, except Vriesea roberto-seidelli) are centered around 0.5, indicating that these taxa are potentially 50:50 hybrids (Figure 3). The remaining hybrids were characterized by a smaller introgression proportion as indicated by their gamma values. Six hybrids had median gamma values between 0.5 and 0.7 and two (Vriesea friburgensis and Zizkaea tuerckheimii) had median values above 0.7 indicating a low level of admixture (Figure 3). Vriesea billbergioides and Tillandsia juncea were identified as the parental species of the hybrid Vriesea roberto-seidelli. All other hybrids had multiple significant triplets involving different P2 and P3, indicating that the parental species of most hybrid taxa could not be unambiguously identified (Table 1 and Supplementary Table 1). When HyDe identifies multiple significant triplets involving several putative parental taxa for a given hybrid, it points to ancestral hybridization which remains detectable in several descendant lineages, even after considerable evolutionary divergence. Although the exact origin of the detected hybrids could not be uncovered, most of them had a signal of admixture biased toward one combination of parental lineages (Table 1). For example, the majority of the significant combinations involved a species from Vriesea s.str and a species from Guzmania in eight out of the nine intergeneric hybrids (Figure 4), while four hybrids of Vriesea had both parental species from Vriesea. s. str in most of their combinations. This prevalence of some lineages in the significant triplets is reflected in Figure 2 (right), where the thickness and darkness of the arrows is proportional to the number of time a genera was identified as a parental lineage for the hybrids in other lineages. This network diagram clearly depicts Guzmania and Vriesea s.str as the main putative parents for Vriesea s.str, Tillandsia, Goudaea, and Werauhia hybrids.

Table 1. Frequency of the possible combinations for the generic origin of the parental species for each hybrid across all significant triplets identified by HyDe.
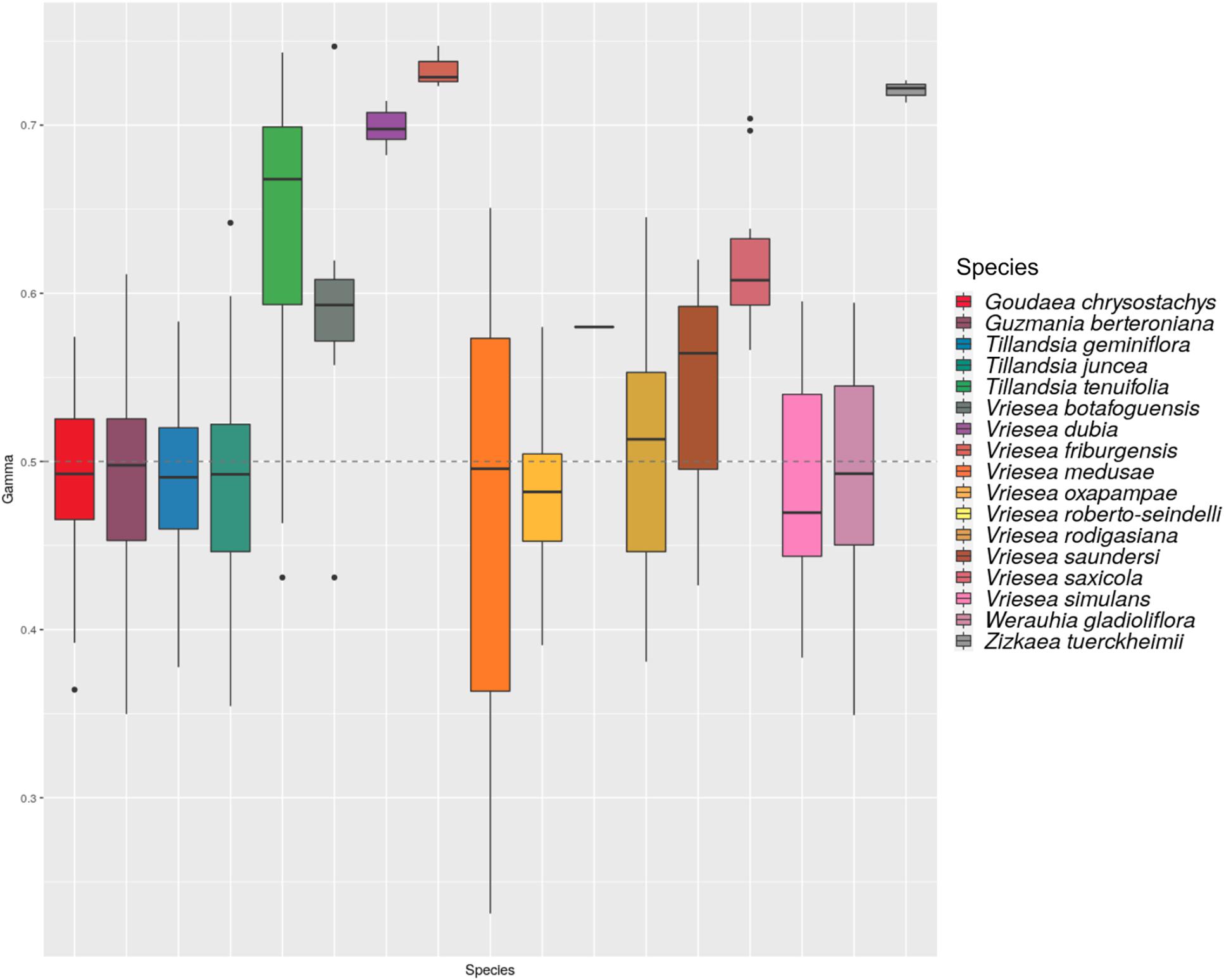
Figure 3. Histogram of the distribution of the gamma statistics across all significant triplets for each detected species. The dashed line indicate the value for 50/50 hybridization.
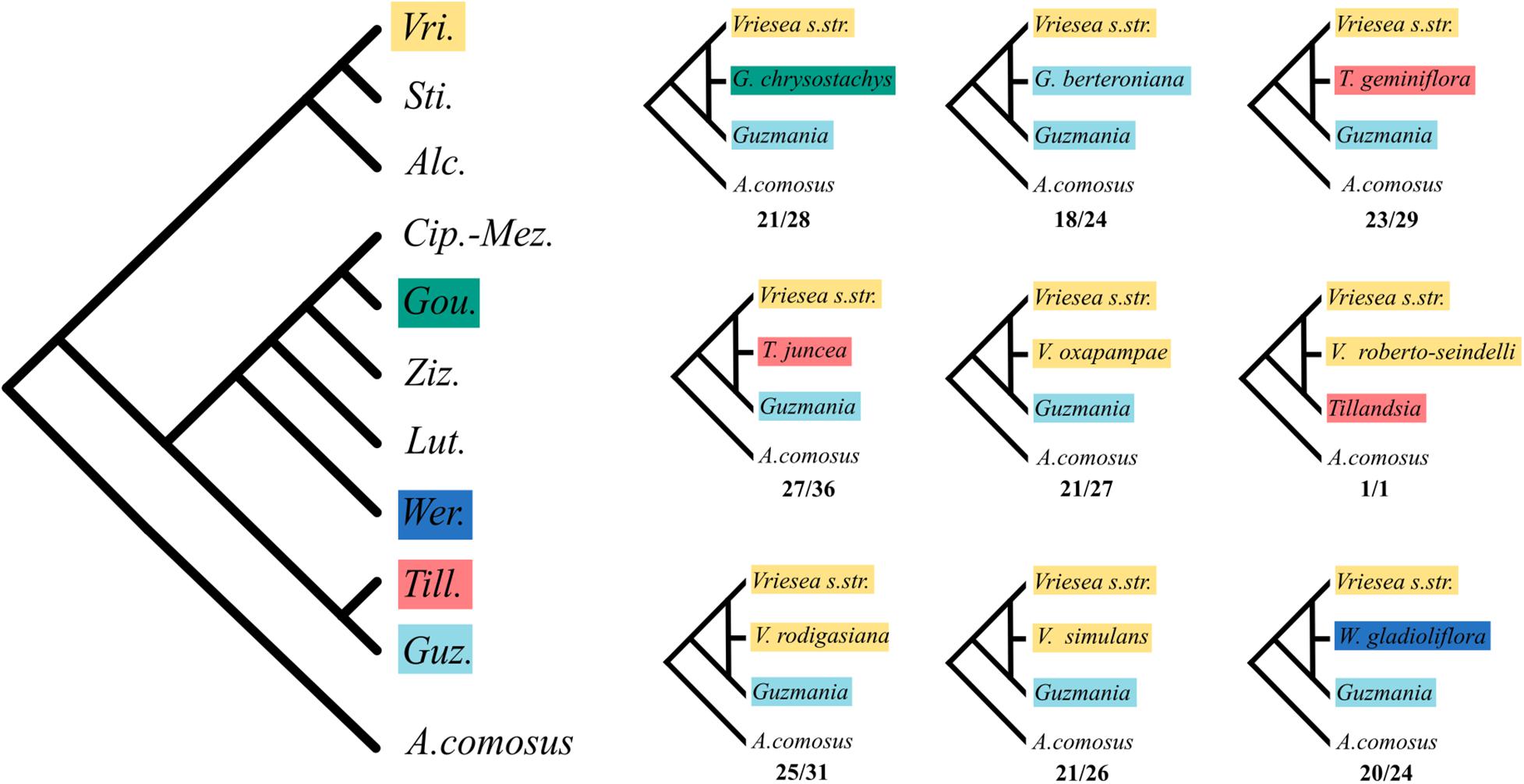
Figure 4. Most likely ancestral intergeneric hybridization events detected by HyDe. Left: Cladogram of the intergeneric phylogenetic relationships within core Tillandsioideae. Colored squares show genera where hybrids have been detected. Right: Most frequent combinations for the generic origin of the nine inter-generic hybrids inferred by HyDe. Numbers below each quartet indicate the number of time the depicted combination was found over the total number of significant triplets for that hybrid.
Discussion
Hybridization as a Widespread Phenomenon in Bromeliaceae
Our study points to widespread hybridization in subfamily Tillandsioideae, both within and among genera. By recovering mitochondrial, chloroplast and nuclear DNA in a single sequencing experiment, we were able to clearly demonstrate incongruent evolutionary histories between the three genomic compartments. Discrepancy between organellar and nuclear phylogenies is common in plants and can be caused either by ILS or hybridization (Naciri and Linder, 2015). By applying a method which uses phylogenetic invariants to detect hybridization in the presence of ILS, we were able to detect a significant level of admixture in the nuclear genome of 17 of the 148 species included in our phylogeny. Four of these (Goudaea chrysostachys, Vriesea dubia, Werauhia gladioliflora, and Zizkaea tuerckheimii) belong to subtribe Cipuropsidinae and two of these originated from ancestral hybridization between the Vriesea s.str. and Tillandsieae lineages (Figure 4). This finding supports the hypothesis that hybridization is responsible for the incongruent placement of subtribe Cipuropsidinae which is recovered as either closely related to the Vrieseinae (in the plastid and mitochondrial phylogenies) or alternatively to the Tillandsieae (in the nuclear phylogeny). In bromeliads, a similar example of cyto-nuclear discordance was found in the genus Puya and was explained by a mixture of ancient and recent hybridization events (Jabaily and Sytsma, 2010; Schulte et al., 2010). Our results add to the growing body of evidence that hybridization may have been a widespread phenomenon and an important driving force in the evolution of bromeliads. Indeed, interspecific and intergeneric artificial crosses of bromeliads are a common horticultural practice (Vervaeke et al., 2004; de Souza et al., 2017) and several studies report the permeable nature of species boundaries among bromeliads (Palma-Silva et al., 2011; Mota et al., 2019). For example, a study on pre-pollination isolating mechanisms in an assemblage of 42 sympatric species of bromeliads in Brazil found that neither microhabitat preferences, flowering phenology nor pollinators contributed to prezygotic reproductive isolation (Wendt et al., 2008). Another study looked at post-pollination barriers among 13 species of bromeliads and found that despite pollen tube growth inhibition being an important mechanism, nearly 25% of interspecific and intergeneric crosses lead to normal pollen tube growth (Matallana et al., 2016). Additionally, three sympatric species of Tillandsia in Mexico showed overlap in flowering time, similarity in reproductive organs morphology and manual crosses among them resulted in high fruit production and viable seeds (Ramírez-Rosas et al., 2020). There is further morphological and genetic evidence for natural introgression or hybrid speciation in several bromeliad genera, including Aechmea (Goetze et al., 2017), Alcantarea (Versieux et al., 2012; Lexer et al., 2016), Fosterella (Paule et al., 2017), Pitcairnia (Palma-Silva et al., 2011; Mota et al., 2020), Puya (Jabaily and Sytsma, 2010; Schulte et al., 2010), Tillandsia (Gardner, 1984; Gonçalves and de Azevêdo-Gonçalves, 2009), and Vriesea (Matos et al., 2016; Zanella et al., 2016; Neri et al., 2018).
Despite this wealth of evidence for a strong hybridization potential in bromeliads, population genetic studies generally have found low levels of interspecific gene flow in extant populations of bromeliads. For example, in Vriesea s.str., gene flow has been reported between V. scalaris and V. simplex (Neri et al., 2018) and between V. carinata and V. incurvata (Zanella et al., 2016) but all species exhibited high genetic structure, a low proportion of admixed individuals in the populations (between 8 and 12%) and low level of introgression. In these cases, the interaction between multiple reproductive barriers such as different reproductive systems, variation in floral traits, temporal flowering differences and low hybrid seed viability may have limited interspecific crosses and helped to maintain species integrity (Zanella et al., 2016; Neri et al., 2017). Similar results have been found in other genera such as Pitcarnia (Palma-Silva et al., 2011; Mota et al., 2020). However, these studies were done at the population level and explicitly targeted recent or ongoing gene-flow within small sympatric populations, whereas we focused on the long-term evolutionary signature of introgression across the whole subfamily. Our results indicate that 11% of the species in our dataset exhibit a signature of ancient hybridization with high levels of admixture. Therefore, while exchange of genetic material seems to be restricted to low level of introgression in the extant populations of bromeliads that have been studied so far, our study provides evidence that gene flow occurred between the ancestors of extant lineages of core Tillandsioideae that have considerably diverged from each other. Whether these events represent cases of hybrid speciation or introgressive hybridization remains to be investigated. Nevertheless, our results suggest that hybridization may have been more frequent in bromeliads than what has been suggested so far. If this is the case, it could have played an essential role in promoting speciation and generating diversity in bromeliads over evolutionary time, similarly to what has been demonstrated in other evolutionary radiations. In fact, the emerging view of bromeliad evolution is that the existence of generally strong, yet occasionally permeable reproductive barriers maintains species cohesion, while allowing the spreading of advantageous alleles (Neri et al., 2017). This is in line with the “speciation with gene flow” concept or the idea of the “porous genome,” in which some parts of the genome can be easily introgressed while genes essential to species cohesion are resistant to gene flow (Wu, 2001). Uniformity of chromosomal numbers in Tillandsioideae may enhance chromosome rearrangements and recombination, promoting speciation in the presence of gene flow (Rieseberg, 2001).
Although our results suggest that hybridization is a widespread phenomenon in Tillandisoideae, it does not rule out the existence of ILS as an additional source of phylogenetic incongruence. In fact, the lower branch support of the phylogenetic tree inferred using a method which models ILS (SVDquartet), compared to a method which does not (RAxML), suggests that ILS is indeed present in the group. Yet, given that HyDe is based on a theoretical frameworks that considers both ILS and hybridization, the presence of ILS is unlikely to have mislead our inference of hybridization. However, we cannot rule out the possibility that other factors related to data acquisition impacted our analyses. First, index hopping, an inherent source of errors linked to sequencing technologies, can potentially lead to mis-assignment of reads between multiplexed samples. In this study, we used a set of 60 dual-indexed Illumina adapters to limit the redundancy of the indexes and reduce this potential error, as recommended by Costello et al. (2018). Ros-Freixedes et al. (2018) also showed that index hopping, when present, has little impact on SNP call accuracy from low-coverage sequence data, and we are therefore confident that this potential bias has a limited impact on our analyses. Bias toward reference allele due to alignment also can have an effect on SNP calling, even if it was shown to be negligible with genome skimming on pigs (Ros-Freixedes et al., 2018). To limit this bias, we used the Tillandsia adpressiflora pseudo-reference genome constructed in de la Harpe et al. (2020) for mapping our samples. The method consisted in incorporating specific variation of a T. adpressiflora individual sequenced at high coverage into the high quality Ananas comosus genome (Ming et al., 2015). This strategy has the advantage of improving the global mapping efficiency of our samples for both reference and alternative alleles, but we cannot exclude that some of our SNP calls were impacted by biases linked to the quality and distance of the reference genome. In addition, because our genome skimming approach favors the detection of the high copy fraction of the genome, our sequence data may include duplicated genes or transposable elements. It is possible that the recovery of alternative paralogs in different lineages can affect the inference of phylogenetic trees and the estimation of the proportion of introgression. A Vriesea de novo genome assembly using long reads sequencing would be very helpful to detect and filter such multicopy genomic regions.
The method that we used to detect introgression has been shown through simulations to perform well in identifying introgressed individuals but may select the wrong parental species in the case of ancestral hybridization, by favoring parental species belonging to the sister clade of the true parent (Kubatko and Chifman, 2019). This is why here we focus on the identification of the hybrids rather than on the exact determination of their origin, which could also be affected the unbalanced taxonomic sampling of our study. Although it is unclear how the limited sampling outside of Vriesea may have impacted the detection of hybridization, a deeper sampling of all Tillandsioid genera would allow to further investigate intergeneric hybridization and confirm that the repeated signal for ancestral hybridization between Vriesea and tribe Tillandsieae is not an artifact due to the lack of intrageneric sampling. Finally, our results suggest that ancient hybrid speciation has occurred in core Tillandsioids, but the analysis we performed does not allow to discriminate between hybrid speciation and introgressive hybridization. A deeper investigation of the exact origin of the hybrids detected in this study would require additional analyses based on whole genome data in order to carry out a proper test of these two scenarios.
Biogeographic History of Tillandsioideae and Opportunities for Hybridization
Kessous et al. (2020) inferred a broad ancestral area for the core Tillandsioideae, spreading across the Atlantic Forest (AF), the Andes and the Chacoan dominion (i.e., the South American “Dry Diagonal,” Neves et al., 2015). The authors hypothesized that tectonic and climatic events during the Miocene (the formation of the Paranean Sea and the Dry Diagonal) likely caused the vicariance between the Andean lineages (Tillandsieae + Cipuropsidinae) and the Vrieseinae in the AF (Kessous et al., 2020). Although the Andean and Amazonian rainforests are at present separated from the AF by the drier biomes of the Dry Diagonal, there is considerable evidence in the literature that these wet forest biomes were in contact during the Miocene and Pleistocene, allowing for migration and biotic exchange (Batalha-Filho et al., 2013; Trujillo-Arias et al., 2017). Our analysis revealed a strong signal of hybridization between Vriesea s.str. and the tribe Tillandsieae in nine species, a finding coherent with the biogeographic history of the group. Indeed, hybridization between Vriesea s.str. and Guzmania is unlikely to be the result of recent hybridization given that these two lineages diverged from each other more than 12 My ago and have their respective centers of diversity in the Brazilian Atlantic Forest and the Andes. It is therefore plausible that hybridization occurred between ancestral populations of different core-tillandsioid genera with overlapping broad distributions or between populations that had geographically diverged between the Andes and the Amazon but were in contact during the Miocene. More recent hybridization events between Vriesea s.str and the Andean lineages are less likely but cannot be ruled out given that Vriesea has dispersed to the Amazonian and Andean regions during the Pleistocene (a few species have a disjunct distribution spanning the Dry Diagonal, with others being restricted to the Andes/Amazon and absent from the AF). In contrast, we did not detect hybridization between Vriesea and its two most closely genera, Alcantarea and Stigmatodon, which are also distributed in the AF and have species occuring in sympatry, nor did we find signal for intrageneric hybridization within these two genera. This result could by explained either by our limited taxonomic sampling, insufficient genomic data, limitation of the method used to detect hybridization, or alternatively by the existence of strong reproductive barriers preventing interspecific crosses among Vriesea, Alcantarea and Stigmatodon. Little is known about Stigmatodon, a recently segregated genus, but studies of Alcantarea have demonstrated the existence of introgression among several Alcantarea species, in spite of high population genetic divergence (Barbará et al., 2009; Lexer et al., 2016). However, because our phylogenetic approach does not include intraspecific sampling and uses only a limited part of the genome, it cannot detect such geographically localized and low level of interspecific gene flow. Despite a difference in style length which could act as a prezygotic reproduction barrier, natural hybridization between Vriesea and Alcantarea cannot be ruled out given that it is at least experimentally feasible under certain conditions (e.g., with Alcantarea as the male donor; de Souza et al., 2017). The main habitat of Alcantarea and Stigmatodon are inselbergs, considered terrestrial islands due to their ecological and spatial isolation which reduces the connectivity between populations (Porembski, 2007). It has been proposed that low level of introgressive hybridization could contribute to the maintenance of the genetic diversity of these isolated populations, thereby balancing the lack of intraspecific gene flow observed in inselberg bromeliad species (Palma-Silva et al., 2011; Mota et al., 2019). Thus, considering that Alcantarea, Stigmatodon and Vriesea are often found in sympatry in the Atlantic Forest inselbergs, hybridization between them seems at least plausible. Further investigation is required to elucidate whether or not gene flow between these lineages occurred at some point during their evolutionary history.
Bromeliads Phylogenomics
Bromeliaceae are known for their low rate of molecular evolution compared to other Poales (Gaut et al., 1992; Smith and Donoghue, 2008), resulting in a lack of resolution in species-level phylogenies reconstructed from a small number of plastid and sometimes nuclear markers (e.g., Sass and Specht, 2010; Versieux et al., 2012; Goetze et al., 2016). In this study, we aimed at circumventing this limitation by using genome skimming, a method which allows the obtention of a large amount of DNA from different genomic compartments without the need for upstream marker development. We obtained thousands of SNPs from the chloroplast, mitochondrial and nuclear genomes for nearly 150 species of Tillandsioideae, representing the largest genomic dataset for bromeliads to date. Using these data to infer a phylogeny, we were able to confidently resolve intergeneric relationships, indicating that the method is well-adapted for intermediate levels of divergence in bromeliads. However, within genera, branch support values ranged from high (Alcantarea) to intermediate (the Stigmatodon clade), to very low (Vriesea s.str.). This lack of power is particularly critical in our chloroplast dataset where only 8.3% of sites are variable despite a high sequencing depth, 53.9x on average, obtained with our genome skimming method. Chloroplast genes have been the most common markers used for phylogenetics analyses in bromeliads so far, but our results suggest that even the full plastome would be unlikely to contain enough information to resolve phylogenetic relationships within Vriesea. Simpson et al. (2017) faced a similar problem using genome skimming approach to obtain plastid genomes for the phylogenomic analysis of a large clade of Boraginaceae. Coissac et al. (2016) suggest that entire plastid genomes obtained with genome skimming would not be enough for “difficult” groups, and that the obtention of hundreds of nuclear loci is likely to be required. They suggested that shallow-pass nuclear loci obtained with genome skimming could provide enough phylogenetic informative data in such cases. Despite considerable genomic data in our study, we obtained low support within the focal clade Vriesea s.str. This is probably due to (1) the substantial amount of missing data (c. 22%) in the alignment of nuclear SNPs, and (2) the fact that only 40% of the nuclear SNPs are parsimony-informative. Indeed, even though 48% of our detected high-quality nuclear sites are polymorphic, SNPs were found mainly at low frequencies (median frequency of 5.9%) and were rarely informative. Hence, while being advantageous in term of cost and time, genome skimming is probably not the most efficient approach for shallow-scale phylogenetic studies in Bromeliaceae, particularly in young and speciose clades such as Vriesea, which diversified into c. 300 species in less than 8 My. Obtaining more informative nuclear SNPs, would require increasing the sequencing depth of each sample and therefore dramatically increasing the cost for large scale phylogenomic studies. For these reasons, we argue that more specific approaches such as target-capture sequencing of low-copy nuclear genes could be the way forward to obtain better-resolved phylogenies of large bromeliad genera such as Guzmania, Tillandsia or Vriesea. Even if the development of such target-capture kits is complex and challenging (de La Harpe et al., 2019; Andermann et al., 2020), it would benefit a wide research community working on many fascinating groups of bromeliads.
Conclusion
By generating an unprecedented genomic dataset for the largest sub-family of Bromeliaceae, our study sheds lights on the evolutionary history of an important floristic component of Neotropical rainforests. We pinpointed the incongruence of the nuclear phylogeny with the chloroplast and mitochondrial phylogenetic trees regarding the position of the subtribe Cipuropsidinae.
Our finding of 17 substantially introgressed taxa, potentially the products of hybrid speciation, in our dataset of 116,478 nuclear SNPs for 148 species of Tillandsioideae suggest that hybridization is a plausible explanation for this incongruence. Furthermore, the signal for hybridization between the ancestral lineages of Vriesea and tribe Tillandsieae suggests that hybridization may be widespread phenomenon in core tillandsioids, and to a greater extent, in bromeliads. Thus, our study adds to the growing body of evidence that hybridization is ubiquitous across this diverse Neotropical plant family and may have fueled the diversification of the most diverse clades of Bromeliaceae such as the core Tillandsioideae and core Bromelioideae. Future genomic studies with deeper sequencing across a wide taxonomic range in bromeliads would likely reveal more hybridization and help to tell apart the respective contribution of hybrid speciation and adaptive introgression to the evolution of bromeliad diversity. Further joint investigation of hybridization at the micro- and macro- evolutionary level will be necessary to clarify the exact mechanisms through which it may have promoted genetic diversity, adaptation and speciation, and ultimately contributed to the adaptive radiation and ecological success of bromeliads.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI (accession: PRJNA554504).
Author Contributions
NS and CL designed the study. TM performed fieldwork and sample collection. MP led the sequencing experiments and post sequencing bioinformatics. TM and MP did the labwork. DK and OL did phylogenetic analyses. OL did hybridization test and led the writing with significant contributions from all co-authors. All authors commented and agreed on the last version of the manuscript.
Funding
This work was funded by the Sinergia grant (CRSII3-147630) from the Swiss National Science Foundation to NS and CL as well as funding from the University of Lausanne. TM was supported by Ph.D. fellowship grants from CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior) and CNPq [Conselho Nacional de Desenvolvimento Científico e Tecnológico—CNPq-SWE (205660/2014-2)/CNPq (142354/2016-3)]. LV received funding from the CNPq (455510/2014-8 and 303794/2019-4). Computational resources were supplied by the project “e-Infrastruktura CZ” (e-INFRA LM2018140) provided within the program Projects of Large Research, Development and Innovations Infrastructures, and by the computing infrastructure (DCSR) of the University of Lausanne.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the Botanical Gardens of the University of Vienna (WU-HBV), Botanical Garden of Rio de Janeiro (RBvb), Marie Selby Botanical Garden (SEL), and Jardin des Serres d’Auteuil (P) for providing plant material, ICMBIO, and IEF-MG for collection permits. TM thanks all the amazing people who collaborated in the fieldwork and other stages of this work.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2021.668281/full#supplementary-material
Supplementary Figure 1 | Phylogenetic tree of core Tillandsioideae inferred with RaxML using the concatenated mitochondrial SNPs.
Supplementary Figure 2 | Phylogenetic tree of core Tillandsioideae inferred with RaxML using the concatenated chloroplast SNPs.
Supplementary Figure 3 | Phylogenetic tree of core Tillandsioideae inferred with RaxML using the concatenated nuclear SNPs.
Supplementary Figure 4 | Phylogenetic tree of core Tillandsioideae inferred with SVDquartet using the concatenated nuclear SNPs.
Supplementary Table 1 | List of all significant triplets identified by HyDe.
Footnotes
References
Andermann, T., Torres Jiménez, M. F., Matos-Maraví, P., Batista, R., Blanco-Pastor, J. L., Gustafsson, A. L. S., et al. (2020). A guide to carrying out a phylogenomic target sequence capture project. Front. Genet. 10:1407.
Barbará, T., Martinelli, G., Palma-Silva, C., Fay, M. F., Mayo, S., and Lexer, C. (2009). Genetic relationships and variation in reproductive strategies in four closely related bromeliads adapted to neotropical ‘inselbergs’: Alcantarea glaziouana, A. regina, A. geniculata and A. imperialis (Bromeliaceae). Ann. Bot. 103, 65–77. doi: 10.1093/aob/mcn226
Barrier, M., Baldwin, B. G., Robichaux, R. H., and Purugganan, M. D. (1999). Interspecific hybrid ancestry of a plant adaptive radiation: allopolyploidy of the Hawaiian silversword alliance (Asteraceae) inferred from floral homeotic gene duplications. Mol. Biol. Evol. 16, 1105–1113. doi: 10.1093/oxfordjournals.molbev.a026200
Batalha-Filho, H., Fjeldså, J., Fabre, P.-H., and Miyaki, C. Y. (2013). Connections between the Atlantic and the Amazonian forest avifaunas represent distinct historical events. J. Ornithol. 154, 41–50. doi: 10.1007/s10336-012-0866-7
Benzing, D. H. (2000). Bromeliaceae: Profile of an Adaptive Radiation. Cambridge: Cambridge University Press.
Blanco-Pastor, J. L., Vargas, P., and Pfeil, B. E. (2012). Coalescent simulations reveal hybridization and incomplete lineage sorting in Mediterranean Linaria. PLoS One 7:e39089. doi: 10.1371/journal.pone.0039089
Blischak, P. D., Chifman, J., Wolfe, A. D., and Kubatko, L. S. (2018). HyDe: a Python package for genome-scale hybridization detection. Syst. Biol. 9, 821–829. doi: 10.1093/sysbio/syy023
Bouchenak-Khelladi, Y., Muasya, A. M., and Linder, H. P. (2014). A revised evolutionary history of poales: origins and diversification: evolutionary history of poales. Bot. J. Linn. Soc. 175, 4–16. doi: 10.1111/boj.12160
Bryant, D., Bouckaert, R., Felsenstein, J., Rosenberg, N. A., and RoyChoudhury, A. (2012). Inferring species trees directly from biallelic genetic markers: bypassing gene trees in a full coalescent analysis. Mol. Biol. Evol. 29, 1917–1932. doi: 10.1093/molbev/mss086
Chifman, J., and Kubatko, L. (2014). Quartet inference from SNP data under the coalescent model. Bioinformatics 30, 3317–3324. doi: 10.1093/bioinformatics/btu530
Chifman, J., and Kubatko, L. (2015). Identifiability of the unrooted species tree topology under the coalescent model with time-reversible substitution processes, site-specific rate variation, and invariable sites. J. Theor. Biol. 374, 35–47. doi: 10.1016/j.jtbi.2015.03.006
Coissac, E., Hollingsworth, P. M., Lavergne, S., and Taberlet, P. (2016). From barcodes to genomes: extending the concept of DNA barcoding. Mol. Ecol. 25, 1423–1428. doi: 10.1111/mec.13549
Costello, M., Fleharty, M., Abreu, J., Farjoun, Y., Ferriera, S., Holmes, L., et al. (2018). Characterization and remediation of sample index swaps by non-redundant dual indexing on massively parallel sequencing platforms. BMC Genomics 19:332.
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
de La Harpe, M., Hess, J., Loiseau, O., Salamin, N., Lexer, C., and Paris, M. (2019). A dedicated target capture approach reveals variable genetic markers across micro- and macro-evolutionary time scales in palms. Mol. Ecol. Resour. 19, 221–234. doi: 10.1111/1755-0998.12945
de la Harpe, M., Paris, M., Hess, J., Barfuss, M. H. J., Serrano-Serrano, M. L., Ghatak, A., et al. (2020). Genomic footprints of repeated evolution of CAM photosynthesis in a Neotropical species radiation. Plant Cell Environ. 43, 2987–3001. doi: 10.1111/pce.13847
de Souza, E. H., Versieux, L. M., Souza, F. V. D., Rossi, M. L., Costa, M. A. P., and Martinelli, A. P. (2017). Interspecific and intergeneric hybridization in Bromeliaceae and their relationships to breeding systems. Sci. Hortic. 223, 53–61. doi: 10.1016/j.scienta.2017.04.027
Dodsworth, S. (2015). Genome skimming for next-generation biodiversity analysis. Trends Plant Sci. 20, 525–527. doi: 10.1016/j.tplants.2015.06.012
Epskamp, S., Cramer, A. O., Waldorp, L. J., Schmittmann, V. D., and Borsboom, D. (2012). qgraph: network visualizations of relationships in psychometric data. J. Stat. Softw. 48, 1–18.
Gardner, C. S. (1984). Natural hybridization in “Tillandsia” subgenus “Tillandsia”. Selbyana 7, 380–393.
Gaut, B. S., Muse, S. V., Clark, W. D., and Clegg, M. T. (1992). Relative rates of nucleotide substitution at the rbcL locus of monocotyledonous plants. J. Mol. Evol. 35, 292–303. doi: 10.1007/bf00161167
Givnish, T. J., Barfuss, M. H. J., Ee, B. V., Riina, R., Schulte, K., Horres, R., et al. (2014). Molecular Phylogenetics and evolution adaptive radiation, correlated and contingent evolution, and net species diversification in Bromeliaceae. Mol. Phylogenet. Evol. 71, 55–78. doi: 10.1016/j.ympev.2013.10.010
Goetze, M., Schulte, K., Palma-Silva, C., Zanella, C. M., Büttow, M. V., Capra, F., et al. (2016). Diversification of Bromelioideae (Bromeliaceae) in the Brazilian Atlantic rainforest: a case study in Aechmea subgenus Ortgiesia. Mol. Phylogenet. Evol. 98, 346–357. doi: 10.1016/j.ympev.2016.03.001
Goetze, M., Zanella, C. M., Palma-Silva, C., Büttow, M. V., and Bered, F. (2017). Incomplete lineage sorting and hybridization in the evolutionary history of closely related, endemic yellow-flowered Aechmea species of subgenus Ortgiesia (Bromeliaceae). Am. J. Botany 104, 1073–1087. doi: 10.3732/ajb.1700103
Gonçalves, C. N., and de Azevêdo-Gonçalves, C. F. (2009). A new hybrid bromeliad from southernmost Brazil, Tillandsia ×baptistana. Novon J. Bot. Nomenclature 19, 353–356. doi: 10.3417/2001154
Gouda, E. J., Butcher, D., and Gouda, C. S. (2018). Encyclopaedia of Bromeliads, Version 4. Utrecht: University Botanic Gardens.
Grant, P. R., and Grant, B. R. (2019). Hybridization increases population variation during adaptive radiation. Proc. Natl. Acad. Sci. USA 116:23216.
Green, R. E., Krause, J., Briggs, A. W., Maricic, T., Stenzel, U., Kircher, M., et al. (2010). A draft sequence of the neandertal genome. Science 328, 710–722.
Harrison, R. G., and Larson, E. L. (2014). Hybridization, introgression, and the nature of species boundaries. J. Heredity 105, 795–809. doi: 10.1093/jhered/esu033
Irisarri, I., Singh, P., Koblmüller, S., Torres-Dowdall, J., Henning, F., Franchini, P., et al. (2018). Phylogenomics uncovers early hybridization and adaptive loci shaping the radiation of Lake Tanganyika cichlid fishes. Nat. Commun. 9:3159.
Jabaily, R. S., and Sytsma, K. J. (2010). Phylogenetics of Puya (Bromeliaceae): placement, major lineages, and evolution of Chilean species. Am. J. Bot. 97, 337–356. doi: 10.3732/ajb.0900107
Joly, S., McLenachan, P. A., and Lockhart, P. J. (2009). A statistical approach for distinguishing hybridization and incomplete lineage sorting. Am. Nat. 174, E54–E70.
Kessous, I. M., Neves, B., Couto, D. R., Paixão-Souza, B., Pederneiras, L. C., Moura, R. L., et al. (2020). Historical biogeography of a Brazilian lineage of Tillandsioideae (subtribe Vrieseinae, Bromeliaceae): the Paranaean Sea hypothesized as the main vicariant event. Bot. J. Linn. Soc. 192, 625–641.
Kircher, M., Sawyer, S., and Meyer, M. (2012). Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res. 40:e3. doi: 10.1093/nar/gkr771
Kozak, K. M., Joron, M., McMillan, W. O., and Jiggins, C. D. (2021). Rampant genome-wide admixture across the Heliconius radiation. Genome Biol. Evol. evab099. doi: 10.1093/gbe/evab099
Kubatko, L. S., and Chifman, J. (2019). An invariants-based method for efficient identification of hybrid species from large-scale genomic data. BMC Evol. Biol. 19:112.
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9:357. doi: 10.1038/nmeth.1923
Leaché, A. D., Banbury, B. L., Felsenstein, J., de Oca, A. N., and Stamatakis, A. (2015). Short tree, long tree, right tree, wrong tree: new acquisition bias corrections for inferring SNP phylogenies. Syst. Biol. 64, 1032–1047. doi: 10.1093/sysbio/syv053
Lexer, C., Marthaler, F., Humbert, S., Barbará, T., de la Harpe, M., Bossolini, E., et al. (2016). Gene flow and diversification in a species complex of Alcantarea inselberg bromeliads. Bot. J. Linn. Soc. 181, 505–520. doi: 10.1111/boj.12372
Loiseau, O., Olivares, I., Paris, M., de La Harpe, M., Weigand, A., Koubínová, D., et al. (2019). Targeted capture of hundreds of nuclear genes unravels phylogenetic relationships of the diverse neotropical Palm Tribe Geonomateae. Front. Plant Sci. 10:864.
Machado, T. M., Loiseau, O., Paris, M., Weigand, A., Versieux, L. M., Stehmann, J. R., et al. (2020). Systematics of Vriesea (Bromeliaceae): phylogenetic relationships based on nuclear gene and partial plastome sequences. Bot. J. Linn. Soc. 192, 656–674. doi: 10.1093/botlinnean/boz102
Marques, D. A., Meier, J. I., and Seehausen, O. (2019). A combinatorial view on speciation and adaptive radiation. Trends Ecol. Evol. 34, 531–544. doi: 10.1016/j.tree.2019.02.008
Matallana, G., Oliveira, P. E., Silva, P. R. R., and Wendt, T. (2016). Post - pollination barriers in an assemblage of Bromeliaceae in south - eastern Brazil. Bot. J. Linn. Soc. 181, 521–531. doi: 10.1111/boj.12406
Matos, J. Z., Juan, A., Agulló, J. C., and Crespo, M. B. (2016). Morphological features, nuclear microsatellites and plastid haplotypes reveal hybridisation processes between two sympatric Vriesea species in Brazil (Bromeliaceae). Phytotaxa 261, 58–74. doi: 10.11646/phytotaxa.261.1.2
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Meier, J. I., Marques, D. A., Mwaiko, S., Wagner, C. E., Excoffier, L., and Seehausen, O. (2017). Ancient hybridization fuels rapid cichlid fish adaptive radiations. Nat. Commun. 8:14363.
Ming, R., VanBuren, R., Wai, C. M., Tang, H., Schatz, M. C., Bowers, J. E., et al. (2015). The pineapple genome and the evolution of CAM photosynthesis. Nat. Genet. 47, 1435–1442. doi: 10.1038/ng.3435
Mota, M. R., Pinheiro, F., Leal, B. S. S., Wendt, T., and Palma-Silva, C. (2019). The role of hybridization and introgression in maintaining species integrity and cohesion in naturally isolated inselberg bromeliad populations. Plant Biol. 21, 122–132. doi: 10.1111/plb.12909
Mota, M. R., Pinheiro, F., Leal, B. S., Sardelli, C. H., Wendt, T., and Palma-Silva, C. (2020). From micro- to macroevolution: insights from a Neotropical bromeliad with high population genetic structure adapted to rock outcrops. Heredity 125, 353–370. doi: 10.1038/s41437-020-0342-8
Naciri, Y., and Linder, H. P. (2015). Species delimitation and relationships: the dance of the seven veils. Taxon 64, 3–16. doi: 10.12705/641.24
Neri, J., Wendt, T., Leles, B., dos Santos, M. F., and Palma-Silva, C. (2017). Variation in reproductive systems facilitates species boundaries of sympatric Vriesea (Bromeliaceae). Bot. J. Linn. Soc. 184, 272–279. doi: 10.1093/botlinnean/box026
Neri, J., Wendt, T., and Palma-Silva, C. (2018). Natural hybridization and genetic and morphological variation between two epiphytic bromeliads. AoB Plants 10:lx061.
Neves, D. M., Dexter, K. G., Pennington, R. T., Bueno, M. L., and Filho, A. T. O. (2015). Environmental and historical controls of floristic composition across the South American dry diagonal. J. Biogeogr. 42, 1566–1576. doi: 10.1111/jbi.12529
Palma-Silva, C., Leal, B. S. S., Chaves, C. J. N., and Fay, M. F. (2016). Advances in and perspectives on evolution in Bromeliaceae. Bot. J. Linn. Soc. 181, 305–322. doi: 10.1111/boj.12431
Palma-Silva, C., Wendt, T., Pinheiro, F., Barbará, T., Fay, M. F., Cozzolino, S., et al. (2011). Sympatric bromeliad species (Pitcairnia spp.) facilitate tests of mechanisms involved in species cohesion and reproductive isolation in Neotropical inselbergs. Mol. Ecol. 20, 3185–3201. doi: 10.1111/j.1365-294x.2011.05143.x
Patterson, N., Moorjani, P., Luo, Y., Mallick, S., Rohland, N., Zhan, Y., et al. (2012). Ancient admixture in human history. Genetics 192, 1065–1093. doi: 10.1534/genetics.112.145037
Paule, J., Wagner, N. D., Weising, K., and Zizka, G. (2017). Ecological range shift in the polyploid members of the South American genus Fosterella (Bromeliaceae). Ann. Bot. 120, 233–243.
Pfennig, K. S. (2021). Biased hybridization and its impact on adaptive introgression. Trends Ecol. Evol. 36, 488–497.
Porembski, S. (2007). Tropical inselbergs: habitat types, adaptive strategies and diversity patterns. Braz. J. Bot. 30, 579–586. doi: 10.1590/s0100-84042007000400004
Ramírez-Rosas, K., Aguirre-Jaimes, A., Ramírez-Morillo, I. M., and García-Franco, J. G. (2020). Floral biology and potential hybridization of three sympatric epiphytic bromeliads in Veracruz, Mexico. Plant Species Biol. 35, 197–209. doi: 10.1111/1442-1984.12279
Rieseberg, L. H. (2001). Chromosomal rearrangements and speciation. Trends Ecol. Evol. 16, 351–358. doi: 10.1016/s0169-5347(01)02187-5
Ros-Freixedes, R., Battagin, M., Johnsson, M., Gorjanc, G., Mileham, A. J., Rounsley, S. D., et al. (2018). Impact of index hopping and bias towards the reference allele on accuracy of genotype calls from low-coverage sequencing. Genet. Sel. Evol. 50:64.
Sass, C., and Specht, C. D. (2010). Phylogenetic estimation of the core Bromelioids with an emphasis on the genus Aechmea (Bromeliaceae). Mol. Phylogenet. Evol. 55, 559–571. doi: 10.1016/j.ympev.2010.01.005
Schulte, K., Silvestro, D., Kiehlmann, E., Vesely, S., Novoa, P., and Zizka, G. (2010). Detection of recent hybridization between sympatric Chilean Puya species (Bromeliaceae) using AFLP markers and reconstruction of complex relationships. Mol. Phylogenet. Evol. 57, 1105–1119. doi: 10.1016/j.ympev.2010.09.001
Schumer, M., Xu, C., Powell, D. L., Durvasula, A., Skov, L., Holland, C., et al. (2018). Natural selection interacts with recombination to shape the evolution of hybrid genomes. Science 360, 656–660. doi: 10.1126/science.aar3684
Seehausen, O. (2004). Hybridization and adaptive radiation. Trends Ecol. Evol. 19, 198–207. doi: 10.1016/j.tree.2004.01.003
Seehausen, O. (2013). Conditions when hybridization might predispose populations for adaptive radiation. J. Evol. Biol. 26, 279–281. doi: 10.1111/jeb.12026
Silvestro, D., Zizka, G., and Schulte, K. (2014). Disentangling the effects of key innovations on the diversification of bromelioideae (Bromeliaceae). Evolution 68, 163–175. doi: 10.1111/evo.12236
Simpson, M. G., Guilliams, C. M., Hasenstab-Lehman, K. E., Mabry, M. E., and Ripma, L. (2017). Phylogeny of the popcorn flowers: use of genome skimming to evaluate monophyly and interrelationships in subtribe Amsinckiinae (Boraginaceae). Taxon 66, 1406–1420. doi: 10.12705/666.8
Smith, L., and Downs, R. (1974). Flora Neotropica Monograph no. 14.(Pitcairnioideae)(Bromeliaceae). New York, NY: Hafner Publishing Company.
Smith, S. A., and Donoghue, M. J. (2008). Rates of molecular evolution are linked to life history in flowering plants. Science 322, 86–89. doi: 10.1126/science.1163197
Smith, S. A., and O’meara, B. C. (2012). treePL: divergence time estimation using penalized likelihood for large phylogenies. Bioinformatics 28, 2689–2690. doi: 10.1093/bioinformatics/bts492
Soltis, P. S., and Soltis, D. E. (2009). The role of hybridization in plant speciation. Annu. Rev. Plant Biol. 60, 561–588. doi: 10.1146/annurev.arplant.043008.092039
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Svardal, H., Quah, F. X., Malinsky, M., Ngatunga, B. P., Miska, E. A., Salzburger, W., et al. (2020). Ancestral hybridization facilitated species diversification in the lake Malawi Cichlid fish adaptive radiation. Mol. Biol. Evol. 37, 1100–1113. doi: 10.1093/molbev/msz294
Swofford, D. L. (2001). Paup∗: Phylogenetic Analysis Using Parsimony (and other Methods) 4.0. B5. Princeton: CiteSeerX.
Trujillo-Arias, N., Dantas, G. P. M., Arbeláez-Cortés, E., Naoki, K., Gómez, M. I., Santos, F. R., et al. (2017). The niche and phylogeography of a passerine reveal the history of biological diversification between the Andean and the Atlantic forests. Mol. Phylogenet. Evol. 112, 107–121. doi: 10.1016/j.ympev.2017.03.025
Versieux, L. M., Barbará, T., Wanderley, M., das, G. L., Calvente, A., Fay, M. F., et al. (2012). Molecular phylogenetics of the Brazilian giant bromeliads (Alcantarea, Bromeliaceae): implications for morphological evolution and biogeography. Mol. Phylogenet. Evol. 64, 177–189. doi: 10.1016/j.ympev.2012.03.015
Vervaeke, I., Wouters, J., Londers, E., Deroose, R., and Proft, M. P. D. (2004). Morphology of artificial hybrids of Vriesea splendens x Tillandsia cyanea and V. splendens x Guzmania lingulata (Bromeliaceae). Ann. Bot. Fennici 1840, 201–208.
Wagner, N. D., Wöhrmann, T., Öder, V., Burmeister, A., and Weising, K. (2015). Reproduction biology and chloroplast inheritance in Bromeliaceae: a case study in Fosterella (Pitcairnioideae). Plant Syst. Evol. 301, 2231–2246. doi: 10.1007/s00606-015-1226-x
Wendt, T., Coser, T. S., Matallana, G., and Guilherme, F. A. G. (2008). An apparent lack of prezygotic reproductive isolation among 42 sympatric species of Bromeliaceae in southeastern Brazil. Plant Syst. Evol. 275, 31–41. doi: 10.1007/s00606-008-0054-7
Wu, C. (2001). The genic view of the process of speciation. J. Evol. Biol. 14, 851–865. doi: 10.1046/j.1420-9101.2001.00335.x
Keywords: Bromeliaceae, genome skimming, phylogenomics, hybridization, Vriesea
Citation: Loiseau O, Mota Machado T, Paris M, Koubínová D, Dexter KG, Versieux LM, Lexer C and Salamin N (2021) Genome Skimming Reveals Widespread Hybridization in a Neotropical Flowering Plant Radiation. Front. Ecol. Evol. 9:668281. doi: 10.3389/fevo.2021.668281
Received: 15 February 2021; Accepted: 30 April 2021;
Published: 28 May 2021.
Edited by:
Danilo M. Neves, Federal University of Minas Gerais, BrazilReviewed by:
Clarisse Palma-Silva, State University of Campinas, BrazilTim Barraclough, University of Oxford, United Kingdom
Copyright © 2021 Loiseau, Mota Machado, Paris, Koubínová, Dexter, Versieux, Lexer and Salamin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Oriane Loiseau, b3JpYW5lLmxvaXNlYXVAdW5pbC5jaA==
†Deceased