 Valeria Alavez
Valeria Alavez Ángela P. Cuervo-Robayo
Ángela P. Cuervo-Robayo Enrique Martínez-Meyer
Enrique Martínez-Meyer Ana Wegier
Ana Wegier- 1Posgrado en Ciencias Biológicas, Universidad Nacional Autónoma de México (UNAM), Mexico City, Mexico
- 2Laboratorio de Genética de la Conservación, Jardín Botánico, Instituto de Biología, Universidad Nacional Autónoma de México (UNAM), Mexico City, Mexico
- 3Comisión Nacional Para el Conocimiento y Uso de la Biodiversidad (CONABIO), Insurgentes Sur-Periférico, Tlalpan Mexico City, Mexico
- 4Departamento de Zoología, Instituto de Biología, Universidad Nacional Autónoma de México (UNAM), Mexico City, Mexico
Mexico is the center of origin and genetic diversity of upland cotton (Gossypium hirsutum L.), the most important source of natural fiber in the world. Currently, wild and domesticated populations (including genetically modified [GM] varieties) occur in this country and gene flow among them has shaped the species’ genetic diversity and structure, setting a complex and challenging scenario for its conservation. Moreover, recent gene flow from GM cultivars to wild Mexican cotton populations has been reported since 2011. In situ conservation of G. hirsutum requires knowledge about the extent of its geographic distribution, both wild and domesticated, as well as the possible routes and mechanisms that contribute to gene flow between the members of the species wild-to-domesticated continuum (i.e., the primary gene pool). However, little is known about the distribution of feral populations that could facilitate gene flow by acting as bridges. In this study, we analyzed the potential distribution of feral cotton based on an ecological niche modeling approach and discussed its implications in the light of the distribution of wild and domesticated cotton. Then, we examined the processes that could be leading to the escape of seeds from the cultivated fields. Our results indicate that the climatic suitability of feral plants in the environmental and geographic space is broad and overlaps with areas of wild cotton habitat and crop fields, suggesting a region that could bridge cultivated cotton and its wild relatives by allowing gene flow between them. This study provides information for management efforts focused on the conservation of wild populations, native landraces, and non-GM domesticated cotton at its center of origin and genetic diversity.
Introduction
Crop ferality can play an important role in gene flow between crops and their wild relatives and introgression from artificially selected sources can have significant evolutionary consequences (Ashiq Rabbani et al., 1998; Berville et al., 2005; Snow and Campbell, 2005; Devaux et al., 2007; Gering et al., 2019). Furthermore, with the growing adoption of genetically modified (GM) crops worldwide, transgenic flow turned into a widely discussed topic and ferality an issue of concern amongst biosafety specialists. Feral plants are commonly known as cultivated taxa whose domestication syndrome has been partially or totally reverted (a process also known as atavism or de-domestication) (Gressel, 2005; Gering et al., 2019). However, recent research indicates that feralization is not simply a “reversal” from domestication, but rather a complex phenomenon that can involve several processes: adaptation to new habitats, novel selection pressures, admixture with wild relatives and other domesticated varieties, and even when feralization restores ancestral phenotypes, novel genetic mechanisms (Gering et al., 2019). Thus, as diverse and complex evolutionary histories shape ferality, three fundamental characteristics common to feral populations can be summarized: (1) they are composed of free-living organisms that are primarily descended from semi-domesticated ancestors that escaped cultivation; (2) they are able to reproduce successfully without intentional human intervention; and (3) they can establish and perpetuate themselves in natural or semi-natural habitats, not necessarily returning to truly “wild” habitats, but rather frequently occurring within disturbed settings (White et al., 2006; Bagavathiannan and van Acker, 2008; Gering et al., 2019). Given the above, feral populations are part of wild-to-domesticated systems that could experience gene flow among their members, especially in areas where they coexist. Gene flow represents an important mechanism for the spread and establishment of domesticated genetic material (including transgenes) into the wild, which has several conservation implications for crop wild relatives, as is the case of upland cotton (Gossypium hirsutum L.) in Mexico.
At its indigenous range (i.e., semi-arid tropics and subtropics of the Caribbean, northern South America, and Mesoamerica) (Brubaker and Wendel, 1994), upland cotton exists as a complex of wild-to-domesticated forms that belong to the primary gene pool of the species (Brubaker and Wendel, 1994; Andersson and de Vicente, 2010). Presently, in Mexico—its center of origin, diversity, and domestication (Ulloa et al., 2005; Burgeff et al., 2014; Mendoza et al., 2017)—cotton occurs as a continuum of cultivated and highly improved varieties, genetically modified varieties, traditionally managed landraces, feral, and wild populations. Predominantly, wild cotton populations are found in coastal habitats—as part of littoral vegetation or derived from it (Fryxell, 1979)—in scattered patches that conform to metapopulation dynamics (Hanski, 1998; Freckleton and Watkinson, 2002; Wegier et al., 2011). Particularly, the natural distribution of the latter has been widely discussed by several authors as a result of a debate regarding the “truly wild” status. Views on this subject range from describing a very restricted distribution of few remaining wild populations at the Yucatán Peninsula and the Caribbean (Coppens d’Eeckenbrugge and Lacape, 2014) to accounting for a broader distribution at coastal habitats along Pacific islands, the Caribbean, Mexican Pacific coasts, the Gulf of Mexico, and Baja California Sur (Fryxell, 1979; Wegier et al., 2011).
As a domesticate, upland cotton has a long history of human management and utilization. In Mexico, evidence indicates that cotton has been cultivated since pre-Hispanic times where it was probably grown in many of the coastal valleys along the Gulf coast in Veracruz, on the Pacific coast of Oaxaca, the coast of Sinaloa, and throughout the lowland Maya region at the Yucatán Peninsula (Mathiowetz, 2020). Moreover, growing areas extended inland toward regions that provided suitable warm temperatures and sufficient water through rainfall or irrigation (e.g., along river valleys), such as the states of Morelos and Oaxaca (Berdan, 1987; Charlton et al., 1991; Hironymous, 2007). Later, during the colonial period, cotton cultivation followed almost the same pattern (Hironymous, 2007); however, between the nineteenth and twentieth centuries, with the modernization of the textile industry, the production grew significantly and new cotton-growing regions emerged (Cerutti and Almaraz, 2013; Rocha-Munive et al., 2018), namely: Comarca Lagunera (Coahuila and Durango); Colorado River Delta and Mexicali valley (Baja California); the Ascensión area, Juárez valley, and the Meoqui region (Chihuahua); San Luis Río Colorado, Caborca, Hermosillo coast, and the valleys of Guaymas, Yaqui, and Mayo (Sonora); and Matamoros (Tamaulipas). Cotton cultivation peaked from 1935 to 1955 (e.g., from a quarter of a million bales collected in 1940, the harvest increased to more than two million in 1955) (Cerutti and Almaraz, 2013), but the large areas of monoculture enabled the emergence and rapid dispersal of pests, leading to a drastic decline due to the high costs associated to extensive applications of insecticides and the evolution of pest resistance. Consequently, since 1996, GM cotton expressing tolerance to herbicides and lepidopteran resistance has been sowed in northern Mexico in the same regions that were established during the nineteenth and twentieth centuries. Today, practically all the cultivated area depends on imported GM seed (Jiménez Martínez and Ayala Angulo, 2020).
Although cotton utilization in Mesoamerica can be traced back before the pre-Classic period (Smith and Stephens, 1971), G. hirsutum has been subjected to incomplete domestication; therefore, individuals can reproduce and persist without human intervention. The species’ semi-domestication coupled with its capacity for long-distance dispersal are the main factors that account for cotton ferality. As in the tribe Gossypieae, dispersal in G. hirsutum is virtually synonymous with seed dispersal (Fryxell, 1979). Several mechanisms are involved in this capacity and are tightly associated with the species’ evolutionary history. For instance, the origin of tetraploid cottons –the phylogenetic group to which G. hirsutum belongs (i.e., G. barbadense, G. tomentosum, G. darwinii, G. mustelinum, and G. hirsutum)- resulted from the union of genomes A and D of the eight diploid genomes described for the genus (i.e., genomes A to G, and K). Although sharing an ancestral African origin, Genomes A and D diverged separately for millions of years –the former in Africa and the latter in America– until a second transoceanic migration led to their allopolyploidization in America. The latter event allowed for the propagation in littoral habitats that differ from the inland arid environments where diploid cottons occur (Fryxell, 1979). Moreover, its dense hairy seeds –a trademark of the Gossypieae and the target trait to cotton domestication – have a central role in natural mechanisms of seed dispersal by: (1) enabling floatation for water dispersal; (2) facilitating wind dispersal; and (3) being an attractive material to nest-building birds (Fryxell, 1979; Arteaga Rojas, 2021). Additionally, activities related to cotton production and utilization involve transporting domesticated seeds through long distances, from the cultivation fields to temporary storage facilities, cotton gins, distribution centers, and final locations (Wegier, 2013). This dynamic has taken place since pre-Columbian times when communities imported the raw materials to spun, wave, and prepare textiles that were used locally or further moved to be offered as tribute (Berdan, 1987).
With such dispersal capabilities in mind, gene flow among G. hirsutum primary gene pool members is feasible, even between distant populations. Wegier et al. (2011) described patterns of historical long-distance gene flow among wild metapopulations in Mexico and demonstrated that recent gene flow, followed by introgressive hybridization, occurred between wild populations and commercial cotton cultivars, through the detection of transgene expression at the former. Considering that cotton volunteers and feral populations usually reach reproductive maturity, produce fruits with seeds, and are very common in areas where cottonseed is supplied as livestock feed (Andersson and de Vicente, 2010), they could also contribute actively to gene flow within the primary gene pool. Cotton ferality can be limited by unintentional factors that hinder plant germination, growth, and reproduction, such as the relative drought of non-managed habitats, roadside weed removal, or cattle grazing. However, if suitable conditions prevail, feral cotton populations can persist for decades, as has happened in several tropical regions such as northern Australia, Vietnam, southern United States, Hawaii, Brazil, and Mexico (Hilbeck et al., 2004; Hawkins et al., 2005; Andersson and de Vicente, 2010).
Given that Mexico is the center of origin and genetic diversity of G. hirsutum and several other important crops to humankind, specific regulatory instruments safeguard its genetic and morphological diversity (i.e., the total gene pool of the crop, including wild relatives, landraces, or varieties), as well as the areas in which they occur, such as the Law of Biosafety of Genetically Modified Organisms—LBOGM (Diario Oficial de la Federación [DOF], 2005), LBOGM regulation (DOF, 2008), and the Cartagena Protocol on Biosafety to the Convention on Biological Diversity (Secretariat of the Convention on Biological Diversity, 2000). Moreover, the IUCN Red List of Threatened Species and the Mexican Official Standard NOM-059-SEMARNAT-2010 (a normative instrument that identifies species or populations at risk) list G. hirsutum as a vulnerable species or subjected to special protection, respectively (DOF, 2019; Wegier et al., 2019). Considering the legal background described above, the release of GM cotton in Mexico must be preceded by environmental risk assessments designed to identify potential adverse effects and estimate the level of risk associated with them. Gene flow to native cotton populations is one of the main concerns and should always be evaluated on a case-by-case basis. Key aspects in this regard are the geographical distribution of wild and highly improved crops, their potential to escape cultivation and establish elsewhere, and the dispersal mechanisms that would allow them to reach these areas. In this study, we aim to describe the potential distribution of feral cotton through an ecological niche modeling approach and discuss its relation to the distribution of wild and domesticated cotton. To that end, we have reviewed and gathered an extensive dataset of occurrences of the three groups, modeled their ecological niches, and assessed their differences and overlap to evaluate how feral cotton may be accounting for gene flow between crops and wild relatives. We hope that our findings can encourage a necessary discussion on the management and conservation of G. hirsutum in the complex and dynamic scenario in which its wild-to-domesticated forms coexist.
Materials and Methods
Occurrence Data
We collated occurrence records for G. hirsutum wild, feral, and highly domesticated populations across their geographic range in Mexico. We examined, gathered, and cleaned the data from the following sources: (1) Wild records, through direct observation during several surveys across the Mexican coastal dunes performed annually over the last 17 years (Wegier, 2007). Populations were considered wild if they were found in the expected littoral habitat and the growth form conformed to a perennial shrub or tree (Fryxell, 1979; Wegier et al., 2011). In addition, we carefully revised plant collections (i.e., XAL, MEXU) to retrieve inaccessible but relevant localities (e.g., Revillagigedo or Tamaulipas) considering geographic and taxonomic accuracy. (2) We registered feral cotton occurrences from field observations and the National Biodiversity Information System (SNIB, for its acronym in Spanish) (CONABIO, 2020). Records were considered feral if they occurred outside cultivation, without apparent human management (i.e., agricultural land use), and away from the habitat described for wild cotton (Gering et al., 2019). Additionally, to obtain records of established populations, we only considered localities reported by two or more authors and/or in different years. (3) In Mexico, practically all the area cultivated with cotton is GM. Therefore, we inferred occurrences for commercial cultivars (i.e., highly domesticated varieties that hereafter we will refer to as “domesticated”) by calculating the centroids of the polygons requested in the GM-cotton release applications submitted from 1995 to 2016 (Burgeff et al., 2014; SIOVM, 2020). After cleaning, we thinned all datasets following a distance-based approach (excluded duplicated records within a grid of 5 × 5 Km) to avoid spatial correlation with the NTBOX package version 0.4.6.1 (Osorio-Olvera et al., 2020) in R version 3.5.0 (R Core Team, 2017). Finally, for modeling and evaluation purposes, we split thinned databases into two sets (i.e., training and testing) of nearly identical size following the checkerboard partition method implemented in the package ENMeval version 0.3.0 (Muscarella et al., 2018). We show the localities used in this study in Figure 1 (wild: n = 175; feral: n = 120; domesticated: n = 278) and in an interactive map at http://rpubs.com/valav/712873.
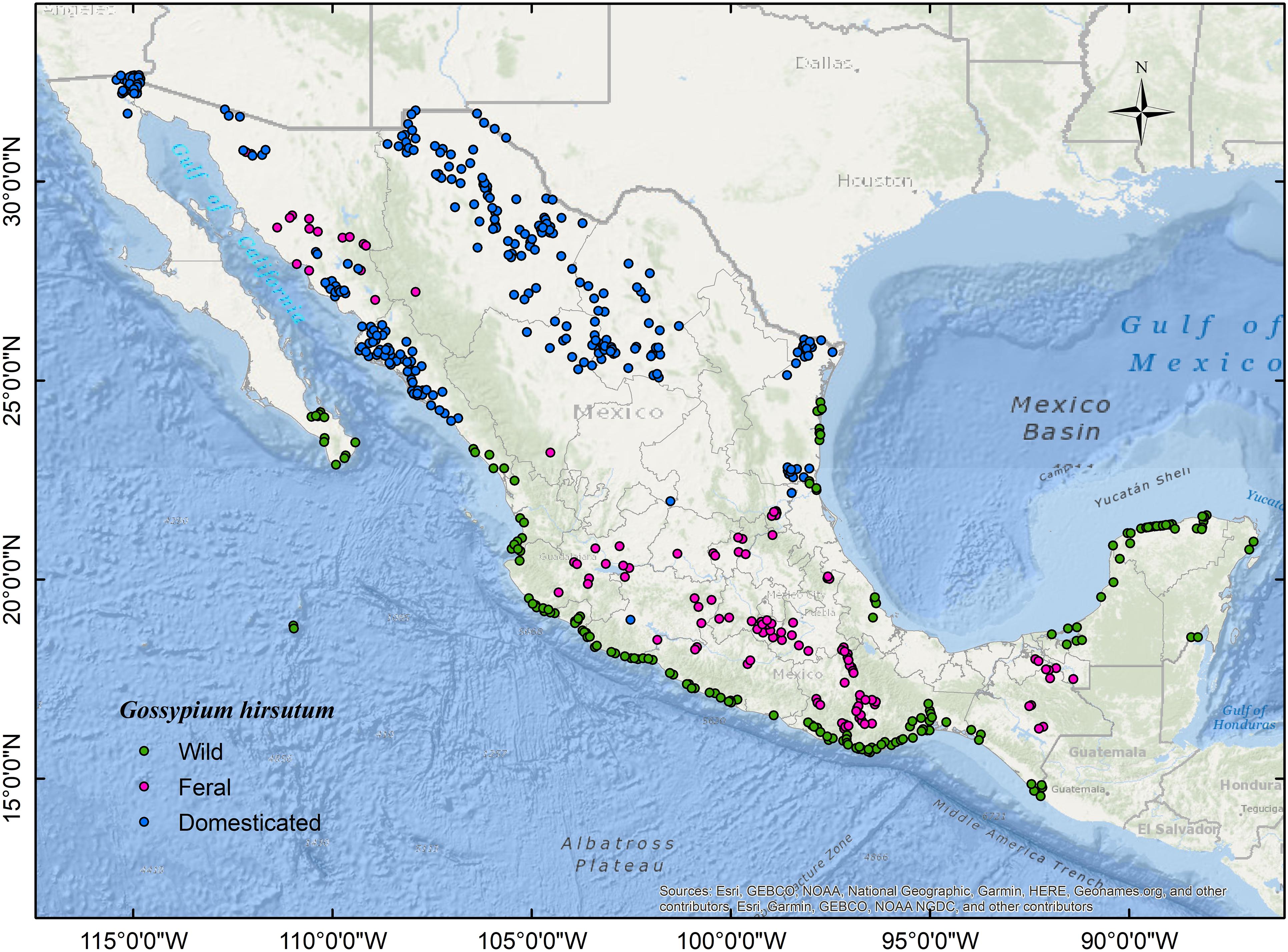
Figure 1. Map of the study area showing occurrence data: wild cotton (n = 175; green), feral cotton (n = 120; magenta), and highly domesticated cultivars, hereafter referred to as “domesticated” (n = 278; blue).
Climatic Variables
The environmental layers used in this study correspond to bioclimatic variables summarizing annual, seasonal, and extreme tendencies derived from monthly temperature and precipitation data for Mexico between 1910 and 2009 at 30” resolution (∼1 km) (Table 1; Cuervo-Robayo et al., 2014). We excluded four variables (i.e., mean temperature of the most humid quarter, mean temperature of the least humid quarter, precipitation of the warmest quarter, and precipitation of the coldest quarter) due to artifacts resulting from the combination of temperature and precipitation data (Escobar et al., 2014). To avoid model overfitting due to multicollinearity among variables, we selected a sub-set of uncorrelated and biologically meaningful variables for each cotton group. For this, we first assessed variables’ contribution in exploratory runs of a Maxent model (Phillips et al., 2006; Supplementary Table 1) by measuring the variable contribution percentage, permutation importance, and model gain through jackknife tests for each cotton group. Then, we calculated pairwise Pearson’s correlation coefficients from the occurrences of the three cotton groups. From these analyses, we excluded highly correlated variables (| r | > 0.85) and retained important variables with the greatest biological relevance for further modeling (Wegier, 2013; Simoes et al., 2020). For wild cotton, we used six variables: annual mean temperature (Bio1), mean diurnal range (Bio2), temperature seasonality (Bio4), maximum temperature of the warmest month (Bio5), temperature annual range (Bio7), and precipitation of the driest quarter (Bio17). For feral cotton, five: isothermality (Bio3), temperature seasonality (Bio4), minimum temperature of the coldest month (Bio6), precipitation of the driest month (Bio14), and precipitation seasonality (Bio15). And also five for the domesticated group: annual mean temperature (Bio1), isothermality (Bio3), temperature seasonality (Bio4), annual precipitation (Bio12), and precipitation of the driest month (Bio14).
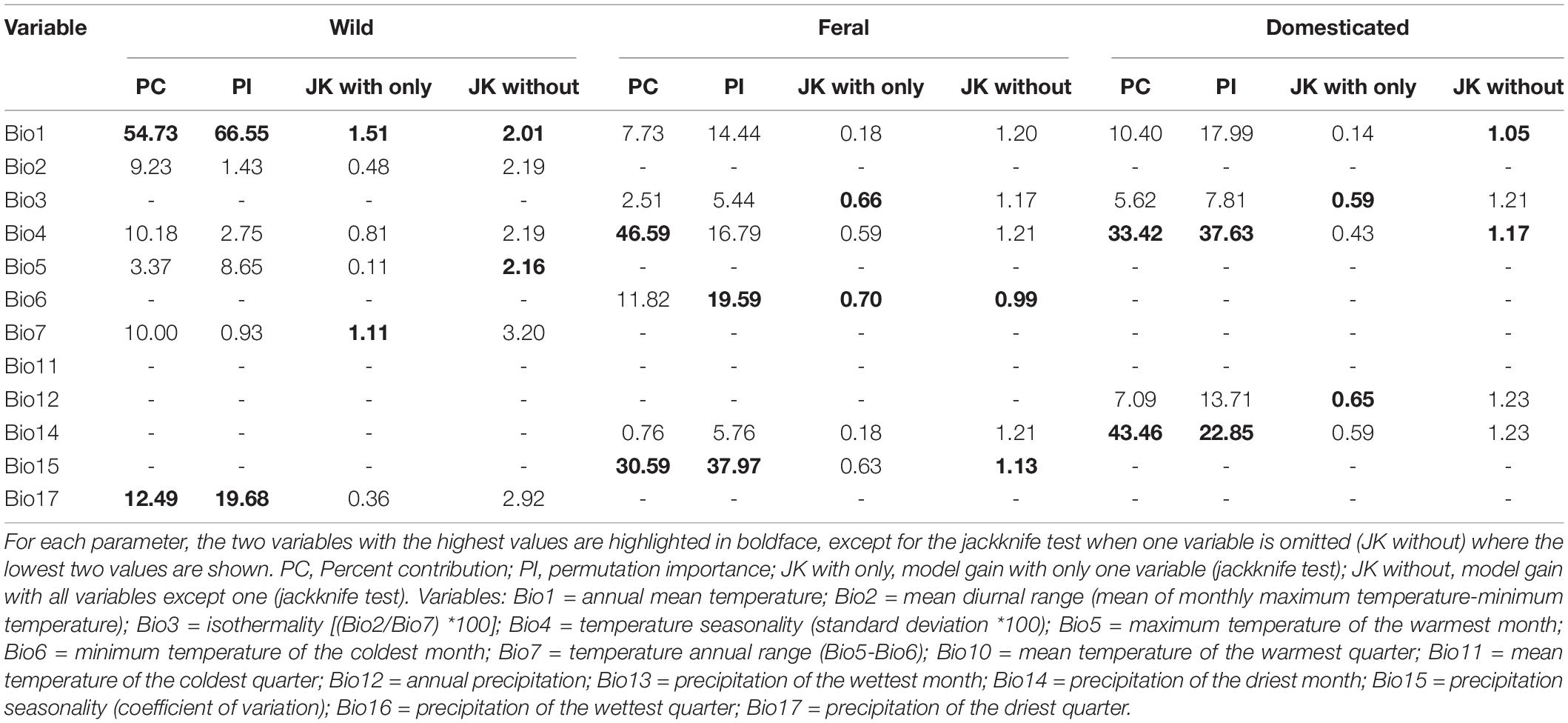
Table 1. Importance of variables to model construction.
The calibration area, or the “M” element of the BAM diagram, refers to areas that have been accessible to the species via dispersal over relevant time periods (Barve et al., 2011; Peterson and Soberón, 2012). Given G. hirsutum long-distance dispersal features, both historical and recent, we considered the geographic extent of Mexico as the calibration area. This approach allows to predict areas that wild, feral, and domesticated cottons could potentially occupy within this range and allow comparisons among these groups.
Ecological Niche Modeling
We modeled the ecological niche of wild, feral, and domesticated cotton using the Maxent algorithm version 3.3.3k (Phillips et al., 2006). Maxent is a machine-learning algorithm that uses the maximum entropy principle to identify a target probability distribution subject to a set of constraints related to the environmental characteristics of occurrences and a sample of the calibration area (Phillips et al., 2004, 2006). To select an optimal combination of Maxent parameters [regularization multipliers (RM) and feature classes (FC), see below], we developed a series of candidate models with the R package ENMeval (Muscarella et al., 2018); thus, we assessed five FC combinations: L, LQ, LQH, LQHP, and LQHPT (where, L: linear; Q: quadratic; H: hinge; P: product, and T: threshold) and tested RM values ranging from 0.5 to 4 in increments of 0.5. We selected the best combination of model parameters according to performance and complexity criteria, namely: area under the ROC curve (AUC) > 0.9, omission rates < 0.10, and the Akaike information criterion (AICc) where delta AIC ≤ 2. Then, models with specific RM and FC settings for each cotton group were run in Maxent (i.e., 10-fold cross-validation models, 20,000 background points, and 1,000 maximum iterations). We selected the average Cloglog output for environmental continuous suitability visualization and reclassified it into a binary map (i.e., presence or absence of suitable conditions) in ArcGIS version 10.2.1 (ESRI, 2015) by applying a threshold value balancing a low omission error and the proportional predicted area. Finally, to assess model performance and significance, we evaluated the AUC ratio of the partial receiver operating characteristic curve (pROC; 1,000 replicates, and E = 0.05), calculated the omission rate, and performed binomial tests with the testing datasets obtained previously (see the occurrence data section above) in NTBOX (Osorio-Olvera et al., 2020).
Niche Overlap, Similarity and Equivalence in the Environmental Space
In order to assess how feral cotton may be accounting for gene flow between crops and wild relatives, we performed niche overlap, similarity, and equivalency analyses using the package “ecospat” (Broennimann et al., 2012; Di Cola et al., 2017) in R version 3.5.0 (R Core Team, 2017). These methods are based on an ordination approach (i.e., PCA) to estimate the occurrence and climatic factor densities along environmental axes (PCA-env) and calculate niche overlap with them. We evaluated niche overlap with the Schoener’s D metric and Hellinger’s I distance (Broennimann et al., 2012), both ranging from 0 (no overlap) to 1 (complete overlap). In addition, we evaluated niche conservation or niche divergence hypotheses through niche equivalence and niche similarity tests (100 permutations for each analyses) and assessed the statistical significance of the measured niche overlap against null model niches taken randomly from the background area. The equivalence analysis tests if the ecological niches are identical (interchangeable): if the estimated niche overlap value falls below the 95% confidence interval of the null model, niche equivalence is rejected. Niche similarity test compares the niche overlap of one range in a randomly drawn background, while keeping the other unchanged. In our study, this process was repeated in either direction (1 ↔ 2): values above or below the 95% confidence interval of the null model support niche conservatism or niche divergence, respectively.
Niche Optimum and Breadth
Additionally, we performed a post hoc test to assess differences in central tendency and statistical dispersion among ecological niches of the three cotton groups when equivalency or similarity analyses were inconclusive (Molina-Henao and Hopkins, 2019). Specifically, we evaluated differences in niche optimum and breadth following the bootstrap resampling approach described by Molina-Henao and Hopkins (2019), as follows: we calculated niche optimum and breadth values for each cotton group as the median and length of the 95% inter-percentile interval along the first two principal components (PCA-env), respectively. Then, we estimated the difference in medians and inter-percentile interval lengths between each group. Afterward, we created a null distribution of differences in optima and breadths for each comparison by pooling the observed presence data and resampling them at random with replacement into two new sets of the same size as the original samples. From these resampled sets, we calculated the median and length of the 95% inter-percentile interval and estimated pairwise differences in optima and breadths from the two sets, generating a null model of differences with these results. We repeated this process 1,000 times for each comparison (i.e., wild-feral, wild-domesticated, and feral-domesticated). If the observed test statistic (i.e., difference in optima and difference in breadths) was higher than the 95% confidence interval of the null models, the null hypothesis was rejected, meaning that niche optimum or breadth were significantly different.
K-Means Clustering
To further assess if wild, feral, and domesticated cotton could be partitioned into different environmental groups, we evaluated the clustering tendencies of the dataset. First, to observe if the described cotton groups also congregated and conformed to corresponding groups in the environmental space, we constructed pairwise bivariate distributions (pair-plots), univariate density distributions in the “Seaborn” package v.0.11.0 (Waskom et al., 2020) in Python 3.4.8 (van Rossum and Drake, 2009), and a principal component analysis with the “FactoMineR” package in R (Husson et al., 2013). We confirmed the clustering tendency by estimating the Hopkins statistic (H) with the “clustertend” package (YiLan and RuTong, 2015) in R. The Hopkins statistic tests the spatial randomness of the data by measuring the probability that a given dataset is generated by a uniform data distribution (i.e., H = 0.5). Thus, if the Hopkins statistic is close to 0, the null hypothesis is rejected, meaning that the data are significantly clusterable (Kassambara, 2017). Afterward, we conducted clustering analyses through the k-means unsupervised algorithm with the “factoextra” package (Kassambara and Mundt, 2017) in R. This method makes inferences from empirical data without previously referring to known labels by grouping within the same cluster objects that are as similar as possible. Briefly, the algorithm randomly selects k objects from the dataset defined as centroids or cluster means; then, the remaining objects are assigned to their closest centroid based on their Euclidean distance from the mean, and a new mean is calculated for each cluster. As centroids are recalculated, observations are reassigned iteratively until convergence (i.e., cluster assignments stop changing). We analyzed four datasets: (1) wild + feral; (2) wild + domesticated; (3) feral + domesticated; and (4) the three groups together. The number of clusters (k) to be generated must be specified before the analyses, so we set k as the expected number of clusters according to the known structure of each dataset (i.e., k = 2 or 3) to assess the agreement between the k-means clusters and our data. To evaluate the goodness-of-fit of our clustering results, we estimated the Silhouette Coefficients (Si) and the corrected Rand Index (CRI) for internal and external validation, respectively, with the “fpc” package (Hennig, 2007) in R. Particularly, the silhouette analysis measures how well an observation is clustered by estimating the average distance between clusters: if Si is close to 1, objects are very well clustered; if Si is close to 0, objects lie between two clusters; and if Si is negative, objects are probably placed in the wrong cluster. On the other hand, the CRI quantifies the agreement between the clustering results and an external reference, in this case, cotton classes, ranging from −1 (no agreement) to 1 (perfect agreement) (Kassambara, 2017).
Results
Ecological Niche Model Predictions
We generated 40 candidate models for each cotton group and selected the parameter combinations that obtained the best performance and complexity estimates (i.e., wild: FC = LQHP, RM = 4; feral: FC = LQ, RM = 0.5; domesticated: FC = LQHP, RM = 3; Supplementary Materials 2). With these settings, all niche models were statistically significant (p < 0.0001∗∗∗) according to both significance evaluations (binomial tests and partial ROC analysis; AUC ratios: wild = 1.84; feral = 1.48; domesticated = 1.65). Wild and domesticated cotton models exhibited good predictive performance (omission error: wild = 0.06; domesticated = 0.078), while the feral model showed a higher omission error (i.e., 0.185). Model outputs indicate differences in climatic suitability and geographic distributions among the three G. hirsutum groups: wild cotton toward the coasts of the Pacific, southern Baja California Sur, and the Yucatán Peninsula, as well as patches along the Gulf of Mexico and Isla Socorro; domesticated cotton with broad suitability in northern Mexico; and feral cotton showing a wide northwest-southeast pattern, predominantly across inland regions (Figures 2, 3).
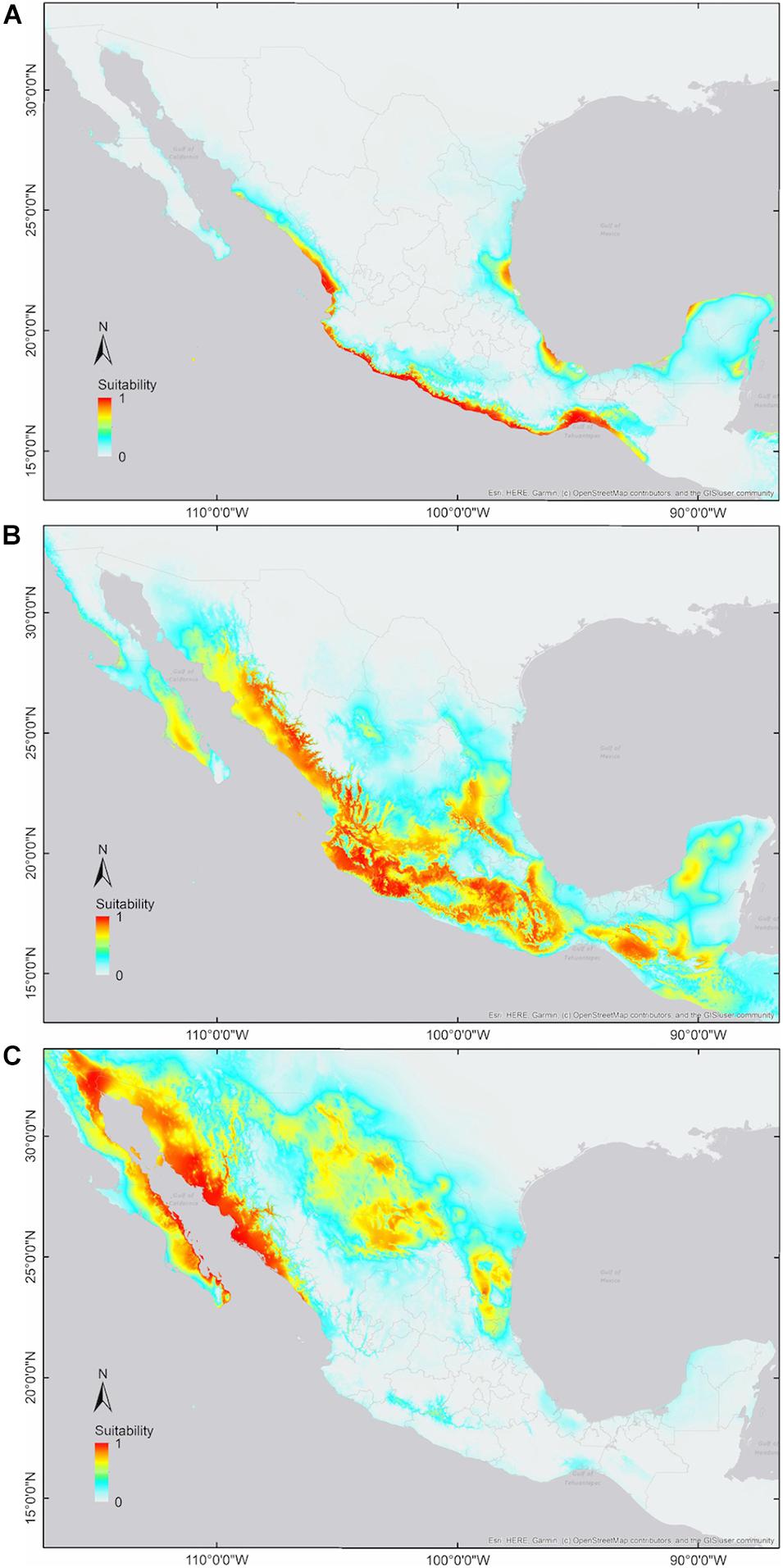
Figure 2. Predicted environmental suitability (continuous output) for members of G. hirsutum wild-to-domesticated complex. (A) Wild cotton; (B) feral cotton; and (C) domesticated cotton. Color range: High suitability (red-orange), medium suitability (yellow-pale green), low suitability (turquoise), and no suitability (no color).
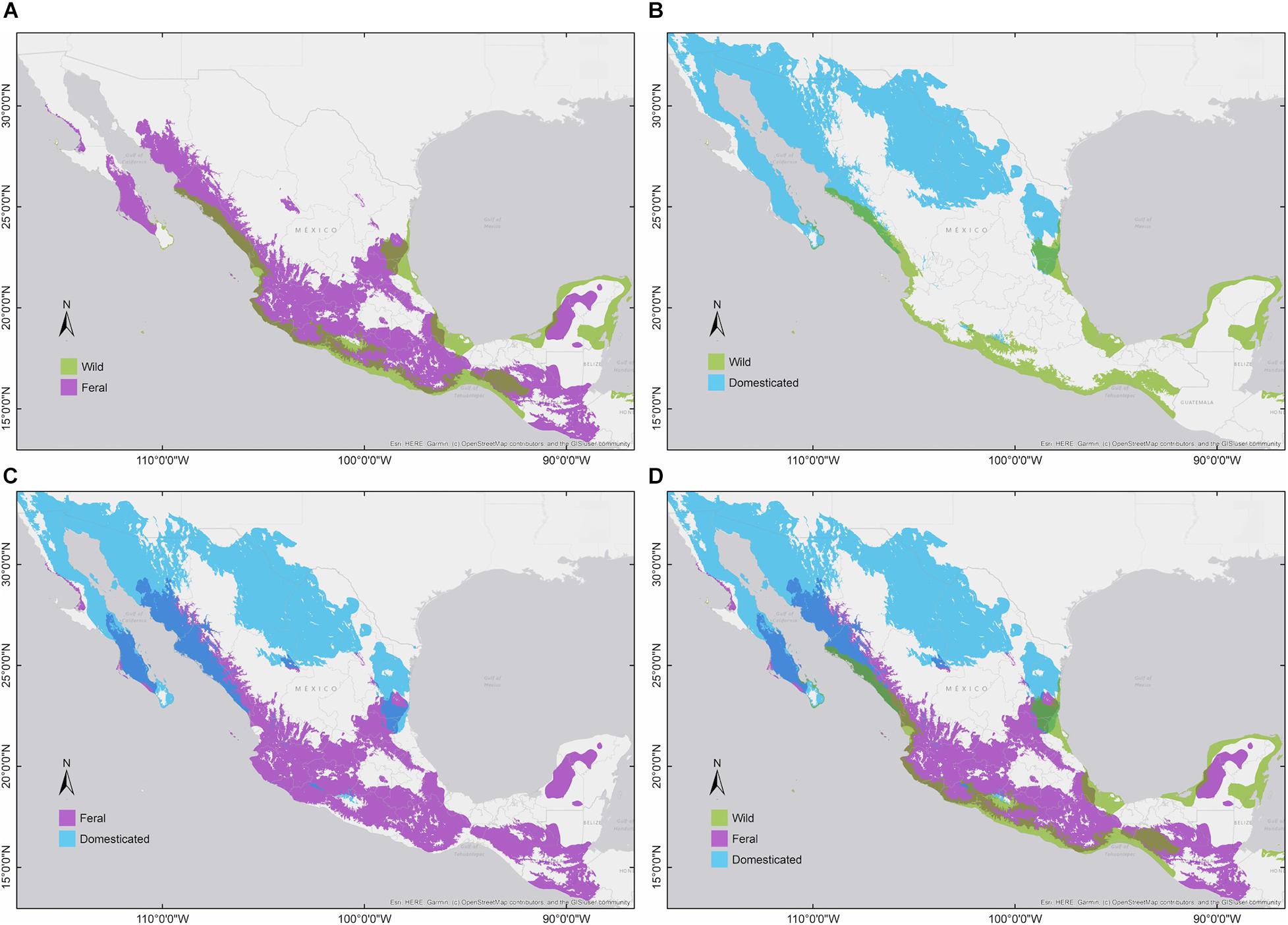
Figure 3. Binary environmental suitability models comparing the extent of the potential distribution of members of G. hirsutum wild-to-domesticated complex (colors indicate suitability: wild = green, feral = magenta, and domesticated = blue). (A) Wild vs. feral; (B) wild vs. domesticated; (C) feral vs. domesticated; and (D) the three cotton groups.
The most important variables for the three cotton models were: (1) Wild: annual mean temperature (Bio1); (2) Feral: temperature seasonality (Bio4), minimum temperature of coldest month (Bio6), and precipitation seasonality (Bio15); and (3) Domesticated: annual mean temperature (Bio1), temperature seasonality (Bio4), annual precipitation (Bio12), and precipitation of driest month (Bio14) (Table 1).
Niche Overlap
The estimates of niche overlap in the environmental space indicate that the three cotton groups overlap to some extent (Table 2). Specifically, feral cotton has a wider overlap with both wild (Schoener’s D = 0.29; Hellinger’s I = 0.54) and domesticated cotton (Schoener’s D = 0.27; Hellinger’s I = 0.46), than the latter pair. However, although wild and domesticated cotton show lower overlap values (Schoener’s D = 0.11; Hellinger’s I = 0.19), their niches slightly share an environmental space proportion. In addition, the results from the equivalency tests suggest that wild, feral, and domesticated niches are not equivalent as the overlap estimates fall below the 95% confidence interval of the null hypothesis and significantly support the alternative hypothesis of niche divergence (Table 3 and Supplementary Figure 1). Similarity tests were not significant for niche conservatism nor niche divergence (Table 3), indicating that niches among cotton groups are not more similar or dissimilar than expected by chance. This indicates that the niches are not significantly conserved and they may differ in measures of central tendency, statistical dispersion, or both; thus, we conducted post hoc analyses comparing niche optimum and breadth (Molina-Henao and Hopkins, 2019).

Table 2. Niche overlap indices.

Table 3. Similarity and equivalence tests p-values for niche conservatism (Co) and divergence (Di) hypotheses between wild (W), feral (F), and domesticated (D) cotton.
Niche Optimum and Breadth
Analyses of niche optimum and breadth show that wild, feral, and domesticated cotton significantly differ in optimum along both PCA axes; therefore, central tendencies of PC values are dissimilar among cotton groups. Conversely, breadth along the first axis is not statistically different for any of the groups, while along the second axis, feral cotton breadth shows a broader inter-percentile range that significantly differs from the other two (Figure 4, Table 4, and Supplementary Figures 2, 3).
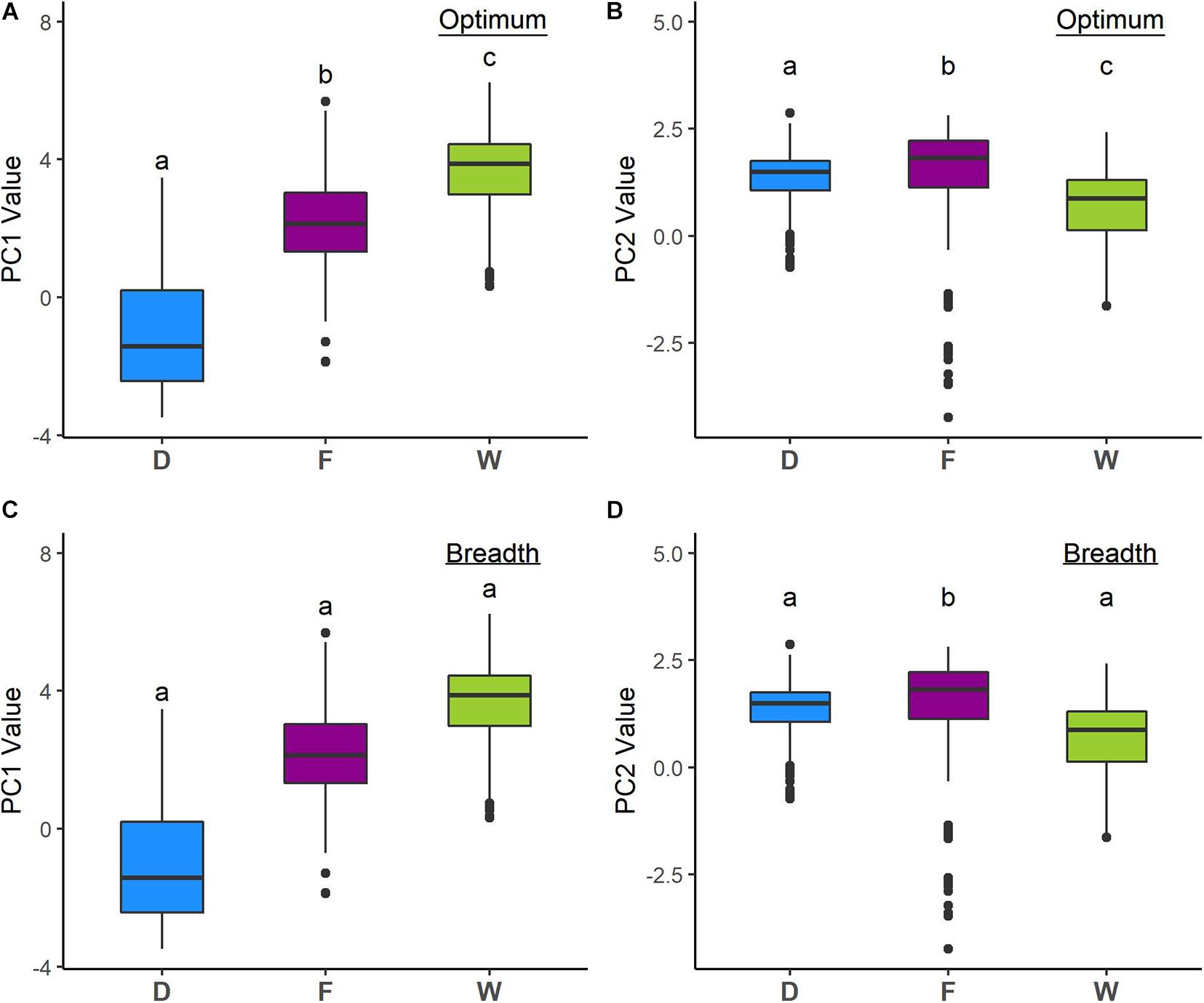
Figure 4. Niche optimum and breadth of the two first principal components (PCA-env). Letters depict statistically significant differences according to bootstrap tests (see Table 4). Optimum corresponds to the median and breadth to the length of the 95% inter-percentile interval of the PCA values along the first two PC axes. (A) PC1 optimum, (B) PC2 optimum, (C) PC1 breadth, and (D) PC2 breadth.
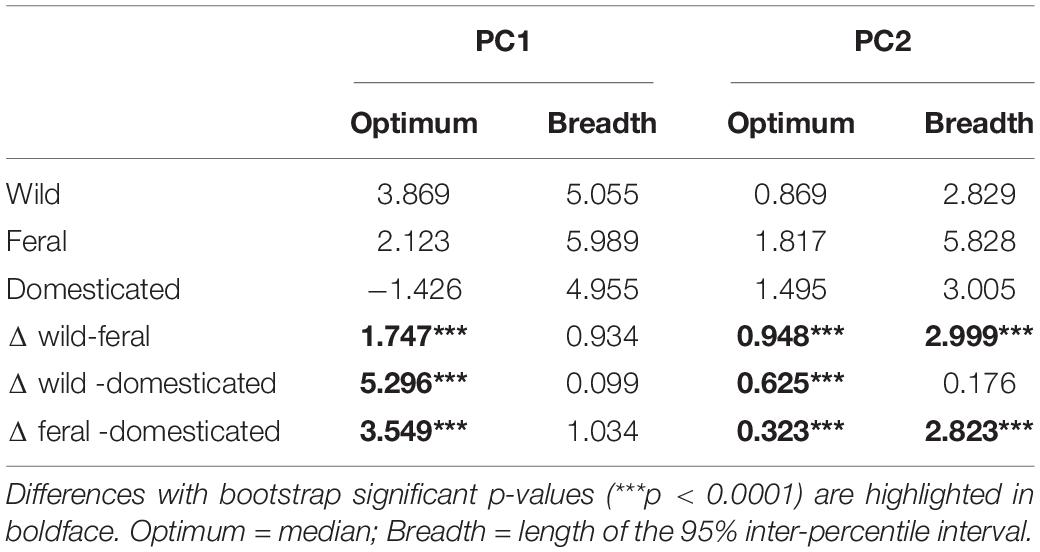
Table 4. Niche optimum, breadth, and differences (Δ) in both parameters among cotton groups.
Clustering
The cotton groups showed clustering tendencies in the pairwise comparison of environmental variables (Supplementary Figure 4) and PCA biplots (Figure 5). For the three cotton groups, the first two components explained 71.2% of the variation in cotton occurrence within the climatic space. PC1 described 53.6% of the variance: Bio7, Bio6, Bio4, Bio11, Bio13, Bio12, Bio16, Bio2, and Bio1 were the most important variables explaining variation on this axis (in decreasing order) with higher contribution values than expected if all variables contributed uniformly. PC2 described 17.6% of the variance with Bio17, Bio14, Bio10, Bio5, Bio15, and Bio1 contributing the most. In both axes temperature and precipitation explained variability; however, in PC1, temperature variables accounted for the higher contributions, while along PC2, precipitation variables did (Supplementary Figure 5). Interestingly, wild, feral, and cultivated cottons formed corresponding groups along the first axis, although not completely separated (Figure 5), with feral cottons laying between the more distant wild and domesticated cottons (Figure 5D).
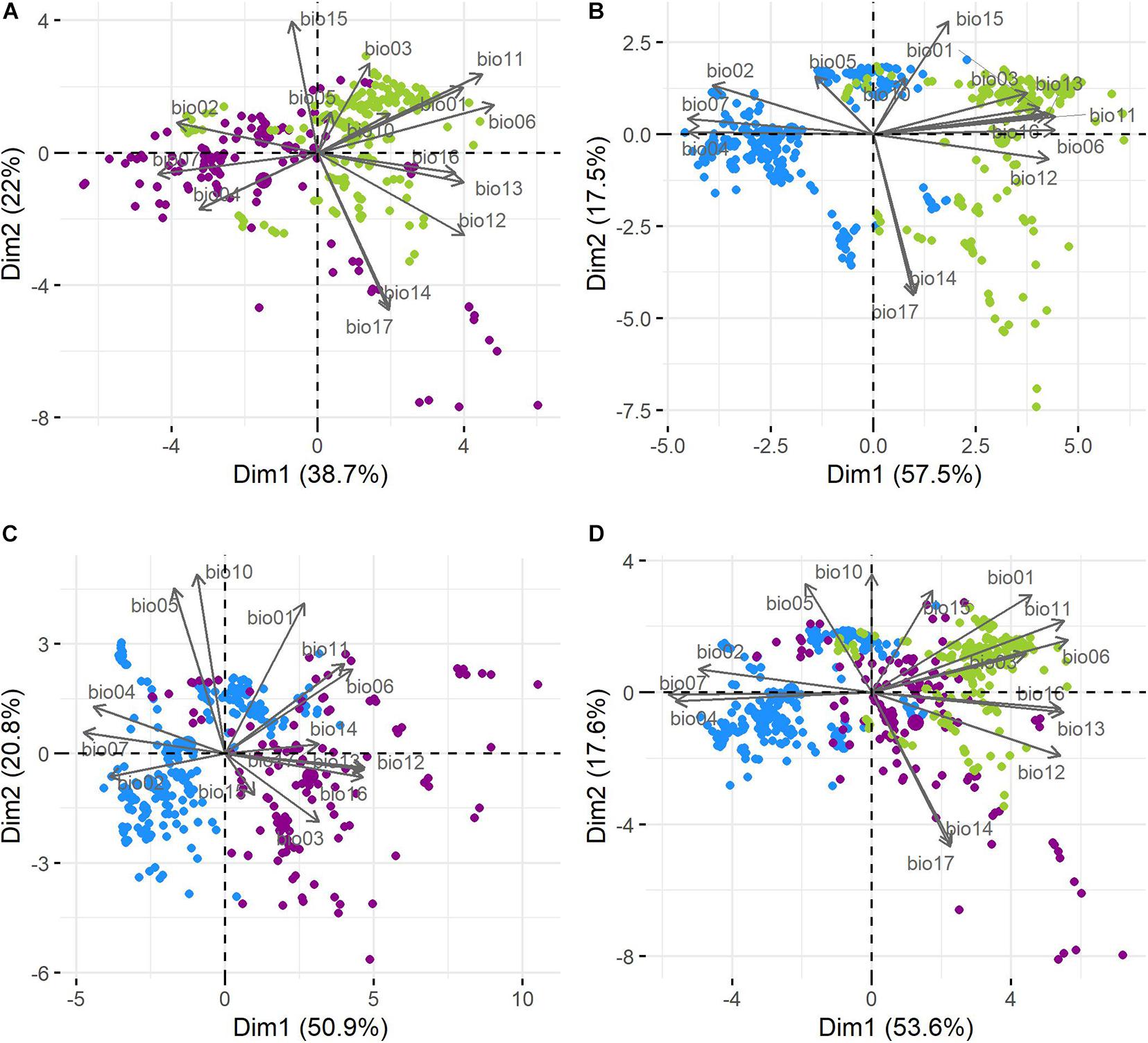
Figure 5. Principal component analysis (biplots). (A) Wild and feral; (B) wild and domesticated; (C) feral and domesticated; and (D) the three cotton groups. Wild = green, feral = magenta, and domesticated = blue.
These observations were supported by Hopkins index values close to 0 for all pairwise comparisons as well as for the whole dataset. Pairwise k-means analysis resulted in clusters that retrieved the expected groups with acceptable agreement (Table 5 and Figure 6), with the wild-domesticated cotton comparison obtaining the highest CRI value (0.736). The analysis with the whole dataset obtained a good CRI (0.474), mainly because the two larger clusters recovered most wild and domesticated groups as expected; however, feral cottons divided among the three resulting clusters, failing to retrieve most of its members in a predominant cluster (Table 5 and Figure 6).
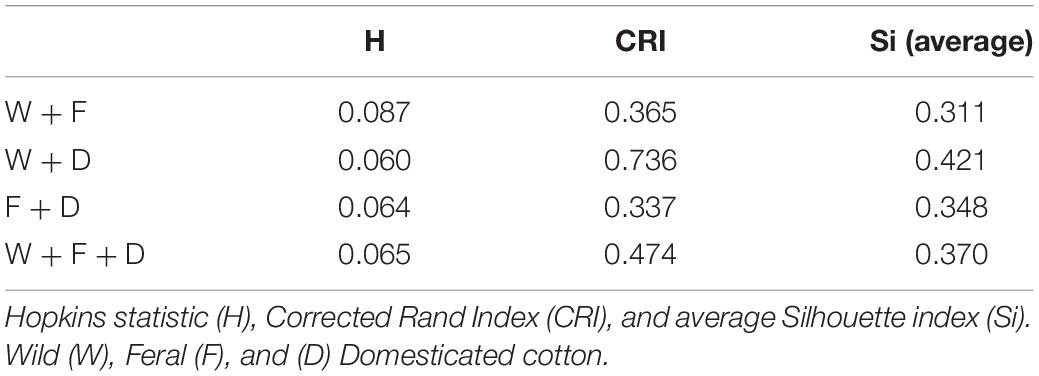
Table 5. Cluster tendency and validation indices for all datasets.
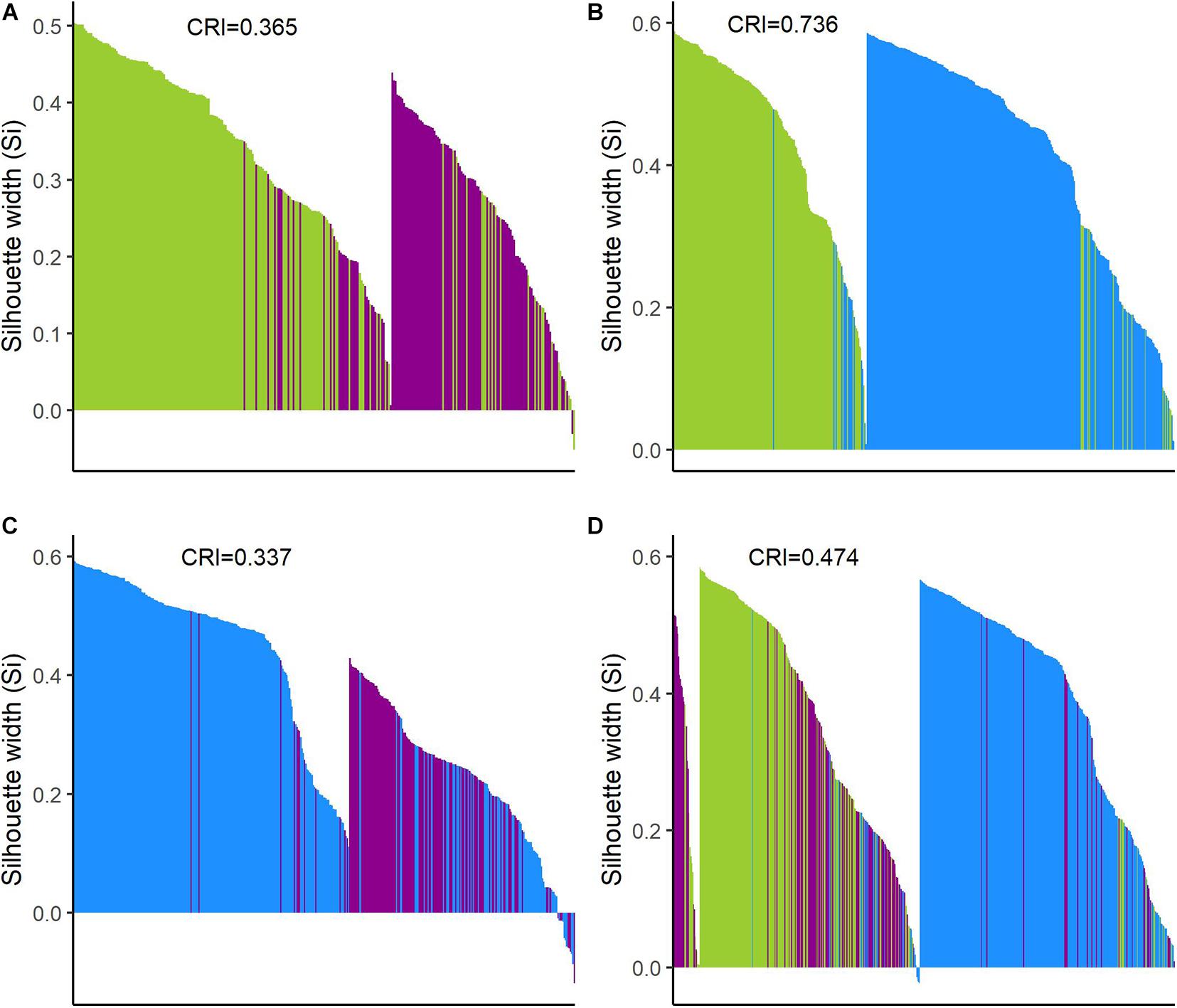
Figure 6. Silhouette plots of k-means clustering analyses. Plots show the Silhouette coefficient (Si) of each observation (bar). Si close to 1: objects are very well clustered; Si close to 0: objects lie between two clusters; and negative Si: objects are probably placed in the wrong cluster. (A) Wild and feral; (B) wild and domesticated; (C) feral and domesticated; and (D) the three cotton groups. Wild = green, feral = magenta, and domesticated = blue.
The Silhouette plots (Figure 6) depict the assignment of each individual to a cluster as well as its Silhouette index. For pairwise analyses, the resulting clusters predominantly recovered the expected groups with some inaccurate assignments; again, the wild-domesticated analysis resulted in the better resolved clusters according to their expected identity (Figures 6B, 7B), while in wild-feral and domesticated-feral clusterings, the predominantly feral cluster showed more inexact assignments (Figures 6A,C, 7A,C). The Silhouette plot with the whole dataset shows a variable degree of cluster resolution (Figures 6D, 7D): a predominantly domesticated cluster with few inexact assignments, a predominantly wild cluster with more inexact assignments than the latter (mostly feral), and a small feral-wild cluster with the highest Silhouette indices belonging to feral individuals. In all analyses, Si values indicate that several points are between clusters meaning that although the clustering tendency exists, some members of the resulting groupings are proximate to each other, as can be seen in the PCA and the pair-plots.
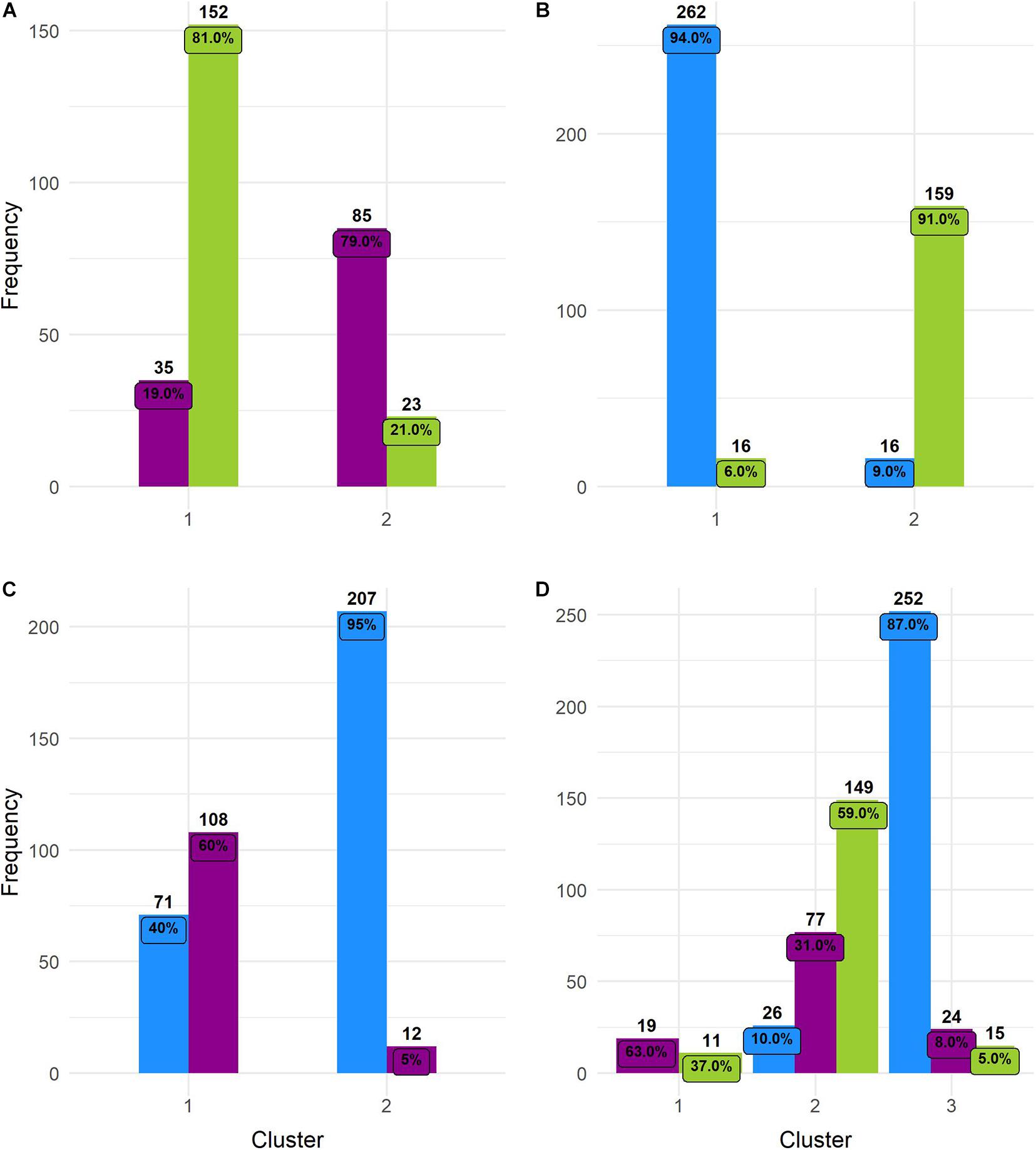
Figure 7. Histograms of k-means clustering results. Numbers above bars = total number of individuals grouped in a particular cluster. Numbers inside bars = percentage. (A) Wild and feral; (B) wild and domesticated; (C) feral and domesticated; and (D) the three cotton groups. Wild = green, feral = magenta, and domesticated = blue.
Discussion
Ecological Niches of the Wild-to-Domesticated Cotton Continuum
Our results show substantial differences in the ecological niches of members of the wild-to-domesticated cotton complex; however, they also highlight that wild, feral, and domesticated cotton niches overlap in the environmental and geographic space. For wild and cultivated cotton, the resulting potential distributions agree with their known occurrences, the former at litoral habitats in the Pacific and Atlantic coasts of Mexico, and the latter at the north of the country, which is consistent with the low omission rates obtained for these models. Moreover, as we gathered a very complete dataset of highly domesticated cotton localities, its potential distribution is wider than previously reported in other studies (Rocha-Munive et al., 2018). Feral potential distribution is broad and extends through northwest-southeast Mexico, thus bridging wild and cultivated distributions, and almost creating a continuum among the three groups along the country.
Pairwise niche equivalence tests suggest that the niches of wild, feral, and domesticated cotton are less equivalent than expected by chance, significantly rejecting these niches are identical. Specifically, no significant p-values were found for the hypothesis of niche conservatism (i.e., complete overlap) and highly significant results were obtained for the divergence hypothesis for any pair of comparisons (Table 3). Indeed, MaxEnt results illustrate that the suitabilities and potential distribution of the three cotton groups are very different (Figures 2, 3) and, overall, temperature is an important factor in determining differences among groups, specifically with regard to tolerance to low temperatures: domesticated cottons exhibit the highest tolerance, although cold temperature affects yield (Constable and Bange, 2015), and feral and wild cottons show intermediate and low tolerance, respectively (Supplementary Figure 4, see distribution densities for Bio1, Bio6, and Bio11; Figure 8 see response curves for Bio1). However, the three groups also exhibit overlap in several areas: wild and domesticated cotton in southern Baja California Sur, Sinaloa, and Tamaulipas (Figure 3B); wild and feral along the Pacific and Atlantic coasts (Figure 3A); and, feral and domesticated in northern Baja California Sur, Sinaloa, Sonora, and Tamaulipas (Figure 3C). Moreover, similarity tests indicated that observed values are not significantly different than the background and although niche optimum analyses revealed significant differences in the central tendency (median) of each group –supporting the conclusion that niches are not identical-, niche breadths were not statistically different in PC1, suggesting that the dispersion of the data is similar in the three groups and may account for the observed overlap and non-significant results of the similarity test. Clustering analyses resulted in comparable findings. The k-means algorithm retrieved the expected groups with an acceptable agreement, supporting correlation within groups and differences between them, especially in pairwise analyses. However, clusters were not fully separated and some occurrences laid between groups, resulting in inaccurate assignments, particularly when the whole dataset was analyzed. In all cases, wrong designations were more frequent in the predominantly feral cluster (Figure 6) but, as our results have shown, this group lays between the wild and domesticated groups (Figures 3D, 5D), which explains this pattern.
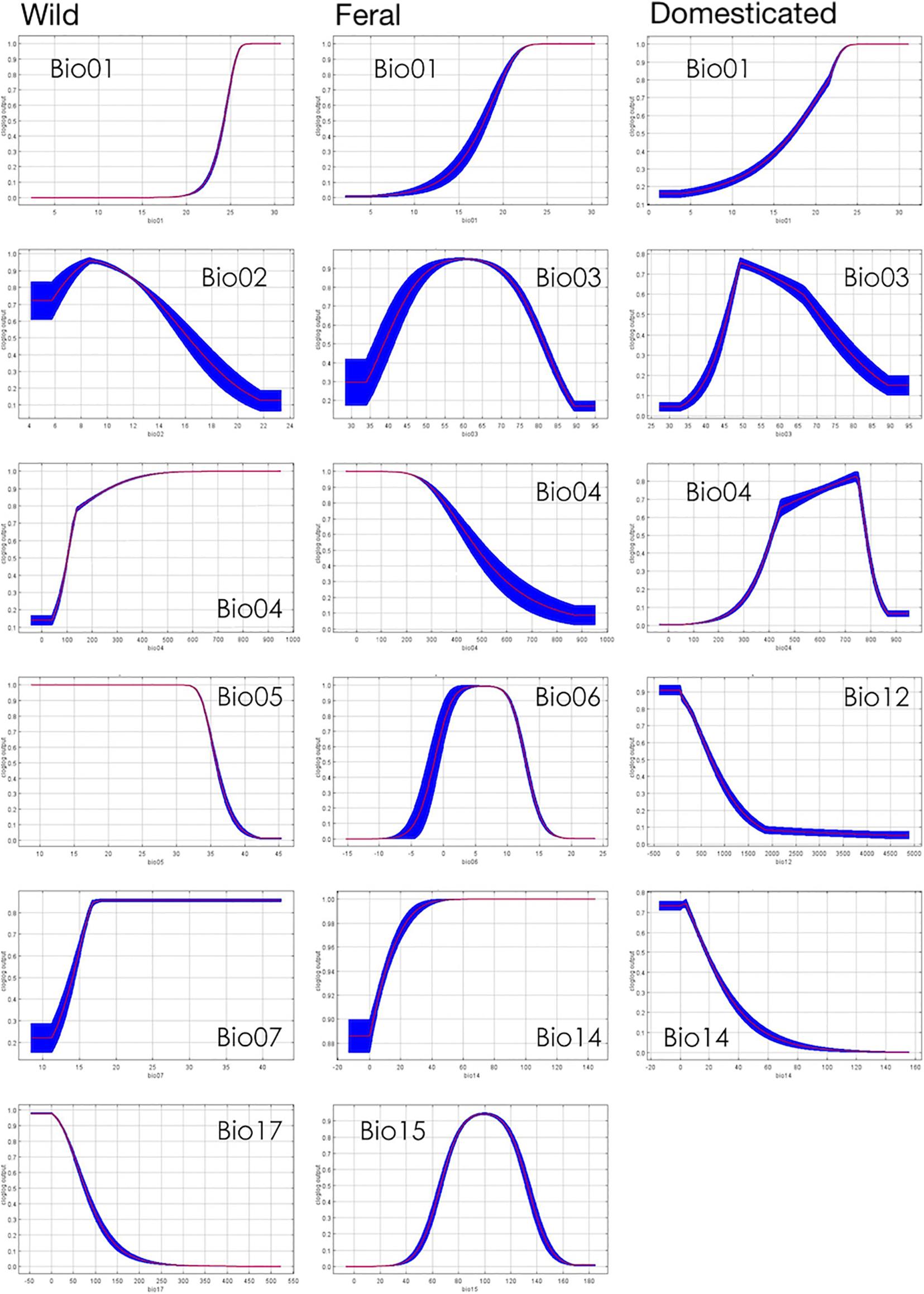
Figure 8. Response curves showing the relationships between suitability and bioclimatic variables.
Notably, the omission rate of the feral model is high despite its width, which reveals that the dataset contains environmentally uncharacteristic localities. This is consistent with its breadth being significantly different in the PC2 and possessing more outliers than any of the other two groups. This could be explained by the heterogeneous nature of cotton ferality. Routes to feralization are diverse and comprise numerous survival strategies that result in genetic and phenotypic variation (Ammann et al., 2005; Gering et al., 2019). For instance, feral populations can be categorized as “endoferal” or “exoferal”: the former originating from a single domesticated lineage, whereas the latter are derived from further admixture with domesticated taxa, wild relatives, or both (Gressel, 2005; Gering et al., 2019). As mentioned above, G. hirsutum has a long history of domestication in Mexico and like other domesticated plants in Mesoamerica, it has been subjected to a diversity of in situ and ex situ management practices, coupled with constant human-mediated seed movement (Casas et al., 2007). These continuous and ongoing processes, along with gene flow and introgression within the primary gene pool, have shaped the evolutionary history of G. hirsutum resulting in complex relationships among the members of the wild-to-domesticated continuum. As a result, feral populations could have emerged from multiple domesticated sources (e.g., pre-Hispanic or post-Columbian cultivars, varieties from the nineteenth to twentieth centuries, past or contemporary incipient domesticates, or current highly domesticated cottons) and experienced admixture from different wild and cultivated lineages at multiple timepoints. Moreover, feralization can produce rapid evolutionary changes due to novel selective pressures and introgression (Mukherjee et al., 2012; Ellstrand et al., 2013; Gering et al., 2019). The consequences of these processes will depend on endo and/or exoferal origins and the genotype–phenotype interactions across new environmental conditions. Thus, feral populations can exhibit a greater range of phenotypes than their domesticated ancestors and also adapt locally to different settings (Hendrickson, 2013; Burger and Ellstrand, 2014). The resulting genetic divergence can cause a shift in the species’ fundamental niche (Mukherjee et al., 2012) while plasticity can be important in colonizing new environments (Gering et al., 2019). Therefore, feral G. hirsutum most likely encompasses mixed populations with different evolutionary histories and origins which, in turn, could comprise wider environmental tolerances and potential ranges.
The feralization process and its population dynamics are still unclear (Meffin et al., 2015). The diverse pathways from which feral populations originate and evolve obscure the elucidation of their evolutionary history. In addition, individual populations may differ in their responses to particular environments and the conditions that allow their establishment may differ from those that determine their persistence (Meffin et al., 2015). Furthermore, local biotic interactions can also play a role in structuring populations. Knowledge on these processes is lacking for feral cotton and this complicates the characterization of its niche. Yet, recognizing these caveats will benefit future modeling approaches dealing with ferality. For instance, community based approaches built on phylogenetic and ecological information could lead to improved niche construction by capturing the environmental responses of particular lineages within the feral group and revealing cryptic niche evolution or local adaptation events (Alvarado-Serrano and Knowles, 2014; Smith et al., 2019).
Origin of Cotton Ferality: Seed Dispersion in Mexico
In Mexico, cotton seeds can escape cultivation through several processes that can be classified into three broad categories: (1) Abiotic factors associated with the climate (e.g., storms, ocean currents, or hurricanes), irrigation channels, and nearby roads; (2) biotic factors such as birds that collect fibers and seeds to build their nests (Arteaga Rojas, 2021) or livestock that feeds on seeds and disperse them; and (3) by human activities associated with cotton production (Wegier et al., 2011; Wegier, 2013). Human-mediated seed movement is of particular interest when looking into ferality sources. For instance, seed leakage occurs during transportation when unsuitable vehicles are used and the lack of route records hinders actions to address this issue. In Mexico, most seeds are distributed as cattle feed after ginning. It should be noted that after passing through the digestive tract of livestock, 3% of the seeds are able to germinate, grow into an adult plant –if suitable environmental conditions prevail–, and subsequently disperse seeds and pollen (Wegier, 2013). Additionally, these seeds are transferred to ranches where they are stored in cool, ventilated places to reduce the likelihood of fire (seeds are flammable due to their high lipid content). These are usually roofed warehouses, often built with branches, with wide ventilation openings, thus allowing both biotic and abiotic seed dispersal (Wegier, 2013). Moreover, cotton is also grown under traditional management systems and home gardens. Within these practices, Mexican locals and small-scale farmers traditionally participate in plant and seed exchange networks that influence seed movement, and their involvement in ferality merits further assessment. In the past few years, cotton has increasingly joined the ornamental plant business and the sale of fully grown cotton plants has progressively become popular across Mexico. With this in mind, genetic evidence reveals that some feral populations could correspond to highly improved plant escapes. According to Wegier et al. (2011), the feral populations that they evaluated had the same single haplotype that was found homogeneously in all cultivated plants. The only exception was the feral population of Morelos, in central Mexico, which was genetically more diverse (Wegier et al., 2011).
Unlike voluntary plants, which rarely last more than one or two seasons, feral populations are self-perpetuating and occur in uncultivated areas (Gressel, 2005; Devaux et al., 2007). Nevertheless, volunteers in field margins can be sources for feral populations because crop ferality is initiated by dispersion to adjacent unmanaged ecosystems. Moreover, if cultivation ceases in previously cultivated fields, semi-domesticated species could still grow in those lands for many years, becoming feral (or ruderal) when agricultural management stops. That could be the case for many feral populations described in this study. Notably, when reviewing the history of localities where feral cotton has been described, several sites corresponded to pre-Columbian inland cotton-growing areas, particularly within and nearby the Aztec Empire (such as the above mentioned Morelos population; Berdan, 1987; Figure 9). Moreover, as cotton and its derived products played a major role on Mesoamerican civilizations’ economy, a dynamic productive chain involving constant transportation of raw cotton and textiles was created around production, spinning, weaving, and trade. As such, cotton has been anciently moving across Mexico due to economic and cultural activities, as have been documented for the Aztec Empire and its tributary provinces (Berdan, 1987), the Mayan region at Yucatán Peninsula (Ardren et al., 2010), Mixtec Oaxaca (King, 2011), and the Huichol Sierra (Mathiowetz, 2020). These productive networks remained during Colonial times; however, when northern cultivars replaced most of the cotton production in Mexico, former cotton growing areas and transportation routes declined. Yet, feral cotton distribution today may have been influenced by these activities and some localities conform to vestiges of pre-Columbian cotton-related activities.
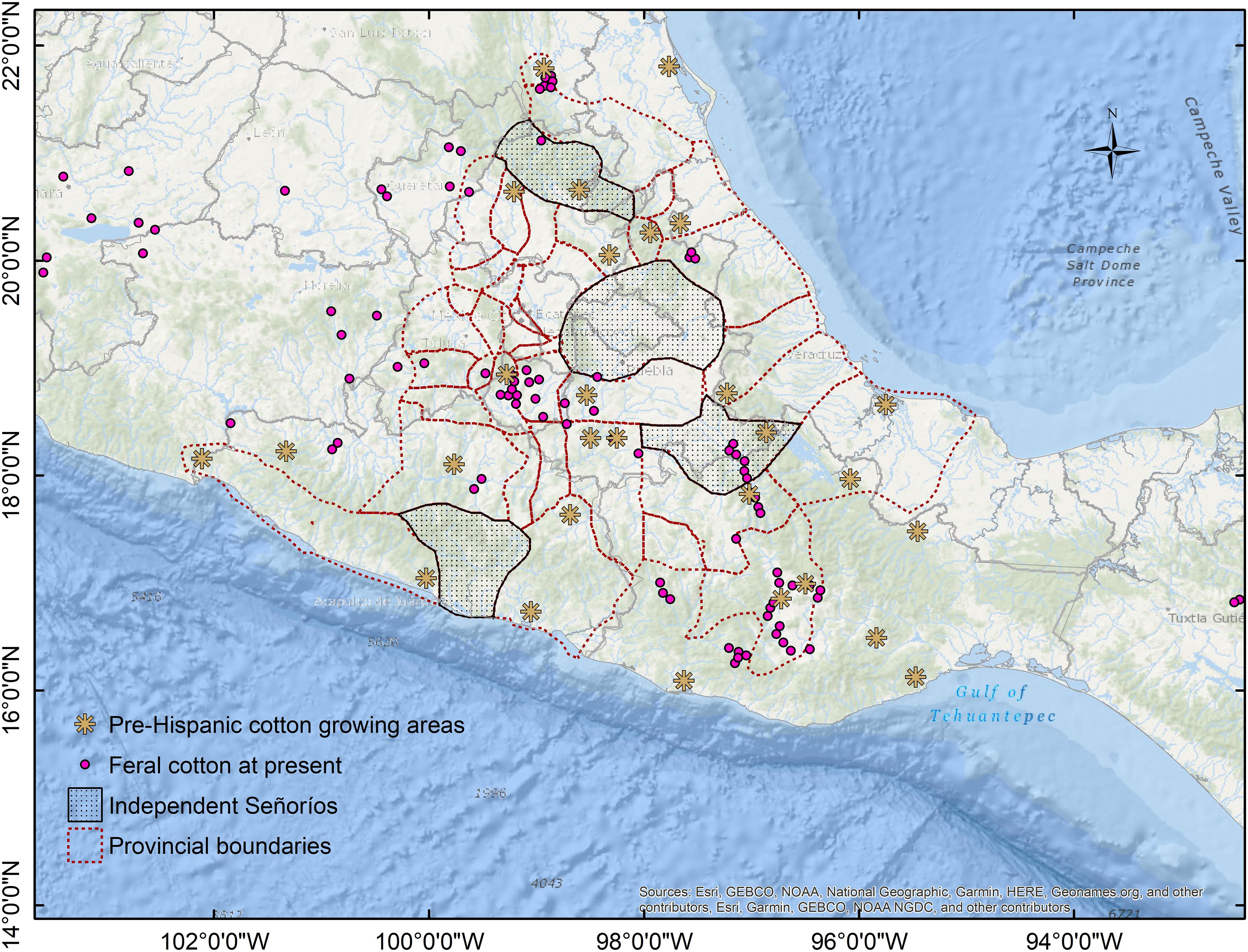
Figure 9. Feral cotton occurrences and pre-Columbian cotton-growing areas within the Aztec Empire and nearby provinces. Modified from Berdan (1987).
Implications on Cotton Conservation in Mexico
Centers of origin, domestication, and genetic diversity are fundamental areas for the conservation of biodiversity and the sustainable use of its genetic resources, particularly of crop wild relatives (Mercer and Perales, 2010; Casas et al., 2019). Therefore, in Mexico, access and management of the G. hirsutum wild-to-domesticated complex must be responsible and considering in situ conservation as a priority, while also recognizing the close relationship between cultural richness and biological diversity. With the advent of GM crops, concerns about gene flow toward wild relatives have been increasing and several international agreements and national legal frameworks have been set to minimize or prevent adverse effects (e.g., Convention on Biological Diversity, Cartagena Protocol on Biosafety, Nagoya-Kuala Lumpur Supplementary Protocol, Mexican Biosafety Law of Genetically Modified Organisms, among others), particularly in areas where GM varieties and wild relatives coexist. As a response, measures involving buffer zones or separation distances between crops and native distributions have been established to reduce admixture. For instance, in Mexico, geographical separation is mandatory for the sowing and release of GM cotton into the environment, and the distribution of wild populations and cultivars is taken into consideration in risk assessments (Rocha-Munive et al., 2018).
In this study, for niche modeling the wild cotton, we chose a “broad distribution” approach for two main reasons: (1) the occurrence of wild forms scattered throughout the Pacific Islands as well as the Caribbean and Mexican coasts suggests wide diffusion via ocean currents (Fryxell, 1979) and, as Sauer (1967) stated, “if lint bearing cottons were naturally present in the New World as sea dispersed pioneers, they were not likely to be confined to Yucatán”; and (2) even if Mexican populations from the Pacific coast would not be regarded as “truly wild” (Coppens d’Eeckenbrugge and Lacape, 2014), they are part of the naturally occurring flora at coastal dunes of the region and harbor valuable genetic diversity (Wegier et al., 2011). Thus, they deserve equal attention to conservation efforts. We have also modeled the ecological niche of domesticated cotton using an extensive set of localities from GM cotton release applications and not only the sites that have permission for commercial sowing (Sandoval Vázquez, 2017). The reasoning for this approach was also twofold: (1) solicited polygons should reflect the environmental requirements preferred for domesticated cotton, allowing us to better characterize its niche; and (2) including more data would portray a scenario of the extent of GM cotton potential distribution if the majority of the applications were approved, which would be of interest for adequate risk assessments. Moreover, we have modeled the environmental niche of feral cotton, a heterogeneous group that occurs in populations scattered throughout Mexico and could be playing a role in gene flow dynamics within G. hirsutum wild-to-domesticated complex. Furthermore, as some of these populations may be vestiges of pre-Hispanic cultivation, their genetic diversity could be very valuable for documenting the domestication history of cotton in Mexico.
The evolutionary processes that originate and maintain cotton’s diversity have led to unique and shared variation. Conserving the populations that harbor that diversity should be an imminent endeavor. From our perspective, an integral approach toward this goal is essential, meaning that both in situ and ex situ conservation are necessary and complementary. For this purpose, understanding that G. hirsutum conforms to a wild-to-domesticated continuum is fundamental, as it recognizes that valuable genetic diversity is contained in wild populations, native landraces, and some feral and domesticated lineages. Gene flow is an evolutionary force that has a significant impact on current diversity. In the wild, a mixed mating system and small population sizes maintain heterogeneity between cotton populations. However, the number of cultivated plants is orders of magnitude greater than wild and ferals. Thus, gene flow from genetically homogeneous or GM sources is biased by their abundance.
Both domesticated and feral potential distributions are wide and overlap with the wild distribution: the former in northern Mexico and the latter throughout the northwest to the southeast. This pattern indicates that feral cotton potential distribution could act as a bridge between the cultivation areas and wild populations, which is highly relevant in terms of biosecurity and conservation (Reagon and Snow, 2006; Bagavathiannan and van Acker, 2009; Gressel, 2015). As discussed above, feral cotton probably includes plants from multiple lineages and knowledge on the origins of these plants could improve future niche modeling approaches. However, their persistent occurrence highlights the importance of taking these plants and their potential distribution into consideration when geographic distance is still regarded as a measure preventing gene flow and admixture. Wegier et al. (2011) first reported gene flow at long distances between cultivated and wild populations of G. hirsutum, through the identification of recombinant proteins in wild populations. Afterward, (Hernández-Terán et al., 2019) confirmed the introgression of transgenes (Cry1Ab/Ac, Cry2Ab, and CP4EPSPS) in wild cotton by enzyme-linked immunosorbent assay (ELISA) and sequencing of Polymerase Chain Reaction (PCR) products. Notably, they also reported transgene presence in domesticated cotton sold in ornamental plant stalls in markets. These studies point out that geographic distancing between cotton crops and wild relatives has not been a successful measure preventing gene flow and subsequent introgression in Mexico. Geographic distancing measures are based on gene flow patterns expected under a model of isolation by distance where dispersal limitation results in genetic differentiation. However, they will not be effective on crops whose gene flow patterns conform to long-distance models, such as cotton (Wegier et al., 2011). At present, GM traits introgressed in wild populations resulted in ecological consequences for cotton plants, communities, and ecosystems (Vázquez-Barrios et al., 2021).
Finally, it is important to realize that the legal custody of GM seeds is lost once the seed is separated from the fiber and it begins to be referred to as “grain,” even though legal responsibility should be binding during all stages of the supply chain, as is stated in the law (DOF, 2008; Wegier, 2013). Indeed, the careful control of cotton seeds before sowing is relaxed after harvest (Rocha-Munive et al., 2018). Moreover, Mexico also imports viable cotton GM seed to meet the demand for livestock feed, however, as these cross-border seeds lack labels, they fall outside the contingency plans of Mexican GMO regulatory institutions. Furthermore, an additional and illegal problem involves the cultivation of “fuzzy seeds.” These constitute the stock that has been ginned and is kept for sowing in the following season without the required permits and without paying rights to the companies that own the patents because the information on their specific GM transformation event is lost in the process.
Given the above, in situ conservation strategies for cotton in Mexico should focus on: (1) preserving populations within the wild-to-domesticated complex that possess significant and unique diversity and the processes that originate and maintain them; (2) limiting gene flow from domesticated cotton to valuable genetic resources; (3) identifying and preventing hazards that threaten wild and feral small population sizes (e.g., habitat loss); (4) establishing active communication with local communities to promote appreciation of their genetic resources; (5) determining feral lineages to identify conservation targets and modern cotton escapees that need regulation; and 6) considering legal, social, cultural, and environmental complexities that occur at centers of origin, domestication, and genetic diversity, among others. It is important to note that different approaches to implementing these measures will depend on the particularities of each of the parts that integrate the wild-to-domesticated complex. Ex situ conservation actions should complement these strategies; for instance, seed and in vitro banks would allow research and germplasm conservation, while botanical garden collections would contribute to education, appreciation, and awareness of genetic resources’ importance.
Conclusion and Future Directions
The conservation of the primary gene pool of G. hirsutum is paramount and gene flow from domesticated cultivars to wild relatives should be prevented. In Mexico, wild, feral, and domesticated cottons occupy non-identical niches while also sharing environmental conditions where they overlap, with feral cotton laying as an intermediate between wild and domesticated forms. Our findings indicate that geographic distance measures in GM release permits are not efficient to avoid transgene flow because wild-to-domesticated cotton niches are not isolated and previous genetic evidence shows that G. hirsutum’s gene flow patterns conforms to a long-distance model (Wegier et al., 2011; Hernández-Terán et al., 2019).
The eco-geographic niche of feral cotton should be considered in the design of future risk assessment and transgene monitoring studies to prevent further transgene flow. To that end, the origin and genetic diversity of feral populations should be investigated to understand the relationships within this heterogeneous group and between other members of the wild-to-domesticated continuum. Phenotypic and genetic studies focused on this goal will be valuable for describing intraspecific variation and will provide deeper insight into the relationships within the primary gene pool. This information will be relevant for cotton evolutionary and domestication studies, and will shed light on the impact these plants have on the gene flow dynamics of the system. Based on that knowledge, biosecurity measures, transgene mitigation strategies, and conservation programs should be reevaluated.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
VA and AW conceived and designed the research, collated and curated the data, and performed fieldwork. VA conducted the analyses, prepared figures and tables, and drafted the manuscript. ÁC-R and EM-M guided niche modeling and spatial analyses. All authors contributed to result interpretation, revised and edited the manuscript, and approved the final draft.
Funding
This work was financially supported by projects UNAM-PAPIIT IN214719 and “Program for the conservation of wild populations of Gossypium hirsutum in Mexico,” DGAP003/WN003/18, funded by CONABIO. This study is part of the Ph.D. thesis of VA, who is a doctoral student from the Posgrado en Ciencias Biológicas, Universidad Nacional Autónoma de México (UNAM) and is supported by CONACYT (scholarship no. 762515).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer, AG-R, declared a shared affiliation, though no collaboration, with the authors to the handling editor
Acknowledgments
We express our gratitude to the Agrobiodiversity Department at CONABIO for their support and advice during the planning and development of this research, especially to Francisca Acevedo, Caroline Burgeff, Oswaldo Oliveros, and María Andrea Orjuela. We are very thankful to Daniel Ortiz and Cuauhtémoc Enríquez for geospatial polygon analysis and to Haven López-Sánchez for valuable comments and suggestions. In addition, we acknowledge the support of all the colleagues, students, and friends that have recorded the presence of cotton plants over the years. Finally, we are truly grateful to all the people at the communities where our work was done; they kindly guided us through the field and shared their knowledge on local flora.
Dedication
This article is dedicated to the memory of Doña Epifania, who always welcomed us during fieldwork in Oaxaca and whose generosity and thoughts about life were truly inspiring.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2021.653271/full#supplementary-material
References
Alvarado-Serrano, D. F., and Knowles, L. L. (2014). Ecological niche models in phylogeographic studies: applications, advances and precautions. Mol. Ecol. Resour. 14, 233–248. doi: 10.1111/1755-0998.12184
Ammann, K., Jacot, Y., and Rufener Al Mazyad, P. (2005). “About the origin of feral crops and their relatives, their detection in historic records,” in Crop Ferality and Volunteerism, ed. J. Gressel (Boca Raton, FL: CRC PressTaylor & Francis Group), 31–44.
Andersson, M. S., and de Vicente, M. C. (2010). Gene Flow Between Crops and Their Wild Relatives. Baltimore, MD: JHU Press.
Ardren, T., Manahan, T. K., Wesp, J. K., and Alonso, A. (2010). Cloth production and economic intensification in the area surrounding Chichen Itza. Lat. Am. Antiq. 21, 274–289. doi: 10.7183/1045-6635.21.3.274
Arteaga Rojas, A. E. (2021). Análisis del Posible uso del Algodón Silvestre (Gossypium hirsutum) Como Material de Construcción en Nidos de Aves en Yucatán y Oaxaca. Bachelor dissertation. Facultad de Ciencias. México: UNAM.
Ashiq Rabbani, M., Murakami, Y., Kuginuki, Y., and Takayanagi, K. (1998). Genetic variation in radish (Raphanus sativus L.) germplasm from Pakistan using morphological traits and RAPDs. Genet. Resour. Crop Evol. 45, 307–316.
Bagavathiannan, M. V., and van Acker, R. C. (2008). Crop ferality: implications for novel trait confinement. Agric. Ecosyst. Environ. 127, 1–6. doi: 10.1016/j.agee.2008.03.009
Bagavathiannan, M. V., and van Acker, R. C. (2009). The biology and ecology of feral alfalfa (Medicago sativa L.) and its implications for novel trait confinement in North America. CRC Crit. Rev. Plant Sci. 28, 69–87. doi: 10.1080/07352680902753613
Barve, N., Barve, V., Jiménez-Valverde, A., Lira-Noriega, A., Maher, S. P., Peterson, A. T., et al. (2011). The crucial role of the accessible area in ecological niche modeling and species distribution modeling. Ecol. Modell. 222, 1810–1819. doi: 10.1016/j.ecolmodel.2011.02.011
Berdan, F. F. (1987). Cotton in Aztec Mexico: production, distribution and uses. Mex. Stud. 3, 235–262. doi: 10.2307/1051808
Berville, A., Muller, M.-H., Poinso, B., and Serieys, H. (2005). “Ferality: risks of gene flow between sunflower and other Helianthus species,” in Crop Ferality and Volunteerism: A Threat to Food Security in the Transgenic Era, ed. J. Gressel (Boca Raton, FL: CRC Press), 209–230. doi: 10.1201/9781420037999.ch14
Broennimann, O., Fitzpatrick, M. C., Pearman, P. B., Petitpierre, B., Pellissier, L., Yoccoz, N. G., et al. (2012). Measuring ecological niche overlap from occurrence and spatial environmental data. Glob. Ecol. Biogeogr. 21, 481–497. doi: 10.1111/j.1466-8238.2011.00698.x
Brubaker, C. L., and Wendel, J. F. (1994). Reevaluating the origin of domesticated cotton(Gossypium hirsutum; Malvaceae) using nuclear restriction fragment length polymorphisms(RFLPs). Am. J. Bot. 81, 1309–1326. doi: 10.1002/j.1537-2197.1994.tb11453.x
Burgeff, C., Huerta, E., Acevedo, F., and Sarukhán, J. (2014). How much can GMO and non-GMO cultivars coexist in a megadiverse country? AgBioForum 17, 90–101.
Burger, J. C., and Ellstrand, N. C. (2014). Rapid evolutionary divergence of an invasive weed from its crop ancestor and evidence for local diversification: rapid evolution and diversification of feral rye. J. Syst. Evol. 52, 750–764. doi: 10.1111/jse.12111
Casas, A., Ladio, A. H., and Clement, C. R. (2019). Editorial: ecology and evolution of plants under domestication in the Neotropics. Front. Ecol. Evol. 7:231. doi: 10.3389/fevo.2019.00231
Casas, A., Otero-Arnaiz, A., Pérez-Negrón, E., and Valiente-Banuet, A. (2007). In situ management and domestication of plants in Mesoamerica. Ann. Bot. 100, 1101–1115. doi: 10.1093/aob/mcm126
Cerutti, M., and Almaraz, A. (2013). Algodón en el Norte de México (1920-1970): Impactos Regionales de un Cultivo Estratégico. Tijuana, BC: El Colegio de la Frontera Norte.
Charlton, T. H., Nichols, D. L., and Charlton, C. O. (1991). Aztec craft production and specialization: archaeological evidence from the city-state of Otumba, Mexico. World Archaeol. 23, 98–114. doi: 10.1080/00438243.1991.9980161
CONABIO (2020). Sistema Nacional de Información Sobre Biodiversidad. Registros de Ejemplares. [Gossypium hirsutum]. Comisión Nacional para el Conocimiento y Uso de la Biodiversidad. Available online at: https://www.snib.mx/ (accessed March 29, 2020).
Constable, G. A., and Bange, M. P. (2015). The yield potential of cotton (Gossypium hirsutum L.). Field Crops Res. 182, 98–106. doi: 10.1016/j.fcr.2015.07.017
Coppens d’Eeckenbrugge, G., and Lacape, J.-M. (2014). Distribution and differentiation of wild, feral, and cultivated populations of perennial upland cotton (Gossypium hirsutum L.) in Mesoamerica and the Caribbean. PLoS One 9:e107458. doi: 10.1371/journal.pone.0107458
Cuervo-Robayo, A. P., Téllez-Valdés, O., Gómez-Albores, M. A., Venegas-Barrera, C. S., Manjarrez, J., and Martínez-Meyer, E. (2014). An update of high-resolution monthly climate surfaces for Mexico. Int. J. Climatol. 34, 2427–2437. doi: 10.1002/joc.3848
Devaux, C., Lavigne, C., Austerlitz, F., and Klein, E. K. (2007). Modelling and estimating pollen movement in oilseed rape (Brassica napus) at the landscape scale using genetic markers. Mol. Ecol. 16, 487–499. doi: 10.1111/j.1365-294x.2006.03155.x
Di Cola, V., Broennimann, O., Petitpierre, B., Breiner, F. T., D’Amen, M., Randin, C., et al. (2017). ecospat: an R package to support spatial analyses and modeling of species niches and distributions. Ecography 40, 774–787. doi: 10.1111/ecog.02671
Diario Oficial de la Federación [DOF] (2005). Ley de Bioseguridad de Organismos Genéticamente Modificados. Available online at: http://www.diputados.gob.mx/LeyesBiblio/pdf/LBOGM_061120.pdf (accessed November 6, 2020).
DOF (2008). Reglamento de la Ley de Bioseguridad de Organismos Genéticamente Modificados. Available online at: https://www.gob.mx/profepa/documentos/reglamento-de-la-ley-de-bioseguridad-de-organismos-geneticamente-modificadosgeneticamente-modificados (accessed November 6, 2020).
DOF (2019). Modificación del Anexo Normativo III, Lista de Especies en Riesgo de la Norma Oficial Mexicana NOM-059-SEMARNAT-2010, Protección Ambiental. Especies Nativas de México de Flora y Fauna Silvestres. Categorías de Riesgo y Especificaciones para su Inclusión, Exclusión o Cambio. Lista de Especies en Riesgo, Publicada el 14 de Noviembre de 2019. Mexico: Diario Oficial de la Federación.
Ellstrand, N. C., Meirmans, P., Rong, J., Bartsch, D., Ghosh, A., de Jong, T. J., et al. (2013). Introgression of crop alleles into wild or weedy populations. Annu. Rev. Ecol. Evol. Syst. 44, 325–345. doi: 10.1146/annurev-ecolsys-110512-135840
Escobar, L. E., Lira-Noriega, A., Medina-Vogel, G., and Townsend Peterson, A. (2014). Potential for spread of the white-nose fungus (Pseudogymnoascus destructans) in the Americas: use of Maxent and NicheA to assure strict model transference. Geospat. Health 9, 221–229. doi: 10.4081/gh.2014.19
ESRI (2015). ArcGIS for Desktop, version 10.2.1: Gis Software. Redlands, CA: Environmental Sytems Research Institute.
Freckleton, R. P., and Watkinson, A. R. (2002). Large-scale spatial dynamics of plants: metapopulations, regional ensembles and patchy populations. J. Ecol. 90, 419–434. doi: 10.1046/j.1365-2745.2002.00692.x
Fryxell, P. A. (1979). The Natural History of the Cotton Tribe (Malvaceae, tribe Gossypieae). College Station, TX: Texas A & M University Press.
Gering, E., Incorvaia, D., Henriksen, R., Conner, J., Getty, T., and Wright, D. (2019). Getting back to nature: feralization in animals and plants. Trends Ecol. Evol. 34, 1137–1151. doi: 10.1016/j.tree.2019.07.018
Gressel, J. (2015). Dealing with transgene flow of crop protection traits from crops to their relatives. Pest Manag. Sci. 71, 658–667. doi: 10.1002/ps.3850
Hawkins, J. S., Pleasants, J., and Wendel, J. F. (2005). Identification of AFLP markers that discriminate between cultivated cotton and the Hawaiian Island Endemic, Gossypium tomentosum Nuttall ex Seeman. Genet. Resour. Crop Evol. 52, 1069–1078. doi: 10.1007/s10722-004-6115-z
Hendrickson, S. L. (2013). A genome wide study of genetic adaptation to high altitude in feral Andean horses of the páramo. BMC Evol. Biol. 13:273. doi: 10.1186/1471-2148-13-273
Hennig, C. (2007). The fpc Package Version 2.2-5. Available online at: http://btr0x2.rz.uni-bayreuth.de/math/statlib/R/CRAN/doc/packages/fpc.pdf (accessed April 7, 2020).
Hernández-Terán, A., Wegier, A., Benítez, M., Lira, R., Sosa Fuentes, T. G., and Escalante, A. E. (2019). In vitro performance in cotton plants with different genetic backgrounds: the case of Gossypium hirsutum in Mexico, and its implications for germplasm conservation. PeerJ 7:e7017. doi: 10.7717/peerj.7017
Hilbeck, A., Andow, D., and Underwood, E. (2004). The GMO guidelines project and a new ecological risk assessment. IOBC Wprs Bull. 27, 93–96.
Hironymous, M. (2007). Santa María Ixcatlán, Oaxaca: From Colonial Cacicazgo to Modern Municipio. Available online at: https://repositories.lib.utexas.edu/bitstream/handle/2152/13258/hironymousm16499.pdf?sequence=2 (accessed November 6, 2020).
Husson, F., Josse, J., Le, S., and Mazet, J. (2013). FactoMineR: Multivariate Exploratory Data Analysis and Data Mining with R. R Package Version 1.
Jiménez Martínez, A., and Ayala Angulo, M. (2020). Algodón Genéticamente Modificado: Riesgos y Solicitudes de Liberación, Vol. 1. Mexico: Diálogos Ambientales, Secretaría de Medio Ambiente y Recursos Naturales.
Kassambara, A. (2017). Practical Guide to Cluster Analysis in R: Unsupervised Machine Learning. New York, NY: STHDA.
Kassambara, A., and Mundt, F. (2017). Factoextra: Extract and Visualize the Results of multivariate Data Analyses. R Package Version, Vol. 1, 337–354.
King, S. M. (2011). Thread production in early postclassic coastal Oaxaca, Mexico: technology, intensity, and gender. Anc. Mesoamerica 22, 323–343. doi: 10.1017/s0956536111000253
Mathiowetz, M. (2020). “Weaving our life: the economy and ideology of cotton in postclassic West Mexico,” in Ancient West Mexicos, ed. E. Joshua (Gainesville, FL: University Press of Florida), 302–348. doi: 10.2307/j.ctvxrpxsc.14
Meffin, R., Duncan, R. P., and Hulme, P. E. (2015). Landscape-level persistence and distribution of alien feral crops linked to seed transport. Agric. Ecosyst. Environ. 203, 119–126. doi: 10.1016/j.agee.2015.01.024
Mendoza, C. P., Del Rosario Tovar Goìmez, M., Gonzalez, Q. O., De Jesuìs Legorreta Padilla, F., and Corral, J. A. R. (2017). Recursos geneìticos del algodoìn en Meìxico: conservacioìn ex situ, in situ y su utilizacioìn. Rev. Mex. Cienc. Agrícolas 7, 5–16. doi: 10.29312/remexca.v7i1.366
Mercer, K. L., and Perales, H. R. (2010). Evolutionary response of landraces to climate change in centers of crop diversity. Evol. Appl. 3, 480–493. doi: 10.1111/j.1752-4571.2010.00137.x
Molina-Henao, Y. F., and Hopkins, R. (2019). Autopolyploid lineage shows climatic niche expansion but not divergence in Arabidopsis arenosa. Am. J. Bot. 106, 61–70. doi: 10.1002/ajb2.1212
Mukherjee, A., Williams, D. A., Wheeler, G. S., Cuda, J. P., Pal, S., and Overholt, W. A. (2012). Brazilian peppertree (Schinus terebinthifolius) in Florida and South America: evidence of a possible niche shift driven by hybridization. Biol. Invasions 14, 1415–1430. doi: 10.1007/s10530-011-0168-7
Muscarella, A. R., Galante, P. J., Soley-Guardia, M., Boria, R. A., Kass, J. M., Uriarte, M., et al. (2018). ENMeval: Automated Runs and Evaluations of Ecological Niche Models.
Osorio-Olvera, L., Lira-Noriega, A., Soberón, J., Peterson, A. T., Falconi, M., Contreras-Díaz, R. G., et al. (2020). ntbox : an r package with graphical user interface for modelling and evaluating multidimensional ecological niches. Methods Ecol. Evol. 11, 1199–1206. doi: 10.1111/2041-210x.13452
Peterson, A. T., and Soberón, J. (2012). Species distribution modeling and ecological niche modeling: getting the concepts right. Nat. Conserv. 10, 102–107. doi: 10.4322/natcon.2012.019
Phillips, S. J., Anderson, R. P., and Schapire, R. E. (2006). Maximum entropy modeling of species geographic distributions. Ecol. Modell. 190, 231–259. doi: 10.1016/j.ecolmodel.2005.03.026
Phillips, S. J., Dudík, M., and Schapire, R. E. (2004). “A maximum entropy approach to species distribution modeling,” in Proceedings of the 21st International Conference on Machine Learning ICML ‘04, (New York, NY: Association for Computing Machinery), 83.
R Core Team (2017). R A Language and Environment for Statistical Computing. Versión 3.4.3. Vienna: R Foundation for Statistical Computing.
Reagon, M., and Snow, A. A. (2006). Cultivated Helianthus annuus (Asteraceae) volunteers as a genetic “bridge” to weedy sunflower populations in North America. Am. J. Bot. 93, 127–133. doi: 10.3732/ajb.93.1.127
Rocha-Munive, M. G., Soberón, M., Castañeda, S., Niaves, E., Scheinvar, E., Eguiarte, L. E., et al. (2018). Evaluation of the impact of genetically modified cotton after 20 years of cultivation in Mexico. Front. Bioeng. Biotechnol. 6:82. doi: 10.3389/fbioe.2018.00082
Sandoval Vázquez, D. (2017). Treinta Años de Transgénicos en México (Compendio Cartográfico). México: Centro de Estudios para el Cambio en el Campo Mexicano.
Sauer, J. D. (1967). Geographic Reconnaissance of Seashore Vegetation Along the Mexican Gulf Coast. Baton Rouge, LA: Louisiana State University Press.
Secretariat of the Convention on Biological Diversity (2000). Cartagena Protocol on Biosafety to the Convention on Biological Diversity: Text and Annexes. Montreal, QC: Secretariat of the Convention of Biological Diversity.
Simoes, M., Romero-Alvarez, D., Nuñez-Penichet, C., Jiménez, L., and Cobos, M. E. (2020). General theory and good practices in ecological niche modeling: a basic guide. Biodivers. Inform. 15, 67–68. doi: 10.17161/bi.v15i2.13376
SIOVM (2020). Sistema de Información de Organismos Vivos Modificados [Live Modified Organisms Information System]. Proyecto GEF- CIBIOGEM/CONABIO. Available online at: http://www.conabio.gob.mx/conocimiento/bioseguridad/doctos/consulta_SIOVM.html (accessed March 29, 2020).
Smith, A. B., Godsoe, W., Rodríguez-Sánchez, F., Wang, H.-H., and Warren, D. (2019). Niche estimation above and below the species level. Trends Ecol. Evol. 34, 260–273. doi: 10.1016/j.tree.2018.10.012
Smith, C. E., and Stephens, S. G. (1971). Critical identification of Mexican archaeological cotton remains. Econ. Bot. 25, 160–168. doi: 10.1007/bf02860076
Snow, A., and Campbell, L. (2005). “Can feral radishes become weeds?,” in Crop Ferality and Volunteerism, ed. J. Gressel (Boca Raton, FL: CRC Press), 193–207. doi: 10.1201/9781420037999.ch13
Ulloa, M., Saha, S., Jenkins, J. N., Meredith, W. R. Jr., McCarty, J. C. Jr., and Stelly, D. M. (2005). Chromosomal assignment of RFLP linkage groups harboring important QTLs on an intraspecific cotton (Gossypium hirsutum L.) Joinmap. J. Hered. 96, 132–144. doi: 10.1093/jhered/esi020
van Rossum, G., and Drake, F. L. (2009). Introduction to Python 3: (Python Documentation Manual Part 1). Scotts Valley, CA: CreateSpace Independent Publishing Platform.
Vázquez-Barrios, V., Boege, K., Sosa-Fuentes, T. G., Rojas, P., and Wegier, A. (2021). Ongoing ecological and evolutionary consequences by the presence of transgenes in a wild cotton population. Sci. Rep. 11:1959. doi: 10.1038/s41598-021-81567-z
Waskom, M., Botvinnik, O., Gelbart, M., Ostblom, J., Hobson, P., Lukauskas, S., et al. (2020). Available online at: mwaskom/seaborn: v0.11.0 (accessed September 2020). doi: 10.5281/zenodo.4019146.
Wegier, A. (2007). Informe Final del Proyecto “Validación de Información de Registros Biológicos y de Mapas de Distribución Puntual y de Los Modelos de Áreas de Distribución Potencial de Las Especies del Género Gossypium en México,” Bajo el Proyecto 0051868. Continuación de la Creación de Capacidades Institucionales y Técnicas para la Toma de Decisiones en Materia de Bioseguridad [Final Report of the Project “Validation of the Information of Biological Registers, Punctual Distribution Maps and of the Potential Distribution Modeled Areas for Species of the Gossypium Genus in Mexico,” Under the Project 0051868. Continuation of Institutional Capacity Building and Decision Making Techniques in Biosafety Topics]. México: Programa de las Naciones Unidas para el Desarrollo México (PNUD), Comisión Intersecretarial de Bioseguridad de los Organismos Genéticamente Modificados (CIBIOGEM).
Wegier, A., Alavez, V., Vega, M., and Azurdia, C. (2019). Gossypium hirsutum. IUCN Red List of Threatened Species. doi: 10.2305/iucn.uk.2019-2.rlts.t71774532a71774543.en
Wegier, A. L. (2013). Diversidad Genética y Conservación de Gossypium hirsutum Silvestre y Cultivado en México. Ph.D. thesis. México DF: Universidad Nacional Autónoma de México.
Wegier, A., Piñeyro-Nelson, A., Alarcón, J., Gálvez-Mariscal, A., Alvarez-Buylla, E. R., and Piñero, D. (2011). Recent long-distance transgene flow into wild populations conforms to historical patterns of gene flow in cotton (Gossypium hirsutum) at its centre of origin. Mol. Ecol. 20, 4182–4194. doi: 10.1111/j.1365-294x.2011.05258.x
White, A. D., Lyon, D. J., Mallory-Smith, C., Medlin, C. R., and Yenish, J. P. (2006). Feral rye (Secale cereale) in agricultural production systems. Weed Technol. 20, 815–823. doi: 10.1614/wt-05-129r1.1
Keywords: upland cotton (Gossypium hirsutum L.), ecological niche modeling ENM, conservation, gene flow, wild-to-domesticated complex
Citation: Alavez V, Cuervo-Robayo ÁP, Martínez-Meyer E and Wegier A (2021) Eco-Geography of Feral Cotton: A Missing Piece in the Puzzle of Gene Flow Dynamics Among Members of Gossypium hirsutum Primary Gene Pool. Front. Ecol. Evol. 9:653271. doi: 10.3389/fevo.2021.653271
Received: 14 January 2021; Accepted: 28 April 2021;
Published: 28 May 2021.
Edited by:
Raymond L. Tremblay, University of Puerto Rico, Puerto RicoReviewed by:
Antonio Gonzalez-Rodriguez, Universidad Nacional Autónoma de México, MexicoJames Frelichowski, United States Department of Agriculture (USDA), United States
Copyright © 2021 Alavez, Cuervo-Robayo, Martínez-Meyer and Wegier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valeria Alavez, di5hbGF2ZXpAY2llbmNpYXMudW5hbS5teA==; Ana Wegier, YXdlZ2llckBpYi51bmFtLm14