 Chunlin Chen
Chunlin Chen Wenjie Yang
Wenjie Yang Jianquan Liu
Jianquan Liu Zhenxiang Xi
Zhenxiang Xi Lei Zhang
Lei Zhang Quanjun Hu
Quanjun Hu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol., 26 February 2021
Sec. Evolutionary and Population Genetics
Volume 9 - 2021 | https://doi.org/10.3389/fevo.2021.638230
Distributional shifts driven by Quaternary climatic oscillations have been suggested to cause interspecific hybridization and introgression. In this study, we aimed to test this hypothesis by using population transcriptomes and coalescent modeling of two alpine none-sister gentians. Previous studies suggested that historical hybridizations occurred between Gentiana siphonantha and G. straminea in the high-altitude Qinghai-Tibet Plateau although both species are not sister to each other with the most recent divergence. In the present study, we sequenced transcriptomes of 33 individuals from multiple populations of G. siphonantha and G. straminea. The two species are well delimited by nuclear genomic SNPs while phylogenetic analyses of plastomes clustered one G. straminea individual into the G. siphonantha group. Further population structure analyses of the nuclear SNPs suggested that two populations of G. siphonantha were admixed with around 15% ancestry from G. straminea. These analyses suggested genetic introgressions from G. straminea to G. siphonantha. In addition, our coalescent-based modeling results revealed that gene flow occurred between the two species since Last Glacier Maximum after their initial divergence, which might have leaded to the observed introgressions. Our results underscore the significance of transcriptome population data in determining timescale of interspecific gene flow and direction of the resulting introgression.
Interspecific hybridization has facilitated the movement of genes (nuclear and/or cytoplasmic) between species, which is increasingly being detected in a variety of animal and plant groups, such as butterflies (Pardo-Diaz et al., 2012), humans (Huerta-Sánchez et al., 2014), poplars (Suarez-Gonzalez et al., 2016), and sunflowers (Owens et al., 2016). Hybridization and the resulting introgression can potentially blur species boundaries, and may drive rare species to extinction through genetic swamping (Huxel, 1999; Todesco et al., 2016; Ma et al., 2019). Such hybridization and introgression primarily occur between closely related species, especially for lineages that have not yet evolved complete reproductive isolation (Confer et al., 2020). For example, hybridization and introgression have been observed between two subspecies that have been diverging in response to ecological selection (Anadón et al., 2015) and between two allopatric sibling species resulted from their secondary contact caused by geographic range expansion (Zamudio and Savage, 2003).
The Qinghai–Tibet Plateau sensu lato (QTPsl; Liang et al., 2018) is the world’s highest and largest plateau, with an average elevation exceeding 4,500 m. For plants occurring in the QTPsl, it has been suggested that introgression may be more common than previously appreciated, particularly since the Last Glacial Maximum (LGM) due to postglacial range expansion (Liu et al., 2014). Most of these studies, however, have relied on few genetic markers. For example, Hu et al. (2016) conducted a population genetic survey of two alpine gentians (Gentiana siphonantha Maxim. ex Kusn. and G. straminea Maxim.) based on one nuclear (ITS) and two plastid loci (trnL-trnF and trnS-trnG). Their results suggested that hybridization and subsequent introgression led to the sharing of a chloroplast haplotype.
Both G. siphonantha and G. straminea belong to the section Cruciata Gaudin (Gentiana L.; Gentianaceae), which includes 22 extant species that diversified during the Pliocene (Zhang et al., 2009). G. siphonantha occurs only in the northeastern QTPsl, while G. straminea is widely distributed throughout the QTPsl. Thus, their natural distribution ranges overlap in the northeastern QTPsl, with a few geographic locations where they occur sympatrically (Hu et al., 2016). A recent phylotranscriptomic study has shown that these two gentians are not sister to each other (Chen et al., 2020), which can be well distinguished based on the shape of the inflorescence, the length of the calyx tube, and the color of the corolla (Figure 1). The flowers of G. siphonantha are crowded into terminal clusters, the calyx tube is 4–6 mm long, and the color of the corolla is dark blue-purple. In contrast, the flowers of G. straminea are arranged in lax cymes, the calyx tube is 15–28 mm long, and the color of the corolla ranges from white–green to yellow–green (Ho and Liu, 2001).
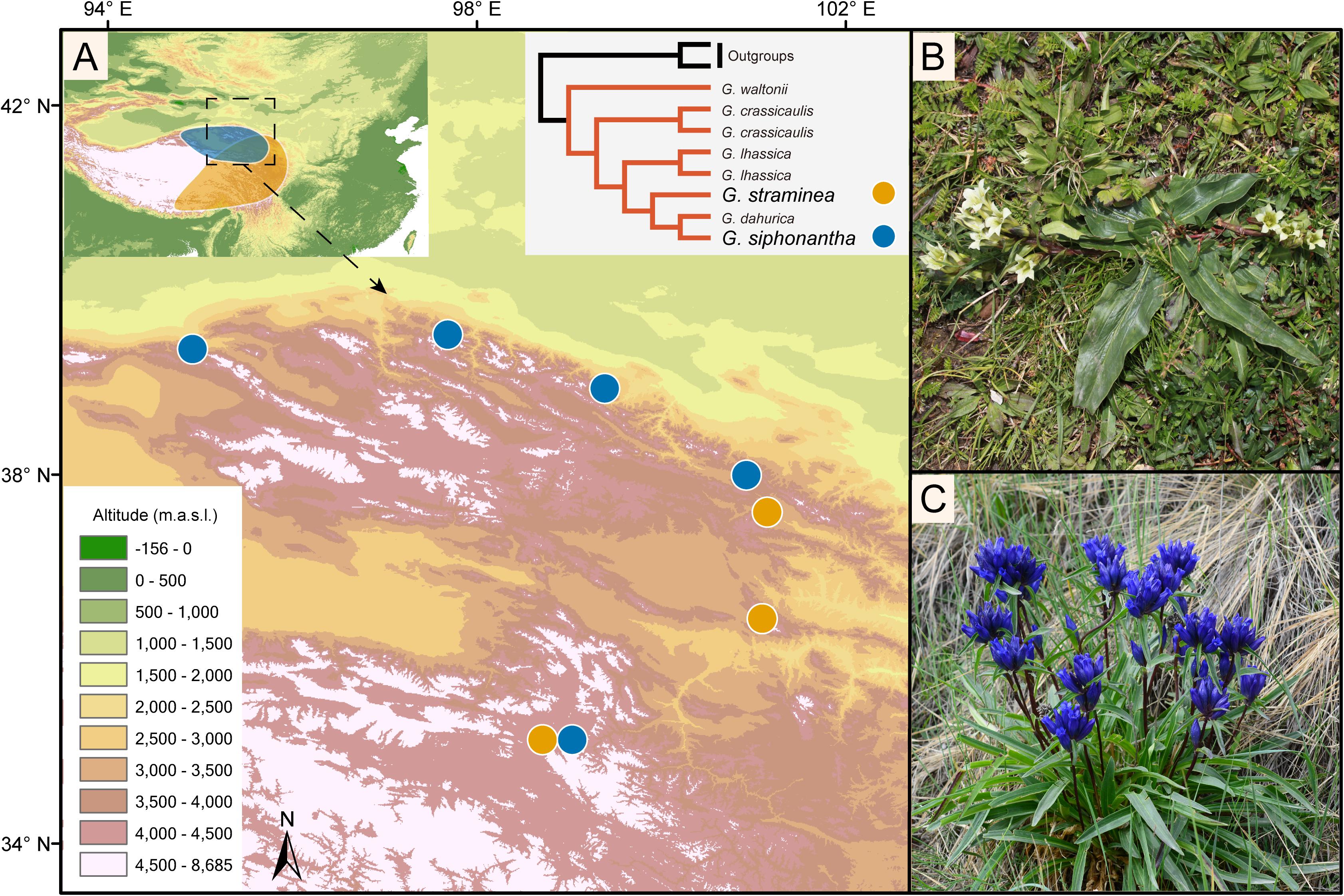
Figure 1. Map showing the geographic distribution of Gentiana siphonantha and G. straminea populations collected and the photos of the two species. (A) The distribution of G. siphonantha and G. straminea. Map on top left shows the natural distribution of the two species (shadowed areas). Species tree on top right shows the phylogenetic position of the two species from Chen et al. (2020); (B) G. straminea; (C) G. straminea.
Previous field studies have demonstrated that the flowers of G. siphonantha and G. straminea are both protandrous and herkogamous to prevent self-pollination (He and Liu, 2004; Duan et al., 2005; Hou et al., 2008). Despite the obvious dissimilarity in floral morphology, a single species of bumblebee (i.e., Bombus sushkini Skorikov) has been identified as the most effective pollinator for both gentians (He and Liu, 2004; Duan et al., 2005; Hou et al., 2008). And there is increasing evidence to suggest that such a pollinator sharing could increase the possibility of gene flow between plant species (Wang et al., 2016; Yan et al., 2017). In addition, a few natural hybrids between G. siphonantha and G. straminea have been reported in their sympatric regions (Li et al., 2008). Population transcriptomics has been widely employed to infer the timing and prevalence of gene flow induced by inter-lineage hybridization (Ru et al., 2018; Ma et al., 2019; Wang et al., 2019; Li et al., 2020). Here, we aimed to address (i) whether gene flow between G. siphonantha and G. straminea can be detected using population transcriptomic data and (ii) whether gene flow between these two species is related to range expansion.
Both diploids and tetraploids have been reported for Gentiana straminea, while only diploids for G. siphonantha (Yuan, 1993). To ensure the accuracy in identifying orthologous genes, we only sampled diploids of G. straminea from three geographic locations with known karyotype information. For G. siphonantha, we sampled individuals from five geographic locations (Figure 1 and Supplementary Table S1). We collected the materials of the two species from one sympatric site and all individuals there were found to have very clear morphological delimitations without morphologically intermediate hybrids as found before in another sympatric site (Li et al., 2008). The collected fresh leaves were immediately frozen in liquid nitrogen and then stored in refrigerator (−80°C) before RNA extraction. Total RNA was isolated using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, United States), and residual DNA was removed using TURBO DNA-free Kit (Thermo Fisher Scientific, Waltham, MA, United States). Double-strand cDNA was then synthesized from total RNA, and sequenced on the Illumina HiSeq X Ten platform with 150 bp paired-end reads. Adapter and low-quality reads were filtered according to Ru et al. (2018) and all sequencing data were deposited in NCBI under BioProject PRJNA682483. Transcriptomes of G. lhassica Burkill and G. waltonii Burkill from the same section, were downloaded and included as outgroups (data retrieved from NCBI Sequence Archive [SRA] SRR9856857 and SRR9856832).
Reference transcriptome was assembled by Trinity v2.8.4 (Grabherr et al., 2011) from the previously sequenced transcriptome of G. straminea (Chen et al., 2020) and only the longest isoforms were retained. The redundancy was evaluated using CD-HIT-EST v4.6.8 (W. Li and Godzik, 2006). For each sequenced individual, clean reads were mapped to the reference transcriptome and plastome of G. officinalis Harry Sm., respectively, using BWA-MEM v0.7.17 (Li, 2013). Duplicates were removed using Picard v2.18.111, and the IndelRealigner module from the Genome Analysis Toolkit v3.8.1 (McKenna et al., 2010) was used to perform local realignment of reads around putative indels. SNPs were then called using mpileup in SAMtools v1.5 (Li et al., 2009). Here, the base quality and mapping quality were set to 20 and 30, respectively, and only those SNPs with a minimum quality score of 10 and a minimum depth of 5× were retained. In addition, SNPs at or within 5 bp from any indels were removed and those with minor allele frequency (MAF) <5% were also removed. Variant sites with significant deviation from Hardy-Weinberg equilibrium (P = 0.001) were excluded.
Maximum likelihood phylogeny was estimated using RAxML (Stamatakis, 2006) with the GTRGAMMA model, and bootstrap support was estimated using 100 bootstrap replicates. Genetic structure of the two species was assessed via ADMIXTURE analysis and principal components analysis (PCA) with the filtered SNPs. We applied PLINK v1.07 (Purcell et al., 2007; Danecek et al., 2011) with parameter—indep-pairwise 50 5 0.4 to reduce the linkage disequilibrium effect. SmartPCA from EIGENSOFT v7.2.1 (Broad Institute of Harvard and Massachusetts Institute of Technology, Cambridge, MA, United States) was used to conduct PCA analysis (Price et al., 2006). The ancestry of each individual was evaluated by ADMIXTURE v1.3.0 (Alexander and Lange, 2011) and the varying group number (K) was set from 1 to 5. The optimal K was determined by lower cross-error validation. Because the individuals of G. siphonantha collected from the two southern sites showed some ancestry from G. straminea, we estimated the genetic differentiation (FST, Weir and Cockerham, 1984) among the northern and the southern individuals of G. siphonantha and G. straminea using Vcftools v0.1.15 (Danecek et al., 2011), respectively. Meanwhile, the genetic differentiations among individuals collected from all eight sampling locations were also estimated.
Fastsimcoal2 was employed to infer the times of divergence and gene flow based on the multidimensional site frequency spectrum for G. siphonantha and G. straminea (Fagundes et al., 2007; Excoffier and Foll, 2011). In order to mitigate the effects induced by selection, we only analyzed the SNPs at fourfold degenerate sites (with no missing data across all individuals). In addition, SNPs within 5 bp of each other and those that significantly deviated (P < 0.05) from Hardy-Weinberg equilibrium were also excluded. We then calculated the mutation rate via fossil-calibrated approach (i.e., μ = D∗g/(2∗T), where D is the observed frequency of pairwise differences between two species, T is the estimated divergence time and g is the generation time for two species). To estimate D, the 1:1 orthologs between Swertia macrosperma C. B. Clarke (Chen et al., 2020) and G. straminea, G. siphonantha, respectively, were identified by Orthofinder version 2.2.3 (Emms and Kelly, 2015), which were followed by sequence alignment via Muscle version 3.8.1 (Edgar, 2004). The fourfold degenerate sites were extracted from the alignments, which was followed by pairwise difference calculation between two species. The divergence time was 30.4 Mya between Swertia and Gentiana (Chen et al., 2020). Based on the Ds and divergence times, estimated mutation rates were S. macrosperma-G. siphonantha, 8.38 × 10–9 mutations per site per generation and S. macrosperma-G. straminea, 7.89 × 10–9 mutations per site per generation, respectively. We used the average of these two mutations rates, that is, 8.13 × 10–9 mutations per site per generation to infer the demographic history. Parameter estimates were obtained via the composite ML approach with 4 hypothesized models: continuous gene flow between the two species since their initial divergence (TDIV) from the common ancestor (Model 1), gene flow between the two species since their initial divergence (TDIV) to a specific time (TCON) in the past (Model 2), the second gene flow between the two species after their initial divergence (TCON) without gene flow (Model 3) and no gene flow between the two species since they diverged (Model 4). Overall ML estimates were obtained from 40 independent runs, with 50,000–100,000 coalescent simulations and 10–40 likelihood maximization algorithm cycles. Akaike’s information criterion and Akaike’s weight of evidence were employed to assess the relative fit of each model (Excoffier et al., 2013).
We also estimated the presence of gene flow using Treemix v.1.13 (Pickrell and Pritchard, 2012). First, the individuals of Gentiana siphonantha and G. straminea collected from eight locations and the two outgroup species were pooled into nine groups (i.e., eight from the respective individuals collected from eight locations and one from the two outgroup species). Then, the presence of different number of migration events (m = 0–5, each with 5 replicates.) were tested with a block size of 100 SNPs (k = 100). Meanwhile, the optimal number of migration events were determined by the R package OptM with the linear method (Fitak, unpublished).
Short reads from 33 individuals were newly generated in this research and the average number of reads per individual was 21,461,975 (ranging from 18,327,655 to 26,411,399). Meanwhile, we also included transcriptomic reads from two species (Gentiana lhsicca and G. waltonii) generated from a previous research as outgroups (Chen et al., 2020). In addition, transcriptomic reads of G. straminea generated from this study were used to assemble the reference transcriptome. The assembled reference transcriptome had a contig N50 of 1,784 bp and 9.90% missing BUSCOs, suggesting a well assembled reference for further analysis.
After filtering procedure described, we retained 698,375 SNPs for population structure analysis of the two species. Over half of all SNPs (383,173) are shared among the individuals of Gentiana siphonantha collected from northern and southern area and G. straminea (Supplementary Figure S1). Phylogenetic analysis of whole transcriptomic SNPs revealed that G. siphonantha and G. straminea forms two strongly supported (i.e., ≥90 bootstrap support) monophyletic clades, respectively (Figure 2). However, one individual of G. straminea clustered with G. siphonantha in our plastome SNP-based phylogenetic analysis (Supplementary Figure S2). Three clusters were identified by PCA analysis, with 4.45 and 2.14% of the total variance explained by the first two components, respectively, among the identified clusters, one consists of all individuals of G. straminea collected, one comprises the individuals of G. siphonantha collected from three northern locations of and the third contains the individuals of G. siphonantha collected from two southern locations that are sympatric or parapatric to G. straminea (Figure 2). Cross-validation error analysis shows that two genetic groups (K = 2) is the optimal classification for the ADMIXTURE analysis (Supplementary Figure S3). When K = 2, G. straminea and the individuals of G. siphonantha collected from northern area comprise two distinct clusters and the southern individuals of G. siphonantha collected shows admixture pattern with 85% of ancestry from G. siphonantha and 15% of ancestry from G. straminea. When K = 3, the individuals of G. straminea formed two distinctive clusters and the individuals of northern G. siphonantha formed the third cluster, while all individuals of southern G. siphonantha contained about 75% ancestry of G. straminea and 25% ancestry from the two clusters of G. straminea (Supplementary Figure S4). Transcriptome-wide differentiation (FST) between two relatively pure cluster for G. siphonantha and G. straminea was greater than that between the southern individuals of G. siphonantha and G. straminea (Figure 3 and Supplementary Figure S5).
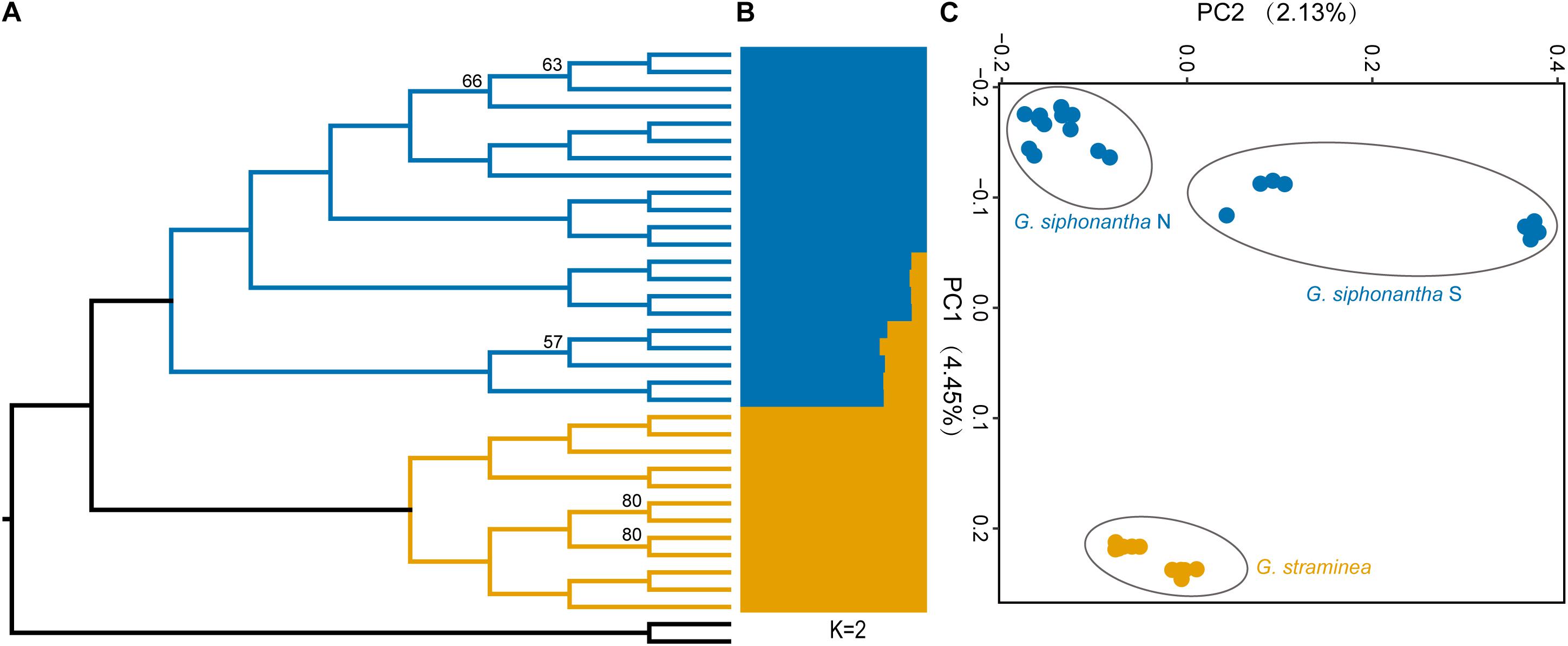
Figure 2. Phylogenetic and population genetic analyses of Gentiana siphonantha and G. straminea based on SNPs. (A) Maximum likelihood- based phylogenetic analysis of the populations of G. siphonantha and G. straminea. (B) Population structure plots with K = 2. (C) Principal component analysis plot of the first two components.
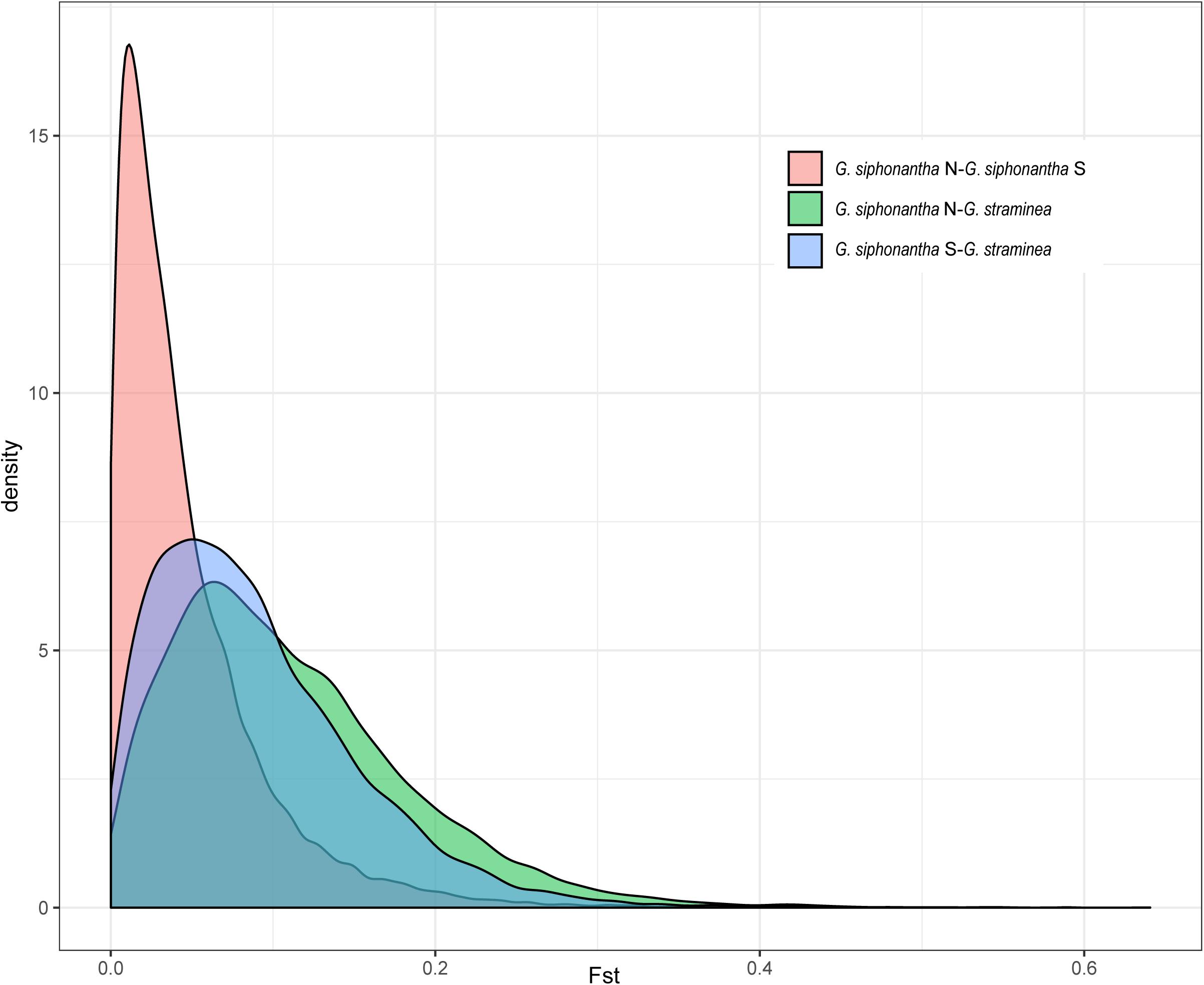
Figure 3. Distribution of genetic differentiation (FST) among the northern and southern populations of Gentiana siphonantha and G. straminea based on nuclear SNP dataset.
Coalescent-based simulations were employed to estimate the levels of gene flow among the two species identified (Excoffier et al., 2013). Of the four candidate migration models (Figure 4) explored, the best-fitting one identified (i.e., maximum Akaike’s weight value, Supplementary Table S2) includes two out of the four possible migration (Figure 4). This best-fit model (model 3) revealed the second gene flow between G. siphonantha and G. straminea since 20,000 years ago after their initial divergence. The estimated gene flow from G. straminea to G. siphonantha was much higher than vice versa (i.e., 7.70 × 10–5 vs. 1.16 × 10–5, Figure 4). The OptM analysis showed that two migration events were the optimal estimation (Supplementary Figure S6A), under which estimation, the migration events were detected from individuals of Gentiana siphonantha collected from one location to individuals of G. straminea collected from two locations (Supplementary Figure S6B).
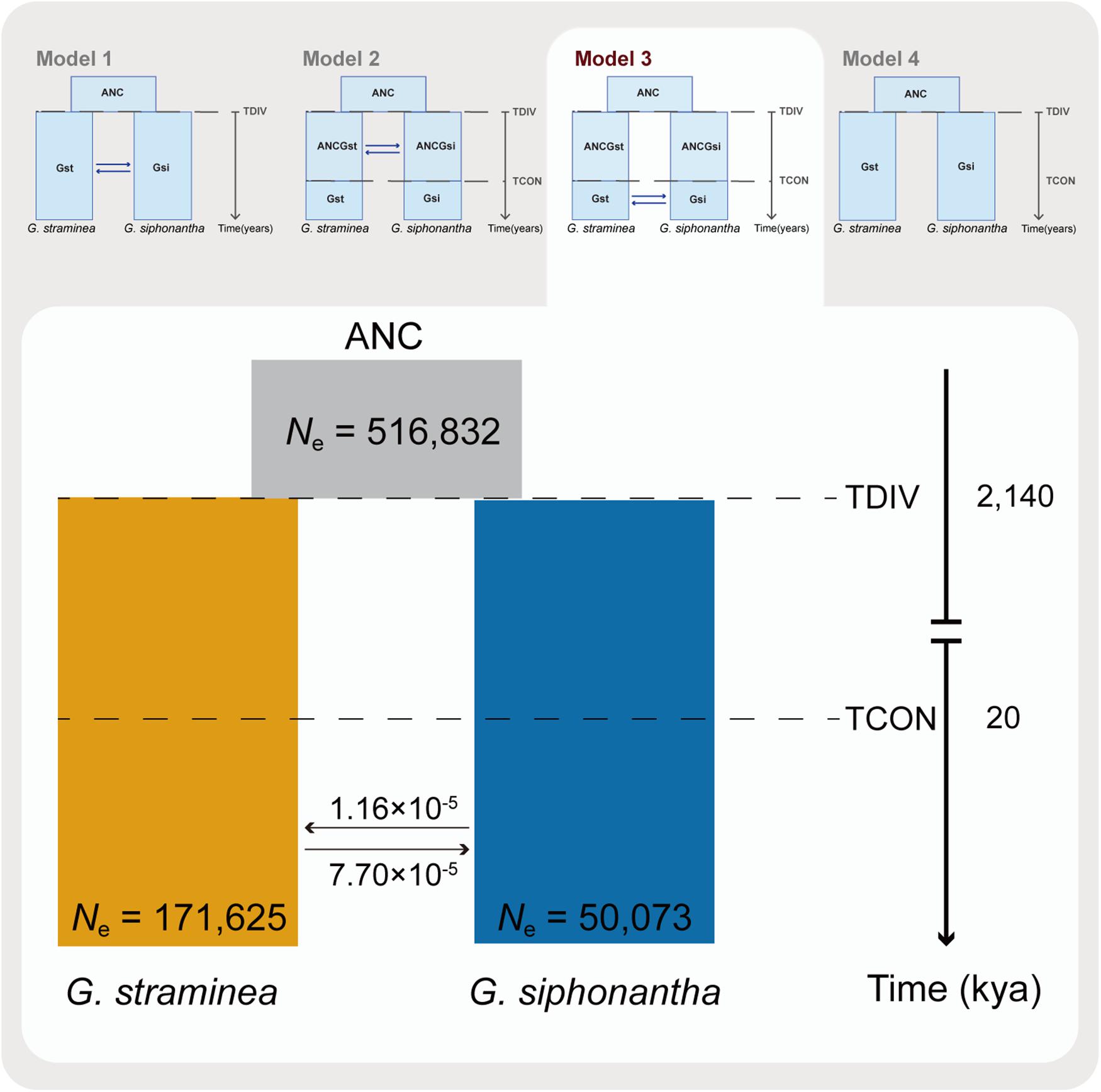
Figure 4. Schematic of demographic history of Gentiana siphonantha and G. straminea. Graphic summary of the models (i.e., model 1: there has been continuous gene flow since TDIV; model 2: there had been gene flow between the two species from TDIV to TCON; model 3: there has been gene flow from TCON to present; Model 4: there has been no gene flow between the two species since TDIV) explored were shown in the upper part, where ANC denotes the ancestor of the two species, TDIV means the time of divergence between the two species, TCON stands for a specific time in the past when there was gene flow between the two species. The best-fitting model was shown in detail in the lower part of the photo, where the values close to arrows indicate migration rate per generation and both the divergence times and estimated population size are also indicated.
A previous research showed that the individuals of Gentiana siphonantha and G. straminea occurred sympatrically or parapatrically might have experienced historical hybridization and gene flow, which could have resulted in genetic homogenization at ITS (Internal Transcribed Spacer DNA) loci and shared cpDNA haplotypes despite well morphological delimitations (Hu et al., 2016). Except for hybridization after speciation due to the second contact, incomplete lineage sorting could also explain these observed genetic variations between and within each morphological units (Wang et al., 2018; Zhang et al., 2019). If hybridization did occur in the contact zone in the current or past of the two species, such introgressions could be clearly detected by population genomic data. We sampled individuals of G. siphonantha that are geographically isolated from G. straminea and that occur sympatrically with G. straminea. Our results clearly support that hybridization did occur between these two non-sister species in the following three aspects. First, our population structure analyses clearly suggested that southern individuals of G. siphonantha collected with overlapped distributions to G. straminea contained genomic introgressions from this species. Second, our plastome-SNP based phylogenetic analysis suggested that one individual of G. straminea with very clear morphological delimitation nested deeply inside G. siphonantha (Supplementary Figure S2). This phylogenetic relationship suggested that hybridization resulted in a plastome introgression from G. straminea to G. siphonantha. Finally, our coalescent simulations of genomic variations within and between two species suggested that continuous gene flow occurred between the two species since 20,000 years ago. This gene flow was caused by the interspecific hybridizations, which might have also leaded to homogenization of the nuclear ITS sequences of the two species found before (Hu et al., 2016). This likely hybridization was also supported by the field pollination observations. In the sympatric distribution of both species, they shared the same pollinators and overlapped flowering time although G. straminea flowers usually early than G. siphonantha (Hou et al., 2008).
In addition, we further found asymmetrical gene flow between the two species, mainly from the widespread G. straminea to the narrowly distributed G. siphonantha. This asymmetry was indicated not only by estimated gene flow between the two species (Figure 4), but also based on genomic composition of the two species: only G. straminea-specific mutations were detected in individuals of G. siphonantha with sympatric or parapatric distributions. In addition, plastomes were maternally inherited in the family Genetianaceae (Lu et al., 2015). One G. straminea individual within the G. siphonantha plastome clade suggests that G. siphonantha had served as the maternal parent during the past hybridization. However, this individual clustered with G. straminea in the nuclear SNP phyloeny, suggesting the repeated backcrossing and strong gene flow with G. straminea. Therefore, it is likely that the widespread G. straminea with a large number of individuals invaded the already occupied region by G. siphonantha and resulted in such an asymmetrical and secondary gene flow after two species had diverged initially without gene flow. Meanwhile, a previous phylogeographic study of the aridity-tolerant G. straminea supports this hypothesis. Phylogeographic analyses and ecological niche modeling demonstrated that this species survived and diversified in the central QTPsl, but expanded to the QTPsl edges during Quaternary glacial oscillations (Lu et al., 2015). Thus, the expanded distribution of G. straminea might have reached the northeast QTPsl where G. siphonantha occurred during the Last Glacial Maximum (LGM, 20,000 year ago). The following overlapped distribution of these two species made it possible for them to hybridize with each other and resulted in the secondary gene flow. In addition, such hybridizations might have also occurred between closely related species or intra-specific lineages of other genera in the QTPsl because of the Quaternary climatic oscillations (Liu et al., 2014). For example, hybridizations occurred and even one hybrid lineage originated in the genus Cupressus when two parental lineages overlapped their distributions during this stage (Li et al., 2020).
Our results suggest evolution and diversification of plants living in high mountains such as Qinghai-Tibet Plateau could be similar to the “flickering connectivity system” (FCS) of the north Andean páramos (Flantua et al., 2019; Muellner-Riehl, 2019). The FCS suggests repeatedly isolate and reconnect adjacent populations potentially accelerating speciation events. Our results indicate the secondary contact and followed hybridization promoted microevolution in mountain habitats, which could have impact on the diverging trajectory of the two species. It should be noted that only individuals from few locations were used for the two species in the present study because of the high cost and unknown ploidies of the unsampled sites for the widespread G. straminea. Further studies with wider sampling range and genome-wide data will be needed to reveal the detailed dynamic settings of evolution and genomic context linked to response to climate changes in Qinghai-Tibet Plateau. Overall, our population genomic analyses based on transcriptomes of 33 individuals from multiple locations of Gentiana siphonantha and G. straminea supported their historical hybridizations since the LGM. These hybridizations leaded to the asymmetrical gene flow from the widespread G. straminea to narrowly distributed G. siphonantha possibly because of their contrasted occurrence frequencies in the field. These findings are consistent with a previous phylogeographic study that G. straminea expanded its distributional range from the central to northeast QTPsl. Our results highlight the significance of population genomic data in determining timescale and direction of gene flow during interspecific historical hybridization.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA682483.
QH, JL, and CC designed the research. CC and LZ carried out the field research and collected the samples. CC and WY analyzed the data. CC, QH, JL, and ZX wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the National Natural Science Foundation of China (31700323 and 31590821).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2021.638230/full#supplementary-material
Alexander, D. H., and Lange, K. (2011). Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinformatics 12:246. doi: 10.1186/1471-2105-12-246
Anadón, J. D., Graciá, E., Botella, F., Giménez, A., Fahd, S., and Fritz, U. (2015). Individualistic response to past climate changes: niche differentiation promotes diverging quaternary range dynamics in the subspecies of Testudo graeca. Ecography 38, 956–966. doi: 10.1111/ecog.01163
Chen, C. L., Zhang, L., Li, J. L., Mao, X. X., Zhang, L. S., Hu, Q. J., et al. (2020). Phylotranscriptomics reveals extensive gene duplication in the subtribe Gentianinae (Gentianaceae). J. Syst. Evol. 1–11. doi: 10.1111/jse.12651
Confer, J. L., Porter, C., Aldinger, K. R., Canterbury, R. A., Larkin, J. L., and Mcneil, D. J. Jr. (2020). Implications for evolutionary trends from the pairing frequencies among golden-winged and blue-winged warblers and their hybrids. Ecol. Evol. 10, 10633–10644. doi: 10.1002/ece3.6717
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
Duan, Y. W., He, Y. P., and Liu, J. Q. (2005). Reproductive ecology of the Qinghai-Tibet Plateau endemic Gentiana straminea (Gentianaceae), a hermaphrodite perennial characterized by herkogamy and dichogamy. Acta Oecol. 27, 225–232. doi: 10.1016/j.actao.2005.01.003
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Emms, D. M., and Kelly, S. (2015). OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16, 1–14. doi: 10.1186/s13059-015-0721-2
Excoffier, L., Dupanloup, I., Huerta-Sánchez, E., Sousa, V. C., and Foll, M. (2013). Robust demographic inference from genomic and SNP data. PLoS Genet. 9:e1003905. doi: 10.1371/journal.pgen.1003905
Excoffier, L., and Foll, M. (2011). fastsimcoal: a continuous-time coalescent simulator of genomic diversity under arbitrarily complex evolutionary scenarios. Bioinformatics 27, 1332–1334. doi: 10.1093/bioinformatics/btr124
Fagundes, N. J. R., Ray, N., Beaumont, M., Neuenschwander, S., Salzano, F. M., Bonatto, S. L., et al. (2007). Statistical evaluation of alternative models of human evolution. Proc. Natl. Acad. Sci. U.S.A. 104, 17614–17619. doi: 10.1073/pnas.0708280104
Flantua, S. G. A., O’Dea, A., Onstein, R. E., Giraldo, C., and Henry, H. (2019). The flickering connectivity system of the North Andean Páramos. J. Biogeogr. 46, 1808–1825. doi: 10.1111/jbi.13607
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
He, Y. P., and Liu, J. Q. (2004). Pollination ecology of Gentiana straminea Maxim.(Gentianaceae), an alpine perennial in the Qinghai-Tibet Plateau. Acta Ecol. Sin. 24, 215–220.
Hou, Q. Z., Meng, L. H., and Yan, H. L. (2008). Pollination ecology of Gentiana siphonantha (Gentianaceae) and a further comparison with its sympatric congener species. J. Syst. Evol. 46, 554–562. doi: 10.3724/SP.J.1002.2008.07063
Hu, Q., Peng, H., Bi, H., Lu, Z., Wan, D., and Wang, Q. (2016). Genetic homogenization of the nuclear ITS loci across two morphologically distinct gentians in their overlapping distributions in the Qinghai-Tibet Plateau. Sci. Rep. 6:34244. doi: 10.1038/srep34244
Huerta-Sánchez, E., Jin, X., Xun, X., Bianba, Z., Peter, B. M., Vinckenbosch, N., et al. (2014). Altitude adaptation in Tibetans caused by introgression of Denisovan-like DNA. Nature 512, 194–197. doi: 10.1038/nature13408
Huxel, G. R. (1999). Rapid displacement of native species by invasive species: effects of hybridization. Biol. Conserv. 89, 143–152. doi: 10.1016/S0006-3207(98)00153-0
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997v2.
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, J., Milne, R. I., Ru, D., Miao, J., Tao, W., Zhang, L., et al. (2020). Allopatric divergence and hybridization within Cupressus chengiana (Cupressaceae), a threatened conifer in the northern Hengduan Mountains of western China. Mol. Ecol. 29, 1250–1266. doi: 10.1111/mec.15407
Li, W., and Godzik, A. (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659. doi: 10.1093/bioinformatics/btl158
Li, X., Wang, L., Yang, H., and Liu, J. Q. (2008). Confirmation of natural hybrids between Gentiana straminea and G. siphonantha (Gentianaceae) based on molecular evidence. Front. Biol. China 3, 470–476. doi: 10.1007/s11515-008-0076-0
Liang, Q., Xu, X., Mao, K., Wang, M., Wang, K., Xi, Z., et al. (2018). Shifts in plant distributions in response to climate warming in a biodiversity hotspot, the Hengduan Mountains. J. Biogeogr. 45, 1334–1344. doi: 10.1111/jbi.13229
Liu, J. Q., Duan, Y. W., Hao, G., Ge, X. J., and Sun, H. (2014). Evolutionary history and underlying adaptation of alpine plants on the Qinghai-Tibet Plateau. J. Syst. Evol. 52, 241–249. doi: 10.1111/jse.12094
Lu, Z., Chen, P., Bai, X., Xu, J., He, X., Niu, Z., et al. (2015). Initial diversification, glacial survival, and continuous range expansion of Gentiana straminea (Gentianaceae) in the Qinghai–Tibet Platea. Biochem. Syst. Ecol. 62, 219–228. doi: 10.1016/j.bse.2015.09.005
Ma, Y. Z., Wang, J., Hu, Q. J., Li, J. L., Sun, Y. S., Zhang, L., et al. (2019). Ancient introgression drives adaptation to cooler and drier mountain habitats in a cypress species complex. Commun. Biol. 2:213. doi: 10.1038/s42003-019-0445-z
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a mapreduce framwork for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Muellner-Riehl, A. N. (2019). Mountains as evolutionary arenas: patterns, emerging approaches, paradigm shifts, and their implications for plant phylogeographic research in the Tibeto-Himalayan region. Front. Plant Sci. 10:195. doi: 10.3389/fpls.2019.00195
Owens, G. L., Baute, G. J., and Rieseberg, L. H. (2016). Revisiting a classic case of introgression: hybridization and gene flow in Californian sunflowers. Mol. Ecol. 25, 2630–2643. doi: 10.1111/mec.13569
Pardo-Diaz, C., Salazar, C., Baxter, S. W., Merot, C., Figueiredo-Ready, W., Joron, M., et al. (2012). Adaptive introgression across species boundaries in Heliconius butterflies. PLoS Genet. 8:e1002752. doi: 10.1371/journal.pgen.1002752
Pickrell, J. K., and Pritchard, J. K. (2012). Inference of population splits and mixtures from genomewide allele frequency data. PLoS Genet. 8:e1002967. doi: 10.1371/journal.pgen.1002967
Price, A. L., Patterson, N. J., Plenge, R. M., Weinblatt, M. E., Shadick, N. A., and Reich, D. (2006). Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909. doi: 10.1038/ng1847
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A. R., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. doi: 10.1086/519795
Ru, D., Sun, Y., Wang, D., Chen, Y., Wang, T., Hu, Q., et al. (2018). Population genomic analysis reveals that homoploid hybrid speciation can be a lengthy process. Mol. Ecol. 27, 4875–4887. doi: 10.1111/mec.14909
Stamatakis, A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22, 2688–2690. doi: 10.1093/bioinformatics/btl446
Suarez-Gonzalez, A., Hefer, C. A., Christe, C., Corea, O., Lexer, C., Cronk, Q. C. B., et al. (2016). Genomic and functional approaches reveal a case of adaptive introgression from Populus balsamifera (balsam poplar) in P. trichocarpa (black cottonwood). Mol. Ecol. 25, 2427–2442. doi: 10.1111/mec.13539
Todesco, M., Pascuai, M. A., Owens, G. L., Ostevik, K. L., Moyers, B. T., Hubner, S., et al. (2016). Hybridization and extinction. Evol. Appl. 9, 892–908. doi: 10.1111/eva.12367
Wang, G., Cannon, C. H., and Chen, J. (2016). Pollinator sharing and gene flow among closely related sympatric dioecious fig taxa. Proc. R. Soc. B 283:20152963. doi: 10.1098/rspb.2015.2963
Wang, K., Lenstra, J. A., Liu, L., Hu, Q., Ma, T., Qiu, Q., et al. (2018). Incomplete lineage sorting rather than hybridization explains the inconsistent phylogeny of the wisent. Commun. Biol. 1:169. doi: 10.1038/s42003-018-0176-6
Wang, T. J., Ru, D. F., Zhang, D., and Hu, Q. J. (2019). Analyses of genome-scale variation reveal divergence of two Sinalliaria species (Brassicaceae) with continuous but limited gene flow. J. Syst. Evol. 57, 268–277. doi: 10.1111/jse.12461
Weir, B. S., and Cockerham, C. C. (1984). Estimating F-statistics for the analysis of population structure. Evolution 38, 1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x
Yan, L. J., Burgess, K. S., Milne, R., Fu, C. N., Li, D. Z., and Gao, L. M. (2017). Asymmetrical natural hybridization varies among hybrid swarms between two diploid Rhododendron species. Ann. Bot. 120, 51–61. doi: 10.1093/aob/mcx039
Yuan, Y. M. (1993). Karyological studies on Gentiana section Cruciata Gaudin (Gentianaceae) from China. Caryologia 46, 99–114. doi: 10.1080/00087114.1993.10797252
Zamudio, K. R., and Savage, W. K. (2003). Historical isolation, range expansion, and secondary contact of two highly divergent mitochondrial lineages in spotted salamanders (Ambystoma maculatum). Evolution 57, 1631–1652. doi: 10.1111/j.0014-3820.2003.tb00370.x
Zhang, D., Hao, G. Q., Guo, X. Y., Hu, Q. J., and Liu, J. Q. (2019). Genomic insight into “sky island” species diversification in a mountainous biodiversity hotspot. J. Syst. Evol. 57, 633–645. doi: 10.1111/jse.12543
Keywords: gene flow, introgression, interspecific hybridization, Gentiana siphonantha, Gentiana straminea
Citation: Chen C, Yang W, Liu J, Xi Z, Zhang L and Hu Q (2021) Population Transcriptomics Reveals Gene Flow and Introgression Between Two Non-sister Alpine Gentians. Front. Ecol. Evol. 9:638230. doi: 10.3389/fevo.2021.638230
Received: 05 December 2020; Accepted: 09 February 2021;
Published: 26 February 2021.
Edited by:
Alison G. Nazareno, Federal University of Minas Gerais, BrazilReviewed by:
Jacqueline Souza Lima, Federal University of Rio Grande do Sul, BrazilCopyright © 2021 Chen, Yang, Liu, Xi, Zhang and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Quanjun Hu, aHVxdWFuanVuQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.