
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol., 26 April 2021
Sec. Behavioral and Evolutionary Ecology
Volume 9 - 2021 | https://doi.org/10.3389/fevo.2021.632590
This article is part of the Research TopicAdvances in the Evolutionary Ecology of TermitesView all 9 articles
 Edimar A. Moreira1†
Edimar A. Moreira1† Gabriela F. Persinoti2†
Gabriela F. Persinoti2† Letícia R. Menezes1Douglas A. A. Paixão2
Letícia R. Menezes1Douglas A. A. Paixão2 Thabata M. Alvarez3
Thabata M. Alvarez3 João P. L. Franco Cairo4†Fabio M. Squina5Ana Maria Costa-Leonardo6
João P. L. Franco Cairo4†Fabio M. Squina5Ana Maria Costa-Leonardo6 Andre Rodrigues7
Andre Rodrigues7 David Sillam-Dussès8
David Sillam-Dussès8 Alberto Arab1*
Alberto Arab1*Lignocellulose digestion in termites is achieved through the functional synergy between gut symbionts and host enzymes. However, some species have evolved additional associations with nest microorganisms that collaborate in the decomposition of plant biomass. In a previous study, we determined that plant material packed with feces inside the nests of Cornitermes cumulans (Syntermitinae) harbors a distinct microbial assemblage. These food nodules also showed a high hemicellulolytic activity, possibly acting as an external place for complementary lignocellulose digestion. In this study, we used a combination of ITS sequence analysis, metagenomics, and metatranscriptomics to investigate the presence and differential expression of genes coding for carbohydrate-active enzymes (CAZy) in the food nodules and the gut of workers and soldiers. Our results confirm that food nodules express a distinct set of CAZy genes suggesting that stored plant material is initially decomposed by enzymes that target the lignin and complex polysaccharides from fungi and bacteria before the passage through the gut, where it is further targeted by a complementary set of cellulases, xylanases, and esterases produced by the gut microbiota and the termite host. We also showed that the expression of CAZy transcripts associated to endoglucanases and xylanases was higher in the gut of termites than in the food nodules. An additional finding in this study was the presence of fungi in the termite gut that expressed CAZy genes. This study highlights the importance of externalization of digestion by nest microbes and provides new evidence of complementary digestion in the context of higher termite evolution.
Termites (Blattodea: Isoptera) are eusocial cockroaches comprising over 3,000 species widespread in tropical and subtropical regions. These insects play an important role in ecosystems through the decomposition of plant lignocellulose (Brune, 2014; Mikaelyan et al., 2015), having a positive impact in nutrient cycling and soil-water dynamics through the construction of their nests and tunnel networks in soil (Neupane et al., 2015; Siebers et al., 2015; Jouquet et al., 2018).
All termites live in nests containing hundreds to thousands of individuals divided in castes (reproductive individuals, workers, and soldiers) that cooperatively conduct the tasks of the colony. Nests protect termites against the environment and predators and offer a place for feeding, reproduction, and nursery (Bignell and Eggleton, 2000). Higher termites (Termitidae) have evolved remarkable nesting strategies including the construction of epigeal mounds which can reach a prominent size in some species (Korb, 2011). Mounds are usually made of micro-aggregates of soil and feces forming porous structures that control the microclimate (Korb, 2003; Singh et al., 2019) and could function as an external reservoir of microorganisms (Fall et al., 2007; Manjula et al., 2014) or even like a fermenting chamber (Korb, 2011; Schmidt et al., 2014). Some authors have hypothesized that externalization of digestion by nest microorganisms, as seen in the subfamilies Macrotermitinae and Sphaerotermitinae (Garnier-Sillam et al., 1989; Aanen et al., 2002), was the first step in the evolution of Termitidae and might have driven the loss of gut protozoans and the acquisition of specialized lignocellulolytic bacteria resulting in higher efficiency in plant decomposition (Brune, 2014; Chouvenc et al., 2021).
Cornitermes cumulans (Kollar, 1932) (Termitidae: Syntermitinae) is a mound-building termite in pastures and savannas of central South America and in some areas its nest density is exceptionally high compared to other termite species (Coles De Negret and Redford, 1982; Buschini, 2006). The hard epigeal mounds are made externally of soil with an internal core of soft carton made from fecal material and soil particles (Redford, 1984). Although feeding in situ has been reported (Coles De Negret and Redford, 1982), C. cumulans is a harvester termite, cutting and transporting grass and litter into its mound. The plant material is then stored in nodules made with feces and saliva (Figure 1A) and subsequently consumed by workers (Lima and Costa-Leonardo, 2007). Food nodules showed a high hemicellulolytic activity possibly mediated by bacteria and fungi. It is possible that the pre-processing of plant material by the action of microbes in the food nodules before gut passage acts as a complementation of biomass digestion in C. cumulans. However, only partial taxonomic information for fungal strains was reported because less than 10% of reads were assigned to order level at the time (Menezes et al., 2018).
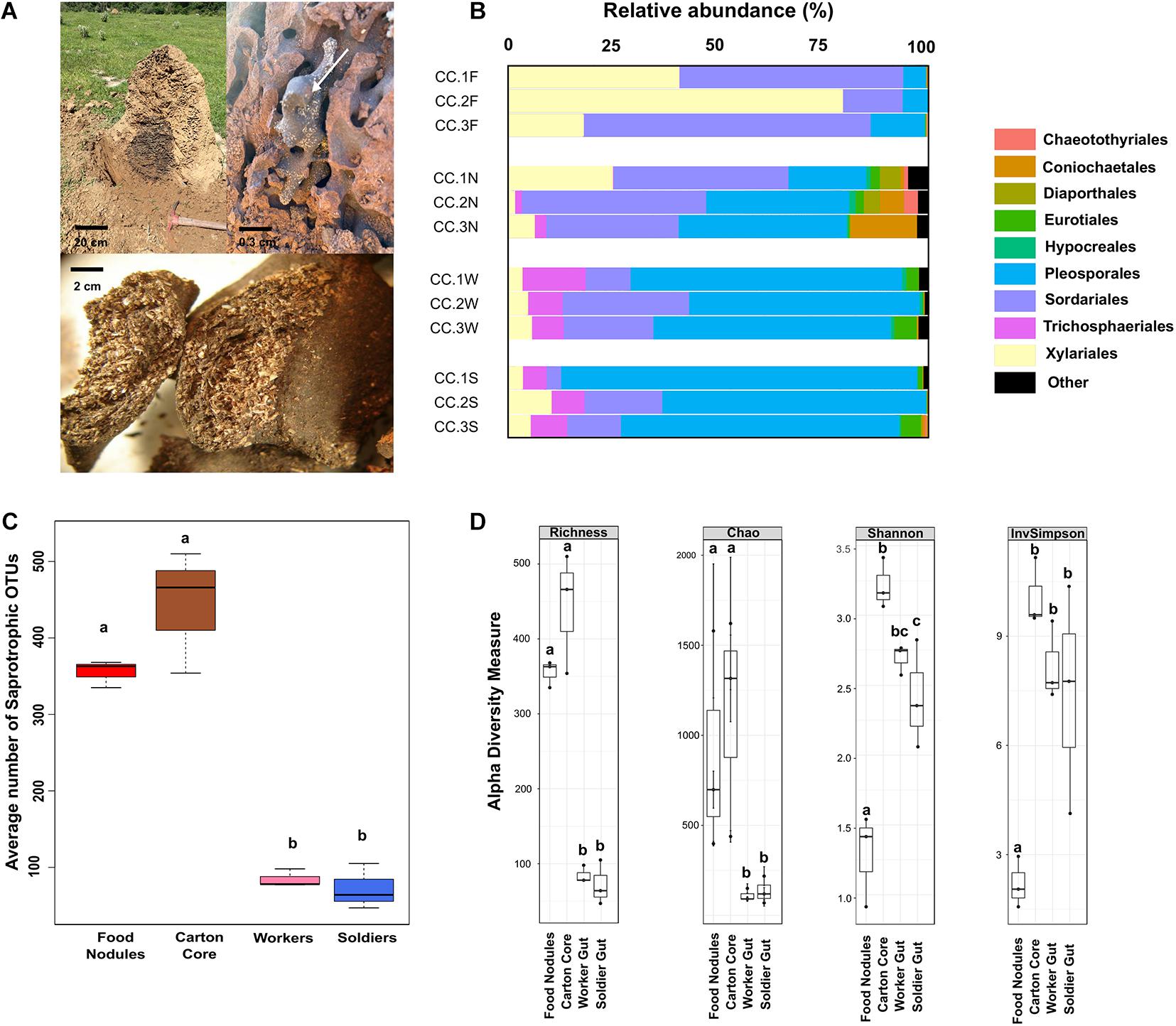
Figure 1. (A) Nest of Cornitermes cumulans showing the central carton core with a food nodule (arrow) and a zoomed picture of the plant material inside a food nodule. (B) Proportion of reads assigned to different fungal orders in gut and nest substrates for each sampled colony. (C) Average number of saprotrophic fungal OTUs associated to each one of the samples. (D) Alpha-diversity indices estimated for fungal ITS amplicons. Graphics report the observed operational taxonomic units (OTUs) richness, Chao1 estimator of richness, and Shannon’s and Simpson’s indices. Boxplots report median, upper and lower quartiles, and maximum and minimum values. Different letters denote significant differences (p-values < 0.05 in GLMM). Samples codes: CC1 = colony 1, CC2 = colony 2, and CC3 = colony 3. F, food nodules; N, carton nest core; W, worker gut; S, soldier gut.
In this study, we assessed the taxa and guilds that differentiate fungal communities associated to the food nodules of C. cumulans by using deep taxonomic and guild assignment of datasets generated through internal transcribed spacer (ITS) sequencing. To investigate the potential complementation of lignocellulose digestion among the termite host, the gut symbionts and the microbiota of food nodules, we then performed metagenomic and metatranscriptomic analyses on this tri-partite system to assess: (i) the repertoire and functional contribution of lignocellulolytic enzymes; (ii) how these enzymes are linked taxonomically to the termite holobiont (host, gut, and nest microbiota); and (iii) the differences of enzyme functionality across sterile castes.
Internal transcribed spacer reads were obtained from raw Illumina datasets of three (3) C. cumulans colonies (CC1, CC2, and CC3) deposited in the European Nucleotide Archive (ENA) with accession no. PRJEB17080 (Menezes et al., 2018). Libraries were processed using the UPARSE pipeline (Edgar, 2013). Paired end reads were first merged using fastq_mergepairs from USEARCH package version 8.1.1803. Reads with a minimum overlap of 50 bp and a maximum expected error of 0.5 were mapped to fungal ITS2 region using ITSx software (Bengtsson-Palme et al., 2013). Reads were further compared to ITS2 UCHIME reference dataset to filter chimera sequences using chimera UCHIME (Edgar et al., 2011), also implemented in USEARCH package. The filtered reads were then subjected to clustering into OTUs (operational taxonomic units) at 97% of sequence similarity according to UPARSE-OUT algorithm. The OTU table was generated by mapping the reads from each sample back to the OTUs. Taxonomic assignment was performed using sintax command as implemented in USEARCH version 10.0.240 software using RDP Warcup training set v2 (Deshpande et al., 2016) database. Fungal genera with sintax cutoff > 0.7 were considered to discriminate taxa that significantly differentiate fungal communities. Relative abundances were calculated as the number of reads per taxon. OTUs were further classified into ecological guilds using FUNGuild, only considering assignments that were either “highly probable” or “probable” (Nguyen et al., 2016). Fungi were then assigned to saprotrophs (undefined saprotrophs and dung saprotrophs), symbiotrophs (endophytic and lichenized fungi) and plant pathogens. We also used publicly available fungal datasets generated through Illumina sequencing of the ITS2 region, that included samples of: (1) soil from pastures located nearby our sampling sites, which commonly contain C. cumulans mounds (de Oliveira et al., 2020); (2) the carton nest core of the sympatric non-food-storing termite Procornitermes araujoi (Syntermitinae) which builds mounds with outer walls made of thin layer of soil with an internal layer lined with fecal material (Supplementary Figure 1; Moreira et al., 2018); (3) different species of bark beetles (Rassati et al., 2019; Seibold et al., 2019); and (4) nests of a leaf-cutting bee (Rothman et al., 2019).
We used R version 3.4.4 (R Core Team, 2018) to conduct analyses using different software packages. Alpha-diversity estimates were calculated using the function plot_richness in the phyloseq package (McMurdie and Holmes, 2013). Rarefaction was assessed using vegan package (Oksanen et al., 2018). Generalized linear mixed models (GLMM) were performed to check for overall significant differences of α-diversity estimates and the total number of OTUs among samples. The type of substrate was included as fixed effects, while the colony number was included as random effect. Multiple comparisons were conducted by general linear hypotheses using the glht-function in the multcomp package (Hothorn et al., 2008). A redundancy analysis (RDA) (999 permutations) of Hellinger-transformed abundance data using the vegan package (Oksanen et al., 2018) was used to evaluate whether nest and gut substrates differ in fungal composition at the OTU level. The anova function from the package vegan were used to test the significance of RDA models and axes, respectively. We used Calypso web-server (Version 8.58), an online platform (Zakrzewski et al., 2017), to construct taxonomic heatmaps and the RDA of fungal metanalysis. The transmission of fungi between nest substrates and the gut of workers and soldiers was evaluated by analyzing the number of intersecting OTUs using the UpSetR package (Conway et al., 2017). We used linear discriminant analysis (LDA) effect size (LEfSe) to detect taxa that significantly differentiate fungal communities (Segata et al., 2011). To assess LEFSe, we used Galaxy web application, an online platform for the evaluation of multiple microbial community composition data (Afgan et al., 2016). The LDA score threshold was 4.0. Relative abundances less than 0.01% of the total reads were omitted from further analysis. We then identified variation in taxa using GLMM. To identify OTUs that were significantly different among samples, we used DESeq2 package (Love et al., 2014). Statistical significance was defined as p ≤ 0.05 in both statistical methods. Only OTUs identified by both LEfSe and DESeq2 were discussed. Plots were constructed with the packages ggplot2 (Wickham, 2016) and ggord (Beck, 2017).
The whole gut of 200 workers and soldiers and 50 mg of nest substrates (the carton nest core and food nodules) from colony CC1 were extracted in 2 ml tubes containing 1 mL of lysis buffer (500 mM NaCl, 50 mM Tris–HCl, pH 8.0, 50 mM EDTA, and 4% sodium dodecyl sulfate (SDS). Gut samples were obtained by anesthetizing termites on ice for 5 min and dissected with fine forceps. All the samples were kept at −20°C until DNA extraction and purification using a bead-beating protocol (Yu and Morrison, 2004). DNA integrity was assessed by agarose gel electrophoresis (1.0 w/v) and the quantification was performed using NanoDrop spectrophotometer by measuring the absorbance at 260 nm. For metagenomic sequencing purposes, a library was constructed, using the Nextera library preparation kit (Illumina), according to the manufacturer’s instructions. The prepared library was validated and quantified using the Agilent bioanalyzer 2100 system with a 12,000 DNA assay kit (Agilent) and Kapa Biosystems next-generation sequencing library qPCR kit (Kapa Biosystems), respectively. Sequencing was performed at LNBR NGS facility (CNPEM – Campinas, São Paulo, Brazil) using an Illumina HiSeq 2500 platform and applying the paired-end protocol (2 × 100-bp paired ends).
Total RNA (10 μg) was extracted from 200 termite guts and 50 mg of the nest substrates of colony CC1 using Trizol reagent protocol (Invitrogen). Trizol/Chloroform step was performed twice. The total RNA was purified using the RNeasy Plant Mini Kit (Qiagen) following manufacturer’s instructions. The quality of RNA was verified using RNAnano chip Bioanalyzer 2100 (Agilent). Good quality RNAs (RIN > 8.0) were submitted to rRNA depletion using the RiboZero rRNA Removal Kit under manufacturer instruction with a slight modification: at depletion step, we used a blend of rRNA removal solution from RiboZero Gold and RiboZero Bacteria kits (1:1) aiming to deplete both prokaryotic and eukaryotic rRNAs. Finally, the depleted RNAs were purified using Ampure XP beads, following the manufacturer’s instructions, and kept at −80°C until library preparation. A total of 50 ng of depleted RNA were used for library preparation using the TruSeq Stranded Total RNA Sample Preparation kit (Illumina), following the manufacturer’s instructions. Quality control, quantification, and sequencing were performed as described before.
Metagenome (MG) and metatranscriptome (MT) raw reads were quality checked using FastQC1 and quality-filtered using Trimmomatic v.0.36 (Bolger et al., 2014). MT reads were also inspected using SortMeRNA to remove rRNA reads, and then both MG and MT reads were taxonomically classified using Kaiju (Menzel et al., 2016). Next, the whole set of MG trimmed reads were de novo co-assembled using IDBA_UD (version 1.1.1) (Peng et al., 2012). Gene prediction and annotation of was performed using Prokka v.1.11 with the meta parameter (Seemann, 2014) and carbohydrate-active enzymes (CAZy) annotation was performed using dbCAN database (Zhang et al., 2018). Taxonomy of predicted protein coding genes were inferred according the easy-taxonomy function of MMseqs2 suite (Steinegger and Söding, 2017) to identify putative bacterial, fungal, and termite-host genes by LCA method. Both, MG and MT reads were mapped to the complete set of predicted genes recovered from the MG de novo assembly using Kallisto v. 0.46.1 (Bray et al., 2016) to estimate genes and transcripts abundance. Normalized expression of protein coding genes was estimated with the Transcripts Per Kilobase Million (TPM) method and used to analyze the abundance of expression of the predicted fungal, bacterial, and host putative CAZy genes. Homology to peptide pattern (Hotpep) was used to infer CAZy function (Busk et al., 2017).
We detected 968 fungal OTUs (2,163,723 sequences from the ITS region) associated to the gut and the nests of C. cumulans. Of these, 579 OTUs (1,370,000 sequences, 63.3% of the total) were annotated to the food nodules, 774 OTUs (739,691 sequences, 34.2%) to the carton nest core and 197 OTUs (54,032 sequences, 2.5%) to the gut of workers and soldiers. Deep taxonomic analyses using the Warcup database successfully assigned 88 and 84% of fungal reads at the order and family level, respectively. Overall, nest and gut samples yielded the phyla Ascomycota (99.9% of sequence reads) and Basidiomycota, representing 13 classes, 42 orders, 98 families, and 212 genera (Supplementary Tables 1, 2). Rarefaction curves indicated adequate sampling of fungi for a valid comparison among fungal communities (Supplementary Figure 2). The average number of OTUs was significantly higher in nest samples, the majority classified as undefined saprotrophs (on average 76.5% of sequence reads) and symbiotrophs (endophytes 4.8% and lichenized 2.5%) of the orders Pleosporales, Sordariales, and Xylariales (Ascomycota) using FUNGuild (Figures 1B,C and Supplementary Figure 3).
Observed richness was significantly lower in gut assemblages (Figure 1D). On the other hand, food nodules and carton core samples of C. cumulans nests did not differ in richness and shared approximately 99% of their sequence reads (293 OTUs), but estimated diversity was significantly lower in food nodules. Constrained ordination showed that community composition of food nodules formed a distinct cluster separated from carton nest core (F = 2.84, df = 1, p = 0.002) and gut samples (F = 6.61, df = 1, p = 0.001) (Figures 2A,B) and was characterized by the abundance of saprotrophic OTUs of Sordariales and Xylariales (Supplementary Tables 3, 4). We then applied LEfSe and DESeq2 in order to determine the variation in the composition of fungal communities and both analyses consistently showed that the saprotroph OTU 1 of Xylariaceae was strongly associated to food nodules (LDA = 5.24; p = 0.04; DESeq2: log2 fold change = −4.32, p = 0.002), whereas the saprotroph OTU 7 of Sordariales (LDA = 4.57; p = 0.04; DESeq2: log2 fold change = 1.15, p < 0.001) and the endophyte OTU 4 (Coniochaetales: Lecythophora) (LDA = 4.62; p = 0.04; DESeq2: log2 fold change = 3.26, p < 0.001) were the most prominent fungal species found in the carton nest core (Figure 2C). No differences were observed among colonies. All the results were confirmed by GLMM (Supplementary Table 5).
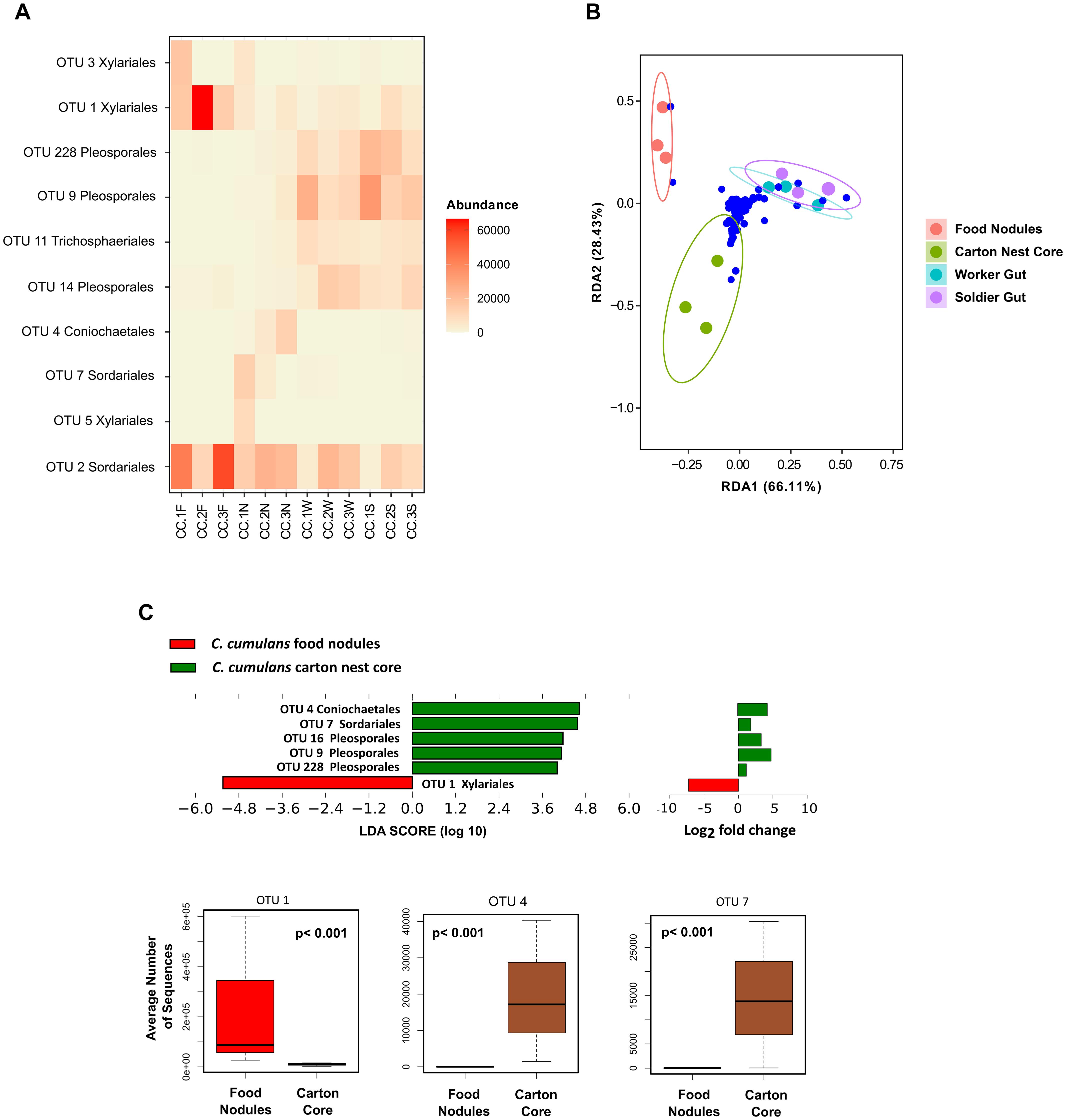
Figure 2. (A) Comparison of the relative abundances (>1%) of fungal OTUs of C. cumulans. (B) Ordination plots showing fungal communities of gut and nest samples after redundancy analysis of Hellinger transformed abundance of OTUs (blue dots) with colony number (colored circles) as conditional variable. RDA1 Nest Substrates; variance = 0.140, p-value = 0.001, and RDA1 Termite Gut; variance = 0.06, p-value = 0.002, permutations tests (n = 999). (C) Linear discriminant analysis (LDA) combined with effect size measurements (LEfSe, score > 4) and DESeq2 (p ≤ 0.05) analyses to identify the fungal OTUs responsible for the differences between nest substrates. Boxplots report median, upper and lower quartiles, and maximum and minimum values of the relative abundance of taxa identified by both DESeq2 and LEfSe analyses. A p-value < 0.05 was considered significant in GLMM analyses. See Figure 1 for samples codes.
The gut mycobiota of workers and soldiers was represented by 11 classes, 26 orders, 52 families, 90 genera, and 197 fungal OTUs (Supplementary Table 1) with the prevalence of the genus Phoma (Pleosporales) (Supplementary Tables 2, 3). Diversity and composition of fungal gut assemblages were not influenced by the type of caste (RDA, F = 0.84; df = 1; p = 0.540) (Figure 2B) and no features with significant differences were found between workers and soldiers. However, we detected a high percentage (between 89 and 99%) of transmission of fungal OTUs between nest substrates and the gut of workers and soldiers. The majority of shared OTUs belongs to Sordariales and Pleosporales. Additionally, workers and soldiers shared between 97.5 and 98.4% of their sequence reads (Supplementary Figure 4).
Analyses of the variation in community structure showed that the composition of Ascomycota fungi differed significantly among termite nests (C. cumulans and P. araujoi), soil samples and substrates associated with two phylogenetically distant insects (RDA, F = 15.47; df = 5; p = 0.001) (Supplementary Figure 5A). The nest substrates of termites formed a cluster, clearly separated from other fungal assemblages. The more prominent genera associated to termite nest substrates, like Daldinia (Xylareaceae) and Lecythophora (Coniochaetaceae), were found in lower abundances in other assemblages. However, Thielavia and Chaetomium (Chaetomiaceae) were similarly abundant in both soil and termite nests. We also found that fungal communities associated to bark beetles and bee nests formed separated clusters with their prominent genera present in lower abundances in termite nests (Supplementary Figure 5B).
The microbial assemblages from colony CC1 were further investigated by metagenomics (MG) and metatranscriptomics (MT) sequencing. Taxonomy analyses of the sequences showed a higher abundance of bacteria over fungi in both nest and gut samples (Figures 3A,B). Actinobacteria and Proteobacteria were dominant in the nest substrates, whereas Firmicutes and Spirochetes were prevalent in the termite gut, confirming our previous results with 16S rRNA sequencing (Menezes et al., 2018). Ascomycota was present in both nest and gut samples. However, a fungus of genus Neocallimastix (Chytridiomycota: Neocallimastigales) was only found in the gut of workers and soldiers. Bacillus (Firmicutes), Treponema (Spirochetes), Streptomyces (Actinobacteria), and Aspergillus (Ascomycota) were the most abundant microbial genera in the samples (Supplementary Tables 6, 7 and Supplementary Figure 6A). We did not obtain RNA from nest core samples; however, the MG showed a similar profile between the carton nest and food nodules (Figure 3A). The MT sequences from fungi were less abundant (Figure 3B), suggesting a lower metabolic activity from fungi over bacteria. Although our data was derived from a single colony (CC1), the fungal taxa responsible for the expression of CAZy transcripts showed similar abundances among the three colonies used in ITS sequencing, for food nodules (H = 0.74; df = 2; p = 0.689; Kruskal–Wallis test) and the gut of workers and soldiers (H = 1.11; df = 5; p = 0.953) (Supplementary Table 2).
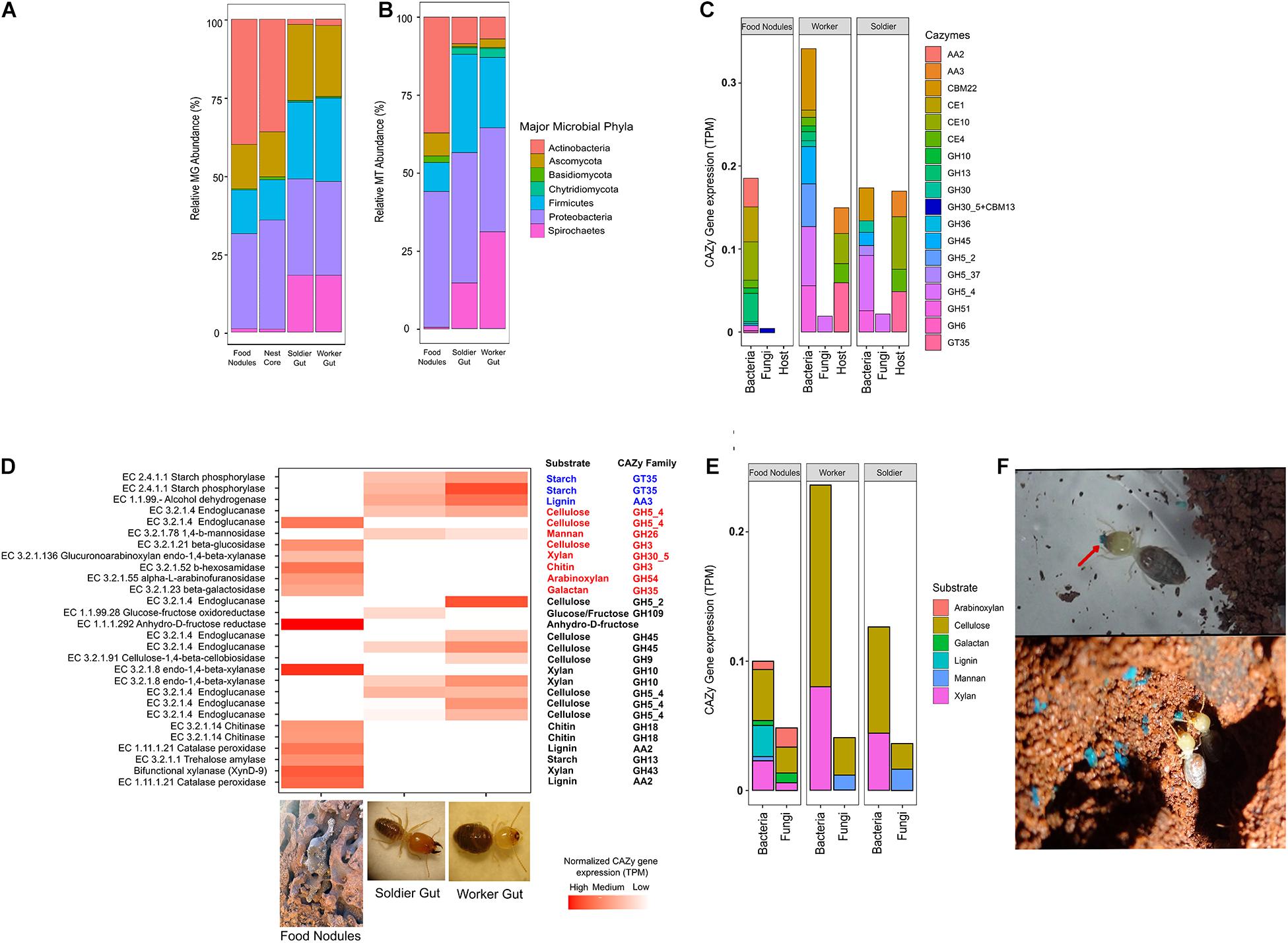
Figure 3. (A) Relative metagenomic (MG) and (B) metatranscriptomic (MT) abundance assigned to major phyla in C. cumulans. (C) Contribution of the termite holobiont (gut, nest microbiota and host) to CAZy expression. (D) Complementary CAZy gene expression and lignocellulolytic activity in food nodules and gut of workers and soldiers. The heatmap shows normalized expressions of protein coding genes of major CAZy families colored according to taxonomy assignment (Blue: termite host; Red: fungi; Black: bacteria). (E) CAZy gene expressions predicted for lignocellulosic substrates in food nodules and gut of workers and soldiers. (F) Detail of the food storage behavior of C. cumulans workers on the nest walls using blue-marked sugar-cane (red arrow).
The MG assembly of food nodules yielded 124,543 bacterial and 3,926 fungal genes. Of these, 213 were assigned to bacterial phyla and 86 to fungal CAZy-coding genes. The majority of transcripts (95%) corresponded to the glycoside hydrolases (GHs) and glycosyl transferases (GTs) (Figure 3C) and more than 60% of CAZy transcripts were specific to the food nodules (Supplementary Figures 6B,C). By using peptide-based annotation, we predicted that the expression of CAZy genes is high for cellulases and hemicellulases. Consequently, the lignocellulosic activity reported previously in these food storage structures (Menezes et al., 2018) is associated to the expression of bacterial and fungal endoglucanases (EC 3.2.1.4), a fungal beta-glucosidases (EC 3.2.1.21), a bacterial xylanase (EC 3.2.1.80), a fungal xylanase (EC 3.2.1.136), and fungal galactosidase (EC 3.2.1.23), and these enzymes were taxonomically assigned to the phyla Proteobacteria and Ascomycota (Supplementary Figure 6A). We also obtained transcripts associated to a catalase-peroxidases (EC 1.11.1.21) and an amylase (EC 3.2.1.1) which were linked to Actinobacteria, and chitinases of bacterial (EC 3.2.1.14) and fungal (EC 3.2.1.52) origin (Supplementary Tables 6, 7 and Figures 3D,E).
The gut microbiota of C. cumulans yielded 88,454 bacterial and fungal 3,370 genes. Of these, 121 were assigned to bacterial and 34 to fungal CAZy. GH5_2 and GH5_4 genes are the highest expressed GH families associated to the gut microbiome. Additionally, the non-catalytic carbohydrate-binding module gene (CBM22) is also abundant. Although the functional lignocellulolytic profile overlapped partially with food nodules (Figure 3D), it was linked to different microbial taxa. We also detected a high expression of transcripts associated with endoglucanase (EC 3.2.1.4), xylanase (EC 3.2.1.8), and endomannanase (EC 3.1.2.78) which were linked to Firmicutes, Chytridiomycota and Ascomycota, respectively (Supplementary Figure 6A). On the other hand, we identified 2,262 genes in the termite C. cumulans, but only 21 CAZy gene transcripts. A starch phosphorylase (EC 2.4.1.1), an alcohol-2-dehydrogenase (EC 1.1.99.-) and the carbohydrate esterases (CE4 and CE10) were predicted in the MG assembly (Supplementary Table 8 and Figure 3D). The total CAZy expression levels associated to cellulose and xylan degradation were higher in the gut of termites, and nearly equally expressed in workers and soldiers (Figure 3E).
Termites rely on a mutualistic association with gut symbionts to decompose the plant biomass; however, the digestive efficiency is achieved by the functional complementation with host endogenous lignocellulases (Tokuda and Watanabe, 2007; Scharf, 2008; Scharf et al., 2011). Moreover, termites also interact with the surrounding soil microbiota and at least two extant lineages (Macrotermitinae and Sphaerotermitinae) engaged in an external association with nest microorganisms, where each component of the termite holobiont has a collaborative role in the decomposition of lignocellulose (Poulsen et al., 2014; Chouvenc et al., 2021).
In this study, we present evidences of complementary digestion by external nest microorganisms in a neotropical grass-harvester termite C. cumulans (Syntermitinae) that store plant material inside its nests (Figure 3F). Our findings revealed that plant biomass (previously cut and packed in the food nodules) is initially decomposed by enzymes that target the lignin and complex polysaccharides as cellulose, xylan, arabinoxylan, and starch. We hypothesized that pre-processing of the plant material reduces the recalcitrance of the lignocellulose before gut passage. Since lignin breakdown by peroxidases needs O2 (Breznak and Brune, 1994), food nodules are a suitable environment for this process. Peroxidases and dehydrogenases of bacterial origin are known to participate in lignin degradation (Pelmont et al., 1989; Tamboli et al., 2011; Chauhan, 2020). The catalase-peroxidase (EC 1.11.1.21) assigned to Actinobacteria in this study was able to oxidase lignin when incubated with lignocellulosic material (de Gonzalo et al., 2016). The final step in the lignocellulose digestion process involves the passage of the pre-processed plant material in the gut of workers where it is again targeted by xylanases, possibly for overcoming any xylan residuals and making the cellulose more accessible to the endoglucanases produced by the gut microbiota. Although we found that endoglucanase transcripts were expressed in the food nodules, our results showed higher expression of cellulases in the gut of termites.
Other CAZy predicted in the gut were the non-catalytic carbohydrate-binding module CBM22 that promotes the association of xylanases and the carbohydrate esterases of C. cumulans (CE4 and CE10) (Sermsathanaswadi et al., 2017) that could be acting in the breakdown of hemicelluloses (Franco Cairo et al., 2016). The presence of chitinases was predicted in the food nodules and linked to bacteria of the phylum Firmicutes (EC 3.2.1.14) and to an ascomycete fungus (EC 3.2.1.52). Chitinases could be related to fungi cell-wall growth or the activity of mycolytic bacteria strains (Solanki et al., 2012) found in the food nodules. Chitinase transcripts were not obtained in the gut of C. cumulans; however, other higher termites are known to produce chitinases which can improve the digestibility of fungal substrates and decaying wood (Poulsen et al., 2014; Hu et al., 2019) or increase the permeability of peritrophic matrix in the midgut for the absorption of nutrients.
Although there is functional overlap between the food nodules and gut, CAZy were linked to different taxa. It could be explained by the different conditions that make these places more suitable to different kind of microorganisms. In the food nodules, cellulose and hemicellulose digestion were linked to Proteobacteria and Ascomycota, whereas catalase activity was assigned to Actinobacteria. On the other hand, lignocellulases in the gut of C. cumulans were linked to Firmicutes, Ascomycota, and Chytridiomycota. All these microbes dominated the core microbiota and are known to produce cellulases, hemicellulases (Couturier et al., 2016; Grieco et al., 2019; Matsuzawa et al., 2019, 2020) and laccases (Murphy et al., 2021). In fungus-growing insects that exploit plant-derived resources, the association between fungi and bacteria result in a multipartite functional metabolism of lignocellulose (Barcoto et al., 2020). The ecological relevance of such microbial interaction in the food nodules of C. cumulans needs to be determined.
Because our data was derived from a single colony, we cannot exclude the possibility of intercolonial variation that may affected the expression levels. Indeed, the relative abundances of some fungi orders of food nodules showed variation among the colonies used for ITS sequencing. However, the taxa responsible for CAZy expression showed similar abundances for bacteria (Menezes et al., 2018) and fungi (this study).
Fungi were less abundant than bacteria in all the samples and their CAZy expression levels associated to cellulose and xylan were also lower. Nevertheless, a noteworthy observation in this study was that the termite gut of C. cumulans is inhabited by fungi that expressed CAZy gene transcripts. One gene transcript, assigned to GH5_4 family and classified as endoglucanase EC 3.2.1.4 was linked to Neocallimastix (Chytridiomycota: Neocallimastigales). This finding suggests this termite possesses a functional mycobiota in its gut that contributes to the lignocellulose digestion. Neocallimastigales comprises anaerobic highly active lignocellulolytic fungi of the gastrointestinal tract of herbivores, which has also been reported in termites (Lee et al., 2015; da Silva et al., 2017).
Most of the fungi associated to the nests of C. cumulans (Syntermitinae) corresponded to saprotrophic Ascomycota, but other guilds within this same fungal phylum are also present in this microenvironment. Like termites, saprotrophic fungi are important plant-litter decomposers in terrestrial ecosystems using lignocellulolytic enzymes (Crowther et al., 2012) and their incidence inside termite nests could be explained by the addition of feces to the internal walls (Brauman, 2000), creating an appropriate environment for the survival and reproduction of these microorganisms (Holt, 1998; Vesala et al., 2019). Lichenized and endophytic fungi were also detected in the nest substrates but in lower abundances. The presence of these fungi has been reported in dead plant material probably consuming organic compounds released by saprotrophic microbes acting on the decomposition of the plant matter (Ottosson et al., 2015), and the mechanisms by which they colonize termite nests are unknown, but they were possibly transported from foraging parties to the mounds by workers. We found a distinct fungal composition of food nodules in comparison to the carton nest core, characterized by the abundance of saprotrophic Xylariales and Sordariales. Members of these fungal orders are known to colonize several substrates, including leaf litter, vertebrate dung, and the interior of termite nests (Okane et al., 2008; Guedegbe et al., 2009). In fungus-growing termites, Xylariales appear to be opportunistic saprotrophs of the stored plant material and fungus combs either due to the failure of microbe suppression mechanisms or after the nests have been abandoned by the termites (Mueller and Gerardo, 2002; Visser et al., 2011).
Approximately 90% of fungal reads were shared between nest fecal material and the gut of C. cumulans. One possible explanation for this is that termites consume the fecal material of their nests, independently of the food storing behavior. Carton feeding has been reported in other termites (Brauman et al., 2000), but the precise function of this behavior is not completely understood. Fungal colonization of lignocellulosic substrates is known to enrich its nitrogen content (Jacobson et al., 2015) and because most wood- and litter-feeding termites have a diet poor in nitrogen (Tayasu et al., 1994), consumption of nest carton could also help increase nitrogen intake by the termites. An alternative hypothesis is that nest fungi were originally gut symbionts deposited by the workers together with their feces and saliva on the nest walls. Since we found small differences in the number and nature of fungi of gut samples between colonies within each termite species, it is possible that the fungus is part of the natural gut symbionts and could function in their nests as a supplementary reserve of lignocellulolytic microbiota for these termites (Nalepa et al., 2001; Fall et al., 2004). The large number of shared microbes and CAZy transcripts between sterile castes is consistent with the transmission of microorganisms among nestmates. Soldiers, larvae, and reproductive castes cannot feed themselves and depend on workers for nutrition through trophallaxis (Machida et al., 2001).
In wood-feeding termites, fungi are known to play facultative interactions by attracting individuals to the food source or by improving colony development; however, the effect of fungi may also depend on the termite species (Birkemoe et al., 2018). For example, it is known that the wood decayed by the brown rot fungus Gloeophyllum trabeum attracts the termites Reticulitermes virginicus, R. flavipes (Rhinotermitidae), and Nasutitermes columbicus (Termitidae) (Esenther et al., 1961; Smythe et al., 1967) because of the presence of a particular alcohol in this decayed wood, (3Z,6Z,8E)-dodeca-3,6,8-trien-1-ol (Matsumura et al., 1968, 1969), known to be the only compound or one of the compounds of the trail pheromones of these termite species (Sillam-Dussès, 2010). Besides the potential benefits of nest fungi, we cannot discard the possibility of fungal entomopathogens or commensals to be present in the termite nest. Some representatives of Hypocreales found in our samples could be potential entomopathogens (Araújo and Hughes, 2016), but they were found in low frequency because they may be suppressed by the abundant Actinobacteria (Chouvenc et al., 2018) or by termite gland secretions (Rosengaus et al., 2004) or these fungi are just dormant in this environment (Hajek, 2017).
In this work we improved ITS sequence analyses to bring a more comprehensive understanding of the fungal communities associated to the nest, gut, and food nodules stored by C. cumulans and we bring additional metagenomic and metatranscriptomic analyses of the microbial communities associated with this termite species. Taken together with our previous analyses of the 16S rRNA bacterial communities and the differential lignocellulolytic activity in food nodules (Menezes et al., 2018), we have provided important new evidence suggesting complementary digestion of the plant material inside food storage structures before the passage through the gut of a Neotropical higher termite species. We have shown that fungi are also CAZy contributors in the termite holobiont. This could explain why the survival of this termite increased significantly when workers were fed with food nodules compared to other plant feedstocks (Janei et al., 2020). From the perspective of termite evolution, this study brings new information about the multicomponent nature of the holobiont in higher termites. The complementary digestive strategy of C. cumulans may resemble the one linking the fungus-growing termites and their mutualistic fungus Termitomyces (Poulsen et al., 2014). The association with nest microorganisms is considered as an evolutionary novelty in higher termites that originated millions of years ago in two sister lineages (Macrotermitinae and Sphaerotermitinae) after transition of gut symbionts in the ancestor of Termitidae (Bucek et al., 2019). However, our findings in C. cumulans supports the hypotheses that externalization of the digestion was an ancestral condition of higher termites that evolved through the incorporation of soil lignocellulolytic microbes into the fecal nest material (Chouvenc et al., 2021). Consequently, this association led to the loss of gut flagellates and the acquisition of novel bacterial gut symbionts before the diversification of Termitidae. Thus, we could speculate that ectosymbiosis with nest microorganisms is a plesiomorphic characteristic of higher termites. The externalization of digestion is also present in phylogenetically related species as Syntermes dirus (unpublished results) (Syntermitinae) and Velocitermes heteropterus (Nasutitermitinae) (Menezes et al., 2018), emphasizing the need to further investigate the ecological relevance of nest microbiota in Termitidae.
The datasets analyzed for this study can be found in the ENA accession no. PRJEB17080, https://www.ebi.ac.uk/ena/browser/view/PRJEB17080.
AA, GP, AC-L, and FS designed the study. EM, LM, TA, and DP performed the experiments. AA and GP analyzed data. AA, GP, JC, AR, and DS-D contributed to new analyses and wrote the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by the São Paulo Research Foundation (FAPESP), grants # 2015/21497-6 and 2018/22839-6 (AA), 2015/50590-4 (FE) and 2016/09950-0 (JC) and the National Council for Scientific and Technological Development (CNPq), grant # 306279/2020-7 (FE). EM and LM were grant fellows of the Universidade Federal do ABC and the Coordination for the Improvement of Higher Education Personnel (CAPES), respectively.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Tiago Carrijo for providing nest pictures. We would also like to thank Brazilian Biorenewables National Laboratory (LNBR/CNPEM) NGS Sequencing Facility for generating the metagenomics and metatranscriptomics sequencing data described here.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2021.632590/full#supplementary-material
Aanen, D. K., Eggleton, P., Rouland-Lefevre, C., Guldberg-Froslev, T., Rosendahl, S., and Boomsma, J. J. (2002). The evolution of fungus-growing termites and their mutualistic fungal symbionts. Proc. Natl. Acad. Sci. U. S. A. 99, 14887–14892. doi: 10.1073/pnas.222313099
Afgan, E., Baker, D., van den Beek, M., Blankenberg, D., Bouvier, D., Čech, M., et al. (2016). The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 44, W3–W10. doi: 10.1093/nar/gkw343
Araújo, J. P. M., and Hughes, D. P. (2016). Diversity of entomopathogenic fungi. Which groups conquered the insect body? Adv. Genet. 94, 1–39. doi: 10.1016/bs.adgen.2016.01.001
Barcoto, M. O., Carlos-Shanley, C., Fan, H., Ferro, M., Nagamoto, N. S., Bacci, M., et al. (2020). Fungus-growing insects host a distinctive microbiota apparently adapted to the fungiculture environment. Sci. Rep. 10:12384. doi: 10.1038/s41598-020-68448-7
Beck, M. W. (2017). ggord: Ordination Plots with ggplot2. R Package Version 1.0.0. Available online at: https://zenodo.org/badge/latestdoi/35334615 (accessed September, 2020).
Bengtsson-Palme, J., Ryberg, M., Hartmann, M., Branco, S., Wang, Z., Godhe, A., et al. (2013). Improved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Methods Ecol. Evol. 4, 914–919. doi: 10.1111/2041-210X.12073
Bignell, D., and Eggleton, P. (2000). “Termites in ecosystems,” in Termites: Evolution, Sociality, Symbioses, Ecology, eds T. Abe, D. E. Bignell, and M. Higashi (Amsterdam: Kluwer Academy), 363–387.
Birkemoe, T., Jacobsen, R. M., Sverdrup-Thygeson, A., and Biedermann, P. H. W. (2018). “Insect-fungus interactions in dead wood systems,” in Saproxylic Insects, ed. M. D. Ulyshen (Berlin: Springer International Publishing).
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brauman, A. (2000). Effect of gut transit and mound deposit on soil organic matter transformations in the soil feeding termite: a review. Eur. J. Soil Biol. 36, 117–125. doi: 10.1016/S1164-5563(00)01058-X
Brauman, A., Bignell, D. E., and Tayasu, I. (2000). “Termites: evolution, sociality, symbioses, ecology,” in Ter- mites: Evolution, Sociality, Symbioses, Ecology, eds T. Abe, D. E. Bignell, and M. Higashi (Dordrecht: Kluwer Academic Publishers), 233–259.
Bray, N. L., Pimentel, H., Melsted, P., and Pachter, L. (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527. doi: 10.1038/nbt.3519
Breznak, J. A., and Brune, A. (1994). Role of the microorganisms in the digestion of lignocellulose by termites. Ann. Rev. Entomol. 39, 453–487.
Brune, A. (2014). Symbiotic digestion of lignocellulose in termite guts. Nat. Rev. Microbiol. 12, 168–180. doi: 10.1038/nrmicro3182
Bucek, A., Šobotník, J., He, S., Shi, M., McMahon, D. P., Holmes, E. C., et al. (2019). Evolution of termite symbiosis informed by transcriptome-based phylogenies. Curr. Biol. 29, 3728–3734.e4. doi: 10.1016/j.cub.2019.08.076
Buschini, M. L. T. (2006). Spation ditribution of nests of Cornitermes cumulans (Isoptera: Termitidae) in a pasture in the municipality of Rio Claro (SP), Brazil. Ambiência 2, 65–72.
Busk, P. K., Pilgaard, B., Lezyk, M. J., Meyer, A. S., and Lange, L. (2017). Homology to peptide pattern for annotation of carbohydrate-active enzymes and prediction of function. BMC Bioinformatics 18:214. doi: 10.1186/s12859-017-1625-9
Chauhan, P. S. (2020). Role of various bacterial enzymes in complete depolymerization of lignin: a review. Biocatal. Agric. Biotechnol. 23:101498. doi: 10.1016/j.bcab.2020.101498
Chouvenc, T., Elliott, M. L., Šobotník, J., Efstathion, C. A., and Su, N.-Y. (2018). The termite fecal nest: a framework for the opportunistic acquisition of beneficial soil streptomyces (Actinomycetales: Streptomycetaceae). Environ. Entomol. 47, 1431–1439. doi: 10.1093/ee/nvy152
Chouvenc, T., Šobotník, J., Engel, M. S., and Bourguignon, T. (2021). Termite evolution: mutualistic associations, key innovations, and the rise of Termitidae. Cell. Mol. Life Sci. 78, 2749–2769. doi: 10.1007/s00018-020-03728-z
Coles De Negret, H. R., and Redford, K. H. (1982). The biology of nine termite species (Isoptera: Termitidae) from the Cerrado of Central Brazil. Psyche (Stuttg.) 89, 81–106.
Conway, J. R., Lex, A., and Gehlenborg, N. (2017). UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics 33, 2938–2940. doi: 10.1093/bioinformatics/btx364
Couturier, M., Tangthirasunun, N., Ning, X., Brun, S., Gautier, V., Bennati-Granier, C., et al. (2016). Plant biomass degrading ability of the coprophilic ascomycete fungus Podospora anserina. Biotechnol. Adv. 34, 976–983. doi: 10.1016/j.biotechadv.2016.05.010
Crowther, T. W., Boddy, L., and Hefin Jones, T. (2012). Functional and ecological consequences of saprotrophic fungus–grazer interactions. ISME J. 6, 1992–2001. doi: 10.1038/ismej.2012.53
da Silva, R. R., Pedezzi, R., and Souto, T. B. (2017). Exploring the bioprospecting and biotechnological potential of white-rot and anaerobic Neocallimastigomycota fungi: peptidases, esterases, and lignocellulolytic enzymes. Appl. Microbiol. Biotechnol. 101, 3089–3101. doi: 10.1007/s00253-017-8225-5
de Gonzalo, G., Colpa, D. I., Habib, M. H. M., and Fraaije, M. W. (2016). Bacterial enzymes involved in lignin degradation. J. Biotechnol. 236, 110–119. doi: 10.1016/j.jbiotec.2016.08.011
de Oliveira, T. B., de Lucas, R. C., de Almeida Scarcella, A. S., Contato, A. G., Pasin, T. M., Martinez, C. A., et al. (2020). Fungal communities differentially respond to warming and drought in tropical grassland soil. Mol. Ecol. 29, 1550–1559. doi: 10.1111/mec.15423
Deshpande, V., Wang, Q., Greenfield, P., Charleston, M., Porras-Alfaro, A., Kuske, C. R., et al. (2016). Fungal identification using a Bayesian classifier and the Warcup training set of internal transcribed spacer sequences. Mycologia 108, 1–5. doi: 10.3852/14-293
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Esenther, G. R., Allen, T. C., Casida, J. E., and Shenefelt, R. D. (1961). Termite attractant from fungus-infected wood. Science 134:50. doi: 10.1126/science.134.3471.50
Fall, S., Hamelin, J., Ndiaye, F., Assigbetse, K., Aragno, M., Chotte, J. L., et al. (2007). Differences between bacterial communities in the gut of a soil-feeding termite (Cubitermes niokoloensis) and its mounds. Appl. Environ. Microbiol. 73, 5199–5208. doi: 10.1128/AEM.02616-06
Fall, S., Nazaret, S., Chotte, J. L., and Brauman, A. (2004). Bacterial density and community structure associated with aggregate size fractions of soil-feeding termite mounds. Microb. Ecol. 48, 191–199. doi: 10.1007/s00248-003-1047-2
Franco Cairo, J. P. L., Carazzolle, M. F., Leonardo, F. C., Mofatto, L. S., Brenelli, L. B., Gonçalves, T. A., et al. (2016). Expanding the knowledge on lignocellulolytic and redox enzymes of worker and soldier castes from the lower termite Coptotermes gestroi. Front. Microbiol. 7:1518. doi: 10.3389/fmicb.2016.01518
Garnier-Sillam, E., Toutain, F., Villemin, G., and Renoux, J. (1989). Études préliminaires des meules originales du termite xylophage Sphaerotermes sphaerothorax (Sjostedt). Insectes Soc. 36, 93–312. doi: 10.1007/BF02224882
Grieco, M. B., Lopes, F. A. C., Oliveira, L. S., Tschoeke, D. A., Popov, C. C., Thompson, C. C., et al. (2019). Metagenomic analysis of the whole gut microbiota in Brazilian termitidae termites Cornitermes cumulans, Cyrilliotermes strictinasus, Syntermes dirus, Nasutitermes jaraguae, Nasutitermes aquilinus, Grigiotermes bequaerti, and Orthognathotermes mirim. Curr. Microbiol. 76, 687–697. doi: 10.1007/s00284-019-01662-3
Guedegbe, H. J., Miambi, E., Pando, A., Houngnandan, P., and Rouland-Lefevre, C. (2009). Molecular diversity and host specificity of termite-associated Xylaria. Mycologia 101, 686–691. doi: 10.3852/08-182
Holt, J. (1998). Microbial activity in the mounds of some Australian termites. Appl. Soil Ecol. 9, 183–187. doi: 10.1016/S0929-1393(98)00073-0
Hothorn, T., Bretz, F., and Westfall, P. (2008). Simultaneous inference in general parametric models. Biometrical J. 50, 346–363. doi: 10.1002/bimj.200810425
Hu, H., da Costa, R. R., Pilgaard, B., Schiøtt, M., Lange, L., and Poulsen, M. (2019). Fungiculture in termites is associated with a mycolytic gut bacterial community. mSphere 4:13. doi: 10.1128/mSphere.00165-19
Jacobson, K., van Diepeningen, A., Evans, S., Fritts, R., Gemmel, P., Marsho, C., et al. (2015). Non-rainfall moisture activates fungal decomposition of surface litter in the namib sand sea. PLoS One 10:e0126977. doi: 10.1371/journal.pone.0126977
Janei, V., Haifig, I., Schönhaus, G. C., and Costa-Leonardo, A. M. (2020). Gut content and laboratory survival of the termite cornitermes cumulans (Isoptera: Termitidae: Syntermitinae) with different diets including nest stored food. Neotrop. Entomol. 49, 677–684. doi: 10.1007/s13744-020-00796-5
Jouquet, P., Chaudhary, E., and Kumar, A. R. V. (2018). Sustainable use of termite activity in agro-ecosystems with reference to earthworms. A review. Agron. Sustain. Dev. 38:3. doi: 10.1007/s13593-017-0483-1
Korb, J. (2003). Thermoregulation and ventilation of termite mounds. Naturwissenschaften 90, 212–219. doi: 10.1007/s00114-002-0401-4
Korb, J. (2011). “Termite mound architecture, from function to construction,” in Biology of Termites: A Modern Synthesis, eds D. Bignell, Y. Roisin, and N. Lo (Dordrecht: Springer). doi: 10.1007/978-90-481-3977-4_13
Lee, S. M., Guan, L. L., Eun, J.-S., Kim, C.-H., Lee, S. J., Kim, E. T., et al. (2015). The effect of anaerobic fungal inoculation on the fermentation characteristics of rice straw silages. J. Appl. Microbiol. 118, 565–573. doi: 10.1111/jam.12724
Lima, J. T., and Costa-Leonardo, A. M. (2007). Recursos alimentares explorados pelos cupins (Insecta: Isoptera). Biota Neotrop. 7, 243–250. doi: 10.1590/S1676-06032007000200027
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Machida, M., Kitade, O., Miura, T., and Matsumoto, T. (2001). Nitrogen recycling through proctodeal trophallaxis in the Japanese damp-wood termite Hodotermopsis japonica (Isoptera, Termopsidae). Insect Sociaux 48, 52–56.
Manjula, A., Sathyavathi, S., Pushpanathan, M., Gunasekaran, P., and Rajendhran, J. (2014). Microbial diversity in termite nest. Curr. Sci. 72, 267–275. doi: 10.18520/cs/v106/i10/1430-1434
Matsumura, F., Coppel, H. C., and Tai, A. (1968). Isolation and identification of termite trail-following pheromone. Nature 219, 963–964. doi: 10.1038/219963a0
Matsumura, F., Tai, A., and Coppel, H. C. (1969). Termite trail-following substance, isolation and purification from reticulitermes virginicus and fungus-infected wood. J. Econ. Entomol. 62, 599–603. doi: 10.1093/jee/62.3.599
Matsuzawa, T., Kameyama, A., Nakamichi, Y., and Yaoi, K. (2020). Identification and characterization of two xyloglucan-specific endo-1,4-glucanases in Aspergillus oryzae. Appl. Microbiol. Biotechnol. 104, 8761–8773. doi: 10.1007/s00253-020-10883-7
Matsuzawa, T., Watanabe, M., Kameda, T., Kameyama, A., and Yaoi, K. (2019). Cooperation between β-galactosidase and an isoprimeverose-producing oligoxyloglucan hydrolase is key for xyloglucan degradation in Aspergillus oryzae. FEBS J. 286, 3182–3193. doi: 10.1111/febs.14848
McMurdie, P. J., and Holmes, S. (2013). Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
Menezes, L., Alvarez, T. M., Persinoti, G. F., Franco, J. P., Squina, F., Moreira, E. A., et al. (2018). Food storage by the savanna termite Cornitermes cumulans (Syntermitinae): a strategy to improve hemicellulose digestibility? Microb. Ecol. 76, 492–505. doi: 10.1007/s00248-017-1128-2
Menzel, P., Ng, K. L., and Krogh, A. (2016). Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. doi: 10.1038/ncomms11257
Mikaelyan, A., Dietrich, C., Köhler, T., Poulsen, M., Sillam-Dussès, D., and Brune, A. (2015). Diet is the primary determinant of bacterial community structure in the guts of higher termites. Mol. Ecol. 24, 5284–5295. doi: 10.1111/mec.13376
Moreira, E. A., Alvarez, T. M., Persinoti, G. F., Paixão, D. A. A., Menezes, L. R., Cairo, J. P. F., et al. (2018). Microbial communities of the gut and nest of the humus- and litter-feeding termite Procornitermes araujoi (Syntermitinae). Curr. Microbiol. 75, 1609–1618. doi: 10.1007/s00284-018-1567-0
Mueller, U. G., and Gerardo, N. (2002). Fungus-farming insects: multiple origins and diverse evolutionary histories. Proc. Natl. Acad. Sci. U. S. A. 99, 15247–15249. doi: 10.1073/pnas.242594799
Murphy, R., Benndorf, R., de Beer, Z. W., Vollmers, J., Kaster, A.-K., Beemelmanns, C., et al. (2021). Comparative genomics reveals prophylactic and catabolic capabilities of actinobacteria within the fungus-farming termite symbiosis. mSphere 6, 1–12. doi: 10.1128/mSphere.01233-20
Nalepa, C. A., Bignell, D. E., and Bandi, C. (2001). Detritivory, coprophagy, and the evolution of digestive mutualisms in Dictyoptera. Insectes Soc. 48, 194–201. doi: 10.1007/PL00001767
Neupane, A., Maynard, D. S., and Bradford, M. A. (2015). Consistent effects of eastern subterranean termites (Reticulitermes flavipes) on properties of a temperate forest soil. Soil Biol. Biochem. 91, 84–91. doi: 10.1016/j.soilbio.2015.08.025
Nguyen, N. H., Song, Z., Bates, S. T., Branco, S., Tedersoo, L., Menke, J., et al. (2016). FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 20, 241–248. doi: 10.1016/j.funeco.2015.06.006
Okane, I., Toyama, K., Nakagiri, A., Suzuki, K., Srikitikulchai, P., Sivichai, S., et al. (2008). Study of endophytic Xylariaceae in Thailand: diversity and taxonomy inferred from rDNA sequence analyses with saprobes forming fruit bodies in the field. Mycoscience 49, 359–372. doi: 10.1007/S10267-008-0440-6
Oksanen, J., Blanchet, F. G., Friendly, M., Kindt, R., Legendre, P., McGlinn, D., et al. (2018). vegan: Community Ecology Package. R Package Version 2.5-2. Available online at: https://CRAN.R-project.org/package=vegan. R Packag (accessed September, 2020).
Ottosson, E., Kubartová, A., Edman, M., Jönsson, M., Lindhe, A., Stenlid, J., et al. (2015). Diverse ecological roles within fungal communities in decomposing logs of Picea abies. FEMS Microbiol. Ecol. 91:fiv012. doi: 10.1093/femsec/fiv012
Pelmont, J., Tournesac, C., Mliki, A., Barrelle, M., and Beguin, C. (1989). A new bacterial alcohol dehydrogenase active on degraded lignin and several low molecular weight aromatic compounds. FEMS Microbiol. Lett. 57, 109–114. doi: 10.1111/j.1574-6968.1989.tb03230.x
Peng, Y., Leung, H. C. M., Yiu, S. M., and Chin, F. Y. L. (2012). IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428. doi: 10.1093/bioinformatics/bts174
Poulsen, M., Hu, H., Li, C., Chen, Z., Xu, L., Otani, S., et al. (2014). Complementary symbiont contributions to plant decomposition in a fungus-farming termite. Proc. Natl. Acad. Sci. U. S. A. 111, 14500–14505. doi: 10.1073/pnas.1319718111
Rassati, D., Marini, L., and Malacrinò, A. (2019). Acquisition of fungi from the environment modifies ambrosia beetle mycobiome during invasion. PeerJ 7:e8103. doi: 10.7717/peerj.8103
R Core Team (2018). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Redford, K. H. (1984). The termitaria of Cornitermes cumulans (Isoptera, Termitidae) and their role in determining a potential keystone species. Biotropica 16, 112–119. doi: 10.2307/2387842
Rosengaus, R. B., Traniello, J. F. A., Lefebvre, M. L., and Maxmen, A. B. (2004). Fungistatic activity of the sternal gland secretion of the dampwood termite Zootermopsis angusticollis. Insectes Soc. 51, 259–264. doi: 10.1007/s00040-004-0749-x
Rothman, J. A., Andrikopoulos, C., Cox-Foster, D., and McFrederick, Q. S. (2019). Floral and foliar source affect the bee nest microbial community. Microb. Ecol. 78, 506–516. doi: 10.1007/s00248-018-1300-3
Scharf, M. E. (2008). Termite digestomes as sources for novel lignocellulases. Society 540–552. doi: 10.1002/bbb
Scharf, M. E., Karl, Z. J., Sethi, A., and Boucias, D. G. (2011). Multiple levels of synergistic collaboration in termite lignocellulose digestion. PLoS One 6:e21709. doi: 10.1371/journal.pone.0021709
Schmidt, A. M., Jacklyn, P., and Korb, J. (2014). ‘Magnetic’ termite mounds: is their unique shape an adaptation to facilitate gas exchange and improve food storage? Insectes Soc. 61, 41–49. doi: 10.1007/s00040-013-0322-6
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60. doi: 10.1186/gb-2011-12-6-r60
Seibold, S., Müller, J., Baldrian, P., Cadotte, M. W., Štursová, M., Biedermann, P. H. W., et al. (2019). Fungi associated with beetles dispersing from dead wood – Let’s take the beetle bus! Fungal Ecol. 39, 100–108. doi: 10.1016/j.funeco.2018.11.016
Sermsathanaswadi, J., Baramee, S., Tachaapaikoon, C., Pason, P., Ratanakhanokchai, K., and Kosugi, A. (2017). The family 22 carbohydrate-binding module of bifunctional xylanase/β-glucanase Xyn10E from Paenibacillus curdlanolyticus B-6 has an important role in lignocellulose degradation. Enzyme Microb. Technol. 96, 75–84. doi: 10.1016/j.enzmictec.2016.09.015
Siebers, N., Martius, C., Eckhardt, K.-U., Garcia, M. V. B., Leinweber, P., and Amelung, W. (2015). Origin and alteration of organic matter in termite mounds from different feeding guilds of the amazon rainforests. PLoS One 10:e0123790. doi: 10.1371/journal.pone.0123790
Singh, K., Muljadi, B. P., Raeini, A. Q., Jost, C., Vandeginste, V., Blunt, M. J., et al. (2019). The architectural design of smart ventilation and drainage systems in termite nests. Sci. Adv. 5:eaat8520. doi: 10.1126/sciadv.aat8520
Smythe, R. V., Coppel, H. C., Lipton, S. H., and Strong, F. M. (1967). Chemical studies of attractants associated with Reticulitermes flavipes and R. virginicus1. J. Econ. Entomol. 60, 228–233. doi: 10.1093/jee/60.1.228
Solanki, M. K., Robert, A. S., Singh, R. K., Kumar, S., Pandey, A. K., Srivastava, A. K., et al. (2012). Characterization of mycolytic enzymes of bacillus strains and their bio-protection role against rhizoctonia solani in tomato. Curr. Microbiol. 65, 330–336. doi: 10.1007/s00284-012-0160-1
Steinegger, M., and Söding, J. (2017). MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028. doi: 10.1038/nbt.3988
Tamboli, D. P., Telke, A. A., Dawkar, V. V., Jadhav, S. B., and Govindwar, S. P. (2011). Purification and characterization of bacterial aryl alcohol oxidase from Sphingobacterium sp. ATM and its uses in textile dye decolorization. Biotechnol. Bioprocess Eng. 16, 661–668. doi: 10.1007/s12257-011-0031-9
Tayasu, I., Sugimoto, A., Wada, E., and Abe, T. (1994). Xylophagous termites depending on atmospheric nitrogen. Naturwissenschaften 81, 229–231. doi: 10.1007/BF01138550
Tokuda, G., and Watanabe, H. (2007). Hidden cellulases in termites: revision of an old hypothesis. Biol. Lett. 3, 336–339.
Vesala, R., Harjuntausta, A., Hakkarainen, A., Rönnholm, P., Pellikka, P., and Rikkinen, J. (2019). Termite mound architecture regulates nest temperature and correlates with species identities of symbiotic fungi. PeerJ 6:e6237. doi: 10.7717/peerj.6237
Visser, A. A., Kooij, P. W., Debets, A. J. M., Kuyper, T. W., and Aanen, D. K. (2011). Pseudoxylaria as stowaway of the fungus-growing termite nest: interaction asymmetry between Pseudoxylaria, Termitomyces and free-living relatives. Fungal Ecol. 4, 322–332. doi: 10.1016/j.funeco.2011.05.003
Wickham, H. (2016). ggplot2 – Elegant Graphics for Data Analysis, Second Edn. New York, NY: Springer-Verlag.
Yu, Z., and Morrison, M. (2004). Improved extraction of PCR-quality community DNA from digesta and fecal samples. Biotechniques 36, 808–812. doi: 10.2144/3605A0808
Zakrzewski, M., Proietti, C., Ellis, J. J., Hasan, S., Brion, M. J., Berger, B., et al. (2017). Calypso: a user-friendly web-server for mining and visualizing microbiome-environment interactions. Bioinformatics 33, 782–783. doi: 10.1093/bioinformatics/btw725
Keywords: Isoptera, nest microrganisms, carbohydrate active enzymes, saprotrophs, food storage, nutrition, Blattodea, Syntermitinae
Citation: Moreira EA, Persinoti GF, Menezes LR, Paixão DAA, Alvarez TM, Cairo JPLF, Squina FM, Costa-Leonardo AM, Rodrigues A, Sillam-Dussès D and Arab A (2021) Complementary Contribution of Fungi and Bacteria to Lignocellulose Digestion in the Food Stored by a Neotropical Higher Termite. Front. Ecol. Evol. 9:632590. doi: 10.3389/fevo.2021.632590
Received: 23 November 2020; Accepted: 01 April 2021;
Published: 26 April 2021.
Edited by:
George Tsiamis, University of Patras, GreeceReviewed by:
Gaku Tokuda, University of the Ryukyus, JapanCopyright © 2021 Moreira, Persinoti, Menezes, Paixão, Alvarez, Cairo, Squina, Costa-Leonardo, Rodrigues, Sillam-Dussès and Arab. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alberto Arab, YWxiZXJ0b2FyYWJAZ21haWwuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.