 Marc Rius
Marc Rius Xavier Turon
Xavier Turon
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
OPINION article
Front. Ecol. Evol., 14 December 2020
Sec. Population, Community, and Ecosystem Dynamics
Volume 8 - 2020 | https://doi.org/10.3389/fevo.2020.595711
This article is part of the Research TopicThe Genomics of Biological InvasionView all 8 articles
Phylogeography is an integrative discipline that aims to understand the geographic ordination of genotypes. In recent decades, phylogeographic approaches have been used to enhance our understanding of both biogeography and landscape genetics across a variety of spatial and temporal scales. By definition, species studied using these approaches need to meet certain assumptions (e.g. mutation and drift need to be at equilibrium). However, artificially dispersed species (i.e. non-indigenous, naturalised and invasive species) often do not comply with these assumptions. Thus, the use of phylogeographic approaches to study these species may lead to erroneous interpretations. Considering that self-denominated phylogeographic studies of invasive species have proliferated in recent years and that genomic tools are now more accessible than ever before, kick starting this debate is particularly timely. We argue herein that invasion scientists must carefully use phylogeographic approaches when studying genomic data obtained from the introduced range. In addition, the assumptions of these phylogeographic approaches need to be explicitly considered when interpreting genomic patterns of invasive species. Finally, we suggest abandoning the use of the term ‘phylogeography' for describing geographically contextualized genomic data from the introduced range to avoid both terminological and methodological confusion.
DNA sequencing has been one of biology's strongest assets since its inception in the last quarter of the 20th century and has extensively accelerated progress in genetic, and more recently, genomic research (Collins et al., 2003). The ability to decipher an organism's DNA has allowed researchers to, for example, understand patterns of population connectivity (Allendorf et al., 2010), unravel evolutionary histories of genotypes around the world (Selkoe et al., 2010; Pérez-Portela et al., 2017) and untangle how evolution shapes ecological communities (Weber et al., 2017). Our ability to sequence DNA has led to the emergence of a plethora of disciplines that have greatly helped research progress, one such discipline being phylogeography.
Phylogeography aims to investigate the relationship among geographic history, biogeography, and the mechanisms driving speciation (Avise et al., 1987; Avise, 2009). This discipline has enabled researchers to conduct phylogenetic analyses of geographically contextualized genomic data, allowing new links between phylogenetics and population genetics. Being hypothesis driven, phylogeography allows testing the role of historical processes (such as vicariance events and environmental changes) in the evolution of species. This has fostered our understanding of the geographic ordination of genotypes and our ability to infer the most likely evolutionary history of many taxa (Avise, 2000). Phylogeographic approaches are normally used in studies of intraspecific genetic lineages (Avise, 2009) and are increasingly adopting a comparative approach (e.g., comparing co-distributed species that have been affected by common climatic fluctuations) to study links between micro- and macro-evolutionary processes at the community level (Dawson, 2014; Pascual et al., 2017; Turon et al., 2020).
Phylogeographic analyses assume long-term natural evolutionary processes (Avise, 2009) and both ancient and modern DNA are routinely considered in these analyses (Navascués et al., 2010). Phylogeographic studies predominantly focus on the Quaternary period (Figure 1), particularly around the last glacial maximum (Beheregaray, 2008). Phylogeography has an explicit genealogical dimension (both spatial and temporal) (Avise, 2009) and includes methods from population genomics (Reitzel et al., 2013), coalescent theory (Nielsen, 2006), and phylogenetics (Riddle et al., 2008). For example, phylogeographic studies often use analytical tools such as haplotype networks, mismatch analyses, genetic differentiation estimators, phylogenies, analyses of molecular variance (AMOVA), gene trees, and coalescence analyses (Knowles, 2003). As a result, phylogeography has been considered one of the most integrative disciplines in biology (Hickerson et al., 2010). The number of phylogeographic studies has grown exponentially in recent years due to the increased accessibility of sequencing techniques (Beheregaray, 2008) and the emergence of genomic tools (e.g., Angeloni et al., 2012; Rellstab et al., 2015; Reyes-Velasco et al., 2018; Tan et al., 2019). This “genomics revolution” has increased the potential for fine-scale resolution of phylogeographic patterns and represents a leap forward in the discipline, albeit some caveats remain [reviewed in Cutter (2013); Bowen et al. (2014)].
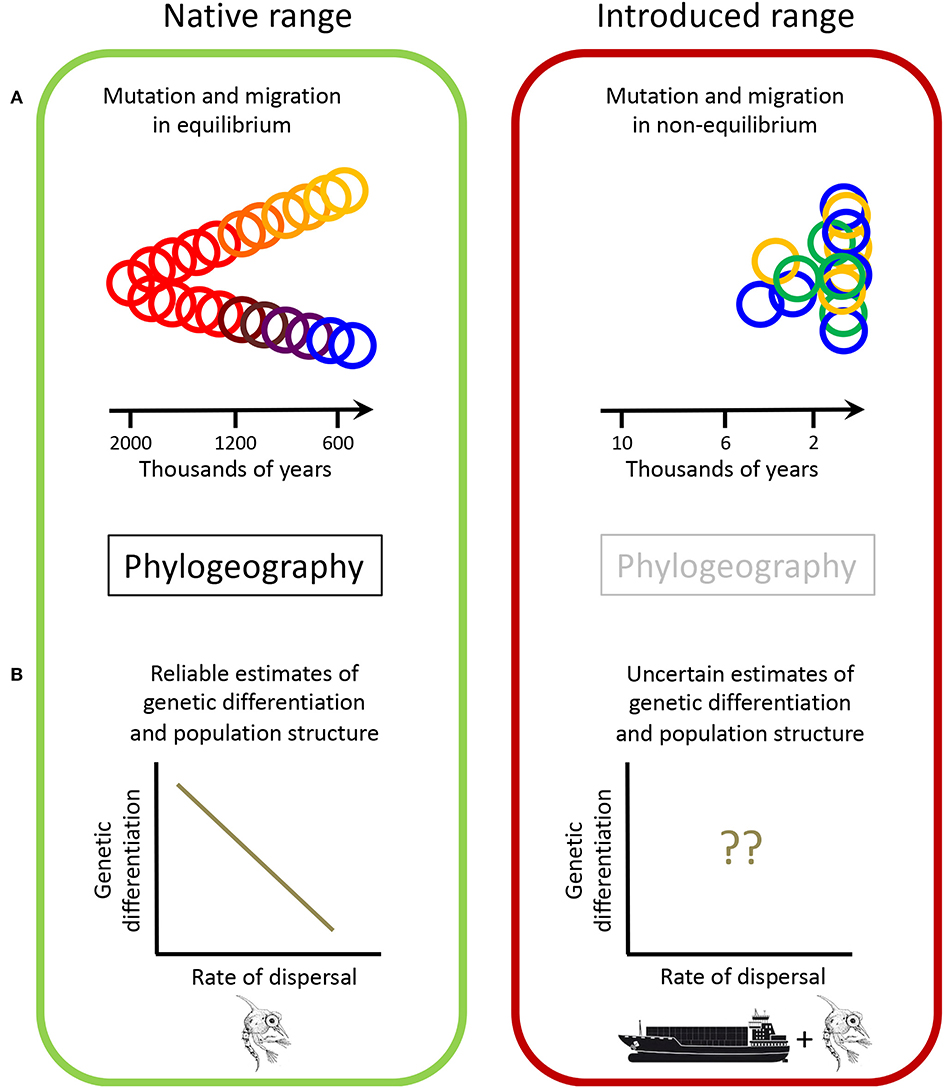
Figure 1. The use of phylogeography to study the geographic ordination of genotypes in native and introduced ranges. The main differences are highlighted considering: (A) Different evolutionary histories and (B) Population structure patterns found in the different range types. The time scale in (A) reflects that most studies of phylogeography focus on the Quaternary period (Beheregaray, 2008). Regarding the introduced range, although humans began navigating the seas ~50,000 years ago (Broome, 1994) and domestication of animals and plants saw its onset 13,000 years ago (Diamond, 1998), most non-indigenous species introductions have occurred in recent times (Carlton, 2009). The green circles (A) indicate admixed genotypes (see details in text). The decapod crustacean zoeae larva (from Weiss, 2017) in (B) represents the natural dispersal capabilities of species, while the ship tanker (from: www.shutterstock.com/image-vector/cargo-vessels-tugboat-oil-platform-isolated-239670235) denotes human-mediated transport of species.
Non-indigenous species have been artificially transported since humans started navigating the seas (Figure 1) but it is well-established that most species introductions have occurred over the last century (Carlton, 2009). The relatively short temporal scale considered in invasion science permits testing hypotheses that are impossible to address in other disciplines (e.g., mechanisms that shape the establishment of new ranges). Thus, biological invasions provide unparalleled experiments in biogeography (Richardson and Pyšek, 2007) that sometimes challenge well-established theories in genetics, ecology and evolutionary biology (Sax et al., 2007). As in other fields, the use of genomic tools has recently enlarged the scope of invasion science [reviewed in Rius et al. (2015a); Viard et al. (2016)].
Tracking genetic lineages of non-indigenous species across different spatial scales is a core tenet in invasion science (Cristescu, 2015). As a result, phylogeography would appear to be a valid approach to study these lineages. Indeed, a Web of Science search undertaken on the 16th of August 2020, using the words “phylogeography” and “biological invasions” showed that the mean number of studies per year between 1996 and 2007 was three studies (standard error of one), while the annual average between 2008 and 2020 was 16 (standard error of 1.5). Thus, there has been in recent years a sharp increase in the use of the term “phylogeography” in studies of non-indigenous species.
Phylogeography relies on a number of assumptions to be met, including the need for mutation rates, migration and genetic drift to be in equilibrium (Fitzpatrick et al., 2012). Such equilibrium is required, for example to detect vicariance events from genomic signatures, to infer an accurate timing of coalescent events or demographic changes, or to provide reliable estimates of migration rates. These assumptions are in general not fulfilled in genomic studies of the introduced range (Figure 1). Genetic equilibrium requires a long timeline to establish, orders of magnitude larger than the time required for most biological invasions to be successful (Carlton, 2009). Thus, studies have argued that phylogeographic inferences made on non-equilibrium introduced populations may be invalid (Fitzpatrick et al., 2012).
Considering that phylogeography and invasion science focus on old and recent processes, respectively, discerning between long-term evolutionary processes and recent range shifts is paramount (Alvarez et al., 2005). In our literature search, only a few studies focused solely on the native range (e.g., Roman and Palumbi, 2004), where the assumptions of phylogeography can be met. In turn, the genomic variation found in the introduced range has mostly evolved in the native range (Davies et al., 1999). Since genealogy-based methods require long evolutionary time scales and gene trees reflect changes that occurred prior to the introduction events, the correct interpretation of the results of these methods can be challenging. Further erroneous conclusions can be obtained if measures of genetic differentiation between populations are used to infer migration within the introduced range (Figure 1). This is because bottleneck effects could be masked in rapidly expanding populations (more so if seeded from different source populations of the native range) and genetic admixture between imported divergent genotypes (Figure 1) could make demographic changes detected using mismatch analyses meaningless.
Genomic tools are often used to infer patterns of spread of non-indigenous species in their introduced range. These inferences need to be carefully interpreted, as some studies have suggested that artificial transport blurs the DNA signatures that are used to study biological invasions (Rius et al., 2012; David, 2018). In addition, studies of genomics patterns of biological invasions increasingly report hybridization between previously isolated genotypes due to human activities (Chunco, 2014; Viard et al., 2020). Although the recent mixing of divergent genotypes may help non-indigenous species cope better with or adapt to novel conditions found in the introduced range (Bourne et al., 2018; Blakeslee et al., 2020), it considerably obscures the interpretation of phylogeographic results.
We expect that invasion scientists will increasingly use genomic tools and improve the temporal collection of samples (e.g., historical/museum collections) to better understand invasion histories (e.g., Lees et al., 2011). Such biological archives will facilitate sharing samples and genomic data amongst research groups, as well as improve predictions of invasion dynamics and future species introductions.
Genomic data are invaluable to address taxonomic issues (common in invasion science) to detect cryptic invasions, to track origins and pathways, to determine if single or multiple introductions have occurred, and to assess hybridization and introgression (Rius et al., 2015b; Viard and Comtet, 2015). These questions are different from the ones commonly addressed in phylogeographic studies, which test hypotheses related to patterns of evolutionary change over space and time. The genomic study of invasive species can hardly include an evolutionary framework and should focus on using analytical tools that allow the description of patterns of genomic variation. A phylogeny or a haplotype network cannot be used to assess evolutionary patterns within the introduced range, but can provide evidence of taxonomic problems and cryptic invasions (Pérez-Portela et al., 2013; Rius and Teske, 2013).
Well-established population genetic analyses (such as FST or AMOVA) are useful in invasion genomics but should only be based on allele frequency changes, not on evolutionary distances between alleles, and cannot be interpreted as explaining population structure as a result of drift and migration. These analyses can provide important insights into invasion scenarios, particularly when used in parallel with reduced-space graphical representations. Other well-suited analytical tools for studies on invasion genomics are non-equilibrium and non-genealogical techniques such as assignment tests, clustering techniques (STRUCTURE and others) and the Approximate Bayesian Computation method (Estoup and Guillemaud, 2010). Furthermore, the increased accessibility of high throughput sequencing techniques, whereby many loci can be genotyped simultaneously and provide effective genome scans (Clark et al., 2010), opens the door for the study of important non-equilibrium processes, such as selection and adaptation, often occurring in introduced populations (Chown et al., 2015; Sherman et al., 2016). Ultimately, the power of invasion genomic is determined by the resolution of the genomic markers used, the strength of the population structure in the native range and the sampling intensity across both native and introduced ranges (Casso et al., 2019; Hudson et al., 2020).
Despite the short time scales and associated idiosyncrasies of biological invasions, the term “phylogeography” is widely used in invasion science. Phylogeography refers to a much narrower set of conditions than those framed by biological invasions. Specifically, the formal use of the term “phylogeography” refers to specific analytical methods used to infer historical processes leading to contemporary geographic distribution of genetic diversity and thus on analysis of historical (in the sense of evolutionary time frames) processes. Thus, caution is needed when using phylogeographic approaches to study biological invasions, and the interpretation of results from the introduced range must explicitly acknowledge the potential shortcomings. These recommendations do not aim to discourage researchers in their use of genetic/genomic data. These data are perfectly valid to address research questions in invasion science (Darling and Mahon, 2011) and provide extremely important information to understand species invasions (Darling and Blum, 2007; Geller et al., 2010; Rius et al., 2015a; Viard and Comtet, 2015; Viard et al., 2016), but both a proper use of the analytical methods and a correct interpretation of results are necessary.
It is becoming increasingly clear that invasion scientists are adopting phylogeographic methods with little consideration of the assumptions that underpin these methods. Phylogeography has become a catch-all name for any study of biological invasions that incorporates population genomics and phylogenies to study geographic ordination of genotypes. Although these methods can provide useful descriptive information, strict phylogeographic inference is generally not possible in introduced populations. The problems associated with the loose use of the term “phylogeography” to describe the reconstruction of invasion histories are thus two-fold: terminological, as the term strictly refers to a narrow set of techniques, and methodological, as these techniques are not well-suited for non-equilibrium situations (Fitzpatrick et al., 2012). Engaging with how the term “phylogeography” and the associated analytical techniques are used is important, as it is safe to assume that the growing number of invasion science studies using the term “phylogeography” presented earlier will only continue to grow as widespread use of high throughput sequencing technology is increasingly utilized by invasion scientists (Rius et al., 2015a).
Invasion science is a discipline where terminology is of vital importance (Blackburn et al., 2011) and is the object of on-going debates (Colautti and Richardson, 2009; Robinson et al., 2016). Here, we argue that invasion scientists must consider the inherent differences between equilibrium and non-equilibrium situations when interpreting genomic data of biological invasions. As DNA techniques are increasingly taken up by invasion scientists (Rius et al., 2015a), it is important to recognize the assumptions associated to each type of analysis. We recommend that the loose use of the term “phylogeography” in invasion science be discontinued and instead terms such as “invasion genetics” or “invasion genomics” be used. We have in recent years witnessed a booming in the use of genomic tools to address research questions in invasion science, and thus setting solid conceptual and terminological bases is particularly timely.
MR conceived the opinion piece and wrote the first draft. XT revised the draft and contributed with new text. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to acknowledge John Darling, James Fordyce and Conrad A. Matthee for insightful discussions while preparing the earlier versions of this manuscript. The ideas developed in this opinion note were partly conceived during the development of the project CTM2017-88080 (MCIU/AEI/FEDER/UE) from the Spanish Government.
Allendorf, F. W., Hohenlohe, P. A., and Luikart, G. (2010). Genomics and the future of conservation genetics. Nat. Rev. Genet. 11, 697–709. doi: 10.1038/nrg2844
Alvarez, N., Mckey, D., Hossaert-Mckey, M., Born, C., Mercier, L., and Benrey, B. (2005). Ancient and recent evolutionary history of the bruchid beetle, Acanthoscelides obtectus say, a cosmopolitan pest of beans. Mol. Ecol. 14, 1015–1024. doi: 10.1111/j.1365-294X.2005.02470.x
Angeloni, F., Wagemaker, N., Vergeer, P., and Ouborg, J. (2012). Genomic toolboxes for conservation biologists. Evol. Appl. 5, 130–143. doi: 10.1111/j.1752-4571.2011.00217.x
Avise, J. (2000). Phylogeography: The History and Formation of Species. Cambridge, MA; London: Harvard University press.
Avise, J. C. (2009). Phylogeography: retrospect and prospect. J. Biogeogr. 36, 3–15. doi: 10.1111/j.1365-2699.2008.02032.x
Avise, J. C., Arnold, J., Ball, R. M., Bermingham, E., Lamb, T., Neigel, J. E., et al. (1987). Intraspecific phylogeography: the mitochondrial DNA bridge between population genetics and systematics. Ann. Rev. Ecol. Syst. 18, 489–522. doi: 10.1146/annurev.es.18.110187.002421
Beheregaray, L. B. (2008). Twenty years of phylogeography: the state of the field and the challenges for the Southern Hemisphere. Mol. Ecol. 17, 3754–3774. doi: 10.1111/j.1365-294X.2008.03857.x
Blackburn, T. M., Pyšek, P., Bacher, S., Carlton, J. T., Duncan, R. P., Jarošík, V., et al. (2011). A proposed unified framework for biological invasions. Trends Ecol. Evol. 26, 333–339. doi: 10.1016/j.tree.2011.03.023
Blakeslee, A. M. H., Manousaki, T., Vasileiadou, K., and Tepolt, C. K. (2020). An evolutionary perspective on marine invasions. Evol. Appl. 13, 479–485. doi: 10.1111/eva.12906
Bourne, S. D., Hudson, J., Holman, L. E., and Rius, M. (2018). Marine Invasion Genomics: Revealing Ecological and Evolutionary Consequences of Biological Invasions. Springer International Publishing.
Bowen, B. W., Shanker, K., Yasuda, N., Malay, M. C. D., von der Heyden, S., Paulay, G., et al. (2014). Phylogeography unplugged: comparative surveys in the genomic era. Bull. Mar. Sci. 90, 13–46. doi: 10.5343/bms.2013.1007
Carlton, J. T. (2009). “Deep invasion ecology and the assembly of communities in historical time,” in Biological Invasions in Marine Ecosystems, eds G. Rilov and J. A. Crooks (Berlin: Springer Berlin Heidelberg), 13–56.
Casso, M., Turon, X., and Pascual, M. (2019). Single zooids, multiple loci: independent colonisations revealed by population genomics of a global invader. Biol. Invasions 21, 3575–3592. doi: 10.1007/s10530-019-02069-8
Chown, S. L., Hodgins, K. A., Griffin, P. C., Oakeshott, J. G., Byrne, M., and Hoffmann, A. A. (2015). Biological invasions, climate change and genomics. Evol. Appl. 8, 23–46. doi: 10.1111/eva.12234
Chunco, A. J. (2014). Hybridization in a warmer world. Ecol. Evol. 4, 2019–2031. doi: 10.1002/ece3.1052
Clark, M. S., Tanguy, A., Jollivet, D., Bonhomme, F., Guinand, B., and Viard, F. (2010). “Populations and pathways: genomic approaches to understanding population structure and environmental adaptation,” in Introduction to Marine Genomics, eds J. M. Cock, K. K. Tessmar-Raible, C. Boyen, and F. Viard (Dordrecht: Springer Netherlands), 73–118.
Colautti, R. I., and Richardson, D. M. (2009). Subjectivity and flexibility in invasion terminology: too much of a good thing? Biol. Invasions 11, 1225–1229. doi: 10.1007/s10530-008-9333-z
Collins, F. S., Green, E. D., Guttmacher, A. E., and Guyer, M. S. (2003). A vision for the future of genomics research. Nature 422, 835–847. doi: 10.1038/nature01626
Cristescu, M. E. (2015). Genetic reconstructions of invasion history. Mol. Ecol. 24, 2212–2225. doi: 10.1111/mec.13117
Cutter, A. D. (2013). Integrating phylogenetics, phylogeography and population genetics through genomes and evolutionary theory. Mol. Phylog. Evol. 69, 1172–1185. doi: 10.1016/j.ympev.2013.06.006
Darling, J. A., and Blum, M. J. (2007). DNA-based methods for monitoring invasive species: a review and prospectus. Biol. Invasions 9, 751–765. doi: 10.1007/s10530-006-9079-4
Darling, J. A., and Mahon, A. R. (2011). From molecules to management: adopting DNA-based methods for monitoring biological invasions in aquatic environments. Environ. Res. 111, 978–988. doi: 10.1016/j.envres.2011.02.001
David, A. A. (2018). Reconsidering panmixia: the erosion of phylogeographic barriers due to anthropogenic transport and the incorporation of biophysical models as a solution. Front. Mar. Sci. 5:280. doi: 10.3389/fmars.2018.00280
Davies, N., Villablanca, F. X., and Roderick, G. K. (1999). Determining the source of individuals: multilocus genotyping in nonequilibrium population genetics. Trends Ecol. Evol. 14, 17–21. doi: 10.1016/S0169-5347(98)01530-4
Dawson, M. N. (2014). Natural experiments and meta-analyses in comparative phylogeography. J. Biogeogr. 41, 52–65. doi: 10.1111/jbi.12190
Diamond, J. (1998). Guns, Germs and Steel: A Short History of Everybody for the Last 13,000 Years. New York, NY: Random House.
Estoup, A., and Guillemaud, T. (2010). Reconstructing routes of invasion using genetic data: why, how and so what? Mol. Ecol. 19, 4113–4130. doi: 10.1111/j.1365-294X.2010.04773.x
Fitzpatrick, B. M., Fordyce, J. A., Niemiller, M. L., and Reynolds, R. G. (2012). What can DNA tell us about biological invasions? Biol. Invasions 14, 245–253. doi: 10.1007/s10530-011-0064-1
Geller, J. B., Darling, J. A., and Carlton, J. T. (2010). Genetic perspectives on marine biological invasions. Ann. Rev. Mar. Sci. 2, 367–393. doi: 10.1146/annurev.marine.010908.163745
Hickerson, M. J., Carstens, B. C., Cavender-Bares, J., Crandall, K. A., Graham, C. H., Johnson, J. B., et al. (2010). Phylogeography's past, present, and future: 10 years after Avise, 2000. Mol. Phylog. Evol. 54, 291–301. doi: 10.1016/j.ympev.2009.09.016
Hudson, J., Johannesson, K., McQuaid, C. D., and Rius, M. (2020). Secondary contacts and genetic admixture shape colonization by an amphiatlantic epibenthic invertebrate. Evol. Appl. 13, 600–612. doi: 10.1111/eva.12893
Knowles, L. L. (2003). The burgeoning field of statistical phylogeography: statistical phylogeography. J. Evol. Biol. 17, 1–10. doi: 10.1046/j.1420-9101.2003.00644.x
Lees, D. C., Lack, H. W., Rougerie, R., Hernandez-Lopez, A., Raus, T., Avtzis, N. D., et al. (2011). Tracking origins of invasive herbivores through herbaria and archival DNA: the case of the horse-chestnut leaf miner. Front. Ecol. Environ. 9, 322–328. doi: 10.1890/100098
Navascués, M., Depaulis, F., and Emerson, B. C. (2010). Combining contemporary and ancient DNA in population genetic and phylogeographical studies. Mol. Ecol. Res. 10, 760–772. doi: 10.1111/j.1755-0998.2010.02895.x
Nielsen, R. (2006). Reducing an intellectual deficit: coalescent theory for (almost) everyone. Evolution 60, 208–209. doi: 10.1111/j.0014-3820.2006.tb01097.x
Pascual, M., Rives, B., Schunter, C., and Macpherson, E. (2017). Impact of life history traits on gene flow: a multispecies systematic review across oceanographic barriers in the Mediterranean Sea. PLoS ONE 12:e0176419. doi: 10.1371/journal.pone.0176419
Pérez-Portela, R., Arranz, V., Rius, M., and Turon, X. (2013). Cryptic speciation or global spread? the case of a cosmopolitan marine invertebrate with limited dispersal capabilities. Sci. Rep. 3:3197. doi: 10.1038/srep03197
Pérez-Portela, R., Rius, M., and Villamor, A. (2017). Lineage splitting, secondary contacts and genetic admixture of a widely distributed marine invertebrate. J. Biogeogr. 44, 446–460. doi: 10.1111/jbi.12917
Reitzel, A. M., Herrera, S., Layden, M. J., Martindale, M. Q., and Shank, T. M. (2013). Going where traditional markers have not gone before: utility of and promise for RAD sequencing in marine invertebrate phylogeography and population genomics. Mol. Ecol. 22, 2953–2970. doi: 10.1111/mec.12228
Rellstab, C., Gugerli, F., Eckert, A. J., Hancock, A. M., and Holderegger, R. (2015). A practical guide to environmental association analysis in landscape genomics. Mol. Ecol. 24, 4348–4370. doi: 10.1111/mec.13322
Reyes-Velasco, J., Manthey, J. D., Bourgeois, Y., Freilich, X., Boissinot, S., and Chiang, T.-Y. (2018). Revisiting the phylogeography, demography and taxonomy of the frog genus Ptychadena in the Ethiopian highlands with the use of genome-wide SNP data. PLoS ONE 13:e0190440. doi: 10.1371/journal.pone.0190440
Richardson, D. M., and Pyšek, P. (2007). Elton, C.S. 1958: the ecology of invasions by animals and plants. London: Methuen. Progr. Phys. Geogr. 31, 659–666. doi: 10.1177/0309133307087089
Riddle, B. R., Dawson, M. N., Hadly, E. A., Hafner, D. J., Hickerson, M. J., Mantooth, S. J., et al. (2008). The role of molecular genetics in sculpting the future of integrative biogeography. Progr. Phys. Geogr. 32, 173–202. doi: 10.1177/0309133308093822
Rius, M., Bourne, S., Hornsby, H. G., and Chapman, M. A. (2015a). Applications of next-generation sequencing to the study of biological invasions. Curr. Zool. 61, 488–504. doi: 10.1093/czoolo/61.3.488
Rius, M., and Teske, P. R. (2013). Cryptic diversity in coastal Australasia: a morphological and mitonuclear genetic analysis of habitat-forming sibling species: cryptic diversity in coastal australasia. Zool. J. Linn. Soc. 168, 597–611. doi: 10.1111/zoj.12036
Rius, M., Turon, X., Bernardi, G., Volckaert, F. A. M., and Viard, F. (2015b). Marine invasion genetics: from spatio-temporal patterns to evolutionary outcomes. Biol. Invasions 17, 869–885. doi: 10.1007/s10530-014-0792-0
Rius, M., Turon, X., Ordóñez, V., and Pascual, M. (2012). Tracking invasion histories in the sea: facing complex scenarios using multilocus data. PLoS ONE 7:e35815. doi: 10.1371/journal.pone.0035815
Robinson, T., Alexander, M., Simon, C., Griffiths, C. L., Peters, K., Sibanda, S., et al. (2016). Lost in translation? Standardising the terminology used in marine invasion biology and updating South African alien species lists. Afr. J. Mar. Sci. 38, 129–140. doi: 10.2989/1814232X.2016.1163292
Roman, J., and Palumbi, S. R. (2004). A global invader at home: population structure of the green crab, Carcinus maenas, in Europe. Mol. Ecol. 13, 2891–2898. doi: 10.1111/j.1365-294X.2004.02255.x
Sax, D., Stachowicz, J., Brown, J., Bruno, J., Dawson, M., Gaines, S., et al. (2007). Ecological and evolutionary insights from species invasions. Trends Ecol. Evol. 22, 465–471. doi: 10.1016/j.tree.2007.06.009
Selkoe, K. A., Watson, J. R., White, C., Horin, T. B., Iacchei, M., Mitarai, S., et al. (2010). Taking the chaos out of genetic patchiness: seascape genetics reveals ecological and oceanographic drivers of genetic patterns in three temperate reef species. Mol. Ecol. 19, 3708–3726. doi: 10.1111/j.1365-294X.2010.04658.x
Sherman, C. D. H., Lotterhos, K. E., Richardson, M. F., Tepolt, C. K., Rollins, L. A., Palumbi, S. R., et al. (2016). What are we missing about marine invasions? filling in the gaps with evolutionary genomics. Mar. Biol. 163:198. doi: 10.1007/s00227-016-2961-4
Tan, H. Z., Ng, E. Y. X., Tang, Q., Allport, G. A., Jansen, J. J. F. J., Tomkovich, P. S., et al. (2019). Population genomics of two congeneric Palaearctic shorebirds reveals differential impacts of Quaternary climate oscillations across habitats types. Sci. Rep. 9:18172. doi: 10.1038/s41598-019-54715-9
Turon, X., Antich, A., Palacín, C., Præbel, K., and Wangensteen, O. S. (2020). From metabarcoding to metaphylogeography: separating the wheat from the chaff. Ecol. Appl. 30:e02036. doi: 10.1002/eap.2036
Viard, F., and Comtet, T. (2015). “Applications of DNA-based methods for the study of biological invasions,” in Biological Invasions in Changing Ecosystems, eds J. Canning-Clode (Warsaw: De Gruyter Open).
Viard, F., David, P., and Darling, J. A. (2016). Marine invasions enter the genomic era: three lessons from the past, and the way forward. Curr. Zool. 62, 629–642. doi: 10.1093/cz/zow053
Viard, F., Riginos, C., and Bierne, N. (2020). Anthropogenic hybridization at sea: three evolutionary questions relevant to invasive species management. Phil. Trans. R Soc. B 375:20190547. doi: 10.1098/rstb.2019.0547
Weber, M. G., Wagner, C. E., Best, R. J., Harmon, L. J., and Matthews, B. (2017). Evolution in a community context: on integrating ecological interactions and macroevolution. Trends Ecol. Evol. 32, 291–304. doi: 10.1016/j.tree.2017.01.003
Keywords: gene trees, genealogy, introduction pathways, landscape genetics, non-equilibrium population, population genomics, seascape genetics
Citation: Rius M and Turon X (2020) Phylogeography and the Description of Geographic Patterns in Invasion Genomics. Front. Ecol. Evol. 8:595711. doi: 10.3389/fevo.2020.595711
Received: 17 August 2020; Accepted: 16 November 2020;
Published: 14 December 2020.
Edited by:
Emiliano Mori, University of Siena, ItalyReviewed by:
Andrea Galimberti, University of Milano-Bicocca, ItalyCopyright © 2020 Rius and Turon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marc Rius, bS5yaXVzQHNvdG9uLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.