 Felipe S. Campos
Felipe S. Campos Ricardo Lourenço-de-Moraes
Ricardo Lourenço-de-Moraes- 1NOVA Information Management School (NOVA IMS), Universidade Nova de Lisboa, Lisbon, Portugal
- 2Programa de Pós-Graduação em Ecologia e Monitoramento Ambiental (PPGEMA), Universidade Federal da Paraíba (UFPB), Rio Tinto, Brazil
The Overlooked Wildlife Spillover in Human-Dominated Ecosystems
The rapid dissemination of severe acute respiratory syndrome coronavirus 2 (SARS-CoV−2) has opened up an environmental dilemma—investigating the relationship between the evolutionary history of coronaviruses (CoVs) and the zoonotic spillover in humans to avoid new rapidly evolving pathogens. To guide politicians in health policy decision-making, scientists have an urgent need to explore how cross-species virus transmission can help prevent pandemics (Zhou et al., 2020). The emergence of new epidemic diseases varies among different taxonomic groups, and the human-made change in natural environments causes eco-evolutionary consequences. Therefore, the alteration of this natural role caused by human pressures on wild species, we label as “ecological fever” —a new One Health perspective from ecology to society. Following the new phylogenies of coronavirus proposed by Gorbalenya et al. (2020) and Zhang et al. (2020), we explore the adaptive evolution of coronaviruses across mammal species and its importance for wildlife conservation. Here, we show reconstructed ancestral states of coronaviruses under maximum-likelihood estimations across an entire class of host organisms (i.e., Mammalia). In this opinion paper, we explore the evolution and cross-species transmission of coronaviruses and highlight the need to preserve natural habitats of wildlife in order to prevent future pandemics.
Phylogenetic Relationships
We reconstruct a full trait-based evolution for a range of coronaviruses (CoVs) through the following steps: (1) According to the new phylogenies of CoVs proposed by Gorbalenya et al. (2020) and Zhang et al. (2020), we build a maximum clade credibility tree based on likelihood ratio tests for the genome tree of 21 CoVs (Bat Hp-BetaCoV, SARSr-CoV BtKY72, SARS-CoV, SARS-CoV PC4–227, Pangolin-CoV, SARSr-CoV RaTG13, SARS-CoV−2, Ro-BatCoV GCCDC1, Ro-BatCoV HKU9, Ei-BatCoV C704, EriCoV, MERS-CoV, Ty-BatCoV HKU4, Pi-BatCoV HKU5, MrufCoV 2JL14, HCoV OC43, ChRCoV HKU24, HCoV HKU1, MHV, HCoV 229E and HCoV NL63); (2) Using the GenBank database (https://www.ncbi.nlm.nih.gov/genbank/), we assess all mammalian hosts (n = 44 species; see Supplementary Table 1) of the 21 CoVs; (3) We create a data matrix to compare the pairwise distribution of the CoVs and the mammalian orders (i.e., Artiodactyla, Carnivora, Chiroptera, Eulipotyphla, Lagomorpha, Pholidota, Primates, and Rodentia), providing ancestral character states under stochastic character mapping analysis—SIMMAP (Bollback, 2006) with 1,000 simulations, accounting for phylogenetic uncertainty; (4) Using the full time-calibrated phylogeny of Mammalia proposed by Upham et al. (2019), we evaluate the proportional phylogenetic relationships of the 21 CoVs across their mammalian hosts. To test phylogenetic signals, we calculate D-statistic for discrete attributes to show the degree of phylogenetic clustering under Brownian expectations, as defined by Fritz and Purvis (2010). We perform all the phylogenetic analysis and the null simulations in R software (R Development Core Team, 2019), using the packages “caper” (Orme et al., 2018) and “phytools” (Revell, 2012); (5) To allow further comparisons between the CoVs evolution and the mammalian phylogeny, we investigate the evolutionary adaptation of coronaviruses under the Phylogenetic or Brownian trait evolution in adaptive radiations of mammals.
Coevolution Coronavirus in Mammal Hosts
Given the strong transmission patterns of the coronavirus infection in mammals, their potential zoonotic transfers rely on the number of phylogenetically distinct hosts. Searching for coronaviruses' phylogenetic signals in infected species, we find differences in species-level host relationships within the full phylogeny of Mammalia proposed by Upham et al. (2019). Using phylogenetic inferences based on ecological trait evolution, we show that 44% of the coronaviruses have zoonotic origins based on a non-random phylogenetic structure, and 56% have their origins expected by Brownian phylogenetic structure under random processes (Figure 1). Mapping the full-length genomes of coronavirus across mammals, we find that they are evolving to adapt to specific mammalian taxa as well as a random evolutionary pattern, suggesting easy transmission across different mammalian species.
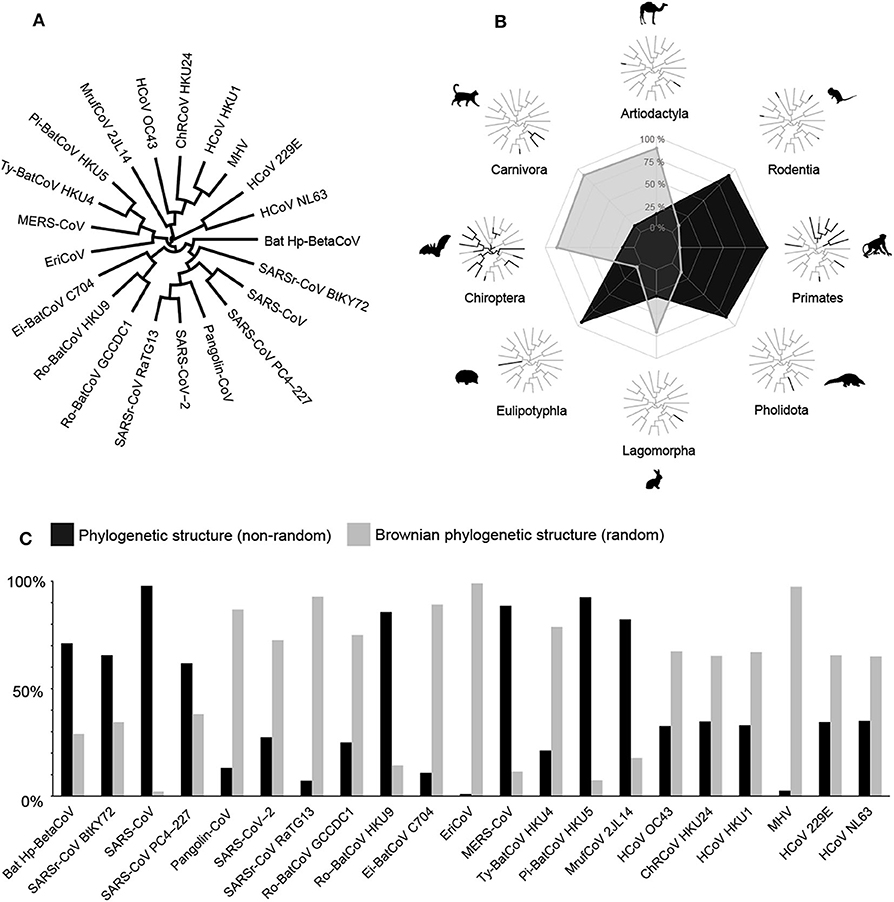
Figure 1. Phylogenetic relationships between coronaviruses and their mammalian hosts. (A) Full genome tree of 21 CoVs. (B) Maximum-likelihood ancestral state reconstructions for eight mammalian host orders by stochastic character mapping (SIMMAP). Radar chart shows the variation (%) between Phylogenetic and Brownian structure evolution of CoVs in mammalian orders. (C) Proportional differences of Phylogenetic and Brownian structure variation (%) in the evolutionary history of CoVs on the time-calibrated phylogeny of Mammalia.
We show that the coronavirus radiation into the orders Chiroptera, Lagomorpha, Carnivora, and Artiodactyla have a Brownian phylogenetic structure, with a random origin within Mammalia. This evolutionary pattern can be explained by the high plasticity of the coronavirus to infect species from these mammalian orders. In this way, they are able to mutate and evolve in new types of hosts, which then become reservoirs of infection (Andersen et al., 2020). Within all mammalian orders, Chiroptera has the highest diversity of coronavirus with a worldwide distribution, occurring in all continents except Antarctica (Calisher et al., 2006). From an ecological perspective, bat-related coronavirus relies on their higher population sizes in the breeding seasons, which increase fecal-oral transmissions and highlight the necessity to keep them protected from human interference in natural habitats (Drexler et al., 2014). In bats, multiple viruses can coexist asymptomatically (Calisher et al., 2006), while in other mammal species, a single virus can cause numerous health problems (Zhou et al., 2020). In the case of the SARS-CoV, we find a Brownian phylogenetic structure for Chiroptera and a phylogenetic structure among the other mammal groups infected as hosts. In Carnivora, our estimates show the SARS-CoV PC4–227 has a phylogenetic structure, with a single host species represented by the common palm civet (Paradoxurus hermaphroditus), suggesting a low capacity to infect other hosts. Recent research on the potential origin of SARS-CoV−2 shows its closely phylogenetic relatedness to Pangolin-CoV and bat SARSr-CoV RaTG13 (Andersen et al., 2020). However, it is already known that SARS-CoV−2 does not originate directly from pangolins (Manis javanica)—indeed infected with Pangolin-CoV (Andersen et al., 2020). Our results suggest that the SARS-CoV−2 has a Brownian phylogenetic structure, and cross-species transmission from an unknown animal species could be acting as an intermediate host among bats, pangolins, and humans (Zhang et al., 2020).
Evolutionary Pathways of Shared Pathogens
On the intermediate host species-relationships, some researchers show that the SARS-CoV initially jumped from bats-to-civets-to-humans (Song et al., 2005). In contrast, the MERS-CoV jumped from bats-to-camels-to-humans (Müller et al., 2014). Moreover, some bat coronavirus shows coevolution with their specific host genera (Drexler et al., 2014). Besides being considered as natural and important coronaviruses reservoirs (Zhou et al., 2020), bats have a key functional role in providing ecosystem services to improve human well-being, such as biological control and pollination (Kunz et al., 2011). Therefore, there is an urgent need to conserve bat species in their natural habitat for keeping humans safe from infectious diseases. Although humans are highly susceptible to infection by SARS-CoV related viruses (Zhou et al., 2020), coronaviruses can co-exist harmoniously in wild environments with bats (Calisher et al., 2006), which are essential to preserve crucial relationships between biodiversity and ecosystem functioning for improved public health outcomes (Kunz et al., 2011).
Implications for Conservation and Future Directions
Accelerated modern human-induced changes in natural environments can have many consequences beyond infectious diseases, species extinction and climate-health issues. Our findings support conservation studies aimed at exploring the eco-evolutionary consequences of environmental change in managing public health. The coronavirus crisis results from the human disregard for biodiversity in their natural habitat and the wildlife trade spillover to spreading diseases. The substantial increase in deforestation of species-rich regions (e.g., Amazon forest and Southeast Asian forests) may be the beginning of a new pandemic. Opportunities for prevention may emerge from ecology, evolution, and conservation science, which can play a key role in defining links between people and nature, having several implications for human well-being in the short-run. For post-pandemic projections, future studies should focus on outbreak prevention through ecological and evolutionary knowledge instead of disease-related health efforts. Using trait-based science across the Tree of Life for mammals, we find evidence on the evolutionary role of coronavirus in human-wildlife relationships.
Data Availability Statement
The raw data used in this paper are available in Supplementary Table 1.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
FSC was supported by the Portuguese Foundation for Science and Technology (FCT, Portugal), under the projects PTDC/CTA-AMB/28438/2017 (Assessment of Ecosystem Services and Biodiversity—ASEBIO) and UIDB/04152/2020 (Information Management Research Center—MagIC). RL was supported by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, Finance Code 001 (CAPES, Brasil).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank L. Rincón, M. Recchia, and A. Szklarczyk for the useful comments on the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2020.575286/full#supplementary-material
References
Andersen, K. G., Rambaut, A., Lipkin, W. I., Holmes, E. C., and Garry, R. F. (2020). The proximal origin of SARS-CoV-2. Nat. Med. 26, 450–452. doi: 10.1038/s41591-020-0820-9
Bollback, J. P. (2006). SIMMAP: stochastic character mapping of discrete traits on phylogenies. BMC Bioinformatics 7:88. doi: 10.1186/1471-2105-7-88
Calisher, C. H., Childs, J. E., Field, H. E., Holmes, K. V., and Schountz, T. (2006). Bats: important reservoir hosts of emerging viruses. Clin. Microbiol. Rev. 19, 531–545. doi: 10.1128/CMR.00017-06
Drexler, J. F., Corman, V. M., and Drosten, C. (2014). Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS. Antiviral Res. 101, 45–56. doi: 10.1016/j.antiviral.2013.10.013
Fritz, S. A., and Purvis, A. (2010). Selectivity in mammalian extinction risk and threat types: a new measure of phylogenetic signal strength in binary traits. Conserv. Biol. 24, 1042–1051. doi: 10.1111/j.1523-1739.2010.01455.x
Gorbalenya, A. E., Baker, S. C., Baric, R. S., de Groot, R. J., Drosten, C., Gulyaeva, A. A., et al. (2020). The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 5, 536–544. doi: 10.1038/s41564-020-0695-z
Kunz, T. H., de Torrez, E. B., Bauer, D., Lobova, T., and Fleming, T. H. (2011). Ecosystem services provided by bats. Ann. N.Y. Acad. Sci. 1223, 1–38. doi: 10.1111/j.1749-6632.2011.06004.x
Müller, M. A., Corman, V. M., Jores, J., Meyer, B., Younan, M., Liljander, A., et al. (2014). MERS coronavirus neutralizing antibodies in camels, eastern Africa, 1983–1997. Emerg. Infect. Dis. 20, 2093–2095. doi: 10.3201/eid2012.141026
Orme, D., Freckleton, R., Thomas, G., Petzoldt, T., Fritz, S., and Isaac, N. (2018). caper: Comparative Analyses of Phylogenetics and Evolution in R. Available online at: https://cran.r-project.org/package=caper
R Development Core Team (2019). R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. Available online at: http://www.R-project.org
Revell, L. J. (2012). phytools: An R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 3, 217–223. doi: 10.1111/j.2041-210X.2011.00169.x
Song, H. D., Tu, C. C., Zhang, G. W., Wang, S. Y., Zheng, K., Lei, L. C., et al. (2005). Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc. Natl. Acad. Sci. U.S.A. 102, 2430–2435. doi: 10.1073/pnas.0409608102
Upham, N. S., Esselstyn, J. A., and Jetz, W. (2019). Inferring the mammal tree: Species-level sets of phylogenies for questions in ecology, evolution, and conservation. PLoS Biol. 17:e3000494. doi: 10.1371/journal.pbio.3000494
Zhang, T., Wu, Q., and Zhang, Z. (2020). Probable pangolin origin of SARS-CoV-2 associated with the COVID-19 outbreak. Curr. Biol. 30, 1346–1351. doi: 10.1016/j.cub.2020.03.022
Keywords: outbreak, coevolution, mammal hosts, biodiversity conservation, one health, pandemics
Citation: Campos FS and Lourenço-de-Moraes R (2020) Ecological Fever: The Evolutionary History of Coronavirus in Human-Wildlife Relationships. Front. Ecol. Evol. 8:575286. doi: 10.3389/fevo.2020.575286
Received: 23 June 2020; Accepted: 07 September 2020;
Published: 15 October 2020.
Edited by:
Vincent Obanda, Kenya Wildlife Service, KenyaReviewed by:
Patrick I. Chiyo, Independent Researcher, Edinburgh, United KingdomMoh A. Alkhamis, Kuwait University, Kuwait
Copyright © 2020 Campos and Lourenço-de-Moraes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Felipe S. Campos, ZmNhbXBvc0Bub3ZhaW1zLnVubC5wdA==
†These authors have contributed equally to this work