 Jin-Hyeop Jeong
Jin-Hyeop Jeong Heesoo Kim
Heesoo Kim Seongho Ryu
Seongho Ryu Won Kim
Won Kim- 1School of Biological Sciences, Seoul National University, Seoul, South Korea
- 2Soonchunhyang Institute of Medi-Bio Science, Soonchunhyang University, Cheonan, South Korea
Introduction
The Pycnogonida, or sea spiders, are essential arthropod taxa for understanding the early evolutionary history of arthropods and relationships between primary clades of this phylum. Studies based on both molecular and morphological data suggest that sea spiders have archaic origins as old as the early Cambrian. They are also considered an early-branching lineage with a deep split origin, which is implied by their incomplete early fossil records and relatively recent diversification of extant members (Dunlop and Selden, 2009; Rota-Stabelli et al., 2013; Sabroux et al., 2019; Lozano-Fernandez et al., 2020). However, their peculiar morphologies such as extremely reduced trunk and 8–12 walking legs containing part of their digestive and reproductive organs are highly derived and very unusual among arthropods (Sabroux et al., 2019). With a combination of the molecular phylogenetic studies using the limited numbers of marker genes, their peculiar adult and developmental morphologies had led the Cormogonida hypothesis (Zrzavý et al., 1998; Giribet et al., 2001; Dunlop and Arango, 2005; Machner and Scholtz, 2010), which places pycnogonids as a sister taxon to all other arthropods, as opposed to the monophyletic Chelicerata hypothesis, which recognizes their sister taxon relationship with the Euchelicerata. Unlike these molecular phylogenetics studies based on a few markers, numerous phylogenomic studies based on thoroughly sampled expressed sequences tags repeatedly designated sea spiders as basalmost chelicerates (Meusemann et al., 2010; Regier et al., 2010; Rehm et al., 2011). The most recent phylogenomic researches incorporating significantly increased taxa and tested loci based on the whole-genome or transcriptome data (Sharma et al., 2014; Ballesteros and Sharma, 2019; Lozano-Fernandez et al., 2019) have brought consensus on pycnogonids firmly nested in the Chelicerata.
These recent phylogenomic studies provided newly sequenced chelicerate genomes or transcriptomes, in addition to invaluable insights on the evolution of Euchelicerata (Sharma et al., 2014; Ballesteros and Sharma, 2019; Lozano-Fernandez et al., 2019). However, studies using such an approach on the Pycnogonida are currently very limited and fail to incorporate a de novo assembled pycnogonid genome (Dietz et al., 2019; Ballesteros et al., 2020). This lack of pycnogonid genome assembly is also a challenging obstacle for studies on the evolution of their distinctive morphology that requires high-quality genome assemblies with reliable annotations (Garb et al., 2018). To the best of our knowledge, no pycnogonid genome has been sequenced to date, and there are only three cases of unpublished flow cytometry reports on pycnogonid species so far (Animal Genome Size Database, https://genomesize.com). We aimed to provide an annotated pycnogonid genome sequence using the PacBio long-read de novo genome sequencing to provide useful information for understanding the evolution of unique pycnogonid morphological and developmental characteristics. Here, we report a draft of the first high quality annotated genome of a common sea spider species, Nymphon striatum, in Korean waters. This is the first de novo sequenced and assembled genome that represents the Class Pycnogonida.
Results
The genome size of N. striatum was estimated at ~607 Mb, while the genome survey result showed relatively high (~1.9%) heterozygosity ratios and prominent heterozygosity peaks clearly separated from the homozygosity peaks (Supplementary Figure 1). The almost equal distributions of non-duplicated (1X) and duplicated (2X) K–mers at the homozygosity peak further supported the highly heterozygous nature of its genome (Supplementary Figure 3) that we speculated the pooling of N. striatum specimens was likely its main cause. The initial contig–level genomic assembly of N. striatum was 1.26 Gb long, of which the total base length exceeded twice those of the genome survey results (Table 1A). Genome assembly assessment using Benchmarking Universal Single-Copy Orthologs (BUSCO) (Simão et al., 2015) also showed that 53.47% of BUSCO genes from the initial assembly were duplicated, which further indicates the presence of numerous redundant haplotypic contigs (Table 1B). Therefore, these haplotypic contigs were reduced by applying Purge Haplotigs (Roach et al., 2018) and Redundans (Pryszcz and Gabaldoón, 2016). The Purge Haplotigs-curated contig–level draft genome demonstrated prominent improvements in its total length, which was reduced to 732.9 Mb, and contig N50 almost doubled to 360.90 Kb, with its number of contigs being reduced to 2,946 (Table 1A). Using arthropod database, the BUSCO assessment of the revised contig–level assembly resulted in a substantial improvement: the percentage of complete, but duplicated BUSCO genes was reduced to 16.79% (Table 1B and Supplementary Figure 2). The K–mer distribution analyses using the K–mer analysis toolkit (Mapleson et al., 2017), further found that the ratios of heterozygous haplotigs (indicated by 2X, or duplicated K–mers) were greatly reduced at the homozygosity peak after running Purge Haplotigs (Supplementary Figure 3). After conducting scaffolding and gap closing, the final version of the scaffold–level draft genome was composed of 1,638 scaffolds with scaffold N50 increased to 701.80 Kb and a minute fraction (0.04%) of N bases (Table 1A). We found that 31.9% of the genomic scaffolds were longer than 500 Kb, and among them, 159 scaffolds were over 1,000 Kb. In particular, only seven scaffolds were shorter than 10 Kb, which indicated that our genome was highly contiguous (Figure 1A).
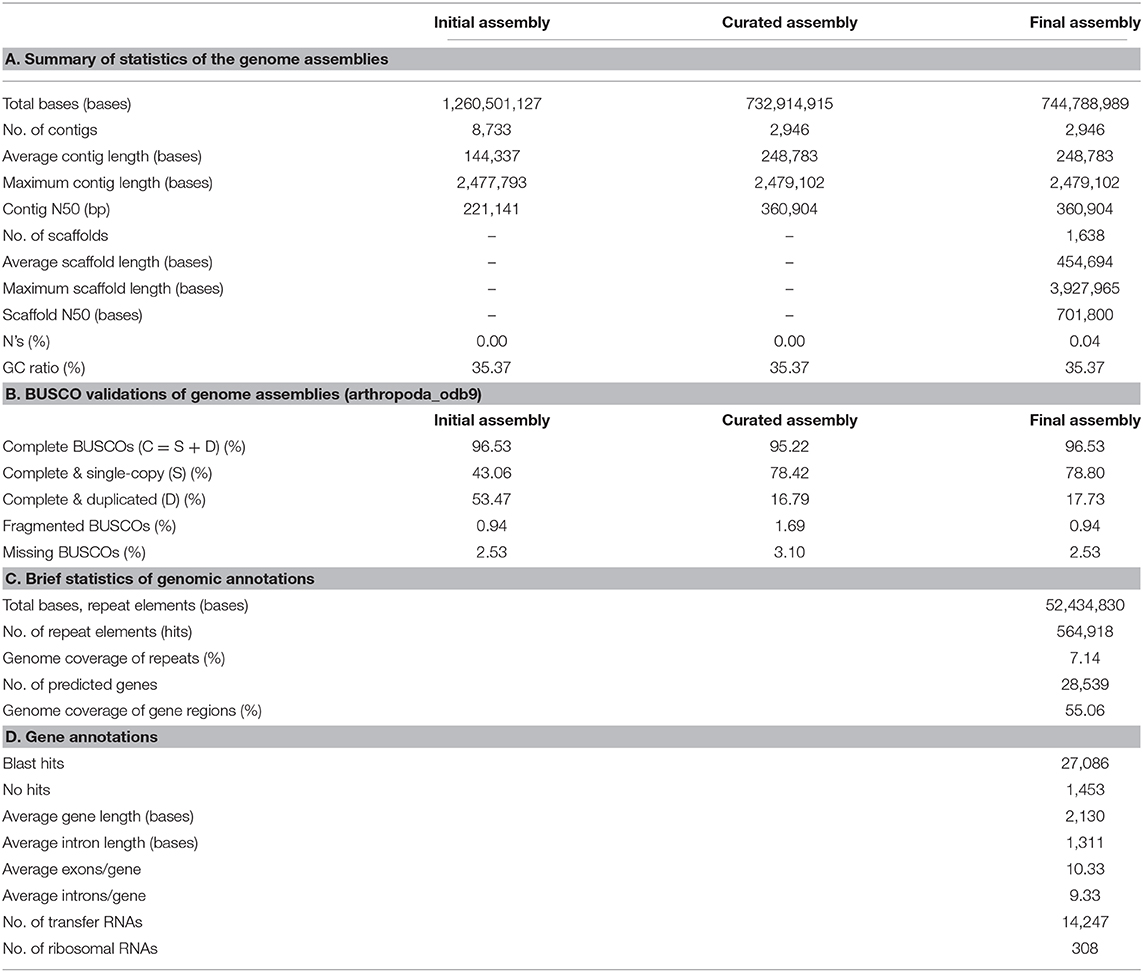
Table 1. Summary of genome assembly and annotation of Nymphon striatum.
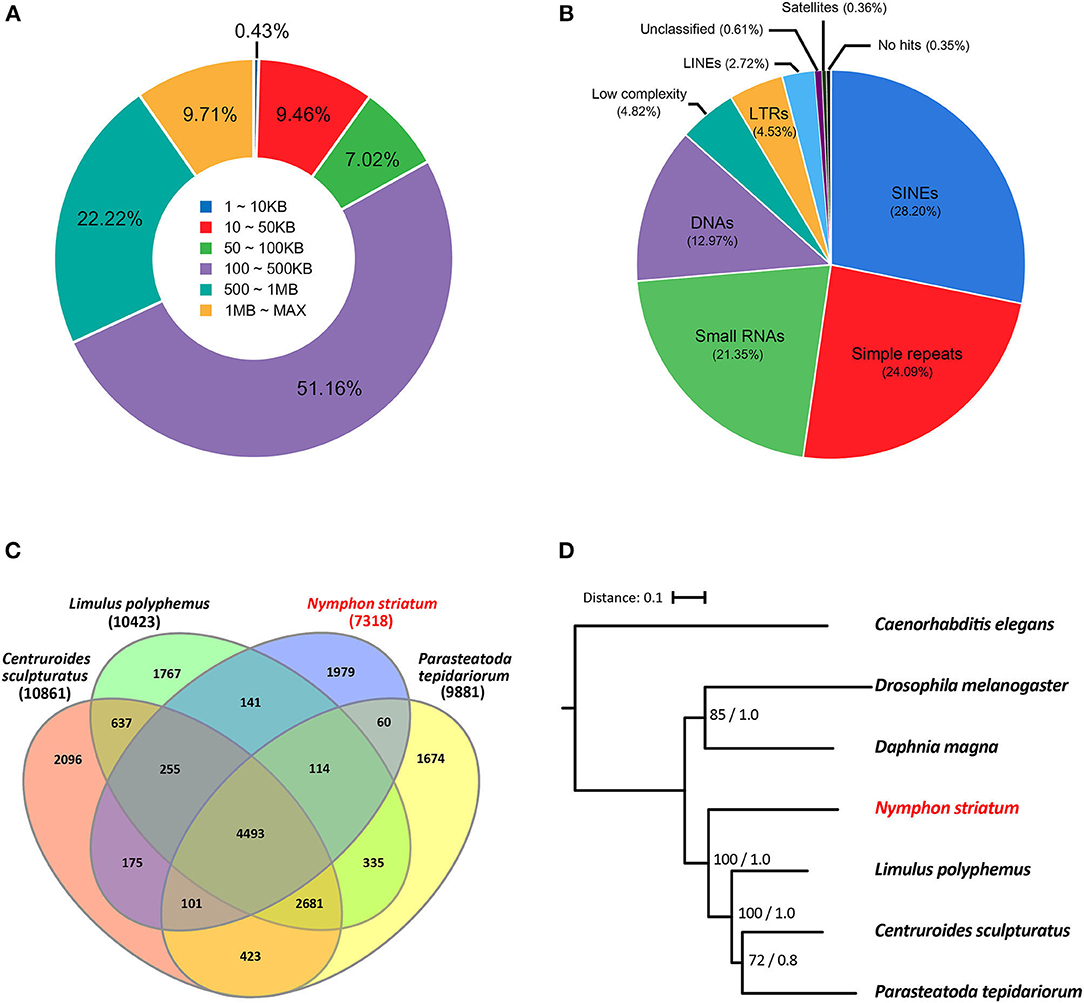
Figure 1. Characteristics of the Nymphon striatum genome assembly. (A) The length distributions of the gap–closed scaffolds; (B) ab initio predicted repetitive elements and their subclass distributions; (C) a Venn diagram of the orthologous clusters among five chelicerate species; (D) the phylogenetic relationship of N. striatum with other six ecdysozoan species; Caenorhabditis elegans was selected as an outgroup taxon for the analysis. For each node, its bootstrap support value and the posterior probability are indicated at the base of the node.
An ab initio repeat element prediction resulted in a total of 7.14% of the genomic sequences annotated as repetitive sequences (Table 1C). Among these repeat elements, short interspersed nuclear elements (SINEs) accounted for 28.20% of the total length of the annotated repeat sequences, and they were recorded as the most enriched repeats from the N. striatum genome. The SINEs were followed by simple repeats (24.09%), small RNAs (21.35%), and non-SINE interspersed elements (20.82%), while satellites (0.36%) occurred the least among the categorized repeat elements (Figure 1B). The final N. striatum genome consisted of 28,539 genes which spanned 56.01% of the total genomic length. Additionally, 14,247 transfer RNA and 308 ribosomal RNA genes were annotated (Table 1D). The summed repetitive element contents of 7.14% and gene span of 51.06% were significantly different when they were compared to those of other chelicerate genomes used in the basic comparative analyses of this current study. The ortholog analysis conducted using six ecdysozoan genomes (Supplementary Table 3) resulted in 7,597 orthologous clusters and 3,888 singletons for N. striatum, with 4,493 orthologous clusters shared within four chelicerate species (Figure 1C). Phylogenetic tree reconstruction strongly supported the earliest-branching position of N. striatum nested in the monophyletic Chelicerata (Figure 1D) with maximum bootstrap support values (100%) and posterior probabilities (1.0). Nevertheless, more terrestrial arachnid taxa are required to be sampled in further analysis to provide a conclusion about a contentious phylogenetic position of horseshoe crabs deeply nested in the Arachnida that the recent phylogenomic studies on the Euchelicerata have debated (Sharma et al., 2014; Ballesteros and Sharma, 2019; Lozano-Fernandez et al., 2019). To sum up, we suggest that the draft N. striatum genome that we assembled is nearly complete and provides the first reference genome representing pycnogonids.
Materials and Methods
Sample Collection and Sequencing
We collected 40 individuals of N. striatum at Sacheon-hang, (37.82609°N, 128.93379°E, at a depth of 32 m, on 2018.07.12., NCBI BioSample accession ID: SAMN13567730), South Korea by SCUBA diving. These 40 sea spiders were brought to the laboratory alive, and then pooled together to compensate for the small size of the organisms and to ensure that the amount and quality of extracted DNA are acceptable for PacBio sequencing. All 40 sea spider individuals were collected from a single population at the same location to minimize the heterozygosity of the sequenced genomic reads. These pooled sea spiders were then buffered with RNAlater™ (Thermo Fisher Scientific, MA, USA) and lysed using QIAzol Reagent (Qiagen, MA, USA) according to the manufacturer's protocols. To isolate the DNA, the lysate was centrifuged according to the QIAzol Reagent protocol. The DNA sample was then extracted from the interphase of the lysate using the MG Genomic DNA Purification kit (Macrogen Inc, Seoul, Korea). The extracted DNA sample was quantified by NanoDrop 1000 spectrometer (Qiagen, MA, USA) and qualified using a 2100 Bioanalyzer (Agilent Technologies, CA, USA).
We generated and sequenced two copies of genomic Illumina Paired–end (PE) libiraries (Illumina HiSeq X Ten, read-length 151 bp, insertion size 350 bp), five different insert–sized Mate pair libraries (Illumina NovaSeq 6000, read-length 101 bp, insertion sizes of 550 bp, 3 kb, 5 kb, 8 kb, 10 kb), and a single PacBio library (PacBio Sequel, SMRTbell 20 kb library) for N. striatum (Supplementary Table 1). These libraries for Illumina and PacBio sequencing were prepared using their respective manufacturer's instructions. The Illumina platforms generated 136.28 Gb of genomic PE raw reads and 287.98 Gb of Mate pair raw reads, and the PacBio Sequel (Pacific Biosciences, CA, USA) platform generated 84.83 Gb of long–read sequences with their N50 estimated as 19.43 Kb.
De novo Assembly of N. striatum Genome
De novo sequenced genomic Illumina reads were assessed using FastQC v0.11.7 (Marçais and Kingsford, 2011) and then underwent adapter trimming and filtering (Q > 30) using our in–house scripts. These genomic PE reads were subjected to the genome survey by Jellyfish v2.2.10 using its configurations of count step (-C -c 3 -s 100000000), merge step (default parameter), histo step (-h 10000000000), and k–mer sizes (17, 21, 25 bp). The HGAP 4 (Chin et al., 2013) assembly application was used to assemble 84.83 Gb of PacBio subreads into contigs with its default operating options for alignment, assembly, consensus, and polishing using the Arrow application. These assembled contigs were error–corrected by mapping filtered genomic PE sequences using default parameters of Pilon v1.21, followed by additional polishing by mapping PacBio reads using SMRT Link (v6.0.0.47841) to obtain consensus genomic contig sequences. To reduce these haplotypic contigs from our initial assembly, Purge Haplotigs and the reduction module of Redundans were applied with their default operating parameters, respectively. The statistics and BUSCO assessments of the two alternatives of curated contig-level assembly were then compared. Since the Purge Haplotgis-curated contig-level genome showed superior statistics of its assemblage (Supplementary Table 2A) and duplicated complete BUSCO gene contents (16.79 vs. 34.8%, Supplementary Table 2B) than those of Redundans curated one, it was selected for the downstream analyses. The K–mer analysis toolkit (Mapleson et al., 2017) was used to validate contig–level assemblies before and after purging haplotigs by mapping the PE reads against the genome assemblies with using default parameters. The Scaffolding Pre-assembled Contigs after Extension (SSPACE, Boetzer et al., 2010) program was used to scaffold haplotig–purged contigs with mate pair reads of five insertion sizes. The gaps between genomic scaffolds were closed using PBJelly (English et al., 2012) and GMcloser (Kosugi et al., 2015). After gap closing, the scaffolds were polished once more using the SMRT Link to finalize the scaffold–level of the draft genome of N. striatum. BUSCO v2 was applied to assess our three versions of genome assemblies at each respective step (Table 1B) using the eukaryote_odb9 and arthropoda_odb9 databases. Repetitive sequences of the draft genome were annotated ab initio and then masked for the gene annotation procedure using RepeatMasker v4.0.6 (Tarailo-Graovac and Chen, 2009) with our customized RepBase library (based on RepBase 24.03), which is available in the online Supplementary Data.
Genomic Annotations
The NCBI reference genome datasets of three chelicerates (Supplementary Table 3) were downloaded for the downstream steps of genomic annotations. De novo transcriptome sequencing was conducted, nevertheless the sequenced transcriptomic reads were abandoned due to their low quality that the pooling procedure of specimens potentially resulted in. Therefore, GeMoMa (Keilwagen et al., 2018) was used to predict open reading frames with homology-based approach, based on the gap-closed N. striatum genome and transcriptome datasets of three reference chelicerate genomes. The Seqping v0.1.33 pipeline (Chan et al., 2017) was used to conduct gene model prediction and annotation, based on the direct evidence of reference protein sets from three chelicerate genomes and ab initio predicted gene models, which were trained using the evidence. Three chelicerate protein sequences were manually curated to reduce redundant copies, and then used as a reference. Two different Seqping associated tools, GlimmerHMM and AUGUSTUS were trained using these protein reference data, and they were used to predict gene models. MAKER2 (v2.31.8) was used to combine the homology–based and ab initio predicted gene models to construct a consensus set of annotated genes (Holt and Yandell, 2011; Chan et al., 2017). Then, these gene models were functionally annotated using BLAST (v2.7.1+) against the bioinformatics databases which were applied for the research of the Amphibalanus amphitrite reference genome (Kim et al., 2019) (Supplementary Materials).
Orthologous Gene Families Analysis and Phylogenomic Assays
Six ecdysozoan reference genome datasets were selected and downloaded (Supplementary Table 3) from NCBI for analyzing orthologous genes using OrthoMCL v2.0.9 (Fischer et al., 2011). Datasets of non–chelicerate species were excluded for increasing the visual legibility of the Venn diagram of analyzed orthologs (Figure 1C). To construct phylogenetic tree, we curated 106 non–redundant orthologous genes that were shared with all seven analyzed genomes. These orthologs were aligned and then BeforePhylo (https://github.com/qiyunzhu/BeforePhylo) was used to concatenate them into a supermatrix. MAFFT was used to obtain an alignment of these concatenated protein sequences (Katoh et al., 2017). To analyze phylogenetic relationship using maximum likelihood and Bayesian inference methods, RAxML 8.2.12 (Stamatakis, 2014) and MrBayes 3.2.7 (Ronquist et al., 2012) were used with 1,000 and 3,000,000 pseudoreplication values, respectively.
Data Availability Statement
The datasets generated for this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI under accession numbers PRJNA595812 and SAMN13567730.
Author Contributions
WK and SR conceived the study. J-HJ and HK performed the bioinformatics analyses and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the Collaborative Genome Program and the Marine Biotechnology Program of the Korea Institute of Marine Science and Technology Promotion (KIMST) funded by the Ministry of Ocean and Fisheries (MOF) (Nos. 20180430 and 20170431) Korea.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We appreciate our colleague at Seoul National University, Damin Lee, about his contribution to collect and identify Nymphon striatum samples for this research.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2020.554164/full#supplementary-material
References
Ballesteros, J. A., Setton, V. W., Santibáñez López, C. E., Arango, C. P., Brenneis, G., Brix, S., et al. (2020). Phylogenomic resolution of sea spider diversification through integration of multiple data classes. Mol. Biol. Evol. mssa228. doi: 10.1093/molbev/msaa228
Ballesteros, J. A., and Sharma, P. P. (2019). A critical appraisal of the placement of Xiphosura (Chelicerata) with account of known sources of phylogenetic error. Syst. Biol. 68, 896–917. doi: 10.1093/sysbio/syz011
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D., and Pirovano, W. (2010). Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579. doi: 10.1093/bioinformatics/btq683
Chan, K. L., Rosli, R., Tatarinova, T. V., Hogan, M., Firdaus-Raih, M., and Low, E. T. L. (2017). Seqping: gene prediction pipeline for plant genomes using self-training gene models and transcriptomic data. BMC Bioinform. 18:29. doi: 10.1101/038018
Chin, C. S., Alexander, D. H., Marks, P., Klammer, A. A., Drake, J., Heiner, C., et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10:563. doi: 10.1038/nmeth.2474
Dietz, L., Dömel, J. S., Leese, F., Mahon, A. R., and Mayer, C. (2019). Phylogenomics of the longitarsal Colossendeidae: the evolutionary history of an Antarctic sea spider radiation. Mol. Phylogenet. Evol. 136, 206–214. doi: 10.1016/j.ympev.2019.04.017
Dunlop, J. A., and Arango, C. P. (2005). Pycnogonid affinities: a review. J. Zool. Syst. Evol. Res. 43, 8–21. doi: 10.1111/j.1439-0469.2004.00284.x
Dunlop, J. A., and Selden, P. A. (2009). Calibrating the chelicerate clock: a paleontological reply to Jeyaprakash and Hoy. Exp. Appl. Acarol. 48:183. doi: 10.1007/s10493-009-9247-1
English, A. C., Richards, S., Han, Y., Wang, M., Vee, V., Qu, J., et al. (2012). Mind the gap: upgrading genomes with Pacific biosciences RS long-read sequencing technology. PLoS ONE 7:e47768. doi: 10.1371/journal.pone.0047768
Fischer, S., Brunk, B. P., Chen, F., Gao, X., Harb, O. S., Iodice, J. B., et al. (2011). Using OrthoMCL to assign proteins to OrthoMCL-DB groups or to cluster proteomes into new ortholog groups. Curr. Protoc. Bioinform. 35, 6.12.1–6.12.19. doi: 10.1002/0471250953.bi0612s35
Garb, J. E., Sharma, P. P., and Ayoub, N. A. (2018). Recent progress and prospects for advancing arachnid genomics. Curr. Opin. Insect Sci. 25, 51–57. doi: 10.1016/j.cois.2017.11.005
Giribet, G., Edgecombe, G. D., and Wheeler, W. C. (2001). Arthropod phylogeny based on eight molecular loci and morphology. Nature 413, 157–161. doi: 10.1038/35093097
Holt, C., and Yandell, M. (2011). MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 12:491. doi: 10.1186/1471-2105-12-491
Katoh, K., Rozewicki, J., and Yamada, K. D. (2017). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 20, 1160–1166. doi: 10.1093/bib/bbx108
Keilwagen, J., Hartung, F., Paulini, M., Twardziok, S. O., and Grau, J. (2018). Combining RNA-seq data and homology-based gene prediction for plants, animals, and fungi. BMC Bioinform. 19, 1–12. doi: 10.1186/s12859-018-2203-5
Kim, J.-H., Kim, H. K., Kim, H., Chan, B. K. K., Kang, S., and Kim, W. (2019). Draft genome assembly of a fouling barnacle, Amphibalanus amphitrite (Darwin, 1854): the first reference genome for Thecostraca. Front. Ecol. Evol. 7:465. doi: 10.3389/fevo.2019.00465
Kosugi, S., Hirakawa, H., and Tabata, S. (2015). GMcloser: closing gaps in assemblies accurately with a likelihood-based selection of contig or long-read alignments. Bioinformatics 31, 3733–3741. doi: 10.1093/bioinformatics/btv465
Lozano-Fernandez, J., Tanner, A. R., Giacomelli, M., Carton, R., Vinther, J., Edgecombe, G. D., et al. (2019). Increasing species sampling in chelicerate genomic-scale datasets provides support for monophyly of Acari and Arachnida. Nat. Commun. 10:2295. doi: 10.1038/s41467-019-12259-6
Lozano-Fernandez, J., Tanner, A. R., Puttick, M. N., Vinther, J., Edgecombe, G. D., and Pisani, D. (2020). A Cambrian–Ordovician terrestrialization of arachnids. Front. Genet. 11:182. doi: 10.3389/fgene.2020.00182
Machner, J., and Scholtz, G. (2010). A scanning electron microscopy study of the embryonic development of Pycnogonum litorale (Arthropoda, Pycnogonida). J. Morphol. 271, 1306–1318. doi: 10.1002/jmor.10871
Mapleson, D., Accinelli, G. G., Kettleborough, G., Wright, J., and Clavijo, B. J. (2017). KAT: a K–mer analysis toolkit to quality control NGS datasets and genome assemblies. Bioinformatics 33, 574–576. doi: 10.1101/064733
Marçais, G., and Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770. doi: 10.1093/bioinformatics/btr011
Meusemann, K., von Reumont, B. M., Simon, S., Roeding, F., Strauss, S., Kück, P., et al. (2010). A phylogenomic approach to resolve the arthropod tree of life. Mol. Biol. Evol. 27, 2451–2464. doi: 10.1093/molbev/msq130
Pryszcz, L. P., and Gabaldoón, T. (2016). Redundans: an assembly pipeline for highly heterozygous genomes. Nucleic Acids Res. 44:e113. doi: 10.1093/nar/gkw294
Regier, J. C., Shultz, J. W., Zwick, A., Hussey, A., Ball, B., Wetzer, R., et al. (2010). Arthropod relationships revealed by phylogenomic analysis of nuclear protein-coding sequences. Nature 463, 1079–1083. doi: 10.1038/nature08742
Rehm, P., Borner, J., Meusemann, K., von Reumont, B. M., Simon, S., Hadrys, H., et al. (2011). Dating the arthropod tree based on large-scale transcriptome data. Mol. Phylogenet. Evol. 61, 880–887. doi: 10.1016/j.ympev.2011.09.003
Roach, M. J., Schmidt, S. A., and Borneman, A. R. (2018). Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinform. 19:460. doi: 10.1186/s12859-018-2485-7
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Rota-Stabelli, O., Daley, A. C., and Pisani, D. (2013). Molecular timetrees reveal a Cambrian colonization of land and a new scenario for ecdysozoan evolution. Curr. Biol. 23, 392–398. doi: 10.1016/j.cub.2013.01.026
Sabroux, R., Audo, D., Charbonnier, S., Corbari, L., and Hassanin, A. (2019). 150-million-year-old sea spiders (Pycnogonida: Pantopoda) of Solnhofen. J. Syst. Palaeontol. 17, 1927–1938. doi: 10.1080/14772019.2019.1571534
Sharma, P. P., Kaluziak, S. T., Pérez-Porro, A. R., González, V. L., Hormiga, G., Wheeler, W. C., et al. (2014). Phylogenomic interrogation of Arachnida reveals systemic conflicts in phylogenetic signal. Mol. Biol. Evol. 31, 2963–2984. doi: 10.1093/molbev/msu235
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Tarailo-Graovac, M., and Chen, N. (2009). Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 25, 4–10. doi: 10.1002/0471250953.bi0410s25
Zrzavý, J., Hypša, V., and Vlášková, M. (1998). “Arthropod phylogeny: taxonomic congruence, total evidence and conditional combination approaches to morphological and molecular data sets,” in Arthropod Relationships. The Systematics Association Special Volume Series, Vol. 55, eds R. A. Fortey, and R. H. Thomas (Dordrecht: Springer), 97–107.
Keywords: Nymphon striatum, sea spider, Chelicerata, draft genome, PacBio sequencing
Citation: Jeong J-H, Kim H, Ryu S and Kim W (2020) The First Pycnogonid Draft Genome of Nymphon striatum. Front. Ecol. Evol. 8:554164. doi: 10.3389/fevo.2020.554164
Received: 21 April 2020; Accepted: 09 October 2020;
Published: 09 November 2020.
Edited by:
Enrique P. Lessa, Universidad de la República, UruguayReviewed by:
Jesus Lozano-Fernandez, Instituto de Biología Evolutiva (IBE), SpainRosa M. Fernandez, Harvard University, United States
Copyright © 2020 Jeong, Kim, Ryu and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Seongho Ryu, cnl1QHNjaC5hYy5rcg==; Won Kim, d29ua2ltQHBsYXphLnNudS5hYy5rcg==