
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol., 29 October 2020
Sec. Behavioral and Evolutionary Ecology
Volume 8 - 2020 | https://doi.org/10.3389/fevo.2020.535715
This article is part of the Research TopicAdvances in Ungulate EcologyView all 22 articles
 Joost F. de Jong1*
Joost F. de Jong1* Pim van Hooft1
Pim van Hooft1 Hendrik-Jan Megens2
Hendrik-Jan Megens2 Richard P. M. A. Crooijmans2
Richard P. M. A. Crooijmans2 Gerard Arjen de Groot3Josephine M. Pemberton4
Gerard Arjen de Groot3Josephine M. Pemberton4 Jisca Huisman4
Jisca Huisman4 Luděk Bartoš5
Luděk Bartoš5 Laura Iacolina6Sip E. van Wieren1Ronald C. Ydenberg7
Laura Iacolina6Sip E. van Wieren1Ronald C. Ydenberg7 Herbert H. T. Prins1
Herbert H. T. Prins1Many ungulate populations have a complex history of isolation and translocation. Consequently, ungulate populations may have experienced substantial reductions in the level of overall gene flow, yet simultaneously have augmented levels of long-distance gene flow. To investigate the effect of this dual anthropogenic effect on the genetic landscape of ungulates, we genotyped 35K SNPs in 47 red deer (Cervus elaphus) of Netherlands, including putative autochthonous relic populations as well as allochthonous populations established in private estates and rewilding areas. We applied FST and ordination analyses to determine the meta-population genetic structure and thereby the occurrence of hybridization. At population level, we investigated levels of inbreeding through individual-based diversity measures, including Runs of Homozygosity. We documented that both spatial genetic structure and within-population genetic variation differed markedly from patterns assumed from present-day abundance and distribution. Notwithstanding the small spatial scale, red deer populations formed distinct genetic clusters, and some had higher genetic similarity to distant than to nearby populations. Moreover, the putative autochthonous relic deer populations had much reduced levels of polymorphism and multi-locus heterozygosity, despite relatively large current population sizes. Accordingly, genomes of these deer contained a high proportion of long (>5 Mb) Runs of Homozygosity. Whereas the observed high levels of inbreeding warrant defragmentation measures, the presence of adjacent autochthonous and allochthonous genetic stocks imply that facilitation of gene flow would cause genetic homogenization. Such distortions of the genetic landscape of ungulates creates management dilemmas that cannot be properly anticipated without baseline genetic monitoring.
In the Anthropocene, ungulates are subject to both population isolation and translocation. Because of eradication and overexploitation, many ungulates occur in small or bottlenecked autochthonous population relics that are further isolated by anthropogenic barriers (Linnell and Zachos, 2011; Deinet et al., 2013; Ripple et al., 2015). Being such iconic elements of the ‘the world around us,’ ungulates have a long history of translocation (Iacolina et al., 2019). Translocations may come in the form of supplementation of existing populations or (re)introduction of new populations, and frequently involve immigrants from non-native, distant stocks (Seddon et al., 2012; Iacolina et al., 2018, 2019). The dual anthropogenic influences of isolation and translocation have contrasting effects on the key evolutionary process of gene flow, as it is both impeded as well as augmented. This means that the genetic landscape (e.g., Söderquist et al., 2017 and Wang et al., 2019) of ungulate populations is altered by inbreeding (i.e., mating among closely related individuals) as well as outbreeding (i.e., mating among distantly related individuals). Consequently, ungulate populations are at risk of inbreeding depression (i.e., lowered fitness of inbred individuals) on the one hand (Ralls et al., 2018) and outbreeding depression (i.e., lowered fitness of hybrids) on the other (Frankham et al., 2011). Furthermore, outbreeding results in loss of genetic integrity and in genetic homogenization, i.e., an increase of genetic similarity of populations (Kolodny et al., 2019).
The occurrence of inbreeding depression in isolated wildlife populations is now well established (Hasselgren and Norén, 2019). This outcome is especially true for ungulates, for which genomic approaches developed for domestic counterparts could be adopted. The genomes of over 20 ungulates have now been assembled (Martchenko et al., 2018). The genomic consequences of population isolation have been investigated in a variety of species, showing that many populations have individuals with genomes containing frequent and long Runs of Homozygosity (e.g., wild boar (Sus scrofa), ibex (Capra ibex), bighorn sheep (Ovis canadensis), Soay sheep (Ovis aries), and red deer (Cervus elaphus); Bérénos et al., 2016; Grossen et al., 2018, 2019; Hasselgren and Norén, 2019). A negative relationship between inbreeding and individual survival and reproductive performance was reported in red deer of the Isle of Rum, Scotland (Huisman et al., 2016). Moreover, in a recent study involving 26 European ibex populations, growth rate appeared to be substantially lower in inbred than in non-inbred populations (Bozzuto et al., 2019).
The general consensus is that in contemporary ungulate populations outbreeding, and the consequences thereof, is less adverse and pressing than inbreeding (Edmands, 2007; Pekkala et al., 2014, Bell et al., 2019). Recently, however, the debate began on whether the consequences of outbreeding are underestimated (Kolodny et al., 2019; Bell et al., 2019). There is equivocal evidence that hybrids may have lowered relative fitness (Marshall and Spalton, 2000; Bell et al., 2019). Such outbreeding depression may arise because of genetic incompatibility and reduced local adaptation. Apart from this, genetic homogenization may reduce the genetic diversity at a species-wide scale (Kolodny et al., 2019), in particular when migrants swamp the genetic variation of the native stock (Bell et al., 2019; Harris et al., 2019). Species exhibit geographic variation in phenotypic traits. Ungulates, in particular, have substantial regional variation in morphology (body size, size of horns and antlers, coat pattern and coloration) and behavior (timing of mating season, vocalization) (e.g., Putman and Flueck, 2011; Castelló, 2016). Underlying this geographic variation in phenotypes may be a complex and often little understood spatial genetic structuring (e.g., Moodley and Bruford, 2007). Translocation and subsequent hybridization of ungulates therefore may have both genetic and conspicuous phenotypic effects.
In practice, ungulate management policies often account poorly for both inbreeding and outbreeding. “Genetic legislation” or guidelines on inbreeding and outbreeding are absent or non-normative (Hoban et al., 2013; Ralls et al., 2018). The main genetic guideline, the 100/1000 Ne rule (or 50/500, Frankham et al., 2014), has gained a foothold, but in practice actual effective population sizes are seldom estimated. Indeed, genetic diversity measures are not incorporated in IUCN assessments (Vitorino et al., 2019). Similarly, the IUCN guidelines on translocation (“Guidelines for Reintroductions and Other Conservation Translocations”) do not contain normative or quantitative targets (IUCN/SSC, 2013). Although recommended (e.g., IUCN/SSC, 2013), genetic monitoring, whether pre- or post-intervention, is rarely applied (Hoban et al., 2013). Considering the long history of diverse anthropogenic impacts, the demographic history of contemporary ungulate populations is often complex. Present-day abundance and distribution may therefore give little indication of genetic status.
The simultaneous risk of inbreeding and outbreeding in contemporary ungulate populations is exemplified by the red deer of Netherlands. After a complex and partially known history of eradication, overexploitation, fragmentation and translocation, red deer of Netherlands now occur in remnant populations with different genetic ancestry. By 1900, the red deer population was likely reduced to perhaps a few tens of individuals, which occurred in the pastoral lands of the Veluwe moraine forest and heaths. To improve hunting opportunities, Dutch royalty and local landlords translocated red deer from across the European continent to estates (Rijk and Pelzers, 1991; van den Hoorn, 1992). Fences around the estates, and later busy provincial and national roads, probably hindered gene flow between the introduced allochthonous estate population and the presumably relic, autochthonous population. Within the framework of the “rewilding movement”, red deer were introduced in a disjunct area of The Oostvaardersplassen around 1990. This area was in a newly reclaimed polder about 30 km removed from the Veluwe area. These deer were translocated from various stocks, predominantly Scotland and Czechia. Across the species range, deer including red deer show considerable phenotypic variation in morphology (body size, antler size and shape) and behavior (e.g., male rutting roars) (Mystkowska, 1966; Geist, 1998, pp. 170-222; Putman and Flueck, 2011; Volodin et al., 2018). This phenotypic variation is partially heritable (Kruuk et al., 2002; Coulson et al., 2003; Flueck and Smith-Flueck, 2011). In Netherlands, hunters maintain that up until today the allochthonous and autochthonous red deer populations differ in timing of mating activity and morphology (in particular, body and antler size; Figure 1). These differences may, however, also be caused by local variation in population density as well as in resource quality the latter because of contrasting soils and management regimes (including forage improvement and supplementary feeding). For example, the Oostvaardersplassen area has very fertile marine clay soils while the Veluwe area is on nutrient-poor leached Pleistocene cover sands.

Figure 1. Difference in the shape of red deer antlers of relic and estate populations of Netherlands. Above: Antlers characteristic for deer of the allochthonous estate Hoge Veluwe (outside) and the autochthonous relic population. The antlers are from deer that are approximately seven years old. The antlers differ, inter alia, in the shape of the crown, which is a fork in the relic population, but tends to form palms in the estate. Below: Antlers collected in the estate Hoge Veluwe in 1985 (left) and 2013 (estimated age of the deer: both eight years). The characteristic shape of tines and palms is retained over generations.
To what extent gene flow may have homogenized the populations with distinct genetic ancestry, or whether anthropogenic barriers still prevent this homogenization is unknown. In addition, the level of inbreeding in the various populations is poorly understood. The various red deer populations underwent bottlenecks or founder effects, and today vary from a few hundred to thousands of animals. All populations accord with Dutch nature legislation based on European directives, which prescribes that to guarantee a ‘favorable conservation status’, ungulate populations should have a minimum size of 150 (assuming a minimum effective population size of 50, and an effective to census population size ratio of 0.33; Groot Bruinderink et al., 2000). The first microsatellite study was suggestive of the existence of separate genetic clusters, but did not detect genetic erosion (Groot et al., 2016). Concerns about inbreeding have triggered defragmentation measures in the past few decades, including the removal of fences and the construction of highway wildlife overpasses. The need for such measures, as well as the potential hybridization consequences, are poorly understood. In Dutch ungulate management, the allochthonous ancestry of some red deer populations is not part of the decision-making process (Spek, 2014).
Here, we report on a case study of the alteration of the genetic landscape of ungulates caused by population isolation and translocation, through analyses of 35K SNPs of red deer populations of Netherlands. Specifically, at meta-population level, we applied genetic ordination to study the occurrence of hybridization and assess its potential. At the population level, we investigated levels of inbreeding through Runs of Homozygosity. We expected a distortion of the null model of Isolation by Distance, as evidenced by (i) distinct genetic clusters at small spatial scales, and (ii) genetic similarity between distant, rather than nearby, populations. Furthermore, we expected (iii) relic autochthonous populations to be inbred, because of their bottlenecked history. In contrast, we expected allochthonous populations, which were established by founders of diverse genetic ancestry, to be outbred. Nontheless, if fences around estates cause isolation, then autochthonous estate populations also would be expected to have relatively low genetic variation. We test the overall hypothesis that genetic status of red deer can be derived from contemporary abundance and distribution patterns in a human-dominated landscape.
We assembled a SNP dataset from red deer populations from Netherlands with Czechia and Scotland as references (Figure 2A and Supplementary Table 1). Within Netherlands, we sampled red deer of the two main populations: the Veluwe and the Oostvaardersplassen (Figure 2C). In the Veluwe, we sampled two allochthonous populations from (formerly) fenced estates (NP Hoge Veluwe, Kroondomeinen), and two relic autochthonous populations, in the southwest (Planken Wambuis) and southeast (Veluwezoom and Deelerwoud, referred to as Veluwezoom). In addition, we sampled deer from the rewilding population at the Oostvaardersplassen. To avoid potential sampling bias, we excluded 1st and 2nd degree relatives (following Anderson et al., 2010, see subsequent calculation). This resulted in the following sample sizes: 15 deer from the Oostvaardersplassen, 13 from the Veluwe estate populations and 19 from the relic Veluwe populations. In additional 10, 36 and 100 samples were used for the reference populations of Czechia, mainland Scotland and Isle of Rum, respectively (Supplementary Table 1). To decrease the bias due to unequal sample size, a maximum of 25 samples from the Scottish populations were randomly selected for some of the analyses (PCoA, rarefaction, FST).
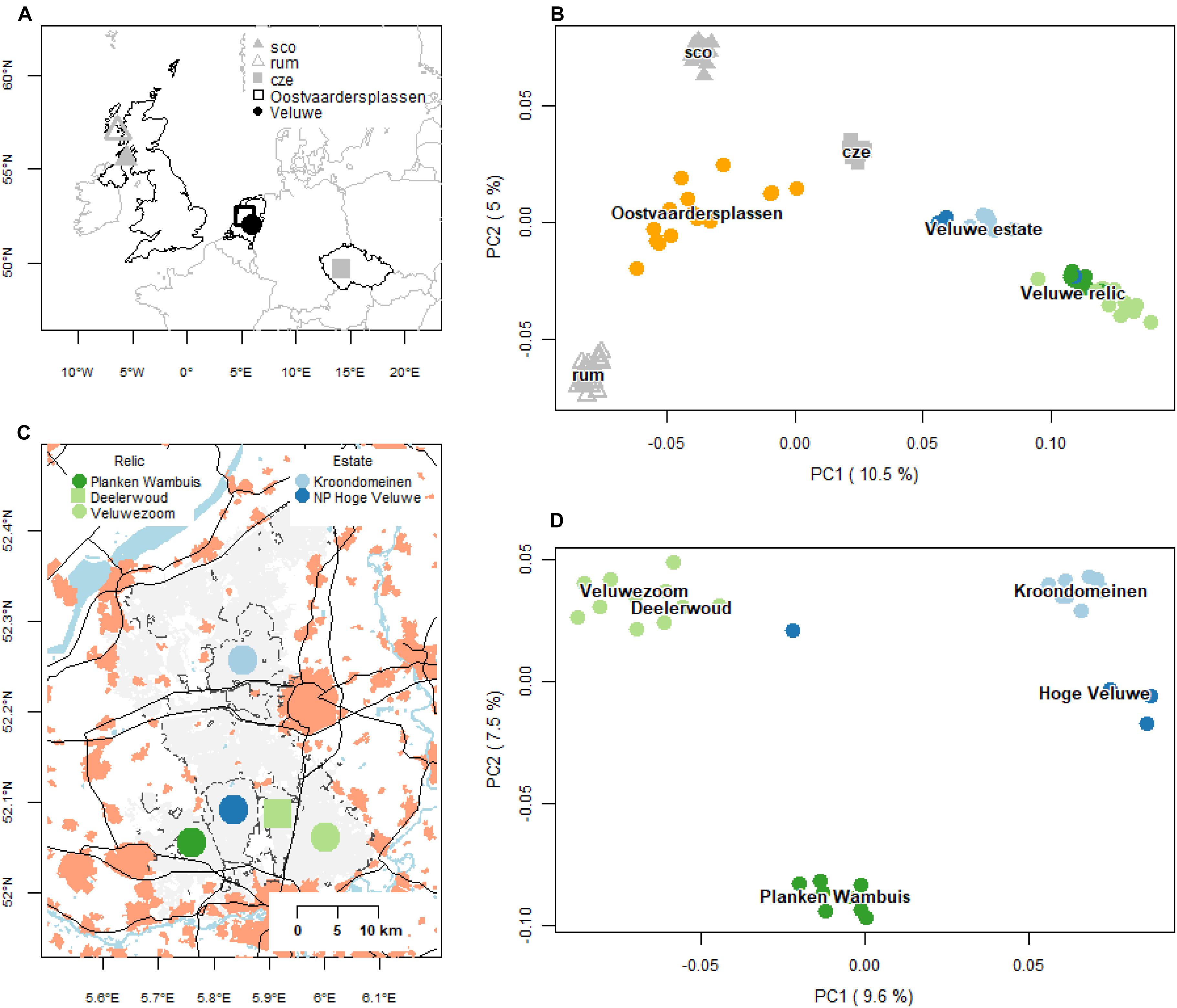
Figure 2. Geography and genetic structure of red deer samples. (A) Geographic distribution of the red deer populations of Netherlands, and reference populations of Czechia and Scotland (mainland and Isle of Rum). Oostvaardersplassen is an allochthonous rewilding population, established in the early 1990s. Kroondomeinen and Hoge Veluwe are allochthonous Veluwe estate populations, established the early 1900s. Planken Wambuis (Southwest Veluwe) and Veluwezoom/Deelerwoud (Southeast Veluwe) are relic autochthonous populations. Abbreviations of the reference populations are: sco, mainland Scotland; rum, Isle of Rum; cze, Czechia. (B) First two axis of a PCoA of pairwise Hamming genetic distances of all populations (based on a maximum of 25 deer per population). Labels are positioned at the median ordination axis scores. (C) Geographic distribution of subpopulations of the focal populations in the Veluwe. Based on demographic history, red deer populations of the Veluwe can be distinguished as either relic populations, i.e., continuously present (color: green) and estate populations, which were founded at the beginning of the 20th century and are fenced (color: blue). Filled light gray areas are forests and heaths, light red areas cities and towns, black lines roads and black dashed lines fences. (D) PCoA biplot of the focal Veluwe populations. Labels are positioned at the median ordination values.
Collection and SNP genotyping of the Scottish deer is described in Senn and Pemberton (2009) and Huisman et al. (2016). Samples of red deer of Netherlands and Czechia were SNP genotyped specifically for this study. These samples, mostly tongue and ear tissue, were obtained shortly after death from animals that were culled for population management purposes, died from traffic collisions or because of natural mortality (the latter in the Oostvaardersplassen). Samples were genotyped with the cervine 50 K Illumina Infinium iSelect HD Custom BeadChip (Brauning et al., 2015; Rowe et al., 2015). Chromosome and chromosome positions of SNP loci were based on the linkage map by Johnston et al. (2017). After quality control, which included filtering on a minimum individual and SNP call rate of 0.98 and a minor allele frequency of > 0.01, 35,522 SNPs remained, of which 33,688 were autosomal. These remaining SNPs had a median density of one SNP per 53Kbp. For all analyses except ROH detection, we excluded SNPs that were in Linkage Disequilibrium (LD; r2 > 0.2) (Anderson et al., 2010). After LD pruning, the number of remaining autosomal SNPs was 27,396 (median spacing of 55Kbp).
Data management and standard genetic analyses were conducted with a combination of the software PLINK1.9 (Purcell et al., 2007) and R (R Core Team, 2019), specifically the package Adegenet (Jombart and Ahmed, 2011). To ensure independence of markers, we pruned SNPs with PLINK using a LD threshold of r2 = 0.2. We used the PLINK pairwise IBD estimator to calculate pi_hat, which is the proportion of IBD between pairs of individuals (Purcell et al., 2007). We did this for various genetic clusters separately because the method assumes that samples do not show population stratification. We pruned individuals such that the maximum estimated pi_hat was 0.1.
To study genetic structure, we estimated genetic dissimilarity at the individual and population level. As individual measure we used the Hamming genetic distance estimator, which we calculated with the R package poppr (Kamvar et al., 2015). The Hamming genetic distance measure is simply the inverse of the proportion of alleles that are shared (i.e., that are Identical-by-State). For dissimilarity at the population level, we calculated the fixation index FST (Weir and Cockerham, 1984) with the R package StAMPP (Pembleton et al., 2013). The FST estimator used by this program is accurate even when sample size is small (minimally 5 individuals), provided that the number of markers is large (∼10,000 loci or more) and differentiation is weak (FST < 0.10) (Willing et al., 2012). Significance of FST-values was tested by bootstrapping loci 1,000 times (Pembleton et al., 2013). To study the main genetic partitioning among samples, we applied multidimensional reduction of the pairwise genetic distance matrices by use of PCoA (Principal Coordinate Analysis) as implemented in the R package Ape (Paradis et al., 2004). Furthermore, we evaluated the most likely number of clusters and estimated individual ancestry coefficients using the least-square method incorporated in the R package LEA (function ‘snmf’; Frichot and Francois, 2015). We used the aforementioned PCoA and LEA ancestry analysis for direct gene flow estimationthrough the proportion of misassignments (Paetkau et al., 2004). The medium-density SNP dataset gave resolution to detect first generation hybrids. Hence, we calculated migration rates for the current and previous generation (i.e., parents). We calculated 95% confidence intervals around migration rates using the binomial distribution (R base package function ‘binom.test’).
To study inbreeding, we calculated Multilocus Heterozygosity. MLH estimates based on 10,000 or more SNPs correlates near to perfect with whole genome MLH estimations (r2 ≈ 0.99 from a ROH study on wolves (Canis lupus), by Kardos et al., 2018). In addition to MLH, we detected Runs of Homozygosity (ROHs). The fraction of the genome that contains ROHs (FROH) has been shown to be the best estimator for inbreeding (Keller et al., 2011; Kardos et al., 2015), especially when sample size is small (Gazal et al., 2014). Moreover, in contrast to other inbreeding estimators, this metric is comparable among different populations and species, provided that similar segment sizes and variation because of recombination rate are considered. Kardos et al. (2018) reported a high correlation between FROH estimates based on whole-genome sequences and estimates based on 10,000 SNP markers. Nevertheless, SNP estimations of FROH tend to be biased upwards. Specifically, for whole genome sequence FROH values of 0.0625, SNP based FROH estimates ranged from 0.05 to 0.10; for whole genome sequences FROH values of 0.125, SNP based FROH estimates ranged from 0.10 to 0.20. Following recommendations of Howrigan et al. (2011) we defined the number and length of ROHs with PLINK1.9 as a tract of at least 50 completely homozygous, moderately pruned SNPs (i.e., LD r2> 0.5, heterozygote loci not allowed); 50 such SNPs is equivalent to approximately 5 Mb. We restricted ourselves to quantification of ROHs longer than 5Mb, so as to minimize the chance of false positives (Howrigan et al., 2011; Kardos et al., 2018). Following McQuillan et al. (2008) and Keller et al. (2011) we distinguished ROHs of > 20 Mb, 10–20 Mb, and 5–10 Mb. Given that the length of ROHs is related to the number of generations since the shared ancestor as 1/2 g M, with g and M representing generations and Morgan, respectively (Howrigan et al., 2011), and assuming that 1 Morgan ≈ 1 Mb, these classes roughly correspond with a shared ancestor < 2, 2–5, and 5–10 generations ago (Ceballos et al., 2018).
PCoA and ancestry analyses showed that the Dutch red deer populations formed discrete genetic clusters, with little to no gene flow (Figures 2B,D,3). Deer of the same source populations had distinct ordination scores and ancestry coefficients. For all population comparisons, FST-values were significantly different from zero, and ranged from 0.04 to 0.15, with the highest values observed in the Veluwe estate populations (P = 0.001; Supplementary Table 2).
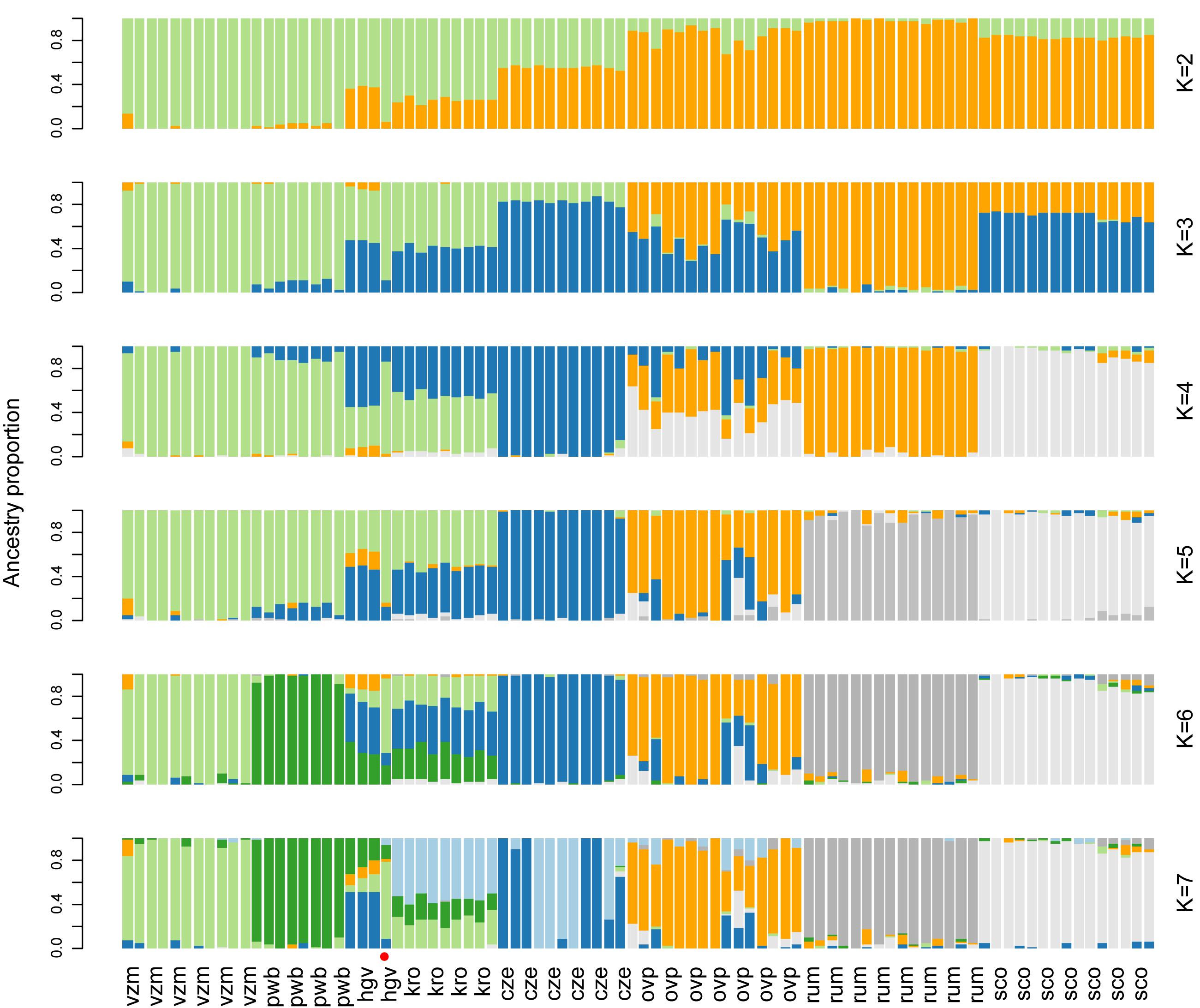
Figure 3. Ancestry analysis of red deer focal and reference populations, at K = 2 to K = 7. The most likely number of K clusters lies within the range 2 to 4 (Supplementary Figure 6). Each vertical bar represents one individual. The red dot indicates the putative migrant. Population abbreviations and sample sizes are as follows: Of the autochthonous relic populations: vzm, Veluwezoom (11); pwb, Planken Wambuis (8); of the allochthonous estate populations: hgv, Hoge Veluwe (4) and kro, Kroondomeinen (9), of the allochthonous rewilding population: ovp, Oostvaardersplassen (15); and the reference populations: cze, Czechia (11), rum, Isle of Rum (15), sco, mainland of Scotland (15). For populations with more than 15 samples, 15 individuals were randomly selected.
Nevertheless, an apparent scattered (rather than a clumped) ordination for deer of the rewilding Oostvaardersplassen population as well as the relic Veluwezoom population occurred (Figure 2B). Oostvaardersplassen deer had relatively high within-population genetic distances, as well as strong variation in genetic distances to other populations (Supplementary Figure 3A) – the latter causing the scatter (Supplementary Figure 3A). In the relic Veluwezoom population, in contrast, deer were genetically similar to each other (low within-population genetic distances), and strongly dissimilar to deer of other populations (high among-population genetic distances) (Supplementary Figure 3B). Here, the scatter was caused by variation in both the within-population genetic distances (low vs. very low) as well as in the among-population genetic distances (high vs. very high) – as illustrated in Supplementary Figure 3D. Furthermore, whereas genomes of deer of Oostvaardersplassen were characterized by a high proportion of heterozygote genotypes, genomes of deer of Veluwezoom had high proportion of genotypes that were homozygous for the major or minor allele (Supplementary Figure 3E). Hence, the scattered ordination of the Oostvaardersplassen and Veluwezoom population were caused by contrasting genetic properties.
Despite the existence of genetic clusters within the Veluwe area, there was a single animal with a dispersal history among the neighboring deer populations of the Veluwe: this animal was sampled within the estate Hoge Veluwe, but had a genetic signature in between its source population and the relic population Veluwezoom (based on the first two axes of PCoA of Veluwe deer, Figure 2D, and the ancestry analysis, Figure 3). Most probably, this individual was the first generation offspring of a disperser. None of the other 34 Veluwe samples indicated dispersal events. Considering zero dispersal events of 35 sampled deer, and one dispersal events of 70 parents, the estimated migration rate was 0.009 (95% CI: 0.000 – 0.052, Binomial exact calculation).
PCoA and ancestry analysis showed that the genetic clusters of Dutch red deer had diverse ancestry. First, the allochthonous Oostvaardersplassen population had higher genetic similarity to Scottish and Czechian reference populations than to other Dutch populations (Figures 2B, 3). The Oostvaardersplassen population additionally showed a high degree of within-population heterogeneity (high within-population Hamming genetic distance, Supplementary Figures 2, 3; and scattered ordination, Figure 2B). Indeed, some individuals were most similar to deer from other populations (Supplementary Figure 4). Second, the autochthonous and allochthonous deer of the Veluwe had a partially shared genetic ancestry (Figure 3; green cluster). A signal of allochthonous ancestry also occurred—the allochthonous estate populations were genetically more similar to each other than to surrounding autochthonous relic populations (PCoA; Figures 2B,D). The allochthonous estate populations had high ancestry scores for the genetic cluster allocated to the Czechia reference population (Figure 3; blue cluster). This genetic signature was small or absent in the autochthonous relic populations Planken Wambuis and Veluwezoom, respectively.
We observed strong variation in the Multilocus heterozygosity (MLH) of red deer of Netherlands. The most heterozygous individual (maximum MLH = 0.40, from the allochthonous rewilding population) had a 54% higher MLH than the least heterozygous individual (minimum MLH = 0.26, from the autochthonous relic population). Median multilocus heterozygosity differed significantly among the Dutch red deer populations (Figure 4A; Kruskal Wallis χ2 = 100.4, d.f. = 7, P < 0.001). Deer from the autochthonous relic populations had a lower median MLH than deer from both the allochthonous rewilding and estate populations, and also lower than deer from European reference populations (Wilcoxon rank pairwise comparison test, P < 0.001, Supplementary Table 3). Among the allochthonous populations, deer from the estate populations had lower median MLH than the rewilding population, and also lower than the two Scottish populations. Furthermore, the autochthonous relic populations, and to a lesser degree also the allochthonous estate populations, had a high proportion of non-segregating SNPs (Figure 3B).
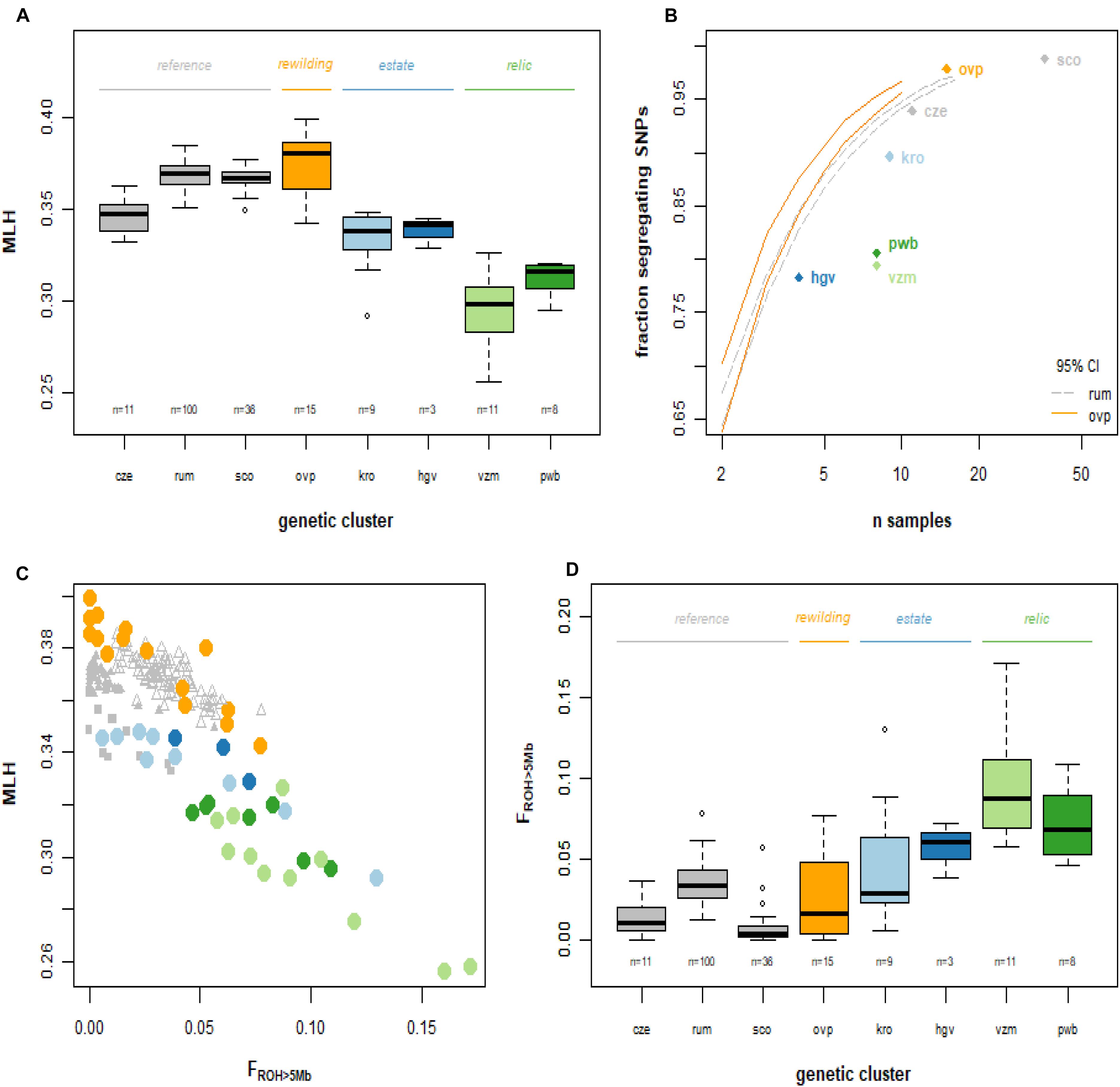
Figure 4. Genetic diversity of the red deer study populations. Abbreviations as in Figures 1, 2. (A) Boxplot of observed multilocus heterozygosity. (B) Fraction of monomorphic SNPs, at population level, in relation to sample size. The relic populations of the Veluwe have relatively many monomorphic SNPs. Lines represent the bootstrapped 95% confidence intervals for Rum (dashed gray line) and the allochthonous Oostvaardersplassen (unbroken, orange line) deer populations (i.e., rarefaction curves). (C) Individual FROH vs. multilocus observed heterozygosity. FROH is the genomic fraction of all ROHs longer than 5 Mb. (D) Fraction of the genome containing ROH segments > 5 Mb, per individual, grouped over genetic clusters. ROH segments with a length of 5 Mb stem from a common ancestor of maximum 10 generations ago.
Multilocus heterozygosity was strongly and positively correlated with the fraction of the genome containing ROH segments larger than 5Mb (FROH>5Mb; Figure 3C; Spearman rank correlation, rs = -0.54, d.f. = 256, P < 0.001). Among the red deer populations of Netherlands, there were significant differences in FROH> 5Mb (Kruskal Wallis χ2 = 106.1, d.f. = 7, P < 0.001; Figure 3D). Notwithstanding substantial individual variation (with maximum FROH>5Mb of 0.08), the rewilding population had the lowest median FROH > 5Mb values. Conversely, red deer of the autochthonous relic populations had a significantly higher median FROH > 5Mb than the allochthonous rewilding population as well as European reference populations (Wilcoxon rank pairwise comparison test, P < 0.05; Supplementary Table 4 and Supplementary Figure 5). Red deer of these two relic populations had a minimum FROH > 5Mb of 0.05, and a median FROH > 5Mb of 0.07 (Planken Wambuis) and 0.09 (Veluwezoom). Most of the detected FROH segments had a length of 5–10 Mb, corresponding to a shared ancestor 5–10 generations back in time (Supplementary Figure 5). Despite having few individuals with high FROH > 5Mb, the median FROH > 5Mb of the allochthonous rewilding and estate deer populations did not differ from reference populations.
Using genome-wide SNP analysis we showed that because of historic and contemporary human impacts, red deer populations of Netherlands differ greatly, and sometimes unexpectedly, in genetic composition. Population isolation (because of anthropogenic barriers) and translocation resulted in adjacent red deer populations having little gene flow and, consequently, discrete genetic clusters with diverse ancestry. Concurrently, red deer populations were shown to vary widely in genetic diversity. In particular, autochthonous relic populations were substantially inbred, despite their relatively large contemporary population size. Altogether, the genetic landscape of red deer of Netherlands can be characterized as a complex mosaic of patches with distinct, uncorrelated properties.
The findings illustrate the substantial distortion that humans can cause to the natural spatial pattern of genetic variation of red deer specifically and ungulates in general. Earlier microsatellite studies indicated that historic and contemporary anthropogenic interventions affect the genetic diversity of red deer populations in, inter alia, Scotland, Iberia, Belgium and Germany (Nussey et al., 2006; Pérez-Espona et al., 2013; Queiros et al., 2014; Hoffmann et al., 2016; Frantz et al., 2017). Recently, there has been a growing attention on the effects of translocation for genetic variation in ungulates (e.g., Iacolina et al., 2018; Gille et al., 2019; Jahner et al., 2019). Our SNP study on the main extant red deer populations of the human-dominated and fine-grained landscape of Netherlands, improves the resolution of our understanding of the human-caused alteration of the genetic landscape. We demonstrate that the genetic differences among deer populations come in the form of sharp discontinuities, and at a spatial scale much smaller than typical male red deer dispersal distances (around 10 km and up to 50 km, Pérez-Espona et al., 2008). This is in contrast to a recent study on red deer and wild boar in the human-dominated landscape of Belgium (Frantz et al., 2017). The study of Frantz et al. (2017) was based on many (thousands) individuals, but few (tens) of markers – the opposite of the study design of our research. Next to showing fine-scale genetic structure, we are the first to apply Runs of Homozygosity as a means to assess genetic effects of human interventions on red deer, revealing hitherto unnoticed substantial inbreeding. Given that in a preceding microsatellite study (Groot et al., 2016) other red deer populations of Northwestern Europe (not included in this research) had much lower levels of genetic diversity than Dutch populations, we presume that SNP genotyping of other red deer populations will reveal similar or even higher levels of inbreeding.
The observed substantial distortion of the genetic landscape of the Dutch red deer populations highlights the limitations of landscape genetics null models of panmixia (gene flow is ubiquitous), Isolation by Distance (gene flow decreases over geographic distance), and even Isolation by Resistance (gene flow is impeded by barriers) (Manel et al., 2003) for managed ungulate populations. None of these models captures the gene flow patterns of populations that are fragmented, because of human interventions, but also supplemented through translocation, practices common for the management of many ungulates (Seddon et al., 2012), prompting the need for the development of specific approaches.
The relevance of historical human interventions was highlighted by our finding that alterations of the genetic landscape may be long-lasting. In the Veluwe the height of the anthropogenic impact is therefore long past. Bottleneck and translocation events date back to beginning of the 20th century, which, assuming a generation time of 7 years (Coulson et al., 1998) is 14 deer generations in the past. Before this study we therefore assumed that gene flow between the putative Veluwe autochthonous and allochthonous Veluwe populations would have resulted in homogenization. This would predict that presently there should be little genetic structure, and that levels of genetic variation would be similar between subpopulations by now. The detected migrant offspring provided evidence that gene flow is not absent., The allochthonous estate populations, however, were shown to still have low genetic similarity to adjacent autochthonous populations. The partial shared ancestry indicates that there has been some admixture between the estate and relic populations (possibly because of few occasional breakouts, for example during World War II, Rijk and Pelzers (1991)), but that at large fences around estates have prevented gene flow and admixture, and hence have maintained the segregation of allochthonous estate and autochthonous relic populations. The significance of this finding is that anthropogenic barriers do not merely cause genetic differentiation – they also may preserve the signatures of historical anthropogenic interventions. Hence, in landscapes with anthropogenic barriers, historic anthropogenic effects on ungulates may be maintained much longer than anticipated.
The implication of our study is that present-day abundance and distribution is a poor predictor of the genetic status of ungulates. The autochthonous relic Veluwezoom population is a good case in point. This population is currently the largest of all the Veluwe subpopulations sampled (spring census since 2000: more than 700 animals; Spek, 2014). Moreover, because of highway overpasses this area should be well connected to the other Veluwe subpopulations. Nevertheless, all diversity indicators show this population to be the most isolated and most inbred. We thus posit that the poor genetic status is a legacy of the historic bottleneck combined with an effective absence of genetic exchange. When we shared our findings about inbreeding with them, managers of the Veluwezoom red deer population were surprised. After all, the main bottleneck is thought to have occurred approximately one century ago, though is poorly documented (Rijk and Pelzers, 1991). Even recent demographic events may quickly be forgotten, as we realized when we contacted a retired employee to discover the origin of the founders of the Oostvaardersplassen red deer population (established early 1990s). Furthermore, the management of the autochthonous Veluwe populations was attempting to follow best conservation genetic practices. The management was not aware that conservation geneticists have recently altered the recommendation of minimum inbreeding effective population size to 100 (Frankham et al., 2014). Second, the management was advised an optimistic Ne/Nc-ratio of 0.33 - a ratio universally adopted by ungulate managers in Netherlands (Groot Bruinderink et al., 2000; Spek, 2014), but one that does not account for historic bottlenecks (Vucetich et al., 1997).
A complicating factor is that genetic consequences of historic anthropogenic impacts may be strong yet difficult to detect. In the autochthonous relic populations Veluwezoom and Planken Wambuis the median observed values of FROH > 5 Mb were larger than 0.0625. Using medium-density SNP data, we may have slightly overestimated FROH > 5 Mb (see section “MATERIALS AND METHODS”). The FROH values are above levels expected for offspring of third order relatives, and among the highest reported for a non-insular ungulate population (Hasselgren and Norén, 2019). Such a level of inbreeding was associated with lowered survival and reproductive performance (lowered calf survival on the Isle of Rum, reduced ibex population growth; Walling et al., 2011; Huisman et al., 2016; Bozzuto et al., 2019). In red deer, inbreeding also may be expressed through the occurrence of overbites (shortened lower jaw, or brachygnathy; Zachos et al., 2007). Typically however, inbreeding depression effects (if any) are subtle, and conditional on stressful environments (Keller and Waller, 2002; Pemberton et al., 2016). Phenotypically, deer of the relic inbred Veluwezoom population do not show signs of inbreeding depression. The population has a high growth rate, deer appear to be in good condition, and health issues are not reported (Spek, 2014). Furthermore, even the preceding microsatellite study, involving nine molecular markers, had not detected inbreeding (Groot et al., 2016).
Our study also pinpointed the dilemma between inbreeding and outbreeding. In the introduced deer of the rewilding area Oostvaardersplassen, we detected inbreeding as well as admixture within the same population, and even in genomes of the same individual. In addition, the finding that autochthonous and allochthonous populations are still differentiated and have different genetic ancestry, shows that in areas with a population translocation history, the option of alleviating inbreeding through facilitation of gene flow would lead to admixture. We agree with the current consensus among conservation geneticists that inbreeding should be treated with more concern than outbreeding (Bell et al., 2019). Nontheless, for ungulates the natural spatial genetic structure is substantially affected by fragmentation and translocation, and may, peculiarly, be further altered by defragmentation measures. In the Veluwe, where interpopulation phenotypic differences are allegedly persistent (Figure 1), it is not unlikely that such admixture will affect antler size and shape of autochthonous populations, and thus have more conspicuous phenotypic effects than inbreeding (though not negative). A potential scenario is swamping of allochthonous genetic variation into the gene pool of autochthonous deer (Bell et al., 2019). Irrespective of whether such phenotypic variation is adaptive in many ungulates, managers may influence the genetic integrity and phenotypic traits such as the shape of horns and antlers and coat coloration. The recent rewilding movement has embraced the concept of ecological substitutes, thereby implicitly ignoring intra- and interspecific phenotypic variation (Lorimer et al., 2015). Yet, many translocations of ungulates have been and are being dictated by aesthetic considerations (Seddon et al., 2012), thereby deliberately modifying local phenotypic variation. The consequence is potential disruption of local adaptation, and homogenization of geographic variation (Gippoliti et al., 2018; Kolodny et al., 2019).
The multitude and diversity of anthropogenic impacts and the consequential genetic complex of the genetic landscape pose a challenge for ungulate managers to effectively account for inbreeding and outbreeding consequences. We argue that for ungulate populations historically and presently subject to anthropogenic impact (i) genetic variation cannot be inferred from contemporary distribution and abundance, and (ii) assessment of genetic status is necessary to enable appropriate management. Given that genetic variation may bear the ‘marks’ of unknown or underestimated anthropogenic effects in the past, we recommend managers of ungulate populations to start with a baseline-assessment of standing genetic variation using modern genomic approaches. Ungulate managers may imagine they can slow down loss of genetic variation sufficiently in a population by ensuring high population census size (with little fluctuation, equal sex ratio, etc.) and by connecting subpopulations in the landscape. Historical impacts on population demography, however, may have had such adverse effects that these positive interventions do not suffice and the poor genetic status of a population is not alleviated (at least not on a time scale of decades). Conversely, managers may be unaware of the presence and extent of allochthonous genetic ancestry, and hence of the potential admixture and homogenization effects of their interventions. For genetic monitoring purposes, we recommend the use of genome-wide SNP data or, if affordable, whole genome sequences. The relatively high costs may, at the present, still constrain sample size and cause a trade-off between population coverage and individual width. Yet, as illustrated by our study, SNP data enable the determination of fine-scale genetic structure, the direct estimation of migration rates, and the precise assessment of genetic status. In particular, genomic approaches facilitate the use of accurate, individual-based measures (e.g., MLH, FROH), rather than indirect measures that are equilibrium-based and averaged over populations (e.g., population-level heterozygosity with a few loci, allelic richness). The high levels of inbreeding in the autochthonous red deer populations underline the argument that continuation to ignore genetic factors may severely hamper conservation efforts. We advocate that genetic monitoring has to be made integral to ungulate management and policy making.
In our study we illustrate that genetic variation of present-day ungulates is much affected by ongoing and historic human interventions, and put forward that this likely effects ecological interactions as well. Whereas fragmentation and the associated loss of genetic variation may reduce adaptive potential to future environmental change, translocation possibly causes distortion of ongoing local adaptation processes. A first step towards understanding the ecology of present-day ungulate populations of human-dominated landscapes is therefore a genetic investigation of the often unknown and complex demographic history.
The data are located on the Dryad repository: https://doi.org/10.5061/dryad.hqbzkh1dp.
Samples of deer of Isle of Rum were collected and used following approval of the Animal Welfare and Ethical Review Body of the University of Edinburgh, and under appropriate United Kingdom Home Office licenses (Johnston et al., 2017). For the other deer samples, an ethical review process was not required, because samples were collected from deer already culled as part of population management programs. The samples of mainland Scotland were collected by hunters of the Forestry Commission Scotland. Red deer samples of Netherlands were collected from animals culled as part of population control programs regulated by Faunabeheereenheid Gelderland and Faunabeheereenheid Flevoland. In Czechia, samples were collected from deer culled according to the law No. 449/2001 Sb. Hence, in Czechia, Netherlands, and Mainland Scotland no animal was specifically killed for this research.
JJ, PH, H-JM, LI, RY, SW, and HP contributed to the conception and design of the study. GG, LB, JH, and JP provided samples. RC coordinated the laboratory genetic analysis. JJ performed the statistical analysis, in consultation with PH, H-JM, JH, JP, RY, SW, and HP. JJ wrote the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
This research was funded by ‘Stichting De Eijk,’ the ‘Koninklijke Nederlandse Jagervereniging,’ and by ‘BIJ12’. Genotyping of the Rum and Mainland Scotland samples was funded by an ERC grant to JP.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The photos of red deer antlers of estates and relic populations is mediated by the park management of National Park Hoge Veluwe. Furthermore, we are thankful for insightful discussions with wildlife managers of the estate populations Kroondomeinen and National Park Hoge Veluwe, and the relic populations Veluwezoom and Planken Wambuis.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2020.535715/full#supplementary-material
Anderson, C. A., Pettersson, F. H., Clarke, G. M., Cardon, L. R., Morris, A. P., and Zondervan, K. T. (2010). Data quality control in genetic case-control association studies. Nat. Protoc. 5, 1564–1573. doi: 10.1038/nprot.2010.116
Bell, D. A., Robinson, Z. L., Funk, W. C., Fitzpatrick, S. W., Allendorf, F. W., Tallmon, D. A., et al. (2019). The exciting potential and remaining uncertainties of genetic rescue. Trends Ecol. Evol. 34, 1070–1079. doi: 10.1016/j.tree.2019.06.006
Bérénos, C., Ellis, P. A., Pilkington, J. G., and Pemberton, J. M. (2016). Genomic analysis reveals depression due to both individual and maternal inbreeding in a free-living mammal population. Mol. Ecol. 25, 3152–3168. doi: 10.1111/mec.13681
Bozzuto, C., Biebach, I., Muff, S., Ives, A. R., and Keller, L. F. (2019). Inbreeding reduces long-term growth of Alpine ibex populations. Nat. Ecol. Evol. 3, 1359–1364. doi: 10.1038/s41559-019-0968-1
Brauning, R., Fisher, P. J., McCulloch, A. F., Smithies, R. J., Ward, J. F., Bixley, M. J., et al. (2015). Utilization of high throughput genome sequencing technology for large scale single nucleotide polymorphism discovery in red deer and Canadian elk. bioRxiv 1, 1. doi: 10.1101/027318
Ceballos, F. C., Joshi, P. K., Clark, D. W., Ramsay, M., and Wilson, J. F. (2018). Runs of homozygosity: windows into population history and trait architecture. Nat. Rev. Genet. 19, 220–234. doi: 10.1038/nrg.2017.109
Coulson, T., Kruuk, L. E. B., Tavecchia, G., Pemberton, J. M., and Clutton-Brock, T. H. (2003). Selection on neonatal traits in red deer. Evolution 57, 2879–2892. doi: 10.1111/j.0014-3820.2003.tb01528.x
Coulson, T. N., Pemberton, J. M., Albon, S. D., Beaumont, M., Marshall, T. C., Slate, J., et al. (1998). Microsatellites reveal heterosis in red deer. Proc. Biol. Sci. 22, 489–495. doi: 10.1098/rspb.1998.0321
Deinet, S., Ieronymidou, C., Burfield, I. J., Foppen, R. P., Collen, B., and Böhm, M. (2013). Wildlife Comeback in Europe: The Recovery of Selected Mammal and Bird Species. London: ZSL.
Edmands, S. (2007). Between a rock and a hard place: evaluating the relative risks of inbreeding and outbreeding depression for conservation and management. Mol. Ecol. 16, 463–475. doi: 10.1111/j.1365-294x.2006.03148.x
Flueck, W. T., and Smith-Flueck, J. M. (2011). Intraspecific phenotypic variation in deer: the role of genetic and epigenetic processes. Anim. Prod. Sci. 51, 365–374. doi: 10.1071/an10169
Frankham, R., Ballou, J. D., Eldridge, M. D. B., Lacy, R. C., Ralls, K., Dudash, M. R., et al. (2011). Predicting the probability of outbreeding depression. Conserv. Biol. 25, 465–475. doi: 10.1111/j.1523-1739.2011.01662.x
Frankham, R., Bradshaw, C. J. A., and Brook, B. W. (2014). Genetics in conservation management: revised recommendations for the 50/500 rules, Red List criteria and population viability analyses. Biol. Conserv. 170, 56–63. doi: 10.1016/j.biocon.2013.12.036
Frantz, A. C., Zachos, F. E., Bertouille, S., Eloy, M. C., Colyn, M., and Flamand, M. C. (2017). Using genetic tools to estimate the prevalence of non-native red deer (Cervus elaphus) in a western European population. Ecol. Evol. 7, 7650–7660. doi: 10.1002/ece3.3282
Frichot, E., and Francois, O. (2015). LEA: an R package for landscape and ecological association studies. Methods Ecol. Evol. 6, 925–929. doi: 10.1111/2041-210x.12382
Gazal, S., Sahbatou, M., Perdry, H., Letort, S., Genin, E., and Leutenegger, A. L. (2014). Inbreeding coefficient estimation with dense SNP data: comparison of strategies and application to HapMap III. Hum. Hered. 77, 49–62.
Gille, D. A., Buchalski, M. R., Conrad, D., Rubin, E. S., Munig, A., Wakeling, B. F., et al. (2019). Genetic outcomes of translocation of bighorn sheep in Arizona. J. Wildl. Manage. 83, 838–854. doi: 10.1002/jwmg.21653
Gippoliti, S., Cotterill, F. P. D., Groves, C. P., and Zinner, D. (2018). Poor taxonomy and genetic rescue are possible co-agents of silent extinction and biogeographic homogenization among ungulate mammals. Biogeographia 33, 41–54.
Groot, G. A. D., Spek, G. J., Bovenschen, J., Laros, I., Meel, T. V., and Jansman, H. A. H. (2016). Herkomst en Migratie van Nederlandse Edelherten en Wilde Zwijnen: een Basiskaart van de Genetische Patronen in Nederland en Omgeving. Wageningen.
Groot Bruinderink, G. W. T. A., Lammertsma, D. R., and Pouwels, R. (2000). De geschiktheid van Natuurgebieden in Noord-Brabant en Limburg als Leefgebied voor Edelhert en Wild Zwijn. Wageningen: Research Instituut voor de Groene Ruimte.
Grossen, C., Biebach, I., Angelone-Alasaad, S., Keller, L. F., and Croll, D. (2018). Population genomics analyses of European ibex species show lower diversity and higher inbreeding in reintroduced populations. Evol. Appl. 11, 123–139. doi: 10.1111/eva.12490
Grossen, C., Guillaume, F., Keller, L. F., and Croll, D. (2019). Accumulation and purging of deleterious mutations through severe bottlenecks in ibex. bioRxiv [Preprint]. doi: 10.1101/605147
Harris, K., Zhang, Y., and Nielsen, R. (2019). Genetic rescue and the maintenance of native ancestry. Conserv. Genet. 20, 59–64. doi: 10.1007/s10592-018-1132-1
Hasselgren, M., and Norén, K. (2019). Inbreeding in natural mammal populations: historical perspectives and future challenges. Mamm. Rev. 49, 369–383. doi: 10.1111/mam.12169
Hoban, S., Hauffe, H., Perez-Espona, S., Arntzen, J. W., Bertorelle, G., Bryja, J., et al. (2013). Bringing genetic diversity to the forefront of conservation policy and management. Conserv. Genet. Resour. 5, 593–598. doi: 10.1007/s12686-013-9859-y
Hoffmann, G. S., Johannesen, J., and Griebeler, E. M. (2016). Population dynamics of a natural red deer population over 200 years detected via substantial changes of genetic variation. Ecol. Evol. 6, 3146–3153. doi: 10.1002/ece3.2063
Howrigan, D. P., Simonson, M. A., and Keller, M. C. (2011). Detecting autozygosity through runs of homozygosity: a comparison of three autozygosity detection algorithms. BMC Genomics 12:460. doi: 10.1186/1471-2164-12-460
Huisman, J., Kruuk, L. E. B., Ellis, P. A., Clutton-Brock, T., and Pemberton, J. M. (2016). Inbreeding depression across the lifespan in a wild mammal population. Proc. Natl. Acad. Sci. U.S.A. 113, 3585–3590. doi: 10.1073/pnas.1518046113
Iacolina, L., Corlatti, L., Buzan, E., Safner, T., and Šprem, N. (2019). Hybridization in European ungulates: a review of the current status, causes and consequences. Mamm. Rev. 49, 45–59. doi: 10.1111/mam.12140
Iacolina, L., Pertoldi, C., Amills, M., Kusza, S., Megens, H. J., Bâlteanu, V. A., et al. (2018). Hotspots of recent hybridization between pigs and wild boars in Europe. Sci. Rep. 8:17372.
IUCN/SSC (2013). Guidelines for Reintroductions and Other Conservation Translocations. Version 1.0. Gland: IUCN Species Survival Commission.
Jahner, J. P., Matocq, M. D., Malaney, J. L., Cox, M., Wolff, P., Gritts, M. A., et al. (2019). The genetic legacy of 50 years of desert bighorn sheep translocations. Evol. Appl. 12, 198–213. doi: 10.1111/eva.12708
Johnston, S. E., Huisman, J., Ellis, P. A., and Pemberton, J. M. (2017). A high density linkage map reveals sexual dimorphism in recombination landscapes in red deer (Cervus elaphus). G3 7, 2859–2870. doi: 10.1534/g3.117.044198
Jombart, T., and Ahmed, I. (2011). adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics 27, 3070–3071. doi: 10.1093/bioinformatics/btr521
Kamvar, Z. N., Brooks, J. C., and Grünwald, N. J. (2015). Novel R tools for analysis of genome-wide population genetic data with emphasis on clonality. Front. Genet. 6:208. doi: 10.3389/fgene.2015.00208
Kardos, M., Åkesson, M., Fountain, T., Flagstad, Ø., Liberg, O., Olason, P., et al. (2018). Genomic consequences of intensive inbreeding in an isolated wolf population. Nat. Ecol. Evol. 2, 124–131. doi: 10.1038/s41559-017-0375-4
Kardos, M., Luikart, G., and Allendorf, F. W. (2015). Measuring individual inbreeding in the age of genomics: marker-based measures are better than pedigrees. Heredity 115, 63–72. doi: 10.1038/hdy.2015.17
Keller, L. F., and Waller, D. M. (2002). Inbreeding effects in wild populations. Trends Ecol. Evol. 17, 230–241. doi: 10.1016/s0169-5347(02)02489-8
Keller, M. C., Visscher, P. M., and Goddard, M. E. (2011). Quantification of inbreeding due to distant ancestors and its detection using dense single nucleotide polymorphism data. Genetics 189, 237–249. doi: 10.1534/genetics.111.130922
Kolodny, O., McLaren, M. R., Greenbaum, G., Ramakrishnan, U., Feldman, M. W., Petrov, D., et al. (2019). Reconsidering the management paradigm of fragmented populations. bioRxiv [Preprint]. doi: 10.1101/649129
Kruuk, L. E. B., Slate, J., Pemberton, J. M., Brotherstone, S., Guinness, F., and Clutton-Brock, T. (2002). Antler size in red deer: heritability and selection but no evolution. Evolution 56, 1683–1695. doi: 10.1111/j.0014-3820.2002.tb01480.x
Linnell, J. D. C., and Zachos, F. E. (2011). “Status and distribution patterns of European ungulates: genetics, population history and conservation,” in Ungulate Management in Europe: Problems and Practices, eds R. Putman, M. Apollonio, and R. Andersen (Cambridge: Cambridge University Press), 12–53. doi: 10.1017/cbo9780511974137.003
Lorimer, J., Sandom, C., Jepson, P., Doughty, C. E., Barua, M., and Kirby, K. J. (2015). Rewilding: science, practice, and politics. Annu. Rev. Environ. Resour. 40, 39–62. doi: 10.1146/annurev-environ-102014-021406
Manel, S., Schwartz, M. K., Luikart, G., and Taberlet, P. (2003). Landscape genetics: combining landscape ecology and population genetics. Trends Ecol. Evol. 18, 189–197. doi: 10.1016/s0169-5347(03)00008-9
Marshall, T. C., and Spalton, J. A. (2000). Simultaneous inbreeding and outbreeding depression in reintroduced Arabian oryx. Anim. Conserv. 3, 241–248. doi: 10.1111/j.1469-1795.2000.tb00109.x
Martchenko, D., Prewer, E., Latch, E. K., Kyle, C. J., and Shafer, A. B. A. (2018). “Population genomics of ungulates,” Population Genomics, ed. O. P. Rajora (Cham: Springer).
McQuillan, R., Leutenegger, A.-L., Abdel-Rahman, R., Franklin, C. S., Pericic, M., Barac-Lauc, L., et al. (2008). Runs of homozygosity in European populations. Am. J. Hum. Genet. 83, 359–372.
Moodley, Y., and Bruford, M. W. (2007). Molecular biogeography: towards an integrated framework for conserving pan-African biodiversity. PLoS One 2:e454. doi: 10.1371/journal.pone.0000454
Mystkowska, E. T. (1966). Morphological variability of the skull and body weight of the red deer. Acta Theriol. 11, 129–194. doi: 10.4098/at.arch.66-5
Nussey, D. H., Pemberton, J., Donald, A., and Kruuk, L. E. B. (2006). Genetic consequences of human management in an introduced island population of red deer (Cervus elaphus). Heredity 97, 56–65. doi: 10.1038/sj.hdy.6800838
Paetkau, D., Slade, R., Burden, M., and Estoup, A. (2004). Genetic assignment methods for the direct, real-time estimation of migration rate: a simulation-based exploration of accuracy and power. Mol. Ecol. 13, 55–65. doi: 10.1046/j.1365-294x.2004.02008.x
Paradis, E., Claude, J., and Strimmer, K. (2004). APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290. doi: 10.1093/bioinformatics/btg412
Pekkala, N., Knott, K. E., Kotiaho, J. S., Nissinen, K., and Puurtinen, M. (2014). The effect of inbreeding rate on fitness, inbreeding depression and heterosis over a range of inbreeding coefficients. Evol. Appl. 7, 1107–1119. doi: 10.1111/eva.12145
Pemberton, J. M., Ellis, P. E., Pilkington, J. G., and Bérénos, C. (2016). Inbreeding depression by environment interactions in a free-living mammal population. Heredity 118, 64–77. doi: 10.1038/hdy.2016.100
Pembleton, L. W., Cogan, N. O., and Forster, J. W. (2013). StAMPP: an R package for calculation of genetic differentiation and structure of mixed-ploidy level populations. Mol. Ecol. Resour. 13, 946–952. doi: 10.1111/1755-0998.12129
Pérez-Espona, S., Hall, R. J., Pérez-Barbería, F. J., Glass, B. C., Ward, J. F., and Pemberton, J. M. (2013). The impact of past introductions on an iconic and economically important species, the red deer of Scotland. J. Hered. 104, 14–22. doi: 10.1093/jhered/ess085
Pérez-Espona, S., Pérez-Espona, F. J., McLeod, J. E., Jiggins, C. D., Gordon, I. J., and Pemberton, J. M. (2008). Landscape features affect gene flow of Scottish highland red deer (Cervus elaphus). Mol. Ecol. 17, 981–996. doi: 10.1111/j.1365-294x.2007.03629.x
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A. R., Bender, D., et al. (2007). PLINK: a toolset for whole-genome association and population-based linkage analysis. Am. J. Hum. Genet. 81, 559–575. doi: 10.1086/519795
Putman, R., and Flueck, W. T. (2011). Intraspecific variation in biology and ecology of deer: magnitude and causation. Anim. Prod. Sci. 51, 277–291. doi: 10.1071/an10168
Queiros, J., Vicente, J., Boadella, M., Gortazar, C., and Alves, P. C. (2014). The impact of management practices and past demographic history on the geneticdiversity of red deer (Cervus elaphus): an assessment of population and individual fitness. Biol. J. Linn. Soc. 111, 209–223. doi: 10.1111/bij.12183
R Core Team (2019). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Ralls, K., Ballou, J. D., Dudash, M. R., Eldridge, M. D. B., Fenster, C. B., Lacy, R. C., et al. (2018). Call for a paradigm shift in the genetic management of fragmented populations. Conserv. Lett. 11:e12412. doi: 10.1111/conl.12412
Rijk, J. D., and Pelzers, E. (1991). Geschiedenis van het edelhert in Nederland. Het Edelhert 26, 1–32. doi: 10.1007/978-94-011-9335-1_1
Ripple, W. J., Newsome, T. M., Wolf, C., Dirzo, R., Everatt, K. T., Galetti, M., et al. (2015). Collapse of the world’s largest herbivores. Sci. Adv. 1:e1400103. doi: 10.1126/sciadv.1400103
Rowe, S. J., Clarke, S. M., Van Stijn, T. C., Hyndman, D. L., Ward, J. F., McEwan, K. M., et al. (2015). Brief communication: developing genomic tools in the New Zealand deer industry. Proc. N.Z. Soc. Anim. Prod. 75, 91–93.
Seddon, P. J., Strauss, W. M., and Innes, J. (2012). “Animal translocations: what are they and why do we do them?,” in Reintroduction Biology: Integrating Science and Management, eds J. G. Ewen, D. P. Armstrong, K. A. Parker, et al. (West Sussex: Wiley-Blackwell).
Senn, H. V., and Pemberton, J. M. (2009). Variable extent of hybridization between invasive sika (Cervus nippon) and native red deer (C. elaphus) in a small geographical area. Mol. Ecol. 18, 862–876. doi: 10.1111/j.1365-294x.2008.04051.x
Söderquist, P., Elmberg, J., Gunnarsson, G., Thulin, C. G., Champagnon, J., and Guillemain, M. (2017). Admixture between released and wild game birds: a changing genetic landscape in European mallards (Anas platyrhynchos). Eur. J. Wildl. Res. 63:98. doi: 10.1007/s10344-017-1156-8
Spek, G. J. (2014). Faunabeheerplan grofwild FBE Gelderland 2014-2019. Arnhem: Faunabeheereenheid Gelderland.
Vitorino, L. C., Borges Souza, U. J., Jardim, T. P. F. A., and Ballesteros-Mejia, L. (2019). Towards inclusion of genetic diversity measures into IUCN assessments: a case study on birds. Anim. Biodivers. Conserv. 42, 317–335. doi: 10.32800/abc.2019.42.0317
Volodin, I. A., Nahlik, A., Tari, T., Frey, R., and Volodina, E. V. (2018). Rutting roars in native Pannonian red deer of Southern Hungary and the evidence of acoustic divergence of male sexual vocalization between Eastern and Western European red deer (Cervus elaphus). Mamm. Biol. 94, 54–65. doi: 10.1016/j.mambio.2018.10.009
Vucetich, J. A., Waite, T. A., and Nunney, L. (1997). Fluctuating population size and the ratio of effective to census population size. Evolution 51, 2017–2021. doi: 10.1111/j.1558-5646.1997.tb05123.x
Walling, C. A., Nussey, D. H., Morris, A., Clutton-Brock, T. H., Kruuk, L. E. B., and Pemberton, J. M. (2011). Inbreeding depression in red deer calves. BMC Evol. Biol. 11:318. doi: 10.1186/1471-2148-11-318
Wang, D., Yao, H., Li, Y., Xu, Y., Ma, X., and Wang, H. (2019). Global diversity and genetic landscape of natural populations and hatchery stocks of largemouth bass Micropterus salmoides across American and Asian regions. Sci. Rep. 9:16697. doi: 10.1038/s41598-019-53026-3
Weir, B. S., and Cockerham, C. C. (1984). Estimating F-statistics for the analysis of population structure. Evolution 38, 1358–1370. doi: 10.2307/2408641
Willing, E. M., Dreyer, C., and van Oosterhout, C. (2012). Estimates of genetic differentiation measured by F(ST) do not necessarily require large sample sizes when using many SNP markers. PLoS One 7:e42649. doi: 10.1371/journal.pone.0042649
Keywords: red deer, ungulate, gene flow, translocation, single nucleotide polymorphisms, genetic monitoring
Citation: de Jong JF, van Hooft P, Megens H-J, Crooijmans RPMA, de Groot GA, Pemberton JM, Huisman J, Bartoš L, Iacolina L, van Wieren SE, Ydenberg RC and Prins HHT (2020) Fragmentation and Translocation Distort the Genetic Landscape of Ungulates: Red Deer in the Netherlands. Front. Ecol. Evol. 8:535715. doi: 10.3389/fevo.2020.535715
Received: 17 February 2020; Accepted: 29 September 2020;
Published: 29 October 2020.
Edited by:
R. Terry Bowyer, University of Alaska Fairbanks, United StatesReviewed by:
Kris Hundertmark, University of Alaska Fairbanks, United StatesCopyright © 2020 de Jong, van Hooft, Megens, Crooijmans, de Groot, Pemberton, Huisman, Bartoš, Iacolina, van Wieren, Ydenberg and Prins. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joost F. de Jong, am9vc3QuZGVqb25nQHd1ci5ubA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.