 Carolina Osuna-Mascaró
Carolina Osuna-Mascaró Jorge Doña
Jorge Doña Kevin P. Johnson2
Kevin P. Johnson2 Manuel de Rojas
Manuel de Rojas- 1Department of Genetics, Faculty of Sciences, University of Granada, Granada, Spain
- 2Illinois Natural History Survey, Prairie Research Institute, University of Illinois at Urbana-Champaign, Champaign, IL, United States
- 3AllGenetics & Biology SL, A Coruña, Spain
- 4Department of Microbiology and Parasitology, Faculty of Pharmacy, University of Sevilla, Seville, Spain
Introduction
Rhinonyssidae (Mesostigmata) is a family of mites with more than 500 described species worldwide (George, 1961; Fain, 1994; Dimov and de Rojas, 2012). Most of these mites are hematophagous endoparasites that inhabit the nasal cavity of passerine birds (Vitzthum, 1935). Specifically, their feeding activity may damage the nasal cavities of birds and may even lead to the death of the hosts (Rhinonyssidosis avium disease) (Dimov, 2011). Also, though yet not investigated, these mites are expected to be reservoirs or vectors of infections such as West Nile fever, Q fever, avian influenza, and Lyme disease (Dimov, 2012; Arabkhazaeli et al., 2016).
However, mainly due to sampling difficulties (e.g., these mites usually can only be collected from dead birds), this host-parasite system is very poorly understood. Almost all species of birds are inhabited by rhinonyssid mites (Walter and Proctor, 2013). Despite their broad distribution, there is almost no genomic information available for mites of this family (De Rojas et al., 2001, 2002; Dimov and de Rojas, 2012), thus hampering studies on host-parasite system.
In this data report, with the aim of obtaining mitochondrial and bacterial information, we performed shotgun whole genome sequencing of two species of Rhinonyssidae (Tinaminyssus melloi and Ptilonyssus chloris) that inhabit the nasal cavity of Columba livia and Chloris chloris, respectively. For both species, we assembled and functionally annotated their mitochondrial genomes. We also carried out a metagenomic analysis (metagenomic classification and genome-resolved metagenomics) of these two mite species to provide bacterial information for these mite taxa for the first time (though note that this study should not be taken as a pathogen survey). Our study presents highly valuable mitogenomic and metagenomic resources for this host-parasite system, with implications for further eco-evolutionary studies.
Sampling, DNA Extraction, and Sequencing
We collected individual mites of Tinaminyssus melloi and Ptilonyssus chloris from freshly dead host individuals by dissecting the nasal cavities of birds under a stereomicroscope. We conserved the mites at −20°C in tubes with 100% ethanol. Then, mites were washed in ethanol (Andrews, 2013) and shipped to AllGenetics & Biology SL for DNA isolation, amplification, and library preparation. Given the low amount of DNA obtained from DNA extractions of single individuals, individual mites of both species were pooled (6 mites for Ptilonyssus chloris and 10 mites for Tinaminyssus melloi) following previous studies on mites' high-throughput sequencing and mitogenome assembly (Esteban et al., 2018; Vizcaíno et al., 2018). The DNA was extracted from each pooled sample (i.e., not from individual mites) using the manufacturer's protocols of Quick-DNA MicroPrep Plus kit (Zymo). The libraries were constructed for each sample using the NextFlex DNA Sample Prep kit (Illumina) strictly following the manufacturer's protocols. The DNA was sheared and tagged at both 5' and 3' ends, and samples were dual-indexed. The constructed libraries were quantified using the Qubit dsDNA HT Assay kit (Thermo Scientific) and quality-checked in an Agilent 2100 Bioanalyzer (Agilent Technologies). Lastly, the libraries were pooled in equimolar amounts according to the Qubit results and sequenced in a fraction of an Illumina MiSeq 300PE (Novogen, China).
Mitochondrial Genome Assembly and Annotation
We assembled the mitochondrial genomes using the MitoZ v2.4 pipeline (Meng et al., 2019). We used the “all” module with default parameters. In brief, MitoZ first filters out raw reads as follows: reads with many ambiguities (>10 N's), low-quality reads (Q ≤ 17), and PCR duplicates. Then, MitoZ de-novo assembles the mitogenome based on a modified version of SOAPdenovo-Trans. We used the quick mode assembly (default), and if any gene was missing after the assembly (see results section “Mitochondrial genome assembly and annotation” of Ptilonyssus chloris), we used the multi-kmer mode for recovering the missing genes that failed in the previous step, as recommended by the developers. Finally, MitoZ filters out the candidate mitogenome sequences using a confidence score and annotates the protein-coding genes, tRNA, and rRNA. Additionally, we used the MITOS Web Server (Bernt et al., 2013) to compare the annotations and to ensure the correct annotation of all the genes. Then, the mitogenomes were plotted with OGDRAW v.1.3.1 (Greiner et al., 2019). Lastly, we evaluated the potential incidence of index swapping (i.e., a recently described sequencing artifact that results in 1 to 10% of reads misassigned to the wrong sample; Esling et al., 2015; Sinha et al., 2017; Owens et al., 2018). Specifically, we mapped all raw reads from Tinaminyssus melloi to the Ptilonyssus chloris mitochondrial genome assembly and, vice versa (i.e., all raw reads from Ptilonyssus chloris to the Tinaminyssus melloi mitochondrial genome assembly). For the read mapping analyses, we used Geneious R.11 mapper (Kearse et al., 2012) with high sensitivity and default parameters.
Gene Rearrangement Analyses
We evaluated the mitochondrial gene order in these taxa, because mitochondrial genome rearrangements have been reported in some mesostigmatid mites species (Li et al., 2019). We used CREx (Bernt et al., 2007), to determine the gene arrangement in the two assembled mite mitochondrial genomes. In particular, we constructed a gene order distance matrix using common intervals for Tinaminyssus melloi and Ptilonyssus chloris and compared both of them (1) with each other, (2) with the mitochondrial gene order of Varroa destructor, an obligate parasite of Apis mellifera (Navajas et al., 2002), and (3) with the established ancestral gene order of the arthropods (Staton et al., 1997).
Metagenomic Analyses: Metagenomic Classification and Genome-Resolved Metagenomics
To characterize the overall taxonomic content of the metagenomes, we used the metagenomic classifier Kaiju (Menzel et al., 2016) with the following parameters: Reference database: nr +euk; Database date: 2017-05-16; SEG low complexity filter: yes; Run mode: greedy; Minimum match length: 11; Minimum match score: 75; Allowed mismatches: 5.
Then, we processed raw reads with the complete metaWRAP v1.1.5 pipeline (Uritskiy et al., 2018) using the recommended databases. These analyses were carried out on a 4 AMD Opteron with 16 2.4 GHz processors and 64 CPU cores and 504 GB RAM, maintained by the UIUC Life Sciences Computing Services (University of Illinois, Urbana, IL, USA). (1) We used the metaWRAP Read_qc module (default parameters) to quality-trim the reads of each sample. (2) Then, as recommended for assembling and binning low-abundance organisms, we co-assembled reads from the two samples with the metaWRAP Assembly module (–use-metaspades option) (Nurk et al., 2017). (3) After the assembly, we binned reads with the metaWRAP Binning module (–maxbin2 –concoct –metabat2 options). (4) We consolidated the resulting bins into a final bin set with both metaWRAP's Bin_refinement module (-c 1 - × 1 options) and the Reassemble_bins module. (5) We quantified the bins (i.e., resulting from the Bin_refinement module) with Salmon (Patro et al., 2017) using the Quant_bins module (default parameters). (6) Finally, we classified bins using the Classify_bins module. This module uses Taxator-tk, which gives highly accurate but conservative classifications (Dröge et al., 2014). Accordingly, we uploaded our final bacterial bin to MiGA for a complementary analysis to determine the most likely taxonomic classification and novelty rank of the bins (Rodriguez et al., 2018). We used the NCBI Genome database (Prokaryotes) for this analysis.
Genome Organization, Structure, and Composition
For Tinaminyssus melloi, we obtained a total of 3,600,154 raw reads. The assembled mitogenome was circular and 14,759 bp in length (mean coverage: 99.3X). The base composition was 39.9% A, 18.9% C, 8.8% of G, 33.0% of T. The GC content was 27.7%. We recovered a total of 37 genes: 13 protein-coding genes (PCGs), 22 tRNA, and 2 rRNA (Figure 1).
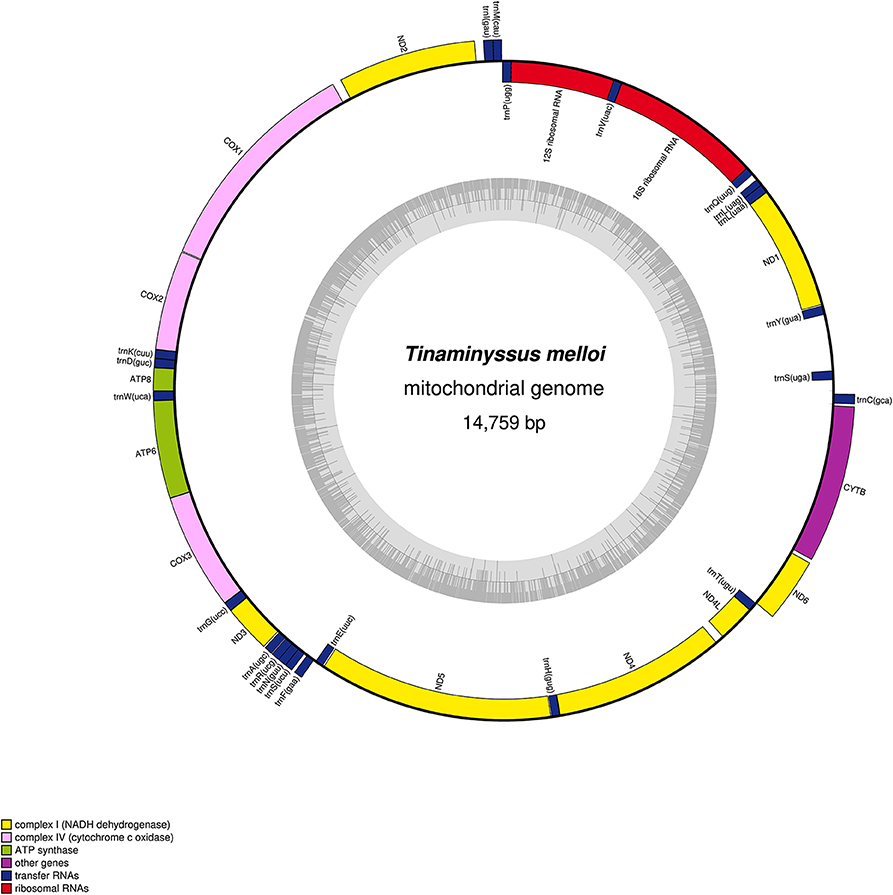
Figure 1. Mitochondrial genome gene map of Tinaminyssus melloi. Gene components are color-coded. The inner circle graph depicts GC content across the genome.
For Ptilonyssus chloris, we obtained a total of 1,554,790 raw reads and assembled a complete circular mitogenome with 14,433 bp in length (mean coverage: 197X). The base composition was 40.4% of A, 6.1% of C, 10.1% of G, and 43.4% of T. The GC content was 16.2%. Using the MitoZ annotation module, we recovered a total of 34 genes, containing 12 protein-coding genes (PCGs), 21 tRNA, and 1 rRNA. However, ND6, trnS, and the l-rRNA genes were missing. These genes were still missing after using the multi-kmer mode recommended for recovering genes missed during the assembly. Nonetheless, using the MITOS annotation Web Server, we found 37 genes in the assembly, including the three genes missed by the MitoZ pipeline (Figure S2).
A very low incidence of index-swapping was found in both assemblies. In particular, only 10 out of 1,800,077 raw reads (<0.001%) mapped to the wrong sample in Ptilonyssus chloris analysis and 13 out of 777,395 raw reads (0.001%) did so in the Tinaminyssus melloi analysis. Therefore, index swapping should not have impacted our assemblies.
The mitochondrial gene order was the same for both mite species (Figure S1). We found that they did not retain the established ancestral mitochondrial organization of arthropods (Staton et al., 1997). In addition, the mitochondrial gene order was also different from that of Varroa destructor (Mesostigmata) (Figure S1). Specifically, compared to Varroa, these mitogenomes contain transpositions in trnY, trnM, trnP, and rrnS, and inversions in trnC, trnY-trnQ-nad1, trnF-trnE, and trnT. Mitochondrial genomes rearranged or highly rearranged in gene order have been previously described and studied in mesostigmatid mite species (Li et al., 2019). Interestingly, to our knowledge, the rhynonissid mitochondrial genome organization plan found here has not been found in other mesostigmatid families.
Metagenomic Composition and Metagenome-Assembled Genomes (MAGS)
For Tinaminyssus melloi, the top three bacterial species in terms of the number of assigned reads were (Figure 2; Tables S1, S2): (1) Clostridioides difficile [n (i.e., number of assigned reads) = 15,019], (2) Ralstonia solanacearum (n = 4,518), and (3) Kingdom Bacteria (n = 3,194). Note that some hits could not be identified at the species level. For Ptilonyssus chloris, the top three bacterial species were: (1) Ralstonia solanacearum (n = 1,609), (2) Clostridioides difficile (n = 1,341), and (3) Delftia (n = 990). Some of these bacteria may be pathogenic [e.g., Clostridioides difficile, which may cause diarrhea and mortality in humans, and also may cause clinical disease in livestock and birds (Lawson et al., 2016; Moono et al., 2016)] but also may have an environmental origin (Zhu et al., 2018). Further studies are needed on the potential role of rhynonissid mites as reservoirs of pathogenic bacteria.
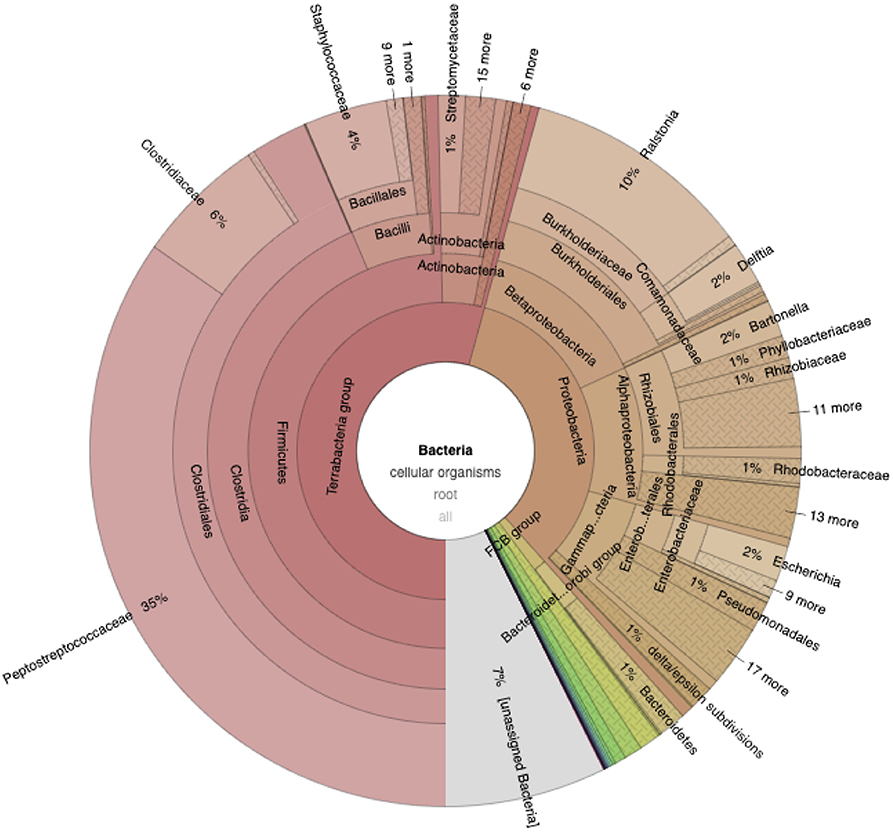
Figure 2. Krona plot showing the relative distribution of Bacteria in Tinaminyssus melloi. Note that while this chart depicts Kaiju results, the taxonomic level displayed differs from that of Tables S1, S2 (see full interactive krona plots for comparisons at the same taxonomic level; Links to the Deposited Data section).
From the genome-resolved metagenomic approach, 1,517,456 and 3,549,318 high-quality reads were used after running the read_qc module of metaWRAP for Tinaminyssus melloi and Ptilonyssus chloris, respectively. We retrieved a single bacterial metagenome-assembled genome (MAG) from the co-assembly. According to CheckM, the MAG was 26% complete (Salmon contig coverage: 26X), with 0.46% contamination, GC = 0.32, N50 = 1,539, and the total length was 382,098 bp. The metaWRAP Classify_bins module classified the MAG as belonging to the family Bartonellaceae. A further taxonomic classification analysis in MiGA revealed highly congruent results. Specifically, the closest related species found was Bartonella bacilliformis KC583 (NC008783; 52.27% AAI, i.e., maximum average amino acid identity). In addition, the MAG was classified as possibly belonging to the family Bartonellaceae (p-value: 0.37). Interestingly, Bartonellaceae taxa have been found consistently associated with mites, though the particular role of these bacteria is still unknown (Kim et al., 2005; Hubert et al., 2017; Doña et al., 2019). We also found the MAG most likely belongs to a species not represented in the MiGA database (p-value: 0.0015) and probably belongs to a genus not represented in the database (p-value: 0.08). Lastly, the MAG was more abundant in Tinaminyssus melloi (68.06 genome copies per million reads) than in Ptilonyssus chloris (1.75 genome copies per million reads), in which this very low abundance require further study to rule out methodological issues (e.g., index-swapping) (Esling et al., 2015; Sinha et al., 2017; Owens et al., 2018). Future studies are needed to fully assemble the genome of this bacterial taxon, ideally with higher coverage to ultimately uncover its potential role in these mites.
Discrepancies between the genome-resolved metagenomic and Kaiju results (e.g., in which a relatively small number of reads was classified as Bartonella) may reflect the already known differences between these two approaches (i.e., assembly-based and read-based analyses), in which many variables affect (e.g., database completeness; Quince et al., 2017). In addition, given that our coverage for the metagenomic assemblies was not very high, many bins belonging to taxa with a higher number of reads in Kaiju may have been discarded throughout the metaWRAP pipeline because of high levels of heterogeneity in the assembled bin/MAGs. Future metagenomic studies with higher coverage are encouraged to provide genomic resources of the whole community of microbial taxa associated with these mites.
Overall, the resources presented here contained useful information for future studies on rhinonyssid mites, a largely neglected group of parasites. In particular, the data provided here can be useful for phylogenomic studies, studies on mitochondrial genome evolution, studies of the microbiomes of parasitic mites, and for studies of disease dynamics.
Links to the Deposited Data
The mitogenome sequences of Tinaminyssus melloi and Ptilonyssus chloris are available at NCBI genome database with the BioProject number PRJNA575688. The high-throughput sequencing data of each sample is available at the Sequence Read Archive SRA under the accession numbers SRR10228588 and SRR10228589. The first draft mitochondrial genome sequences for both species with all genes annotated has been submitted to GenBank under the accessions numbers: MN557820 and MN557819. An interactive version of Krona plots can be found at: https://figshare.com/s/8f6df0887c7ca918813a (private link for review). The metagenome-assembled genome is available at NCBI (submission ID: SUB6386351).
Data Availability Statement
The names of the repository/repositories and accession number(s) can be found above in the Links to the deposited data section.
Author Contributions
CO-M, JD, and MR conceived this study. CO-M and JD analyzed the data. KJ supervised the research and provided logistical support. RE managed and uploaded data to specific repositories. CO-M wrote the first draft, which was revised by all other authors.
Funding
This work was supported by a V plan propio de investigación de la Universidad de Sevilla, Spain grant to MR, and the NSF DEB-1239788, DEB-1342604, and DEB-1855812 to KJ.
Conflict of Interest
RE was employed by the company AllGenetics & Biology SL.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2020.00142/full#supplementary-material
References
Andrews, E. S. (2013). Analyzing arthropods for the presence of bacteria. Curr. Protoc. Microbiol. 28, 1E−6E. doi: 10.1002/9780471729259.mc01e06s28
Arabkhazaeli, F., Madani, S. A., and Ghavami, S. (2016). Outbreak of an unusual tracheal mite, Ptilonyssus morofskyi (Acarina: Rhinonyssidae), in canaries (Serinus canaria) with concurrent infection with staphylococcus aureus and Macrorhabdus ornithogaster. J. Avian Med. Surg. 30, 269–274. doi: 10.1647/2015-088
Bernt, M., Donath, A., Jühling, F., Externbrink, F., Florentz, C., Fritzsch, G., et al. (2013). MITOS: improved de novo metazoan mitochondrial genome annotation. Mol. Phylogene. Evol. 69, 313–319. doi: 10.1016/j.ympev.2012.08.023
Bernt, M., Merkle, D., Ramsch, K., Fritzsch, G., Perseke, M., Bernhard, D., et al. (2007). CREx: inferring genomic rearrangements based on common intervals. Bioinformatics 23, 2957–2958. doi: 10.1093/bioinformatics/btm468
De Rojas, M., Mora, M., Ubeda, J., Cutillas, C., Navajas, M., and Guevara, D. (2002). Phylogenetic relationships in rhinonyssid mites (Acari: Rhinonyssidae) based on ribosomal DNA sequences: insights for the discrimination of closely related species. Parasitol. Res. 88, 675–681. doi: 10.1007/s00436-002-0647-y
De Rojas, M., Mora, M. D., Ubeda, J. M., Cutillas, C., Navajas, M., and Guevara, D. C. (2001). Phylogenetic relationships in rhinonyssid mites (Acari: Rhinonyssidae) based on mitochondrial 16S rDNA sequences. Exp. Appl. Acarol. 25, 957–967. doi: 10.1023/A:1020651214274
Dimov, I., and de Rojas, M. (2012). One new species of nasal mites of the genus Vitznyssus (Rhinonyssidae) from the Leningrad province, Russia. J. Acarol. Soc. Jap. 21, 125–130. doi: 10.2300/acari.21.125
Dimov, I. D. (2012). “Epizootological study of spreading of the Rhinonyssid mites in nasal cavities of birds from northwest Russia,” in Proceedings Second International Epizootiology Days (Belgrade), 176–181.
Doña, J., Proctor, H., Serrano, D., Johnson, K. P., Oploo, A. O. V., Huguet-Tapia, J. C., et al. (2019). Feather mites play a role in cleaning host feathers: New insights from DNA metabarcoding and microscopy. Mol. Ecol. 28, 203–218. doi: 10.1111/mec.14581
Dröge, J., Gregor, I., and McHardy, A. C. (2014). Taxator-tk: precise taxonomic assignment of metagenomes by fast approximation of evolutionary neighborhoods. Bioinformatics 31, 817–824. doi: 10.1093/bioinformatics/btu745
Esling, P., Lejzerowicz, F., and Pawlowski, J. (2015). Accurate multiplexing and filtering for high-throughput amplicon-sequencing. Nucleic Acids Res. 43, 2513–2524. doi: 10.1093/nar/gkv107
Esteban, R., Doña, J., Vierna, J., Vizcaíno, A., Serrano, D., and Jovani, R. (2018). The complete mitochondrial genome of the feather mite trouessartia rubecula jablonska, 1968 (Astigmata: Analgoidea: Trouessartiidae). Mitochondrial DNA Part B 3, 652–654. doi: 10.1080/23802359.2018.1476072
Fain, A. (1994). Adaptation, specificity and host-parasite coevolution in mites (Acari). Int. J. Parasitol. 24, 1273–1283. doi: 10.1016/0020-7519(94)90194-5
George, J. E. (1961). The nasal mites of the genus Ptilonyssus (Acarina: Rhinonyssidae) occurring in some North American passeriform birds. J. Kansas Entomol. Soc. 34, 105–132.
Greiner, S., Lehwark, P., and Bock, R. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3. 1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64. doi: 10.1093/nar/gkz238
Hubert, J., Erban, T., Kopecky, J., Sopko, B., Nesvorna, M., Lichovnikova, M., et al. (2017). Comparison of microbiomes between red poultry mite populations (Dermanyssus gallinae): predominance of Bartonella-like bacteria. Microb. Ecol. 74, 947–960. doi: 10.1007/s00248-017-0993-z
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kim, C. M., Kim, J. Y., Yi, Y. H., Lee, M. J., Cho, M. R., Shah, D. H., et al. (2005). Detection of Bartonella species from ticks, mites and small mammals in Korea. J. Vet. Sci. 6, 327–334. doi: 10.4142/jvs.2005.6.4.327
Lawson, P. A., Citron, D. M., Tyrrell, K. L., and Finegold, S. M. (2016). Reclassification of clostridium difficile as clostridioides difficile (Hall and O'Toole 1935) Prévot 1938. Anaerobe 40, 95–99. doi: 10.1016/j.anaerobe.2016.06.008
Li, W. N., Shao, R., Zhang, Q., Deng, W., and Xue, X. F. (2019). Mitochondrial genome reorganization characterizes various lineages of mesostigmatid mites (Acari: Parasitiformes). Zool. Scripta 48, 679–689. doi: 10.1111/zsc.12369
Meng, G., Li, Y., Yang, C., and Liu, S. (2019). MitoZ: a toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 47:e63. doi: 10.1093/nar/gkz173
Menzel, P., Ng, K. L., and Krogh, A. (2016). Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 7:11257. doi: 10.1038/ncomms11257
Moono, P., Foster, N. F., Hampson, D. J., Knight, D. R., Bloomfield, L. E., and Riley, T. V. (2016). Clostridium difficile infection in production animals and avian species: a review. Foodbor. Pathog. Dis. 13, 647–655. doi: 10.1089/fpd.2016.2181
Navajas, M., Conte, Y. L., Solignac, M., Cros-Arteil, S., and Cornuet, J. M. (2002). The complete sequence of the mitochondrial genome of the honeybee ectoparasite mite varroa destructor (Acari: Mesostigmata). Mol. Biol. Evol. 19, 2313–2317. doi: 10.1093/oxfordjournals.molbev.a004055
Nurk, S., Meleshko, D., Korobeynikov, A., and Pevzner, P. A. (2017). metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834. doi: 10.1101/gr.213959.116
Owens, G. L., Todesco, M., Drummond, E. B., Yeaman, S., and Rieseberg, L. H. (2018). A novel post hoc method for detecting index switching finds no evidence for increased switching on the Illumina HiSeq X. Mol. Ecol. Res. 18, 169–175. doi: 10.1111/1755-0998.12713
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A., and Kingsford, C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14:417. doi: 10.1038/nmeth.4197
Quince, C., Walker, A. W., Simpson, J. T., Loman, N. J., and Segata, N. (2017). Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 35:833. doi: 10.1038/nbt.3935
Rodriguez, L. M., Gunturu, S., Harvey, W. T., Rosselló-Mora, R., Tiedje, J. M., Cole, J. R., et al. (2018). The Microbial Genomes Atlas (MiGA) webserver: taxonomic and gene diversity analysis of archaea and bacteria at the whole genome level. Nucleic Acids Res. 46, W282–W288. doi: 10.1093/nar/gky467
Sinha, R., Stanley, G., Gulati, G. S., Ezran, C., Travaglini, K. J., Wei, E., et al. (2017). Index switching causes “spreading-of-signal” among multiplexed samples in Illumina HiSeq 4000 DNA sequencing. BioRxiv [Preprint]. doi: 10.1101/125724
Staton, J. L., Daehler, L. L., and Brown, W. M. (1997). Mitochondrial gene arrangement of the horseshoe crab Limulus polyphemus L.: conservation of major features among arthropod classes. Mol. Biol. Evol. 14, 867–874. doi: 10.1093/oxfordjournals.molbev.a025828
Uritskiy, G. V., DiRuggiero, J., and Taylor, J. (2018). MetaWRAP-a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 6:158. doi: 10.1186/s40168-018-0541-1
Vitzthum, H. G. (1935). Milben aus der Nasenhöhle von Vögeln. J. Ornithol. 83, 563–587. doi: 10.1007/BF01905801
Vizcaíno, A., Doña, J., Vierna, J., Marí-Mena, N., Esteban, R., Mironov, S., et al. (2018). Enabling large-scale feather mite studies: an Illumina DNA metabarcoding pipeline. Exp. Appl. Acarol. 76, 81–97. doi: 10.1007/s10493-018-0288-1
Walter, D. E., and Proctor, H. C., (eds.). (2013). “Animals as habitat,” in Mites: Ecology, Evolution & Behaviour (Dordrecht: Springer), 341–422.
Keywords: gene order, genome assembly, genome-resolved metagenomics, mitochondria, parasites
Citation: Osuna-Mascaró C, Doña J, Johnson KP, Esteban R and de Rojas M (2020) Complete Mitochondrial Genomes and Bacterial Metagenomic Data From Two Species of Parasitic Avian Nasal-Mites (Rhinonyssidae: Mesostigmata). Front. Ecol. Evol. 8:142. doi: 10.3389/fevo.2020.00142
Received: 23 October 2019; Accepted: 24 April 2020;
Published: 19 May 2020.
Edited by:
Mariana Mateos, Texas A&M University, United StatesReviewed by:
Pavel Klimov, University of Michigan, United StatesDmitry Yurievich Sherbakov, Limnological Institute (RAS), Russia
Copyright © 2020 Osuna-Mascaró, Doña, Johnson, Esteban and de Rojas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manuel de Rojas, ZGVyb2phc0B1cy5lcw==