 Anthony I. Cognato1*
Anthony I. Cognato1* Gina Sari2
Gina Sari2 Sarah M. Smith1Roger A. Beaver3
Sarah M. Smith1Roger A. Beaver3 You Li4
You Li4 Jiri Hulcr4
Jiri Hulcr4 Bjarte H. Jordal5Hisashi Kajimura6Ching-Shan Lin7
Bjarte H. Jordal5Hisashi Kajimura6Ching-Shan Lin7 Thai Hong Pham8Sudhir Singh9Wisut Sittichaya10
Thai Hong Pham8Sudhir Singh9Wisut Sittichaya10- 1Department of Entomology, Michigan State University, East Lansing, MI, United States
- 2Indonesia Agricultural Quarantine Agency, Jakarta, Indonesia
- 3Retired, Chiang Mai, Thailand
- 4School of Forest Resources and Conservation, University of Florida, Gainesville, FL, United States
- 5University Museum of Bergen, University of Bergen, Bergen, Norway
- 6Graduate School of Bioagricultural Sciences, Nagoya University, Nagoya, Japan
- 7Department of Entomology, National Taiwan University, Taipei, Taiwan
- 8Vietnam National Museum of Nature and Graduate School of Science and Technology, Vietnam Academy of Science and Technology, Hanoi, Vietnam
- 9Forest Entomology Division, Forest Research Institute, New Forest, Dehradun, Uttarakhand, India
- 10Department of Pest Management, Faculty of Natural Resources, Prince of Songkla University, Hat Yai, Thailand
DNA holds great potential for species identification and efforts to create a DNA database of all animals and plants currently contains >7.5 million sequences representing ~300,000 species. This promise of a universally applicable identification tool suggests that morphologically based tools and taxonomists will soon not have utility. Here we demonstrate that DNA-based identification is not reliable without the contributions of taxonomic experts. We use ambrosia beetles (Xyleborini), which are known for great diversity as well as global invasions and damage, as a test case. Recent xyleborine introductions have caused major economic and ecological losses, thus timely species identifications of new invaders are necessary. This need is hampered by a paucity of identification tools and a fauna that is only moderately documented. To help alleviate deficiencies in their identification, we created COI and CAD DNA barcode databases (490 and 429 specimens), representing over half of the known fauna of Southeast Asia (165/316 species). Taxonomic experts identified species based on original descriptions and type specimens. Tree, distance, and iterative methods were used to assess the identification and delimitation of species. High intra- and interspecific COI distances were observed for congeneric species and attributed to the beetle's inbreeding system. Neither of the two markers provided 100% identification success but with the neighbor-joining tree-based method, 80% of species were identified by both genes. As for species delimitation, an obvious barcode gap between intra- and interspecific differences was not observed. Correspondence between distance-based groups and morphology-based species was poor. In a demonstration of iterative taxonomy, we constructed parsimony-based phylogenies using COI and CAD sequences for two genera. Although not all clades were resolved or supported, we provided better explanations for species boundaries in light of morphological and DNA sequence differences. Confident species identifications demonstrated <3% COI and <1% CAD difference and recognition of new species became more probable when there was >10–12% COI and/or >2–3% CAD. Involvement of taxonomic experts from the start of this project was essential for the creation of a stable foundation for the DNA identification of xyleborine species. In general, their role in DNA barcoding cannot be underestimated and is further discussed.
Introduction
Xyleborine ambrosia beetles (Curculionidae: Scolytinae) occur throughout the world's forests with most of the diversity in the moist tropics where they comprise the majority of the scolytine diversity (Browne, 1961; Wood and Bright, 1992; Hulcr et al., 2015). These beetles exhibit two conspicuous life history traits: they cultivate symbiotic fungi for food in tunnels that they bore into recently dead trees (and their parts), and they are haplodiploid and highly inbred with female-skewed sex ratios averaging 13:1 (Kirkendall, 1993; Cooperband et al., 2016; Castro et al., 2019). These traits have allowed these beetles to colonize the world and gave them their infamous reputation as potential invasive species (Jordal et al., 2001; Gohli et al., 2016; Brockerhoff and Liebhold, 2017). One female with her fungal food stored in specialized body cavities (mycangia) can start a new population after establishing a fungal garden and laying an unfertilized egg which develops into a haploid son. After developing into an adult, the male mates with his mother to produce diploid eggs which develop into females. The adult females mate with their brother and then emerge from the natal nest to beget other families (Kirkendall, 1993; Wood, 2007). These traits allowed multiple lineage radiations on both remote Pacific islands as well as continents from at least 15 million years ago (Cognato et al., 2011, 2018; Jordal and Cognato, 2012). As a result, Xyleborini are the largest and most diverse scolytine tribe, representing nearly 20% of all described species (Hulcr et al., 2015; Gohli et al., 2017). Global trade and the use of wood products as ballast and crating have contributed to an accelerated rate of dispersal of these beetles in many parts of the world (Haack and Rabaglia, 2013; Cognato et al., 2015; Gohli et al., 2017; Meurisse et al., 2018). The first recored introduction of a xyleborine species in the US dates to 1817, but the rate of introduction accelerated with 17 new out of the total 28 exotic species in just the last 30 years (Haack and Rabaglia, 2013; Smith and Cognato, 2015; Gomez et al., 2018; Hoebeke et al., 2018). A subset of these species has also been introduced into Europe in the last two decades (Kirkendall and Faccoli, 2010; Dodelin, 2018).
Most introduced species have an apparently benign effect on the environment because most non-native species occupy woody debris unused by the meager native Holarctic xyleborine fauna (Wood, 1982; Knížek, 2011; Hulcr et al., 2017). However recent findings suggest that the native wood decay fungus community may be displaced by a non-native fungus proliferated by a non-native xyleborine (Hulcr et al., 2018). In addition, three species, Euwallacea fornicatus, Euwallacea perbrevis, and Xyleborus glabratus, and their associated fungi have caused economic and ecological destruction to US orchards and natural forests. These species threaten the multi-million dollar avocado industry and have already altered the ecology of natural landscapes with the loss of millions of red bay trees (Eskalen et al., 2012; Boland, 2016; Carrillo et al., 2016; Hughes et al., 2017).
The introduction of exotic xyleborines presents a serious threat to native forests and much time and funding has been invested at the national level in the US and Europe to detect non-native beetles (Kirkendall and Faccoli, 2010; Rabaglia et al., 2019). The faunas of Europe and America north of Mexico are well-known but a taxonomic impediment concerning tropical xyleborines challenges these efforts by hindering the identification of unknown specimens. Few species identification keys exist for the faunas of the New and Old World tropics where xyleborines are most speciose and this limited knowledge of their diversity hampers the ability to identify species (Kirkendall and Faccoli, 2010). It is estimated that only 75% of the Southeast Asian and 25% of the South American faunas have been described so far (Wood and Bright, 1992; Hulcr et al., 2015; Smith et al., 2017). Even with taxonomic tools, the small and subtle morphological differences that define many xyleborine species make it difficult for non-experts to accurately identify species (Cognato et al., 2015; Gomez et al., 2018; Hoebeke et al., 2018; Smith et al., 2019). Identification of immature stages to species or genus presents the greatest challenge even for experts. This taxonomic impediment could be remedied in part by the creation of a DNA database based on expertly identified specimens, as with other wood-boring beetles (Wu et al., 2017).
At the start of molecular systematics, the use of molecules, especially DNA, for the identification of species was recognized (e.g., Nanney, 1982; Sperling and Hickey, 1994; Foster et al., 2004). The franchise of “DNA barcoding” popularized the use of a ~700 nucleotide section of mitochondrial cytochrome c oxidase subunit I gene (COI), amplified and sequenced with universal primers (Folmer et al., 1994), to identify most animal species (Hebert et al., 2003a). This rapid proliferation of sequences and application to most taxa demonstrated that many species could be distinguished from related species with obvious reproductive barriers (Hebert et al., 2003b; Sperling, 2003). However, the best use of these data to identify species i.e., tree-based and DNA sequence match identification, was debated (Meier et al., 2006; Taylor and Harris, 2012). Although DNA barcoding was initially invisioned for species identification, diagnosticians readily suggested, and sometimes declared new species for non-monophyletic species recovered in neighbor-joining trees and those that transgressed the 2% barcoding gap (e.g., Hebert et al., 2003b; Barrett and Hebert, 2005; Zahiri et al., 2017). Thus, DNA barcoders trespassed into the field of taxonomy. Delimitation of species based solely on phenetic measures and disgaurd of basic taxonomic prinicples caused much controversy and response from the systematics community (e.g., Will and Rubinoff, 2004; DeSalle et al., 2005; Ebach and Holdrege, 2005; Prendini, 2005; Will et al., 2005; Brower, 2006; Cognato, 2006). Major objections included taxonomy based on one DNA locus, the use of a standardized barcoding gap, neighbor-joining analysis, and the absence of taxonomic expertise in the delimitation of species (see Prendini, 2005 for review). However, approached scientifically with deposition of vouchers, adequate sample size, and the phylogenetic/systematic framework, DNA barcoding data can identify species and contribute to the discovery of new taxa (e.g., Schindel and Miller, 2005; Packer et al., 2009b; Adamski and Miller, 2015; Taft and Cognato, 2017; Gibbs, 2018; DeSalle and Goldstein, 2019).
Issues with the implementation of DNA barcoding still exist for certain taxa (Taylor and Harris, 2012). The universal COI PCR primers fail to amplify DNA for some groups of taxa or particular species within groups (e.g., Hebert et al., 2004; Ward et al., 2005; Smith and Cognato, 2014). This has led to modifications of the original PCR primers to capture the barcoding region, to the use of different primer pairs to capture a partial barcoding region, or to the abandonment of the barcoding region (e.g., Jordal and Kambestad, 2014; Smith and Cognato, 2014). However, nearly all barcoding projects use the fragment as designated by Hebert et al. (2003a). Different evolutionary rates within some highly divergent or conserved taxa hamper identification because of non-uniform nucleotide differences and challenge the use of a standard barcoding gap to distinguish species (Hebert et al., 2003b; Cognato, 2006). In addition, taxa with non-sexual or inbreeding mating may defy standard species concepts as they do based on morphology. Issues with heteroplasmy and pseudogenes (numts) can also decrease the accuracy of identification with the use of the COI barcoding region and mitochondrial DNA in general (Song et al., 2008; Magnacca and Brown, 2010; Moulton et al., 2010; Jordal and Kambestad, 2014). The adoption of different genes for identification can help to alleviate these COI barcoding region issues for some taxa (e.g., Foster et al., 2013).
Taxonomic experts have been underutilized in developing DNA barcodes. Among DNA barcoding studies, either taxonomists are ignored (e.g., Lait and Hebert, 2018), mentioned only as identifiers (e.g., Kekkonen and Hebert, 2014), or called upon to interpret the taxonomic implications of the resulting data in subsequent studies (e.g., Barrett and Hebert, 2005). The exclusion of taxonomists or explicit taxonomic methodology for DNA barcoding studies can yield suspect conclusions or irreproducible results (e.g., Hebert et al., 2004; Chang et al., 2014). Also the discovered “new species” only add to the taxonomic impediment if the species are not formally described (e.g., Brower, 2010; Pinheiro et al., 2019). Incorporation of taxonomists from the start of a DNA barcoding project would alleviate many of the mentioned issues, as observed in the more informative barcoding studies (e.g., Trewick, 2008; Packer et al., 2009a).
Although there are potential issues and limitations of DNA barcoding using COI, preliminary data suggest the feasibility of identification and delimiting xyleborine species (Dole et al., 2010; Cognato et al., 2011, 2015, 2019; Jordal and Kambestad, 2014; Stouthamer et al., 2017; Gomez et al., 2018). Studies of a few closely related species of different genera demonstrated: (1) The universal or scolytine specific barcoding COI primers produce PCR products and DNA sequences for most species; (2) non-monophyletic species; (3) high intraspecific nucleotide difference (> 10% as compared to 2–3% for outbreeding scolytines) (4) the use of nuclear genes as alternative diagnostic loci; and (5) the results of a few studies identified new species (Gomez et al., 2018; Cognato et al., 2019). In addition, there are currently overlapping generations of scolytine taxonomists that can identify specimens to species and can interpret the DNA results in reference to these identifications.
In this study, we develop a DNA identification foundation for 165 species of 316 Southeast (SE) Asian xyleborines representing more than half the known species. The goal is to create a DNA barcode resource in conjunction with the historically most comprehensive taxonomic revision of the fauna (Smith et al., in preparation), intended to serve as a model taxonomic product where DNA barcodes and morphological systematics are iteratively used and in mutual support. Another intent is to integrate fundamental biosystematics with direct application: species of this fauna are the most often intercepted wood borers at US ports-of-entry, therefore diagnosticians need a dataset of authenticated DNA sequences as an identification tool (Haack and Rabaglia, 2013). Anticipating the issue of high COI nucleotide divergence we tested the species identification potential of an alternative locus—in this case CAD. Although, any other gene locus could potentially provide species diagnostic DNA sequences such as, 28S rDNA, preliminary rDNA data suggested that the species level nucleotide variation of this locus was not consistant among scolytine taxa (Jordal and Kambestad, 2014; Cognato et al., 2019). We compare tree-based and DNA match methods for the identification of species and demonstrate the use of DNA barcodes for the discovery of species. We demonstrate that the use of COI and CAD can help the identification and delimitation of xyleborine species and discuss the role of the taxonomist in the creation of a DNA barcoding database.
Materials and Methods
Specimens
Specimens were collected from various localities in SE Asia via excision of the beetles infesting wood or from ethanol baited flight interception traps. A total of 508 individuals representing 33 genera and 258 species with more than half from SE Asia (165) were included in this study (Supplementary Table 1). Specifically, 490 and 429 individuals were included in the COI and CAD datasets, respectively. The head and pronotum were removed and placed in a 1.5 ml microfuge vial for the extraction of DNA. DNA extraction followed using Qiagen tissue extraction kit and protocol (Qiagen Ltd., Hilden, Germany). Pinned vouchers were deposited at the A.J. Cook Arthropod Research Collection, Michigan State University. Specimens were identified to species based on comparison to type specimens and published descriptions by SMS, RAB, and AIC. We consider these morphologically-based identifications null hypotheses of species limits.
DNA Amplification and Sequencing
DNA sequences of partial Cytochrome c Oxidase subunit I (COI) mtDNA were generated with primers LCO1490: 5′-GGTCAACAAATCATAAAGATATTGG-3′ and HCO2198: 5′-TAAACTCAGGGTGACCAAAAAATCA-3′ (Folmer et al., 1994). Each 25 ml PCR reaction contained 4.5 ml template DNA; 2.5 ml buffer; 1 ml MgCl2; 0.5 ml dNTPs; 0.75 ml each primer; 0.25 ml of hot star taq and the reactions were subjected to the PCR thermal protocol listed in Hebert et al. (2003a). When PCR failed, a primer pair designed for scolytines was used (1495b: 5′-AACAAATCATAAAGATATTGGRAC-3′ and rev750: 5′-GAAATTATNCCAATTCCTGG-3′; Smith and Cognato, 2014). PCR amplification protocol consisted of 15 min denaturation at 95°C and 38 cycles at 94°; 50°C each for 30 s and 72°C 45 s, followed by a 5 min extension at 72°C.
Sequences of the nuclear protein coding gene (CAD) gene were generated with forward primers CADforB2 5′ GARAARGTNGCNCCNAGTATGGC-3′ (Jordal et al., 2011) or CADfor4 5′ TGGAARGARGTBGARTACGARGTGGTYCG-3′ (Danforth et al., 2006) and the reverse primer apCADrev1mod 5′ GCCATYRCTCBCCTACRCTYTTCAT-3′ (Danforth et al., 2006). Each 25 ml PCR reaction contained 4.5 ml template DNA; 2.5 ml buffer; 1 ml MgCl2; 0.5 ml dNTPs; 0.75 ml each primer; 0.25 ml of hot star taq. PCR amplification protocol consisted of 15 min denaturation at 95°C and 35 cycles at 94°C for 30 s; 55°C for 30 s and 72°C for 1 min, followed by a 5 min extension at 72°C.
PCR products were electrophoresed and visualized on 1.5% TAE agarose gel stained with ethidium bromide. PCR products were purified of excess primers and unincorporated nucleotides using ExoSAP-IT™ following the manufacturer's protocol (Thermo Fisher Scientific). Sequencing of the purified PCR products occurred at the Research Technology Support Facility at Michigan State University using BigDye Terminator v.1.1 (Applied Biosystems, Foster City, CA, USA) cycle sequencing kit and visualized on an ABI 3730 or 3700 (Applied Biosystems). The DNA sequences were compiled and inspected with Sequencer 4.7 (Gene Codes, Ann Arbor, MI, USA). Sequences were assessed for potential pseudogenes following the recommendations of Jordal and Kambestad (2014). Consensus sequences derived from the forward and reverse sequences were used in subsequent analyses and deposited in Genbank (Supplementary Table 1).
Taxon Identification
For the tree-based method, COI and CAD sequences were assembled in separate NEXUS files using the software PAUP version 4.0a (build 161) (Swofford, 2002). Previously published sequences were also included from studies in which we could verify the species status of vouchers (Cognato et al., 2011, 2015, 2019). These specimens provided a global context as many of these species occurred outside the study area. Neighbor-joining trees were calculated using uncorrected “p”–distances. We used “p”–distance instead of Jukes-Cantor (Jukes and Cantor, 1969) or Kumura-2 (Kimura, 1980) models of nucleotide substitution because these models do not affect the interspecific distance among closely related species thus not benefiting the identification of species (Srivathsan and Meier, 2012). The number of monophyletic species and genera were recorded.
DNA sequence match methods rely only on DNA sequence similarity without reliance on the clustering of sequences in a neighbor-joining tree (Saitou and Nei, 1987). This is advantageous because it avoids the pitfalls of neighbor-joining analysis (DeSalle and Goldstein, 2019) and includes percent sequence difference criterion in species identification. Using the TaxonDNA software (Meier et al., 2006), we calculated DNA sequence match for the COI and CAD sequences and recorded the number of successful, ambiguous, and misidentifications of species. We varied the analyses by including best match, best close match, and all species barcode criteria. Best match criterion is the most relaxed given the query sequence needs to match only one sequence without regard to percent similarity. For the best close match criterion, the query sequence needs to match a threshold percent similarity observed in 95% conspecifics. The chosen threshold percent similarities for the genes were traditional barcodes gaps (2 and 3%) and approximate barcode gaps based on the empirical data. The species barcode criterion is similar to the best close match method but the query sequence needs to match all conspecific sequences as top matches.
Taxon Discovery
We used Automatic Barcode Gap Discovery (ABGD) and TaxaDNA to identify COI and CAD barcode gaps among species (Meier et al., 2006; Puillandre et al., 2012). Although other means for assessing barcode gaps exist (such as, Ratnasingham and Hebert, 2013), these methods provide assessment of multiple gap values and models of nucleotide evolution. We used TaxaDNA software to cluster sequences based on the barcode gaps and determined the number of violations of the predetermined taxonomy based on morphology and comparison to type specimens. Different barcode gap values were assessed with ABGD software (http://wwwabi.snv.jussieu.fr/public/abgd/abgdweb.html, accessed 9 August 2019) where Pmin = 0.001, Pmax = 0.1, Steps = 10, and the relative gap width (X) = 1.0 for both genes. Preferred groups of sequences were selected based on an intermediate value of P after an initial steep decline in number of estimated groups (Puillandre et al., 2012).
For two genera we provide examples of the application of iterative taxonomy (Yeates et al., 2011) to deliberate species limits. Based on monophyletic genera as found in the CAD NJ-tree, we created NEXUS files for the species of Ambrosiophilus and Euwallacea which included COI and CAD sequences for the corresponding species. For these data sets, we performed maximum parsimony analyses using a heuristic search with 100 random stepwise additions. Non-parametric bootstrap (Felsenstein, 1985) values were calculated for all generic datasets with 500 pseudoreplicates using a heuristic search with simple stepwise additions. These results were discussed in reference to morphological characters typically used to diagnose species (Hulcr et al., 2007).
Results
PCR and Sequencing
The PCR primer pairs do not reliably amplified the target locus for COI and CAD. The COI primers 1495b and rev750 and CAD primers ApCADfor4 and CADrev1mod amplified the target loci most often. The combination of COI and CAD primer pairs had 88 and 72% success rates, respectively. COI sequences showed no double peaks in chromatograms, however double peaks were observed in some in CAD chromatograms, which we attributed to allelic variation. These nucleotide positions were labeled with an appropriate ambiguity code.
Taxon Identification
In the tree based identification method, monophyly of genera, and species was not found for all taxa in COI and CAD neighbor-joining trees (Table 1 and Supplementary Figures 1, 2). However, of the ~65% of species that were represented >1 sequence, 80% of the species were identified for both genes. CAD neighbor-joining tree resolved more monophyletic genera (17) as compared to the COI neighbor-joining tree (7) which is expected given the observed high COI nucleotide substitution rate (see below). Fifty percent of the COI sequences were successfully clustered with the same species, while 14% did not and 35% had an ambiguous placement. Fifty-two percent of the CAD sequences were successfully clustered with the same species, while 11% did not and 39% had an ambiguous placement.
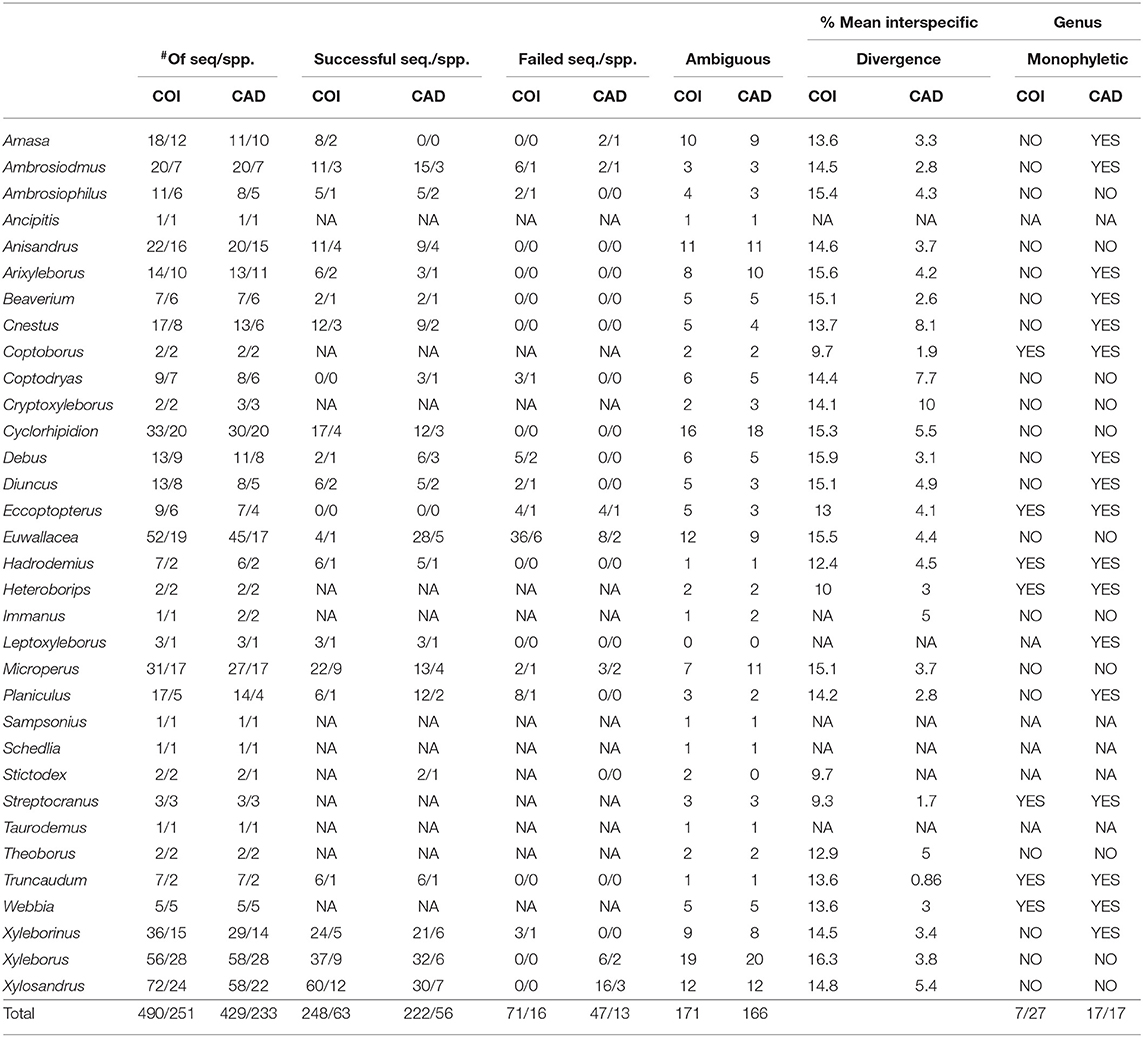
Table 1. Tree based identification: monophyly for xyleborine species and genera found in the neighbor-joining analyses.
The DNA sequence match identification did not perform as well as the tree based identification (Table 2). For both genes, best match of sequences performed the worst with 54–60% successful identifications but also with 35–40% misidentifications. For COI, the all-species barcode criterion was the most stringent and only 25 and 34% of identifications were successful at 3 and 9% thresholds respectfully. For COI, the best close match performed the best at 9% threshold with 55% successful identification as compared to 42% successful identification at a 3% threshold. The number of ambiguous and misidentified sequences was below 3%. At 2 and 3% thresholds for CAD, success with the best close match and all species barcode criteria was similar to COI however, ambiguous, and misidentification of sequence ranged from 4 to 49%.
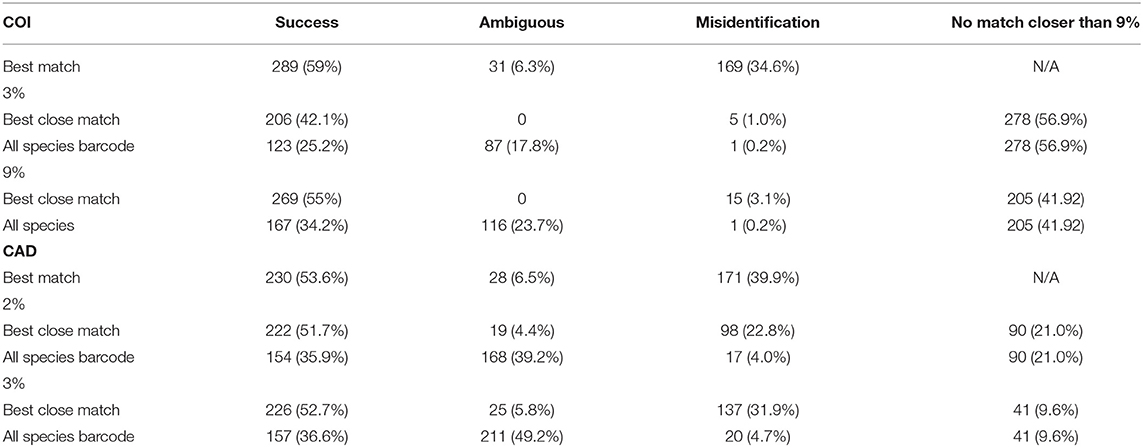
Table 2. Identification success with various similarity thresholds using DNA match method.
Average interspecific difference for congenerics ranged 9.3–16.3% for COI and 0.86–10% for CAD (Table 1). Most genera with <13.6% COI interspecific difference were monophyletic while the association between interspecific difference and monophyly was not consistent. Intraspecific differences averaged 8.34% (most <10%) and 1.26% (most <2%) for COI and CAD, respectively (Figure 1).
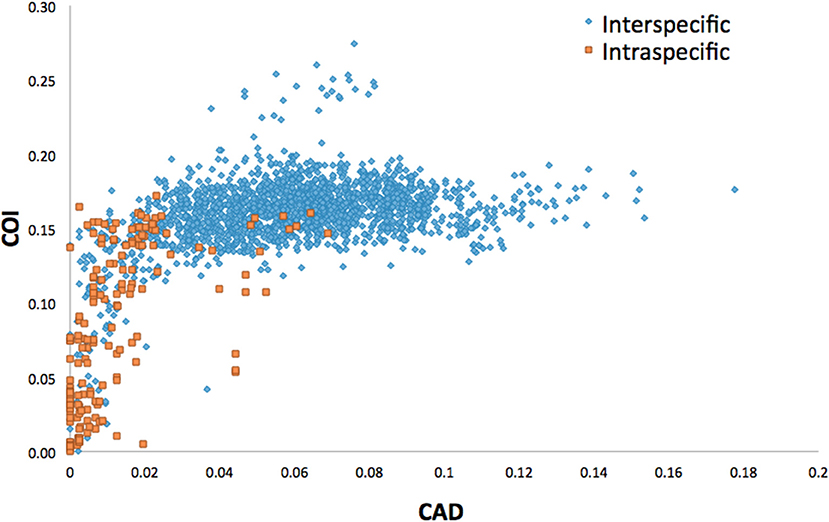
Figure 1. Pairwise uncorrected “p” intra- and interspecific distances for COI and CAD.
Taxon Discovery
Barcode gaps between interspecific and intraspecific differences for COI and CAD were not distinct (Figures 2, 3). These differences greatly overlapped between 12 and 17% for COI, and 1 and 3% for CAD (Figures 2, 3). TaxonDNA analyses found minimum of DNA cluster threshold violations at 9% for COI and 3% for CAD, respectively (Table 3). The ABGD analyses did not find any gaps in the distribution of sequence differences for both genes. The correspondence between ABGD groups and taxonomically recognized species was poor. The species were divided into 394 and 251 groups for COI (P = 0.00278) and CAD (P =0.0017), respectively which consisted of mostly over split species while in other cases different species were grouped together.
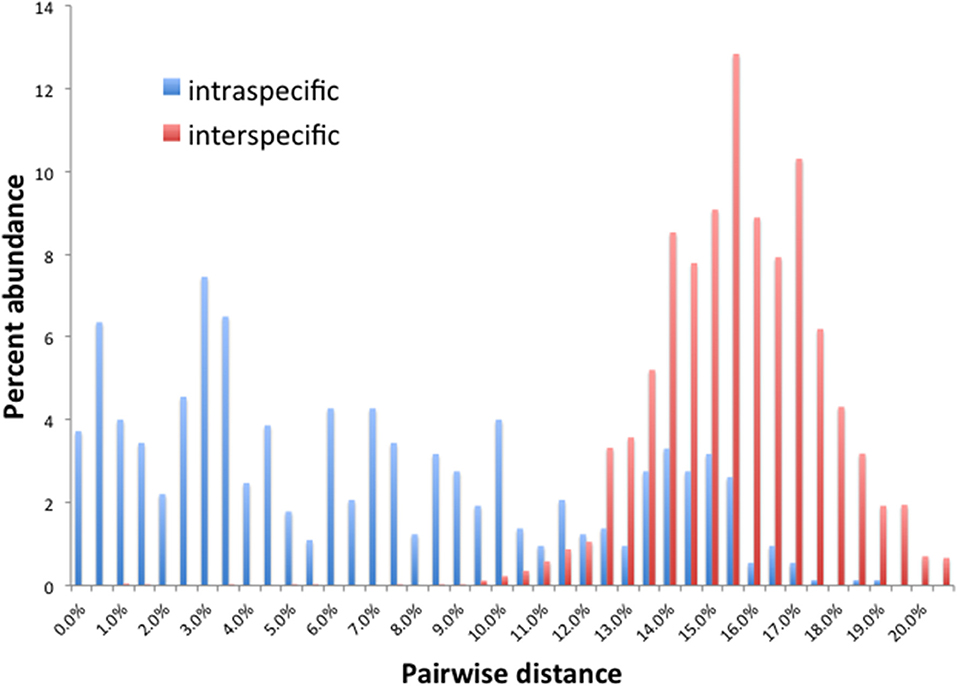
Figure 2. Frequency of uncorrected “p” intra- and interspecific COI distances.
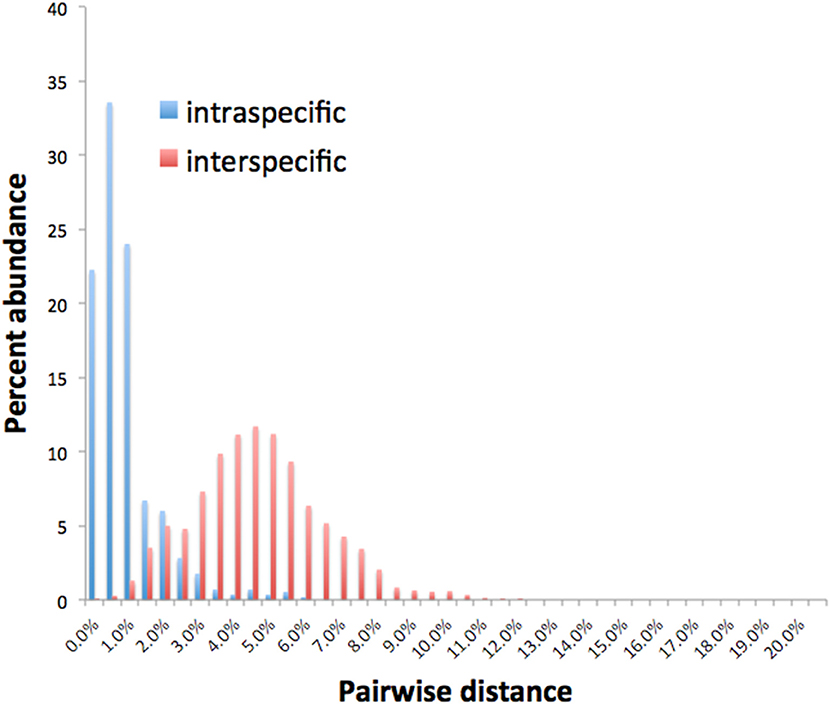
Figure 3. Frequency of uncorrected “p” intra- and interspecific CAD distances.
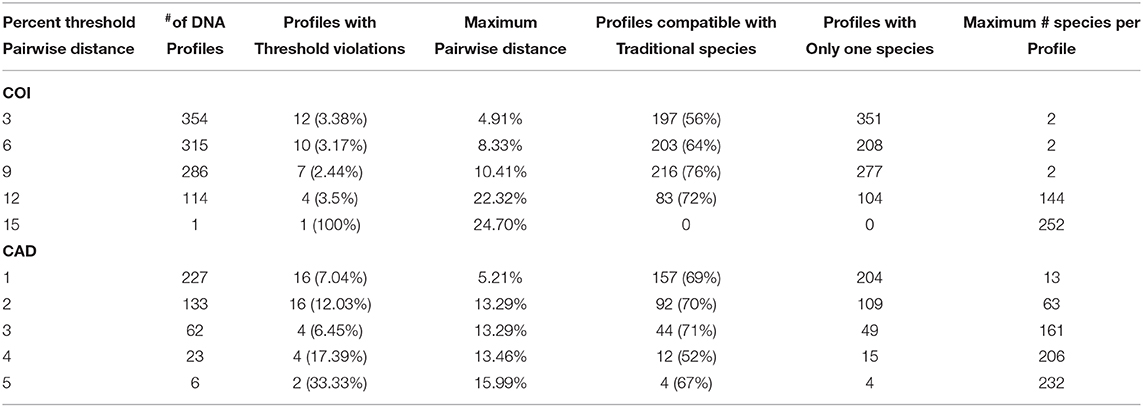
Table 3. DNA clusters based on a pair-wise distance thresholds for interspecific sequence comparisons.
Iterative Taxonomy
Parsimony analysis found one most parsimonious tree for 11 individuals of Ambrosiophilus which was represented by five A. osumiensis specimens (Figure 4). The clade containing all A. osumiensis individuals and two internal clades had bootstrap values above 95%. All other clades had lower bootstrap values (<70%). Percentage COI and CAD sequence difference among the A. osumiensis individuals range from 3.5–7.5 and 1.2–2.7%. Compared to its sister species A. subnepotulus, the COI sequence difference ranged 12.9–15.8% (A. subnepotulus CAD was missing from the dataset). Total interspecific COI and CAD sequence differences ranged 15.3–20.2 and 3.6–7.9%, respectfully. Considerable morphological differences occur among the clades of A. osumiensis. Such variation occurs in the shape of the pronotum; in the minute structure of the elytral declivity and pronotal disc; interstriae width; strial puncture size; number and size of tubercles on declivital interstriae 2; antennal club type (Hulcr et al., 2007); amount of elytral vestiture; and body size, with individuals differing up to 0.9 mm in length (0.5 mm or less is typical, Smith, unpublished).
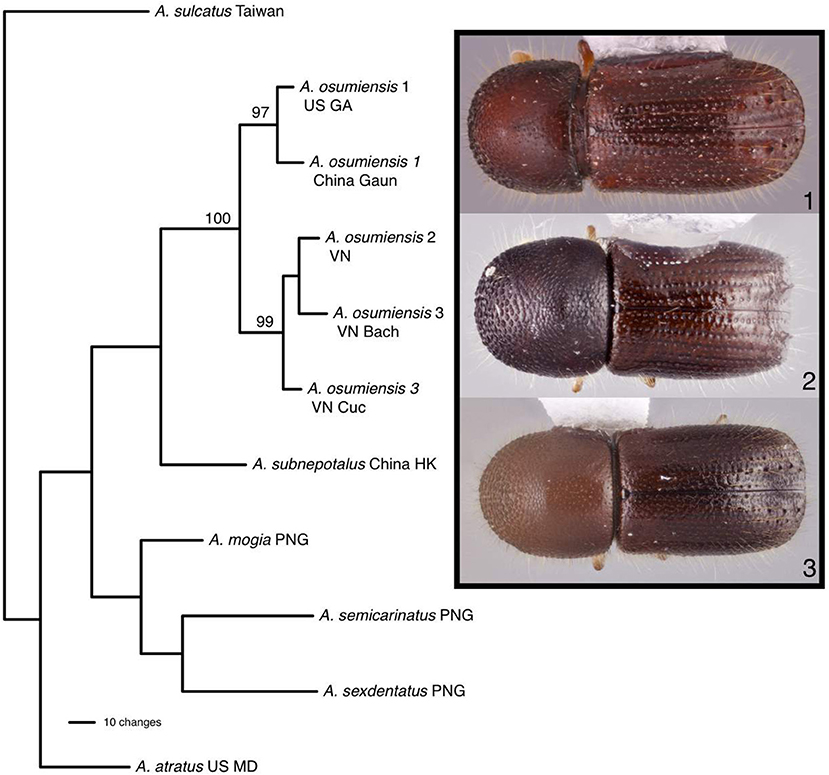
Figure 4. The one most parsimonious tree found for Ambrosiophilus species based on COI and CAD data. Numbers indicate bootstrap values. Habitus images depict morphological variation observed among tubercles of the elytra.
Parsimony analysis found 2,475 most parsimonious trees for 57 individuals of Euwallacea (Figure 5). Twenty species were included with seven species represented by more than one individual. Only Euwallacea fornicatus, Euwallacea interjectus, Euwallacea velatus, and Euwallacea wallacei were monophyletic. The COI and CAD sequence difference among the Euwallacea fornicatus individuals ranged 1.4–3.2 & 0.0–0.7% and between the sister-species E. kuroshio, 9.8–10.9 & 1.2–1.7%. Euwallacea interjectus was subdivided by two internal clades (A, B, & C) with bootstrap values above 95. Overall percentage COI and CAD sequence difference among the E. interjectus individuals ranged from 0.3–15.7 to 0.4–2.6%, respectfully. However, within clades A, B, and C 2.0, 2.7, and 0.3–3.7% COI sequence differences were observed, respectively. For CAD sequence differences only one comparison was observed for clade A (0.5%) while a range sequence differences (0.2–0.8%) was observed for clade C. Euwallacea andamanensis, Euwallacea funereus, Euwallacea similis, and Euwallacea semirudis, were not monophyletic and intraspecific COI and CAD sequence difference ranged 1.0–16.8 & 0.0–4.4%. Many Euwallacea species demonstrated little to no morphological variation in characters typically used to diagnose xyleborine species, particularly in the sculpturing of the elytral declivity.
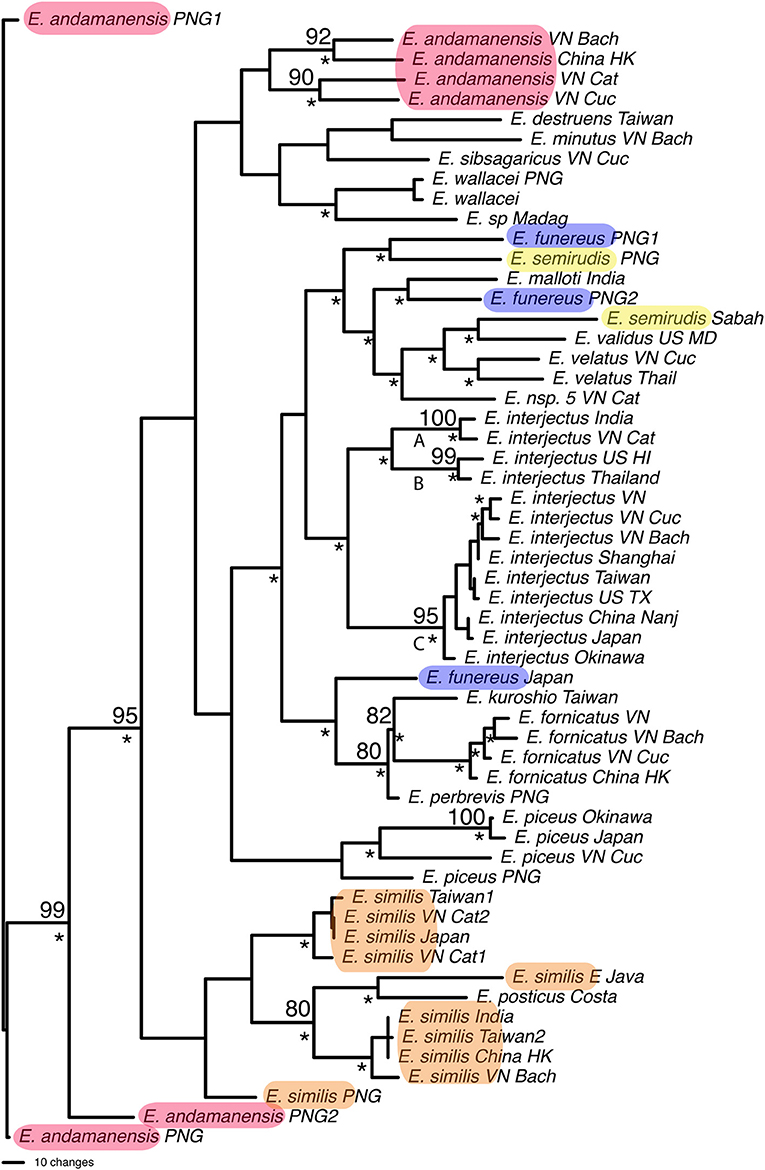
Figure 5. One of 2,475 most parsimonious trees found for Euwallacea species based on COI and CAD data. Numbers indicate bootstrap values. *Clade found in the strict consensus of most parsimonious trees. Highlighted species are not monophyletic.
Discussion
This study is the first to describe the application of COI and CAD DNA sequences for the identification and delimitation of xyleborine ambrosia beetles based on the largest sampling of species, to date, representing nearly all genera. The most striking observation is the prevalent high amount of COI intraspecific and interspecific pairwise differences which also was observed in earlier studies of limited xyleborine species (Figures 1–3) (Dole et al., 2010; Cognato et al., 2011). There are many reasons for high intraspecific COI sequence differences including unrecognized putative cryptic species, poorly defined species boundaries, effects of Wolbachia infection, and pseudogenes (Funk and Omland, 2003; Rubinoff et al., 2006). Most of the morphologically defined species for all genera exhibit 10–12% difference; thus, we contend that this observation is not the result of rampant cryptic speciation that one would expect given a 2% standard sequence divergence between species as promoted by the Barcode initiative (Hebert et al., 2003b; Ashfaq and Hebert, 2016). Cryptic species are evident at intraspecific differences ~13%, such as in the E. fornicatus species complex and other lineages (Gomez et al., 2018; Cognato et al., 2019; Smith et al., in preparation). Our sequence data shows no evidence of Wolbachia or pseudogenes. The uncommon haplodiploid mating system of Xyleborini may provide the best explanation for the high intraspecific COI sequence differences. The diploid female/haploid male sex-ratio is skewed on average 13:1 and ranges from 2:1 to 83:1 (French and Roeper, 1975; Beaver and Browne, 1979; Kirkendall, 1993; Cooperband et al., 2016; Castro et al., 2019). A female has an apparent greater chance of reproducing compared to diploid-diploid species because if unmated she lays a haploid egg which produces a male. She mates with her son to produce diploid daughters. Thereby, a single COI nucleotide mutation can be amplified to population levels in a short time (e.g., 13 female offspring each for 12 generations = ~23 × 1012 in a year, assuming all live and reproduce). Similarly high levels of intra- and interspecific COI differences have been observed among inbreeding bark-feeding scolytines with female skewed sex ratios (Kambestad et al., 2017). In comparison, the CAD intraspecific nucleotide differences were less for most pairwise intraspecific and interspecific comparisons at <2% and as most as 10%, respectively (Figure 3). It is unknown if these sequence differences are unexpectedly high like the COI differences because a comparable dataset of pairwise intraspecific values does not exist for diploid- diploid scolytine groups. However, these values may be as expected for single copy nuclear genes given that xyleborines may experience uncommon interfamilial matings which could maintain a minimal amount of gene flow within a species (Storer et al., 2017).
No one method clearly identified or delimited species. A barcode gap was not evident between intra- and interspecific COI and CAD sequences differences no matter the method used. While TaxonDNA analyses found DNA cluster thresholds (9% for COI and 3% for CAD) near or within the observed overlap of intra- and interspecific differences (Figures 2, 3), ABGD split most species into multiple groups. The tree-based NJ analysis performed better where monophyly and an approximate percentage DNA sequence difference helped to recognize species boundaries. Even better was the iterative approach highlighted for two genera where monophyly was rigorously tested in a parsimony framework and association between the clades and diagnostic morphological characters were examined by taxonomic experts (Figures 4, 5).
These authoritative DNA databases of >400 sequences of COI and CAD are stable foundations for the improved systematics of SE Asian xyleborine ambrosia beetles. However, they currently have limitations in the identification and delimitation of species as is the case for most other DNA identification databases (e.g., Ekrem et al., 2007). Correct determinations are limited to the included 161 of 316 known SE Asian species. Identifications will improve with time as the database grows with the addition of the missing known species. However, the addition of undescribed species is also expected as under-collected regions are sampled. Approximately 30% of the SE Asian xyleborine fauna remains undiscovered, so far (Smith, Beaver, Cognato, pers. observation). In addition, this study exposes taxonomic issues concerning polyphyly of some species and monophyletic species with variable morphology (see discussion below). Both situations suggest that further data is needed to test species limits. Delimitation and description of new and problematic species will be necessary in order to continue the accuracy of this identification database.
This study highlights three taxonomic scenarios that are expected as this database grows. (1) Ambrosiophilus osumiensis exemplifies the scenario of a monophyletic species with variable morphology (Figure 4). Differences in the number, position, and size of tubercles of the elytral declivity have been used to delimit xyleborine species (Hulcr et al., 2007; Wood, 2007). However some species were suspected as geographically variants of the same species (Hulcr and Cognato, 2013) and only recently the validity of some suspect species has been investigated in a phylogenetic context (e.g., Cognato et al., 2015; Gomez et al., 2018). The morphological variation illustrated for Ambrosiophilus osumiensis (Figure 4) was previously presumed diagnostic for three species (Ambrosiophilus metanepolulus, Ambrosiophilus nodulosus, Ambrosiophilus osumiensis) but given that only a maximum 7.5% COI and 2.7% CAD difference occurs among individuals, they are now considered one species (Smith et al., unpublished). Potentially these A. osumiensis morphotypes could represent valid species. Investigation of pre and/or post mating barriers in a phylogenetic context of a more widely sampled A. osumiensis individuals would aid in discerning the species validity of the A. osumiensis morphotypes (as in Cooperband et al., 2015). (2) Euwallacea exemplifies a situation where little to no morphological difference occurs among polyphyletic species or monophyletic species in which subclades exhibit >10–12% COI and 2–3% CAD difference (Figure 5). The E. fornicatus species complex has recently received much taxonomic attention given their pest status and that different lineages impart various levels of economic damage. Although qualitative diagnostic characters were not observed, consistent quantitative characters, and morphometric analysis were congruent with lineages that demonstrated >10% COI difference compared to each other (Stouthamer et al., 2017; Gomez et al., 2018; Smith et al., 2019). In addition, potential pre- and post-mating reproductive barriers and fidelity with different symbiotic fungal strains support the validity of the recognized species (Kasson et al., 2013; Cooperband et al., 2015, 2017). Cryptic species may riddle Euwallacea given the > 12% COI difference observed in species like E. interjectus and polyphyly of others (Figure 5). Their species status will remain unknown until detailed morphometric and biological analyses can be conducted. (3) A recently published study on Xyleborus glabratus demonstrates an ideal situation where monophyly, molecular difference, and morphological variation coalesce to support the recognition of new species (Cognato et al., 2019). Upon discovery in the field SMS and AIC initially hypothesized that the included specimens were X. glabratus but upon inspection in the laboratory species level morphological diagnostic characters of the elytral declivity were noted. These characters associated with monophyletic groups and >14% COI and >1.5% CAD differences (Figure 6). Two species were described and much morphological difference within X. glabratus was documented. A lineage of X. glabratus with 9% COI difference was not described as a species because of the lack of morphological diagnostic characters. This study and others (Cognato and Sun, 2007; Kambestad et al., 2017; Gomez et al., 2018) are examples of the decision process for the recognition of scolytine species in the context of morphology and molecular phylogenies.
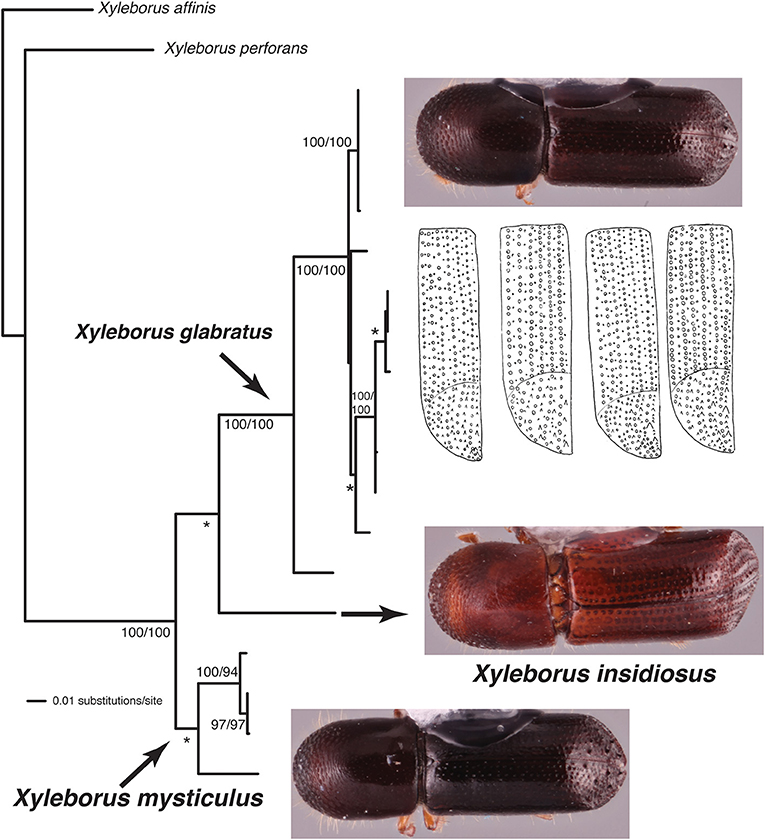
Figure 6. Phylogeny of Xyleborus glabratus redrawn from Cognato et al. (2019). Habitus images depict intra- and interspecific morphological variation observed among granules of the elytra. Numbers at nodes indicate parsimony/likelihood bootstrap values and *bootstrap values <70%.
Based on the presented DNA databases and the case studies, we recommend the following conservative guidelines for the identification and delimitation of xyleborine taxa. (1) Confident identifications demonstrate <3 and <1% pairwise uncorrected “p” COI and/or CAD difference between an unknown and a named barcode DNA sequence. (2) Delimitation of new species becomes more probable when pairwise uncorrected “p” COI and/or CAD differences increase beyond 10–12 and 2–3%, respectively. These values are most useful for the naïve diagnostician or when specimens lack additional morphological diagnostic characters (such as, larvae). Indeed, there are cases were species can be identified with confidence when pairwise difference exceeds these pairwise percentages, for example, X. glabratus (Cognato et al., 2019) or when species fall near (or below) expected interspecific pairwise percentages. These cases will usually be evident with a sample size that includes a representative genotypic variation for the species. When in doubt, a taxonomic expert should review these cases using systematic methodology.
The taxonomic experts for this study (SMS, RAB, and AIC) have ~75 years of combined experience in the identification and delimitation of scolytine species using both morphological and phylogenetic inference. Their initial morphologically-based (null) species hypotheses (i.e., identifications) were informed by this experience, the study of type specimens, and original species descriptions of all SE Asian species. Yet for several species, for example A. osumiensis, they reassessed the morphology-based identifications based on the COI/CAD phylogeny. In some cases this resulted in a broader morphological-based species concept and in other cases, the delimitation of new species (as in, Cognato et al., 2019). This iterative process [similar to reciprocal illumination (Hennig, 1966)] treats species as hypotheses of evolutionary lineages, which are tested with morphological, phylogenetic, and/or molecular evidence (Hey, 2006; Yeates et al., 2011). At this stage most of the species included in this study have diagnostic morphological characters, are monophyletic, and/or demonstrate >10 and >2% sequence difference for COI and CAD. The inclusion of more specimens and DNA sequence of different genes in subsequent phylogenetic studies will test species limits and likely improve the delimitation of xyleborine species especially for the highlighted problematic species (e.g., Cognato et al., 2020).
Involvement of taxonomic experts during the process of creating a DNA database for species identification is critical for a solid taxonomic foundation. Without their initial identification, followed by tests of and deliberation of species boundaries, the database would be incomplete and misleading; that is, DNA barcodes identified only to higher taxa or that are misidentified to species. For example, in the BoLD public database ~10% of the ~7000 Scolytinae specimens are not identified to species and ~6% are only identified to subfamily (http://v4.boldsystems.org, accessed 5 September 2019). These values are relatively good given that less than half of the sequences in Genbank (including BoLD data) are named to species (Page, 2016). The accuracy of species identifications in BoLD is difficult to assess because either vouchers are not imaged or the image quality does not allow for species identification. Also the specimen identifiers are not indicated and if the identifier is named then their taxonomic experience is unrecorded. The citations of the authoritative reference(s) used to make species identifications are mostly lacking. Although the BoLD system allows for the revision of identifications, the above missing information hampers peer-review of species names associated with DNA barcodes. Peer- review of taxonomic identifications is critical to the scientific process inherent in species identification. For example, relying on only a 2–3% percent sequence divergence standard for estimating species diversity, Ashfaq and Hebert (2016) suggested an unexpectedly high estimate of cryptic arthropod pest species. This estimate ignored the accuracy of the species determinations, limited sample size of COI haplotypes, and the biology of the pest. In one case, Xylosandrus crassiusculus, our data clearly shows that it is a highly variable (i.e., COI haplotypes) monophyletic species and not three potential cryptic species (Ashfaq and Hebert, 2016). Taking these steps to improve species identification and verification of species in current global databases will improve accuracy of the DNA barcodes (Wu et al., 2017) and applications to biodiversity assessment or the testing of ecological hypotheses (e.g., Caesar et al., 2006; Cognato and Caesar, 2006; Miller et al., 2016).
The initial DNA barcoding movement predicted an end to traditional taxonomy (Hebert et al., 2003a; Sperling, 2003; Smith, 2005; Will et al., 2005; Brower, 2006) and along with a call for DNA taxonomy, the taxonomist's role in these enterprises was uncertain (Tautz et al., 2003; Blaxter, 2004). In 16 years, DNA barcoding publications have proliferated and millions of DNA barcodes have been generated (Taylor and Harris, 2012; DeSalle and Goldstein, 2019). Despite this overwhelming zeal for barcoding, taxonomists remained relevant and advocates of DNA barcoding have welcomed more interaction with taxonomists (e.g., Miller, 2007; Packer et al., 2009a; Miller et al., 2016; Zahiri et al., 2017). For example, DNA barcoding funding helped stop a decline in traditional taxonomy in Canada but productivity had not returned to pre-decline levels of 1980 (Packer et al., 2009a). As already acknowledged, thousands of taxonomists are needed to describe newly collected morphological distinct species as well as species discovered as the result of DNA barcoding (Wheeler et al., 2012). Although taxonomists' involvement in DNA barcoding studies is essential for a reliable identification system and improved understanding of biodiversity, the monetary support future taxonomists is uncertain. For example, the recent $180 million global investment in DNA barcoding aims to discover two million new species; however, the number of traditional taxonomists employed to help with this endeavor is not apparent (BioScan, https://ibol.org/, accessed 16 September 2019; Pennisi, 2019). One would hope that as with past funding of DNA barcoding, this initiative will have a positive impact on training taxonomists and taxonomic publications (Packer et al., 2009a). If funding has not been allocated for taxonomists, then $180 million will only result in a backlog of “DNA barcode species” that will need further study and potentially formal description (Pinheiro et al., 2019).
Creation of a DNA database for species identification is not trivial. It relies on authoritatively identified specimens for use in the generation of DNA barcodes. Misidentified specimens result in a misleading DNA identification tool. For this reason, taxonomists should be part of barcoding ventures from beginning to end so to establish null hypotheses of species boundaries and to interpret non-monophyletic species and/or lineages with unexpected high sequence differences deemed as “DNA barcode species.” The taxonomist could then quickly address these “DNA barcode species” by comparison of morphology or inclusion in a rigorously reconstructed multi-gene phylogeny so to test the “DNA barcode species” and to describe validated species. This study exemplifies this approach. Through an iterative process we tested our initial morphologically based species identifications with DNA barcodes (sequences from COI and CAD in this case) and then re-examined our identifications with additional specimens, morphological characters, and additional genes. Some “DNA barcode species” were validated and some were synonymized with known species. We will not contribute to the taxonomic impediment because this DNA barcode project occurred within the context of a traditional taxonomic review of the SE Asian xyleborine fauna and descriptions of new species will soon be published (Smith et al., in preparation). We believe that DNA barcodes are best delivered as an outcome of taxonomic reviews, revisions, or monographs. Indeed one could approach the discovery and description of new species with the DNA barcodes first followed by morphological and phylogenetic study (Puillandre et al., 2012; Kekkonen and Hebert, 2014; Miller et al., 2016; DeSalle and Goldstein, 2019), especially in cases where a taxonomic expert does not exist for the higher taxon. But it could take years for an expert to test the validity of the “DNA barcode species” if she is not vested in the initial project (Fontaine et al., 2012). Thus, it is prudent to include the taxonomic expert throughout a DNA barcoding project because (1) the resulting DNA barcodes will be tied to authoritatively identified species which increases the scientific value of future biodiversity research, (2) new species will be described faster (e.g., <4 years for species discovered in this study), and (3) other taxonomic tools and information may be produced (e.g., illustrated morphological keys and distribution maps). If a taxonomist for a particular taxon does not exist, then the barcoding project should take the opportunity to train an expert for the orphaned taxon through the employment of existing taxonomists as mentors of the new generation (as in, Rodman and Cody, 2003). By adopting a modern systematic approach, one that analyses all available data in phylogenetic context so to improve taxonomy (Will et al., 2005; Yeates et al., 2011; DeSalle and Goldstein, 2019), the barcoding initiative could make a more meaningful impact on our understanding of biodiversity.
Data Availability Statement
The data generated for this study were deposited in GenBank (Supplementary Table 1) and NEXUS files can be found at www.canr.msu.edu/hisl/.
Author Contributions
AC conceived the study and wrote the initial manuscript. AC, SMS, YL, JH, HK, C-SL, TP, SS, and WS collected or provided access to specimens. SMS, RB, and AC identified specimens. GS and AC generated and analyzed sequence data. AC, JH, BJ, and SMS revised drafts of the manuscript. AC and JH funded various aspects of this research. All authors read, commented on, and approved the final version of the manuscript.
Funding
This research was supported by USDA-APHIS Cooperative Agreement Award (16-8130-0666-CA), NSF (DEB-1256663), NSF-PEET (DEB-0328920), USDA Forest Service Early Detection Rapid Response program cooperative agreement (11-DG-11420004-257) and the Ernst Mayr Travel Grant in Animal Systematics (Harvard University) to AC. In addition grants from the JSPS KAKENHI #17H03831 to HK, USDA-APHIS Farm Bill section 10007, the USDA Forest Service, the National Science Foundation and the Florida Forest Service to JH supported various aspects of this study.
Disclaimer
The authors are solely responsible for the writing of this paper.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the curators of the following institutions who allowed access to or the loan of type specimens from the collections in their care: Dirk Ahrens (ZFMK), Max Barclay (NHML), Lutz Behne (SDEI), Johannes Bergsten (SMNH), James Boone (BPBM), Michel Brancucci and Isabelle Zürcher-Pfander (NHMB), Lourdes Chamorro (NMNH), Chris Grinter (CASC), Jiri Hájek (MNHP), Matthias Hartmann (NKME), Martin Husemann and Thure Dalsgaard (UHZM), Pol Limbourg (IRSNB), Katrina Menard (OMNH), Otto Merkl (HNHM), Hélène Perrin (MNHN), Roberto Poggi and Maria Tavano (MCG), Harald Schillhammer (NHMW), Kyle Schnepp (FSCA), Wichai Srisuka (QSBG), David Szymroszczyk (MIZ), Joachim Willers (MFNB), Hiraku Yoshitake (NIAES).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2020.00027/full#supplementary-material
Supplementary Figure 1. Neighbor-joining tree based on pairwise uncorrected “p” distances for COI for 490 Xyleborini specimens.
Supplementary Figure 2. Neighbor-joining tree based on pairwise uncorrected “p” distances for CAD for 429 Xyleborini specimens.
Supplementary Table 1. Xyleborini specimens included in this study.
References
Adamski, D., and Miller, S. E. (2015). Two new yellow-banded sister species of Syntomaula Meyrick (Lepidoptera: Gelechioidea: Cosmopterigidae) from Papua New Guinea associated with Rubiaceae. J. Lepidopter. Soc. 69, 307–316. doi: 10.18473/lepi.69i4.a6
Ashfaq, M., and Hebert, P. D. N. (2016). DNA barcodes for bio-surveillance: regulated and economically important arthropod plant pests. Genome 59, 933–945. doi: 10.1139/gen-2016-0024
Barrett, R. D. H., and Hebert, P. D. N. (2005). Identifying spiders through DNA barcodes. Can. J. Zool. 83, 481–491. doi: 10.1139/z05-024
Beaver, R. A., and Browne, F. G. (1979). The scolytidae and platypodidae (Coleoptera) of Penang, Malaysia. Orient. Insects 12, 575–624. doi: 10.1080/00305316.1978.10432538
Blaxter, M. L. (2004). The promise of DNA taxonomy. Philos. Trans. R. Soc. B. 359, 669–679. doi: 10.1098/rstb.2003.1447
Boland, J. M. (2016). The impact of an invasive ambrosia beetle on the riparian habitats of the Tijuana River Valley, California. Peer J. 4:e2141. doi: 10.7717/peerj.2141
Brockerhoff, E. G., and Liebhold, A. M. (2017). Ecology of forest insect invasions. Biol. Invasions. 19, 1–19. doi: 10.1007/s10530-017-1514-1
Brower, A. V. Z. (2006). Problems with DNA barcodes for species delimitation: ‘Ten species’ of Astraptes fulgerator reassessed (Lepidoptera: Hesperiidae). System. Biodivers. 4, 127–132 doi: 10.1017/S147720000500191X
Brower, A. V. Z. (2010). Alleviating the taxonomic impediment of DNA barcoding and setting a bad precedent: names for ten species of ‘Astraptes fulgerator’ (Lepidoptera: Hesperiidae: Eudaminae) with DNA-based diagnoses. System. Biodivers. 8, 485–491. doi: 10.1080/14772000.2010.534512
Browne, F. G. (1961). The biology of Malayan Scolytidae and Platypodidae. Malayan Forest Records 22, 1–255.
Caesar, R. M., Sörensson, M., and Cognato, A. I. (2006). Integrating DNA data and traditional taxonomy to streamline biodiversity assessment: an example from edaphic beetles in the Klamath ecoregion, California, USA. Diversity Distribut. 12, 483–489. doi: 10.1111/j.1366-9516.2006.00237.x
Carrillo, D., Cruz, L., Kendra, P., Narvaez, T., Montgomery, W., Monterroso, A., et al. (2016). Distribution, pest status and fungal associates of Euwallacea nr. fornicatus in Florida avocado groves. Insects 7:55. doi: 10.3390/insects7040055
Castro, J., Smith, S. M., Cognato, A. I., Lanfranco, D., Martinez, M., and Guachambala, M. (2019). Life cycle and development of Coptoborus ochromactonus Smith and Cognato (Coleoptera: Curculionidae: Scolytinae). J. Econ. Entomol. 112, 729–735. doi: 10.1093/jee/toy403
Chang, H., Liu, Q., Hao, D., Liu, Y., An, Y., and Yang, X. (2014). DNA barcodes and molecular diagnostics for distinguishing introduced Xyleborus (Coleoptera: Scolytinae) species in China. Mitochondr. DNA 25, 63–69. doi: 10.3109/19401736.2013.779260
Cognato, A. I. (2006). Standard percent DNA sequence difference does not predict species boundaries. J. Econ. Entomol. 99, 1037–1045. doi: 10.1093/jee/99.4.1037
Cognato, A. I., and Caesar, R. M. (2006). Will DNA barcoding advance efforts to conserve biodiversity more efficiently than traditional taxonomic methods?: Introduction. Front. Ecol. Environ. 4, 268–269. doi: 10.1890/1540-9295(2006)004[0268:WDBAET]2.0.CO;2
Cognato, A. I., Hoebeke, E. R., Kajimura, H., and Smith, S. M. (2015). History of the exotic ambrosia beetles Euwallacea interjectus and Euwallacea validus (Coleoptera: Curculionidae: Xyleborini) in the United States. J. Econ. Entomol. 108, 1129–1135. doi: 10.1093/jee/tov073
Cognato, A. I., Hulcr, J., Dole, S. A., and Jordal, B. H. (2011). Phylogeny of haplo–diploid, fungus-growing ambrosia beetles (Curculionidae: Scolytinae: Xyleborini) inferred from molecular and morphological data. Zool. Scr. 4, 174–186. doi: 10.1111/j.1463-6409.2010.00466.x
Cognato, A. I., Jordal, B. H., and Rubinoff, D. (2018). Ancient “Wanderlust” leads to diversification of endemic hawaiian Xyleborus species (Coleoptera: Curculionidae: Scolytinae). Insect Syst. Diver. 2:1. doi: 10.1093/isd/ixy005
Cognato, A. I., Smith, S.M., and Beaver, R. A. (2020). Two new genera of Oriental xyleborine ambrosia beetles (Coleoptera, Curculionidae: Scolytinae). Zootaxa 4722, 540–554. doi: 10.11646/zootaxa.4722.6.2
Cognato, A. I., Smith, S. M., Li, Y., Pham, T. H., and Hulcr, J. (2019). Genetic variability among native Xyleborus glabratus Eichhoff populations native to Southeast Asia and the description of two related species. J. Econ. Entomol. 112, 1274–1284. doi: 10.1093/jee/toz026
Cognato, A. I., and Sun, J. H. (2007). DNA based cladograms augment the discovery of a new Ips species from China (Coleoptera: Curculionidae: Scolytinae). Cladistics 23, 539–551. doi: 10.1111/j.1096-0031.2007.00159.x
Cooperband, M., Cossé, A., Stouthamer, R., Carrillo, D., and Jones, T. (2017). Attractants of ambrosia beetles in the Euwallacea fornicatus species complex. Pheromones and other semiochemicals in integrated production. IOBC-WPRS Bull. 126, 89–92. doi: 10.7287/peerj.preprints.3175
Cooperband, M. F., Stouthamer, R., Carillo, D., Eskalen, A., Thibault, T., Cossé, A. A., et al. (2016). Biology of two members of the Euwallacea fornicatus species complex (Coleoptera: Curculionidae: Scolytinae), recently invasive in the U.S.A., reared on an ambrosia beetle artificial diet. Agric. For. Entomol. 18, 223–237. doi: 10.1111/afe.12155
Cooperband, M. F., Stouthamer, R., Carrillo, D., and Cossé, A. (2015). “A crossing study to evaluate invasive Euwallacea near fornicatus populations in California and Florida,” in 26th USDA Interagency Research Forum on Invasive Species, Vol. 26, K. A. McManus and K. W. Gottschalk (Washington, DC: US Forest Service), 62.
Danforth, B. N., Fang, J., and Sipes, S. D. (2006). Analysis of family level relationships in bees (Hymenoptera: Apiformes) using 28S and two previously unexplored nuclear genes: CAD and RNA polymerase II. Mol. Phylogenet. Evol. 39, 358–372. doi: 10.1016/j.ympev.2005.09.022
DeSalle, R., Egan, M. G., and Siddall, M. (2005). The unholy trinity: taxonomy, species delimitation and DNA barcoding. Philos. Trans. R. Soc. B. 360, 1905–1916. doi: 10.1098/rstb.2005.1722
DeSalle, R., and Goldstein, P. (2019). Review and Interpretation of Trends in DNA Barcoding. Front. Ecol. Evol. 7:302. doi: 10.3389/fevo.2019.00302
Dodelin, B. (2018). Espèce Invasive Nouveau Pour la Faune de France Scolyte Cyclorhipidion fukiense Installé en Europe. Available online at: https://entomodata.wordpress.com/2018/04/24/cyclorhipidion-fukiense-installe-en-europe/
Dole, S. A., Jordal, B. H., and Cognato, A. I. (2010). Polyphyly of Xylosandrus reitter inferred from nuclear and mitochondrial genes (Coleoptera: Curculionidae: Scolytinae). Mol. Phylogenet. Evol. 54, 773–782. doi: 10.1016/j.ympev.2009.11.011
Ebach, M. C., and Holdrege, C. (2005). More taxonomy, not DNA barcoding. Bioscience 55, 822–823. doi: 10.1641/0006-3568(2005)055[0823:MTNDB]2.0.CO;2
Ekrem, T., Willassen, E., and Stur, E. (2007). A comprehensive DNA sequence library is essential for identification with DNA barcodes. Mol. Phylogenet. Evol. 43, 530–542. doi: 10.1016/j.ympev.2006.11.021
Eskalen, A., Gonzalez, A., Wang, D. H., Twizeyimana, M., Mayorquin, J. S., and Lynch, S. C. (2012). First report of a Fusarium sp. and its vector tea shot hole borer (Euwallacea fornicatus) causing Fusarium dieback on avocado in California. Plant Dis. 96:1070. doi: 10.1094/PDIS-03-12-0276-PDN
Felsenstein, J. (1985). Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x
Folmer, O., Black, M., Hoeh, W., Lutz, R., and Vrijenhoek, R. (1994). DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 3, 294–299.
Fontaine, B., Perrard, A., and Bouchet, P. (2012). 21 years of shelf life between discovery and description of new species. Curr. Biol. 22:R944. doi: 10.1016/j.cub.2012.10.029
Foster, B. T., Cognato, A. I., and Gold, R. E. (2004). DNA-based identification of the eastern subterranean termite, Reticulitermes flavipes (Isoptera: Rhinotermitidae). J. Econ. Entomol. 97, 95–101. doi: 10.1093/jee/97.1.95
Foster, P. G., Bergo, E. S., Bourke, B. P., Oliveira, T. M. P., Nagaki, S. S., Sant'Ana, D. C., et al. (2013). Phylogenetic analysis and DNA-based species confirmation in Anopheles (Nyssorhynchus). PLoS ONE 8:e54063. doi: 10.1371/journal.pone.0054063
French, J. R., and Roeper, R. A. (1975). Studies on the biology of the ambrosia beetle Xyleborus dispar (F) (Coleoptera: Scolytidae). Zeitschr. Angew. Entomol. 78, 241–247. doi: 10.1111/j.1439-0418.1975.tb04178.x
Funk, D. J., and Omland, K. E. (2003). Species-level paraphyly and polyphyly: Frequency, causes, and consequences, with insights from animal mitochondrial DNA. Annu. Rev. Ecol. Evol. Syst. 34, 397–423. doi: 10.1146/annurev.ecolsys.34.011802.132421
Gibbs, J. (2018). DNA barcoding a nightmare taxon: assessing barcode index numbers and barcode gaps for sweat bees. Genome 61, 21–31. doi: 10.1139/gen-2017-0096
Gohli, J., Kirkendall, L. R., Smith, S. M., Cognato, A. I., Hulcr, J., and Jordal, B. H. (2017). Biological factors contributing to bark and ambrosia beetle species diversification. Evolution 71, 1258–1272. doi: 10.1111/evo.13219
Gohli, J., Selvarajah, T., Kirkendall, L. R., and Jordal, B. H. (2016). Globally distributed Xyleborus species reveal recurrent intercontinental dispersal in a landscape of ancient worldwide distributions. BMC Evol. Biol. 16:37. doi: 10.1186/s12862-016-0610-7
Gomez, D. F., Skelton, J., Steininger, M. S., Stouthamer, R., Rugman-Jones, P., Sittichaya, W., et al. (2018). Species delineation within the Euwallacea fornicatus complex revealed by morphometric and phylogenetic analyses. Insect Syst. Diver. 2, 1–11. doi: 10.1093/isd/ixy018
Haack, R. A., and Rabaglia, R. J. (2013). “Exotic bark and ambrosia beetles in the USA: potential and current invaders” in Potential Invasive Pests of Agricultural Crop Species, ed J. Pena (Wallingford: CAB International), 48–74. doi: 10.1079/9781845938291.0048
Hebert, P. D. N., Cywinska, A., Ball, S. L., and deWaard, J. R. (2003a). Biological identifications through DNA barcodes. Proc. R. Soc. London Ser. B. 270, 313–321. doi: 10.1098/rspb.2002.2218
Hebert, P. D. N., Penton, E. H., Burns, J. M., Janzen, D. H., and Hallwachs, W. (2004). Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proc. Natl. Acad. Sci. U.S.A. 101, 14812–14817. doi: 10.1073/pnas.0406166101
Hebert, P. D. N., Ratnasingham, S., and deWaard, J. R. (2003b). Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proc. R. Soc. London Ser. B. 270, S96–S99. doi: 10.1098/rsbl.2003.0025
Hennig, W. (1966). Phylogenetic Systematics. Translated by D. D. Davis and R. Zangerl. Urbana: University of Illinois Press.
Hey, J. (2006). On the failure of modern species concepts. Trends Ecol. Evol. 21, 447–450. doi: 10.1016/j.tree.2006.05.011
Hoebeke, E. R., Rabaglia, R. J., KníŽek, M., and Weaver, J. S. (2018). First records of Cyclorhipidion fukiense (Eggers) (Coleoptera: Curculionidae: Scolytinae: Xyleborini), an ambrosia beetle native to Asia, in North America. Zootaxa 4394, 243–250. doi: 10.11646/zootaxa.4394.2.7
Hughes, M. A., Riggins, J. J., Koch, F. H., Cognato, A. I., Anderson, C., Formby, J. P., et al. (2017). No rest for the laurels: symbiotic invaders causes unprecedented damage to southern USA forests. Biol. Invasions 19, 2143–2157. doi: 10.1007/s10530-017-1427-z
Hulcr, J., Atkinson, T. H., Cognato, A. I., Jordal, B. H., and McKenna, D. D. (2015). “Morphology, taxonomy and phylogenetics of bark beetles” in Bark Beetles: Biology and Ecology of Native and Invasive Species, eds F. E. Vega and R. W. Hofstetter (San Diego, CA: Elsevier Inc.), 41–84. doi: 10.1016/B978-0-12-417156-5.00002-2
Hulcr, J., Black, A., Prior, K., Chen, C.-Y., and Li, H. F. (2017). Studies of ambrosia beetles (Coleoptera: Curculionidae) in their native ranges help predict invasion Impact. Fla. Entomol. 100, 257–261. doi: 10.1653/024.100.0219
Hulcr, J., and Cognato, A. I. (2013). Xyleborini of New Guinea: A Taxonomic Monograph. Thomas Say Publications in Entomology. Maryland: Entomological Society of America.
Hulcr, J., Dole, S. A., Beaver, R. A., and Cognato, A. I. (2007). Cladistic review of generic taxonomic characters in Xyleborini (Coleoptera: Curculionidae: Scolytinae). Syst. Entomol. 32, 568–584. doi: 10.1111/j.1365-3113.2007.00386.x
Hulcr, J., Skelton, J., Johnson, A. J., Li, Y., and Jusino, M. A. (2018). Invasion of an inconspicuous ambrosia beetle and fungus may alter wood decay in Southeastern North America. PeerJ. Preprints 6:e27334v1. doi: 10.7287/peerj.preprints.27334v1
Jordal, B. H., Beaver, R. A., and Kirkendall, L. R. (2001). Breaking taboos in the tropics: incest promotes colonization by wood-boring beetles. Glob. Ecol. Biogeogr. 10, 345–357. doi: 10.1046/j.1466-822X.2001.00242.x
Jordal, B. H., and Cognato, A. I. (2012). Molecular phylogeny of bark and ambrosia beetles reveals multiple origins of fungus farming during periods of global warming. BMC Evol. Biol. 12:133. doi: 10.1186/1471-2148-12-133
Jordal, B. H., and Kambestad, M. (2014). DNA barcoding of bark and ambrosia beetles reveals excessive NUMTs and consistent east-west divergence across Palearctic forests. Mol. Ecol. Resour. 14, 7–17. doi: 10.1111/1755-0998.12150
Jordal, B. J., Sequeira, A., and Cognato, A. I. (2011). The age and phylogeny of wood boring weevils and the origin of subsociality. Mol. Phylogenet. Evol. 59, 708–724. doi: 10.1016/j.ympev.2011.03.016
Jukes, T. H., and Cantor, C. R. (1969). “Evolution of protein molecules” in Mammalian Protein Metabolism, ed H. N. Munro (New York, NY: Academic Press), 21–132. doi: 10.1016/B978-1-4832-3211-9.50009-7
Kambestad, M., Kirkendall, L. R., Knutsen, I. L., and Jordal, B. H. (2017). Cryptic and pseudo-cryptic diversity in the world's most common bark beetle—Hypothenemus eruditus. Organ. Divers. Evol. 17, 633–652. doi: 10.1007/s13127-017-0334-6
Kasson, M. T., O'Donnell, K., Rooney, A. P., Sink, S., Ploetz, R. C., Ploetz, J. N., et al. (2013). An inordinate fondness for Fusarium: phylogenetic diversity of fusaria cultivated by ambrosia beetles in the genus Euwallacea on avocado and other plant hosts. Fungal Genet. Biol. 56, 147–157. doi: 10.1016/j.fgb.2013.04.004
Kekkonen, M., and Hebert, P. D. N. (2014). DNA barcode-based delineation of putative species: efficient start for taxonomic workflows. Mol. Ecol. Resour. 14, 706–715. doi: 10.1111/1755-0998.12233
Kimura, M. (1980). A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J. Mole. Evol. 16, 111–120. doi: 10.1007/BF01731581
Kirkendall, L. R. (1993). “Ecology and evolution of biased sex ratios in bark and ambrosia beetles” in Evolution and Diversity of Sex Ratios in Insects and Mites, eds. D. L. Wrensch and M. A. Ebbert (New York, NY: Chapman and Hall), 235–345. doi: 10.1007/978-1-4684-1402-8_8
Kirkendall, L. R., and Faccoli, M. (2010). Bark beetles and pinhole borers (Curculionidae, Scolytinae, Platypodinae) alien to Europe. Zookeys 56, 227–251. doi: 10.3897/zookeys.56.529
Knížek, M. (2011). “Scolytinae” in Catalogue of Palaearctic Coleoptera, Vol. 7. Curculionoidea I, eds I. Löbl and A. Smetana (Stenstrup, Apollo Books), 204–251.
Lait, L. A., and Hebert, P. D. N. (2018). Phylogeographic structure in three North American tent caterpillar species (Lepidoptera: Lasiocampidae): Malacosoma americana, M. californica, and M. disstria. PeerJ. 6:e4479. doi: 10.7717/peerj.4479
Magnacca, K. N., and Brown, M. J. F. (2010). Mitochondrial heteroplasmy and DNA barcoding in Hawaiian Hylaeus (Nesoprosopis) bees (Hymenoptera: Colletidae). BMC Evol. Biol. 10:174. doi: 10.1186/1471-2148-10-174
Meier, R., Shiyang, K., Vaidya, G., and Ng, P. K. L. (2006). DNA barcoding and taxonomy in Diptera: a tale of high intraspecific variability and low identification success. Syst. Biol. 55, 715–728. doi: 10.1080/10635150600969864
Meurisse, N., Rassati, D., Hurley, B. P., Brockerhoff, E. G., and Haack, R. A. (2018). Common pathways by which non-native forest insects move internationally and domestically. J. Pest Sci. (2004) 92, 13–27. doi: 10.1007/s10340-018-0990-0
Miller, S. E. (2007). DNA barcoding and the renaissance of taxonomy. Proc. Natl Acad. Sci. U.S.A. 104, 4775–4776. doi: 10.1073/pnas.0700466104
Miller, S. E., Hausmann, A., Hallwachs, W., and Janzen, D. H. (2016). Advancing taxonomy and bioinventories with DNA barcodes. Phil. Trans. R. Soc. B. 371:20150339. doi: 10.1098/rstb.2015.0339
Moulton, M. J., Song, H., and Whiting, M. F. (2010). Assessing the effects of primer specificity on eliminating numt coamplification in DNA barcoding: a case study from Orthoptera (Arthropoda: Insecta). Mol. Ecol. Resour. 10, 615–627. doi: 10.1111/j.1755-0998.2009.02823.x
Packer, L., Gibbs, J., Sheffield, C. S., and Hanner, R. (2009b). DNA barcoding and the mediocrity of morphology. Mol. Ecol. Resour. 9, 42–50. doi: 10.1111/j.1755-0998.2009.02631.x
Packer, L., Grixti, J. C., Roughley, R. E., and Hanner, R. (2009a). The status of taxonomy in Canada and the impact of DNA barcoding. Can. J. Zool. 87, 1097–1110. doi: 10.1139/Z09-100
Page, R. D. M. (2016). DNA barcoding and taxonomy: dark taxa and dark texts. Phil. Trans. R. Soc. B. 371:20150334. doi: 10.1098/rstb.2015.0334
Pennisi, E. (2019). $180 million DNA ‘barcode’ project aims to discover 2 million new species. Science doi: 10.1126/science.aay2877. [Epub ahead of print].
Pinheiro, H. T., Moreau, C. S., Daly, M., and Rocha, L. A. (2019). Will DNA barcoding meet taxonomic needs? Science 365, 873–874. doi: 10.1126/science.aay7174
Prendini, L. (2005). Comment on “Identifying spiders through DNA barcodes”. Can. J. Zool. 83, 498–504. doi: 10.1139/z05-025
Puillandre, N., Lambert, A., Brouillet, S., and Achaz, G. (2012). ABGD, automatic Barcode Gap discovery for primary species delimitation. Mol. Ecol. 21, 1864–1877. doi: 10.1111/j.1365-294X.2011.05239.x
Rabaglia, R. J., Cognato, A. I., Hoebeke, E. R., Johnson, C. W., LaBonte, J. R., Carter, M. E., et al. (2019). Early detection and rapid response a 10-year summary of the USDA Forest Service program of surveillance for non-native bark and ambrosia beetles. Am. Entomol. 65, 29–42. doi: 10.1093/ae/tmz015
Ratnasingham, S., and Hebert, P. D. N. (2013). A DNA-Based registry for all animal species: the Barcode Index Number (BIN) system. PLoS ONE 8:e66213. doi: 10.1371/journal.pone.0066213
Rodman, J. E., and Cody, J. H. (2003). The taxonomic impediment overcome: NSF's Partnerships for Enhancing Expertise in Taxonomy (PEET) as a model. Syst. Biol. 52, 428–435. doi: 10.1080/10635150309326
Rubinoff, D., Cameron, S., and Will, K. (2006). A genomic perspective on the shortcomings of mitochondrial DNA for “barcoding” identification. J. Hered. 97, 581–594. doi: 10.1093/jhered/esl036
Saitou, N., and Nei, M. (1987). The neighbor-joining method a new method for reconstructing phylogenetic trees. Mole. Biol. Evol. 4, 406–425.
Schindel, D. E., and Miller, S. E. (2005). DNA barcoding a useful tool for taxonomists. Nature 435:17. doi: 10.1038/435017b
Smith, S. M., and Cognato, A. I. (2014). A taxonomic monograph of Nearctic Scolytus Geoffroy (Coleoptera, Curculionidae, Scolytinae). Zookeys 450, 1–182. doi: 10.3897/zookeys.450.7452
Smith, S. M., and Cognato, A. I. (2015). Ambrosiophilus peregrinus n. sp., an exotic ambrosia beetle discovered in Georgia, U.S.A. (Coleoptera: Curculionidae: Scolytinae). Coleopter. Bull. 69, 213–220. doi: 10.1649/0010-065X-69.2.213
Smith, S. M., Gomez, D. F., Beaver, R. A., Hulcr, J., and Cognato, A. I. (2019). Reassessment of the species in the Euwallacea fornicatus (Coleoptera: Curculionidae: Scolytinae) complex after the rediscovery of the ‘lost’ type specimen. Insects 10:261. doi: 10.3390/insects10090261
Smith, S. M., Petrov, A. V., and Cognato, A. I. (2017). Beetles (Coleoptera) of Peru: a survey of the Families. Curculionidae: Scolytinae. Coleopter. Bull. 71, 77–94. doi: 10.1649/0010-065X-71.1.77
Smith, V. S. (2005). DNA Barcoding: Perspectives from a “Partnerships for Enhancing Expertise in Taxonomy” (PEET) Debate. Syst. Biol. 54, 841–844. doi: 10.1080/10635150500354894
Song, H., Buhay, J. E., Whiting, M. F., and Crandall, K. A. (2008). Many species in one: DNA barcoding overestimates the number of species when nuclear mitochondrial pseudogenes are coamplified. Proc. Natl. Acad. Sci. U.S.A. 105, 13486–13491. doi: 10.1073/pnas.0803076105
Sperling, F. A. (2003). DNA barcoding: deus ex machina. Newslett. Biol. Survey Canada (Terrestrial Arthropods) 22, 50–53.
Sperling, F. A., and Hickey, D. A. (1994). Mitochondrial DNA sequence variation in the spruce budworm species complex (Choristoneura: Lepidoptera). Mol. Biol. Evol. 11, 656–665.
Srivathsan, A., and Meier, R. (2012). On the inappropriate use of Kimura-2-Parameter (K2P) divergences in the DNA – barcoding literature. Cladistics 28, 190–194. doi: 10.1111/j.1096-0031.2011.00370.x
Storer, C., Peyton, A., McDaniel, S., Jordal, B., and Hulcr, J. (2017). Cryptic genetic variation in an inbreeding and cosmopolitan pest, Xylosandrus crassiusculus, revealed using ddRADseq. Ecol. Evol. 7, 10974–10986. doi: 10.1002/ece3.3625
Stouthamer, R., Rugman-Jones, P., Thu, P. Q., Eskalen, A., Thibault, T., Hulcr, J., et al. (2017). Tracing the origin of a cryptic invader: phylogeography of the Euwallacea fornicatus (Coleoptera: Curculionidae: Scolytinae) species complex. Agr. Forest Entomol. 19, 366–375. doi: 10.1111/afe.12215
Swofford, D. L. (2002). PAUP*: Phylogenetic Analysis Using Parsimony (*and Other Methods), 4.0b10. Sunderland, MA: Sinauer Associates.
Taft, W. H., and Cognato, A. I. (2017). Recognition of a new species of Carmenta from New Mexico supported by morphology and mitochondrial cytochrome oxidase I data (Lepidoptera: Sesiidae: Sesiinae: Synanthedonini). Zootaxa 4337, 436–444. doi: 10.11646/zootaxa.4337.3.8
Tautz, D., Arctander, P., Minelli, A., Thomas, R. H., and Vogler, A. P. (2003). A plea for DNA taxonomy. Trends Ecol. Evol. 18, 70–74. doi: 10.1016/S0169-5347(02)00041-1
Taylor, H. R., and Harris, W. E. (2012). An emergent science on the brink of irrelevance: a review of the past 8 years of DNA barcoding. Mol. Ecol. Resour. 12, 377–388. doi: 10.1111/j.1755-0998.2012.03119.x
Trewick, S. A. (2008). DNA Barcoding is not enough: mismatch of taxonomy and genealogy in New Zealand grasshoppers (Orthoptera: Acrididae). Cladistics 24, 240–254. doi: 10.1111/j.1096-0031.2007.00174.x
Ward, R. D., Zemlak, T. S., Innes, B. H., Last, P. R., and Hebert, P. D. N. (2005). DNA barcoding Australia's fish species. Phil. Trans. R. Soc. B. 360, 1847–1857. doi: 10.1098/rstb.2005.1716
Wheeler, Q. D., Knapp, S., Stevenson, D. W., Stevenson, J., Blum, S. D., Boom, B. M., et al. (2012). Mapping the biosphere: exploring species to understand the origin, organization and sustainability of biodiversity. Syst. Biodivers. 10, 1–20. doi: 10.1080/14772000.2012.665095
Will, K. W., Mishler, B. D., and Wheeler, Q. D. (2005). The perils of DNA barcoding and the need for integrative taxonomy. Syst. Biol. 54, 844–851. doi: 10.1080/10635150500354878
Will, K. W., and Rubinoff, D. (2004). Myth of the molecule: DNA barcodes for species cannot replace morphology for identification and classification. Cladistics 20, 47–55. doi: 10.1111/j.1096-0031.2003.00008.x
Wood, S. L. (1982). The bark and ambrosia beetles of North and Central America (Coleoptera: Scolytidae), a taxonomic monograph. Great Basin Naturalist Memoirs 8, 1–1359.
Wood, S. L. (2007). Bark and Ambrosia Beetles of South America (Coleoptera, Scolytidae). M. L. Bean Life Science Museum. Provo, UT: Brigham Young University.
Wood, S. L., and Bright, D. E. (1992). A catalog of Scolytidae and Platypodidae (Coleoptera), part 2: taxonomic index. Great Basin Naturalist Memoirs 13, 1–1533.
Wu, Y., Trepanowski, N. F., Molongoski, J. J., Reagel, P. F., Lingafelter, S. W., Nadel, H., Myers, S. W., et al. (2017). Identification of wood-boring beetles (Cerambycidae and Buprestidae) intercepted in trade-associated solid wood packaging material using DNA barcoding and morphology. Sci. Rep. 7:40316. doi: 10.1038/srep40316
Yeates, D. H., Seago, A., Nelson, L., Cameron, S. L., Joseph, L., and Trueman, J. W. H. (2011). Integrative taxonomy, or iterative taxonomy? Syst. Entomol. 36, 209–217. doi: 10.1111/j.1365-3113.2010.00558.x
Keywords: CAD, COI, DNA barcoding, species delimitation, species identification
Citation: Cognato AI, Sari G, Smith SM, Beaver RA, Li Y, Hulcr J, Jordal BH, Kajimura H, Lin C-S, Pham TH, Singh S and Sittichaya W (2020) The Essential Role of Taxonomic Expertise in the Creation of DNA Databases for the Identification and Delimitation of Southeast Asian Ambrosia Beetle Species (Curculionidae: Scolytinae: Xyleborini). Front. Ecol. Evol. 8:27. doi: 10.3389/fevo.2020.00027
Received: 04 October 2019; Accepted: 31 January 2020;
Published: 26 February 2020.
Edited by:
David S. Thaler, Universität Basel, SwitzerlandReviewed by:
Michael J. Raupach, Bavarian State Collection of Zoology, GermanyQuentin Wheeler, SUNY College of Environmental Science and Forestry, United States
Copyright © 2020 Cognato, Sari, Smith, Beaver, Li, Hulcr, Jordal, Kajimura, Lin, Pham, Singh and Sittichaya. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anthony I. Cognato, Y29nbmF0b0Btc3UuZWR1