 Stefanie Wagner
Stefanie Wagner Christophe Plomion
Christophe Plomion Ludovic Orlando
Ludovic Orlando
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol. , 31 January 2020
Sec. Paleoecology
Volume 8 - 2020 | https://doi.org/10.3389/fevo.2020.00011
This article is part of the Research Topic Applied Uses of Ancient DNA View all 14 articles
The ultra-short DNA molecules still preserved in archeological remains can provide invaluable genetic information about past individuals, species, and communities within the half to 1-million-year time range. The sequence data are, however, generally affected by post-mortem DNA damage and include specific patterns of nucleotide mis-incorporations, which can help data authentication. Recent work in ancient mammals has shown that such patterns can also help assess past levels of DNA methylation in CpG contexts. Despite pioneering work in barley and sorghum, ancient epigenetic marks have received limited attention in plants and it remains unknown whether ancient epigenetic signatures can be retrieved in any of the three main sequence contexts (CG, CHG, and CHH). To address this question, we extended a statistical methylation score originally proposed to trace cytosine methylation in mammal sequence data to accommodate the three methylation contexts common in plants. We applied this score to a range of tissues (wood, cobs, and grains) and species (oak, maize, and barley), spanning both desiccated and waterlogged archeological samples. Ancient sequence data obtained for USER-treated DNA extracts yielded methylation scores on par with DNA methylation levels of modern organellar and nuclear genomes. At the quantitative level, scores were (1) positively correlated to post-mortem cytosine deamination, and (2) replicated relative contributions of CG, CHG, and CHH contexts to DNA methylation assessed by bisulfite DNA sequencing of modern plant tissues. This demonstrates that genuine DNA methylation signatures can be characterized in ancient plant remains, which opens new avenues for investigating the plant evolutionary response to farming, pollution, epidemics, and changing environmental conditions.
Ancient DNA research has witnessed a true revolution over the last decade, shifting from the sequencing of single genomes from ancient anatomically modern humans (Rasmussen et al., 2010) and extinct hominins (Green et al., 2010; Reich et al., 2010) to now multiple thousands of genomes, including from non-human animals and plants (Brunson and Reich, 2019) and their pathogens (Spyrou et al., 2018, 2019). This has provided invaluable insights into the human past (Prüfer et al., 2014; Vernot and Akey, 2014; Narasimhan et al., 2019) and at the same time ancient genome time series have started revealing biological changes underlying plant and animal domestication, uncovering genomic turnovers that were unexpected from patterns of genetic variation present in modern genomes (Fonseca et al., 2015; Gaunitz et al., 2018; Frantz et al., 2019; Verdugo et al., 2019).
In addition to providing snapshots of DNA variation in the past, ancient DNA data can also provide insights into ancient epigenetic marks, leveraging degradation reactions that naturally affect DNA molecules after death (Hanghøj and Orlando, 2018). This could potentially inform on evolutionary changes in expression regulatory networks (Gokhman et al., 2014; Pedersen et al., 2014), age pyramids in ancient population (Hanghøj et al., 2016), as well as social and stress exposure. While accurate ancient chromatin compaction maps can be inferred from differential DNA decay within and outside of nucleosomes (Pedersen et al., 2014; Hanghøj et al., 2016), DNA cytosine methylation represents the epigenetic mark that received most of the scholar attention thus far in ancient DNA research. Early work was based on bisulfite sequencing (Llamas et al., 2012; Smith et al., 2015) but this approach requires amounts of DNA material generally not available in archeological specimens. Alternative experimental methodologies, including enrichment of DNA extracts through methyl binding domains (Smith et al., 2014; Seguin-Orlando et al., 2015), have thus been developed. They, however, also showed limitations owing to the ultra-fragmented and degraded nature of ancient DNA molecules (Seguin-Orlando et al., 2015). Therefore, ancient DNA methylation marks are generally not directly measured, but statistically inferred from patterns of nucleotide mis-incorporations along the genome.
The most powerful approach leverages ancient DNA sequence data retrieved after treatment of the DNA extracts with an enzymatic mix (USER) that excises Uracil nucleotidic bases (U) present in ancient DNA templates, and cleaves the DNA at the resulting abasic site (Briggs et al., 2010). Post-mortem deamination rates are known to be inflated toward the termini of ancient DNA molecules, due to the presence of overhanging ends (Briggs et al., 2007). These degradation reactions affect C residues regardless of their methylation state, transforming those unmethylated C residues into Uracils, and those methylated into Thymines (Pedersen et al., 2014). In the absence of USER treatment, U residues are maintained in the pool of ancient DNA molecules entering into DNA library preparation and sequencing. As a result, C → T mis-incorporations during sequencing will take place at all C residues affected by post-mortem deamination, be methylated (and transformed into T residues) or not (transformed into U residues, and sequenced as T residues). Following USER treatment, however, ancient DNA templates are cleaved at U residues, which almost excludes the incorporation of templates containing U residues into DNA libraries. Here, C → T mis-incorporations are introduced only at those methylated C that were affected by post-mortem deamination (and transformed into T residues). Simple scores, linearly related to the average counts of CpG → TpG mutations, have thus been proposed to infer regional levels of DNA methylation in ancient genome (Gokhman et al., 2014; Pedersen et al., 2014). Therefore, scores calculated from sequence data generated following USER-treatment should be lower than those measured in the absence of USER-treatment. This framework has been recently extended, including statistical models of DNA damage, to account for sequencing errors and sequence variation within the sequenced individual (Hanghøj et al., 2019).
Statistical inference of ancient DNA methylation has so far been only applied to mammals (mainly hominins, horses, and aurochsen; Hanghøj et al., 2016) and no studies have explored whether the same principles could help to obtain DNA methylation profiles in ancient plant remains, despite a growing wealth of sequence data in ancient crops, such as maize (Fonseca et al., 2015; Ramos-Madrigal et al., 2016; Vallebueno-Estrada et al., 2016; Swarts et al., 2017; Kistler et al., 2018) and barley (Mascher et al., 2016), and other cultivated (e.g., sorghum, Smith et al., 2019; grape, Ramos-Madrigal et al., 2019) or uncultivated (e.g., oak, Wagner et al., 2018) plants. This is likely due to the complex methylation and demethylation machinery present in higher plants, which in contrast to mammals, affects three main different sequence contexts: CG (or CpG), CHG, and CHH [where H refers to Adenine (A), C, or T], with relative methylation rates generally following the sequence CG > CHG > CHH (Feng et al., 2010; Niederhuth et al., 2016; Takuno et al., 2016; Bartels et al., 2018).
In this study, we extended the methylation score originally proposed by Pedersen et al. (2014) to accommodate the three sequence contexts underlying DNA methylation in plants and assessed, for the first time, whether DNA methylation information can be statistically inferred from ancient DNA data retrieved from a range of tissues (seeds, cobs, and wood) and species (barley, maize, and oak). The retrieval of signatures both qualitatively and quantitatively in line with expectations from plant DNA methylation profiles in modern plant tissues supports the validity of the approach implemented. This opens new avenues for future ancient DNA research in plants combining both genome and epigenetic time series to retrace the evolutionary response of plants to farming, pollution, epidemics, and changing environmental conditions, including global warming and increasing droughts.
In the absence of DNA methylation maps for ancient plants, we reasoned that the validity of putative ancient DNA methylation signatures could be gauged through their ability to replicate genome-wide patterns characterized in modern plants. This follows the rationale first applied to mammals and contrasting C deamination rates within and outside CpGs, (almost) unmethylated mitochondrial DNA vs. nuclear DNA, hypomethylated CpG islands vs. hypermethylated CG-rich promoters, and more (Pedersen et al., 2014; Hanghøj et al., 2016). More specifically, we extended the Ms statistics of normalized C → T mutation counts to other contexts than CG (i.e., CHG and CHH) (Figure 1) and tested two predictions. First, we tested whether overall Ms levels calculated from nuclear DNA were significantly greater than those estimated from chloroplast DNA, a locus known for an almost complete lack of DNA methylation (Cokus et al., 2008; Feng et al., 2010). Second, we tested the expected sequence of higher DNA methylation rates in CG contexts relative to CHG and CHH contexts (Feng et al., 2010; Takuno et al., 2016; Bartels et al., 2018). Both predictions are expected to be valid and specific to sequence data generated following USER treatment of raw ancient DNA extracts, should Ms scores reflect ancient DNA methylation signatures. The putative DNA methylation signatures retrieved should also be greater at read ends where post-mortem C deamination is faster than in the central region of sequence reads, owing to the overhanging nature of ancient DNA molecules (Briggs et al., 2007).
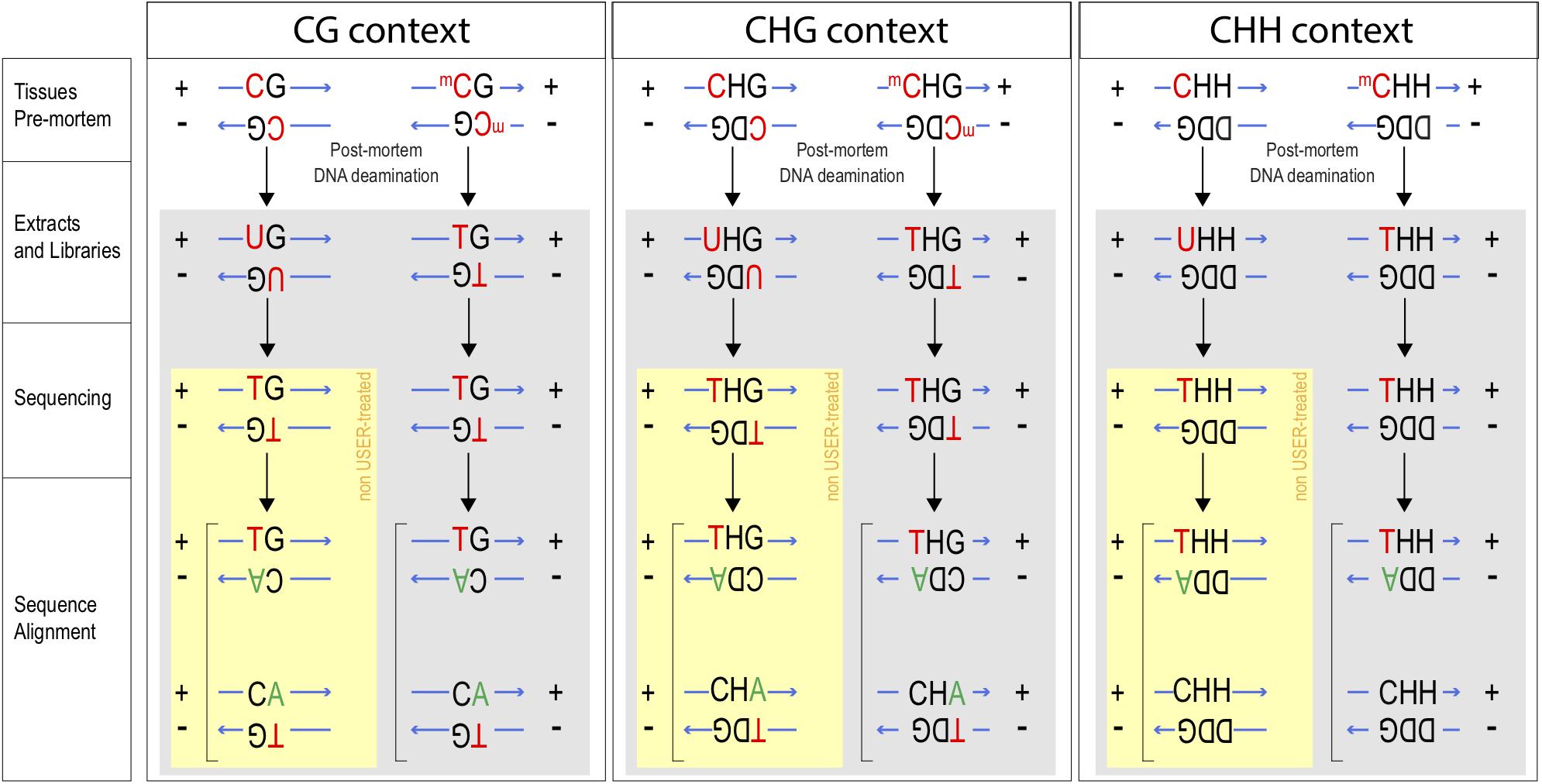
Figure 1. Tracking methylation in CG, CHG, and CHH contexts. After death, unmethylated Cytosines (C) become deaminated into Uracil (U), whereas methylated Cytosines (mC) become Thymines (T). In the absence of USER treatment, Us enter into library preparation and sequencing. C → T mis-incorporation will be observed for deaminated residues from methylated (C → T) and unmethylated Cytosine residues (C → U, sequenced as T; yellow box). If USER treatment is applied, U is excised prior to sequencing and C → T conversions can be used as a proxy for those Cs that were methylated pre-mortem. Base mis-incorporations highlighted in red and green. Ms ratios were calculated for raw extracts and USER treated extracts for the three plant methylation contexts CG, CHG, and CHH (where H refers to A, C, or T and D to T, G, or A).
Post-mortem C deamination rates were calculated within CG contexts following Pedersen et al. (2014). The calculation was extended to CHG contexts, by scanning CAG, CCG, and CTG individually and to CHH contexts by scanning for CAA, CAC, CAT, CCA, CCC, CCT, CTA, CTC, and CTT. Overall, we therefore summed C → T mis-incorporation rates observed at the first position of a total of 12 sequence contexts. For reads aligned against the negative strand of the reference genome, we focused instead on G → A mutations affecting the last position of those contexts. This is so as C deamination on the negative strand replaces the methylated C residue into a T residue, which is then sequenced and read as an A residue when aligned to the positive strand (Figure 1). Let note + sequences aligned to the positive strand, and – those aligned against the negative strand, keeping orientation so as to match BAM alignments (i.e., sequence mapping against the negative strand are displayed in the same orientation as the positive strand). A total count of those ancient C residues that were methylated and present in a CG context is thus provided by the cumulated sum of CG+ → TG+ and CG− → CA− mis-incorporations, where CG+ and CG− refer to the reference genome sequence at a given position, and TG+ and CA− to that sequenced in the ancient specimen. Methylation rates are estimated by normalizing this numerator denominator of all counts where the sequence data show and lack evidence of DNA deamination, i.e. CG+ → CG+, CG+ → TG+, GC− → GT−, and GC− → GC−, thus providing so-called Ms ratios (Pedersen et al., 2014). Now extending to CAG contexts, the numerator can easily be written as the sum of CAG+ → TAG+ and CAG− → CAA− counts, while the denominator becomes the sum of CAG+ → CAG+, CAG+ → TAG+, CAG− → CAA−, and CAG− → CAG− counts. Figure 1 provides a breakdown of the different counts considered in all three main sequence contexts. Read alignments including indels were disregarded to avoid potential inflation of substitution counts on their flanking nucleotides in case of mis-alignment.
Note that a number of processes other than just DNA methylation, such as sequencing errors and the presence of sequence polymorphism in the individual sequenced, contribute to the counts considered. Similarly, in the presence of high post-mortem C deamination rates, the conversion of methylated C residues into T residues is expected to be inflated. Therefore, the Ms ratio calculated do not scale across individuals and can only be used to compare putative methylation levels within (rather than between) individuals.
Previously published Illumina sequence data were retrieved from public data repositories for 12 oak, 6 maize, and 2 barley ancient samples representing a range of preservation conditions, DNA deamination rates, species, and tissues (Table 1 and Supplementary Table S1; Mascher et al., 2016; Swarts et al., 2017; Wagner et al., 2018). Raw sequence data were processed using PALEOMIX (Schubert et al., 2014), which automatically handles adapter trimming, alignment, duplicate removal, and filtering of low-quality read alignments. We used the PALEOMIX parameters specified in Wagner et al. (2018), and the haploid version of the Quercus robur nuclear (750 Mb) and chloroplast1 reference genomes for oak data (Plomion et al., 2016, 2018; Leroy et al., 2017). For maize and barley, the softmasked version of the respective reference genomes (including nuclear and organelle DNA) were used for read alignments. Those references are available on the Ensembl Plants Database2 (maize: B73 reference genome v4; barley: IBSC reference genome v2) (Schnable et al., 2009; International Barley Genome Sequencing Consortium et al., 2012; Kersey et al., 2018) and represent a nuclear genome size of 2.4 and 5.3 Gb, respectively. Mapping statistics are provided in Supplementary Tables S2, S3. For sequence data generated in the absence of USER treatment, ancient DNA damage patterns and quantification were carried out through PALEOMIX, using the methodology implemented in MapDamage (Jónsson et al., 2013).
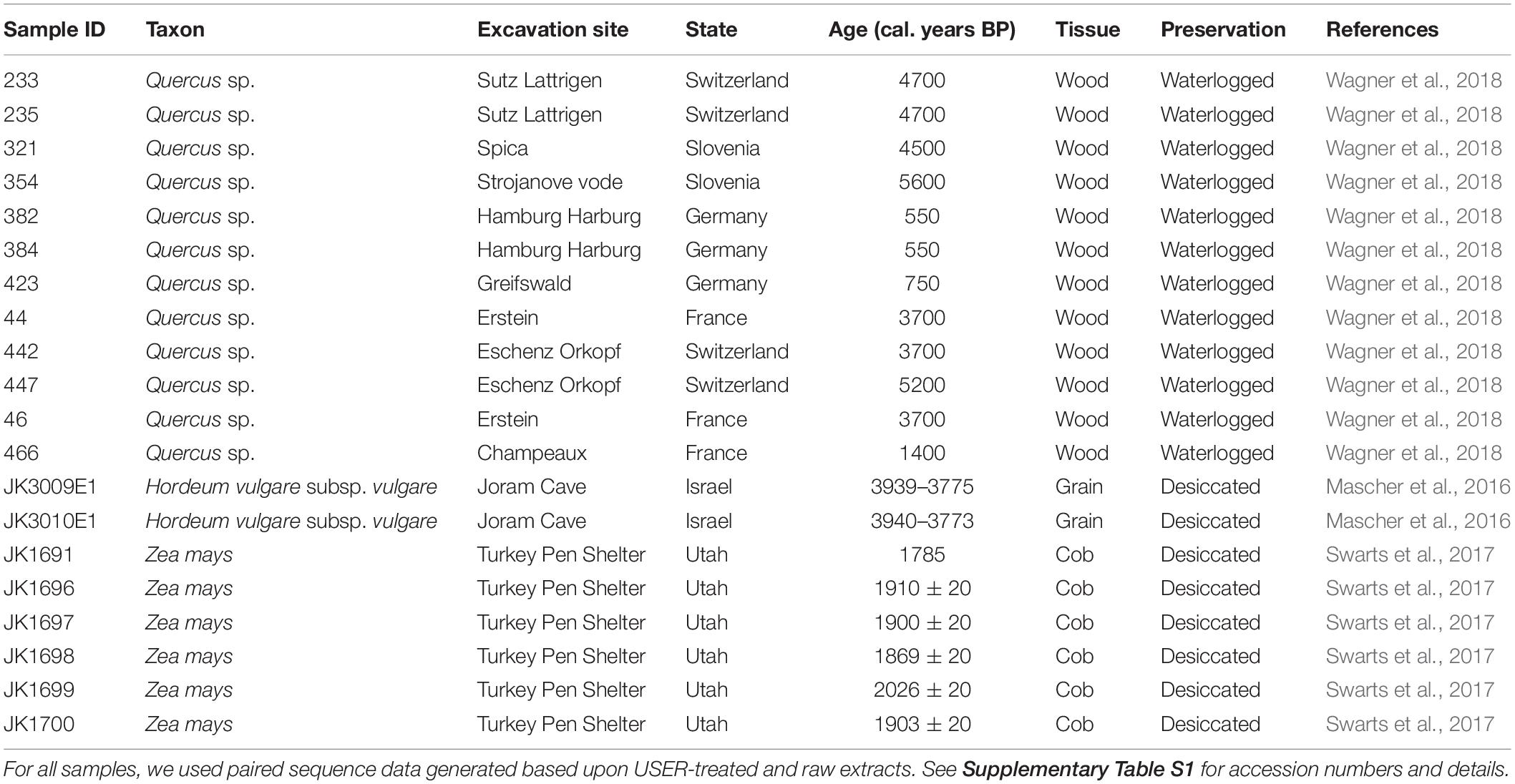
Table 1. Summary of samples analyzed in this study.
We calculated Ms ratios for the 20 ancient specimens where paired sequence data were available for both USER-treated and raw DNA extracts (Figure 2). We confirmed that the Ms ratios obtained from USER-treated data of all three species examined were consistently lower than those obtained in the absence of USER treatment. This hold true for both chloroplast DNA and nuclear DNA, although it was most pronounced for the former (1.2–21-fold vs. 1.1–4-fold reduction). This is in line with the (almost) absence of C methylation along the chloroplast DNA, which considerably limits the amount of deaminated methylated C residues available for sequencing. The same was true for the mitochondrial genome, which could be calculated for maize and barley, which also showed values of Ms ratios post-USER treatment lower to those calculated on nuclear DNA data. This demonstrates that Ms ratios as calculated in this study can recapitulate the known methylation difference between the chloroplast and nuclear genomes.
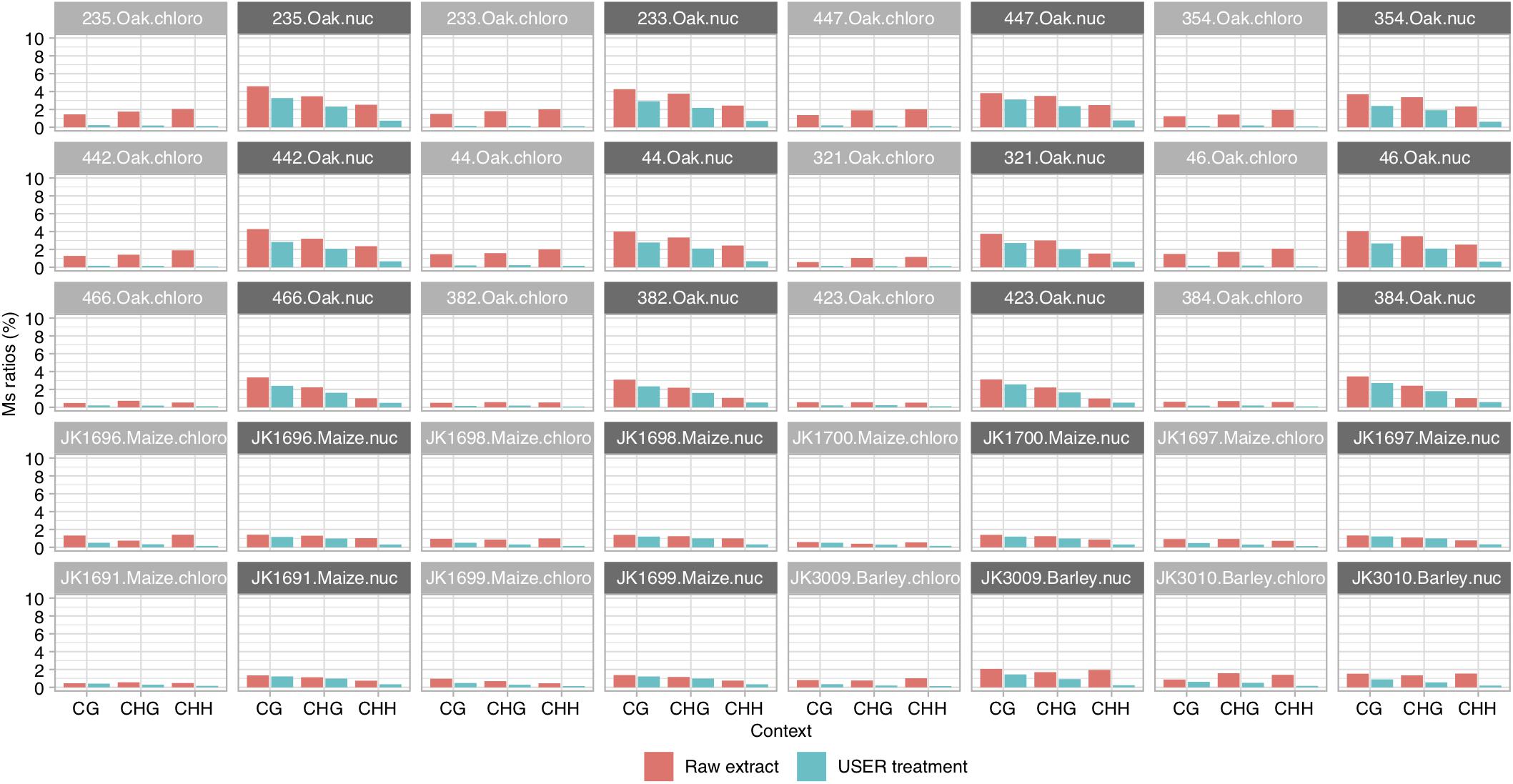
Figure 2. Ms ratios for CG, CHG, and CHH contexts. Results are shown for paired sequence data of USER-treated and raw extracts of ancient oak, maize, and barley samples, for chloroplast and nuclear DNA, respectively. Scores are reported for full read length. For Ms ratios computed for outer and inner read regions, see Supplementary Figure S1.
We next calculated Ms ratios within the main three sequence methylation contexts present in plants (CG, CHG, and CHH) in order to test whether we could recapitulate the generally predominant contribution of CG methylation over both other contexts and the generally minimal contribution of CHH contexts (Feng et al., 2010; Takuno et al., 2016; Bartels et al., 2018). In modern oak, bisulfite sequencing data obtained from buds of European oak trees (Quercus petraea and Q. robur) showed that CG contexts contribute to a fraction of 54.4% of DNA methylation marks in the nuclear genome, while CHG contexts contribute to 39.5% and CHH to 6.1% (unpublished, data kindly provided by Stéphane Maury, ANR EPITREE3). In all 12 ancient oak wood samples investigated here, we found that the sequence context providing the highest Ms ratios calculated on USER-treated data (thus contributing the most to DNA methylation) was consistently the CG context, while CHG and CHH provided the second most and least important values, respectively (Figures 2, 3). Normalizing individual Ms ratios by the sum of all three Ms ratios in each sample calculated provided relative estimates of the respective contribution of each sequence context to DNA methylation (Figure 3). We found Ms ratios supporting 48.6–54.1% methylation within CG contexts, and 35.0–38.9 and 10.9–12.5% within CHG and CHH contexts, respectively, in line with bisulfite DNA sequencing measures on modern oak buds. This suggests that the calculation of Ms ratios proposed in this study can retrieve genuine, quantitative estimates of DNA methylation from ancient plant DNA sequence data. For maize, previous work on unfertilized ears showed that CG contexts contributed to 52.0% of the DNA methylation marks present in the nuclear genome, while CHG and CHH contexts represented a fraction of 44.7 and 3.3%, respectively (Gent et al., 2013). The estimates retrieved within CG (47.3–48.0%) and CHG (38.6–39.2%) contexts from ancient maize cobs were slightly inferior, due to an increased contribution of CHH contexts (12.7–13.5%, Figure 3). This may pertain to marginal differences in the DNA methylation profiles of the tissues compared. Similarly, the estimates retrieved for the relative contribution of CHH contexts to nuclear DNA methylation marks was slightly higher (8.9 and 12.1%) than that directly measured through bisulfite sequencing of barley (1.1%) on mixed samples of seedling, leaf, and apex tissues (unpublished, Benjamin Stich, personal communication) (54.0 and 55.7 vs. 58.2%; and 33.8 and 35.4 vs. 40.7%).
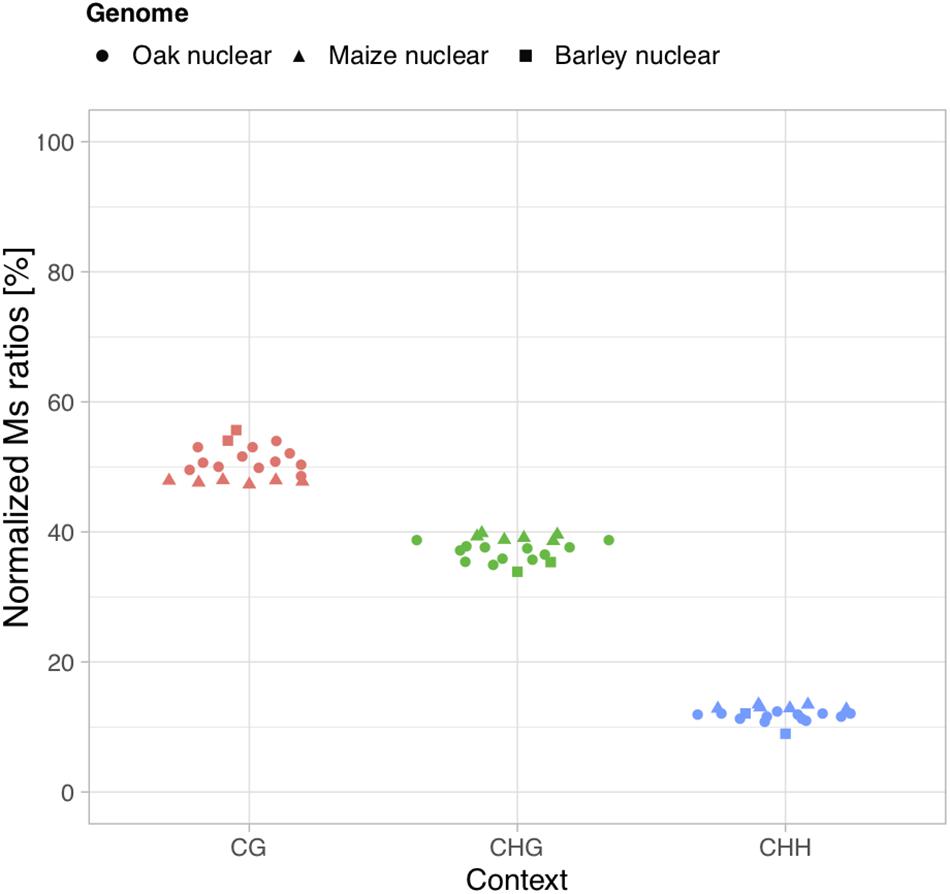
Figure 3. Relative contributions of CG, CHG, and CHH contexts to DNA methylation. Normalized Ms ratios are shown for nuclear DNA analyzed from sequencing data generated based upon USER-treated extracts of 12 ancient oak, 6 maize, and 2 barley samples.
The capacity of Ms ratios to track ancient C methylation marks does not only result from the removal of any possible contribution of unmethylated C residues to sequencing mis-incorporations. It also comes from the deamination reactions that transform methylated C residues into Thymines after death. Therefore, Ms ratios are expected to be maximized in situations where post-mortem C deamination is maximized, for example at read ends. We confirmed this expectation by using sequence data obtained following USER-treatment and contrasting Ms ratios within the first and last 10 read positions on the one hand, and on the remaining positions on the other hand (Supplementary Figure S1). Additionally, we found that post-mortem C deamination rates within overhanging ends, as estimated using mapDamage2 (Jónsson et al., 2013), were positively correlated with Ms ratios. This demonstrates that the Ms ratios calculated in this study provide a measure of the intensity of C deamination reactions.
Altogether, Ms ratios both respond to increased C deamination post-mortem reactions and recapitulate known methylation features between organellar and nuclear DNA as well as between different methylation sequence contexts (CG, CHG, and CHH). This was thus far only described for CG contexts in animals. The extension of Ms ratio calculation to tri-nucleotide context proposed here for the first time, thus provide genuine proxies for inferring plant DNA methylation levels in the past. The ancient sequence data for the three species investigated here is not sufficient to generate precise genome-wide DNA methylation maps and identify differentially methylated regions in ancient and modern samples. The data we used allowed to explore the relative contributions of each individual sequence context to CHG (i.e., CAG, CCG, and CTG) and CHH (i.e., CAA, CAC, CAG, CCA, CCC, CCG, CTA, CTC, and CTG) methylation (Figure 4). We considered that Ms ratios obtained within CG contexts on USER-treated data represented an arbitrary but convenient score of 100%. We then normalized the Ms ratios of each other sequence context examined relative to this value to assess their respective contributions to DNA methylation. We found that all three sequence motifs underlying CHG contexts contributed evenly to DNA methylation (Figure 4). The same was true for the nine sequence motifs underlying CHH contexts, suggesting that the cellular enzymatic machinery responsible for DNA methylation shows no strong preference for any particular sequence motif.
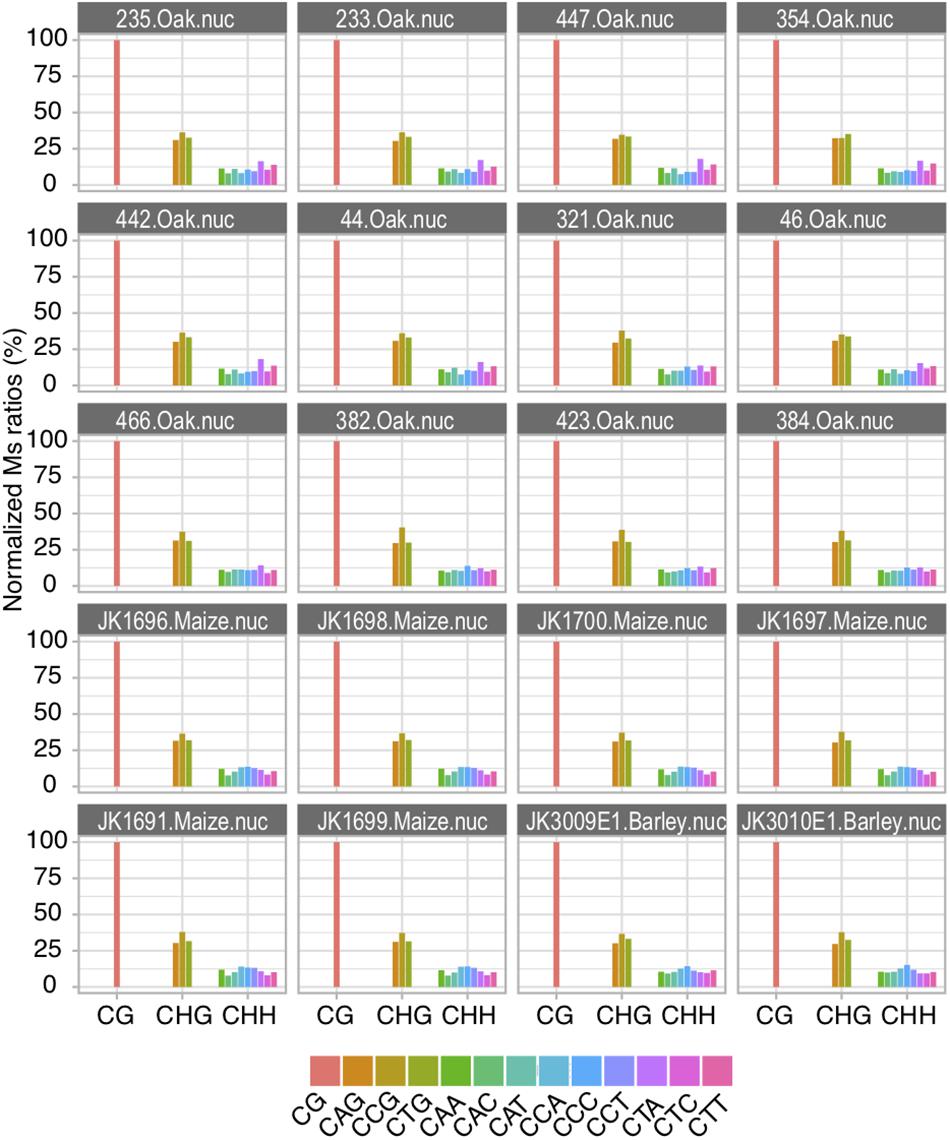
Figure 4. Relative contribution of individual tri-nucleotide sequences to methylation in CHG and CHH contexts. Normalized Ms ratios scored for full read length are shown for nuclear DNA analyzed from sequencing data generated based upon USER-treated extracts.
A variety of ancient DNA studies have demonstrated that epigenetic marks can be revealed in the fossilized tissues from ancient individuals, either through experimental assays directly targeting methylated C residues or indirect statistical analyses leveraging patterns of sequencing errors predominantly affecting these sites [see Hanghøj and Orlando (2018) for a review]. To the best of our knowledge, only two of previous studies were applied to ancient plant DNA material (Smith et al., 2014, 2019). In the first, the authors made use of proteins showing strong affinity to methylated CG to enrich ancient barley DNA extracts for methylated fragments. They also applied standard bisulfite sequencing to quantify genome-wide average of DNA methylation levels, a measure that can reflect the exposure of host cells to viruses (e.g., Boyko et al., 2007). In the second study, the authors have applied a software package originally designed to track methylation at CpG sites in ancient mammal genomes (Hanghøj et al., 2016) to archeological sorghum DNA samples from Egypt spanning a full time series (1,800–100 years BP) (Smith et al., 2019). No information was gathered outside the CG context despite other sequence contexts that contribute to DNA methylation in plants known to be characteristic and highly dynamic in response to intrinsic and extrinsic stress factors (Feng et al., 2010; Takuno et al., 2016; Bartels et al., 2018).
In this study, we demonstrate for the first time that the methodology originally proposed to retrieve DNA methylation information from ancient mammal tissues within CpG contexts (Pedersen et al., 2014) can be extended to tri-nucleotide contexts to retrieve DNA methylation signatures within CG, CHG, and CHH contexts. Several lines of evidence support the validity of the approach proposed here. At the qualitative level, Ms ratios calculated on sequence data generated following USER-treatment of raw ancient DNA extracts replicate the known levels of DNA methylation of organellar and nuclear genomes. At the quantitative level, such Ms ratios are (1) positively correlated to levels of post-mortem C deamination rates, (2) inferior to those estimated on the sequence data produced in the absence of USER-treatment, and (3) replicate the relative contributions of CG, CHG, and CHH contexts to DNA methylation. The latter suggests similar kinetics of cytosine DNA deamination after death in all three contexts. However, direct experimental evidence remains necessary to assess whether methylated C residues show similar deamination when present on CG, CHG, and CHH sequence contexts. Finding support for differential decay would impact the method proposed here, as it would exhibit non-uniform sensitivity for inferring DNA methylation levels across the genome. Direct comparisons directly contrasting specific genomic regions (e.g., promoters), without controlling for similar base compositional effects (e.g., similar fractions of CG, CHG, and/or CGG contexts), would then be flawed.
Ancient DNA studies focusing on plant remains have made significant contributions to the fields of anthropology and biology (Palmer et al., 2012; Allaby et al., 2015; Brown et al., 2015; Estrada et al., 2018; Brunson and Reich, 2019; Pont et al., 2019), with important projects reconstructing the genomic history of maize, grape, and barley domestication (Fonseca et al., 2015; Mascher et al., 2016; Ramos-Madrigal et al., 2016, 2019; Vallebueno-Estrada et al., 2016; Swarts et al., 2017; Kistler et al., 2018). The findings reported here suggest that the ever-increasing amounts of sequencing data underlying such projects will soon open for an evaluation of the epigenetic impact of domestication. By extension, when other taxa and other archeological and environmental contexts will be included, the production of larger ancient sequence datasets will help researchers characterize the epigenetic response of plant species to many other selective pressures, including climate change, pollution, and more (Allaby et al., 2015; Gutaker and Burbano, 2017). Unless candidate loci of interest or predefined sets of CG, CHG, and CHH loci are specifically targeted (e.g., through in solution DNA capture, Ramos-Madrigal et al., 2019), reaching this goal will, however, require massive sequencing efforts. It is crucial to keep in mind that (1) ancient DNA analyses are destructive, (2) archeological material represent a finite resource, and that (3) C deamination inflate sequencing errors and can, thus, impact downstream analyses. In our opinion, focusing sequencing efforts on DNA libraries prepared from ancient DNA extracts treated with the USER enzyme mix will mitigate these issues and will reduce analytical costs, as both genomic and epigenetic information could be revealed from the same sequence data. As we observed that the correlation between the Ms ratios and expected methylation levels at different markers was lost in the absence of USER treatment (Figure 2), we expect that the power to retrieve both types of information from the sequence data obtained on DNA libraries prepared from raw DNA extracts will be limited.
Previous work has shown that signatures that could inform about gene expression, such as nucleosome positioning along genes and their phasing across cells, could be obtained from the sequencing data underlying ancient mammal genomes (Hanghøj et al., 2016). Whether such signatures could also be collected in plants remains unknown and will require further research. Future work should also focus on improving the methodology proposed here, following what recently done for mammals where statistical models, such as the one implemented in the DamMet package (Hanghøj et al., 2019), have been developed to account for the contribution of other factors than DNA methylation to C → T mis-incorporations. This should reduce the impact of sequencing errors and non-reference variants present in the individual sequenced, and thus improve the accuracy of DNA methylation inference. Extending DamMet to tri-nucleotide contexts will, however, be challenging as the number of underlying genotypes could become rapidly intractable and machine learning approaches may prove more powerful to disentangle nucleotide mis-incorporations pertaining to post-mortem deamination reactions affecting methylated C residues from those resulting from all other processes contributing to sequencing errors. Once the methodology will have been refined, and following the amount of sequence data that were found necessary for retrieving accurate DNA methylation estimates in mammals, we anticipate that ancient genomes characterized at a minimal sequencing depth of 20–25× may be used for in depth studies on ancient plant methylomes. Interestingly, such datasets have already started to be produced in plants (e.g., Mascher et al., 2016: Sample JK3014, average read depth 20×).
In this study, we extended a statistical procedure to uncover ancient DNA methylation signatures in the three sequence contexts present in plants. We applied our procedure on previously published ancient DNA data generated using NGS for oak waterlogged oak wood (Quercus sp.), desiccated maize cobs (Zea mays), and desiccated barley grains (Hordeum vulgare subsp. vulgare) and obtained signatures in line with modern plant methylomes. This opens new avenues for designing evolutionary studies on the rich plant material preserved in herbaria and in the archeological record.
All the sequencing data analyzed in this study have been previously published and are available for download from the European Nucleotide Archive under Accession Nos. PRJEB21188 (https://www.ebi.ac.uk/ena/data/search?query=PRJEB21188), PRJEB20529 (https://www.ebi.ac.uk/ena/data/search?query=PRJEB20529), and PRJEB12197 (https://www.ebi.ac.uk/ena/data/search?query=+PRJEB12197).
LO conceived and designed the experiments, analyzed, and interpreted the data. LO wrote the manuscript with inputs from SW and CP. SW prepared the figures.
This work was supported by the Initiative d’Excellence Chaires d’Attractivité, Université de Toulouse (OURASI), and the Villum Fonden miGENEPI research project. CP has received funding from the ANR (epitree project, ANR-17-CE32-0009-01, 2018–21). LO has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Program (Grant Agreement No. 681605-PEGASUS).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Antoine Kremer and the Advanced Grant Project TREEPEACE (No. FP7-339728) for supporting this work and Stéphane Maury and Benjamin Stich for sharing unpublished modern plant methylome data.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2020.00011/full#supplementary-material
Allaby, R. G., Gutaker, R., Clarke, A. C., Pearson, N., Ware, R., Palmer, S. A., et al. (2015). Using archaeogenomic and computational approaches to unravel the history of local adaptation in crops. Philos. Trans. R. Soc. Lond. B Biol. Sci. 370:20130377. doi: 10.1098/rstb.2013.0377
Bartels, A., Han, Q., Nair, P., Stacey, L., Gaynier, H., and Mosley, M. (2018). Dynamic DNA methylation in plant growth and development. Int. J. Mol. Sci 19:2144. doi: 10.3390/ijms19072144
Boyko, A., Kathiria, P., Zemp, F. J., Yao, Y., Pogribny, I., and Kovalchuk, I. (2007). Transgenerational changes in the genome stability and methylation in pathogen-infected plants: virus-induced plant genome instability. Nucleic Acids Res. 35, 1714–1725. doi: 10.1093/nar/gkm029
Briggs, A. W., Stenzel, U., Johnson, P. L. F., Green, R. E., Kelso, J., Prüfer, K., et al. (2007). Patterns of damage in genomic DNA sequences from a Neandertal. Proc. Natl. Acad. Sci. U.S.A. 104, 14616–14621. doi: 10.1073/pnas.0704665104
Briggs, A. W., Stenzel, U., Meyer, M., Krause, J., Kircher, M., and Pääbo, S. (2010). Removal of deaminated cytosines and detection of in vivo methylation in ancient DNA. Nucleic Acids Res. 38:e87. doi: 10.1093/nar/gkp1163
Brown, T., Cappellini, E., Kistler, L., Lister, D., Oliveira, H., Wales, N., et al. (2015). Recent advances in ancient DNA research and their implications for archaeobotany. Veg. Hist. Archaeobot. 24, 207–214. doi: 10.1007/s00334-014-0489-4
Brunson, K., and Reich, D. (2019). The promise of paleogenomics beyond our own species. Trends Genet. 35, 319–329. doi: 10.1016/j.tig.2019.02.006
Cokus, S. J., Feng, S., Zhang, X., Chen, Z., Merriman, B., Haudenschild, C. D., et al. (2008). Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452, 215–219. doi: 10.1038/nature06745
Estrada, O., Breen, J., Richards, S. M., and Cooper, A. (2018). Ancient plant DNA in the genomic era. Nat. Plants 4, 394–396. doi: 10.1038/s41477-018-0187-9
Feng, S., Cokus, S. J., Zhang, X., Chen, P.-Y., Bostick, M., Goll, M. G., et al. (2010). Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. U.S.A. 107, 8689–8694. doi: 10.1073/pnas.1002720107
Fonseca, R. R., Smith, B. D., Wales, N., Cappellini, E., Skoglund, P., and Fumagalli, M. (2015). The origin and evolution of maize in the Southwestern United States. Nat. Plants 1:14003. doi: 10.1038/nplants.2014.3
Frantz, L. A. F., Haile, J., Lin, A. T., Scheu, A., Geörg, C., Benecke, N., et al. (2019). Ancient pigs reveal a near-complete genomic turnover following their introduction to Europe. Proc. Natl. Acad. Sci. U.S.A. 116, 17231–17238. doi: 10.1073/pnas.1901169116
Gaunitz, C., Fages, A., Hanghøj, K., Albrechtsen, A., Khan, N., Schubert, M., et al. (2018). Ancient genomes revisit the ancestry of domestic and Przewalski’s horses. Science 360, 111–114. doi: 10.1126/science.aao3297
Gent, J. I., Ellis, N. A., Guo, L., Harkess, A. E., Yao, Y., Zhang, X., et al. (2013). CHH islands: de novo DNA methylation in near-gene chromatin regulation in maize. Genome Res. 23, 628–637. doi: 10.1101/gr.146985.112
Gokhman, D., Lavi, E., Prüfer, K., Fraga, M. F., Riancho, J. A., Kelso, J., et al. (2014). Reconstructing the DNA methylation maps of the Neandertal and the Denisovan. Science 344, 523–527. doi: 10.1126/science.1250368
Green, R. E., Krause, J., Briggs, A. W., Maricic, T., Stenzel, U., Kircher, M., et al. (2010). A draft sequence of the Neandertal genome. Science 328, 710–722. doi: 10.1126/science.1188021
Gutaker, R. M., and Burbano, H. A. (2017). Reinforcing plant evolutionary genomics using ancient DNA. Curr. Opin. Plant Biol. 36, 38–45. doi: 10.1016/j.pbi.2017.01.002
Hanghøj, K., and Orlando, L. (2018). “Ancient epigenomics,” in Paleogenomics: Genome-Scale Analysis of Ancient DNA, eds C. Lindqvist and O. P. Rajora, (Cham: Springer International Publishing), 75–111. doi: 10.1007/13836_2018_18
Hanghøj, K., Renaud, G., Albrechtsen, A., and Orlando, L. (2019). DamMet: ancient methylome mapping accounting for errors, true variants, and post-mortem DNA damage. Gigascience 8:giz025. doi: 10.1093/gigascience/giz025
Hanghøj, K., Seguin-Orlando, A., Schubert, M., Madsen, T., Pedersen, J. S., Willerslev, E., et al. (2016). Fast, accurate and automatic ancient nucleosome and methylation maps with epiPALEOMIX. Mol. Biol. Evol. 33, 3284–3298. doi: 10.1093/molbev/msw184
International Barley Genome Sequencing Consortium, Mayer, K. F. X., Waugh, R., Brown, J. W. S., Schulman, A., and Langridge, P. (2012). A physical, genetic and functional sequence assembly of the barley genome. Nature 491, 711–716. doi: 10.1038/nature11543
Jónsson, H., Ginolhac, A., Schubert, M., Johnson, P. L. F., and Orlando, L. (2013). mapDamage2.0: fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 29, 1682–1684. doi: 10.1093/bioinformatics/btt193
Kersey, P. J., Allen, J. E., Allot, A., Barba, M., Boddu, S., Bolt, B. J., et al. (2018). Ensembl genomes 2018: an integrated omics infrastructure for non-vertebrate species. Nucleic Acids Res. 46, D802–D808. doi: 10.1093/nar/gkx1011
Kistler, L., Maezumi, S. Y., Gregorio de Souza, J., Przelomska, N. A. S., Malaquias Costa, F., Smith, O., et al. (2018). Multiproxy evidence highlights a complex evolutionary legacy of maize in South America. Science 362, 1309–1313. doi: 10.1126/science.aav0207
Leroy, T., Roux, C., Villate, L., Bodénès, C., Romiguier, J., Paiva, J. A. P., et al. (2017). Extensive recent secondary contacts between four European white oak species. New Phytol. 214, 865–878. doi: 10.1111/nph.14413
Llamas, B., Holland, M. L., Chen, K., Cropley, J. E., Cooper, A., and Suter, C. M. (2012). High-resolution analysis of cytosine methylation in ancient DNA. PLoS One 7:e30226. doi: 10.1371/journal.pone.0030226
Mascher, M., Schuenemann, V. J., Davidovich, U., Marom, N., Himmelbach, A., Hübner, S., et al. (2016). Genomic analysis of 6,000-year-old cultivated grain illuminates the domestication history of barley. Nat. Genet. 48, 1089–1093. doi: 10.1038/ng.3611
Narasimhan, V. M., Patterson, N., Moorjani, P., Rohland, N., Bernardos, R., and Mallick, S. (2019). The formation of human populations in South and Central Asia. Science 365:eaat7487. doi: 10.1126/science.aat7487
Niederhuth, C. E., Bewick, A. J., Ji, L., Alabady, M. S., Kim, K. D., Li, Q., et al. (2016). Widespread natural variation of DNA methylation within angiosperms. Genome Biol. 17:194.
Palmer, S. A., Smith, O., and Allaby, R. G. (2012). The blossoming of plant archaeogenetics. Ann. Anat. 194, 146–156. doi: 10.1016/j.aanat.2011.03.012
Pedersen, J. S., Valen, E., Velazquez, A. M. V., Parker, B. J., Rasmussen, M., Lindgreen, S., et al. (2014). Genome-wide nucleosome map and cytosine methylation levels of an ancient human genome. Genome Res. 24, 454–466. doi: 10.1101/gr.163592.113
Plomion, C., Aury, J.-M., Amselem, J., Alaeitabar, T., Barbe, V., Belser, C., et al. (2016). Decoding the oak genome: public release of sequence data, assembly, annotation and publication strategies. Mol. Ecol. Resour. 16, 254–265. doi: 10.1111/1755-0998.12425
Plomion, C., Aury, J.-M., Amselem, J., Leroy, T., Murat, F., Duplessis, S., et al. (2018). Oak genome reveals facets of long lifespan. Nat. Plants 4, 440–452. doi: 10.1038/s41477-018-0172-3
Pont, C., Wagner, S., Kremer, A., Orlando, L., Plomion, C., and Salse, J. (2019). Paleogenomics: reconstruction of plant evolutionary trajectories from modern and ancient DNA. Genome Biol. 20:29. doi: 10.1186/s13059-019-1627-1
Prüfer, K., Racimo, F., Patterson, N., Jay, F., Sankararaman, S., Sawyer, S., et al. (2014). The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 505, 43–49. doi: 10.1038/nature12886
Ramos-Madrigal, J., Runge, A. K. W., Bouby, L., Lacombe, T., Samaniego Castruita, J. A., Adam-Blondon, A.-F., et al. (2019). Palaeogenomic insights into the origins of French grapevine diversity. Nat. Plants 5, 595–603. doi: 10.1038/s41477-019-0437-5
Ramos-Madrigal, J., Smith, B. D., Moreno-Mayar, J. V., Gopalakrishnan, S., Ross-Ibarra, J., Gilbert, M. T. P., et al. (2016). Genome sequence of a 5,310-year-old maize cob provides insights into the early stages of maize domestication. Curr. Biol. 26, 3195–3201. doi: 10.1016/j.cub.2016.09.036
Rasmussen, M., Li, Y., Lindgreen, S., Pedersen, J. S., Albrechtsen, A., Moltke, I., et al. (2010). Ancient human genome sequence of an extinct Palaeo-Eskimo. Nature 463, 757–762. doi: 10.1038/nature08835
Reich, D., Green, R. E., Kircher, M., Krause, J., Patterson, N., Durand, E. Y., et al. (2010). Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature 468, 1053–1060. doi: 10.1038/nature09710
Schnable, P. S., Ware, D., Fulton, R. S., Stein, J. C., Wei, F., Pasternak, S., et al. (2009). The B73 maize genome: complexity, diversity, and dynamics. Science 326, 1112–1115. doi: 10.1126/science.1178534
Schubert, M., Ermini, L., Der Sarkissian, C., Jónsson, H., Ginolhac, A., Schaefer, R., et al. (2014). Characterization of ancient and modern genomes by SNP detection and phylogenomic and metagenomic analysis using PALEOMIX. Nat. Protoc. 9, 1056–1082. doi: 10.1038/nprot.2014.063
Seguin-Orlando, A., Gamba, C., Sarkissian, C. D., Ermini, L., Louvel, G., Boulygina, E., et al. (2015). Pros and cons of methylation-based enrichment methods for ancient DNA. Sci. Rep. 5:11826. doi: 10.1038/srep11826
Smith, O., Clapham, A. J., Rose, P., Liu, Y., Wang, J., and Allaby, R. G. (2014). Genomic methylation patterns in archaeological barley show de-methylation as a time-dependent diagenetic process. Sci. Rep. 4:5559. doi: 10.1038/srep05559
Smith, O., Nicholson, W. V., Kistler, L., Mace, E., Clapham, A., Rose, P., et al. (2019). A domestication history of dynamic adaptation and genomic deterioration in Sorghum. Nat Plants 5, 369–379. doi: 10.1038/s41477-019-0397-9
Smith, R. W. A., Monroe, C., and Bolnick, D. A. (2015). Detection of cytosine methylation in ancient DNA from five native american populations using bisulfite sequencing. PLoS One 10:e0125344. doi: 10.1371/journal.pone.0125344
Spyrou, M. A., Keller, M., Tukhbatova, R. I., Scheib, C. L., Nelson, E. A., Andrades Valtueña, A., et al. (2019). Phylogeography of the second plague pandemic revealed through analysis of historical Yersinia pestis genomes. Nat. Commun. 10:4470. doi: 10.1038/s41467-019-12154-0
Spyrou, M. A., Tukhbatova, R. I., Wang, C.-C., Valtueña, A. A., Lankapalli, A. K., Kondrashin, V. V., et al. (2018). Analysis of 3800-year-old Yersinia pestis genomes suggests Bronze Age origin for bubonic plague. Nat. Commun. 9:2234. doi: 10.1038/s41467-018-04550-9
Swarts, K., Gutaker, R. M., Benz, B., Blake, M., Bukowski, R., Holland, J., et al. (2017). Genomic estimation of complex traits reveals ancient maize adaptation to temperate North America. Science 357, 512–515. doi: 10.1126/science.aam9425
Takuno, S., Ran, J.-H., and Gaut, B. S. (2016). Evolutionary patterns of genic DNA methylation vary across land plants. Nat. Plants 2:15222. doi: 10.1038/nplants.2015.222
Vallebueno-Estrada, M., Rodríguez-Arévalo, I., Rougon-Cardoso, A., Martínez González, J., García Cook, A., Montiel, R., et al. (2016). The earliest maize from San Marcos Tehuacán is a partial domesticate with genomic evidence of inbreeding. Proc. Natl. Acad. Sci. U.S.A. 113, 14151–14156. doi: 10.1073/pnas.1609701113
Verdugo, M. P., Mullin, V. E., Scheu, A., Mattiangeli, V., Daly, K. G., Maisano Delser, P., et al. (2019). Ancient cattle genomics, origins, and rapid turnover in the Fertile Crescent. Science 365, 173–176. doi: 10.1126/science.aav1002
Vernot, B., and Akey, J. M. (2014). Resurrecting surviving Neandertal lineages from modern human genomes. Science 343, 1017–1021. doi: 10.1126/science.1245938
Keywords: ancient DNA, DNA methylation, genomics, post-mortem DNA damage, oak, maize, barley
Citation: Wagner S, Plomion C and Orlando L (2020) Uncovering Signatures of DNA Methylation in Ancient Plant Remains From Patterns of Post-mortem DNA Damage. Front. Ecol. Evol. 8:11. doi: 10.3389/fevo.2020.00011
Received: 06 November 2019; Accepted: 16 January 2020;
Published: 31 January 2020.
Edited by:
Michael David Martin, Norwegian University of Science and Technology, NorwayReviewed by:
Logan Kistler, Smithsonian Institution, United StatesCopyright © 2020 Wagner, Plomion and Orlando. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ludovic Orlando, bHVkb3ZpYy5vcmxhbmRvQHVuaXYtdGxzZTMuZnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.