 Alba Frias-De-Diego
Alba Frias-De-Diego Manuel Jara
Manuel Jara Luis E. Escobar
Luis E. Escobar
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol. , 25 October 2019
Sec. Biogeography and Macroecology
Volume 7 - 2019 | https://doi.org/10.3389/fevo.2019.00406
This article is part of the Research Topic Diseases at the Wildlife-Human Interface: Tracking Pathogen Evolution and Predicting Future Change View all 5 articles
Papillomaviruses (PV) are associated with epithelial malignancies in animals, including cancer in humans. Limited knowledge exists regarding the evolutionary history of non-human PV. We assessed the phylogeography of PV with emphasis in wildlife hosts. We explored the phylogenetic, geographic, and environmental relationships of PV and hosts applying Bayesian inference and spatial analyses of virus and hosts. We found that the available wildlife PV data support previous reports on the higher incidence of fibropapillomatosis over carcinoma in humans and wildlife, being mammals the most common host. We also found geographic bias on the available wildlife papillomavirus (WPV) information toward the Northern Hemisphere, which may have influenced our results to show Europe as the most likely origin of the available WPV lineages. Therefore, we highlight the need for detailed studies on the presence of WPV in regions and species not included in this study (e.g., reptiles from the tropics) to better inform sites of WPV origin, susceptible species, and spillover potential. Future studies of the clinical and subclinical occurrence, distribution, and phylogenetic signatures of WPV may help to understand the spread, virulence, and epidemiology of PV in general. From an evolutionary perspective, our overview suggests that WPV are a promising host-pathogen system to untangle questions regarding co-evolution due to its large geographic distribution and occurrence in a wide variety of aquatic and terrestrial wildlife species.
Papillomaviruses (PV) are small, non-enveloped DNA viruses known to produce lesions on the skin (warts) and mucous membranes (condylomas) of various species (Houten et al., 2001; Villiers et al., 2004; Dyne et al., 2018). Cottontail rabbits were the first reported animal hosts of PV (Shope and Hurst, 1933), and rabbit PV was the first DNA virus known to be connected to tumor-growth (Orth et al., 1977). PV hosts are widely varied (Campo, 2002; Wosiacki et al., 2005; Diniz et al., 2009; Herbst et al., 2009; Araldi et al., 2014; García-Pérez et al., 2014), but are mostly represented in mammals and birds (Shah et al., 2010). PV isolates are commonly described as “PV types” (Villiers et al., 2004), some of which are linked to the development of epithelial malignancies and some lineages of cancer in humans, which has highlighted PV's medical and epidemiological importance (Van Doorslaer et al., 2015). Most of the effort to understand the evolutionary history of PV has been focused on human papillomaviruses (HPV) and PVs in domestic animals (Howley, 1986; Villiers, 1989; Houten et al., 2001; Filippis et al., 2002; Diniz et al., 2009; Orlando et al., 2011). Only a few studies are available on wildlife hosts (e.g., Bravo et al., 2010; Gottschling et al., 2011), showing the considerable gaps of knowledge regarding our understanding of PV ecology in the wild.
There are over 120 described HPV, from which 30% relate to cancer (Burchell, 2018). Cancer-related HPV are linked to >90% of anal and cervical cancers, ~70% of vaginal and vulvar cancers, and >60% of penile cancer cases worldwide (Centers for Disease Control and Prevention, 2018). In the United States, ~41,000 cases of HPV-related carcinoma cases were reported between 2010 and 2015 (Centers for Disease Control and Prevention, 2018). HPV can be categorized as low or high-risk for cancer development. For example, HPV16 and HPV18 lineages have been described as high-risk lineages in humans (Liu et al., 2018; Ozcagli et al., 2018; Wong and Klapperich, 2018) associated with ~5% of all the cancer cases worldwide, and ~3% of all cancers in United States (Jemal et al., 2013; Plummer et al., 2015).
Disease phylogeography explores the association between biogeographic and phylogenetic features of pathogens (e.g., bacteria, virus, protozoa). Phylogeography has been employed in the study of biodiversity (Domínguez-Domínguez and Vázquez-Domínguez, 2009) to understand the interaction between demographic, genealogic, and environmental processes with species-lineages (Avise, 2000; Domínguez-Domínguez and Vázquez-Domínguez, 2009). Phylogeographic approaches are generally used to assess the impact of historical events on the genetic structure of populations, helping to understand speciation events and the configuration and distribution of extant species (Avise, 2000; Vázquez-Domínguez, 2007; Domínguez-Domínguez and Vázquez-Domínguez, 2009). Disease phylogeography has made major contributions to epidemiology via studies of co-evolution between hosts and viruses, in which viruses and hosts reciprocally affect each other's evolution (Webster et al., 2002; Streicker et al., 2010), reconstructions of disease emergence and spread (Mutreja et al., 2011), and assessments of the historical evolution of epidemics (Cohen, 2000; Altizer et al., 2003).
Phylogeographic approaches for public health can be useful to understand the origins, dispersal, and establishment of pathogens through time and populations with reconstructions of transmission. This allows the prediction of future outbreaks via pathogen spillover—i.e., pathogen transmission between different host species (Shah et al., 2010). Geographical barriers can isolate pathogens facilitating the generation of new species (Meinilä et al., 2004) (i.e., vicariance), forcing virus adaptation to available hosts for a long-term relationship stimulating mutual adaptation—i.e., co-evolution (Shah et al., 2010). Ancestral pathogens can evolve into different lineages over time forming new species, strains, serotypes, or other epidemiological classifications. Some of these lineages have varying virulence across host species. For instance, pathogens in original hosts can be primarily avirulent or virulent only under specific conditions such as immunosuppression. Contrarily, severe symptoms can emerge in newer hosts (Morand et al., 2015).
Our understanding of PV phylogeography come from studies focused mostly on HPV lineages and domestic animals, neglecting potential variation in non-human PV (Ho et al., 1993; García-Vallvé et al., 2005; Gottschling et al., 2007; Pimenoff et al., 2016; Van Doorslaer and McBride, 2016). This species-biased research limits our understanding of the evolutionary history of PV and our abilities to predict and control future PV spillover in humans and animals. Limited availability of non-human papillomavirus information could be behind the limited research on the phylogeography of non-human PV of wildlife origin. Similarly, the burden and distribution of non-human PVs have not been explored in detail. Therefore, we assessed the phylogeography of PV in wildlife hosts, or wildlife papillomavirus (WPV). This study explored the phylogenetic, host, geographic, and environmental relationships of WPV in a multi-scale ecological assessment. There have been previous studies on the origin and evolution of PV that include non-human species (e.g., Van Doorslaer, 2013; Van Doorslaer et al., 2017; Willemsen and Bravo, 2019). We complement this information by focusing analyses on whole genome sequences from WPV, and environmental, and geographic reconstructions. Our goal was to respond to the questions: Are WPV found under tractable and quantifiable phylogenetic, geographic, and environmental conditions? Can we reconstruct the evolutionary history of the virus? And, can the available data reveal ecological patterns of WPV and its hosts? Exploring these questions can help elaborate new hypotheses of WPV evolution to guide future research in disease ecology and biogeography. A potential new hypothesis may include identification of the origins and paths of dispersal of WPV, which can be assessed using well designed phylogeographic analysis with data not yet available.
We reviewed PV records in animals from scientific literature using the keywords “wildlife” + “papillomavirus” + “genome” in the Web of Science and Scopus databases to collect the most available reports of genetic information worldwide to understand the evolutionary history of the virus. Because our exploration in the Web of Science and Scopus resulted in a limited amount of studies (Web of Science n = 7; Scopus n = 31), we gathered complementary scientific publications in Google Scholar using the same keywords, reviewing studies manually to include only literature matching our inclusion criteria. Our review criteria included PV records with GenBank identification, geographic origin, host species, and reported symptomatology (e.g., asymptomatic, fibropapilloma, carcinoma). Reports without reliable location were removed from the analysis. To better assess the human-wildlife interface (i.e., geographic and temporal overlap between both humans and wildlife, resulting in potential exchange of pathogens (Gortazar et al., 2015), we selected reports with complete genome sequences stored in the GenBank, complemented with PV genomes of human and domestic animals to reconstruct the phylogenetic relationships among wild and domesticated PV lineages.
Sequences were aligned using Mega X (Kumar et al., 2018). Given that the sequences analyzed belonged to the same virus species, we assumed a constant mutation rate and applied the Unweighted Pair Group Method with Arithmetic Mean for the alignment of the sequences, as recommended by the developer (Kumar et al., 2018). Phylogenetic trees were generated by a discrete phylogeography estimation by Bayesian inference through Markov chain Monte Carlo (MCMC), implemented in BEAST v2.5.0 (Bouckaert et al., 2014), through a Hasegawa-Kishino-Yano (HKY) model to account for occurrence of nucleotides at different frequencies, transitions, and transversions occurring at different rates with discrete gamma rate heterogeneity amongst sites (Smith et al., 2009; Sana et al., 2018). Our whole genome matrix was partitioned, following previously described nucleotide positions 1–3702 and 3703–7077 (Gottschling et al., 2007). The statistical uncertainty in parameter values across the sampled tree is given by the 95% highest probability density (HPD) values. All analyses were developed for 200 million generations, sampling every 10,000th generation and removing 10% as chain burn-in following recommendations of the developer (Bouckaert et al., 2014). All the Markov chain Monte Carlo analyses were investigated using Tracer software v1.7 (Rambaut et al., 2018) to ensure adequate effective sample sizes (ESS) (>200), which were obtained for all parameters. The final tree was summarized and visualized via Tree Annotator v. 2.3.0 and FigTree 1.4.3, respectively (included in BEAST v2.5.0) (Rambaut and Drummond, 2016; Rambaut, 2017).
To reconstruct WPV phylogeographic radiation, we characterized the geographic distribution of hosts of the reported PV cases considered in this study. We identified the present-day distribution of each host at the continental-level (i.e., Africa, Asia, Europe, North America, South America, Oceania, and Antarctica) due to uncertainties found in the site locations of WPV reports. Host distributions were based on the species' geographic range defined by the International Union for Conservation of Nature (IUCN) (Cosiaux et al., 2016). To assess the most likely geographic origin of the available WPV, we employed Bayesian Binary Markov Chain Monte Carlo (BBM) method analysis to reconstruct the ancestral regions occupied by PV, as previously used in other studies (Durães-Carvalho et al., 2015; Lauron et al., 2015; Forni et al., 2018). BBM was implemented using the software Reconstruction of Ancestral States in Phylogenies, RASP v3.2 (Yu et al., 2015). RASP calculates the probability that a certain parameter representing the origin of a sample was obtained randomly, assessing the quality of the input data. BBM offers a statistical procedure for inferring ancestral states, including geographic distributions at ancestral nodes. BBM assessment uses a full hierarchical Bayesian approach combining the likelihood of the data observed and the probability of events to happen in the order that they did (Ronquist, 2004). We chose to use BBM because it allows null character status information for a portion of input sequences and reduces the uncertainty in the reconstructions of ancestral areas, by reducing the risks of type I error (rejecting the true frequency of monophyletic groups). This attaches a high confidence in large numbers of correct internodes and increases the accuracy of the Bayesian posterior probabilities (Larget and Simon, 1999; Alfaro et al., 2003; Ronquist, 2004).
To account for phylogenetic uncertainty in this analysis, Markov Chain Monte Carlo (MCMC) analysis was conducted simultaneously for one million generations using 10 chains, sampling every 100 generations while the first 25% of generations were discarded as burn-in. Fixed (JC) + G (Jukes-Cantor + Gamma) were used with null root distribution and the temperature was set to 0.1, facilitating the most efficient chain-swapping (Huelsenbeck and Ronquist, 2005). Finally, lineages through time analysis (LTT) was implemented in Tracer (Rambaut et al., 2018) and performed on the previously obtained phylogenetic tree to plot how fast PV lineages have originated over time and assess whether lineage diversification was gradual, or whether it occurred earlier or more recently in time.
To reconstruct the distributions of WPV host species, we revised the geographic ranges of each species for which we found WPV reports according to the IUCN Red List open-access repository (Cosiaux et al., 2016). To assess how many hosts could potentially gather in a specific geographical place, we calculated the host species richness by using spatial analysis in macroecology (SAM) software (Rangel et al., 2010). We created a GIS grid shapefile of the distribution of all hosts species using 50 km spatial scale. We calculated species richness for each taxonomic class to assess species' geographic overlaps. Final maps were created using ArcGIS v10.5 (Environmental Systems Research Institute, 2017).
We used NicheA software to determine the environmental space occupied by WPV and wildlife hosts (Qiao et al., 2016). Specifically, we mapped the distribution of viruses and hosts in environmental dimension. To generate the environmental dimension, we developed a principal component analysis in NicheA based on 19 satellite-derived global bioclimatic variables (2000s decade; 2.5 arcmin resolution) from MERRAclim with information of global temperature and humidity (Vega et al., 2017). The environmental distribution of WPV was estimated using a convex polyhedron calculated around the WPV records and raster of host ranges in terms of the first three principal components (Qiao et al., 2016). We quantified the environmental overlap between WPV and wildlife hosts by taxonomic class using the Jaccard similarity index (Jaccard, 1912). This index is used in ecology to assess the overlap between two organisms in terms of species traits, use of resources, geographic distribution, temporal distribution, and environmental distribution (see Jaccard, 1912; Escobar et al., 2015).
A total of 92 PV records were used to reconstruct the distributions of viruses and host species (Supplementary Table S1). Most samples originated from Europe and North America, presenting notable differences between the numbers of lineages described and their frequency and type of symptomatology. Fibropapilloma was the most frequent form of lesion reported in all continents, and North America and Europe reported all forms of lesions (i.e., carcinoma, fribropapilloma, asymptomatic) (Figure 1A). Records of asymptomatic hosts were mainly found in Europe. A considerable number of WPV records originated from veterinary reports and captive wildlife species (see Supplementary Table S1). The distribution of WPV hosts clustered in the northern Hemisphere (Figure 1B), with strong richness of hosts mammals in North America and Europe. Reports of PV in birds occurred in most continents except Africa and Australia. PVs in bats were reported in Asia and North America, and only North America and Europe reported PVs in rabbits and reptiles (Figure 1B). Marine and terrestrial mammals were the group of hosts covering most of the geographic ranges used by WPV hosts, although the different classes of hosts presented different geographic ranges (Figure 2). The WPV-infected mammals were reported more often in the northern Hemisphere, with less samples coming from the Neotropic, Africa, and Oceania. Bird hosts gathered mostly in Europe and the coastal areas of the Antarctic. There were only three reports of reptiles, one snake and two marine turtle species, all with broad geographic distributions.
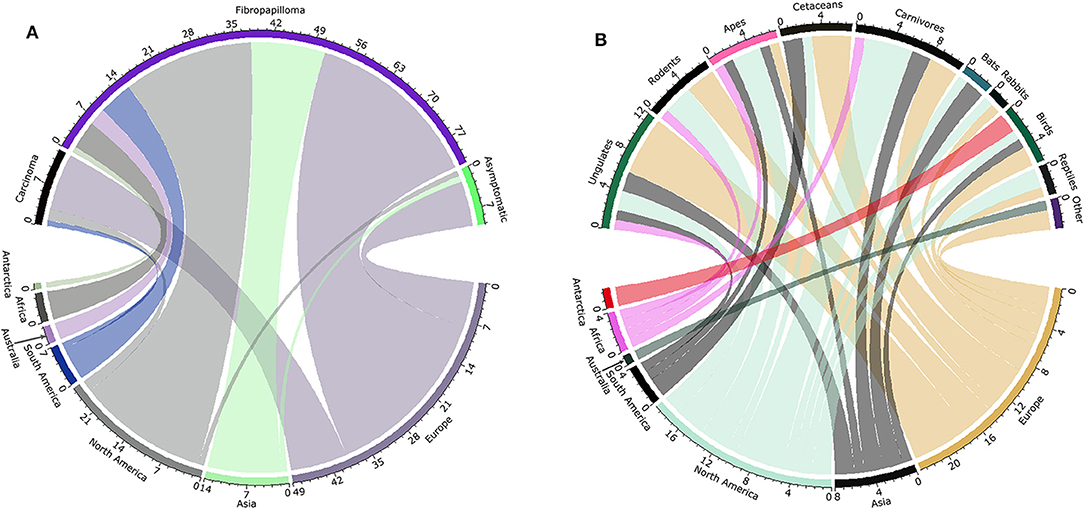
Figure 1. Wildlife papillomavirus (WPV) reports by continent, symptomatology, and host. (A) Frequency of WPV symptoms (black: carcinoma, purple: fibropapilloma, green: asymptomatic) by region (olive: Antarctic, dark gray: Africa, blue: South America, light gray: North America: light green: Asia, gray: Europe). (B) Frequency of reports by continent (red: Antarctica; pink: Africa; olive: Australia; black: South America; light green: North America; black: Asia; yellow: Europe) and host group (green: ungulates; black: rodents; pink: apes; black: cetaceans; black: carnivores; blue: bats; black: rabbits; dark green: birds; black: reptiles; purple: Other species, i.e., marsupials, manatees). Both chord diagrams show the high frequency of reports from the northern Hemisphere (North America and Europe).
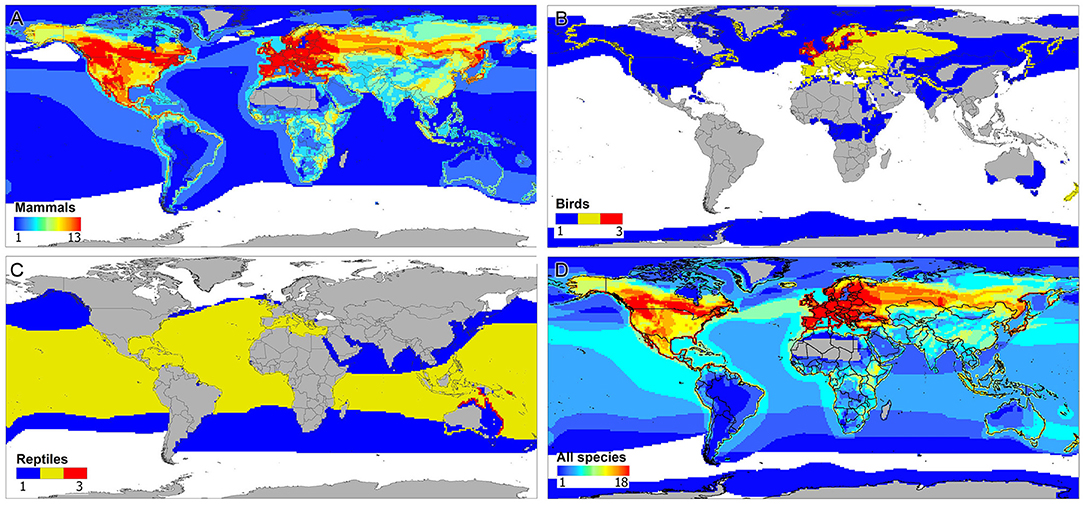
Figure 2. Geographic distribution of wildlife hosts of papillomavirus (WPV). Maps show areas with high (red), moderate (yellow), and low (blue) numbers of WPV hosts species. (A) Mammals. Note that WPV-infected mammals have been reported in the Americas, Europe, Asia, Africa, and Oceania. According to the observations, hotspots of host mammals is expected in the northern Hemisphere. (B) Birds. Note that WPV-infected birds have been reported in coastal areas of the Antarctic and Europe, with considerable overlap of species in northern Europe. (C) Reptiles. Only three reports were found for WPV in reptiles, two turtle species and one snake. (D) Overall distribution of WPV hosts with WPV genomes reported in the literature. Note that most field records of WPV-infected animals are clustered in the northern Hemisphere, resulting in a high richness of WPV hosts in the United States and Canada in North America and in central-western Eurasia.
The phylogenetic relationships revealed significant clusters of WPV from hosts sharing evolutionary history (e.g., from the same class). That is, PV lineages found to be phylogenetically related tended to infect hosts from the same clade (Figure 3). However, these associations were not perfect (see Figure 3). The addition of human papillomavirus (HPV16 and HPV18) to the phylogeny revealed a cluster of PV from apes and humans. Ancestral reconstruction and LTT analyses, based on the data available, show that the most likely origin of the common ancestor of the analyzed lineages is Europe, followed by North America and Africa (Figure 4A), from which WPV gradually diverged to the current lineages (Figure 4B).
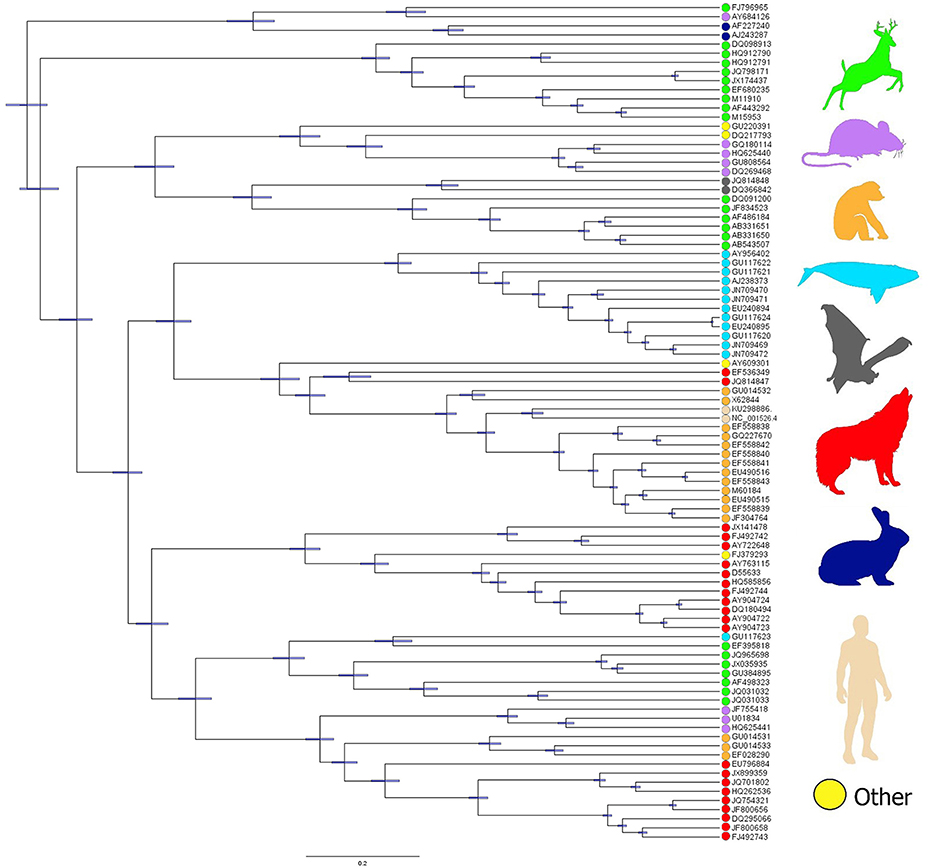
Figure 3. Phylogenetic distribution of WPV lineages in mammals. Dots denote the host species reported according to each WPV lineage. Green: ungulates; purple: rodents; orange: apes: light blue: cetaceans; gray: bats; red: carnivores; dark blue: rabbits (lagomorphs); light pink: humans; yellow: other hosts (FJ379293: hedgehog; AY609301: manatee; GU220391: marsupial).
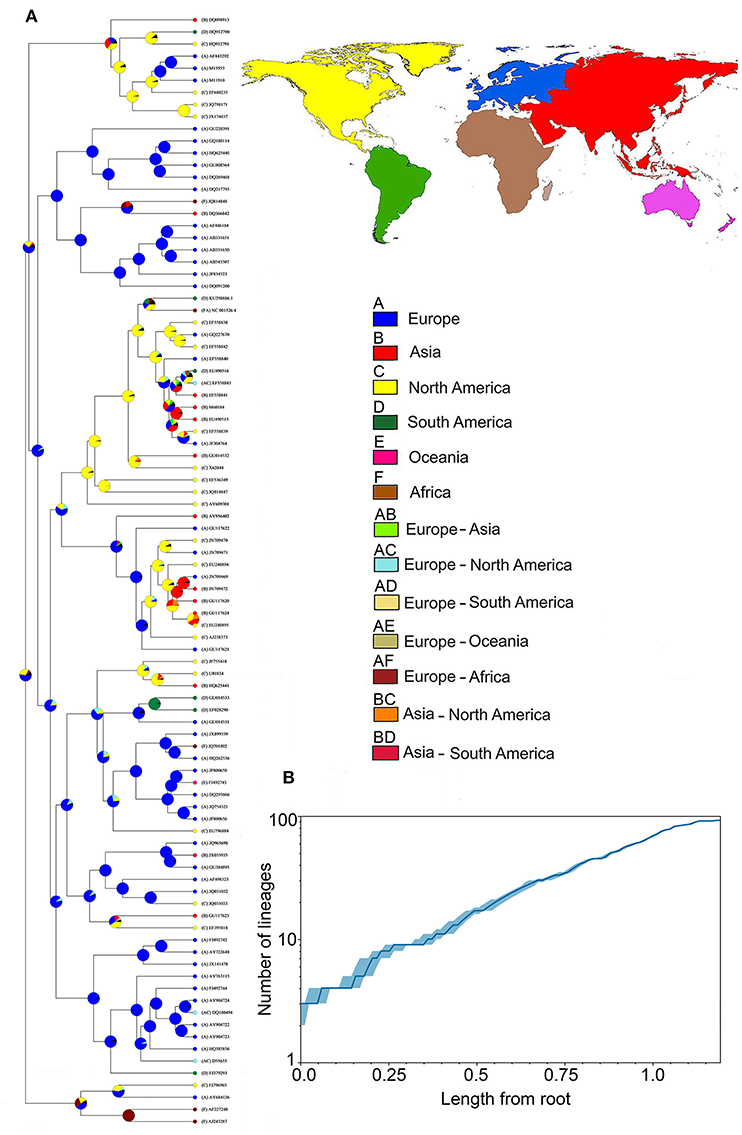
Figure 4. Phylogeography of papillomavirus in wildlife (WPV). (A) Reconstruction of the evolutionary history of mammalian WPV; Tree tips: GenBank ID numbers of the analyzed samples; Nodes: most likely origin of the lineages at that time-point, being (a) blue: Europe; (b) red: Asia; (c) yellow: North America; (d) green: South America; (e) fuchsia: Oceania; and (f) brown: Africa. The combination of two continents includes: (ab) light green: Europe/Asia; (ac) light blue: Europe/North America; (ad) beige: Europe/South America; (ae) light brown: Europe/Oceania; (af) terracotta: Europe/Africa; (bc) orange: Asia/North America; (bd) light red: Asia/South America; (B) Rate of lineage diversification (i.e., number of appearing lineages over time; solid line) showing the magnitude of WPV geographic spread (x axis) in relation to the appearance of new lineages (y axis). The median estimated of the number of lineages through time is represented by the dark blue. The 95% highest posterior density confidence intervals are marked in light blue.
Environmental space occupied by reported WPV cases and their wildlife hosts suggested that WPV infections in reptiles are mostly seen in a specific subset of environmental conditions, and that host species in this class occupy territories with environmental conditions beyond those where WPV infections have been recorded (Figure 5). Contrarily, WPV infections in birds and mammals were seen in environmental conditions that overlapped broadly with the space occupied by hosts. This suggests that the environmental range where WPV have been recorded from mammals and birds was similar to the entire environmental range of their host species. These results were supported by the similarity analyses, which revealed that reptiles had a slightly lower similarity observed (Jaccard Index = 0.23) between the environments occupied by the reported WPV and their host as compared with birds and mammals (Jaccard Index = 0.29 and 0.28, respectively) (Figure 5). Thus, WPV in mammals were found throughout the whole geographic range and environments occupied by their hosts.
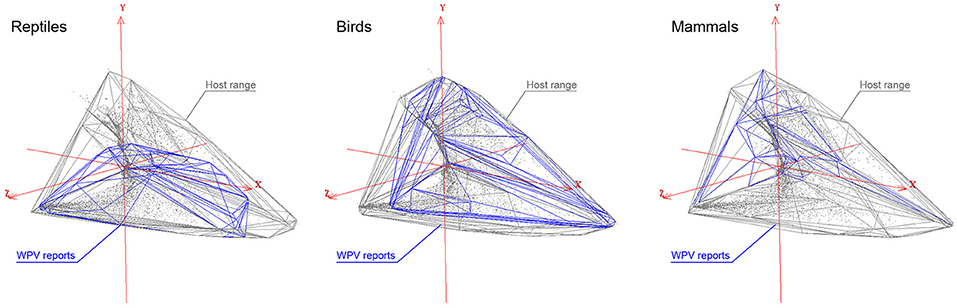
Figure 5. Environmental distribution of the WPV hosts. The first three principal components (X, Y, Z axes) from the original bioclimatic data representing the three-dimensional environmental space (red lines). Distribution of WPV hosts (gray convex polyhedron) and WPV-reports (blue convex polyhedron) are displayed in the environmental space. From left to right: reptiles, birds, and mammals. Gray dots represent single environmental conditions available in the world.
We assessed eco-phylogenetic patterns of wildlife papillomavirus (WPV). We were able to reconstruct evolutionary, distributional, and ecological signatures of PV lineages using available data to date. Results suggest Europe as the most likely geographic origin of the available lineages. Evolutionary linkages between virus and hosts show linkages between WPV similarity and hosts species infected. The phylogeny of PV did not mirror the host phylogeny, aligned with patterns found in previous studies. The coarse scale reconstruction of WPV distribution identified consistent environmental conditions where WPV can be found, and WPV lineages, geographic locations, and hosts species that deserve future surveillance.
Ancestral reconstruction analyses are widely used to understand the origin and evolution of pathogens (Bargues et al., 2006; Bourhy et al., 2008; Paraskevis et al., 2013; Hayman et al., 2016). Here, available WPV reports allowed us to produce a preliminary overview of the evolutionary history and distribution of papillomaviruses in wildlife species, and to reveal the remarkable gaps of data and surveillance. Our ecological and evolutionary approach explored WPV coevolution and spillover, geographic range of virus and host, and plausible origins that would not be considered in classic epidemiological studies (Galvani, 2003; Faria et al., 2011). A better understanding of the ecology and evolution of WPV may help to anticipate outbreaks in wildlife species and will us help to understand the potential emergence of new pathogenic WPV lineages in humans and livestock (Inhorn and Brown, 1990; Price, 2002; Nasir and Campo, 2008; Trewby et al., 2014).
The ancestral reconstruction revealed a high likelihood of diversification and appearance of new lineages consistently and gradually over time, suggesting that WPV diversification would be linked to host diversification, in agreement with a previous assessment (Shah et al., 2010). This host diversification, along with domestication and the ongoing globalization of PV hosts, could explain the similarity between the homogeneity of genomes collected from different parts of the globe, which poses an analogy to the invasive processes described by Scheele et al. (2019), where they found high genetic diversity in the areas of origin of the studied pathogen but low diversity (i.e. homogenization) on the invaded areas (Scheele et al., 2019). The fact that WPV and their wildlife hosts can be found across broad environmental conditions (Figure 5) suggests that the virus could be infecting wildlife hosts distributed in the southern Hemisphere in areas and species beyond those available to us, at least in the host classes evaluated here (Figures 2, 5). Thus, more efforts are necessary to fill taxonomic and geographic gaps by collecting data from (i) novel species to understand potential WPV ability to infect a wider variety of hosts, (ii) surveillance efforts in neglected systems such as marine, freshwaters, and the Arctic ecosystems, and (iii) sampling neglected locations where known hosts occur, which will facilitate the identification of virus-host codivergence (vicariance) and potential variations in virulence across hosts' populations and geographic regions.
A limited number of wildlife host-species from a broad range of taxa suggests the existence of undiscovered WPV, especially from mammals phylogenetically close to the species included in this study, aligned with reports for other pathogens (Olival et al., 2017). From a clinical point of view, a potential driver of WPV linked to fibropapilloma can be associated to the biased sampling of symptomatic individuals, impeding the assessment of WPV subclinical circulation. It has been previously described that some species are often infected by a different number of PV belonging to a variety of taxonomic groups (García-Vallvé et al., 2005), which, along with the presence of spillover events, can be one of the key factors for WPV transmission success. For example, Nasir and Campo (2008) described in detail how Bovine papillomavirus (BPV) can induce benign tumors in cattle (its main host) but also fibroblastic tumors in equids (Nasir and Campo, 2008), spillover that has lately been corroborated by Trewby et al. (2014) via the analysis of its evolutionary history and cross-species link with equids. However, despite the propensity of sister species to participate in PV spillover, PV phylogenies do not necessarily mirror the phylogenies of their hosts (Chan et al., 1995; García-Vallvé et al., 2005). Thus, a more comprehensive survey of PV lineages in specific wildlife species of broad distributions would increase the understanding on WPV transmission and the likely scenarios of spillover to domesticated animals. As anecdotal note, during the search for WPV records we found clinical and pathological PV reports from diverse wildlife species, however, genetic analyses were generally absent. Routine genetic characterizations of WPV and data sharing is a next challenge for PV research.
The global geographic distribution of WPV (Figures 1, 2), its considerable environmental tolerances (Figure 5), and the enormous range of taxa reported to be infected and symptomatic (Figure 1B and Supplementary Table S1), support the idea of WPV as an ecological generalist pathogen, making it suitable to study spillover and co-evolution patterns in the animal kingdom.
Other pathogens like rabies and avian influenza have served to understand co-evolutionary forces of viruses and hosts (Streicker et al., 2010, 2012; Dijk et al., 2015). Thus, PV may be a model system to understand the ecology and evolution of infectious diseases due to its broad range host classes, distributional areas, and environments colonized compared to other viruses. In this sense, WPV can infect a broad range of species including marine and terrestrial mammals, birds, and reptiles across continents and across impressive ecological gradients (Supplementary Table S1). Our exploration may be useful to promote generation of WPV data to better inform veterinary epidemiology, regarding potential emergence of novel WPV in wildlife or livestock and identify scenarios and locations of likely WPV transmission. Enhanced WPV surveillance can also help to elucidate patterns of virus evolution, host adaptation, and virulence. From an ecological perspective, WPV brings an opportunity to study a single pathogen colonizing diverse ecosystems, taxa, and regions, which is urgently needed in disease biogeography to better understand how global change can impact disease transmission.
We found that WPV hosts reported on the available data have a global geographic distribution. Similarly, WPV infections observed in mammals and birds were seen in nearly the complete host's range. The capacity of some WPV lineages to overlap almost the entire host distribution suggests the potential of WPV to infect novel species present in environments and geographic areas overlapping the distribution of known hosts. This was supported by finding WPV mammal hosts in aquatic and terrestrial ecosystems, and from birds present in all the continents, including Antarctic regions (Figure 2). We argue that, because multiple WPV can co-infect a single host (Shah et al., 2010), it is possible that sympatric host species could be co-infected with diverse WPV, facilitating the generation of novel papilloma lineages, or that a single WPV lineage could spill over different host taxa and result in a successful establishment of the virus in the new host, as in the case of bovine PV (BPV1) transmission from cattle to horses (Nasir and Campo, 2008; Trewby et al., 2014). This spillover and establishment phenomena has been observed for other zoonotic viruses of wildlife origin in spillover events from wildlife to humans (e.g., Hendra virus and Nipah virus) with establishment of the virus in the naïve species (Plowright and Eby, 2015). These complex and usually unnoticed spillover events could trigger the emergence of novel pathogens through the combination of potential uninfected hosts (humans, wildlife, and livestock), infected host reservoirs, and environmental conditions suitable to maintain the infection (Wu et al., 2018; Johnson et al., 2019). Zoonotic viruses' most common reservoirs are rodents, primates, bats, and domestic animals (Alexander et al., 2018), whose taxonomy and geographical location have been described as the main pillars for these spillover events to occur (Wu et al., 2018), which can also be the main drivers of spillover in WPV.
Our results revealed two forms of potential sampling bias, one expressed as a high number of reports from regions in the northern Hemisphere and another related to symptomatic individuals from studies reporting WPV that produce skin lesions (Villiers, 1989; Villiers et al., 2004; Stevens et al., 2010; Rector and Van Ranst, 2013). We note that our results were restricted to samples available to us and may vary as the number and distribution of available reports increase (Cusimano and Renner, 2010). However, some patterns detected by this study could be a stepping-stone for the assessment of ecological and anthropogenic factors facilitating or limiting WPV spread. Reconstructions of the areas where pathogens could or could not occur have been developed for other infectious diseases, such as Influenza, Filovirus, and Bluetongue viruses, among others (Peterson et al., 2004; Peterson, 2006; Charland et al., 2009; Pioz et al., 2012), facilitating surveillance efforts and pathogen discovery. Importantly, the possibility of the cross-infection between host species (i.e., virus spillover) cannot be ruled out in captive hosts. Thus, our exploration of WPV hosts could guide future surveillance efforts in the wildlife-domestic interface.
An interesting finding is the fact that WPV demonstrates the ability to infect and cause lesions across diverse wildlife species. This highlights the possibility of WPV spreading through naïve populations of the species reported here, which could be of particular concern in the conservation of some species already threatened due to genetic isolation and habitat loss (e.g., apes) (Goossens et al., 2006). This finding may generate new research questions, including whether virulence decreases in the most ancestral host of the virus and whether reducing sampling bias could modify some of the patterns detected in this preliminary overview. Our preliminary maps of WPV distribution can also help identify risk areas for future WPV surveillance, and most importantly, the regions where more surveillance effort is necessary (e.g., tropics). Thus, we argue that a main contribution of this study is the identification of data limitations to identify robust macroecological and phylogeographic signatures of wildlife papillomavirus. Future research should also explore the environmental drivers of transmission, circulation, and virulence of WPV at the local level to determine the most suitable landscape conditions where WPV spillover may occur (Pearman et al., 2008; Peterson, 2014).
Finally, this study represents a preliminary global exploration of the phylogeographic structure of papillomavirus in wildlife, including their geographic distribution to increase our understanding on the recent expansions of virus lineages, the recent appearance of spillover events, and the likely path toward PV origin and evolution. Because it has been suggested that WPV tumors emerge under conditions of stress in the host (Pereira et al., 2003), PV surveillance in free-ranging wildlife may serve as a measure of ecosystem health and as an early warning system of environmental pollution and global change effects over wildlife.
All datasets generated for this study are included in the manuscript/Supplementary Files.
AF-D-D contributed to the design of the project, performed the data collection, the phylogenetic analyses, and wrote the manuscript. MJ performed the spatial and ecological niche analyses and wrote the manuscript. LE contributed to the design of the project, supervised the methodology, and wrote the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Authors thank Paige Van de Vuurst and Steven N. Winter for their helpful comments and suggestions. Comments from the reviewers and editor were extremely helpful and greatly improved the content of this manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2019.00406/full#supplementary-material
Alexander, K., Carlson, C., Lewis, B., Getz, M. E., Madhav, V. M., Eubank, S. G., et al. (2018). “The ecology of pathogen spillover and disease emergence at the human- wildlife-environment interface,” in The Connections Between Ecology and Infectious Disease, eds C. Hurst (Cham:Springer), 267–298. doi: 10.1007/978-3-319-92373-4_8
Alfaro, M. E., Zoller, S., and Lutzoni, F. (2003). Bayes or bootstrap? A simulation study comparing the performance of Bayesian Markov chain Monte Carlo sampling and bootstrapping in assessing phylogenetic confidence. Mol. Biol. Evol. 20, 255–266. doi: 10.1093/molbev/msg028
Altizer, S., Harvell, D., and Friedle, E. (2003). Rapid evolutionary dynamics and disease threats to biodiversity. Trends Ecol. Evol. 18, 589–596. doi: 10.1016/j.tree.2003.08.013
Araldi, R. P., Carvalho, R. F., Melo, T. C., Diniz, N. S., Sant'Ana, T. A., Mazzuchelli-de-Souza, J., et al. (2014). Bovine papillomavirus in beef cattle: first description of BPV-12 and putative type BAPV8 in Brazil. Genet. Mol. Res. 13, 5644–5653. doi: 10.4238/2014.July.25.20
Avise, J. (2000). Phylogeography: The History and Formation of Species. Cambridge. Harvard University Press.
Bargues, M. D., Klisiowicz, D. R., Panzera, F., Noireau, F., Marcilla, A., Perez, R., et al. (2006). Origin and phylogeography of the Chagas disease main vector Triatoma infestans based on nuclear rDNA sequences and genome size. Infect. Genet. Evol. 6, 46–62. doi: 10.1016/j.meegid.2005.01.006
Bouckaert, R., Heled, J., Kühnert, D., Vaughan, T., Wu, C. H., Xie, D., et al. (2014). BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 10:e1003537. doi: 10.1371/journal.pcbi.1003537
Bourhy, H., Reynes, J. M., Dunham, E. J., Dacheux, L., Larrous, F., Huong, V., et al. (2008). The origin and phylogeography of dog rabies virus. J. Gen. Virol. 89, 2673–2681. doi: 10.1099/vir.0.2008/003913-0
Bravo, I. G., Sanjosé, S., and Gottschling, M. (2010). The clinical importance of understanding the evolution of papillomaviruses. Trends Microbiol. 18, 432–438. doi: 10.1016/j.tim.2010.07.008
Burchell, A. N. (2018). One step closer in understanding the natural history of anal infection with high-risk human papillomavirus. J. Infect. Dis. 218, 1015–1017. doi: 10.1093/infdis/jiy277
Campo, M. S. (2002). Animal models of papillomavirus pathogenesis. Virus Res. 89. 249–261. doi: 10.1016/S0168-1702(02)00193-4
Centers for Disease Control Prevention. (2018). Cancers associated with human papillomavirus, United States, 2011–2015. Available online at: https://www.cdc.gov/cancer/hpv/pdf/USCS-DataBrief-No4-August2018-508.pdf (accessed March 20, 2019).
Chan, S. Y., Delius, H., Halpern, A. L., and Bernard, H. U. (1995). Analysis of genomic sequences of 95 papillomavirus types: uniting typing, phylogeny, and taxonomy. J. Virol. 69, 3074–3083.
Charland, K. M., Buckeridge, D. L., Sturtevant, J. L., Melton, F., Reis, B. Y., Mandl, K. D., et al. (2009). Effect of environmental factors on the spatio-temporal patterns of influenza spread. Epidemiol. Infect. 137, 1377–1387. doi: 10.1017/S0950268809002283
Cohen, M. L. (2000). Changing patterns of infectious disease. Nature 406, 762–767. doi: 10.1038/35021206
Cosiaux, A., Gardiner, L., and Couvreur, T. (2016). The IUCN Red List of Threatened Species. Available online at: https://www.iucnredlist.org/ (accessed March 20, 2019).
Cusimano, N., and Renner, S. S. (2010). Slowdowns in diversification rates from real phylogenies may not be real. Syst. Biol. 59, 458–464. doi: 10.1093/sysbio/syq032
Dijk, J. G., Fouchier, R. A., Klaassen, M., and Matson, K. D. (2015). Minor differences in body condition and immune status between avian influenza virus-infected and noninfected mallards: a sign of coevolution? Ecol. Evol. 5, 436–449. doi: 10.1002/ece3.1359
Diniz, N., Melo, T. C., Santos, J. F., Mori, E., Brando, P. E., Richtzenhain, L. J., et al. (2009). Simultaneous presence of bovine papillomavirus in blood and in short-term lymphocyte cultures from dairy cattle in Pernambuco, Brazil. Genet. Mol. Res. 8, 1474–1480. doi: 10.4238/vol8-4gmr668
Domínguez-Domínguez, O., and Vázquez-Domínguez, E. (2009). Filogeografía: aplicaciones en taxonomía y conservación. Anim. Biodivers. Conserv. 32, 59–70.
Durães-Carvalho, R., Caserta, L. C., Barnabé, A. C., Martini, M. C., Simas, P. V., Santos, M. M., et al. (2015). Phylogenetic and phylogeographic mapping of the avian coronavirus spike protein-encoding gene in wild and synanthropic birds. Virus Res. 201, 101–112. doi: 10.1016/j.virusres.2015.03.002
Dyne, E. A., Henley, S. J., Saraiya, M., Thomas, C. C., Markowitz, L. E., and Benard, V. B. (2018). Trends in human papillomavirus–associated cancers — United States, 1999–2015. Morb. Mortal. Wkly Rep. 67, 918–924. doi: 10.15585/mmwr.mm6733a2
Environmental Systems Research Institute (2017). ArcGIS 10.5.1. Available at: https://www.arcgis.com/ (accessed March 20, 2019).
Escobar, L. E., Ryan, S. J., Stewart-Ibarra, A. M., Finkelstein, J. L., King, C. A., Qiao, H., et al. (2015). A global map of suitability for coastal Vibrio cholerae under current and future climate conditions. Acta Trop. 149, 202–211. doi: 10.1016/j.actatropica.2015.05.028
Faria, N. R., Suchard, M. A., Rambaut, A., and Lemey, P. (2011). Toward a quantitative understanding of viral phylogeography. Curr. Opin. Virol. 1, 423–429. doi: 10.1016/j.coviro.2011.10.003
Filippis, V. R., Ayala, F. J., and Villarreal, L. P. (2002). Evidence of diversifying selection in human papillomavirus type 16 E6 but not E7 oncogenes. J. Mol. Evol. 55, 491–499. doi: 10.1007/s00239-002-2344-y
Forni, D., Cagliani, R., Clerici, M., and Sironi, M. (2018). Origin and dispersal of Hepatitis E virus. Emerg. Microb. Infect. 7:11. doi: 10.1038/s41426-017-0009-6
Galvani, A. P. (2003). Epidemiology meets evolutionary ecology. Trends Ecol. Evol. 18, 132–139. doi: 10.1016/S0169-5347(02)00050-2
García-Pérez, R., Ibáñez, C., Godínez, J. M., Aréchiga, N., Garin, I., Pérez-Suárez, G., et al. (2014). Novel papillomaviruses in free-ranging Iberian bats: no virus–host co-evolution, no strict host specificity, and hints for recombination. Genome Biol. Evol. 6, 94–104. doi: 10.1093/gbe/evt211
García-Vallvé, S., Alonso, A., and Bravo, I. G. (2005). Papillomaviruses: different genes have different histories. Trends Microbiol. 13, 514–521. doi: 10.1016/j.tim.2005.09.003
Goossens, B., Chikhi, L., Ancrenaz, M., Lackman-Ancrenaz, I., and Andau, P. (2006). Genetic signature of anthropogenic population collapse in orangutans. PLoS Biol. 4:e25. doi: 10.1371/journal.pbio.0040025
Gortazar, C., Diez-Delgado, I., Barasona, J. A., Vicente, J., Fuente, J., Boadella, M., et al. (2015). The wild side of disease control at the wildlife-livestock-human interface: a review. Front. Vet. Sci. 1:27. doi: 10.3389/fvets.2014.00027
Gottschling, M., Göker, M., Stamatakis, A., Bininda-Emonds, O. R., Nindl, I., and Bravo, I. G. (2011). Quantifying the phylodynamic forces driving papillomavirus evolution. Mol. Biol. Evol. 28, 2101–2113 doi: 10.1093/molbev/msr030
Gottschling, M., Stamatakis, A., Nindl, I., Stockfleth, E., Alonso, Á., and Bravo, I. G. (2007). Multiple evolutionary mechanisms drive papillomavirus diversification. Mol. Biol. Evol. 24, 1242–1258. doi: 10.1093/molbev/msm039
Hayman, D. T., Fooks, A. R., Marston, D. A., and Garcia, R. J.. (2016). The global phylogeography of Lyssaviruses - Challenging the “out of Africa” hypothesis. PLoS Negl. Trop. Dis. 10:e0005266. doi: 10.1371/journal.pntd.0005266
Herbst, L. H., Lenz, J., Van Doorslaer, K., Chen, Z., Stacy, B. A., Wellehan, J. F., et al. (2009). Genomic characterization of two novel reptilian papillomaviruses, Chelonia mydas papillomavirus 1 and Caretta caretta papillomavirus 1. Virology 383, 131–135. doi: 10.1016/j.virol.2008.09.022
Ho, L., Chan, S. Y., Burk, R. D., Das, B. C., Fujinaga, K., Icenogle, J. P., et al. (1993). The genetic drift of human papillomavirus type 16 is a means of reconstructing prehistoric viral spread and the movement of ancient human populations. J. Virol. 67, 6413–6423.
Houten, V. M., Snijders, P. J., Van Den Brekel, M. W., Kummer, J. A., Meijer, C. J., Van Leeuwen, B., et al. (2001). Biological evidence that human papillomaviruses are etiologically involved in a subgroup of head and neck squamous cell carcinomas. Int. J. Cancer 93, 232–235. doi: 10.1002/ijc.1313
Huelsenbeck, J. P., and Ronquist, F. (2005). “Bayesian analysis of molecular evolution using MrBayes,” in Statistical Methods in Molecular Evolution, eds R. Nielsen (New York, NY: Springer), 183–232. doi: 10.1007/0-387-27733-1_7
Inhorn, M., and Brown, P. (1990). The anthropology of infectious disease. Annu. Rev. Anthropol. 19, 89–117. doi: 10.1146/annurev.an.19.100190.000513
Jaccard, P. (1912). The distribution of the flora in the Alpine zone. N. Phytol. 11, 37–50. doi: 10.1111/j.1469-8137.1912.tb05611.x
Jemal, A., Simard, E. P., Dorell, C., Noone, A.-M., Markowitz, L. E., Kohler, B., et al. (2013). Annual report to the nation on the status of cancer, 1975-2009, featuring the burden and trends in human papillomavirus (HPV)-associated cancers and HPV vaccination coverage levels. J. Natl. Cancer Inst. 105, 175–201. doi: 10.1093/jnci/djs491
Johnson, E. E., Escobar, L. E., and Zambrana-Torrelio, C. (2019). An ecological framework for modeling the geography of disease transmission. Trends Ecol. Evol. 34, 655–668. doi: 10.1016/j.tree.2019.03.004
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Larget, B., and Simon, D. (1999). Markov chain Monte Carlo algorithms for the Bayesian analysis of phylogenetic trees. Mol. Biol. Evol. 16, 750–759. doi: 10.1093/oxfordjournals.molbev.a026160
Lauron, E. J., Loiseau, C., Bowie, R. C. K., Spicer, G. S., Smith, T. B., Melo, M., et al. (2015). Coevolutionary patterns and diversification of avian malaria parasites in African sunbirds (Family Nectariniidae). Parasitology, 142, 635–647. doi: 10.1017/S0031182014001681
Liu, T., Zhang, H., Yang, X., Li, X., Shi, Y., Niu, W., et al. (2018). Study on expression of p16 and human papillomavirus 16 and 18 (E6) in OLP and its malignant transformation. Pathol. Res. Pract. 214, 296–302. doi: 10.1016/j.prp.2017.09.014
Meinilä, M., Kuusela, J., Ziętara, M. S., and Lumme, J. (2004). Initial steps of speciation by geographic isolation and host switch in salmonid pathogen Gyrodactylus salaris (Monogenea: Gyrodactylidae). Int. J. Parasitol. 34, 515–526. doi: 10.1016/j.ijpara.2003.12.002
Morand, S., Krasnov, B. R., and Littlewood, D. T. (2015). “Evolutionary ecology meets phylogenetics,” in Parasite Diversity and Diversification, ed L. A. Weinert (Cambridge: Cambridge University Press), 150–181. doi: 10.1017/CBO9781139794749
Mutreja, A., Kim, D. W., Thomson, N. R., Connor, T. R., Lee, J. H., Kariuki, S., et al. (2011). Evidence for several waves of global transmission in the seventh cholera pandemic. Nature 477, 462–465. doi: 10.1038/nature10392
Nasir, L., and Campo, M. S. (2008). Bovine papillomaviruses: their role in the aetiology of cutaneous tumours of bovids and equids. Vet. Dermatol. 19, 243–254. doi: 10.1111/j.1365-3164.2008.00683.x
Olival, K. J., Hosseini, P. R., Zambrana-Torrelio, C., Ross, N., Bogich, T. L., and Daszak, P. (2017). Host and viral traits predict zoonotic spillover from mammals. Nature 546, 646–650. doi: 10.1038/nature22975
Orlando, P. A., Gatenby, R. A., Giuliano, A. R., and Brown, J. S. (2011). Evolutionary ecology of human papillomavirus: trade-offs, coexistence, and origins of high-risk and low-risk types. J. Infect. Dis. 205, 272–279. doi: 10.1093/infdis/jir717
Orth, G., Breitburd, F., Favre, M., and Croissant, O. (1977). Papillomaviruses: possible role in human cancer. Origins Hum. Cancer 4, 1043–1068.
Ozcagli, E., Biri, A., Dinc, B., and Sardas, S. (2018). How does infection with human papillomavirus 16 and 18 impact on DNA damage and repair in cervical cells and peripheral blood? OMICS J. Integr. Biol. 22, 332–336. doi: 10.1089/omi.2018.0065
Paraskevis, D., Magiorkinis, G., Magiorkinis, E., Ho, S. Y., Belshaw, R., Allain, J. P., et al. (2013). Dating the origin and dispersal of Hepatitis B virus infection in humans and primates. Hepatology 57, 908–916. doi: 10.1002/hep.26079
Pearman, P. B., Guisan, A., Broennimann, O., and Randin, C. F. (2008). Niche dynamics in space and time. Trends Ecol. Evol. 23, 149–158. doi: 10.1016/j.tree.2007.11.005
Pereira, D. B., Antoni, M. H., Danielson, A., Simon, T., Efantis-Potter, J., Carver, C. S., et al. (2003). Life stress and cervical squamous intraepithelial lesions in women with human papillomavirus and human immunodeficiency virus. Psychosom. Med. 65, 427–434. doi: 10.1097/01.PSY.0000041620.37866.89
Peterson, A. T. (2006). Ecological niche modelling and understanding the geography of disease transmission. Vet. Ital. 43, 393–400.
Peterson, A. T. (2014). Disease Transmission Risk: Enriching Models Using Biogeography and Ecology. Baltimore, MD: Johns Hopkins University Press.
Peterson, A. T., Bauer, J. T., and Mills, J. N. (2004). Ecologic and geographic distribution of filovirus disease. Emerg. Infect. Dis. 10:40. doi: 10.3201/eid1001.030125
Pimenoff, V. N., de Oliveira, C. M., and Bravo, I. G. (2016). Transmission between archaic and modern human ancestors during the evolution of the oncogenic human papillomavirus 16. Mol. Biol. Evol. 34, 4–19. doi: 10.1093/molbev/msw214
Pioz, M., Guis, H., Crespin, L., Gay, E., and Calavas, D. (2012). Why did bluetongue spread the way it did? Environmental factors influencing the velocity of bluetongue virus serotype 8 epizootic wave in France. PLoS ONE 7:e43360. doi: 10.1371/journal.pone.0043360
Plowright, R., and Eby, P. (2015). Ecological dynamics of emerging bat virus spillover. Proc. R. Soc. B 282:20142124. doi: 10.1098/rspb.2014.2124
Plummer, M., Martel, C., De Vignat, J., Ferlay, J., Bray, F., and Franceschi, S. (2015). Global burden of cancers attribuible to infections in 2008: a review and syntetic analysis. Lancet Oncol. 13, 607–6015. doi: 10.1016/S1470-2045(12)70137-7
Price, P. W. (2002). Macroevolutionary Theory on Macroecological Patterns. Cambridge:Cambridge University Press. doi: 10.1017/CBO9780511615030
Qiao, H., Peterson, A. T., Campbell, L. P., Soberón, J., Ji, L., and Escobar, L. E. (2016). NicheA: creating virtual species and ecological niches in multivariate environmental scenarios. Ecography 39, 805–813. doi: 10.1111/ecog.01961
Rambaut, A. (2017). FigTree-Version 1.4. 3, a Graphical Viewer of Phylogenetic Trees. Available online at: http://tree.bio.ed.ac.uk/software/figtree/ (accessed October 3, 2018).
Rambaut, A., and Drummond, A. (2016). TreeAnnotator v. 2.4. 3. Available online at: http://beast.community/treeannotator (accessed October 3, 2018).
Rambaut, A., Drummond, A. J., Xie, D., Baele, G., and Suchard, M. A. (2018). Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst. Biol. 67, 901–904. doi: 10.1093/sysbio/syy032
Rangel, T. F., Diniz-Filho, J. A. F., and Bini, L. M. (2010). SAM: a comprehensive application for spatial analysis in macroecology. Ecography 33, 46–50. doi: 10.1111/j.1600-0587.2009.06299.x
Rector, A., and Van Ranst, M. (2013). Animal papillomaviruses. Virology 445, 213–223. doi: 10.1016/j.virol.2013.05.007
Ronquist, F. (2004). Bayesian inference of character evolution. Trends Ecol. Evol. 19, 475–481. doi: 10.1016/j.tree.2004.07.002
Sana, S., Williams, C., Hardouin, E. A., Blake, A., Davison, P., Pegg, J., et al. (2018). Phylogenetic and environmental DNA insights into emerging aquatic parasites: implications for risk management. Int. J. Parasitol. 48, 473–481. doi: 10.1016/j.ijpara.2017.11.002
Scheele, B. C., Pasmans, F., Skerratt, L. F., Berger, L., Martel, A., Beukema, W., et al. (2019). Amphibian fungal panzootic causes catastrophic and ongoing loss of biodiversity. Science 363, 1459–1463. doi: 10.1126/science.aav0379
Shah, S. D., Doorbar, J., and Goldstein, R. A. (2010). Analysis of host-parasite incongruence in papillomavirus evolution using importance sampling. Mol. Biol. Evol. 27, 1301–1314. doi: 10.1093/molbev/msq015
Shope, R. E., and Hurst, E. W. (1933). Infectious papillomatosis of rabbits with a note on the histopathology. J. Exp. Med. 58, 607–624. doi: 10.1084/jem.58.5.607
Smith, G. J. D., Vijaykrishna, D., Bahl, J., Lycett, S. J., Worobey, M., Pybus, O. G., et al. (2009). Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza a epidemic. Nature 459, 1122–1125. doi: 10.1038/nature08182
Stevens, H., Rector, A., and van Ranst, M. (2010). Multiply primed rolling-circle amplification method for the amplification of circular DNA viruses. Cold Spring Harbor Protoc. 2010:pdb-prot5415. doi: 10.1101/pdb.prot5415
Streicker, D. G., Altizer, S. M., Velasco-Villa, A., and Rupprecht, C. E. (2012). Variable evolutionary routes to host establishment across repeated rabies virus host shifts among bats. Proc. Natl. Acad. Sci. U.S.A. 109, 19715–19720. doi: 10.1073/pnas.1203456109
Streicker, D. G., Turmelle, A. S., Vonhof, M. J., Kuzmin, I. V., McCracken, G. F., and Rupprecht, C. E. (2010). Host phylogeny constrains cross-species emergence and establishment of rabies virus in bats. Science 329, 676–679. doi: 10.1126/science.1188836
Trewby, H., Ayele, G., Borzacchiello, G., Brandt, S., Campo, M. S., Del Fava, C., et al. (2014). Analysis of the long control region of bovine papillomavirus type 1 associated with sarcoids in equine hosts indicates multiple cross-species transmission events and phylogeographical structure. J. Gen. Virol. 95:2748. doi: 10.1099/vir.0.066589-0
Van Doorslaer, K. (2013). Evolution of the papillomaviridae. Virology 445, 11–20. doi: 10.1016/j.virol.2013.05.012
Van Doorslaer, K., DeSalle, R., Einstein, M. H., and Burk, R. D. (2015). Degradation of human PDZ-proteins by human alphapapillomaviruses represents an evolutionary adaptation to a novel cellular niche. PLoS Pathog. 11:e1004980. doi: 10.1371/journal.ppat.1004980
Van Doorslaer, K., and McBride, A. A. (2016). Molecular archeological evidence in support of the repeated loss of a papillomavirus gene. Sci. Rep. 6:33028. doi: 10.1038/srep33028
Van Doorslaer, K., Ruoppolo, V., Schmidt, A., Lescroël, A., Jongsomjit, D., Elrod, M., et al. (2017). Unique genome organization of non-mammalian papillomaviruses provides insights into the evolution of viral early proteins. Virus Evol. 3:vex027. doi: 10.1093/ve/vex027
Vázquez-Domínguez, E. (2007). “Filogeografía y vertebrados,” in Ecología Molecular, eds L. E. Eguiarte, V. Souza and X. Aguirre (México: Secretaría de Medio Ambiente y Recursos Naturales; Instituto Nacional de Ecología; Universidad Nacional Autónoma de México; Comisión Nacional para el Conocimiento y Uso de la Biodiversidad, 441–466.
Vega, G. C., Pertierra, L. R., and Olalla-Tárraga, M. Á. (2017). MERRAclim, a high-resolution global dataset of remotely sensed bioclimatic variables for ecological modelling. Sci. Data 4:170078. doi: 10.1038/sdata.2017.78
Villiers, E. M., Fauquet, C., Broker, T. R., Bernard, H. U., and Zur Hausen, H. (2004). Classification of papillomaviruses. Virology 324, 17–27. doi: 10.1016/j.virol.2004.03.033
Webster, J. P., Levin, B. R., Woolhouse, M. E., Webster, J. P., Domingo, E., Charlesworth, B., et al. (2002). Biological and biomedical implications of the co-evolution of pathogens and their hosts. Nat. Genet. 32:569. doi: 10.1038/ng1202-569
Willemsen, A., and Bravo, I. G. (2019). Origin and evolution of papillomavirus (onco). genes and genomes. Philos. Trans. R. Soc. B 374:20180303. doi: 10.1098/rstb.2018.0303
Wong, W. S., and Klapperich, C. M. (2018). A Multiplexed human papillomavirus 16 and 18 diagnostic chip for cervical Cancer in limited-resource settings. J. Glob. Oncol. 2018:11s. doi: 10.1200/jgo.18.30000
Wosiacki, S. R., Barreiro, M. A., Alfieri, A. F., and Alfieri, A. A. (2005). Semi-nested PCR for detection and typing of bovine papillomavirus type 2 in urinary bladder and whole blood from cattle with enzootic haematuria. J. Virol. Methods 126, 215–219. doi: 10.1016/j.jviromet.2005.01.021
Wu, Z., Lu, L., Du, J., Yang, L., Ren, X., Liu, B., et al. (2018). Comparative analysis of rodent and small mammal viromes to better understand the wildlife origin of emerging infectious diseases. Microbiome 6:178. doi: 10.1186/s40168-018-0554-9
Keywords: dispersal, evolution, global, lineage, papillomavirus, phylogeography, viral epidemiology, wildlife
Citation: Frias-De-Diego A, Jara M and Escobar LE (2019) Papillomavirus in Wildlife. Front. Ecol. Evol. 7:406. doi: 10.3389/fevo.2019.00406
Received: 20 November 2018; Accepted: 09 October 2019;
Published: 25 October 2019.
Edited by:
Peter Convey, British Antarctic Survey (BAS), United KingdomReviewed by:
Genoveva Rodriguez-Castaneda, Saint Francis University, United StatesCopyright © 2019 Frias-De-Diego, Jara and Escobar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luis E. Escobar, ZXNjb2JhcjFAdnQuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.