 Elena Zaikova1
Elena Zaikova1 David S. Goerlitz2Scott W. Tighe3Nicole Y. Wagner1Yu Bai1Brenda L. Hall4
David S. Goerlitz2Scott W. Tighe3Nicole Y. Wagner1Yu Bai1Brenda L. Hall4 Julie G. Bevilacqua1Margaret M. Weng5Maya D. Samuels-Fair5
Julie G. Bevilacqua1Margaret M. Weng5Maya D. Samuels-Fair5 Sarah Stewart Johnson1,6*
Sarah Stewart Johnson1,6*- 1Department of Biology, Georgetown University, Washington, DC, United States
- 2Georgetown University Medical Center, Georgetown University, Washington, DC, United States
- 3Advanced Genomics Lab, University of Vermont Cancer Center, Burlington, VT, United States
- 4School of Earth and Climate Sciences, University of Maine, Orono, ME, United States
- 5Department of Earth and Planetary Science, Washington University in St. Louis, St. Louis, MI, United States
- 6Science, Technology, and International Affairs Program, Georgetown University, Washington, DC, United States
Buried upslope from the modern lakes in the McMurdo Dry Valleys of Antarctica are relict lake deposits embedded in valley walls. Within these relict deposits, ancient microbial mats, or paleomats, have been preserved under extremely arid and cold conditions since the receding of larger paleolakes thousands of years ago, and now serve as a sheltered niche for microbes in a highly challenging oligotrophic environment. To explore whether paleomats could be repositories for ancient lake cells or were later colonized by soil microbes, determine what types of metabolic pathways might be present, analyze potential gene expression, and explore whether the cells are in a vegetative or dormant state, we collected paleomat samples from ancient lake facies on the northern slopes of Lake Vanda in Wright Valley in December 2016. Using a gentle lysis technique optimized to preserve longer molecules, combined with a polyenzymatic treatment to maximize yields from different cell types, we isolated high-molecular weight DNA and RNA from ancient paleomat samples. Community composition analysis suggests that the paleomat community may retain a population of indigenous mat cells that may flourish once more favorable conditions are met. In addition to harboring a diverse microbial community, paleomats appear to host heterotrophs in surrounding soils utilizing the deposits as a carbon source. Whole genome long-read PacBio sequencing of native DNA and Illumina metagenomic sequencing of size-sorted DNA (>2,500 nt) indicated possible cell viability, with mat community composed of bacterial taxa. Metagenome assemblies identified genes with predicted roles in nitrogen cycling and complex carbohydrate degradation, and we identified key metabolic pathways such as stress response, DNA repair, and sporulation. Metatranscriptomic data revealed that the most abundant transcripts code for products involved in genetic information processing pathways, particularly translation, DNA replication, and DNA repair. Our results lend new insight into the functional ecology of paleomat deposits, with implications for our understanding of cell biology, Antarctic microbiology and biogeography, and the limits of life in extremely harsh environments.
Introduction
Antarctica is uniquely positioned to investigate fundamental questions about life in extreme environments. Within the McMurdo Dry Valleys, microbes have adapted to some of the most physically and chemically challenging conditions on Earth (Cary et al., 2010). Previous studies have characterized communities in rock and soil samples in the hyper arid and cold conditions of the Dry Valleys, and have found that the intense environmental pressure creates specialized communities (Pointing et al., 2009). Yet, the Dry Valleys support more diverse microbial life than was previously believed (Lee et al., 2012). Additional studies have also begun to assess the effects of climate change on these extreme and highly specialized environments (Chan et al., 2013). Its high latitude produces extreme solar radiative states, with high UV flux in the summer and unabated darkness in the winter. The terrain is scoured by severe katabatic winds, and mean annual air temperatures between −14.8°C and −30°C (Doran et al., 2002). The annual precipitation in snowfall is only 3–50 mm water equivalent, making it one of the driest deserts on the planet (Fountain et al., 2010).
Despite these challenges, a wide array of microbial life inhabits the Dry Valleys, much of which was unknown before the advent of molecular methods. This is particularly the case within the refugia of a dozen ice-covered lakes containing permanently unfrozen water or brine along the valley floors (Van Trappen et al., 2002; Taton et al., 2003; Karr et al., 2005; Murray et al., 2012). The lakes are thought to be the remnants of larger glacial lakes that once occupied the Dry Valleys (Doran et al., 1994), and geologic features associated with these paleolakes have been noted throughout the area (Doran et al., 1998; Hendy et al., 2000; Hall et al., 2002).
One such lake, Lake Vanda, is permanently ice-covered and meromictic (Canfield and Green, 1985; Spigel and Priscu, 1998; Green and Lyons, 2009). It has a maximum depth of about 75 m; the upper 50 m of the water column are cool, oxic, and oligotrophic, while the lower 20 m of the lake bottom are composed of a warm, hypersaline and anoxic brine (Canfield and Green, 1985; Green et al., 1998; Green and Lyons, 2009). Similar to other lakes in the Dry Valleys, Lake Vanda within Wright Valley is thought to be the remnant of a larger glacial lake, Glacial Lake Wright (Hall et al., 2001). Benthic microbial mats currently line the bottom of Lake Vanda, and moat mats form seasonally in meltwater at the ice's edge (Love et al., 1983; Wharton et al., 1983; Zhang et al., 2015). Throughout Wright Valley, ancient microbial mats have been detected in association with relict shorelines and deltas (Hall et al., 2010). The preservation of these features is excellent, as erosion of landforms associated with lake level change is thought to be minimal (Hall et al., 2010). The paleomat record embedded in the valley walls above Lake Vanda spans ~30,000 years. Within 10–20 meters of the current shoreline, along facies dating to ~2,000 years old, such paleomat deposits are particularly prevalent when compared to nearby sites (See Figure 1).

Figure 1. Sampling site location and sample appearance. (A) Map of Antarctica, with the area containing the McMurdo Dry Valleys circled in blue. (B) Image of Lake Vanda and surroundings in Wright Valley; site where the mats were collected is indicated by the yellow oval. (C) Image of paleomat in excavated sampling site, with a whirlpack for scale. The mat was collected ~5 cm below surface. (D) Schematic of paleomat distribution in areas where they are found. (E) Close-up of Lake Vanda paleomat.
Over millennial time scales, these remnant mats are an important locus of biotic activity in the Dry Valleys. Moorhead et al. (1999) pointed to four potential sources of organic matter in the soils of the Dry Valleys: in situ primary production, extraction from endolithic communities, aeolian transport of organic matter from nearby lacustrine environments, and erosion of exposed sediments from ancient lake beds. Of these sources, the largest contributor seems to be erosion of exposed sediments from ancient lakebeds. A comparison of Miers Valley and Beacon Valley found soil from Miers Valley to harbor diverse microorganisms while microbial activity was largely absent within soils from Beacon Valley (Wood et al., 2008a,b). The authors tentatively attributed the difference to the presence of a modern lake in Miers Valley and the absence of lacustrine systems in Beacon Valley.
However, cells can survive over long time periods under conditions of desiccation and extreme cold. Indeed, freeze-drying has long been used as a method to store and transport microbial strains in culture collections (Morgan et al., 2006), and viable cells have been indicated in permanently frozen environments over tens of thousands (Soina and Vorobyova, 2005; Bergholz et al., 2009) and even hundreds of thousands year timescales (Johnson et al., 2007). A cultivation-based study from similar paleomat deposits from the Dry Valleys found that select strains of microbial cells could be revived from paleomat deposits that had been stored at room temperature (Antibus et al., 2012a). Although the authors of the study could not fully exclude the possibility of colonization by soil bacteria, ancient mat samples displayed low abundance and diversity of cultivable bacteria, which would be expected due to a loss of viability over time (Antibus et al., 2012a). A paired study assessing the persistence of 16S rRNA from the same samples also found that DNA abundance and integrity declined with sample age over several millennia (Antibus et al., 2012b). Additionally, extracellular DNA has been found to comprise about 40% of the DNA in soil samples, and this relic DNA can confound the determination of microbial diversity, resulting in inflated estimates (Carini et al., 2016).
Here we present a description of the microbial community composition, metagenomics and metatranscriptomics of a desiccated microbial mat from the Wright Valley. The overarching goal of this work was to investigate whether paleomats may serve as “seed banks” of stress-resistant organisms over long timescales, while also providing habitat and a source of carbon to modern soil-dwelling microbes. Our specific aims were to survey the microbial cells associated with Lake Vanda paleomat deposits to: (1) assess if community structure and taxonomy is different within the relic mat material from that found in surrounding soils and whether it is similar to the composition of modern moat mats, (2) analyze what types of metabolic pathways might be present in the relic mat metagenome, and (3) determine what gene transcripts could be recovered from the relic mat, and (4) discern if the cells in the relic mat are surviving via dormancy, a favored explanation for the persistence of microbes in harsh conditions (Jørgensen, 2011), particularly in response to extreme cold (Abyzov et al., 1998) and desiccation (Lebre et al., 2017). For the purposes of this study, modern mats are defined as microbial mats inhabiting the benthic portion of present-day Lake Vanda, and paleomats are defined as thin, desiccated mats buried upslope from the present lake and carbon-dated to millennial time-scales.
One obstacle to undertaking genomics work in Antarctica, however, is that DNA fragments may be preserved over long timescales. The hyper-arid, exceptionally cold, and salty conditions associated with the Dry Valleys paleolakes are optimal for preserving ancient DNA (Smith et al., 2001; Willerslev and Cooper, 2005; Hofreiter et al., 2015). Thus, the detection of genes or gene fragments using molecular methods may simply indicate the presence of well-preserved ancient DNA, not necessarily the viability or intactness of the parent organism. This has been especially problematic for next-generation sequencing techniques, which rely on short fragments of DNA (150–300 bp in length). The DNA fragment lengths associated with ancient dead biomass, even in cold environments, are typically < 300 bp in length (Hofreiter et al., 2015). The longest ancient DNA sequence ever generated from a dead specimen on ancient timescales is 1,042 bp, from an exquisitely preserved 9,000-year-old Antarctic sample (Lambert et al., 2002). Therefore, longer fragments of DNA would be indicative of intact cells rather than eDNA or aDNA.
For this study, we assessed the Lake Vanda paleomat, surrounding soil, and modern mat bacterial community using amplicon sequencing of the taxonomic marker 16S rRNA gene to compare the composition of the paleomat, surrounding soil and modern mat bacterial communities. To assist in discriminating highly fragmented ancient DNA from DNA derived from viable organisms, we focused on the recovery and sequencing of significantly longer read lengths (>2,500 nt). We leveraged Illumina metagenomic sequencing of whole and long-fragment fractions of DNA in combination with continuous long-read, third-generation Pacific Biosciences sequencing of native DNA to characterize the putative function and structure of the paleomat community. In addition, we isolated and sequenced RNA from the Lake Vanda paleomat to investigate potential transcriptional activity, which truly dormant cells do not undertake, and to identify which genes are expressed, thus providing insight into cell survival strategies.
Materials and Methods
Sample Collection
All samples were collected in December 2016 (austral summer), at ~11 am. Tyvek suits and nitrile gloves were worn to minimize contamination. Paleomat samples were collected from beneath ~5 cm of soil ~500 ft upshore of Lake Vanda (S 77°31.149′ E 161° 38.315). Sterile, DNA-free copper utensils (ashed at 550°C overnight) were used to excavate the site, and multiple sample replicates were collected, a subset of which were collected into sterile cryotubes that were immediately placed into a Taylor Wharton cryoshipper charged with liquid nitrogen, and the remainder was placed into sterile Whirl-Pak bags (Nasco, Fort Atkinson, WI, USA), and immediately placed on dry ice. Soil adjacent to the paleomat samples, ~20 cm away and judged by eye to be free of paleomat fragments, was collected in the same manner in multiple replicates. The modern mat sample was collected from a moat mat at the near edge of Lake Vanda (S 77° 31.168′ E 161° 38.381′), from beneath ~5 cm of meltwater. Following helicopter transport to McMurdo Station, samples were either extracted (in the case of the MiniSeq run) or maintained at a temperature of −80°C for ~1 month, then shipped on dry ice via air transport to Georgetown University, where they were maintained at a temperature of −80°C until additional processing and analysis. Because of limited access to near-shore modern mats due to ice-cover and lean biomass concentrations within the surrounding soil, sufficient amounts of high quality, high molecular weight DNA suitable for metagenomics could not be obtained, these two samples were analyzed just for amplicon-based 16S rRNA gene sequencing. Similarly, to attain μg-quantities of high molecular weight DNA from paleomat samples, several extractions from multiple replicates were pooled.
Scanning Electron Microscopy
Scanning Electron Microscopy was performed by the UVM Microscopy Imaging Core Facility using the following procedure: briefly, the mat samples were suspended in Karnovsky's fixative (2.5% glutaraldehyde/2% formaldehyde in 0.1 M cacodylate buffer) and rinsed three times with 0.1 M cacodylate buffer followed by post-fixation with 1% osmium tetroxide (OsO4) in 0.1 M cacodylate buffer for 30 min at 4°C. A final rinse of the sample was done with 0.1 M cacodylate buffer, followed by serial ethanol dehydration and critical point drying in CO2. Samples were finally sputter-coated with either gold/palladium and imaged using a JEOL JSM-6060 scanning electron microscope (JEOL, Peabody, MA) operating at 10–15 kV.
δ13C and 14C
To assess the age of material collected, paleomat samples were analyzed for δ13C and 14C by stable isotope ratio mass spectrometry (SIRMS) and accelerator mass spectrometry (AMS), respectively, at the National Ocean Sciences Accelerator Mass Spectrometry (NOSAMS) Facility at the Woods Hole Oceanographic Institution in Woods Hole, Massachusettes. Data was collected using the Pelletron Tandem 500 kV AMS system (Povinec et al., 2009; Roberts et al., 2010). The measured δ13C value—a function of taxonomy, productivity, and organic carbon burial—was −17.46%0. Radiocarbon ages were calculated using the Libby half-life of 5,568 years using the convention described by Stuiver and Polac (1977); Stuiver (1980). This calculated carbon date, 2,150 ± 15 years, and refined with a 2-sigma calibration suing the CALIB program and the INTCAL 2013 dataset (Reimer et al., 2013), yielding a 99.6% probability that the calendar-year age lies between 2,040 and 2,148 years BP.
Nucleic Acid Isolation and Sequencing
DNA and RNA were extracted from cells using protocols to maximize lysis efficiency and molecular weight of the molecules isolated.
DNA Extraction and Sequencing
DNA was isolated from ancient paleomat samples collected from Lake Vanda using multiple DNA isolation protocols, using the ZymoBIOMICS DNA Miniprep Kit (Zymo Research, Irvine, CA) or MoBio PowerSoil DNA Isolation kit (Qiagen, USA) with protocol modifications, dependent on the downstream application. Each individual extraction was performed on ~200 mg of starting material.
For 16S rRNA sequencing on the Illumina MiniSeq platform, DNA from mat and soil samples was extracted using physical lysis by bead-beating in the FastPrep-24 Classic Instrument (MP Biomedicals, Santa Ana, CA, USA) with a speed of 6 m/s for 40 s. Following cell lysis, DNA was purified using the ZymoBIOMICS DNA Miniprep Kit (Zymo Research, Irvine, CA) following manufacturer's protocol. The DNA was eluted in DNase/RNase-free water and stored at −80°C. Library preparation, pooling, and MiniSeq sequencing were performed at the University of Illinois at Chicago Sequencing Core (UICSQC). Briefly, genomic DNA was PCR-amplified using a primer set targeting the V4 region of bacterial 16S rRNA gene—CS1_515F (5′- GTGCCAGCMGCCGCGGTAA-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′)—and a two-stage PCR protocol (Bybee et al., 2011; Naqib et al., 2018). Samples were pooled in equal volume, fragments smaller than 300 bp were removed from the pooled library, which was then sequenced on the Illumina MiniSeq platform together with a phiX spike-in, and resulting in 150 bp PE reads.
For shotgun sequencing on the Illumina MiniSeq platform, DNA was extracted using the MoBio PowerSoil DNA Isolation kit (Qiagen, USA) according to the manufacturer's protocol, with the exception of DNA elution in 50 μL of nuclease-free water instead of the reagent provided in the kit (solution C6). The extracted DNA was stored at −80°C. A whole genome shotgun metagenomic sequencing library were constructed from 50 ng of total gDNA using the Nextera XT DNA Library Preparation Kit (Illumina USA) according to the manufacturer's instructions. The library was sequenced on the Illumina MiniSeq system using paired-end 150 bp read lengths and the MiniSeq High Output Reagent Kit (300-cycles) at the Albert P. Crary Science and Engineering Center, McMurdo Station, Antarctica.
An additional DNA preparation was isolated using enzymatic digestion. Briefly, paleomat samples were incubated with MetaPolyzyme (Sigma-Aldrich, USA), a mix of five enzymes (achromopeptidase, chitinase, lyticase, lysostaphin, lysozyme, and mutanolysin) designed to target bacteria and fungi (Tighe et al., 2017). For this DNA preparation, the bead-beating step was omitted to maximize the yield of high molecular weight DNA. DNA extraction from cell lysates was carried out using a ZymoBIOMICS DNA Miniprep Kit following manufacturer's protocol, and the resulting DNA was eluted in DNAase/RNase-free water and stored at −80°C. DNA from this set of extractions was used for whole genome shotgun using the whole DNA fraction, as well as size-selected (>2,500-bp) gDNA fraction. The sequencing libraries were prepared using the MiSeq Reagent Kit v3 according to the manufacturer's instructions, and were sequenced at UICSQC on Illumina MiSeq, producing 2 × 300 bp reads.
DNA was isolated for Pacbio sequencing using enzymatic digestion, and the ZymoBIOMICS DNA Miniprep Kit following manufacturer's protocol with modifications as described above. The sequencing library was prepared following the Pacific Biosciences 2 kb SMRTBell Template Preparation and Sequencing protocol with an additional bead-based template cleanup using 0.5X AMPure PB beads to eliminate smaller-sized fragments. A final library QC was performed with a Qubit fluorometer to determine library concentration and an Agilent Bioanalyzer 2100 (Agilent Genomics) using the DNA 12000 reagent kit to determine the size distribution of the library. The prepared sequence library was loaded onto a single Sequel v2.1 SMRTcell at a concentration of 6 pM, following the PacBio diffusion loading protocol and including a polymerase-bound complex cleanup. One 600-min movie was taken of the SMRTcell.
In all extraction protocols unless otherwise specified, DNA concentration, quality, molecular weight and fragment size distribution were determined via the Agilent Bioanalyzer 2100 using the HS DNA Assay (Agilent, USA). Spectrofluorometry using a Qubit High Sensitivity DNA Assay (Thermo Scientific, United States) was used to quantify the amount of gDNA present in the extractions. In both assessments the manufacturer's protocols were followed.
RNA Extraction and Sequencing
Total RNA was isolated from ~500 mg of ancient paleomat material collected from Lake Vanda (WA3A) using the Zymo Direct-zol RNA MiniPrep kit (Zymo Research Corp., USA) according to the manufacturer's instructions. Briefly, the paleomat material was mechanically disrupted on dry ice in the original collection tube using a sealed, disposable RNase-free spatula. TRIzol reagent (500 μl) was added directly to the sample, which was subsequently vortexed and centrifuged at 12,000 × g for 30 s to remove particulates, and the supernatant was transferred to an RNAse-free tube. Cell lysis in TRIzol was repeated for the remaining paleomat material to provide a total of 1,000 μl supernatant, which was then mixed with one volume of 100% ethanol and loaded onto a Zymo-Spin IIC Column. The RNA sample was treated in-column with 5 U of DNase I, washed and eluted in 20 ul of RNase-free water. RNA yield and integrity were determined via fluorometric quantitation using the Qubit RNA HS Assay Kit (Thermo Scientific, United States), and the Agilent Bioanalyzer 2100 using the RNA 6000 Nano Kit (Agilent, USA) (Figure S1). Sequencing libraries were constructed from 2 ng of total RNA using the SMARTer Stranded Total RNA-Seq Kit v2-Pico library preparation kit (Takara Bio, USA) according to the manufacturer's instructions using protocol option 2 (without prior RNA fragmentation). Library preparation included both an RT (-) control (using paleomat RNA as template, but excluding the SMARTScribe Reverse Transcriptase enzyme), and a negative control using water as template. Both controls resulted in no library construction, confirming the absence of any contaminating gDNA. The final RNASeq (cDNA) library was sequenced on the Illumina MiSeq System using paired-end 300 bp read lengths and the MiSeq Reagent Kit v3 (600-cycle) (Illumina, Inc, USA) at the Genomics and Epigenomics Shared Resource at Georgetown University Medical Center.
qRT-PCR
The RNA extraction was tested for gDNA contamination using qRT-PCR performed on a C1000 thermalcycler fitted with a CFX96 detection module following the method described in Zaikova et al. (2010). The target gene, 16S rRNA, was amplified using either bacteria-specific (27F, 5′-AGAGTTTGATCMTGGCTCAG-3′) or archaea-specific (20F, 5′-TTCCGGTTGATCCYGCCRG-3′) forward primers and the universalreverse primer 519R (5′-GNTTTACCGCGGCKGCTG-3′). A total of 20 μL reaction consisting of 2 μL template, 0.6 μL of each reverse and forward primer, 10 μL of SsoAdvanced Universal SYBR green Supermix (Bio-Rad, USA) and 6.8 μL of nuclease-free water was prepared for each sample. Water was used as the template for the negative control reaction. Serial dilutions across 7 orders of magnitude of chromosomal copy number (107 to 101) of custom gblocks DNA oligos based on cosmopolitan gram-negative and gram-positive 16S bacterial sequences, and Thaumarchaeal and Euryarchaeal 16S archaeal sequences (IDT, USA) were used as the standard bacterial and archaeal PCR controls. Thermal cycle parameters included an initial denaturation at 98°C for 3 min, followed by 35 cycles of 98°C for 15 s, primer annealing at 55°C (for bacteria) or 65°C (for archaea) for 30 s, an extension at 72°C for 15 s followed by a melt curve after 35 cycles. The melt curve was performed from 65 to 95°C with 0.5°C increments.
Sequence Analysis
Microbiome Composition Analysis
Bacterial 16S rRNA gene sequences were processed using the dada2 R package (Callahan et al., 2016) implemented on R version 3.5.0 through RStudio version 1.1.447 (R Core Team, 2013; RStudio Team, 2016). First, demultiplexed, raw reads were trimmed and filtered to remove phiX sequences, primer sequences and low-quality bases, and reads and error rates were estimated for each sample. Identical sequences were then dereplicated into unique sequences, and their corresponding abundance was noted for each sample. Forward and reverse reads were merged, chimeric sequences were removed and an abundance distribution table for each of the unique merged sequences—exact sequence variants (ESVs)—was constructed. Taxonomy was assigned using a naive Bayesian classifier method implementation in dada2, with Silva v128 as the reference database (Quast et al., 2013). Alpha diversity was calculated using the phyloseq R package (McMurdie and Holmes, 2013), distributions of relative taxonomic abundances were visualized with Krona (Ondov et al., 2011), and a Venn diagram comparing ESV distribution among the three samples was produced in R. Since only three samples were examined, and no biological replicates were sequenced, we could not carry out comparative statistical analyses.
Metagenomic Read Assembly and Annotation
Illumina Data Assembly, Metagenomic Binning, and Annotation.
Adapters, sequencing artifacts and poor-quality bases were trimmed from Illumina reads, and the trimmed reads were subsequently filtered for quality and length using BBDuk (Bushnell, 2014). The trimmed, filtered reads were assembled into contigs and scaffolds using MEGAHIT (Li et al., 2015, 2016). BWA MEM (Li, 2013) was used to map reads back to the assembly and samtools (Li, 2011) was used to convert between formats and calculate coverage. MetaBAT2 (Kang et al., 2015) was used to place sequences into genomic bins, using parameters optimized for sensitivity. CheckM (Parks et al., 2015) was used to assess binning quality and ensure that no sequence appeared in more than 1 bin. Following metagenomic binning, 17 MAGs with low contamination, <5%, and high completion, >90%, were produced. Of these, 9 were obtained from the MiniSeq assembly, 6 from the size-selected and 1 from whole fraction MiSeq assemblies. Average nucleotide identity was calculated using JSpeciesWS using blast+ and MUMmer approaches (Richter et al., 2016). As the results using the two methods were highly similar, only the blast-based result is reported here.
Metagenomic assemblies and high-quality genomic bins were annotated ab initio using Prokka version 1.12 (Seemann, 2014). The amino acid sequences of predicted open reading frames (ORFs) within each dataset were analyzed using KAAS to map the KO numbers to the KEGG GENES database (Moriya et al., 2007). Taxonomy of the ORFs identified by Prokka was assigned using Kraken (Wood and Salzberg, 2014). Gene assignments, taxonomy and KEGG pathway distributions weresummarized, analyzed and plotted using R v 3.5.0.
PacBio Sequence Assembly and Annotation.
In order to build the PacBio assembly, Bamtools 2.4.1 (Barnett et al., 2011) was used to convert the CCS (Circular Consensus Sequence) processed subreads from bam format to a fasta file. No additional filtering was performed. Canu 1.6 was used to build the PacBio assemblies. The taxa present in both the CCS-corrected reads and CCS-corrected contigs were identified with Kraken 1.0 and visualized in Krona, and functional annotation was done using Prokka, for consistency with analyses of Illumina metagenomic data. With the exception of taxonomic distribution, all other outputs were visualized and summarized using R.
Transcriptome Analysis
RNA reads were trimmed and filtered for length and quality using BBDuk. Reads passing QC were mapped to contigs produced by assembly of Illumina and PacBio metagenomic sequences. To identify reads that represented subunits of the rRNA gene, reads were mapped to the small subunit (16S rRNA and 18S rRNA) and large subunit (23S rRNA and 28S rRNA) of the rRNA genes available in the Silva v 132 database (Quast et al., 2013). Reads that did not map to rRNA genes were extracted using samtools and used to for transcriptome analysis. Non-rRNA reads were then mapped to the Prokka-predicted genes and assemblies again using bwa. Alignments were assessed using samtools and R. Transcripts per kilobase million (TPM) calculation was performed in R. KEGG category expression was based on predicted genes that had >10 reads mapped and the summed corresponding TPM. As with metagenomic KEGG distributions, the same predicted gene can participate in more than one pathway, so the total exceeds the number of genes predicted.
Sequence Accession Numbers
Metagenomic, metatranscriptomic and SSU rRNA amplicon sequences reported in this study were deposited in the GenBank Sequence Read Archive with accession numbers SRR8217969 - SRR8217976 and project accession PRJNA506221. Metagenomic assemblies, annotations and ESV table are available upon request.
Results
Paleomat Sample Appearance and Age
The Lake Vanda paleomat deposits sampled appeared as desiccated, thin, beige flakes (Figure 1), present in regular layers about 5 cm below the surface, and less structured layers at ~20 cm below surface. The age of the Lake Vanda paleomats studied here was calibrated to be ~2,100 ± 50 years (see Materials and Methods, Pathway Analysis and Survival Strategies), though the age of the mat and the age of the cells may differ, as viable cells present may only contribute a small fraction of the total carbon. The measured δ13C value of the paleomat was −17.46%0, most likely indicative of a primarily heterotrophic community.
Scanning electron microscopy imaging of the paleomat revealed the presence of several types of intact cells within the sheet-like mat flakes (Figure 2A). The mat material appeared layered and quite porous in texture (Figure 2B), and was covered by what appeared to be filamentous cells (Figure 2C). The filaments were ~1.5 μm wide and were associated with coccus cells, which were ~0.8 μm in diameter and often appeared in clusters (Figures 2D,E). In addition to filaments and coccus cells, rod-shaped cells were also observed in the sample, as were coryneform (club-shaped) cells that were observed in mat material grooves (Figure 2F). The coryneform cells had distinct banding on the cell surface and were ~1 μm long and ~0.4 μm wide.

Figure 2. Scanning electron microscopy images of paleomat collected upslope from Lake Vanda. (A) Low magnification of desiccated paleomat flake. (B) Paleomat flake attached to porous sediment grain. (C) A close-up of porous grain with attached microbial filaments indicated by the arrow. (D) Coccoid cells (a) and filamentous microbial cells (b). (E) Close up showing filaments, rod and coccus cells (indicated by arrow). (F) Rod-shaped or club-shaped cells (indicated by arrow).
Sequencing Datasets Summary
To characterize the community structure of the Lake Vanda paleomat microbial community as well as the functional potential encoded by the genomes of organisms and their transcriptional activity, we generated 16S rRNA gene amplicon, four metagenomic, and one metatranscriptome datasets (summarized in Table S1). Bacterial composition and distribution in the paleomat, modern mat and soil surrounding the paleomat was explored using 16S rRNA gene sequencing. For each of the three samples, paired-end reads across the V4 region of the gene were generated, for a total of 305,452 reads for the modern mat, 3,63,111 reads for the paleomat, and 96,133 reads for the surrounding sand. After trimming, quality filtering, chimera, singleton, and unassigned ESV removal, 93% of modern mat reads, 92% of sand reads, and 80% of paleomat reads were used in composition analysis and description. These reads formed 964 ESVs: 476 in the modern mat, 481 in the paleomat, and 106 ESVs in the soil sample.
In order to investigate whether short eDNA and aDNA fragments would impact sequencing results, metagenomic data was generated using two different extraction methods, with differences in DNA fragments sequenced and sequencing platform, as detailed in Materials and Methods. The DNA sequenced on the PacBio platform, as well as size-selected (>2,500 bp DNA fragments) and whole DNA fractions sequenced on the MiSeq platform were all prepared from the same DNA extraction, whose average fragment length was ~5,000 bp. The MiniSeq run, performed in Antarctica, yielded 23,883,870 150-bp paired-end reads, which were assembled into 276,991 contigs with an N50 value of 1,350 bp. The MiSeq whole DNA fraction run (300-bp paired-end) produced fewer reads−6,095,791–and assembled into 2,37,640 shorter contigs (N50 of 677 bp). The MiSeq size-selected sequencing run produced 16,527,393 300-bp paired-end reads, which resulted in 8,58,609 contigs with an N50 of 918 bp. PacBio sequencing and assembly resulted in 44,521 contigs with total length 322 Mbp, however, many reads were short (~7,88,000 reads comprising ~2.8 Gb data) and could not be assembled. Sequence data from DNA isolated using either mechanical lysis (bead-beating) or enzymatic digestion with MetaPolyzyme identified highly similar taxonomic diversity, suggesting that the different DNA isolation methods lysed the same population of cells, and sequencing captured the full representation of the microbial diversity present in the paleomat samples. In addition, we did not identify any pronounced differences in gene identity and frequencies between whole DNA isolations and size-selected fractions, indicating the negligible contribution of eDNA and/or aDNA to the metagenomics sequence data. RNA sequencing yielded 20,249,504 paired-end reads, with an average length of 76 nt. Following trimming, filtering, phiX and artifact removal, >80 % of the reads were used for transcriptome analysis.
Bacterial Community Composition of the Paleomat, Surrounding Soils, and Modern Mat
The paleomat, surrounding soil, and modern mat were distinct in bacterial composition, yet 12 ESVs were common between the three samples (Figure 3A). These shared ESV were not represented by abundant sequences in the modern mat (< 0.01–1% reads each), were slightly more abundant in the paleomat (0.01–3% reads in each ESV), and were moderately abundant in the soil surrounding the paleomat (0.5–1% reads for most, with an ESV identified as unclassified Frankiales that represented 19.5% of the reads). Of these 12 ESVs, 8 were Actinobacterial sequences, 5 of which were Micrococcales taxa, 2 were Propionibacteriales taxa, and 1 Frankiales ESV. The other 4 ESVs were a Deinococcus-Thermus sequence (Truepera sp.), a Chloroflexi taxon, and two Bacteriodetes sequences (Pontibacter and Segetibacter). The majority of ESVs identified in the modern mat (90% ESVs) and paleomat (83%) were unique and not detected in either of the other samples. However, these ESVs represented a different proportion of sequence abundance, with the modern mat unique ESVs representing 87% of the reads in the sample, whereas the unique paleomat ESVs comprised 46% of paleomat 16S reads. In the surrounding soil dataset, the unique (not shared) ESVs represented 46% of the soil ESVs but only 14% of the reads in the sample. Although the paleomat sample share similar numbers of ESVs with either the modern mat or the surrounding soil, the abundances of those ESVs in the paleomat dataset were not equivalent, as ~33% reads were in ESVs shared with surrounding soil, and just 3% of reads by abundance were in ESVs common between the two mat samples. These observations indicate the modern mat harbored a community that was the most distinct from the other samples, and that, although the paleomat bacterial community was distinct, it was more similar to that of the soil sample than the modern mat. Additionally, the modern mat community was the most diverse, and the surrounding soil community was the least diverse (Figure 3A).
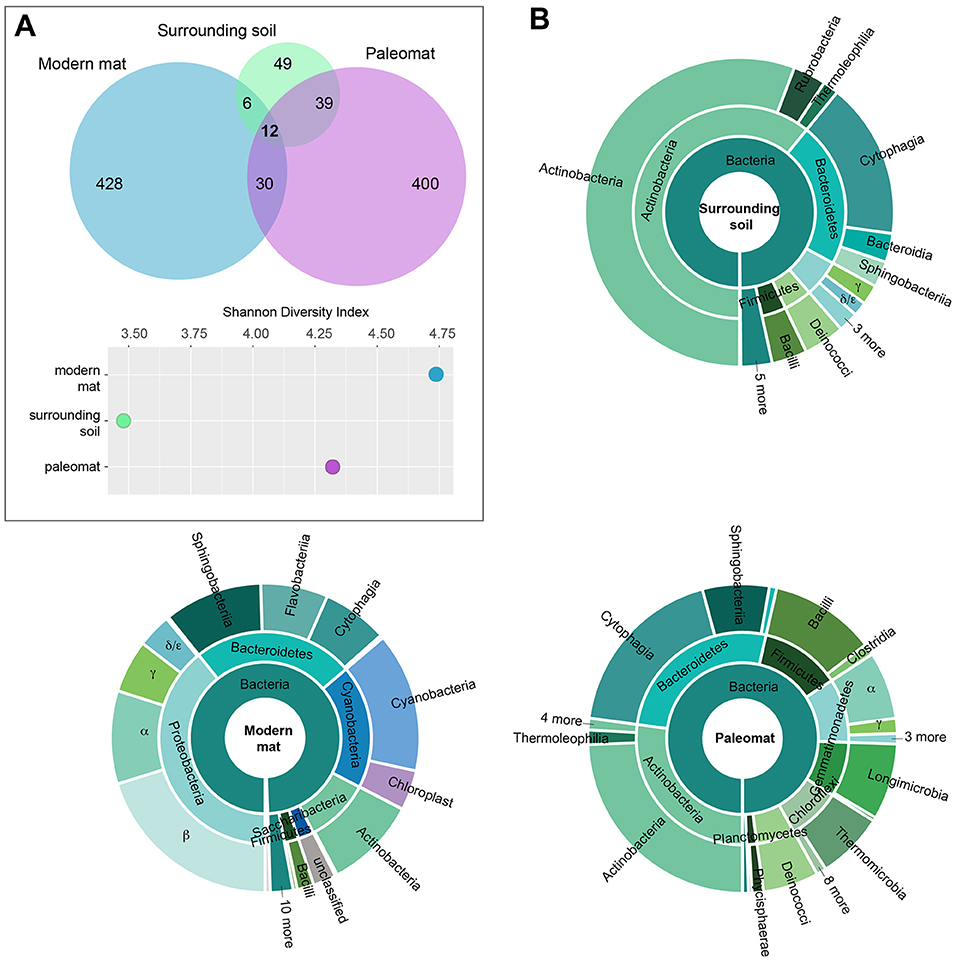
Figure 3. Bacterial community composition of modern mat, paleomat, and soil surrounding the paleomat based on 16S rRNA gene Exact Sequence Variants (ESVs). (A) Top: ESV distribution among samples is indicated by the Venn diagram, with circles scaled according to the number of ESVs identified for the sample. Bottom: Shannon's Diversity Index for each of the samples. (B) Taxonomic composition of ESVs in the 16S rRNA gene data from Lake Vanda modern mat, paleomat and soil surrounding the paleomat. Colors are the same throughout.
Despite some similarity in composition and presence of a few shared ESVs, the distribution, relative abundances, and taxonomic identities of ESVs differed among the samples (Figure 3B). The paleomat was dominated by Actinobacteria, Bacteroidetes, Firmicutes, Proteobacteria, Gammatimonadetes, Chloroflexi, and Deinococcus-Thermus. The most abundant phylum in the paleomat, Actinobacteria, was dominated by Actinomycetales. Bacteroidetes showed a large abundance of Cytophagales and Saprospirales. Bacilli were prevalent among the Firmicutes, and to a lesser degree, Clostridia. Within the proteobacterial members of the paleomat bacterial community, Alphaproteobacteria were the most dominant member, containing abundant Sphingomonadales and Rhodobacterales. Proteobacteria, Bacteroidetes, Cyanobacteria, and Actinobacteria dominated the modern Lake Vanda moat mat bacterial community (Figure 3B). Proteobacteria were represented primarily by Betaproteobacterial taxa, followed by Alphaproteobacteria and Gammaproteobacteria. Bacteroidetes contained a roughly even distribution of Sphingobacteriales, Flavobacteriales, and Cytophagales. Cyanobacteria were primarily represented by Synechococcophycideae, and to a lesser extent sequences that were identified as possible chloroplast 16S.
The surrounding soil, on the other hand, was dominated by Actinobacteria, with taxa belonging to this phylum containing ~60% of the reads. Other numerically-important phyla of the surrounding soil bacterial community were the Bacteroidetes, Proteobacteria, Deinococcus-Thermus, and Firmicutes. These phyla were also the dominant soil community members in McKelvey Valley, where they mediate nitrogen transformation and play key roles in carbon cycling (Chan et al., 2013).
Taxonomic Composition of the Paleomat Metagenome
To identify the constituents of the paleomat microbial community, we assigned taxonomy to contigs and scaffolds assembled from each Lake Vanda paleomat metagenomic sequence dataset. The taxonomic assignment results indicated that in all cases, most sequences could not be assigned to any known taxon, and were unclassified (Figure 4). There was no discernable difference in patterns of community structure between different DNA isolation methods and sequencing approaches,with only minor observed differences. Although sequences generated on the PacBio platform had a lower proportion (55% compared to 88–90% for Illumina) of unclassified sequences, we recovered the same taxa with similar patterns as with Illumina short-read sequencing (Figure 4). In all four datasets, of the contig sequences that could be assigned to a particular taxonomic group, most were affiliated with Actinobacteria. The five most abundant, based on sequence count, Actinobacterial subdivions consisted of Micrococcineae, with strains belonging to genera that can degrade hydrocarbons and recalcitrant substrates, Frankineae, Streptomycineae, Corynebacterineae, and Propionibacterineae. The next-most abundant phylum was Proteobacteria, mostly Alphaproteobacteria (Rhizobiales, Sphingomonadales, Rhodobacterales, Caulobacterales, Rhodospirillales), Gammaproteobacteria (Xanthomonadales and Pseudomonadales), Betaproteobacteria (Burkholderiaceae, Comamonadaceae, Rhodocyclaceae), and Deltaprotobacteria, consisting of Myxococcales. The remaining bacterial phyla, including Firmicutes, Bacteroidetes, Deinococcus-Thermus, and Planctomycetes among others, were each represented by <1% of all assembled sequences in each dataset.
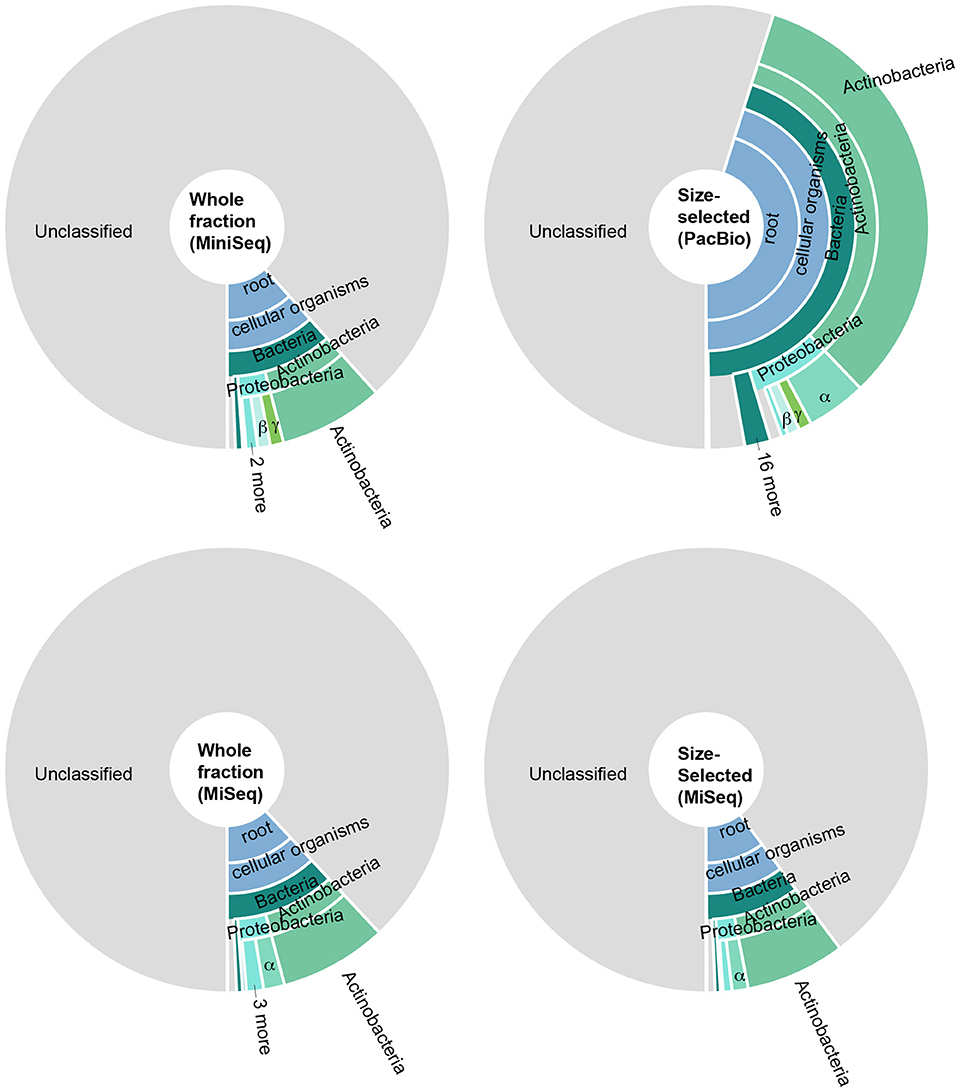
Figure 4. Taxonomic distribution of predicted open reading frames in metagenomic data generated by sequencing (i) the whole DNA fraction on an Illumina MiniSeq (top left), (ii) size-selected fraction on the PacBio system (top right), (iii) whole DNA fraction on an Illumina MiSeq (bottom left), and (iv) the size-selected fraction on an Illumina MiSeq (bottom right). In all cases, most predicted genes could not be assigned to any taxonomy; of the genes that could be assigned, most were Actinobacteria and Proteobacteria. Colors are the same throughout.
Taxonomic Composition of the Transcriptionally-Active Component of the Paleomat Community
To identify non-ribosomal rRNA reads with the metatranscriptome dataset, 16,246,265 paired-end reads passing QC and filtering were mapped to known small subunit rRNA and large subunit rRNA sequences in the Silva database (Quast et al., 2013). This represented >92% RNASeq reads, indicating that <8% reads were likely mRNA. The rRNA reads were removed from downstream transcriptome analyses. Of the remaining reads, 64% mapped to (and were properly paired) the MiniSeq assembly contigs, 57% mapped to the PacBio assembly contigs, 69% mapped to the MiSeq size-selected assembly, and 53% mapped to the MiSeq whole DNA fraction assembly contigs. Mapping of RNASeq reads to the Silva database provided valuable information into potentially active members of the community based on rRNA gene expression. Over 99% of RNASeq-detected rRNA genes with >10 mapped reads were bacterial (Figure S2), and over 75% of these were Actinobacterial taxa, the majority of which were Micrococcales. There were an additional 50 bacterial taxa with mapped reads (Figure S2), however, some assignments, particularly those with only a few mapped reads, may be spurious as the reference database included the full-length genes, including conserved regions, which are present in all bacteria taxa and difficult to reliably identify with short reads.
Transcripts per kilobase million (TPM) values indicated that sequences aligning to Actinobacteria were most abundant within expressed contigs (Figure 5). In decreasing order of expression abundance, the most common organisms within this phylum belong to Micrococcales, Corynbacteriales, Streptomycetales, Propionibacteriales, Micromonosporales, Pseudonocardiales, Streptosporangiales, Frankiales, Geodermatophilales, and seven other orders with total abundances of <1% each. The next most-highly expressed phylum were the Proteobacteria, particularly the Alphaproteobacterial order Sphingomonadales (including the genera Sphingobium, Sphingomonas, Novosphigobium, and Sphingopyxis and pigmented photoheterotroph Erythrobacter), Rhizobiales (including the nitrogen-fixing Bradyrhizobium, Rhizobium, and Azorhizobium), Rhodobacterales (Rhodobacter, which can also fix nitrogen), Rhodospirillales, Gammaproteobacteria (Xanthomonadaceae), Betaproteobacteria (Burkholderiales) and Deltaproteobacteria. Also well-represented in the active community were the Gram-negative, non-spore-forming Bacteroidetes—mostly Cytophagaceae, Cyclobacteriaceae and Flavobacteriaceae—as well as the Firmicutes, grouped primarily into Clostridiales and Bacillales.
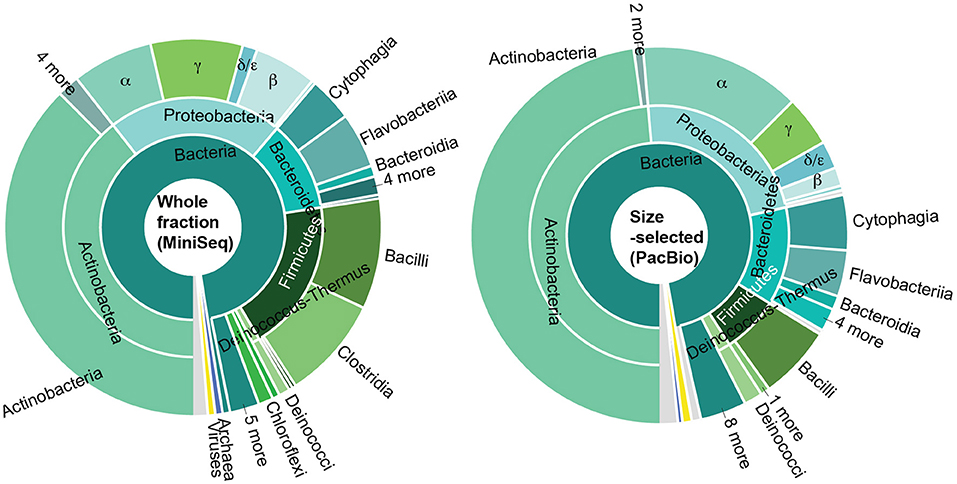
Figure 5. Taxonomic distribution based on abundance of RNASeq reads (sum of TPM values) for communities represented in the whole fraction MiniSeq and size-selected PacBio datasets.
Functional Genes in Lake Vanda Paleomat
As observed with taxonomic composition, the types of genes that were identified in each of the metagenome datasets were highly similar (Figure 6). In all four metagenomic datasets, the KEGG system with the most number of ORFs was Metabolism, particularly Amino Acid Metabolism (including amino acid related enzymes and amino acid biosynthesis and degradation) and Carbohydrate Metabolism (most ORFs in glycolysis, TCA cycle, pentose phosphate cycle, pyruvate, butanoate and propanoate metabolisms, glyoxylate, and dicarboxylate metabolism, amino sugar and nucleotide sugar metabolism). Metabolism of Cofactors and Vitamins (including CoA, biotin and folate biosynthesis, nicotinate and nicotinamide metabolism, as well as porphyrin and chlorophyll metabolism), nucleotide (purine and pyrimidine) metabolism and Energy Metabolism (oxidative phosphorylation, carbon fixation, photosynthesis, methane metabolism, and nitrogen and sulfur metabolisms) were encoded by >1% ORFs in each metagenome (Figure 6B). Within the Genetic Information Processing system, most ORFs were associated with translation and DNA replication and repair. Most ORFs mapping to Cellular Processes were associated with transport and catabolism, including exosome and prokaryotic defense systems, and Cellular Community–quorum sensing and biofilm formation. Predicted ORFs within Environmental Information Processing were associated with membrane transport (ABC and other transporters and secretion system) and signal transduction, primarily two-component systems that enable bacteria to sense and respond to environmental cues. Additionally, predicted ORFs mapped to Organismal and Human Diseases systems, however, 95% of the annotations also mapped to other systems, namely Metabolism, Genetic Information Processing and Cellular Processes. The remaining 5% of annotations mapping to these two systems consisted of genes known to play roles in other functions, for example serpins and nitric oxide reductase, as well as antibiotic resistance genes. A full list of KO numbers and their KEGG system assignments is provided in Supplementary Information as a tsv file.
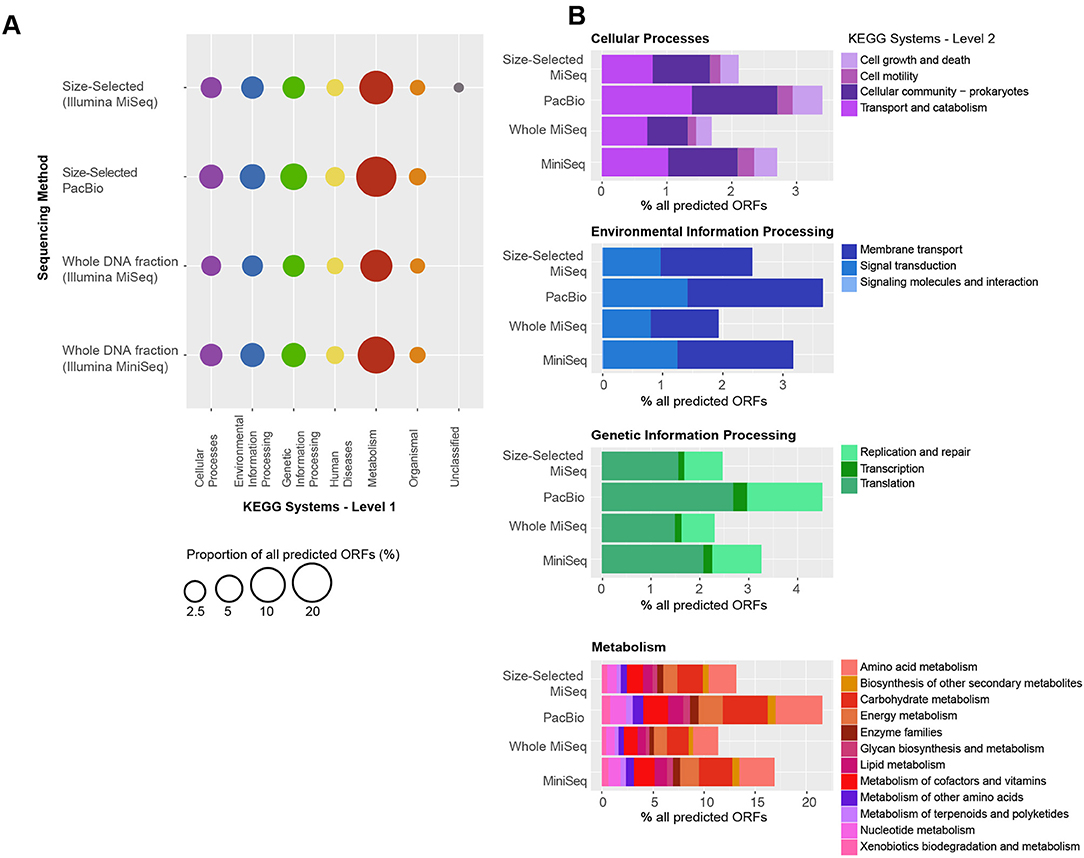
Figure 6. Functional gene distribution, as a percent of all the predicted genes in each sequencing dataset. (A) The KEGG systems are indicated on the x-axis, and the sequencing approach is shown on the y-axis. Many genes mapped to more than one function and to more than one category. All genes mapping to “Organismal” and “Human Diseases” also mapped to other systems, including “Metabolism,” “Genetic Information Processing” and “Cellular Processes.” A full list of KO numbers and their KEGG system assignments is provided in the supplement. (B) Distribution of KEGG subsystems for “Cellular Processes,” “Environmental Information Processing,” “Genetic Information Processing,” and “Metabolism.” In each case, the y-axis is the same as in (A) and the percent of the total genes in each KEGG subsystems is indicated by the x-axis.
Expressed Functions in Lake Vanda Paleomat Community
The KEGG system with highest-expressed, at the RNA-level, predicted ORFs was Genetic Information Processing, mainly translation and ribosome-associated genes, although DNA replication and repair were also transcribed (Figure 7). Metabolism was the second-highest expressed system, and consisted of lipid metabolism, carbohydrate metabolism, energy metabolism (nitrogen and methane), lipopolysaccharide biosynthesis, and xenobiotics degradation. Lipid and fatty acid biosynthesis gene transcription, together with expression at the RNA level of membrane proteins suggest that membrane fluidity and integrity may be actively maintained in at least some members of the paleomat community. Additionally, quorum sensing genes were expressed in the microbial community, as were two-component systems, which are important response systems to changing environmental conditions.
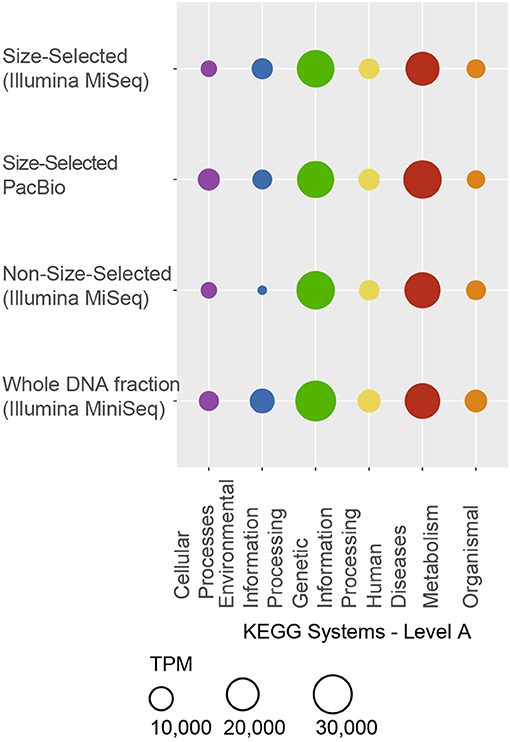
Figure 7. Distirbution of functional genes represented in the transcriptome. KEGG systems are indicated on the x-axis, and the sequencing approach is shown on the y-axis. Many genes mapped to more than one function and to more than one category. Similarly to Figure 6, all genes mapping to “Organismal” and “Human Diseases” also mapped to other systems. A full list of KO numbers with mapped RNA reads is provided in the supplement. Genes with roles in pathways within the “Genetic Information Processing” and “Metabolism” systems were the most abundantly expressed.
Among the most-highly expressed predicted protein-coding genes—coding DNA sequence (CDS) ORFs—were hypothetical proteins and antitoxin genes. Antitoxins form part of toxin-antitoxin (TA) systems, where the toxin is a stable protein that is counteracted by the antitoxin, which can be either a protein or non-coding DNA sequence, and is expressed at a higher level than the toxin. TA systems can perform important cell functions, such as stress response, apoptosis, cell cycle arrest, cell wall biosynthesis and membrane integrity. Other highly expressed transcripts included stress response proteins, Sec-independent protein translocase protein TatAy, cold shock proteins as well as ribosomal proteins, transcription and elongation factors (Figure S3). These observations suggest that the members of the community detected by RNA transcribe the necessary machinery for protein expression, and thus activity, within the Lake Vanda paleomat.
Metagenome Assembled Genome Taxonomic Identity, Function, and Expression
To determine which specific members of the community encoded specific functions, we binned metagenomic sequences to recover metagenome-assembled genomes (MAGs). Closely-related bacterial species may have divergent biological roles, therefore identification of which genes likely came from which genome is a useful tool to explore the genomic potential of, and difference between, species in the absence of pure cultures. A total of 16 high quality (>90% completion, <5% contamination) MAGs were identified in the Illumina-sequenced metagenome assemblies (Figure 8A). Due to similarity of community composition among all datasets, only MAGs from the MiniSeq whole DNA fraction and MiSeq size-selected fraction were used for downstream analyses. These MAGs were comprised of four bacterial phyla, Actinobacteria, Bacteroidetes (Cellulophaga algicola and Cytophagales), Proteobacteria (Alphaproteobacteria, Sphingomonadales, and Xanthomonadaceae) and Firmicutes (Bacilli) (Figure 8B). Average nucleotide identity (ANI) was used to compare the MAGs to each other to identify any MAGs that were likely from the same population. Three pairs of MAGs (one from either sequencing approach) were highly similar, >99% ID, suggesting that they represented the same genomic population (Figure 8B).
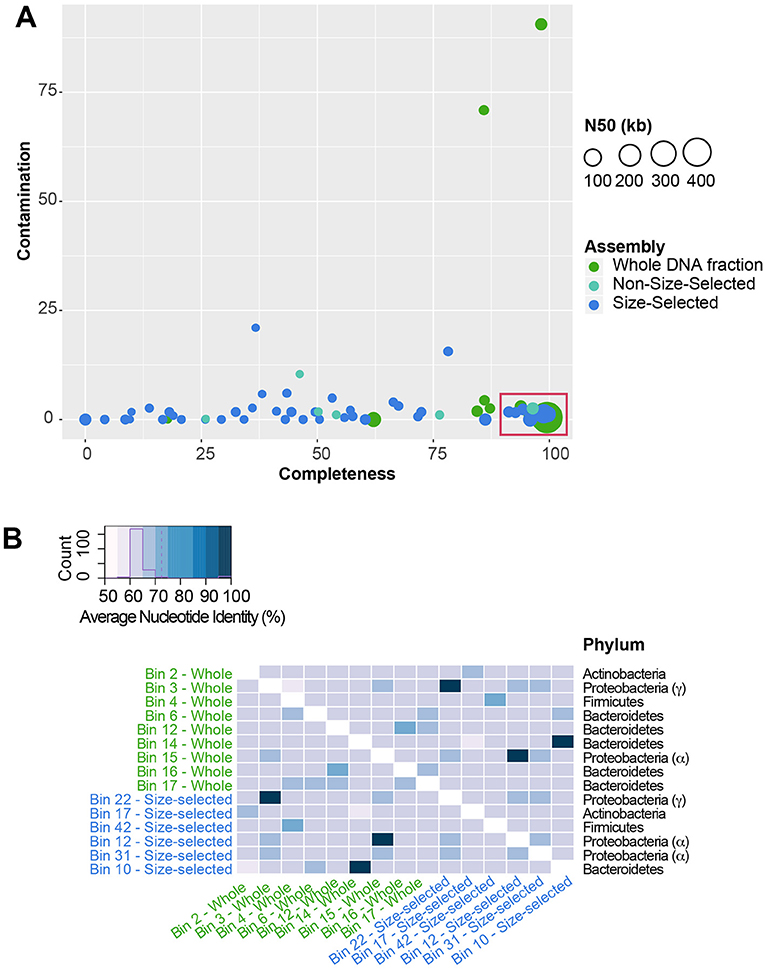
Figure 8. Metagenome-assembled genomes (MAG) within the Illumina-sequenced datasets. (A) Plot showing MAG bin contamination on the y-axis vs. estimated bin completeness on the x-axis. Dots are scaled by the N50 value of the contigs and scaffolds in each MAG, and are color-coded by the sequencing approach. High-quality MAGs used for downstream analysis are indicated by the red box. (B) Average Nucleotide Identity (ANI) of MAGs. Three pairs of MAGs share >99% ANI, indicating that they are likely from the same population of cells.
The MAGs identified in the Lake Vanda paleomat varied in % GC content, ranging from 38 to 67%, coding density (82–90%) and number of genes encoded (Table S2). In addition, MAGs varied in the number of genes expressed, with up to 160 predicted ORFs that had mapped RNA reads (Figure 9). A full list of predicted genes expressed at the RNA level in MAGs is provided in Figure S4. The 50 most highly expressed CDS ORFs showed that the different MAGs express different sets of genes, and that in all MAGs hypothetical proteins were among the most highly-expressed ORFs (Figure 9). The expressed genes that could be functionally annotated share homology with genes involved in stress response, ribosome and transcription, cell cycle regulation, and membrane transport, and likely serve similar functions in the Lake Vanda paleomat.
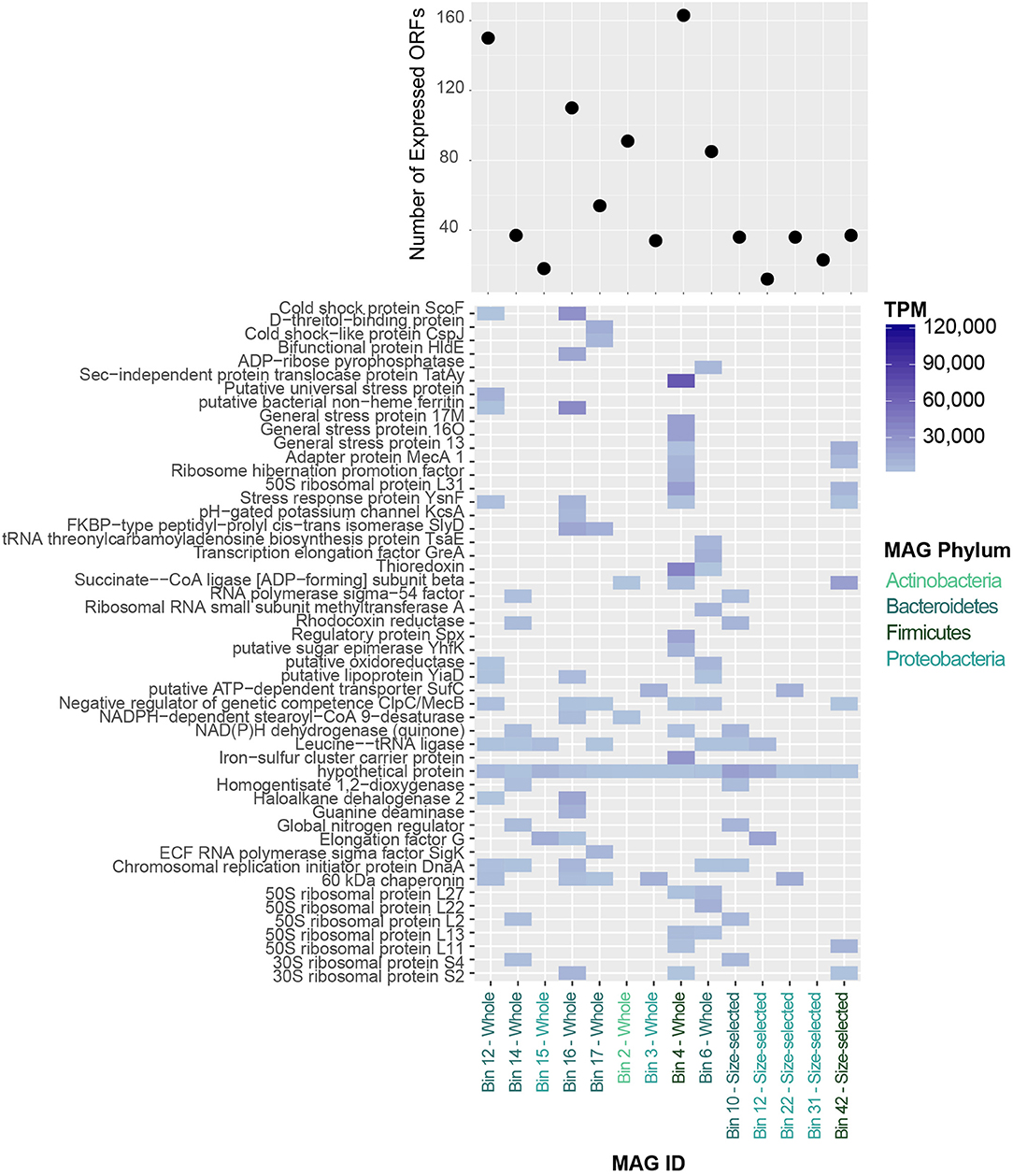
Figure 9. (Top) Number of expressed protein-coding genes in Lake Vanda MAGs. (Bottom) Heatmap of the 50 most highly-expressed protein-coding genes in Lake Vanda MAGs. MAG identities are color-coded according to Phylum-level taxonomy.
Transcribed Genes in Lake Vanda Paleomat MAGs
Understanding which mechanisms organisms express, and therefore probably utilize, to cope with environmental stress in uncultivated organisms is made possible by mapping transcriptomic reads to individual genomes or genomic bins i.e., MAGs. The two Xanthomonadaceae MAGs, whole fraction bin 3 and size-selected bin 22, likely represent the same population of cells due to the similarity of their genomic characteristics. Of the 2,911 genes encoded by the whole fraction bin 3 MAG, only 34 had mapped RNA reads. DNA repair may play an important role in this organism as protein recA and recB were detected in the transcriptome. Additionally, 60 kDa chaperonin groL, which functions in proper assembly and refolding of unfolded polypeptides that are produced under stress conditions. In addition to proper protein folding and DNA repair, this MAG expresses penicillin-binding protein 1A, which is involved in peptidoglycan, and therefore cell wall, biogenesis.
The Alphaproteobacteria MAGs consisted of two distinct populations, the Sphingomonadales (whole fraction bin 15 and size-selected bin 12) and Alphaproteobacteria (size-selected bin 31). The Sphingomonadales MAG expressed just a few genes, 18 of 2,585, which included elongation factor G and chaperone protein ClpB, which processes protein aggregates as part of a stress-induced multi-chaperone system. The size-selected bin 31 MAG only had 23 predicted genes that were detected by transcriptomics. These included regulatory protein BlaR1, which is needed for beta-lactamase induction, as well as the blue light- and temperature-regulated antirepressor BluF, a photoreceptor protein induced at low temperatures and blue light irradiation.
The Actinomycateles MAG, whole fraction bin 2, contained 4,143 proteins, 91 of which were detected in the transcriptome. Among the expressed putative genes were 60 kDa chaperonin 2 and chaperone proteins DnaJ 1 and DnaK, transcription regulators and elongation factors, as well as DNA gyrase subunits A and B. This organism may be sensitive to oxygen concentrations, as it also expresses the hypoxic response protein hrp1, which is induced in response to hypoxia or low levels of nitric oxide and carbon monoxide.
The Bacilli MAGs were not from the same population of cells, sharing ~72% ANI. The whole fraction bin 4 had the highest number of expressed predicted genes, 163, and had low GC content. Among the transcribed predicted genes were general stress proteins yocK, yflT, yugI_2, ysnF, yfkM, and the universal stress protein TeaD, as was OpuCC that helps cells cope with environmental stress. In addition to thioredoxin, ribosome proteins and transcriptional regulation genes, this MAG expressed regulatory protein Spx that may play a role in down-regulating genes involved in growth and negatively affecting competence and sporulation during periods of extreme stress. Other genes with roles in sporulation inhibition (Adapter protein MecA 1) and negative regulation of competence (ClpC/MecB) were also expressed, as was the response regulator Spo0A. These cells were likely in the vegetative state, as Vegetative protein 296 (sufC) and cell division proteins were expressed. Components of the UvrABC DNA repair system were also detected in the transcriptome of this MAG. Similarly, the second Bacilli MAG also contained Adapter protein MecA 1, ClpC/MecB, general stress proteins and UvrABC system components.
The Bacteroidetes MAGs, whole fraction bin 14 and size-selected bin 10, were identified as Cellulophaga algicola, and shared >99% ANI. This organism was very low GC (38%) and contained ~3,300 predicted genes, 37 of which were detected in the transcriptome. The expressed predicted genes included the global nitrogen regulator ntcA_1, gyrB and DNA repair proteins, macrolide export ATP-binding/permease protein MacB, which has roles in energy generation and antibiotic resitance, azurin, and BarA, the kinase component of the two-component regulatory system UvrY/BarA involved carbon metabolism regulation.
The other four Bacteroidetes bins all were identified as Cytophagales, but did not share high ANI with each other. Among the predicted genes detected in the transcriptome for these MAGs were ClpC/MecB, macrolide export ATP-binding/permease protein MacB, oxidoreductases (including fructose, etc.), multidrug resistance protein MdtB, outer membrane proteins, chaperonin and chaperone proteins, stress proteins, cold shock proteins and metal (cobalt, zinc, cadmium) resistance enzymes. Putative genes with roles in reactive oxygen tolerance were also expressed. DNA repair genes including RecA, RadA, DNA gyrase subunits A and B, G/U mismatch-specific DNA glycosylase, an enzyme that excises ethenocytosine and uracil and is required for DNA damage lesion repair, as well as the modification methylase DpnIIA, which protects DNA from cleavage, were also detected in the transcriptome.
Discussion
The bacterial community composition of the paleomat appears to represent a continuum between indigenous mat cells and late colonization by soil microbes, distinct from the community prevalent in the modern moat mat, whose constituent organisms are exposed to significantly different conditions and nutrient availability than those in the paleomat sample. A portion of the OTUs in the 16S rRNA analysis reside within modern mat communities whereas a significant subset also represent soil contributions, though it should be noted that paleomat fragments, too small to be visible, could have been extracted as part of the surrounding soils. Although we could not perform robust statistical comparisons across samples due to a lack of biological replication, our results provide a foundational description of the composition, genomic content and transcription of microbial communities in an Antarctic paleomat, providing a first look into the ecology of these systems.
A variety of DNA extraction techniques, including bead beating and the use of a Metapolyzyme pre-treatment with lysing enzymes to induce spheroplast formation, were used to ensure full lysis of recalcitrant endospores. Extraction methods omitting mechanical lysis (bead-beating) and including Metapolyzyme digestion produced DNA with the highest molecular weight when compared to other methods tested, as determined by visualizing the purified DNA using an Agilent BioAnalyzer. This high molecular weight DNA could then be size-selected using BluePippin or gel-based approaches. Although different from the 16S rRNA gene sequencing results in terms of relative proportions of some bacterial phyla, possibly due to differences in genome sizes and coverage, there were broad similarities in the four metagenomic data sets, despite different sequencing platforms and different extraction techniques (bead beating, chemical lysis), suggesting that the same populations of cells were lysed and sequenced. Furthermore, since there was little difference between size-selected and whole DNA fractions, it appears that the amount of eDNA and/or aDNA present in the sample was negligible or too degraded for sequencing library construction, indicating that the results likely represent a viable community of intact cells.
Lake Vanda Paleomat Community Is Comprised of Bacterial Phyla
A subset of the organisms recovered were similar to those in the Antibus et al. (2012a) cultivation study targeting similar paleomats, suggesting that the bacterial groups identified sourced from within the paleomat are viable. The cultivable bacteria found by Antibus et al. (2012a) in modern mats include members of the Firmicutes, Actinobacteria, Proteobacteria, Cyanobacteria, and Deinococcus-Thermus. In contrast, cultivable bacteria in ancient samples were restricted to Firmicutes, Actinobacteria, and Bacteroidetes (Antibus et al., 2012a). In our study, among organisms associated with gene transcripts, were Actinobacteria, Proteobacteria, Firmicutes, and Bacteroidetes, as well as low levels of Chloroflexi, Cyanobacteria, Actinobacteria, Deinococcus-Thermus, Planctomycetes, Acidobacteria, Pseudothermotoga, Dictyoglomi, Chlorobia, Deferribacteres, Spirochaetes, as well as members of multiple candidate divisions. Similarly, a recent mesocosm study using soil sourced from near Lake Fryxell and aimed at characterizing active taxa, showed that Bacteria, particularly Actinobacteria, Firmicutes, and Proteobacteria, were the dominant members of the community, albeit at lower proportions than in our samples (Buelow et al., 2016). These results indicate that bacteria with diverse metabolic functions, including heterotrophy, autotrophy and roles in nitrogen cycling, are likely able to survive in a non-dormant state within the paleomat community.
In addition to differences inherent between cultivation-based and molecular detection approaches, one reason for the discrepancy in observed potentially active taxa may be due to the fact that samples used by Antibus et al. (2012a) were sourced from older facies (up to 26 k year) than the Lake Vanda paleomats we studied, ~2 k year. Another reason may be that culturing only recovered a subset of the viable cells in the paleomats. While culturing is an important approach to studying cell survival, the vast majority of microbes have not been cultured, and may be resistant to isolation in a laboratory setting due to their as-yet unknown physiological requirements (Schloss and Handelsman, 2005). In a follow-up study of 16S rRNA gene PCR products from bulk DNA, Antibus et al. (2012b) found Firmicutes, Bacteroidetes, Chloroflexi, Cyanobacteria, Actinobacteria, and Actinobacteria from the same microbial mat samples, sourced either from viable cells or preserved genetic material.
In our data set, Actinobacteria, which have been previously detected in modern and ancient permafrost environments (e.g., Kochkina et al., 2001; Yergeau et al., 2010) were the most abundant among clades with mapping gene transcripts, potentially because of the presence of DNA repair at low temperature (Johnson et al., 2007). Corynebacteria, one of the most abundant orders in the Lake Vanda paleomat, are pleomorphic bacteria whose shape depends on surrounding conditions. While usually rod-shaped, Corynebacteria appear distinctively club-shaped during certain stages of the life cycle. They are non-spore-forming, non-motile, organotrophs, and are either aerobic or facultatively anaerobic. Micrococcales are diverse shapes—small cocci and rods—and members of this order, for example Arthrobacter, which is expressed in the Lake Vanda paleomat, can switch between a coccus (stationary phase) and rod-shape (exponential growth). Arthrobacter cocci are resistant to desiccation and starvation. The cells revealed in our scanning electron microscope images of the Lake Vanda paleomat are consistent with the morphologies of both of these abundant non-spore-forming orders (See Figure 2).
Among the Proteobacteria with mapping gene transcripts, Cyclobacteriacea had genes related to carbohydrate metabolism, quorum sensing, antibiotic resistance, and carotenoid biosynthesis. Cytophagaceae are heterotrophic bacteria, whose members are primarily aerobes with a respiratory metabolism. Most of them are rod-shaped, but some exhibit filamentation, produce pigments and utilize polysaccharides and proteins. Flavobacteriaceae most are aerobic with a respiratory metabolism, most are rod-shaped, but some exhibit long filaments, and utilize macromolecules such as polysaccharides and proteins.
We also detected gene transcripts related to members of the Firmicutes phylum, which can exist in a metabolically active state as well as form spores that are resistant to desiccation and extreme conditions. We detected Clostridia, which are anaerobic, mostly saprophytic organisms, as well as Bacillus bacteria, which are mainly aerobic. Despite their spore-forming capability, the gene transcripts mapping to Firmicutes suggest the organisms were not dormant at the time of collection. Since samples were collected into a −190°C liquid nitrogen-primed cryoshipper, transferred directly into a −80°C freezer, then processed on dry ice before a rapid addition of Trizol, it seems unlikely outgrowth could have been triggered. This result is reinforced by the two Firmicutes MAGs, size-selected bin 42 (MiSeq) and whole fraction bin 4 (MiniSeq). They are both Bacilli, but with an ANI of ~72% ID, they are not from the same population of cells. Recovered gene transcripts include those associated the down-regulation of sporulation as well as genes focused on stress response and thiol stability.
Pathway Analysis and Survival Strategies
In additional to taxonomy, our metagenomic and metatranscriptomic data provide insights into functional ecology of the paleomat, both as a community and as MAGs. Different taxa appear to use different strategies, indicated by the observation that different MAGs are associated with different gene transcripts. Indeed, the only annotation that was detected across different MAGs was “hypothetical protein,” which includes multiple predicted protein sequences. Yet, many of the genes perform similar functions, allowing for some common themes to emerge.
Stress Response and DNA Repair Are Key Functions in Paleomat Community
Starvation and nutrient scarcity are two of the main challenges faced by the cells in the Antarctic Dry Valleys. Indeed, mesocosm transcriptomes of organisms in Dry Valleys soils appear to be adapted to low nutrient availability, as the most abundant transcripts were associated with carbon and nitrogen cycling (Buelow et al., 2016). While Antarctic conditions are generally very oligotrophic, it may be that the abundance of organic carbon in the relic mats prevents dormancy from being a dominant survival strategy, despite the extreme desiccation and extreme temperatures. Most members of the community are heterotrophs, as revealed by putative function at metagenome level and expressed function at the metatranscriptome level. However, a few autotrophs are present, and the full Calvin-Benson-Bassham cycle is encoded in metagenomes. Similar observations of dominance by heterotrophic organisms, but presence of autotrophs have been made in Dry Valleys soil communities (Chan et al., 2013; Buelow et al., 2016), suggesting common strategies are utilized by diverse organisms. Additionally, a few organisms, such as Rhodobacter, also have physiological plasticity—the ability to grow autotrophically as well as heterotrophically under different environmental conditions. Whether the specific strains identified by sequencing are capable of this metabolic plasticity remains to be determined.
Amino acid uptake is a response and adaptation to starvation. The gene encoding carbon starvation protein A, cstA, was found in the Lake Vanda paleomat transcriptome. The CstA protein is induced in response to carbon starvation and works by facilitating and increasing peptide uptake (Schultz and Matin, 1991; Rasmussen et al., 2013). Moreover, the presence of cstA gives organisms the ability to survive and continue growing in a low-nutrient environment without forming a spore (Marshall, 1977). Within our data, this gene was primarily associated with Actinobacteria and Proteobacteria, two of the major phyla in our sample. Additionally, ABC transporters involved amino acid uptake were present in the metagenomes. Interestingly, the expressed ABC transporters were similar to those involved in exoprotein transport and sugar (galactose) uptake. However, amino acid transport permeases, oligopeptide transport ATP-binding protein OppF, and dipeptide transport system permease protein DppB, which can be used for heme uptake (Létoffé et al., 2006) were expressed in the paleomat transcriptome. Our transcriptome data suggests that the organisms in the community are adept at taking up nutrients from their environment.
In addition to resisting environmental stresses, organisms in harsh environments must compete for scarce resources. Metagenomic sequences primarily associated with Actinobacteria and Proteobacteria and several MAGs encoded and expressed genes with roles in antibiotic resistance. In Lake Vanda, competition between bacteria within their harsh habitat is likely the leading cause for the evolution of antibiotic production and resistance, offering resistant organisms a competitive advantage (Tam et al., 2015; Wei et al., 2015). In many environments, especially pristine environments poor in nutrients and with extreme conditions contain antibiotic resistance genes that are unlikely to have resulted from anthropogenic contribution to antibiotic resistance.
Universal stress protein, general stress proteins, OpuCC, and stress response proteins were all present in the paleomat transcriptome. Universal stress proteins are induced under high stress conditions, including nutrient starvation and exposure to oxidants and DNA-damaging agents, such as UV, to provide general stress protection via mechanisms independent of other stress responses (Gustavsson, 2002; Tkaczuk et al., 2013). The gene encoding the stress response protein Ysnf was present and expressed in several MAGs. YnsF is dependent on the transcription factor Sigma B (Prágai and Harwood, 2002), which is abundantly present in the metagenome. Presence of this transcription factor usually points to an enhanced response to phosphate starvation, allowing the organism to survive under conditions of phosphate-deprivation (Prágai and Harwood, 2002). Sigma B regulates gene response to phosphate starvation by limiting the metabolic consequences of phosphate deprivation (Allenby et al., 2005). Other stress response genes, rpoE and rpoH, both of which encode sigma factors (Jensen-Cain and Quinn, 2001), are present in the metagenome. Although no other genomic or transcriptomic data is available for other Antarctic paleomats, a GeoChip study of soil organisms from McKelvey Valley also indicated the importance of sigma factors necessary for various environmental stresses, including osmotic shock, desiccation) cold- and heat-shock, and starvation (Chan et al., 2013). RpoH is responsible for sigma 32 and plays a role in the activation of chaperones and DNA repair enzymes (Gruber et al., 2003). RpoE encodes sigma factor 24, and is essential in some Firmicutes for sporulation (Kirk et al., 2014). Therefore, the paleomat community members detected in the transcriptome express the genes necessary to resist environmental stresses and enable their survival.
Among some of the most highly-expressed genes in our dataset were chaperone cold shock proteins. These proteins function to respond to rapid temperature downward shift to help cells adapt to new conditions (reviewed in Keto-Timonen et al., 2016). Downward temperature changes cause changes in promoter activity during translation, suggesting that ribosomes play an important role in sensing temperature drops and eliciting a cold-shock response (Vanbogelen et al., 1990; Jones and Inouye, 1994, 1996; Graumann and Marahiel, 1998; Phadtare and Inouye, 2008). In the paleomat transcriptome, many of the expressed genes were associated with the ribosome and ribosome maturation. Of the nine members of the Csp family identified so far, two are constitutively expressed at physiological temperatures, one is induced under nutrient deficient conditions, and four are sensitive to cold shocks (Phadtare and Inouye, 2008). Interestingly, other chaperone proteins implicated in cold and heat shock responses also play essential roles in the folding of most proteins and are therefore required for survival even at physiological temperatures (Gragerov et al., 1992; Langer et al., 1992).
Chaperonins and protein chaperone proteins, including DnaK and DnaJ were well-represented in the Lake Vanda paleomat transcriptome. Chaperones play an important role in proper protein folding and homeostasis maintenance under thermal stress conditions and hyperosmotic shock by preventing aggregation of misfolded proteins. The chaperones DnaK, DnaJ, GrpE, and GroEL act in a sequential manner to recognize a growing polypeptide, stabilize an intermediate folding state, hydrolyzes ATP, and catalyzes the formation of the protein's native state. Although these proteins function to fold many proteins, including ones unrelated to cold shock tolerance, studies have shown that at least one of the GroE genes was essential for growth at low temperatures (Fayet et al., 1989). Additionally, chaperone proteins can act to re-establish membrane fluidity. Their importance in this role is highlighted by the expression of lipid and fatty acid biosynthesis pathway genes expressed in our dataset.
Besides stress and starvation response, DNA repair was a key function expressed in the transcriptome. The UvrABC that forms part of the prokaryotic nucleotide excision repair system recognizes and fixes DNA damage lesions (Truglio et al., 2006). This type of repair system removes DNA damage caused by UV radiation. We also detected components of base excision repair system that repairs endogenous DNA damage—deamination, alkylation, oxidation, and single-stranded breaks—throughout the life cycle (Krokan et al., 2013). Additionally, we found RNA-expressed genes that are essential for DNA maintenance and recombinational repair. The RecA protein functions in recombinational DNA repair and is regulated through the SOS response, which was present in the metagenome (Cox, 2007). RadA also functions in recombinational DNA repair, where it repairs DNA breaks. Taken together, the functions of genes expressed in the Lake Vanda microbial mat community act to cope with starvation, environmental stress including cold and osmotic shock, and DNA damage either endogenous or due to UV exposure.
Future Work
Given the nature of the RNA we recovered in this study, we cannot exclude the possibility that the RNA is ancient, preserved in the paleomat. Dormancy, for instance, might protect RNA through bundling with protein or polyhydroxybutyrate (Ackermann et al., 1995) within a recalcitrant endospore. We saw little evidence for dormancy, however, or evidence the presence of aDNA or eDNA in the paleomat, which has a far longer half-life than RNA. Protein isolation and metaproteomics would determine what levels of proteins and peptides are present in the paleomat, confirming the processes of transcription and translation.
Expanding this analysis to other, older paleomats in the Dry Vallys is also an intriguing direction for future work. It is possible that over longer time scales, low-level DNA repair and other gene transcripts would be less prevalent as more and more organisms form spores. Sequencing platforms like Oxford Nanopore Technologies' MinION, which has recently been demonstrated in the Dry Valleys as a proof of principle for future in situ analyses (Johnson et al., 2017), and PacBio may also open new ways to interrogate DNA damage and repair via base modification detection. Base modifications, like cytosine methylation, are directly detectable in PacBio data by measuring kinetic variation during base incorporation (Marx, 2016) and have also been detected in nanopore sequencing data (Simpson et al., 2017). As DNA does not have to be amplified before sequencing, this method opens the door to analyzing rare sequence variants in situ. While not yet demonstrated, deamination events may be detectable too. Over time, in the absence of DNA repair, the genomes of truly dormant organisms accumulate damage due to spontaneous chemical reactions. The most common form of hydrolytic DNA damage involves deamination of one of the three nucleic acids containing an amine group, which in the case of the base cytosine generates uracil or its analogs (Shapiro and Hofreiter, 2014). The subsequent pairing of uracil with adenine leads to the observation of characteristic C->T/G->A transitions. If large numbers of deamination events were prevalent in the recovered DNA of spore-forming organisms, it would suggest true dormancy, that DNA repair processes had been limited.
Conclusion
Paleomats in the Dry Valleys of Antarctica may play a key role in Antarctic biogeography, but they have never been studied with metagenomics or metatranscriptomics. The results of this study provide an exploratory look at the genomic potential and possible transcriptional activity of an Antarctic paleomat, and suggest that Dry Valleys paleomats may contain multiple types of viable cells, potentially sourced from indigenous mat communities, followed by later colonization by soil-dwelling microbes. We recovered long continuous strands of DNA from the Lake Vanda paleomat, significantly longer than those recovered from dead organisms on ancient timescales, along with RNA transcripts and several near-complete MAGs. Despite the extremely cold and arid ambient conditions, dormancy does not appear to be a prevalent cell survival strategy, even for spore-forming members of the Firmicutes. This may be because of the relatively abundant relic carbon associated with the ancient microbial mats, but further work is called for.
These paleomats may serve as “seed banks,” outposts for life over long timescales, at the same time as being sheltered nutrient-rich niches for soil-dwelling microbes in the oligotropic terrain of the Dry Valleys. Viable organisms from paleomats may have been reintroduced to benthic and moat mat communities via aeolian transport or during lake level fluctuations, which have occurred many times since the last glacial maximum (Hall et al., 2010). By elucidating the resilience of life in cold, hyper-arid conditions, our results may have implications for biotechnology applications involving stress response and DNA repair as well as providing new insight into Antarctic microbiology and biogeography, and the limits of life in extremely harsh environments.
Author Contributions
EZ, DG, ST, and SJ conceived of the study. BH advised on Antarctic geology and study design. EZ, DG, ST, YB, and SJ performed field work in Antarctica. EZ, DG, ST, YB, and NW performed research. EZ analyzed the data. EZ, DG, ST, YB, NW, BH, JB, MW, MS-F, and SJ wrote the paper.
Funding
This work was supported by NSF Office of Polar Programs Award PLR-1620976 to SJ at Georgetown University. The Genomics and Epigenomics Shared Resource at Georgetown University Medical Center is partially supported by NIH/NCI grant P30-CA051008.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We give special thanks to the Illumina Corporation, specifically Mostafa Ronaghi, for dedicated support of Antarctic research and for supplying necessary instrumentation and reagents while at the Crary Lab at McMurdo Station. We are grateful to Millipore Sigma for early access to Metapolyzyme for DNA extraction. Much appreciated technical contributions were made by the University of Vermont Microscopy Imaging Facility and the Advanced Genome Technology Lab. The University of Wisconsin–Madison Biotechnology Center provided outstanding PacBio sequencing support and advised on data analysis. Members of the Extreme Microbiome Project provided extremely helpful technical input. Last but not least, we would like to thank the U.S. Antarctic Program and the support staff at McMurdo Station, particularly the individuals who work at the Crary Lab, HeloOPS, and MacOPS.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fevo.2019.00001/full#supplementary-material
References
Abyzov, S., Mitskevich, I., and Poglazova, M. (1998). Microflora of the deep glacier horizons of Central Antarctica. Microbiology 67, 451–458.
Ackermann, J., Müller, S., Lösche, A., Bley, T., and Babel, W. (1995). Methylobacterium rhodesianum cells tend to double the DNA content under growth limitations and accumulate PHB. J. Biotechnol. 39, 9–20. doi: 10.1016/0168-1656(94)00138-3
Allenby, N. E. E., O'Connor, N., Prágai, Z., Ward, A. C., Wipat, A., and Harwood, C. R. (2005). Genome-wide transcriptional analysis of the phosphate starvation stimulon of Bacillus subtilis. J. Bacteriol. 187, 8063–8080. doi: 10.1128/JB.187.23.8063-8080.2005
Antibus, D. E., Leff, L. G., Hall, B. L., Baeseman, J. L., and Blackwood, C. B. (2012a). Cultivable bacteria from ancient algal mats from the McMurdo Dry Valleys, Antarctica. Extremophiles 16, 105–1. doi: 10.1007/s00792-011-0410-3
Antibus, D. E., Leff, L. G., Hall, B. L., Baeseman, J. L., and Blackwood, C. B. (2012b). Molecular characterization of ancient algal mats from the McMurdo Dry Valleys, Antarctica. Antar. Sci. 24, 139–146. doi: 10.1017/S0954102011000770
Barnett, D. W., Garrison, E. K., Quinlan, A. R., Strömberg, M. P., and Marth, G. T. (2011). Bamtools: A C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 27, 1691–1692. doi: 10.1093/bioinformatics/btr174
Bergholz, P. W., Bakermans, C., and Tiedje, J. M. (2009). Psychrobacter arcticus 273-4 uses resource efficiency and molecular motion adaptations for subzero temperature growth. J. Bacteriol. 191, 2340–2352. doi: 10.1128/JB.01377-08
Buelow, H. N., Winter, A. S., Van Horn, D. J., Barrett, J. E., Gooseff, M. N., Schwartz, E., et al. (2016). Microbial community responses to increased water and organic matter in the arid soils of the McMurdo Dry Valleys, Antarctica. Front. Microbiol. 7:1040. doi: 10.3389/fmicb.2016.01040
Bushnell, B. (2014). “BBMap: A Fast, Accurate, Splice-Aware Aligner,” in Conference: 9th Annual Genomics of Energy Environment Meeting (Walnut Creek, CA).
Bybee, S., Bracken-Grissom, H., Haynes, B. D., Hermansen, R. A., Byers, R. L., et al. (2011). Targeted amplicon sequencing (TAS): a scalable next-gen approach to multilocus, multitaxa phylogenetics. Genome Biol. Evol. 3, 1312–1323. doi: 10.1093/gbe/evr106
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J., and Holmes, S. P. (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Canfield, D. E., and Green, W. J. (1985). The cycling of nutrients in a closed-basin antarctic lake: Lake Vanda. Biogeochemistry 1, 233–256. doi: 10.1007/BF02187201
Carini, P., Marsden, P. J., Leff, J. W., Morgan, E. E., Strickland, M. S., and Fierer, N. (2016). Relic DNA is abundant in soil and obscures estimates of soil microbial diversity. Nat. Microbiol. 2:16242. doi: 10.1038/nmicrobiol.2016.242
Cary, S. C., McDonald, I. R., Barrett, J. E., and Cowan, D. A. (2010). On the rocks: the microbiology of Antarctic Dry Valley soils. Nat. Rev. Microbiol. 8, 129–38. doi: 10.1038/nrmicro2281
Chan, Y., Van Nostrand, J. D., Zhou, J., Pointing, S. B., and Farrell, R. L. (2013). Functional ecology of an Antarctic Dry Valley. Proc. Natl. Acad. Sci. U.S.A. 110, 8990–8995. doi: 10.1073/pnas.1300643110
Cox, M. M. (2007). Regulation of bacterial RecA protein function. Crit. Rev. Biochem. Mol. Biol. 42, 41–63. doi: 10.1080/10409230701260258
Doran, P. T., McKay, C. P., Clow, G. D., Dana, G. L., Fountain, A. G., Nylen, T., et al. (2002). Valley floor climate observations from the McMurdo dry valleys, Antarctica, 1986-2000. J. Geophys. Res. Atmos. 107, ACL 13-1-ACL 13-12. doi: 10.1029/2001JD002045
Doran, P. T., Wharton, R. A., Des Marais, D. J., and McKay, C. P. (1998). Antarctic paleolake sediments and the search for extinct life on Mars. J. Geophys. Res. E Planets 103, 28481–28493. doi: 10.1029/98JE01713
Doran, P. T., Wharton, R. A., and Lyons, W. B. (1994). Paleolimnology of the McMurdo Dry Valleys, Antarctica. J. Paleolimnol.10, 85–114. doi: 10.1007/bf00682507
Fayet, O., Ziegelhoffer, T., and Georgopoulos, C. (1989). The groES and groEL heat shock gene products of Escherichia coli are essential for bacterial growth at all temperatures. J. Bacteriol. 171, 1379–85. doi: 10.1128/jb.171.3.1379-1385.1989
Fountain, A. G., Nylen, T. H., Monaghan, A., Basagic, H. J., and Bromwich, D. (2010). Snow in the McMurdo Dry Valleys, Antarctica. Int. J. Climatol. 30, 633–642. doi: 10.1002/joc.1933
Gragerov, A., Nudler, E., Komissarova, N., Gaitanaris, G. A., Gottesman, M. E., and Nikiforov, V. (1992). Cooperation of GroEL/GroES and DnaK/DnaJ heat shock proteins in preventing protein misfolding in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 89, 10341–10344. doi: 10.1073/pnas.89.21.10341
Graumann, P. L., and Marahiel, M. A. (1998). A superfamily of proteins that contain the cold-shock domain. Trends Biochem. Sci. 23, 286–90. doi: 10.1016/S0968-0004(98)01255-9
Green, W. J., Canfield, D. E., and Nixon, P. (1998). “Cobalt cycling and fate in Lake Vanda,” in Ecosystem Dynamics in a Polar Desert, Vol 72, ed J. C. Priscu (Washington, DC: American Geophysical Union), 205–215.
Green, W. J., and Lyons, W. B. (2009). The saline lakes of the McMurdo Dry Valleys, Antarctica. Aquat. Geochem. 15, 321–348. doi: 10.1007/s10498-008-9052-1
Gruber, S., Haering, C. H., and Nasmyth, K. (2003). Chromosomal cohesin forms a ring. Cell 112:765. doi: 10.1016/S0092-8674(03)00162-4
Gustavsson, N. (2002). The universal stress protein paralogues of Escherichia coli are co-ordinately regulated and co-operate in the defence against DNA damage. Mol. Microbiol. 43, 107–117. doi: 10.1046/j.1365-2958.2002.02720.x
Hall, B. L., Denton, G. H., Fountain, A. G., Hendy, C. H., and Henderson, G. M. (2010). Antarctic lakes suggest millennial reorganizations of Southern Hemisphere atmospheric and oceanic circulation. Proc. Natl. Acad. Sci. U.S.A. 107:21355–21359. doi: 10.1073/pnas.1007250107
Hall, B. L., Denton, G. H., and Overturf, B. (2001). Glacial Lake Wright, a high-level antarctic lake during the LGM and early holocene. Antar. Sci. 13, 53–60. doi: 10.1017/S0954102001000086
Hall, B. L., Denton, G. H., Overturf, B., and Hendy, C. H. (2002). Glacial Lake Victoria, a high-level antarctic lake inferred from lacustrine deposits in Victoria Valley. J. Q. Sci. 17, 697–706. doi: 10.1002/jqs.691
Hendy, C. H., Sadler, A. J., Denton, G. H., and Hall, B. L. (2000). Proglacial lake-ice conveyors: a new mechanism for deposition of drift in polar environments. Geografiska Annaler Series A Phys Geogr. 82, 249–270. doi: 10.1111/j.0435-3676.2000.00124
Hofreiter, M., Paijmans, J. L., Goodchild, H., Speller, C. F., Barlow, A., Fortes, G. G., et al. (2015). The future of ancient DNA: technical advances and conceptual shifts. BioEssays 37, 284–293. doi: 10.1002/bies.201400160
Jensen-Cain, D. M., and Quinn, F. D. (2001). Differential expression of sigE by Mycobacterium tuberculosis during intracellular growth. Microb. Pathoge. 30, 271–278. doi: 10.1006/mpat.2001.0431
Johnson, S. S., Hebsgaard, M. B., Christensen, T. R., Mastepanov, M., Nielsen, R., Munch, K., et al. (2007). Ancient bacteria show evidence of DNA repair. Proc. Natl. Acad. Sci. U.S.A. 104, 14401–14405. doi: 10.1073/pnas.0706787104
Johnson, S. S., Zaikova, E., Goerlitz, D. S., Bai, Y., and Tighe, S. W. (2017). Real-time DNA sequencing in the Antarctic Dry Valleys using the Oxford Nanopore sequencer. J. Biomol. Tech. 28, 2–7. doi: 10.7171/jbt.17-2801-009
Jones, P. G., and Inouye, M. (1994). The cold-shock response—a hot topic. Mol. Microbiol. 11, 811–818. doi: 10.1111/j.1365-2958.1994.tb00359.x
Jones, P. G., and Inouye, M. (1996). RbfA, a 30S ribosomal binding factor, is a cold-shock protein whose absence triggers the cold-shock response. Mol. Microbiol. 21, 1207–1218. doi: 10.1111/j.1365-2958.1996.tb02582.x
Jørgensen, B. B. (2011). Deep subseafloor microbial cells on physiological standby. Proc. Natl. Acad. Sci. U.S.A. 108, 18193–18194. doi: 10.1073/pnas.1115421108
Kang, D. D., Froula, J., Egan, R., and Wang, Z. (2015). MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 3:e1165. doi: 10.7717/peerj.1165
Karr, E. A., Sattley, W. M., Rice, M. R., Jung, D. O., Madigan, M. T., and Achenbach, L. A. (2005). Diversity and distribution of sulfate-reducing bacteria in permanently frozen Lake Fryxell, McMurdo Dry Valleys, Antarctica. Appl. Env. Microbiol. 71, 6353–6359. doi: 10.1128/AEM.71.10.6353-6359.2005
Keto-Timonen, R., Hietala, N., Palonen, E., Hakakorpi, A., Lindström, M., and Korkeala, H. (2016). Cold shock proteins: a minireview with special emphasis on Csp-family of enteropathogenic Yersinia. Front. Microbiol. 7:1151. doi: 10.3389/fmicb.2016.01151
Kirk, D. G., Zhang, Z., Korkeala, H., and Lindström, M. (2014). Alternative sigma factors SigF, SigE, and SigG are essential for sporulation in Clostridium botulinum ATCC 3502. Appl. Environ. Microbiol. 80, 5141–5150. doi: 10.1128/AEM.01015-14
Kochkina, G. A., Ivanushkina, N. E., Karasev, S. G., Gavrish, E. Y., Gurina, L. V., Evtushenko, L. I., et al. (2001). Survival of micromycetes and actinobacteria under conditions of long-term natural cryopreservation. Microbiology 70, 356–364. doi: 10.1023/A:1010419831245
Krokan, H. E., Bjoras, M., and Base excision repair. (2013). Cold spring harbor perspect. Biology 5:a012583. doi: 10.1101/cshperspect.a012583
Lambert, D. M., Ritchie, P. A., Millar, C. D., Holland, B., Drummond, A. J., and Baroni, C. (2002). Rates of evolution in ancient DNA from Ade'lie penguins. Science 295, 2270–2273. doi: 10.1126/science.1068105
Langer, T., Lu, C., Echols, H., Flanagan, J., Hayer, M. K., and Hartl, F. U. (1992). Successive action of DnaK, DnaJ and GroEL along the pathway of chaperone-mediated protein folding. Nature 356, 683–689. doi: 10.1038/356683a0
Lebre, P. H., De Maayer, P., and Cowan, D. A. (2017). Xerotolerant bacteria: surviving through a dry spell. Nat. Rev. Microbiol. 15, 285–296. doi: 10.1038/nrmicro.2017.16
Lee, C. K., Barbier, B. A., Bottos, E. M., McDonald, I. R., and Cary, S. C. (2012). The inter-valley soil comparative survey: the ecology of dry valley edaphic microbial communities. ISME J. 6, 1046–1057. doi: 10.1038/ismej.2011.170
Létoffé, S., Delepelaire, P., and Wandersman, C. (2006). The housekeeping dipeptide permease is the Escherichia coli heme transporter and functions with two optional peptide binding proteins. Proc. Natl. Acad. Sci. U.S.A. 103, 12891–12896. doi: 10.1073/pnas.0605440103
Li, D., Liu, C. M., Luo, R., Sadakane, K., and Lam, T. W. (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Li, D., Luo, R., Liu, C. M., Leung, C. M., Ting, H. F., Sadakane, K., et al. (2016). MEGAHIT v1.0: a fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 102, 3–11. doi: 10.1016/j.ymeth.2016.02.020
Li, H. (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993. doi: 10.1093/bioinformatics/btr509
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv [Preprint]. arXiv:1303.3997.
Love, F. G., Simmons, J.r. G. M, Parker, B. C., Wharton, R. A. Jr., and Seaburg, K.G. (1983). Modern conophyton-like microbial mats discovered in Lake Vanda, Antarctica. Geomicrobiology 3, 33–48. doi: 10.1080/01490458309377782
Marx, V. (2016). Genetics: profiling DNA methylation and beyond. Nat. Methods 13, 119–122. doi: 10.1038/nmeth.3736
McMurdie, P. J., and Holmes, S. (2013). phyloseq: an R Package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8:e61217. doi: 10.1371/journal.pone.0061217
Moorhead, D. L., Doran, P. T., Fountain, A. G., Lyons, W. B., McKnight, D. M., Priscu, J. C., et al. (1999). Ecological legacies: impacts on ecosystems of the McMurdo Dry Valleys. BioScience 49, 1009–1019. doi: 10.2307/1313734
Morgan, C. A., Herman, N., White, P. A., and Vesey, G. (2006). Preservation of micro-organisms by drying; A review. J. Microbiol. Methods 66, 183–193. doi: 10.1016/j.mimet.2006.02.017
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C, and Kanehisa, M. (2007). KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–185. doi: 10.1093/nar/gkm321
Murray, A. E., Kenig, F., Fritsen, C. H., McKay, C. P., Cawley, K. M., Edwards, R., et al. (2012). Microbial life at −13°C in the brine of an ice-sealed Antarctic lake. Proc. Natl. Acad. Sci. U.S.A. 109, 20626–20631. doi: 10.1073/pnas.1208607109
Naqib, A., Poggi, S., Wang, W., Hyde, M., Kunstman, K., and Green, S. J. (2018). “Making and sequencing heavily multiplexed, high-throughput 16S ribosomal RNA gene amplicon libraries using a flexible, two-stage PCR protocol,” in Gene Expression Analysis, eds N. Raghavachari and N. Garcia-Reyero (New York, NY: Humana Press), 149–169.
Ondov, B. D., Bergman, N. H., and Phillippy, A. M. (2011). Interactive metagenomic visualization in a Web browser. BMC Bioinform. 12:385. doi: 10.1186/1471-2105-12-385
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Phadtare, S., and Inouye, M. (2008). “Cold-shock proteins,” in Psychrophiles: From Biodiversity to Biotechnology, eds R. Margesin, F. Schinner, J. C. Marx, and C. Gerday (Berlin; Heidelberg: Springer).
Pointing, S. B., Chan, Y., Lacap, D. C., Lau, M. C. Y., Jurgens, J. A., and Farrell, R. L. (2009). Highly specialized microbial diversity in hyper-arid polar desert. Proc. Natl. Acad. Sci. 106, 19964–19969. doi: 10.1073/pnas.0908274106
Povinec, P. P., Litherland, A. E., and von Reden, K. F. (2009). Developments in Radiocarbon Technologies: from the libby counter to compound-specific AMS analyses. Radiocarbon 51, 45–78. doi: 10.1017/S0033822200033701
Prágai, Z., and Harwood, C. R. (2002). Regulatory interactions between the Pho and sigma(B)-dependent general stress regulons of Bacillus subtilis. Microbiology 148(Pt 5), 1593–1602. doi: 10.1099/00221287-148-5-1593
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
R Core Team (2013). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing. Available online at: http://www.R-project.org/
Rasmussen, J. J., Vegge, C. S., Frøkiær, H., Howlett, R. M., Krofelt, K. A., Kelly, D. J., et al. (2013). Campylobacter Jejuni carbon starvation protein A (CstA) is involved in peptide utilization, motility and agglutination, and has a role in stimulation of dendritic cells. J. Med. Microbiol. 62(Pt_8), 1135–1143. doi: 10.1099/jmm.0.059345-0
Reimer, P. J., Bard, E., Bayliss, A., Beck, J. W., Blackwell, P. G., Ramsey, C. B., et al. (2013). IntCal13 and Marine13 radiocarbon age calibration curves 0–50,000 Years cal BP. Radiocarbon 55, 1869–1887. doi: 10.2458/azu_js_rc.55.16947
Richter, M., Rosselló-Móra, R., Glöckner, F. O., and Peplies, J. (2016). JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 32, 929–931. doi: 10.1093/bioinformatics/btv681
Roberts, M. L., Burton, J. R., Elder, K. L., Longworth, B. E., McIntyre, C. P., von Reden, K. F., et al. (2010). A high-performance 14C Accelerator Mass Spectrometry system. Radiocarbon 52, 228–235. doi: 10.1017/S0033822200045252
RStudio Team (2016). RStudio: Integrated Development for R. Boston, MA: RStudio, Inc. Available online at: http://www.rstudio.com/
Schloss, P. D., and Handelsman, J. (2005). Metagenomics for studying unculturable microorganisms: cutting the gordian knot. Genome Biol. 6:229. doi: 10.1186/gb-2005-6-8-229
Schultz, J. E., and Matin, A. (1991). Molecular and functional characterization of a carbon starvation gene of Escherichia Coli. J. Mol. Biol. 218, 129–140. doi: 10.1016/0022-2836(91)90879-B
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shapiro, B., and Hofreiter, M. (2014). A paleogenomic perspective on evolution and gene function: new insights from ancient DNA. Science 343:1236573. doi: 10.1126/science.1236573
Simpson, J. T., Workman, R. E., Zuzarte, P. C., David, M., Dursi, L. J., and Timp, W. (2017). Detecting DNA cytosine methylation using nanopore sequencing. Nat. Methods 14, 407–410. doi: 10.1038/nmeth.4184
Smith, C. I., Chamberlain, A. T., Riley, M. S., Cooper, A., Stringer, C. B., and Collins, M. J. (2001). Neanderthal DNA: not just old but old and cold? Nature 10, 771–772. doi: 10.1038/35071177
Soina, V. S., and Vorobyova, E. A. (2005). “Adaptation of bacteria to the terrestrial permafrost environment,” in Origins: Genesis, Evolution and Diversity of Life, ed J. Seckbach (Dordrecht: Springer Netherlands), 427–444.
Spigel, R. H., and Priscu, J. C. (1998). “Physical limnology of McMurdo Dry Valley lakes,” in Ecosystem Dynamics in a Polar Desert, Vol 72, ed J. C. Priscu (Washington, DC: American Geophysical Union), 153–187.
Stuiver, M. (1980). Workshop on 14C data reporting. Radiocarbon 22, 964–966. doi: 10.1017/S0033822200010389
Stuiver, M., and Polac, H. A. (1977). Discussion: reporting of 14C data. Radiocarbon 19, 355–363. doi: 10.1017/S0033822200003672
Tam, H. K., Wong, C. M. V. L., Yong, S. T., Blamey, J., and Gonzalez, M. (2015). Multiple-antibiotic-resistant bacteria from the Maritime Antarctic. Polar Biol. 38, 1129–1141. doi: 10.1007/s00300-015-1671-6
Taton, A., Grubisic, S., Brambilla, E., De Wit, R., and Wilmotte, A. (2003). Cyanobacterial diversity in natural and artificial microbial mats of Lake Fryxell (McMurdo Dry Valleys, Antarctica): a morphological and molecular approach. Appl. Environ. Microbiol. 69, 5157–5169. doi: 10.1128/AEM.69.9.5157-5169.2003
Tighe, S., Afshinnekoo, E., Rock, T. M., McGrath, K., Alexander, N., McIntyre, A., et al. (2017). Genomic methods and microbiological technologies for profiling novel and extreme environments for the extreme microbiome project (XMP). J. Biomol. Tech. 28, 31–39. doi: 10.7171/jbt.17-2801-004
Tkaczuk, K. A. L., Shumilin, I., Chruszcz, M., Evdokimova, E., Savchenko, A., and Minor, W. (2013). Structural and functional insight into the universal stress protein family. Evol. Appl. 6, 434–449. doi: 10.1111/eva.12057
Truglio, J. J., Croteau, D. L., Van Houten, B., and Kisker, C. (2006). Prokaryotic nucleotide excision repair: the UvrABC system. Chem. Rev. 106, 233–252. doi: 10.1021/cr040471u
Van Trappen, S., Mergaert, J., Van Eygen, S., Dawyndt, P., Cnockaert, M. C., and Swings, J. (2002). Diversity of 746 heterotrophic bacteria isolated from microbial mats from ten Antarctic lakes. System. Appl. Microbiol. 25, 603–610. doi: 10.1078/07232020260517742
Vanbogelen, R. A., Hutton, E. M., and Neidhardt, F. C. (1990). Gene-Protein database of Escherichia coli K - 12: Edition 3. Electrophoresis 11, 1131–1166. doi: 10.1002/elps.1150111205
Wei, S., Higgins, C., Adriaenssens, E., Cowan, D., and Pointing, S. (2015). Genetic signatures indicate widespread antibiotic resistance and phage infection in microbial communities of the McMurdo Dry Valleys, East Antarctica. Polar Biol. 38, 919–925. doi: 10.1007/s00300-015-1649-4
Wharton, J.r. R. A, Parker, B. C., and Simmons Jr, G.M. (1983). Distribution, species composition and morphology of algal mats in antarctic dry valley lakes. Phycologia 22, 355–365. doi: 10.2216/i0031-8884-22-4-355.1
Willerslev, E., and Cooper, A. (2005). Ancient DNA. Proc. R. Soc. London B Biol. Sci. 272, 3–16. doi: 10.1098/rspb.2004.2813
Wood, D. E., and Salzberg, S. L. (2014). Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 15:R46. doi: 10.1186/gb-2014-15-3-r46
Wood, J. R., Rawlence, N. J., Rogers, G. M., Austin, J. J., Worthy, T. H., and Cooper, A. (2008a). Coprolite deposits reveal the diet and ecology of the extinct New Zealand megaherbivore moa (Aves, Dinornithiformes). Q. Sci. Rev. 27, 2593–2602. doi: 10.1016/j.quascirev.2008.09.019
Wood, S. A., Rueckert, A., Cowan, D. A., and Cary, S. C. (2008b). Sources of edaphic cyanobacterial diversity in the Dry Valleys of Eastern Antarctica. ISME J. 2, 308–20. doi: 10.1038/ismej.2007.104
Yergeau, E., Hogues, H., Whyte, L. G., and Greer, C. W. (2010). The functional potential of high Arctic permafrost revealed by metagenomic sequencing, qPCR and microarray analyses. ISME J. 4, 1206–1214. doi: 10.1038/ismej.2010.41
Zaikova, E., Walsh, D. A., Stilwell, C. P., Mohn, W. W., Tortell, P. D., and Hallam, S. J. (2010). Microbial community dynamics in a seasonally anoxic fjord: saanich inlet, British Columbia. Environ. Microbiol. 12, 172–91. doi: 10.1111/j.1462-2920.2009.02058.x
Keywords: Antarctica, cell survival, DNA repair, dormancy, extremophiles, metagenomics, stress response, transcriptome
Citation: Zaikova E, Goerlitz DS, Tighe SW, Wagner NY, Bai Y, Hall BL, Bevilacqua JG, Weng MM, Samuels-Fair MD and Johnson SS (2019) Antarctic Relic Microbial Mat Community Revealed by Metagenomics and Metatranscriptomics. Front. Ecol. Evol. 7:1. doi: 10.3389/fevo.2019.00001
Received: 15 July 2018; Accepted: 03 January 2019;
Published: 23 January 2019.
Edited by:
Alison Elizabeth Murray, Desert Research Institute (DRI), United StatesReviewed by:
Anne D. Jungblut, Natural History Museum, United KingdomJamie S. Foster, Space Life Science Laboratory, University of Florida, United States
Stephen Brian Pointing, Yale-NUS College, Singapore
Copyright © 2019 Zaikova, Goerlitz, Tighe, Wagner, Bai, Hall, Bevilacqua, Weng, Samuels-Fair and Johnson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sarah Stewart Johnson, c2FyYWguam9obnNvbkBnZW9yZ2V0b3duLmVkdQ==