 Erik Ersmark1,2*
Erik Ersmark1,2* Cornelya F. C. Klütsch3
Cornelya F. C. Klütsch3 Yvonne L. Chan1
Yvonne L. Chan1 Mikkel-Holger S. Sinding4Steven R. Fain5
Mikkel-Holger S. Sinding4Steven R. Fain5 Natalia A. Illarionova6Mattias Oskarsson7Mathias Uhlén7
Natalia A. Illarionova6Mattias Oskarsson7Mathias Uhlén7 Ya-ping Zhang8
Ya-ping Zhang8 Love Dalén1†
Love Dalén1† Peter Savolainen7†
Peter Savolainen7†- 1Department of Bioinformatics and Genetics, Swedish Museum of Natural History, Stockholm, Sweden
- 2Department of Zoology, Stockholm University, Stockholm, Sweden
- 3Biology Department, Trent University, Peterborough, ON, Canada
- 4Centre for GeoGenetics, Natural History Museum of Denmark, Copenhagen University, Copenhagen, Denmark
- 5National Forensics Laboratory, U. S. Fish and Wildlife Service, Ashland, OR, USA
- 6Laboratory of Microevolution and Domestication of Mammals, A. N. Severtsov Institute of Ecology and Evolution, Moscow, Russia
- 7Science for Life Laboratory, Department of Gene Technology, KTH Royal Institute of Technology, Solna, Sweden
- 8Laboratory of Cellular and Molecular Evolution, and Molecular Biology of Domestic Animals, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, China
The global distribution of the gray wolf (Canis lupus) is a complex assembly consisting of a large number of populations and described subspecies. How these lineages are related to one another is still not fully resolved, largely due to the fact that large geographical regions remain poorly sampled both at the core and periphery of the species' range. Analyses of ancient wolves have also suffered from uneven sampling, but have shown indications of a major turnover at some point during the Pleistocene-Holocene boundary in northern North America. Here we analyze variation in the mitochondrial control region in 122 contemporary wolves from some of the less studied populations, as well as six samples from the previously unstudied Greenland subspecies (Canis l. orion) and two Late Pleistocene samples from Siberia. Together with the publicly available control region sequences of both modern and ancient wolves, this study examines genetic diversity on a wide geographical and temporal scale that includes both Eurasia and North America. We identify 13 new haplotypes, of which the majority is found in northern and eastern Asia. The results show that the Greenland samples are all represented by one haplotype, previously identified in North American wolves, among which this population seems to trace its maternal lineage. The phylogeny and network analyses show a wide spatial distribution of several lineages, but also some clusters with more distinct geographical affiliation. In North America, we find support for an end-Pleistocene population bottleneck through coalescent simulations under an approximate Bayesian framework in contrast to previous studies that suggested an extinction-replacement event. However, we find no support for a similar bottleneck in Eurasia. Overall, this global analysis helps to clarify our understanding of the complex history for wolves in Eurasia and North America.
Introduction
The gray wolf (Canis lupus) exhibits a tremendous ecological flexibility with respect to different environments, ranging from the Arctic tundra to the deserts and dry shrub lands of the Middle East. This iconic canid was one of the most widely distributed large terrestrial mammals in the Late Quaternary with a historical range that covered most of the northern hemisphere (Nowak, 2003), and its range has expanded even further alongside humans as the domestic dog (C. lupus familiaris). In addition to domestication, humans have impacted the wolf considerably by restricting its habitat through active persecution, which has resulted in a dramatic decrease in population sizes, especially over the last two centuries (Boitani, 2003; Leonard et al., 2005). Shrinking habitats overlapping with closely related dogs and coyotes (Canis latrans) have also led to numerous occurrences of hybridization (Andersone et al., 2002; Lucchini et al., 2004; Fain et al., 2010; vonHoldt et al., 2011, 2013). Along with recent turnovers, these characteristics and events make the phylogeographic history of the wolf in many ways difficult to disentangle.
The divergence between wolves and coyotes most likely took place in America at some point between 4.5 and 1.8 million years ago (Mya; Nowak, 2003). However, the more recent time point seems more plausible, considering that a sudden expansion of the genus Canis, sometimes referred to as the “wolf event,” took place at the beginning of the Pleistocene (~2.5–1.8 Mya; Azzaroli, 1983; Wang et al., 2010). This expansion was most likely facilitated by intense continental glaciations, which created open landscapes including the “mammoth steppe biome” (Azzaroli, 1983; Azzaroli et al., 1988).
According to the fossil record, the wolf C. lupus ssp. appeared in Europe around 800 thousand years ago (kya) during the Middle Pleistocene and in the mid-latitudes of North America around 100 kya, where its ancestor previously had gone extinct (Wang et al., 2010). The wolf appears to have been well-established in Europe from around 400 kya and onwards (Meloro et al., 2007). Older records are known only from Siberia and Alaska (Beringia), leading to the assumption that wolves originated somewhere in this region, whence they spread all across the Holarctic (Wang et al., 2010).
Despite the wide geographical distribution and complex evolutionary history of the gray wolf, most genetic analyses to date have concentrated on specific geographical regions (Randi et al., 2000; Aggarwal et al., 2007; Pilot et al., 2010; Weckworth et al., 2010) and/or short fragments of the mitochondrial DNA (mtDNA; Vila et al., 1999; Valiere et al., 2003; Sharma et al., 2004; Jedrzejewksi et al., 2005). Many of these studies have thus suffered from limited geographical coverage and/or insufficient resolution at the genetic level. Further, large areas have remained scarcely sampled, such as Russia/Siberia, China, and the Middle East; regions holding large interconnected populations that potentially contain high genetic diversity. There are also remote regions where wolves have not yet been studied in terms of genetic differentiation and diversity. Among these are the extreme north—home to phenotypically distinct arctic subspecies, which occur in the Canadian arctic (Canis l. arctos) and on Greenland (Canis l. orion; Pocock, 1935; Wozencraft, 2005).
By applying ancient DNA techniques to subfossil remains, many studies have sought to reconstruct the wolf's phylogeographic history, often specifically in relation to its domestication by humans (Verginelli et al., 2005; Germonpré et al., 2009; Skoglund et al., 2011; Thalmann et al., 2013). Like numerous studies on modern wolves, these have generally lacked coverage in terms of geography and/or genetic material, often to a great extent. Samples of ancient wolf remains have mainly been collected from sites in Europe and Alaska, leaving out much of the historical distribution (Stiller et al., 2006; Leonard et al., 2007; Germonpré et al., 2009), and a majority of these samples have exclusively been targeted for a short but variable fragment of the mitochondrial control region (CR; Verginelli et al., 2005; Stiller et al., 2006).
Despite these limitations, ancient DNA studies have provided important insights into the population dynamics in certain regions. In a study on Late Pleistocene wolves from eastern Beringia (Alaska) a population turnover was detected at the Pleistocene-Holocene transition, where a diverse group of haplotypes (haplogroup 2) seemed to have been replaced by a more distinct and monophyletic group (haplogroup 1), which also represents the modern wolves in North America (Leonard et al., 2007). Interestingly, the former group was morphologically described as a robust ecomorph, possibly adapted to large megafaunal prey. The division into these two genetic groups can also be applied to the larger Eurasian population, where both groups are still present in contemporary wolves. However, following the pattern in North America, all Late Pleistocene European specimens have been shown to fall within haplogroup 2 (Pilot et al., 2010; Thalmann et al., 2013), which might suggest that a population turnover occurred in Eurasia as well. Although only present at low frequency today, haplogroup 2 exhibits a great deal of diversity, especially when ancient specimens are included (Pilot et al., 2010). This is also mirrored by indications of past morphological diversity observed both in North America and Europe, which suggests that the wolf has suffered a general decrease, not only in genetic but also morphological diversity across its range (Leonard et al., 2007; Germonpré et al., 2009).
In addition to the issue of the wolf's relationship to the modern dog, there have been several debated uncertainties concerning specific wolf populations and their status in terms of subspecies, hybrids, or even distinct species. It has for instance recently been suggested that wolves from the Great Lakes region of Canada and the United States (Canis l. lycaon) constitutes a hybrid between gray wolves and coyotes or even a species on its own (Fain et al., 2010; vonHoldt et al., 2011, 2013, 2016).
To provide a more comprehensive phylogeograpy of the gray wolf, we have used CR-sequences to study the mtDNA diversity of wolves on a worldwide scale. We specifically aimed to include more modern samples from previously less studied areas, as well as to collate and analyze ancient sequences globally in order to assess the relationships between wolf populations and how they have changed over time. We also sampled wolves from the Great Lakes states Michigan and Minnesota in order to evaluate if this population shows more sharing of haplotypes with wolf or coyote. Finally, to formally test the proposed population turnover in North America, as well as the possibility of a similar turnover in Eurasia, we analyzed the data using serial coalescent simulations.
Materials and Methods
Samples
Tissue samples (n = 128) from wolves were collected from across the range including Scandinavia, Russia/Siberia, Iran, China, North America, and Greenland (Table S1). With the exception of two captive Mexican wolves, all samples were derived from wild animals. Of the six samples from Greenland, two were dated to the fifth and the early eighteenth century, respectively (Table S1). Two Late Pleistocene wolf remains were also analyzed, both taken from permafrost sites on the Taimyr Peninsula in Siberia. After positive DNA screening, these samples were radiocarbon dated at the Oxford Radiocarbon Accelerator Unit (ORAU), yielding approximate ages of 35 and 42 thousand calibrated radiocarbon years before present (kyr BP). For the sake of consistency, all subsequent radiocarbon dates are presented in this (calibrated) form (Tables S1, S3). In summary, a total of 130 novel samples were analyzed (Table S1). For more information regarding samples included in the current study, see Tables S1, S2.
Laboratory Methods
For the modern samples, DNA was extracted from blood and tissue using Proteinase K and organic solvents (Sambrook et al., 1989), or from hairs (Hopgood et al., 1992). PCR amplification of a 582 bp long fragment of the hypervariable domain (HVR1) of the mitochondrial control region was performed with a set of primers used in a previous study (Savolainen et al., 2002). This specific region has commonly been applied in several previous phylogenetic studies on the wolf and was thus targeted to facilitate inclusive comparisons. PCR products were sequenced using the primers above with BigDye Terminator chemistry on ABI 377 and 3700 instruments (Applied Biosystems Inc.).
Both extraction and pre-PCR preparation of the historical and ancient samples was performed at a specifically designated laboratory with high standards of sterility at the Swedish Museum of Natural History in Stockholm, and the Centre for Geogenetics at the Natural History Museum of Denmark. DNA was extracted using a silica-based method, where ca. 50 mg bone powder from each sample was incubated overnight under motion at 55°C in 715 μl extraction buffer (0.45 M EDTA, 0.1 M UREA, 150 μg proteinase K). The samples were then centrifuged at 2300 rpm for 5 min and the supernatants were collected and concentrated using 30K MWCO Vivaspin filters (Sartorius). Purification and elution was performed following Brace et al. (2012).
Taking the fragmented state of ancient DNA into account, six partially overlapping sequences of the mitochondrial control region were amplified to cover a total of 686 bp (including primers), matching the fragment targeted for the modern samples; MS_wolf1- dogdl5 (148 bp; Leonard et al., 2005), MS_wolf2 (205 bp), MS_wolf3 (211 bp), MS_wolf4 (175 bp), MS_wolf5 (149 bp), and MS_wolf6 (132 bp). Primer sequences are listed in Table S4.
Polymerase chain reactions (PCRs) were set up using; 1 mM MgCl2 (Qiagen), 0.2 mM dNTPs, 0.2 μM of each primer, 1X PCR-buffer (Qiagen), 0.1 mg/ml BSA, 2 units of Hotstar Taq (Qiagen) and 2 μl of DNA extract, making a total volume of 25 μl/reaction. Amplifications started with a 10 min denaturation step at 95°C, followed by 55 cycles of denaturation at 95°C for 30 s, annealing for 30 s at 58–62°C, followed by extension at 72°C for 30 s. A final extension step at 72°C for 7 min was included at the end of the procedure. Confirming successful amplifications was done using gel electrophoresis, by applying 5 μl PCR product on a 1.5% agarose gel prepared with fluorescent GelRed (Biotium Inc.). The gel was run in 0.5X TBE buffer (50 ml) and inspected with UV-light. The PCR-products (20 ul) were further purified with EXO-SAP enzymes (5 ul; Thermo Fisher Scientific). Sequencing reactions were then performed using the BigDye Terminator kit ver.1.1 (Applied Biosystems Inc.), and the products were purified with the DyeEx 96 Kit (Qiagen). An ABI 3130xl Genetic analyzer (Applied Biosystems Inc.) was used for the final sequencing analysis.
To easily detect contamination and minimize the risk of cross-contamination, all extractions were made in small series with regular inclusions of blank samples. The same procedure was applied for the PCR preparation, and a minimum of two PCR products were sequenced for every sample to confirm its authenticity and detect possible discrepancies caused by post-mortem DNA damage (Hofreiter et al., 2001).
Alignment and Data Set Establishment
All sequences obtained from the extracted samples were aligned and edited using the software BioEdit 7.2.3 (Hall, 1999; http://www.mbio.ncsu.edu/BioEdit/bioedit.html) and Geneious 7.1.3 (Kearse et al., 2012). In addition to the samples extracted and sequenced by the authors, currently available sequences from GenBank were downloaded for wolves (C. lupus; Table S2). However, since available and published CR-sequences varied to a great extent both in overlap and length, two alignments were initially constructed; one covering the complete stretch of 582 bp (alignment A, n = 314) and a second where all sequences <347 bp were excluded (alignment B, n = 335). The delimitation of the second alignment was made in order to allow for comparison with as many ancient wolf sequences as possible, without cutting the alignment too short and thereby losing information. A third alignment was finally made where all ancient sequences available were included, but which was restricted to a mere 57 bp (alignment C, n = 366). The number of sequences from ancient samples within each alignment was A, n = 10; B, n = 31; and C, n = 62. Published sequences from ancient samples younger than 30 kyr BP that were labeled as uncertain regarding affinity to wolf or dog or as “doglike” were not included (Verginelli et al., 2005; Thalmann et al., 2013). Sequences containing uncertainties at polymorphic sites, and thereby making assignments to known haplotypes or identification of new ones impossible, were also excluded. Uncertainties at non-polymorphic sites, only occurring in a few sequences were accepted. This standard was set since the latter neither provided information for haplotype identification nor were phylogenetically informative.
The targeted sequence in the first two alignments included several insertion-deletions (indels), which were taken into account for the haplotype assignment, but excluded in the following phylogenetic analyses, rendering alignment A and B to cover 558 and 317 bp, respectively. Within the alignments the geographical origin of the samples were indicated at least to the level of continent or region: North America, Europe, south-west Asia, northern plus eastern Asia, India, Japan (Hokkaido), and the Himalayas (including Tibet).
Haplotype Assignment for Alignment A
During analysis, considerable confusion on haplotype designation was revealed. This was due to the varying sequence length used in different studies and lack of unitary designations for new and already known haplotypes. Here, the sequence from Björnerfeldt et al. (2006) was chosen as a reference, because it represents one of the longest mitochondrial sequences (16,730 bp) in this data set (Björnerfeldt et al., 2006). Consequently, this sequence was trimmed to match our longest alignment (A) and named Clu1 in order to establish a new nomenclature for the gathered wolf sequences; new haplotypes were named accordingly and published haplotypes were renamed (Table S2). Additionally, the novel haplotypes in the wolves were compared to all available dog and coyote sequences in order to identify haplotypes shared among (sub) species (Pang et al., 2009). The program DnaSP 5.10 (Librado and Rozas, 2009) was used to identify identical haplotypes in alignment A.
Phylogenetic Analysis
A Bayesian phylogenetic analysis was also performed on alignment A using BEAST 1.8.0 (Drummond and Rambaut, 2007) and applying the HKY + Γ model of nucleotide substitution without partitioning the alignment, which was selected under the AIC criterion in jModelTest (Posada, 2008). Further, we assumed the constant population size setting as a coalescent tree prior, which is suitable for trees describing the relationships between individuals within the same population/species (Drummond and Rambaut, 2007). The posterior distribution of nodes, divergence times, and substitution rates were estimated by Markov chain Monte Carlo (MCMC), where samples were drawn every 1000 MCMC steps from a total of 30 million steps, following a discarded burn-in of 3 million steps. Convergence to the stationary distribution and sufficient sampling were checked by inspection of posterior samples and ESS values. Additionally, analyses were run twice to test for stability and convergence of MCMC chains in plots of posterior log likelihoods in Tracer v1.5.2 (Drummond and Rambaut, 2007). Since radiocarbon dates were available as internal calibration points the “Estimate” option was used with no prior on the substitution rate.
Network Analyses
Since bifurcated phylogenetic trees may not accurately mirror the multi-furcated, reticulated relationships among intraspecific haplotypes (Posada and Crandall, 2001), network analyses were performed: Median-joining networks were constructed using the software PopART 1.0 (Leigh et al., 2012) for alignment A, B, and C excluding indels, and temporal comparisons were made between the Late Pleistocene and modern samples in the former, as well as using five time layers within the set of ancient samples (alignment C). Since alignment A had a limited representation of sequences from ancient samples (n = 10) and analyses were subsequently focused on the shorter alignments.
Coalescent Simulations
To test the population turnover hypothesized in previous studies (Leonard et al., 2007; Pilot et al., 2010), coalescent simulations were carried out on two geographically delimited datasets representing samples from Eurasia and North America. This division was based on the separation of the two landmasses by the Bering Strait in the early Holocene (Hu et al., 2010). Alignment B was chosen for testing the simulations, since the other two alignments were either lacking enough ancient samples (alignment A) or were too restricted in length (alignment C).
Three alternative demographic histories were compared; a constant population size through time, a population bottleneck, and a split model designed to test the possibility of an extinction and replacement of divergent ecotypes (i.e., haplogroups 2 and 1; Leonard et al., 2007) or whether the haplotypes could have originated in situ (Leonard et al., 2007). Priors for the models were as follows: (1) constant model with a constant population size (Nemod) drawn from a uniform prior U (50,000–1,000,000) through time, (2) bottleneck model where a population (Neanc) drawn from a uniform prior U (50,000–1,000,000) decreased instantaneously to a smaller size (Nebot) drawn from a uniform prior U (1000–50,000) at a time (tbot) drawn from a uniform prior U (4333–4000) generations ago and then expanded exponentially to a population size (Nemod) drawn from a uniform prior U (50,000–1,000,000), and finally (3) split model in which a single population (Neanc = (Ne1_mod + Ne2_mod)) was split at a time (tsplit) U (13,333–516,666) generations ago into two populations; the first (Ne1_mod) sampled independently from a uniform prior of U (25,000–500,000) and the second (Ne2_mod) sampled independently from a uniform prior of U (25,000–500,000) with a migration rate (m1_2) U (0.001–0.1) between the two populations.
The prior for the mutation rate of U [1.04577 × 10−5–4.0855 × 10−5 (mutation rate for the analyzed sequence per generation)] was based on a rate of 1.1–4.3%/million years (Pang et al., 2009) and the K2P (Kimura two-parameter; Kimura, 1980) mutation model was used with 0.957% transitions and a gamma shape parameter of 0.0860 with 6 rate classes estimated using ModelTest 3.7 (Posada and Crandall, 1998). The timing of the bottleneck as well as the split time was inferred from the turnover, mutation rate and divergence between the two haplogroups reported in Leonard et al. (2007), calculated with a generation time of 3 years (Mech and Seal, 1987). Coalescent simulations (1 million iterations of each model) were performed using BayeSSC (Anderson et al., 2005) and segregating sites, nucleotide diversity, Tajima's D, and pairwise Fst were calculated for each simulation and used for model choice, cross-validation, Bayes factors, rejection, and local linear regression adjustment, which were carried out in R using the R package abc.R (Csillery et al., 2012) based on the 1000 closest Euclidean distances between simulated and observed summary statistics. A PCA was performed on simulations for each model prior to analysis to check the appropriateness of the simulations and models for producing summary statistics close to the observed and pseudo-observed datasets (PODs) confirmed that the constant, bottleneck, and split model could be differentiated given our sampling and dataset.
Results
Control Region Diversity
In total, we found 114 different wolf haplotypes among 314 sequences in alignment A. Thirteen of these were reported the first time; four from North America, five from Siberia, one from Europe (Russia), one from Iran, and two from China (Table 1 and Table S1; GenBank accession numbers: KX898307-KY124130). The two ancient wolf specimens from Siberia both represented new unique haplotypes, whereas the six samples from Greenland all belonged to a single haplotype (Clu53) previously found among North American wolves. Three haplotypes were identified in wolves sampled from the Great Lakes region (Canis l. lycaon), one of which was shared with coyotes (Table S1). After initial trials of haplotype clustering, these wolves were repeatedly placed in the most basal clade and showed a kinship closer to the coyote, which supports its proclaimed status as a hybrid or a separate species (Fain et al., 2010; vonHoldt et al., 2011). Consistent with a hybrid origin, a recent whole-genome sequence study demonstrated extensive gray wolf and coyote introgression in wolves from the Great lakes region (vonHoldt et al., 2016). After this apparent affiliation was revealed, the samples of eastern Canadian wolves were left out from the subsequent analyses.

Table 1. Genetic diversity according to geographical region based on alignment A excluding indels (558 bp); Hd = haplotype diversity, π = nucleotide diversity.
In alignment B, 90 haplotypes were found among 335 sequences and were assigned with haplotype numbers. As a result of the shorter sequence length several haplotypes that were distinct in alignment A collapsed into larger ones in this shorter alignment. One was even shared by both ancient and modern samples when indels were removed (haplotype nr. 8 and 9; Clu8, Clu9, Clu10, Clu22, Clu108, and Clu109). Alignment C contained 71 haplotypes among 366 sequences.
Bayesian Analysis (BEAST)
The Bayesian analysis in BEAST showed convergence of posterior likelihoods between runs. For all parameters of interest, the effective sample sizes (ESS) were higher than 200 (as recommended in the BEAST manual), suggesting stabilization and good mixing of the MCMC chains.
The phylogeny revealed an overall pattern where clades represented samples from multiple locations, even from geographical regions distant to one another (Figure 1: I–XIX). There were also several recurrently supported clades, which showed considerable geographical unity (III, VII, XIII, XVI, and XVIII). The most basal clades were represented by two lineages of Himalayan and Indian wolves, known for their early divergence among wolf lineages as has previously been shown (Sharma et al., 2004; Aggarwal et al., 2007; Meng et al., 2009). Within the clade including the Himalayan wolves was also a distinct subclade (Clu76) with nine wolves from China and Mongolia (Figure 1). In the remaining phylogeny, all ancient wolves were confined among the basal clades and the two new samples from Siberia aligned next to two of these groups (XVIII and XV).
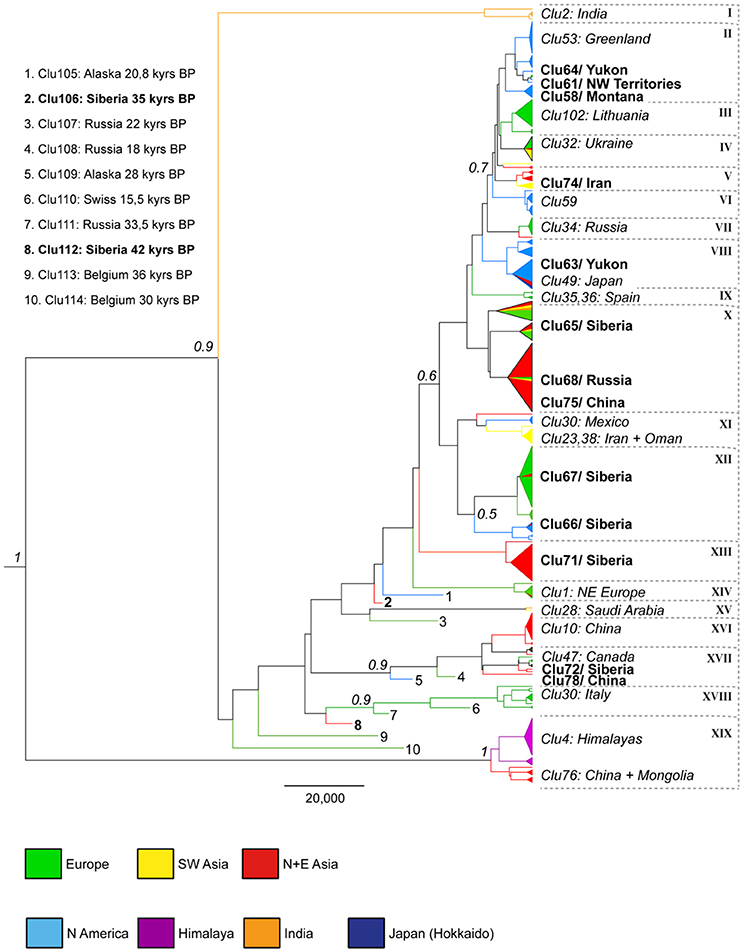
Figure 1. Maximum clade credibility tree based on alignment A from the Bayesian analysis (BEAST) using the posterior distribution from 30 million sampled trees. Nodes with posteriors above 50% are indicated. Key regions/haplotypes are indicated and Late Pleistocene samples are represented with numbers 1–10 and listed in the figure. Novel haplotypes are displayed in bold and clades are denoted (I–XIX).
Among North American wolves, three groupings (II, VI, VIII) were discernable clearly at the upper half of the tree, largely matching the recent findings of clusters of wolf haplotypes in Canada and Alaska (Weckworth et al., 2011). The arctic wolves from Greenland were grouped within the largest group representing American wolves at the top of the tree (II). Two American haplotypes stand out by their more basal position in the phylogeny. One represents Mexican wolves (XI/Clu30), which are currently only found in small captive populations in the US, and which previously have been shown to form a genetically distinct group (Vila et al., 1999). The other is that of a wolf from Vancouver Island in south-western Canada. However, this latter haplotype (Clu47) is also shared by dogs, suggesting a recent hybridization, which has also been previously reported from this region (Munoz-Fuentes et al., 2010).
European samples are found throughout the phylogeny, but also form distinct groups. Two notable examples consist of Spanish (IX/Clu35, 36) and Italian wolves (XVIII/Clu30), which are found widely apart in the phylogeny. Furthermore, the Spanish haplotypes (Clu35, 36) from the former group are unique to the Iberian Peninsula.
The Asian samples are widely spread in the phylogeny as well, especially the ones representing northern and eastern Asia, which are found all across both trees and networks. Three of the new haplotypes reported from this area originated from the easternmost regions; Clu66 from the Chukotka Peninsula, Clu67 from Khakassia and the Amur region and Clu72 from the North Korean border, and they all show a great diversity with close affiliation to wolves on all three continents. The novel haplotype Clu72 also included a distinct 11 bp insertion, not previously reported.
Network Analyses
The networks in many ways mirrored the phylogeny with haplotypes containing wolves from several regions and some clusters showing a geographical affiliation. The clearest delimitation is visible in the network based on alignment A (Figure S1). Much less resolution was displayed in the other networks, since these were based on shorter alignments (Figures 2, 3). Even though the two haplogroups identified by Leonard et al. (2007) could not be clearly distinguished, the temporal comparison of networks based on alignments A and B revealed a pattern where all ancient wolves were clustered in one part of the network (Figure 2 and Figure S1). This supports the indications from previous studies of a past extended diversity that was lost, at least in Europe, Siberia and Alaska, and which overlapped only to a limited extent with that seen in modern wolves. Temporal changes within the ancient samples were also apparent in the network of alignment C, where the constellations of ancient haplotypes are fairly delimited and haplotype continuity is decreasing through time (Figure 3). Among the ancient samples, a decline in haplotype numbers is observed at the beginning of the Holocene. This could reflect a bottleneck event or the lack of available samples from North American wolves in the Holocene (Figure 3; 10–1 kyrs BP).
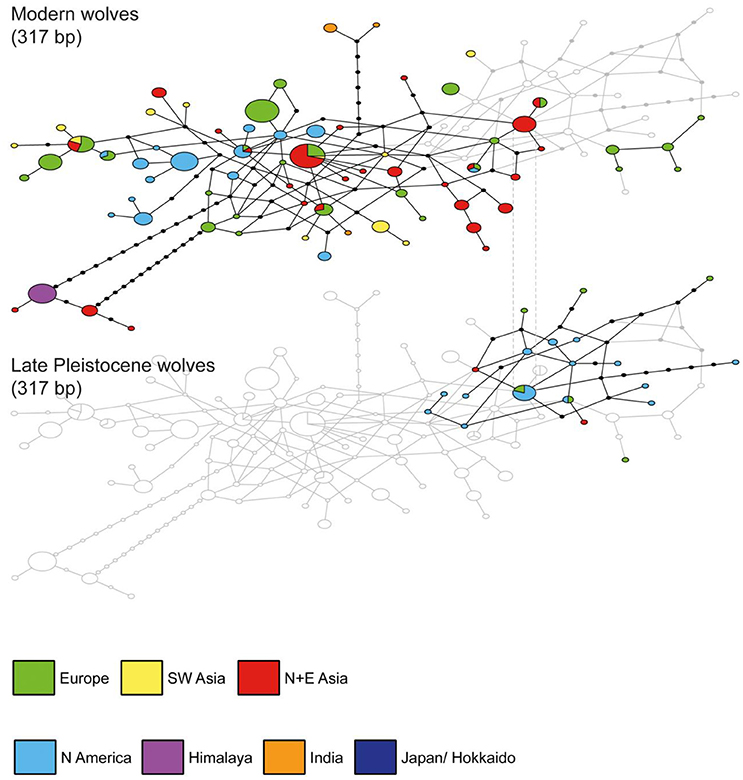
Figure 2. Median joining network analysis for wolf haplotypes based on alignment B excluding indels (317 bp) and including a temporal comparison (Late Pleistocene vs. modern). Haplotypes (ellipses) are separated by the number of mutational steps indicated by continuous lines and black ellipses represent missing haplotypes.
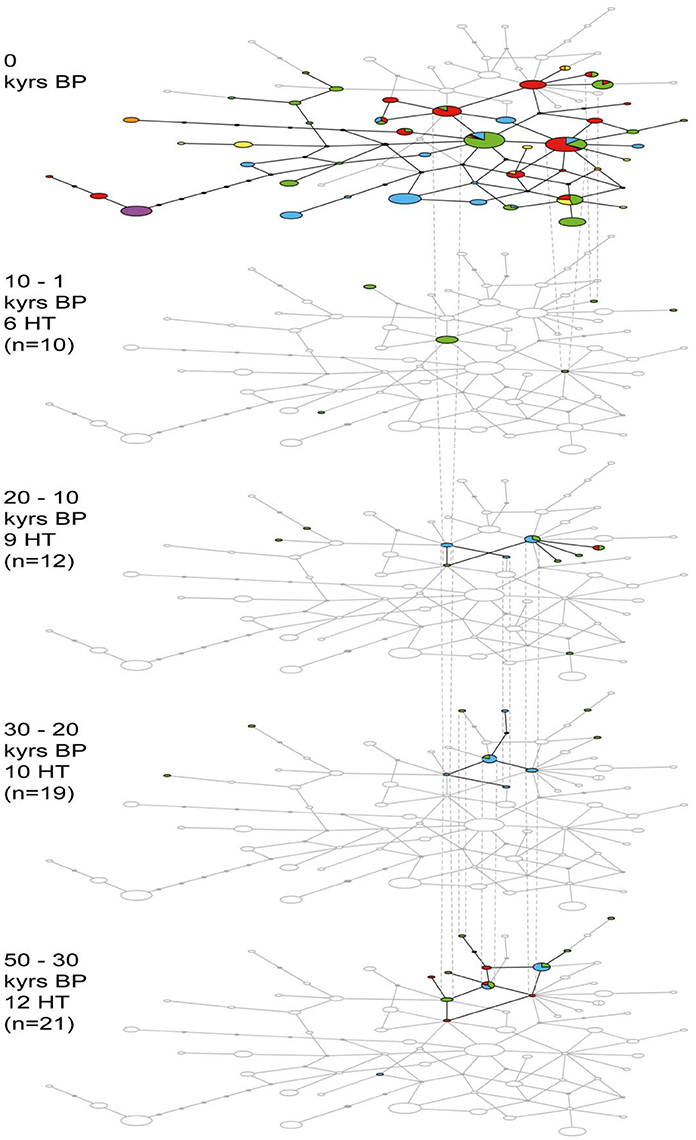
Figure 3. Median joining network analysis for ancient wolf haplotypes (HT) identified for alignment C as well as all modern samples trimmed to match this alignment (excluding indels; 57 bp). Temporal comparisons of five consecutive time periods are presented covering the Late Pleistocene and Holocene. Number of mutational steps is indicated by continuous lines, black ellipses represent missing haplotypes. Colors correspond to geographical region (see Figures 1, 2).
Coalescent Simulations
We found substantial evidence for a population bottleneck taking place in North America. By evaluating the results with the “neural net” method (Csillery et al., 2012), applying a tolerance of 0.001 and 3000 posterior samples, Bayes Factors for: bottleneck vs. constant = 7.92, bottleneck vs. split = 14.99 and posterior model probability for bottleneck = 0.8383. Estimates of the size of the bottleneck were ~4% of the ancient Ne. (Bottleneck size Ne mode = 4027, 95% Confidence Interval 1673–51,171, declining from an ancient Ne = 100,000, 95% Confidence Interval 66,492–242,525, and re-expanding to a modern estimate of 364,000, 95% Confidence Interval 154,645–818,409). For Eurasia, none of the three models (constant, bottleneck, or split) gained substantial support by model choice over the other.
Discussion
By analyzing an expanded global dataset of wolves for the mtDNA control region, we increased the total number of identified haplotypes and also identified specific clades/clusters, which were better characterized by this addition. This emphasizes the importance of dense sampling of wide-spread species in order to cover as much of the genetic variation as possible. Most new haplotypes were discovered in Siberia and China, which together with Europe contained the highest degree of diversity (Table 1). Samples from these regions were also widely distributed in the networks and the phylogeny (Figures 1–3).
Despite their remote location, the arctic wolves from Greenland did not represent a new or unique haplotype. Instead they all belonged to a haplotype (Clu53) shared with other North American wolves and placed within the largest of these clades (II) suggesting that the maternal lineage of contemporary Greenland wolves has its origin in North America. This is well in agreement with a proposed pattern of recurring colonization of Greenland by wolves from the Canadian arctic (i.e., Ellesmere Island; Dawes et al., 1986). Much like the polar bear, the evolutionary history, and possible genetic differentiation of this subspecies could better be explored with genomic markers (Hailer et al., 2012; Cahill et al., 2013).
The network analyses demonstrated that the ancient wolf samples constitute a significant proportion of the global diversity, which was almost entirely lost in North America, and severely diminished in Europe. It is also apparent that all ancient samples fall within one half of the phylogeny and the network based on alignments A and B, thereby constituting lineages different from most of the contemporary wolves. Accordingly, all ancient haplotypes defined in alignment A were unique and not found within the modern dataset (Figure S1). Both of the ancient samples from Siberia analyzed here (Clu106 and Clu112) grouped among the other ancient wolves, confirming this pattern.
A division of both contemporary and ancient wolves into two distinct haplogroups, as described in previous studies (Leonard et al., 2007; Pilot et al., 2010), is not well supported or clearly delimited in our results. However, the pattern of ancient haplotypes disappearing in North America supports a turnover taking place there, which is in line with Leonard et al. (2007), where a demise of the haplogroup containing all Late Pleistocene wolves was suggested. Instead of detecting an extinction-replacement, the coalescent simulations provided a significant support for a bottleneck scenario taking place around the Pleistocene-Holocene boundary around 12,000 years ago in North America. In contrast, the lack of support for any of the simplified population histories for Eurasia may suggest a more diverse demographic history across this landmass, where the process leading to the decline of many of the ancient lineages might have been slower and more ambiguous. In Europe, several recent studies have indeed found discrepant results for the population dynamics in wolves, where mitochondrial data indicated a recent increase in population size, whereas data based on nuclear SNPs instead showed a continuous decline, starting already in the Late Pleistocene (Pilot et al., 2010, 2014). The latter dataset also suggested a similar pattern of decrease in North America (Pilot et al., 2014) which is in line with the current study.
One aspect to keep in mind in relation to the proposed demographic changes is the uneven temporal and geographical distribution of ancient wolf samples, on which this and other studies are based. The Late Pleistocene samples have so far been restricted to Europe, Alaska, and Siberia, leaving out most of Asia. We are thus lacking data on the ancient diversity of Asian wolf populations, which precludes reliable assessments of demographic events and phylogeography across much of this continent. Our coalescent simulations are also inherently limited by the number of sequences as well as variation within and between models that are tested, and can only be evaluated in light of these specific factors.
The paleontological record implies a continuous presence of wolves across the northern hemisphere stretching through the last glacial maximum (LGM), and shows little indication of the wolf's range being significantly restricted during glaciations (Sommer and Benecke, 2005; Leonard et al., 2007). In North America, the continental ice sheets formed an effective barrier during long time periods, which most likely isolated southern wolf populations from those in Beringia (Alaska; Leonard et al., 2007; Weckworth et al., 2010). This is also supported by clear morphological differences observed between Beringian wolves and wolves south of the ice sheets during the Late Pleistocene (Leonard et al., 2007), and the genetic distinction of the Mexican wolves seen today might indeed derive from a past isolation in a southern refugium during the LGM (Leonard et al., 2005; Shafer et al., 2010; Weckworth et al., 2011).
In Eurasian wolves, there are few decisive signs of population structure shaped by glacial refugia during the LGM, something which has been suggested for other mammals (Taberlet and Bouvet, 1994; Stewart et al., 2010). One example is the Italian wolf population, which is clearly distinct, and positioned close to the ancient wolves in the phylogeny. Other studies have estimated that this population was in fact isolated for thousands of generations in the Italian Peninsula (Lucchini et al., 2004). However, more recent factors like hunting and restrictions to gene flow might also have severely decreased genetic diversity and caused genetic drift in this population (Valdiosera et al., 2007). Interestingly, genomic data from a recent study revealed genetic distinctiveness in wolves from both from the Italian and the Iberian peninsulas, explaining it as a result of isolation during the LGM (vonHoldt et al., 2011). In order to shed more light on the genetic singularity of these populations, additional analyses of ancient samples would be needed.
One behavioral feature of the wolf which can potentially counter the establishment of long term phylogeographic structures is its dispersal capabilities. Wolves are extremely mobile and migration of several 1000 kms is common for both sexes (Fritts, 1983; Mech et al., 1995; Ciucci et al., 2009). This is reflected by clades that comprise wolves from two or three continents together (IV, VIII, X, XI). Another aspect of detecting population structures from genetic diversity is the type of genetic material studied, for instance autosomal vs. mitochondrial markers, as well as restricted mitochondrial sequences vs. complete mitochondrial genomes. A previous study on modern wolves based on a short (230 bp) CR sequence failed to detect clear population structures globally (Vila et al., 1999). A number of recent studies analyzing mitochondrial genomes and autosomal SNPs of wolves worldwide have been more successful in this respect, finding significant differentiation between wolf populations of Europe, Asia, North America, and even within the continents (vonHoldt et al., 2011; Thalmann et al., 2013; Fan et al., 2016; Koblmüller et al., 2016). Although restricted to the mitochondrial control region, our analyses did recover most of the same clusters and clades that were revealed from datasets of almost complete mitochondrial genomes (Thalmann et al., 2013; Koblmüller et al., 2016). However, despite the larger sample set, the comparably shorter sequence length used precluded confirmation of the same topology with equally substantial support. The clades within the phylogeny should thus be interpreted with some caution, given their generally low support values.
In contrast to phylogenies based on mitochondrial markers, a phylogeny based on autosomal SNPs differed in a significant way, as it displayed distinct geographical patterns, where the most basal split separated wolves in North America from those in Eurasia (Fan et al., 2016). The discrepancy of this result compared to ours and to the phylogenies based on mitochondrial markers in general (Thalmann et al., 2013; Koblmüller et al., 2016) is of interest. A likely explanation is that mitochondrial lineages in wolves can be highly affected by the animal's wide dispersal as well as incomplete lineage sorting, which has to be considered when results from these different methods are compared (Funk and Omland, 2003; Toews and Brelsford, 2012). Discordance is in fact a general problem when comparing genetic studies on wolves, even when similar markers are used. We specifically encountered this dilemma when searching for published data that often did not overlap with our sequences. In order to facilitate comparisons of data and avoid the confusion of novel haplotype names, a harmonization of methods, and markers is necessary (de Groot et al., 2016).
With the use of autosomal markers, some studies have also managed to discern patterns on more restricted geographical scales (Weckworth et al., 2011; Pilot et al., 2014). In many instances these population patterns, which have shown little correlation with geographical barriers, have instead been explained by ecological specializations in different populations (Carmichael et al., 2001; Geffen et al., 2004; Stronen et al., 2012). A pattern of differentiation along a North-South axis has for instance been observed in European wolves (Stronen et al., 2013), and recently, signs of diversifying selection in terms of body size has been discovered in the same population (Pilot et al., 2014). These findings are much in concordance with the proposed causes to the turnover seen in North America, explained in terms of a population of larger and more specialized wolves going extinct along with other megafauna (Leonard et al., 2007).
Sampling of modern wolves has several limitations, mainly due to practical difficulties, decreasing numbers, and local wolf populations going extinct. Recent bottlenecks have also hampered the ability to achieve an overview of the phylogeography of the past. However, remains of wolves are common both in paleontological and historical collections, and to improve our understanding of the population dynamics of the gray wolf, analyses of more samples are needed to cover both the distant as well as the more recent past. One question of specific interest concerns the origin of the direct ancestors of the lineages that dominate contemporary populations, a question which might be solved by analyzing additional ancient wolf remains from areas south of the Late Pleistocene “mammoth steppe” in Eurasia and North America.
Author Contributions
PS, LD, CK, MU, and YZ designed the study. CK, NI, MO, EE, and M-HS carried out DNA extractions and sequencing. SF, NI, and M-HS collected samples. CK and EE performed the computational analyses, evaluated the results and drafted the manuscript together with LD and PS. YC performed the Bayesian simulations. All authors contributed to the interpretation of the results and took part in writing the manuscript.
Funding
This work was supported by grants from the Swedish Research Council, the Carl Trygger Foundation, the Swedish Kennel Club, as well as logistic field support from the Swedish Polar Research Secretariat. PS is a Research Fellow of the Royal Swedish Academy of Sciences, supported by a grant from the Knut and Alice Wallenberg Foundation. YC is a holder of a European Commission Marie Curie Fellowship (FP7-PEOPLE-2011-IIF-301572).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank everyone contributing with samples for this study. Arman Ardalan is especially acknowledged for helping with the Iranian samples, as is Sofie Wagenius for help with identifying Late Pleistocene wolf remains, and Kristian Gregersen for providing access to Greenlandic specimens. This work was supported by grants from the Swedish Research Council, the Carl Trygger Foundation, the Swedish Kennel Club, as well as logistic field support from the Swedish Polar Research Secretariat. YC is a holder of a European Commission Marie Curie Fellowship (FP7-PEOPLE-2011-IIF-301572). Lastly, we thank Prof. M. Thomas P. Gilbert for his financial support of M-HS's work. The findings and conclusions in this article are those of the author(s) and do not necessarily represent the views of the U. S. Fish and Wildlife Service.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fevo.2016.00134/full#supplementary-material
References
Aggarwal, R. K., Kivisild, T., Ramadevi, J., and Singh, L. (2007). Mitochondrial DNA coding region sequences support the phylogenetic distinction of two Indian wolf species. J. Zool. Syst. Evol. Res. 45, 163–172. doi: 10.1111/j.1439-0469.2006.00400.x
Anderson, C. N., Ramakrishnan, U., Chan, Y. L., and Hadly, E. A. (2005). Serial SimCoal: a population genetics model for data from multiple populations and points in time. Bioinformatics 21, 1733–1734. doi: 10.1093/bioinformatics/bti154
Andersone, Z., Lucchini, V., Randi, E., and Ozolins, J. (2002). Hybridisation between wolves and dogs in Latvia as documented using mitochondrial and microsatellite DNA markers. Mamm. Biol. 67, 79–90. doi: 10.1078/1616-5047-00012
Azzaroli, A. (1983). Quaternary mammals and the “end-Villafranchian” dispersal event - A turning point in the history of Eurasia. Palaeogeogr. Palaeoclimatol. Palaeoecol. 44, 117–139. doi: 10.1016/0031-0182(83)90008-1
Azzaroli, A., Degiuli, C., Ficcarelli, G., and Torre, D. (1988). Late pliocene to early mid-pleistocene mammals in Eurasia - faunal succession and dispersal events. Palaeogeogr. Palaeoclimatol. Palaeoecol. 66, 77–100. doi: 10.1016/0031-0182(88)90082-X
Björnerfeldt, S., Webster, M. T., and Vilá, C. (2006). Relaxation of selective constraint on dog mitochondrial DNA following domestication. Genome Res. 16, 990–994. doi: 10.1101/gr.5117706
Boitani, L. (2003). “Wolf conservation and recovery,” in Wolves: Behavior Ecology and Conservation, eds L. D. Mech and L. Boitani (Chicago, IL: Chicago University Press), 317–340.
Brace, S., Palkopoulou, E., Dalén, L., Lister, A. M., Miller, R., Otte, M., et al. (2012). Serial population extinctions in a small mammal indicate Late Pleistocene ecosystem instability. Proc. Natl. Acad. Sci. U.S.A. 109, 20532–20536. doi: 10.1073/pnas.1213322109
Cahill, J. A., Green, R. E., Fulton, T. L., Stiller, M., Jay, F., Ovsyanikov, N., et al. (2013). Genomic evidence for island population conversion resolves conflicting theories of polar bear evolution. PLoS Genet. 9:e1003345. doi: 10.1371/journal.pgen.1003345
Carmichael, L. E., Nagy, J. A., Larter, N. C., and Strobeck, C. (2001). Prey specialization may influence patterns of gene flow in wolves of the Canadian Northwest. Mol. Ecol. 10, 2787–2798. doi: 10.1046/j.0962-1083.2001.01408.x
Ciucci, P., Reggioni, W., Maiorano, L., and Boitani, L. (2009). Long-distance dispersal of a rescued wolf from the northern apennines to the western alps. J. Wild. Manage. 73, 1300–1306. doi: 10.2193/2008-510
Csillery, K., Francois, O., and Blum, M. G. B. (2012). abc: an R package for approximate Bayesian computation (ABC). Methods Ecol. Evol. 3, 475–479. doi: 10.1111/j.2041-210X.2011.00179.x
Dawes, P. R., Elander, M., and Ericson, M. (1986). The wolf (Canis lupus) in Greenland: a historical review and present status. Arctic 39, 119–132. doi: 10.14430/arctic2059
de Groot, G. A., Nowak, C., Skrbinšek, T., Andersen, L. W., Aspi, J., Fumagalli, L., et al. (2016). Decades of population genetic research reveal the need for harmonization of molecular markers: the grey wolf C anis lupus as a case study. Mamm. Rev. 46, 44–59. doi: 10.1111/mam.12052
Drummond, A. J., and Rambaut, A. (2007). BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7:214. doi: 10.1186/1471-2148-7-214
Fain, S. R., Straughan, D. J., and Taylor, B. F. (2010). Genetic outcomes of wolf recovery in the western Great Lakes states. Conserv. Genet. 11, 1747–1765. doi: 10.1007/s10592-010-0068-x
Fan, Z., Silva, P., Gronau, I., Wang, S., Armero, A. S., Schweizer, R. M., et al. (2016). Worldwide patterns of genomic variation and admixture in gray wolves. Genome Res. 26, 163–173. doi: 10.1101/gr.197517.115
Fritts, S. H. (1983). Record dispersal by a wolf from Minnesota. J. Mammal. 64, 166–167. doi: 10.2307/1380772
Funk, D. J. Omland, K. E. (2003). Species-level paraphyly and polyphyly: frequency, causes, and consequences, with insights from animal mitochondrial dna insights from animal mitochondrial, DNA. Annu. Rev. Ecol. Evol. Syst. 34, 397–423. doi: 10.1146/annurev.ecolsys.34.011802.132421
Geffen, E., Anderson, M. J., and Wayne, R. K. (2004). Climate and habitat barriers to dispersal in the highly mobile grey wolf. Mol. Ecol. 13, 2481–2490. doi: 10.1111/j.1365-294X.2004.02244.x
Germonpré, M., Sablin, M. V., Stevens, R. E., Hedges, R. E. M., Hofreiter, M., Stiller, M., et al. (2009). Fossil dogs and wolves from Palaeolithic sites in Belgium, the Ukraine and Russia: osteometry, ancient DNA and stable isotopes. J. Archaeol. Sci. 36, 473–490. doi: 10.1016/j.jas.2008.09.033
Hailer, F., Kutschera, V. E., Hallström, B. M., Klassert, D., Fain, S. R., Leonard, J. A., et al. (2012). Nuclear genomic sequences reveal that polar bears are an old and distinct bear lineage. Science 336, 344–347. doi: 10.1126/science.1216424
Hall, T. (1999). Bioedit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41, 95–98.
Hofreiter, M., Jaenicke, V., Serre, D., von Haeseler, A., and Pääbo, S. (2001). DNA sequences from multiple amplifications reveal artifacts induced by cytosine deamination in ancient DNA. Nucleic Acids Res. 29, 4793–4799. doi: 10.1093/nar/29.23.4793
Hopgood, R., Sullivan, K. M., and Gill, P. (1992). Strategies for automated sequencing of human mitochondrial DNA directly from PCR products. Biotechniques 13, 82–92.
Hu, A., Meehl, G. A., Otto-Bliesner, B. L., Waelbroeck, C., Han, W., Loutre, M. F., et al. (2010). Influence of Bering Strait flow and North Atlantic circulation on glacial sea-level changes. Nat. Geosci. 3, 118–121. doi: 10.1038/ngeo729
Jedrzejewksi, W., Branicki, W., Veit, C., Medugorac, I., Pilot, M., Bunevich, A. N., et al. (2005). Genetic diversity and relatedness within packs in an intensely hunted population of wolves Canis lupus. Acta Theriol. 50, 3–22. doi: 10.1007/BF03192614
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kimura, M. (1980). A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide-sequences. J. Mol. Evol. 16, 111–120. doi: 10.1007/BF01731581
Koblmüller, S., Vilà, C., Lorente-Galdos, B., Dabad, M., Ramirez, O., Marques-Bonet, T., et al. (2016). Whole mitochondrial genomes illuminate ancient intercontinental dispersals of grey wolves (Canis lupus). J. Biogeogr. 43, 1728–1738. doi: 10.1111/jbi.12765
Leigh, J., Bryant, D., and Steel, M. (2012). PopArt. Dept. of Mathematics and StatisticsUniversity of Otago, Otago.
Leonard, J. A., Vilà, C., Fox-Dobbs, K., Koch, P. L., Wayne, R. K., and Van Valkenburgh, B. (2007). Megafaunal extinctions and the disappearance of a specialized wolf ecomorph. Curr. Biol. 17, 1146–1150. doi: 10.1016/j.cub.2007.05.072
Leonard, J. A., Vilà, C., and Wayne, R. K. (2005). Legacy lost: genetic variability and population size of extirpated US grey wolves (Canis lupus). Mol. Ecol. 14, 9–17. doi: 10.1111/j.1365-294X.2004.02389.x
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Lucchini, V., Galov, A., and Randi, E. (2004). Evidence of genetic distinction and long-term population decline in wolves (Canis lupus) in the Italian Apennines. Mol. Ecol. 13, 523–536. doi: 10.1046/j.1365-294X.2004.02077.x
Mech, L. D., Fritts, S. H., and Wagner, D. (1995). Minnesota Wolf Dispersal to Wisconsin and Michigan. Am. Midl. Nat. 133, 368–370. doi: 10.2307/2426402
Mech, L. D., and Seal, U. S. (1987). Premature reproductive activity in wild wolves. J. Mammal. 68, 871–873. doi: 10.2307/1381570
Meloro, C., Raia, P., and Barbera, C. (2007). Effect of predation on prey abundance and survival in Plio-Pleistocene mammalian communities. Evol. Ecol. Res. 9, 505–525. doi: 10.1080/19401730902852968
Meng, C., Zhang, H., and Meng, Q. C. (2009). Mitochondrial genome of the Tibetan wolf. Mitochondrial DNA 20, 61–63. doi: 10.1080/19401730902852968
Munoz-Fuentes, V., Darimont, C. T., Paquet, P. C., and Leonard, J. A. (2010). The genetic legacy of extirpation and re-colonization in Vancouver Island wolves. Conserv. Genet. 11, 547–556. doi: 10.1007/s10592-009-9974-1
Nowak, R. M. (2003). Wolf evolution and taxonomy. Wolves Behav. Ecol. Conserv. 239–258. doi: 10.7208/chicago/9780226516981.001.0001
Pang, J. F., Kluetsch, C., Zou, X. J., Zhang, A. B., Luo, L. Y., Angleby, H., et al. (2009). mtDNA data indicate a single origin for dogs South of Yangtze River, less than 16,300 years ago, from numerous wolves. Mol. Biol. Evol. 26, 2849–2864. doi: 10.1093/molbev/msp195
Pilot, M., Greco, C., vonHoldt, B., Jedrzejewska, B., Randi, E., Jedrzejewski, W., et al. (2014). Genome-wide signatures of population bottlenecks and diversifying selection in European wolves. Heredity 112, 428–442. doi: 10.1038/hdy.2013.122
Pilot, M., Branicki, W., Jedrzejewski, W., Goszczyński, J., Jedrzejewska, B. B., Dykyy, I., et al. (2010). Phylogeographic history of grey wolves in Europe. BMC Evol. Biol. 10:104. doi: 10.1186/1471-2148-10-104
Pocock, R. I. (1935). “The races of canis lupus,” in Proceedings of the Zoological Society of London (London), 647–686.
Posada, D. (2008). jModelTest: phylogenetic model averaging. Mol. Biol. Evol. 25, 1253–1256. doi: 10.1093/molbev/msn083
Posada, D., and Crandall, K. A. (1998). MODELTEST: testing the model of DNA substitution. Bioinformatics 14, 817–818. doi: 10.1093/bioinformatics/14.9.817
Posada, D., and Crandall, K. A. (2001). Intraspecific gene genealogies: trees grafting into networks. Trends Ecol. Evol. 16, 37–45. doi: 10.1016/S0169-5347(00)02026-7
Randi, E., Lucchini, V., Christensen, M. F., Mucci, N., Funk, S. M., Dolf, G., et al. (2000). Mitochondrial DNA variability in Italian and East European wolves: detecting the consequences of small population size and hybridization. Conserv. Biol. 14, 464–473. doi: 10.1046/j.1523-1739.2000.98280.x
Sambrook, J., Fritsch, E. F., and Maniatis, T. (eds.). (1989). Molecular Cloning: A Laboratory Manual/J. New York, NY: Cold Spring Harbor Laboratory Press.
Savolainen, P., Zhang, Y. P., Luo, J., Lundeberg, J., and Leitner, T. (2002). Genetic evidence for an East Asian origin of domestic dogs. Science 298, 1610–1613. doi: 10.1126/science.1073906
Shafer, A. B., Cullingham, C. I., Côtè, S. D., and Coltman, D. W. (2010). Of glaciers and refugia: a decade of study sheds new light on the phylogeography of northwestern North America. Mol. Ecol. 19, 4589–4621. doi: 10.1111/j.1365-294X.2010.04828.x
Sharma, D. K., Maldonado, J. E., Jhala, Y. V., and Fleischer, R. C. (2004). Ancient wolf lineages in India. Proc. Biol. Sci. 271(Suppl.), S1–S4. doi: 10.1098/rsbl.2003.0071
Skoglund, P., Götherström, A., and Jakobsson, M. (2011). Estimation of population divergence times from non-overlapping genomic sequences: examples from dogs and wolves. Mol. Biol. Evol. 28, 1505–1517. doi: 10.1093/molbev/msq342
Sommer, R., and Benecke, N. (2005). Late-Pleistocene and early Holocene history of the canid fauna of Europe (Canidae). Mamm. Biol. 70, 227–241. doi: 10.1016/j.mambio.2004.12.001
Stewart, J. R., Lister, A. M., Barnes, I., and Dalén, L. (2010). Refugia revisited: individualistic responses of species in space and time. Proc. R. Soc. B Biol. Sci. 277, 661–671. doi: 10.1098/rspb.2009.1272
Stiller, M., Green, R. E., Ronan, M., Simons, J. F., Du, L., He, W., et al. (2006). Patterns of nucleotide misincorporations during enzymatic amplification and direct large-scale sequencing of ancient DNA. Proc. Natl. Acad. Sci. U.S.A. 103, 14977. doi: 10.1073/pnas.0605327103
Stronen, A. V., Jedrzejewska, B., Pertoldi, C., Demontis, D., Randi, E., Niedzialkowska, M., et al. (2013). North-South differentiation and a region of high diversity in european wolves (Canis lupus). PLoS ONE 8:e76454. doi: 10.1371/journal.pone.0076454
Stronen, A. V., Schumaker, N. H., Forbes, G. J., Paquet, P. C., and Brook, R. K. (2012). Landscape resistance to dispersal: simulating long-term effects of human disturbance on a small and isolated wolf population in southwestern Manitoba, Canada. Environ. Monit. Assess. 184, 6923–6934. doi: 10.1007/s10661-011-2469-9
Taberlet, P., and Bouvet, J. (1994). Mitochondrial DNA polymorphism, phylogeography, and conservation genetics of the brown bear Ursus arctos in Europe. Proc. Biol. Sci. 255, 195–200. doi: 10.1098/rspb.1994.0028
Thalmann, O., Shapiro, B., Cui, P., Schuenemann, V. J., Sawyer, S. K., Greenfield, D. L., et al. (2013). Complete mitochondrial genomes of ancient canids suggest a european origin of domestic dogs. Science 342, 871–874. doi: 10.1126/science.1243650
Toews, D. P. L., and Brelsford, A. (2012). The biogeography of mitochondrial and nuclear discordance in animals. Mol. Ecol. 21, 3907–3930. doi: 10.1111/j.1365-294x.2012.05664.x
Valdiosera, C. E., García, N., Anderung, C., Dalén, L., Crégut-Bonnoure, E., Kahlke, R. D., et al. (2007). Staying out in the cold: glacial refugia and mitochondrial DNA phylogeography in ancient European brown bears. Mol. Ecol. 16, 5140–5148. doi: 10.1111/j.1365-294X.2007.03590.x
Valiere, N., Fumagalli, L., Gielly, L., Miquel, C., Lequette, B., Poulle, M. L., et al. (2003). Long-distance wolf recolonization of France and Switzerland inferred from non-invasive genetic sampling over a period of 10 years. Anim. Conserv. 6, 83–92. doi: 10.1017/S1367943003003111
Verginelli, F., Capelli, C., Coia, V., Musiani, M., Falchetti, M., Ottini, L., et al. (2005). Mitochondrial DNA from prehistoric canids highlights relationships between dogs and South-East European wolves. Mol. Biol. Evol. 22, 2541–2551. doi: 10.1093/molbev/msi248
Vila, C., Amorim, I. R., Leonard, J. A., Posada, D., Castroviejo, J., Petrucci-Fonseca, F., et al. (1999). Mitochondrial DNA phylogeography and population history of the grey wolf canis lupus. Mol. Ecol. 8, 2089–2103. doi: 10.1046/j.1365-294x.1999.00825.x
vonHoldt, B. M., Cahill, J. A., Fan, Z., Gronau, I., Robinson, J., Pollinger, J. P., et al. (2016). Whole-genome sequence analysis shows that two endemic species of North American wolf are admixtures of the coyote and gray wolf. Sci. Adv. 2:e1501714. doi: 10.1126/sciadv.1501714
vonHoldt, B. M., Pollinger, J. P., Earl, D. A., Knowles, J. C., Boyko, A. R., Parker, H., et al. (2011). A genome-wide perspective on the evolutionary history of enigmatic wolf-like canids. Genome Res. 21, 1294–1305. doi: 10.1101/gr.116301.110
vonHoldt, B. M., Pollinger, J. P., Earl, D. A., Parker, H. G., Ostrander, E. A., and Wayne, R. K. (2013). Identification of recent hybridization between gray wolves and domesticated dogs by SNP genotyping. Mamm. Genome 24, 80–88. doi: 10.1007/s00335-012-9432-0
Wang, X., Tedford, R. H., and Antón, M. (2010). Dogs: Their Fossil Relatives And Evolutionary History. New York, NY: Columbia University Press.
Weckworth, B. V., Dawson, N. G., Talbot, S. L., Flamme, M. J., and Cook, J. A. (2011). Going coastal: shared evolutionary history between coastal british columbia and southeast alaska wolves (Canis lupus). PLoS ONE 6:e19582. doi: 10.1371/journal.pone.0019582
Keywords: Canis lupus, phylogeography, mtDNA, turnover, control region
Citation: Ersmark E, Klütsch CFC, Chan YL, Sinding M-HS, Fain SR, Illarionova NA, Oskarsson M, Uhlén M, Zhang Y-P, Dalén L and Savolainen P (2016) From the Past to the Present: Wolf Phylogeography and Demographic History Based on the Mitochondrial Control Region. Front. Ecol. Evol. 4:134. doi: 10.3389/fevo.2016.00134
Received: 22 July 2016; Accepted: 09 November 2016;
Published: 02 December 2016.
Edited by:
Badri Padhukasahasram, Illumina, USAReviewed by:
Klaus-Peter Koepfli, Smithsonian Conservation Biology Institute, USAJouni Aspi, University of Oulu, Finland
Copyright © 2016 Ersmark, Klütsch, Chan, Sinding, Fain, Illarionova, Oskarsson, Uhlén, Zhang, Dalén and Savolainen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Erik Ersmark, RXJpay5lcnNtYXJrQG5ybS5zZQ==
†These authors have contributed equally to this work.