
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Ecol. Evol., 27 July 2016
Sec. Phylogenetics, Phylogenomics, and Systematics
Volume 4 - 2016 | https://doi.org/10.3389/fevo.2016.00089
 Yajuan Cheng1
Yajuan Cheng1 Kai Zhou1
Kai Zhou1 Mike W. Humphreys2
Mike W. Humphreys2 John A. Harper2
John A. Harper2 Xiao Ma1
Xiao Ma1 Xinquan Zhang1*
Xinquan Zhang1* Haidong Yan1
Haidong Yan1 Linkai Huang1
Linkai Huang1The species within the Lolium/Festuca grass complex have dispersed and colonized large areas of temperate global grasslands both naturally and by human intervention. The species within this grass complex represent some of the most important grass species both for amenity and agricultural use worldwide. There has been renewed interest by grass breeders in producing hybrid combinations between these species and several countries now market Festulolium varieties as a combination of genes from both genera. The two genera have been differentiated by their inflorescence structure, but controversy has surrounded the taxonomic classification of the Lolium-Festuca complex species for several decades. In order to better understand the complexities within the Lolium/Festuca complex and their genetic background, the phylogeny of important examplers from the Lolium-Festuca complex were reconstructed. In total 40 taxa representing the Festuca and Lolium species with Vulpia myuros and Brachypodium distachyon as outgroups were sampled, using two non-coding intergenic spacers (trnQ-rps16, trnH-psbA) and one coding gene (rbcL). Maximum parsimony (MP), Bayesian inference (BI) analyses based on each partition and combined plastid DNA dataset, and median-jointing network analysis were employed. The outcomes strongly suggested that the subgen. Schedonorus has a close relationship to Lolium, and it is also proposed to move the sect. Leucopoa from subgen. Leucopoa to Subgen. Schedonorus and to separate sect. Breviaristatae from the subgen. Leucopoa. We found that F. californica could be a lineage of hybrid origin because of its intermediate placement between the “broad-leaved” and “fine-leaved” clade.
As one of the largest subtribes from the tribe Poeae (Pooideae, Poaceae), Loliinae encompasses nine genera (Festuca, Lolium, Vulpia, Nardurus, Loliolium Krecz, and Bobr, Scleropoa, Cutandia Willk., Sphenopus, and Bellardiochloa Chiov; Tzvelev, 1982; Soreng and Davis, 2000). Among the genera, Festuca is large and complex having more than 600 species with multiple ploidy levels ranging from diploid (2n = 2x = 14) up to dodecaploid (2n = 12x = 84) whereas Lolium is a small genus with 10 recognized diploid species (Clayton and Renvoize, 1986; Loureiro et al., 2007). The two genera, Festuca and Lolium include a number of important grasses used as pasture, fodder, and amenity purposes. The Lolium genera include the two widely cultivated temperate grass species, L. multiflorum (annual or Italian ryegrass) and L. perenne (perennial ryegrass) which are characterized by rapid growth and high forage quality. There are more than 3000 cultivars grown around the world with many hybrids naturally or artificially produced (Cai et al., 2011). The Festuca genus also includes two agriculturally important forage crop species, tall fescue (F. arundinacea) and meadow fescue (F. pratensis). They differ from ryegrasses, having larger, deeper root systems and greater water and nutrient-use-efficiency, and generally higher stress tolerance than the ryegrasses. Another important group of Festuca species are the fine-leaved fescues which are valued for their forage, turf and ornamental use. Red fescue (F. rubra L.) and sheep fescue (F. ovina L.) are valued for their narrow leaves which minimize water loss and provide improved drought tolerance (Rognli et al., 2010).
The majority of species within the Lolium/Festuca grass complex are heterogeneous and largely obligate outbreeders; they are highly diverse in their growth ontogeny, morphology, and their adaptations to onsets of both climatic and edaphic stress. As a consequence they have dispersed and colonized large areas of temperate global grasslands (Humphreys et al., 2006). As a group, the Festuca-Lolium complex comprises species that are closely allied and are partially interfertile.
The two genera can be easily differentiated by their inflorescence structure, and the taxonomic classification of some Lolium and Festuca species are controversial. A number of taxonomic revisions have proposed placing the “broad-leaved” fescues (Festuca subgen. Schedonorus) into Lolium (Darbyshire, 1993) by natural phenomenon and through experimental evidence. This includes the regular occurrence of spontaneous hybridization between species of Festuca subgen. Schedonorus and chasmogamous species of Lolium which on the other hand only rarely occurs between the major “fine-leaved” fescues and Lolium (Stace, 1975; Barker and Stace, 1982). Furthermore, analysis of morphological data sets (Stebbins, 1956), DNA restriction site variation (Darbyshire and Warwick, 1992), and seed protein (Buliñska-Radomska and Lester, 1988) suggest placing Lolium and Festuca subgen. Schedonorus together as one lineage. In contrast, others have suggested that the broad leaf fescues be separated into a new genus called Schedonorus (Soreng and Terrell, 1997). Aside from the controversial relationship between “broad-leaved” fescues and Lolium species, the Festuca genus per se is also complex with the taxanomic placements of its specific subgenera, sections, and species quite intricate. For instance, sect. Breviaristatae was considered as separate from subgen. Leucopoa (Tzvelev, 1971; Clayton and Renvoize, 1986), However, Soreng et al. (1990) found that F. slerophylla of sect. Leucopoa had a close relationship with F. arundinacea of subgen. Schedonorus based on the chloroplast DNA restriction site variation. By the same method, Darbyshire and Warwick (1992) discovered that examplers of sect. Breviaristatae had no phylogenetic affinity with examplers of sect. Leucopoa, subgen. Leucopoa. Therefore, they suggested that sect. Leucopoa from subgen. Leucopoa be moved to subgen. Schedonorus. F. mairei St. Yves, which is one of the key species in the evolution of polyploid fescues (Buliñska-Radomska and Lester, 1988) was first categorized into section Scariosae of subgen. Festuca (Stammers et al., 1995), but later was proposed to be reclassified into subgen. Schedonrus (Torrecilla and Catalán, 2002).
A better understanding of phylogenetic relationships within the Festuca-Lolium complex species would not only be very useful for future species conservation and for improved collection knowledge, but would also greatly assist future forage grass breeding programs. To ensure future grassland resilience and sustainable forage production for livestock agriculture, it has been considered as an increasingly important strategy to hybridize Lolium and Festuca species in order to gain and combine the complementary attributes of both. As Lolium x Festuca interspecific species' hybrids grass varieties are marketed under their own category termed Festulolium and provide a source of reliable, nutrient-use-efficient, and productive fodder for ruminants (Humphreys et al., 2014). Increased understanding of the phylogenetic relationships between the Lolium/Festuca species and of how the polyploid fescues and their adaptive benefits have evolved can benefit plant breeders and thereby accelerate the development of Festulolium breeding programs to better provide increased forage resilience sufficient to combat climate change.
Analysis of the phylogenetic relationships within the Festuca-Lolium complex encompassed the biological technology revolution from macro morphology to micro genetic level. Previous methods include chloroplast DNA (cpDNA) electrophoresis (Lehväslaiho et al., 1987; Soreng et al., 1990; Darbyshire and Warwick, 1992), RAPD (random amplification of polymorphic DNA) technology (Stammers et al., 1995; Wiesner et al., 1995), ITS (internal transcribe spacer) sequences of nuclear rDNA (Charmet et al., 1997; Gaut et al., 2000; Torrecilla and Catalán, 2002), and sequences of chloroplast trnL-F region (Catalán et al., 2004; Torrecilla et al., 2004). Despite the large genomic resources available for most intensively cultivated species of the Festuca-Lolium complex, conjoined analyses with chloroplast spacers as well as chloroplast genes have not been employed for the analysis of phylogeny among the Festuca-Lolium complex. The chloroplast has highly-conserved genes which are elementary to plants and are variable and informative regions over a long time scale. The use of cpDNA can also analyse the maternal source genome donor and has been applied successfully in the phylogenetic analysis of many taxa (Shaw et al., 2007; Sun, 2007; Nock et al., 2011).
In the present study, we sampled 42 taxa, including 28 Festuca taxa, 12 Lolium taxa and 2 related but out-group species (Vulpia myuros, Brachypodium distachyon). Chosen taxa have been identified by their morphology, they are representatives of broad-leaved and narrow-leaved species, these species are significant because of their importance in agricultural and amenity use and were therefore deemed the most important for this phylogenetic study. DNA sequence data from chloroplast spacers (trnQ-rps16, trnH-psbA) and chloroplast gene (rbcL) were used to resolve phylogenetic relationships among the Festuca-Lolium complex. The main objectives were to: (1) construct the plastid phylogeny of Festuca-Lolium complex using two non-coding intergenic spacers and one coding gene, and compare with the previous analyses; (2) explore the maternal donors of the polyploid species of fescues.
A total of forty taxa were sampled from the Lolium-Festuca complex comprising 28 Festuca taxa corresponding to 3 subgenera, 7 sections, and one subsection, and 12 taxa of Lolium. Vulpia myuros and Brachypodium distachyon were included as out-groups based on previous phylogenetic studies of Loliinae (Inda et al., 2008). The taxa names, accessions numbers, ploidy level, origin and abbreviations are listed in Table 1. All the seed materials with PI were generously provided by the National Plant Germplasm System of USDA. The seeds were first germinated in petri dishes and then the strong seedlings were transferred to pots. Morphological observation and weeding were regularly undertaken in order to ensure the plant purity. Mitotic analyses of root tips were made to verify the ploidy level of each accession.
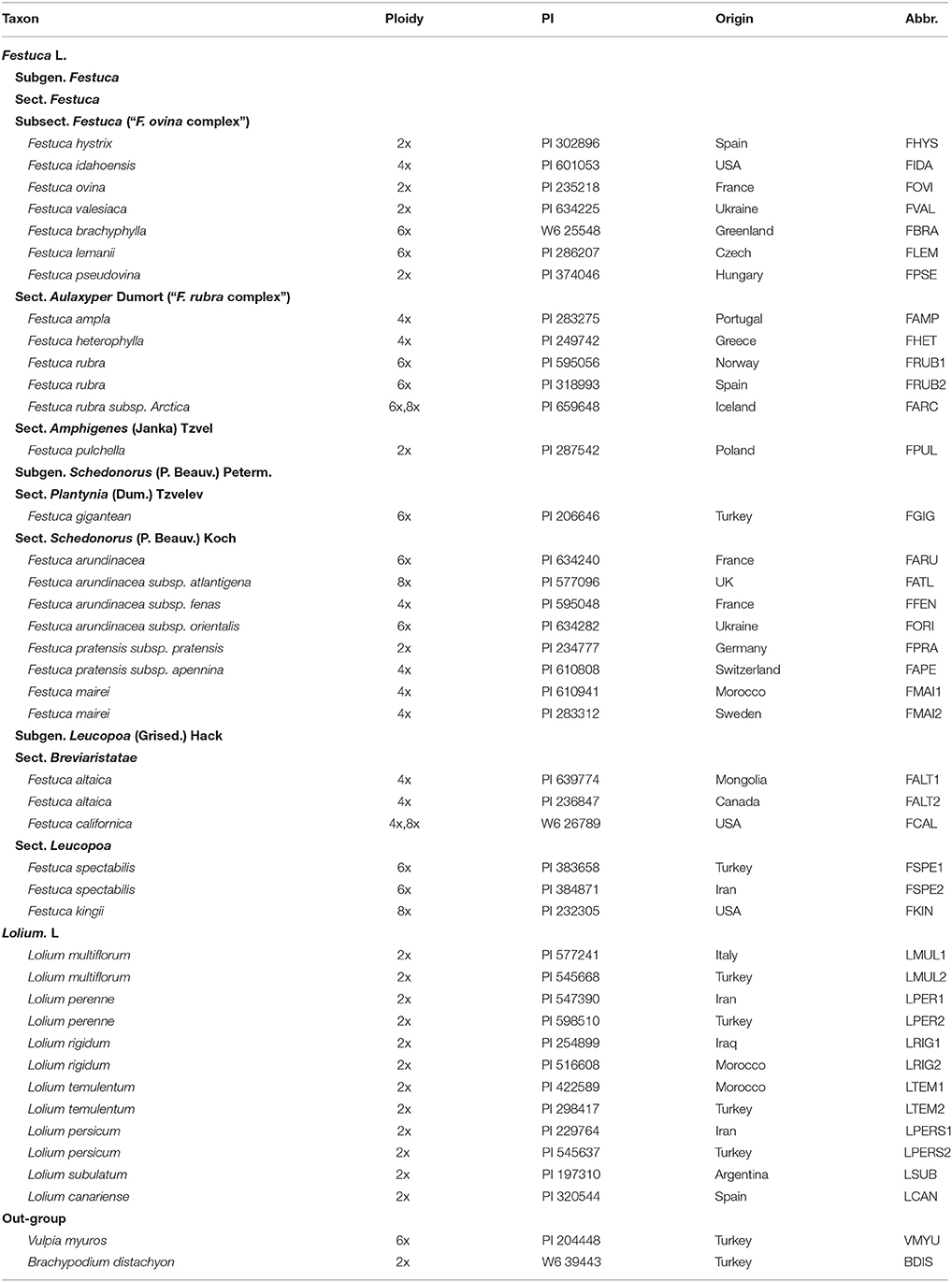
Table 1. List of Lolium/Festuca examplers and the outgroup used in this study.
Genomic DNA was extracted from freeze-dried leaf tissue of each accession by the standard CTAB (cetyl-trimethylammonium bromide) method (Doyle and Doyle, 1987) using the TIANcombi DNA PCR Kit (Beijing, China). One individual was sampled for DNA extraction, and five DNA samples were prepared for each taxon. The quality and concentration of the DNA were assessed by NanoVue Plus spectrophotometry produced by General Electric Company and checked on a 1% agarose-gel. The chloroplast trnH-psbA gene and rbcL were amplified with the universal primers (Table 2). The PCR (polymerase chain reaction) was performed in a final volume of 50 μl, containing 4 μl template DNA with the concentration of 10 ng/μl, 4 μl primer with a concentration of 0.01 mmol/μl, 25 μl 2 × Premix Taq (TaKaRa) with 0.4 mM dNTPs of each nucleotide, 3 mM MgCl2 buffer, 1.25 U Taq DNA polymerase with pigment included, and addition of ddH2O to the final volume. The PCR amplification programs started with a 4 min initial denaturation step at 94°C; followed by 35 cycles of 1 min denaturation at 94°C, 1 min annealing (55°C for rbcL and 50°C for trnH-psbA), and 1.5 min extension at 72°C; ending with a final extension step at 72°C for 10 min. The PCR products were checked in a 1% agarose gel and purified with the AxyPrep DNA kit, and then qualified samples were sent to the Majorbio Company (Shanghai, China) for sequencing. Generally, 3–5 PCR products were sent for sequencing from each taxon.
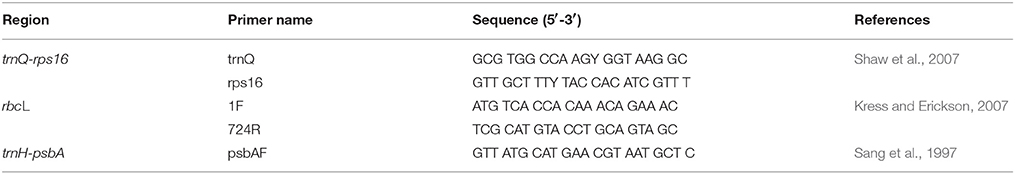
Table 2. Details of primer pairs used to amplify the trnQ-rps16, trnH-psbA, and rbcL gene.
The returned sequences data were initially spliced by the DNASTAR Seqman (Swindell and Plasterer, 1997) and aligned by the BioEdit ver. 7.0.9 (Hall, 1999). The nucleotide sites' information and the nucleotide frequencies were calculated by MEGA software ver. 5.02 (Tamura et al., 2011). The software package DAMBE ver. 5.5.29 (Xia and Xie, 2001) was used to assess the substitution saturation by plotting pairwise rates of transitions and transversions against a correct genetic distance under F84 model.
In order to evaluate the divergence and relationship among taxa, number of sites (n), number of variable site (s), haplotype diversity (Hd) (Nei and Li, 1979), Tajumas's π (Tajima, 1989), Watterson's θw (Watterson, 1975) were calculated, Neutrality test was also performed by the Tajima's and Fu and Li's D statistic (Tajima, 1989; Fu and Li, 1993). All the parameters above were conducted by DnaSP ver. 5.10 (Librado and Rozas, 2009).
Phylogenetic analysis of each partition and the combined plastid DNA dataset (trnQ-rps16, trnH-psbA, rbcL) were created by maximum parsimony (MP) and Bayesian inference (BI). Maximum parsimony (MP) analyses were implemented in PAUP* v.4.0b10 (Swofford, 2002). All characters were treated as unweighted and unordered, gaps were treated as “missing.” The heuristic search option using the Tree Bisection-Reconnection (TBR) branch swapping and MUL-Tree option on, 10 replicates of random addition sequence with the stepwise addition option was employed to obtain the most parsimonious trees. The consensus tree option was set as “retain groups with frequency>50%”. Topological robustness MP analysis was evaluated by bootstrap analysis using a full heuristic search with 1000 replicates (Felsenstein, 1985) each with simple addition sequence.
Bayesian inference was carried out in MrBayes v.3.1.2 (Huelsenbeck and Ronquist, 2001). It performs Bayesian phylogenetic analysis of information from different data partition or the combined dataset. The optimal evolutionary model used for different data matrixes were estimated by jModelTest v.2.1.7 (Posada, 2008; Darriba et al., 2012) was used to determine the using the Akaike information criterion (AIC). The best-fit model was TPMluf+G for trnQ-rps16 data, TIM1+I+G for trnH-psbA data, TIM2+G for rbcL, and TPMluf+I+G was chosen as the most appropriate for combined data analysis. Four MCMC (Markov Chain Monte Carlo) chain (one cold and three heated) were run for 200,000 generations for trnQ-rps16 data, 600,000 generations for trnH-psbA data, and 120,000 generations for rbcL and combined data, each sampling every 10 generations. The analysis was continued until the standard deviation of split frequencies below 0.01. The first 5000, 15,000, and 3000 trees were stationary discarded as “burn-in” for trnQ-rps16 data, trnH-psbA data and rbcL or combined data, respectively (determined empirically from the log-likelihood values using Tracer V1.4; Rambaut and Drummond, 2013). The remaining trees were employed to construct the 50%-majority rule consensus trees and frequencies of clades were evaluated by posterior probabilities (PP).
A Network representation may be more appropriate than the tree presentation when the existence of reticulate evolution such as gene transfer, hybridization, and recombination. The median-jointing (MJ) network analysis can reveal the relationships between ancestral and derived haplotypes which was first employed to discuss the human mtDNA variation (Bandelt et al., 2000). Compared with other graph construction approaches (minimum-spanning network and statistical parsimony), the MJ network using the maximum parsimony (MP) method can provide the best measure of the true genealogy (Cassens et al., 2005). The MJ network was constructed by the Network 4.6.1.3 program (Fluxus Technology Ltd), the test of recombination was performed by the GARD Processor method with the HyPhy package (Pond and Muse, 2005). Based on the result of the test, the combination of the three plastid DNA datasets was used to generate the MJ network because of the absence of recombination signal in alignment (Log Likelihood = −4126.04; AIC = 8430.02).
The length of trnQ-rps16, trnH-psbA and rbcL sequences were 700, 579, and 632 bp respectively, in the final aligned sequences of 40 taxa excluding the out-groups. The specific sites' information and nucleotide frequencies were shown in Table 3. Both the tests of substitution saturation for trnQ-rps16, trnH-psbA spacers and rbcL gene under the F84 model showed a basically linear regression which demonstrated no saturation effects among the mutation of different sequences (see Supplementary Figures 1–3).
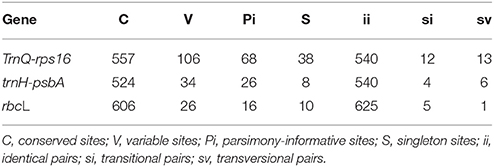
Table 3. Features of the matched data matrix for trnQ-rps16, trnH-psbA, and rbcL gene sequences.
The nucleotide diversity information containing the number of sites (n), number of variable site (s), haplotype diversity (Hd), the average pairwise diversity (π), and the diversity based on the number of segregating sites (θw) of trnQ-rps16, trnH-psbA spacers, and rbcL gene were calculated. The neutrality test results showed negative for the three sequences which might be because of the genetic bottleneck. Above values were displayed in Table 4.
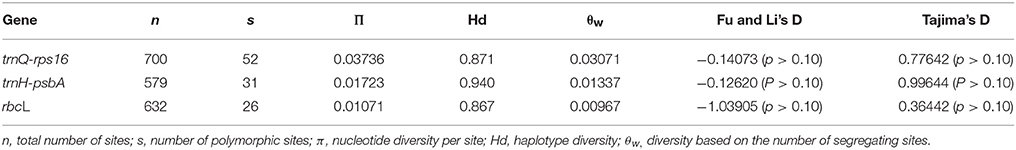
Table 4. Estimates of nucleotide diversity and test statistics for trnH-psbA, trnQ-rps16, and rbcL gene sequences data sets.
All 126 sequences have been submitted to the database of NCBI (National Center for Biotechnology Information), the accession numbers are from KT43895 to KT439068 (see Supplementary Excel, Datasheet).
The aligned trnQ-rps16 sequences produced a total of 697 characters, of which 109 were variable and 75 were parsimony-informative. The parsimony analysis for trnQ-rps16 sequences resulted in 215 most parsimonious trees (tree length = 215; consistency index = 0.9395; retention index = 0.9791; rescale consistency index = 0.9199). The 50% MP majority-rule consensus tree was identical to the tree obtained from BI except for some nodes presenting different statistical support. The tree shown in Figure 1 was MP tree with bootstrap support (BS) above the branches and posterior probabilities (PP) of BI tree below the branches. According to the tree, the two major clades had been strongly supported, mainly corresponding to the width of blades. The first clade included the narrow-leaved fescues and Vulpia myuros, the second clade contained all the Lolium samples and the broad-leaved Festuca taxa.
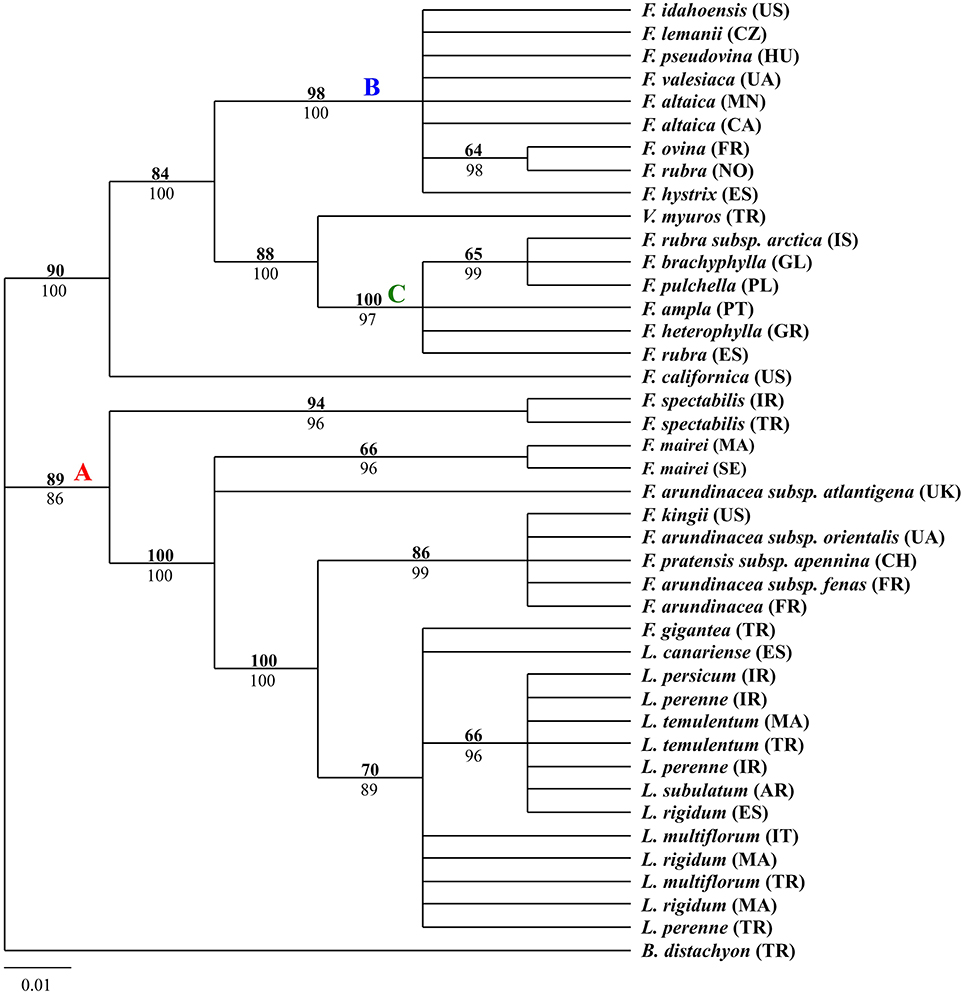
Figure 1. Fifty-percent majority-rule BI tree inferred from the chloroplast non-coding intergenic spacers trnQ-rps16 sequences of Festuca/Lolium examplers. The number above and below the braches indicate boot strap values ≥50% and Bayesian posterior probability values ≥90%.
The total character of the aligned trnH-psbA sequences was 585, of which 27 characters were variable and parsimony-informative. The parsimony analysis for trnH-psbA sequences resulted in 91 most parsimonious trees (tree length = 190; consistency index = 0.3000; retention index = 0.5994; rescale consistency index = 0.1798). The 50% MP majority–rule consensus tree and the tree from BI were different but poorly resolved because of the limited difference among the sequences. The trees are shown in Supplementary Figure 4.
The aligned rbcL sequences yielded a total of 632 characters with 26 variable characters and 16 informative characters among which the parsimony analysis for trnH-psbA sequences resulted in 48 most parsimonious trees (tree length = 51; consistency index = 0.8235; retention index = 0.9511; rescale consistency index = 0.7832). The 50% MP majority-rule consensus tree was highly congruent to the tree obtained from BI except for some nodes presenting different statistical support. The tree showed in Figure 2 was MP tree with bootstrap support (BS) above the branches and posterior probabilities (PP) of BI tree below the branches. The tree outline was different form the other two trees. There were two major clades which had been well supported, one large clade including most of the taxa and one small clade including F. ampla, F. rubra subsp. arctica, F. brachyphylla, F. heterophylla, F. pulchella, and F. rubra.
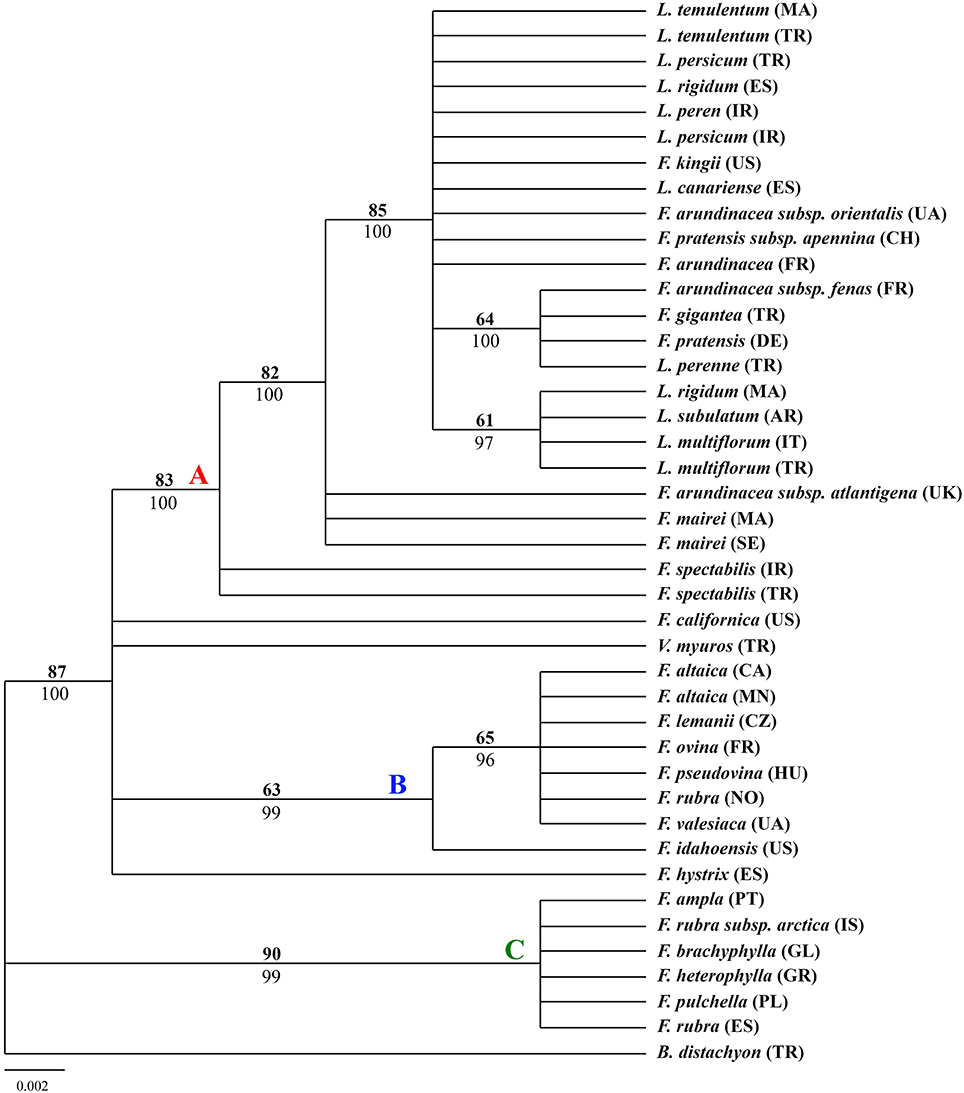
Figure 2. Fifty-percent majority-rule BI tree inferred from the chloroplast gene rbcL sequences of Festuca/Lolium examplers. The number above and below the braches indicate boot strap values ≥50% and Bayesian posterior probability values ≥90%.
Of 1926 total characters within the combined data set of the three plastid DNA regions, 135 characters were variable, and 128 characters were informative. The cladistics parsimony search yielded 363 most parsimonious trees with the tree length of 368 steps, a consistency index (CI) of 0.7853, retention index (RI) of 0.9349, and rescale consistency index (RC) of 0.7342. The 50% MP majority-rule consensus tree was largely incongruent to the tree obtained from BI analyses (Figure 3). According to the classification, the 50% MP tree was mainly influenced by the sequences of the rbcL and the BI tree was largely affected by the trnQ-rps16 sequences data. The two major clades were highly supported in the BI tree rather than in the MP tree.
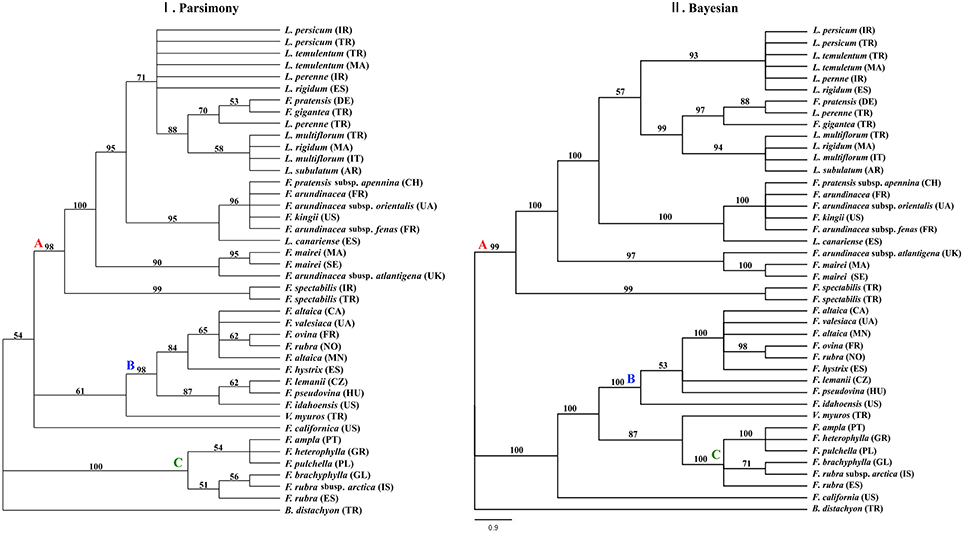
Figure 3. Phylogenies inferred from the combined dataset of Festuca/Lolium examplers using maximum parsimony and Bayesian inference. Fifty-percent majority-rule BI tree inferred from the combined dataset of Festuca/Lolium examplers. (I) The MP majority rule consensus tree with the numbers at nodes indicating bootstrap values ≥50%. (II) The 50% majority rule consensus tree from the Bayesian inference with the numbers at nodes indicating Bayesian posterior probability values ≥90%.
In order to get better insights into the number of haplotypes of the combined sequences and their relations, a median-joint network was employed. Each circular network node represents a single sequence haplotype, with node size being proportional to the number of isolates with the haplotype. Mv (median vectors representing missing intermediates) reveals unsampled nodes inferred by MJ network analysis, and the number along the branches shows the number of mutations. 33 haplotypes were derived from 40 taxa which revealed higher levels of haplotype diversity of the combined sequence data (Figure 4). MJ analysis generally grouped according to the clades shown by the phylogenies of the combined data. The taxa were grouped into three clades, and the clade I and clade II could be considered as one group, and clade I and II were 46 and 35 mutational steps from clade III, respectively. All the Lolium taxa were nested with all the broad-leaved Festuca taxa.
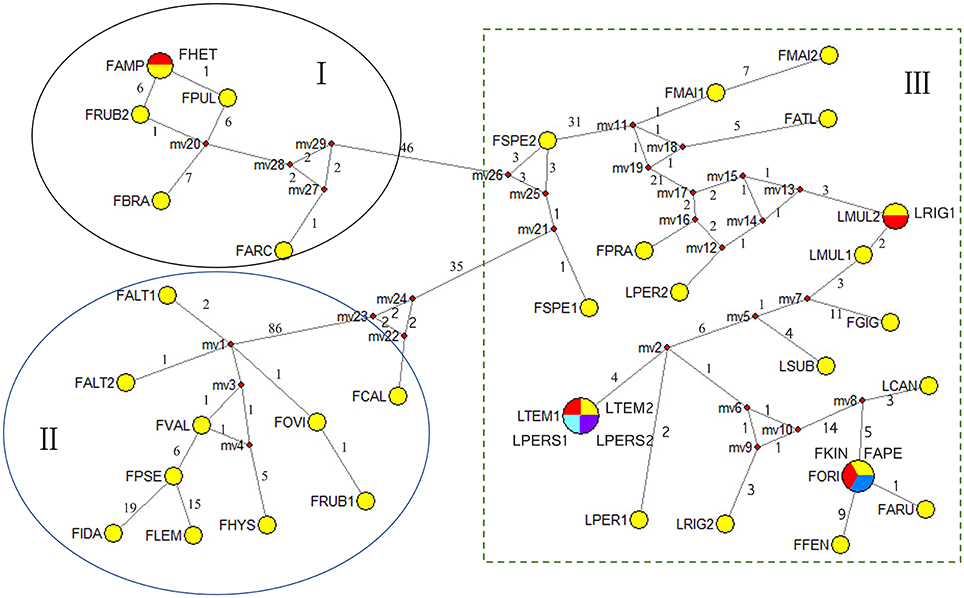
Figure 4. Median-joining networks of the combined data of the Festuca/Lolium examplers. Haplotypes are represented by circles. Numbers along branches indicate number of mutational changes between nodes. Abbreviations of species names are listed in Table 1.
From the combined analyses of the chloroplast spacers trnQ-rps16, trnH-psbA, the chloroplast gene rbcL, and the combined data set and the MJ network of the combined data, the dendrograms achieved all clearly demonstrate three obvious clades. The largest clade (A) contained all the Lolium taxa and included the broad-leaved fescues including all the samples of subgenus Schedonrus (F. arundinacea, F. pratensis, F. mairei and F. gigantea) and samples from sect. Leucopoa of Subgenus Leucopoa (F. spectabilis F. kingii). A smaller clade (B) included all the samples from subsect. Festuca including F. valesiaca, F. ovina, F. hystrix, F. lemanii, F. pseudovina, F. idahoensis as well as F. altaica and F. rubra (NO). The only species of subsect. Festuca not included within this clade is F. brachyphlla. A smaller third clade (C) included samples from sect. Aulaxyper and sect. Amphigenes of subgenus Festuca (F. ampla, F. heterophylla, F. rubra (ES), F. rubra subsp. arctica, F. pulchella) and F. brachyphylla. In the dendrogram of trnQ-rps16 sequences and BI tree of the combined data set, clade B and C with F. californica formed one clade, while in the dendrogram of rbcL sequences and MP tree of the combined data set, a new clade was made up by clade A, clade B, and F. californica.
As all the Lolium species were nested closely with the broad-leaved Festuca species in clade A (Figures 1–4), it could be concluded that the broad-leaved fescues have closer relationship to Lolium grass species than to the fined-leaved fescues. There has been debate about the classification of subgen. Schedonorus. It was suggested that the subgen. Schedonorus (broad-leaved fescues) be included within Lolium (Darbyshire, 1993) despite the obvious differences in their inflorescence morphology (raceme for Festuca and spica for Lolium), whilst others have suggested a split of the subgen. Schedonorus into an independent genus, Schedonorus (Soreng and Terrell, 1997). According to the result demonstrated within the current study, the former classification seems more reasonable, in other words, subgen. Schedonorus has a close relationship to Lolium. Among all the examplers of Lolium, only L. canariense, which is found mainly on poor land in maritime condition (Loos, 1994), has a closer relationship to broad-leaved fescues than to other Lolium species. Two representatives of sect. Leucopoa (F. kingii and F. spectabilis) were placed in the clade A with representatives from subgen. Schedonorus and Lolium, while for the representatives of Sect. Breviaristatae, F. altaica taxa were attached to clade B and F. california has developed as an individual group. Similar results were achieved previously where the phylogenetic relationships among the Festuca-Lolium complex were described using SRAP markers (Cheng et al., 2015) and in earlier studies (Catalán et al., 2004, 2007; Inda et al., 2008). From the current and previous research, we strongly propose that the sect. Leucopoa should be moved from subgen. Leucopoa to Subgen. Schedonorus or into a separate sect. Breviaristatae from the subgen. Leucopoa. According to the strict consensus tree and Bayesian 50% MR consensus tree inferred from ITS and trnL-F sequences, representatives of sect. Breviaristatae (F. altaica and F. californica) have an intermediate placement between the “broad-leaved” and “fine-leaved” clade (Catalán et al., 2004). In the current study, the lineage of F. californica could indicate a hybrid origin due to its intermediate placement between the “broad-leaved” (A) and “fine-leaved” clade (B).
Cytological investigations at the Institute for Biological, Environmental, and Rural Sciences (IBERS) using genomic in situ hybridization (GISH) and as total genomic DNA probes, candidate Festuca species have established the close ancestry of F. pratensis, F. arundinacea var. glaucescens (also known as F. arundinacea subsp. fenas) and F. arundinacea (Humphreys et al., 1995). From all the dendrograms in this study, F. arundinacea, F. arundinacea subsp. fenas, F. pratensis subsp. apennina, F. arundinacea subsp. orientalis, and F. kingii were all gathered closely within one group and thus might share the same ancestry. In the MJ network, F. pratensis subsp. apennina, F. arundinacea subsp. orientalis, and F. kingii shared the same haplotype, and they are also closely associated to F. arundinacea, F. arundinacea subsp. fenas. The North African fescue species F. mairei and F. arundinacea subsp. atlantigena were classified into one group which showed their close relationship. F. mairei was once been placed in sect. Scariosae of subgen. Festuca using a RAPD analysis (Stammers et al., 1995). However, based on the previous SRAP analysis (Cheng et al., 2015) and the research presented in this paper, it is considered as more accurate to place F. mairei within the subgen. Schedonorus.
In all clade B studies herein, F. brachyphylla was separated from other examplers of the Subsect. Festuca of Subgen. Festuca and was associated closer to representatives of Sect. Aulaxyper and Sect. Amphigenes. F. brachyphylla used to be considered as an arctic-alpine counterpart to the more temperate-montane F. ovina, which both belong to section Festuca, but the delimitation of F. brachyphylla and F. ovina has been considered as controversial with some authors having concluded the taxa of F. brachyphylla as a subspecies of F. ovina (Cronquist et al., 1977) whilst other authors have included both taxa in a widely defined F. brachyphylla (Fjellheim et al., 2001). In the present study, F. brachyphylla was differentiated from F. ovina, as shown previously by the same authors using SRAPs markers (Cheng et al., 2015) and also from a Bayesian tree of Loliinae which used trnTF and ITS data (Inda et al., 2008). It is proposed that the F. brachyphylla taxa be considered as a separate entity form Subsect. Festuca. In addition, F. altaica of sect. Breviaristatae was found closely aligned to subsect. Festuca known as the “F. ovina complex.”
In clade C, F. pulchella of Sect. Amphigenes had a close relationship with sect. Aulaxyper known as “F. rubra complex.” Two examplers of F. rubra were clustered within two different groups, it might be because of the large latitude difference. From the MJ network, F. ampla shared the same haplotype with F. heterophylla which indicated a close relationship between them.
In conclusion, as the two major genera of the grass family, the Lolium and Festuca taxa can be considered to have expanded to become the predominant temperate grassland of the world. From a taxonomical perspective, it is essential to classify the different species into their correct sections or subsections of subgenera as well as to clarify the relationships of some important species. From an agricultural perspective, it has become increasingly important to hybridize Lolium and Festuca species in order to gain the attributes of both. Current synthetic Festulolium hybrids are frequently genetically unstable, and so it is important to understand how stable polyploids within the Festuca taxa have evolved from their progenitor species. In the present work, from the analyses of three plastid DNA data, either from the MP trees or BI trees, from the single or combined data, it is shown that the subgen. Schedonorus shares a close relationship with the majority of Lolium grasses. The phylogenetic tree can guide the parents chosen for hybrid breeding. It is recommended that F. mairei should be included within the subgen. Schedonorus. F. californica could have a lineage of hybrid origin because of its intermediate placement between the “broad-leaved” and “fine-leaved” clades. Furthermore, it is suggested that F. brachyphylla should be treated as a separate entity form the “F. ovina complex.” The results add more information and understanding into species evolution within the Lolium/Festuca complex.
YC performed the experiments, analyzed the data and wrote the manuscript. KZ and HY guided the bioinformatics analyses, XZ organized the funding and participated in the samples collecting, XM guided the manuscript writing, JH, and MH provided helpful comments and language editing on the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank National Plant Germplasm System of United States Department of Agriculture (USDA) for kindly supplying the seeds. This work was supported by the National Basic Research Program of China (973 Program) (2014CB138705), the Earmarked Fund for the Modern Agro-Industry Technology Research System (CARS-35-05), and the National Natural Science Foundation of China (NSFC 31372363).
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fevo.2016.00089
Bandelt, H. J., Macaulay, V., and Richards, M. (2000). Median networks: speedy construction and greedy reduction, one simulation, and two case studies from human mtDNA. Mol. Phylogenet. Evol. 16, 8–28. doi: 10.1006/mpev.2000.0792
Barker, C., and Stace, C. (1982). Hybridisation in the genera Vulpia and Festuca: the production of artificial F1 plants. Nord. J. Bot. 2, 435–444. doi: 10.1111/j.1756-1051.1982.tb01206.x
Buliñska-Radomska, Z., and Lester, R. (1988). Intergeneric relationships of Lolium, Festuca, and Vulpia (Poaceae) and their phylogeny. Plant Syst. Evol. 159, 217–227. doi: 10.1007/BF00935973
Cai, H., Stewart, A., Inoue, M., Yuyama, N., and Hirata, M. (2011). “Lolium,” in Wild Crop Relatives: Genomic and Breeding Resources, ed C. Kole (Berlin; Heidelberg: Springer), 165–173.
Cassens, I., Mardulyn, P., and Milinkovitch, M. C. (2005). Evaluating intraspecific “network” construction methods using simulated sequence data: do existing algorithms outperform the global maximum parsimony approach? Syst. Biol. 54, 363–372. doi: 10.1080/10635150590945377
Catalán, P., Torrecilla, P., López-Rodríguez, J. A., Müller, J., and Stace, C. A. (2007). A systematic approach to subtribe Loliinae (Poaceae: Pooideae) based on phylogenetic evidence. Aliso J. Syst. Evol. Bot. 23, 380–405. doi: 10.5642/aliso.20072301.31
Catalán, P., Torrecilla, P., Rodrıìguez, J. Á. L., and Olmstead, R. G. (2004). Phylogeny of the festucoid grasses of subtribe Loliinae and allies (Poeae, Pooideae) inferred from ITS and trnL–F sequences. Mol. Phylogenet. Evol. 31, 517–541. doi: 10.1016/j.ympev.2003.08.025
Charmet, G., Ravel, C., and Balfourier, F. (1997). Phylogenetic analysis in the Festuca-Lolium complex using molecular markers and ITS rDNA. Theor. Appl. Genet. 94, 1038–1046. doi: 10.1007/s001220050512
Cheng, Y., Ma, X., Zhou, K., Humphreys, M. W., and Zhang, X. Q. (2015). Phylogenetic analysis of Festuca–Lolium complex using SRAP markers. Genet. Resour. Crop Evol. 63, 7–18. doi: 10.1007/s10722-015-0324-5
Clayton, W. D., and Renvoize, S. A. (1986). Genera Graminum. Grasses of the World. Kew Bulletin Additional Series, 13. London: Her Majesty's Stationery Office.
Cronquist, A., Holmgren, A. H., Holmgren, N. H., Reveal, J. L., and Holmgren, P. K. (1977). Intermountain flora. Vascular plants of the Intermountain West, USA Vol. 6. The monocotyledons. New York, NY: Columbia University.
Darbyshire, S. J. (1993). Realignment of Festuca subgenus Schedonorus with the genus Lolium (Poaceae). Novon 3, 239–243. doi: 10.2307/3391460
Darbyshire, S. J., and Warwick, S. I. (1992). Phylogeny of North American Festuca (Poaceae) and related genera using chloroplast DNA restriction site variation. Can. J. Bot. 70, 2415–2429. doi: 10.1139/b92-300
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9, 772–772. doi: 10.1038/nmeth.2109
Doyle, J. J., and Doyle, J. L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15.
Felsenstein, J. (1985). Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783–791. doi: 10.2307/2408678
Fjellheim, S., Elven, R., and Brochmann, C. (2001). Molecules and morphology in concert. II. The Festuca brachyphylla complex (Poaceae) in Svalbard. Am. J. Bot. 88, 869–882. doi: 10.2307/2657039
Fu, Y.-X., and Li, W.-H. (1993). Statistical tests of neutrality of mutations. Genetics 133, 693–709.
Gaut, B., Tredway, L., Kubik, C., Gaut, R., and Meyer, W. (2000). Phylogenetic relationships and genetic diversity among members of the Festuca-Lolium complex (Poaceae) based on ITS sequence data. Plant Syst. Evol. 224, 33–53. doi: 10.1007/BF00985265
Hall, T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl. Acids Symp. Ser. 41, 95–98.
Huelsenbeck, J. P., and Ronquist, F. (2001). MRBAYES: bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755. doi: 10.1093/bioinformatics/17.8.754
Humphreys, M., Thomas, H., Morgan, W., Meredith, M., Harper, J., Thomas, H., et al. (1995). Discriminating the ancestral progenitors of hexaploid Festuca arundinacea using genomic in situ hybridization. Heredity 75, 171–174. doi: 10.1038/hdy.1995.120
Humphreys, M. W., O'Donovan, S. A., Farrell, M. S., Gay, A. P., and Kingston−Smith, A. H. (2014). The potential of novel Festulolium (2n= 4x= 28) hybrids as productive, nutrient−use−efficient fodder for ruminants. Food Energy Secur. 3, 98–110. doi: 10.1002/fes3.50
Humphreys, M. W., Yadav, R. S., Cairns, A. J., Turner, L. B., Humphreys, J., and Skøt, L. (2006). A changing climate for grassland research. New Phytologist 169, 9–26. doi: 10.1111/j.1469-8137.2005.01549.x
Inda, L. A., Segarra-Moragues, J. G., Müller, J., Peterson, P. M., and Catalán, P. (2008). Dated historical biogeography of the temperate Loliinae (Poaceae, Pooideae) grasses in the northern and southern hemispheres. Mol. Phylogenet. Evol. 46, 932–957. doi: 10.1016/j.ympev.2007.11.022
Kress, W. J., and Erickson, D. L. (2007). A two-locus global DNA barcode for land plants: the coding rbcL gene complements the non-coding trnH-psbA spacer region. PLoS ONE 2:e508. doi: 10.1371/journal.pone.0000508
Lehväslaiho, H., Saura, A., and Lokki, J. (1987). Chloroplast DNA variation in the grass tribe Festuceae. Theor. Appl. Genet. 74, 298–302. doi: 10.1007/BF00274710
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Loos, B. P. (1994). The Genus Lolium: Taxonomy and Genetic Resources. Wageningen: Centrum voor Plantenveredelings-en Reproduktieonderzoek DLO.
Loureiro, J., Kopecký, D., Castro, S., Santos, C., and Silveira, P. (2007). Flow cytometric and cytogenetic analyses of Iberian Peninsula Festuca spp. Plant Syst. Evol. 269, 89–105. doi: 10.1007/s00606-007-0564-8
Nei, M., and Li, W.-H. (1979). Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. U.S.A. 76, 5269–5273. doi: 10.1073/pnas.76.10.5269
Nock, C. J., Waters, D. L., Edwards, M. A., Bowen, S. G., Rice, N., Cordeiro, G. M., et al. (2011). Chloroplast genome sequences from total DNA for plant identification. Plant Biotechnol. J. 9, 328–333. doi: 10.1111/j.1467-7652.2010.00558.x
Pond, S. L. K., and Muse, S. V. (2005). “HyPhy: hypothesis testing using phylogenies,” in Statistical Methods in Molecular Evolution, ed R. Nielsen (New York, NY: Springer), 125–181.
Posada, D. (2008). jModelTest: phylogenetic model averaging. Mol. Biol. Evol. 25, 1253–1256. doi: 10.1093/molbev/msn083
Rambaut, A., and Drummond, A. J. (2013). Tracer v1.6. Available online at: http://beast.bio.ed.ac.uk/Tracer
Rognli, O. A., Saha, M. C., Bhamidimarri, S., and van der Heijden, S. (2010). “Fescues,” in Fodder Crops and Amenity Grasses, eds B. Boller, U. K. Posselt, and F. Veronesi (New York, NY: Springer), 261–292.
Sang, T., Crawford, D., and Stuessy, T. (1997). Chloroplast DNA phylogeny, reticulate evolution, and biogeography of Paeonia (Paeoniaceae). Am. J. Bot. 84, 1120–1120. doi: 10.2307/2446155
Shaw, J., Lickey, E. B., Schilling, E. E., and Small, R. L. (2007). Comparison of whole chloroplast genome sequences to choose non-coding regions for phylogenetic studies in angiosperms: the tortoise and the hare III. Am. J. Bot. 94, 275–288. doi: 10.3732/ajb.94.3.275
Soreng, R., and Davis, J. (2000). “Phylogenetic structure in Poaceae subfamily Pooideae as inferred from molecular and morphological characters: misclassification versus reticulation,” in Grasses: Systematics and Evolution, eds S. W. Jacobs and J. Everett (Melbourne: CSIRO Publishing), 61–74.
Soreng, R., Davis, J., and Doyle, J. (1990). A phylogenetic analysis of chloroplast DNA restriction site variation in Poaceae subfam. Pooideae. Plant Syst. Evol. 172, 83–97. doi: 10.1007/BF00937800
Soreng, R., and Terrell, E. (1997). Taxonomic notes on Schedonorus, a segregate genus from Festuca or Lolium, with a new nothogenus, x Schedololium, and new combinations. Phytologia 83, 85–88.
Stace, C. A. (1975). Hybridization and the Flora of the British Isles. London; New York, NY; San Francisco, CA: Academic Press.
Stammers, M., Harris, J., Evans, G., Hayward, M., and Forster, J. (1995). Use of random PCR (RAPD) technology to analyse phylogenetic relationships in the Lolium/Festuca complex. Heredity (Edinb). 74, 19–27. doi: 10.1038/hdy.1995.3
Stebbins, G. L. (1956). Taxonomy and the evolution of genera, with special reference to the family Gramineae. Evolution 10, 235–245. doi: 10.2307/2406009
Sun, G. (2007). Genetic diversity of rbcL gene in Elymus trachycaulus complex and their phylogenetic relationships to several Triticeae species. Genet. Resour. Crop Evol. 54, 1737–1746. doi: 10.1007/s10722-006-9183-4
Swindell, S. R., and Plasterer, T. N. (1997). “Seqman,” in Sequence Data Analysis Guidebook, ed S. Swindell (Totowa, NJ: Humana Press), 75–89.
Swofford, D. L. (2002). PAUP*. Phylogenetic analysis using parsimony (* and other methods). Version 4beta 10. Sunderland, MA: Sinauer Associates.
Tajima, F. (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595.
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., and Kumar, S. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739. doi: 10.1093/molbev/msr121
Torrecilla, P., and Catalán, P. (2002). Phylogeny of broad-leaved and fine-leaved Festuca lineages (Poaceae) based on nuclear ITS sequences. Syst. Bot. 27, 241–251. doi: 10.1043/0363-6445-27.2.241
Torrecilla, P., José-Angel, López-Rodríguez, and Catalán, P. (2004). Phylogenetic relationships of Vulpia and related genera (Poeae, Poaceae) based on analysis of ITS and trnL-F sequences. Ann. Missouri Bot. Gard. 91, 124–158. Available online at: http://www.jstor.org/stable/3298573
Tzvelev, N. (1971). On the taxonomy and phylogeny of genus Festuca L. of the USSR Flora. I. The system of the genus and the main trends of evolution. Bot. Zhurn 56, 1252–1262.
Tzvelev, N. (1982). Poryadok zlaki (Poales). Semeystvo zlaki (Poaceae, ili Gramineae). Zhizn Rasteniy v Shesti Tomakh 6, 341–378.
Watterson, G. (1975). On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7, 256–276. doi: 10.1016/0040-5809(75)90020-9
Wiesner, I., Samec, P., and Našinec, V. (1995). Identification and relationships of cultivated accessions from Lolium-Festuca complex based on RAPD fingerprinting. Biol. Plant. 37, 185–195. doi: 10.1007/BF02913208
Keywords: phylogeny, Festuca, Lolium, trnQ-rps16, trnH-psbA, rbcL
Citation: Cheng Y, Zhou K, Humphreys MW, Harper JA, Ma X, Zhang X, Yan H and Huang L (2016) Phylogenetic Relationships in the Festuca-Lolium Complex (Loliinae; Poaceae): New Insights from Chloroplast Sequences. Front. Ecol. Evol. 4:89. doi: 10.3389/fevo.2016.00089
Received: 25 January 2016; Accepted: 14 July 2016;
Published: 27 July 2016.
Edited by:
Anthony I. Cognato, Michigan State University, USAReviewed by:
Dmitry Yurievich Sherbakov, Limnological Institute (Siberian Branch of the Russian Academy of Sciences), RussiaCopyright © 2016 Cheng, Zhou, Humphreys, Harper, Ma, Zhang, Yan and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinquan Zhang, emhhbmd4cUBzaWNhdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.