 Hongmei Li-Byarlay
Hongmei Li-Byarlay
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Ecol. Evol., 19 May 2016
Sec. Evolutionary Ecology of Social Behaviour
Volume 4 - 2016 | https://doi.org/10.3389/fevo.2016.00057
This article is part of the Research TopicGenetic effects on social traits: empirical studies from social animalsView all 7 articles
The social arthropods are characterized by a caste-based division of labor that may be influenced by epigenetic effects. One of the most important and widely studied epigenetic mechanisms is DNA methylation. Advances in understanding of social insect genomes, including epigenetic marks, make it possible to assess the role of DNA methylation in social caste development and social behavior. In this mini review, I summarize and interpret recent findings regarding DNA methylation and discuss how DNA methylation might influence evolution of sociality. In particular, I focus on enzymes associated with DNA methylation, the functions of DNA methylation in caste determination, behavioral gene regulation, and the effects of DNA methylation on learning and memory. Finally, I highlight current challenges and predict future breakthroughs in the field of socioepigenomics.
Social insects are considered superorganisms because of their complex organization in reproduction, behavior, and evolution (Hölldobler and Wilson, 2009). Social insects such as bees, wasps, ants, and termites must produce different adult morphologies as well as different reproductive and behavioral systems to maintain their social structure and division of labor (Wilson, 1979; Evans and Wheeler, 2000; Hartfelder et al., 2015).
When responding to environmental conditions, social insects display phenotypic plasticity based on the same genome (Moczek, 2010). Epigenetics refers to modifications of DNA bases, alterations to DNA-associated histones, and remodeling of chromatin that influence or regulate gene activities, rather than altering the DNA sequence. These modifications are caused by external or environmental factors, and they change the gene transcription and regulation in cells (Bird, 2007). In addition, epigenetics is thought to be one plausible mechanism explaining gene and environment interactions (Liu et al., 2008).
The most well studied epigenetic mechanisms include methylation of the cytosine base (5-methylcytosine, or 5mC), histone posttranslational modifications, chromatin remodeling, and noncoding RNAs (Negre et al., 2011; Dunham et al., 2012). These mechanisms play major roles in transcriptional regulation, genomic imprinting, and the silencing of repetitive DNA elements (Wolffe and Matzke, 1999; Waterland and Jirtle, 2003; Jirtle and Skinner, 2007). Also, DNA cytosine methylation has emerged as a key regulator in shaping animal social behavior (Miller, 2010). The processes of cytosine methylation and demethylation are important mediators in memory formation and behavioral plasticity.
In this mini review, I summarize current knowledge on the roles of DNA methylation (5mC) in social behavior, caste determination, social evolution, and learning and memory. In addition, I discuss the controversies and complications of previous studies, and provide suggestions regarding experimental design and data collection processes.
The key enzymes affecting DNA cytosine methylation are DNA methyltransferase 1 (DNMT1) and 3 (DNMT3). DNMT1 maintains DNA methylation during DNA replication in daughter cells and shows a strong preference for hemimethylated DNA (Goll and Bestor, 2005). This characteristic indicates that DNMT1-mediated methylation is an epigenetic mechanism that sustains the status of methylation marks from the one replication cycle to the next (Jaenisch and Bird, 2003). In contrast, DNMT3 is involved in de novo methylation, which means unmethylated DNA is modified by adding new methylation sites (Goll and Bestor, 2005). Both are thought to be required for a functional methylation system. In insects, the first orthologs of DNMT1 (a and b) and DNMT3 were identified in honey bees (Apis mellifera; Wang et al., 2006). As more and more genomes and transcriptomes were sequenced, more social insects were found to contain functional methylation systems. Figure 1 summarizes current knowledge on the distribution of DNMT1 and DNMT3 in social insects as compared with humans (Wang et al., 2006, 2014; Bonasio et al., 2012; Kocher et al., 2013; Terrapon et al., 2014; Kapheim et al., 2015; Patalano et al., 2015). DNMT2 was previously linked to cytosine methylation, but now it is known to methylate transfer RNA (Goll et al., 2006; Marbaniang and Vogel, 2016).
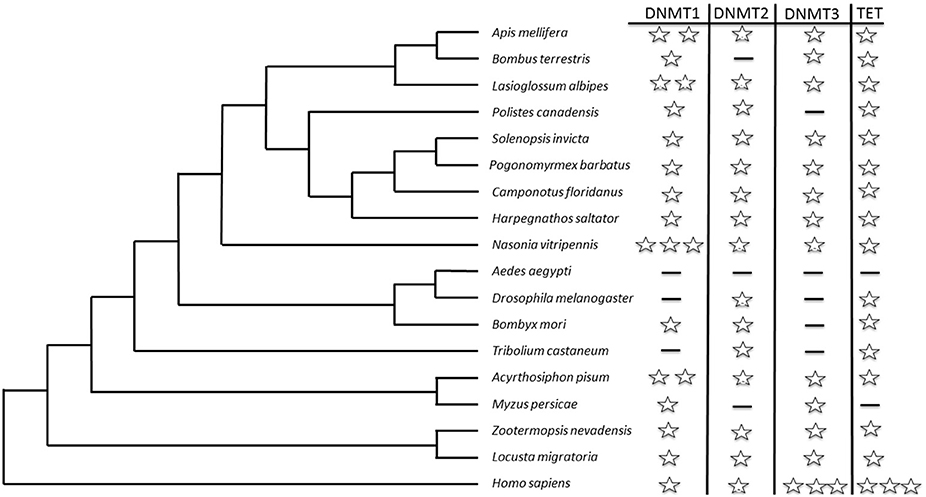
Figure 1. An ultrametric tree displaying current knowledge of the homologs of cytosine methylation-related enzymes in different social insect groups, and comparison with those in humans (Homo sapiens). Each star represents one copy of the enzyme in the genome. A dash indicates that either no copy was identified in the genome or the gene has an unknown status based on the literature and the National Center for Biotechnology Information gene database (http://www.ncbi.nlm.nih.gov/gene/?term=). The tree branch lengths are not scaled to match the true evolutionary status of each group. Only the tree topology is correct.
Working in parallel with methylation, demethylation is an important process that maintains the dynamics of DNA methylation in honey bee cells. Ten-eleven translocation (TET) dioxygenases can oxidize 5mC to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine, and 5-carboxylcytosine (Law and Jacobsen, 2010; Kohli and Zhang, 2013; Wojciechowski et al., 2014). Transcripts of TET have been detected in different stages of honey bee development (embryos, larvae, and adults) and in different tissues (ovaries, brains, and antennae; Wojciechowski et al., 2014; Rasmussen and Amdam, 2015). TET is also widely distributed in different social insect groups, including bees and ants (Figure 1). In addition, TET may be associated with gene regulation via alternative mRNA splicing (Cingolani et al., 2013). TET catalyzes the formation of 5hmC not only in DNA, but also in RNA (Fu et al., 2014). A recent study in Drosophila melanogaster showed that the 5hmC in the RNA is mainly distributed in the coding sequences (Delatte et al., 2016). Methylations in adenine residues (6 mA) of DNA and messenger RNA in eukaryotes have been described (Zhang et al., 2015; Dominissini et al., 2016).
DNA cytosine methylation functions to repress gene expression at the promoter regions in vertebrates. In addition, intragenic methylation of cytosine may modulate alternative promoter use (Maunakea et al., 2010) and alternative exon inclusion (Shukla et al., 2011; Yearim et al., 2015). In insects, cytosine methylation is sparse and is found mainly within the gene body (exons and introns), which is conserved between animals and plants (Ehrlich et al., 1982; Dolinoy et al., 2006; Zemach et al., 2010). Great interest in social evolution spurred rapid developments in genome research, and cytosine methylation was soon discovered in social bees, ants, termites, and aphids (Hunt et al., 2010; Smith et al., 2011; Suen et al., 2011; Wurm et al., 2011; Bonasio et al., 2012; Glastad et al., 2013; Kocher et al., 2013; Amarasinghe et al., 2014; Terrapon et al., 2014; Kapheim et al., 2015). Social genomics created unique opportunities for decoding the molecular bases of social evolution and behavioral plasticity.
Empirical evidence has shown that evolutionary conservation of DNA methylation plays a role in reproductive caste determination in honey bees (Kucharski et al., 2008; Lyko et al., 2010; Foret et al., 2012), bumble bees (Amarasinghe et al., 2014), and ants (Bonasio et al., 2012; Libbrecht et al., 2016). Studies of social epigenetics and socioepigenomics and their roles in behavioral ecology, behavioral genetics, and evolution have been reviewed extensively in the literature (Lyko and Maleszka, 2011; Drewell et al., 2012, 2014; Patalano et al., 2012; Weiner and Toth, 2012; Libbrecht et al., 2013; Dolezal and Toth, 2014; Duncan et al., 2014; Herb, 2014; Maleszka, 2014; Welch and Lister, 2014; Yan et al., 2014, 2015; Bonasio, 2015; Breiling and Lyko, 2015; Isles, 2015; Meloni, 2015; Mukherjee et al., 2015; Rehan and Toth, 2015; Ruden et al., 2015; Wang and Li-Byarlay, 2015; Glastad et al., 2015a).
The first breakthrough in determining the function of DNA methylation was the discovery that silencing DNA methyltransferase 3 (DNMT3) changes the larval developmental outcome from worker phenotype to queen-like phenotype (Kucharski et al., 2008). The provision of royal jelly to the brood is the key environmental factor determining the queen–worker caste difference (Kamakura, 2011; Wang and Li-Byarlay, 2015). Also, a dietary phytochemical may alter caste-related gene expression (Mao et al., 2015). Since this breakthrough, multiple studies have investigated the role of DNA methylation or other epigenetic marks in the queen–worker caste determination of honey bees (Lyko et al., 2010; Spannhoff et al., 2011; Foret et al., 2012; Herb et al., 2012).
There are controversies in whether DNA cytosine methylation pattern is different between queen and workers of honeybees. Lyko et al. (2010) claimed that 561 differentially methylated genes (DMGs) could be identified between adult queen and worker brains, and (Foret et al., 2012) reported 2399 DMGs between the larvae of queens and workers. Studies by Shi et al. (2013) found even more DMGs between queen and worker larvae. However, Herb et al. (2012) identified no DMGs between queen and worker adult honey bees. A recent study from Libbrecht et al. (2016) added more thoughts on the statistical analysis of methylomic experiments.
On the basis of previous studies, several factors should be considered when comparing genomic, transcriptomic, and methylomic studies. First, the experiments of next-generation sequencing studies should be carefully designed. Research budget limitations may compromise the replication of sequencing in “omics” experimental designs (Todd et al., 2016); however, field-related experiments in ecological and evolutionary studies often require large sample sizes to obtain reliable power in statistical analyses. A recent investigation of clonal ants (Cerapachys biroi) with four biological replicates per condition indicated no DMGs in the brain tissues between ants in the reproductive phase and those in the brood-care phase (Libbrecht et al., 2016).
Second, the developmental age and tissue of the samples collected are different. DNA cytosine methylation may display different temporal patterns and dynamics across different developmental stages and tissues, according to mammalian epigenetic studies (Heyn et al., 2012; Numata et al., 2012). In studies by Lyko et al. (2010), Herb et al. (2012) and Shi et al. (2013), the ages of queens and workers differed from those in previous studies, so the later results might not be comparable with earlier work.
Third, the technology implemented to detect genome-wide DNA methylation marks are different, including whole-genome bisulfite sequencing (Li-Byarlay et al., 2013), methylated DNA immunoprecipitation (Mohn et al., 2009), pyrosequencing of bisulfite-treated DNA (Tost and Gut, 2007), methyl-cytosine phosphate guanine (CpG)-binding domain sequencing (Decock et al., 2016), or methylation-sensitive amplified fragment length polymorphism (Xu et al., 2000).
Last, genotypic/alleleic differences between samples that may result in cis-mediated allele specific methylation or epialleles should be considered. Recently, evidence revealed that sequence variation can generate differential methylation in social insects (Maleszka, 2016; Remnant et al., 2016; Wedd et al., 2016). The change of DNA methylation particularly in genetically heterogeneous samples can be influenced where epialleles differ in their methylation status. In such cases it will be very difficult to identify whether methylation is due to a different developmental or behavioral phenomenon, or simply due to sequence variation that results in a change in methylation. Therefore, genetic influences on methylation need to be considered in future studies reporting differential methylation due to behavior or environment. These factors should be addressed in further studies. Standardized and better methods in the future should leverage the power of sequencing and genome-wide analysis to provide a comprehensive view of methylation patterns at single-base resolution across the genome.
Distinctive differences are also found within the worker caste of eusocial hymenoptera. For example, differences in age and morphology can be observed in some ant species (Hölldobler and Wilson, 1990). DNA cytosine methylation is thought to regulate the phenotypic plasticity of size in the carpenter ant (Alvarado et al., 2015). In a honey bee colony, an age-related division of labor is displayed among young nurse worker bees doing in-hive tasks, which mature to become foragers worker bees doing food collection out of the hives. In addition, the forager bee are flexible and can revert to nurse bee behavior when the original nurse bees are removed from the colony. Herb et al. (2012) showed that cytosine methylation marks changed when foragers reverted to performing nurse bee tasks. This study was also the first to link a behavioral phenotype with a change in epigenetic marks in social insects. These studies indicated that DNA cytosine methylation plays key roles in caste determination and the division of labor in social insects.
The methylation pattern changes through different developmental stages in bees and ants. A study by Drewell et al. (2014) indicated that cytosine methylation was higher in the egg than in the sperm of honey bees, and methylation in both the egg and sperm was higher than in adult drones. The larval stage may have a higher level of methylation than the adult stage (Shi et al., 2013). These studies showed a dynamic cycle of methylation throughout the development of the honey bee, which is important for studying transgenerational effects on imprinted genes. In the eggs of Camponotus floridanus and Harpegnathos saltator ants, the raw number of methylated cytosines was much higher than in honey bees (Bonasio et al., 2012). The pattern of methylation is also revealed in the embryo, larvae, and adult stages (Bonasio et al., 2012). In studies of Pogonomyrmex barbatus ants (Smith et al., 2012), using restriction enzyme-based methods, found that virgin queens had higher levels of DNA methylation than did workers. Also, cytosine methylation and the epigenome change in the brain respond to a complicated social environment (Lockett et al., 2012). These reports suggest that DNA methylation may be a conserved molecular mechanism that regulates the development of social bees and ants.
Not all social insects display equal levels of DNA cytosine methylation. In primitively eusocial wasps, such as Polistes dominula and Polistes canadensis, recent studies have indicated even lower genome-wide methylation than in bees and ants and a lack of the key enzyme DNMT3 (Patalano et al., 2015; Standage et al., 2016), even though previous reports indicated potential cytosine methylation (Kronforst et al., 2008; Weiner et al., 2013). This unexpected phenomenon suggests that DNA cytosine methylation may reprogram gene expression and molecular evolution in social wasps in a different way than in other social insects.
In contrast to the high level of DNA cytosine methylation in vertebrates, genome-wide DNA cytosine methylation in social insects is low (Zemach et al., 2010). A vertebrate study (Bird, 1985) showed that a cytosine-guanine (CG) dinucleotide island occurred in the genome because of global or near-global methylation. Social insects lack a high level of global methylation; therefore, CG islands were not expected and might not have been observed. DNA methylation functions to repress gene expression at the CG sites in plant and mammal gene promoters (Law and Jacobsen, 2010). The distribution pattern of methylation in social insect genomes is not as great in the promoter regions as in the gene bodies, which is a more conserved pattern among animals and plants (Ball et al., 2009; Zemach et al., 2010; Feng S. et al., 2010; Glastad et al., 2011; Jones, 2012).
Further studies have revealed that cytosine methylation, plays a key role in gene regulation via alternative splicing (Lyko et al., 2010; Cingolani et al., 2013; Li-Byarlay et al., 2013). Molecular mechanistic evidence from in vitro molecular and cellular experiments demonstrated a plausible mechanism, namely, that cytosine methylation inhibits a DNA-binding protein (CCCTC-binding factor) from binding on the target exon, which leads to the exon being excluded or skipped during the co-transcription process (Shukla et al., 2011). As another possible mechanism, DNA methylation regulated alternative splicing through the heterochromatin protein 1 system and the histone of H3k9me3 in in vitro studies (Yearim et al., 2015). Furthermore, in the exon-skipping pattern of honey bees, less cytosine methylation tended to result in the marked exon being excluded or skipped (Figure 2A). Besides exon skipping, cytosine methylation affects intron retention events. If an intron has more cytosine methylation marks, that intron tends to be retained after splicing (Figure 2B). Both exon skipping and intron retention are examples of complex alternative splicing in the social insect genome (Li-Byarlay et al., 2013).
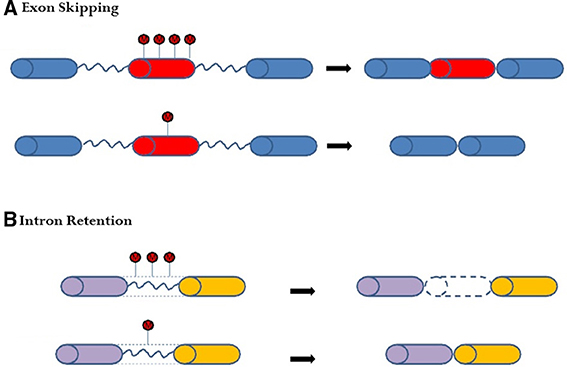
Figure 2. Changes in DNA cytosine methylation on the gene body may lead to alternative splicing in honey bees. (A) In exon-skipping events, less methylation in the middle exon (in red) of the dnmt3 knockdown genome may lead to skipping. (B) In intron-retention events, less methylation on the intron (blue line) may lead to intron exclusion in the dnmt3 knockdown genome. More methylation on the intron may lead to a retained intron in the matured messenger RNA in the control genome.
DNA cytosine methylation could affect alternative gene splicing and might depend on the guanine-cytosine (GC) architecture of the intron–exon structure (Gelfman et al., 2013). Evidence gathered thus far has shown that DNA cytosine methylation could play a different role in co-transcriptional alternative splicing in social insects compared with human cells.
Cytosine methylation is also linked to chromatin states, which affect the coding sequence evolution in social insects (Glastad et al., 2015b). Previous studies have shown that DNA methylation is mirrored by histone modifications in insect genomes (Nanty et al., 2011; Hunt et al., 2013; Glastad et al., 2015c; Yearim et al., 2015). In addition, the transcription factors binding profiles can regulate gene expression by predicting the histone modifications (Benveniste et al., 2014). Therefore, interactions may occur between transcription factors and the epigenetic machinery.
For many years the honeybee has been used as model organism for studying social behavior, social interactions in a complex community, and the process of learning (Von Frisch, 1967; Menzel and Muller, 1996). DNA cytosine methylation is critical for learning and memory, as demonstrated by several animal models in vertebrates (Levenson et al., 2006; Miller and Sweatt, 2007; Miller et al., 2008; Feng J. et al., 2010; Zovkic et al., 2013; Biergans et al., 2015). The first report to show that cytosine methylation plays a key role in the learning and memory processes of honey bees appeared in 2010. Lockett et al. (2010) found that inhibiting DNMT3 decreases the process of storage in memory retention, changes the extinction depending on the treatment time, and induces dnmt3 gene expression after training. In addition, we know that in bees, cytosine methylation is involved in associative long-term memory formation but is not required for short-term memory formation (Biergans et al., 2012). Biergans et al. (2015) also found that dnmt3 gene expression increased after topical treatments of DNMT inhibitors. In addition to gene expression, levels of cytosine methylation in memory-associated genes reveal the molecular basis of memory formation and maintenance.
Researchers are building on current and previous knowledge in the field of epigenetics in social insects, and faster development in the genome and epigenome of social insects has never occurred. Because of their disparate, alternative, environmentally inducible phenotypes, social insects are undoubtedly one of the best groups in which to study epigenetic control of gene regulation, behavioral epigenetics, physiological epigenetics, genome evolution, and neurobiology. Changes in epigenetic marks can be used as a proxy to elucidate environmentally driven phenotypic plasticity and genomic imprinting. Indeed, recent genomic analyses of imprinted genes have opened the door for studies on the potential roles of epigenetics in social insects (Gibson et al., 2015; Kocher et al., 2015; Galbraith et al., 2016).
Balanced experimental design in biological replicates are crucial. When next-generation sequencing experiments are designed, three or more biological replicates should be considered in order to have reproducible research outcomes and sufficient power for statistical analyses. Given the cost of sequencing technology and the cost of library preparation are dropped, the trend for the future will be to have more biological replicates in an experimental design.
The future scientific goal for studies of DNA methylation and other chemical modifications will be to capitalize on recent insights into the molecular and genomic mechanisms operating in behavioral plasticity to create social evolution. More molecular experimental manipulations, such as genome editing, should be used to elucidate the molecular basis of social behavior and the biochemical components underlying the ecological and environmental conditions. By taking advantage of collections of behavioral, ecological, and evolutionary phenotypes, we will be able to use social insect models to study innovative hypotheses in social epigenetics and neuroepigenetics.
The author confirms being the sole contributor of this work and approved it for publication.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
I would like to thank Drs. Karl Glastad, Fred Gould, Brendan Hunt, Parry Kietzman, Margarita Lopez-Uribe for their critical comments to improve the manuscript. HL was supported by a National Research Council Research Associateship, and the W. M. Keck Center for Behavioral Biology at North Carolina State University.
Alvarado, S., Rajakumar, R., Abouheif, E., and Szyf, M. (2015). Epigenetic variation in the Egfr gene generates quantitative variation in a complex trait in ants. Nat. Commun. 6, 6513. doi: 10.1038/ncomms7513
Amarasinghe, H. E., Clayton, C. I., and Mallon, E. B. (2014). Methylation and worker reproduction in the bumble-bee (Bombus terrestris). Proc. Biol. Sci. 281:20132502. doi: 10.1098/rspb.2013.2502
Ball, M. P., Li, J. B., Gao, Y., Lee, J. H., Leproust, E. M., Park, I. H., et al. (2009). Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat. Biotechnol. 27, 361–368. doi: 10.1038/nbt.1533
Benveniste, D., Sonntag, H.-J., Sanguinetti, G., and Sproul, D. (2014). Transcription factor binding predicts histone modifications in human cell lines. Proc. Natl. Acad. Sci. U.S.A. 111, 13367–13372. doi: 10.1073/pnas.1412081111
Biergans, S. D., Galizia, C. G., Reinhard, J., and Claudianos, C. (2015). Dnmts and Tet target memory-associated genes after appetitive olfactory training in honey bees. Sci. Rep. 5:16223. doi: 10.1038/srep16223
Biergans, S. D., Jones, J. C., Treiber, N., Galizia, C. G., and Szyszka, P. (2012). DNA methylation mediates the discriminatory power of associative long-term memory in honeybees. PLoS ONE 7:e39349. doi: 10.1371/journal.pone.0039349
Bird, A. P. (1985). CpG-rich islands and the function of DNA methylation. Nature 321, 209–213. doi: 10.1038/321209a0
Bonasio, R. (2015). The expanding epigenetic landscape of non-model organisms. J. Exp. Biol. 218, 114–122. doi: 10.1242/jeb.110809
Bonasio, R., Li, Q., Lian, J., Mutti, N. S., Jin, L., Zhao, H., et al. (2012). Genome-wide and caste-specific DNA methylomes of the ants Camponotus floridanus and Harpegnathos saltator. Curr. Biol. 22, 1755–1764. doi: 10.1016/j.cub.2012.07.042
Breiling, A., and Lyko, F. (2015). Epigenetic regulatory functions of DNA modifications: 5-methylcytosine and beyond. Epigenet. Chromatin 8, 1. doi: 10.1186/s13072-015-0016-6
Cingolani, P., Cao, X. Y., Khetani, R. S., Chen, C. C., Coon, M., Sammak, A., et al. (2013). Intronic non-CG DNA hydroxymethylation and alternative mRNA splicing in honey bees. BMC Genomics 14:666. doi: 10.1186/1471-2164-14-666
Decock, A., Ongenaert, M., Van Criekinge, W., Speleman, F., and Vandesompele, J. (2016). DNA methylation profiling of primary neuroblastoma tumors using methyl-CpG-binding domain sequencing. Sci. Data 3:160004. doi: 10.1038/sdata.2016.4
Delatte, B., Wang, F., Ngoc, L. V., Collignon, E., Bonvin, E., Deplus, R., et al. (2016). Transcriptome-wide distribution and function of RNA hydroxymethylcytosine. Science 351, 282–285. doi: 10.1126/science.aac5253
Dolezal, A. G., and Toth, A. L. (2014). Honey bee sociogenomics: a genome-scale perspective on bee social behavior and health. Apidologie 45, 375–395. doi: 10.1007/s13592-013-0251-4
Dolinoy, D. C., Weidman, J. R., Waterland, R. A., and Jirtle, R. L. (2006). Maternal genistein alters coat color and protects Avy mouse offspring from obesity by modifying the fetal epigenome. Environ. Health Perspect. 114, 567–572. doi: 10.1289/ehp.8700
Dominissini, D., Nachtergaele, S., Moshitch-Moshkovitz, S., Peer, E., Kol, N., Ben-Haim, M. S., et al. (2016). The dynamic N1-methyladenosine methylome in eukaryotic messenger RNA. Nature 530, 441–446. doi: 10.1038/nature16998
Drewell, R. A., Bush, E. C., Remnant, E. J., Wong, G. T., Beeler, S. M., Stringham, J. L., et al. (2014). The dynamic DNA methylation cycle from egg to sperm in the honey bee Apis mellifera. Development 141, 2702–2711. doi: 10.1242/dev.110163
Drewell, R. A., Lo, N., Oxley, P. R., and Oldroyd, B. P. (2012). Kin conflict in insect societies: a new epigenetic perspective. Trends Ecol. Evol. 27, 367–373. doi: 10.1016/j.tree.2012.02.005
Duncan, E. J., Gluckman, P. D., and Dearden, P. K. (2014). Epigenetics, plasticity, and evolution: how do we link epigenetic change to phenotype? J. Exp. Zool. B Mol. Dev. Evol. 322, 208–220. doi: 10.1002/jez.b.22571
Dunham, I., Kundaje, A., Aldred, S. F., Collins, P. J., Davis, C., Doyle, F., et al. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. doi: 10.1038/nature11247
Ehrlich, M., Gama-Sosa, M. A., Huang, L.-H., Midgett, R. M., Kuo, K. C., Mccune, R. A., et al. (1982). Amount and distribution of 5-methylcytosine in human DNA from different types of tissues or cells. Nucleic Acids Res. 10, 2709–2721. doi: 10.1093/nar/10.8.2709
Evans, J. D., and Wheeler, D. E. (2000). Expression profiles during honeybee caste determination. Genome Biol. 2:research0001. doi: 10.1186/gb-2000-2-1-research0001
Feng, J., Zhou, Y., Campbell, S. L., Le, T., Li, E., Sweatt, J. D., et al. (2010). Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 13, 423–430. doi: 10.1038/nn.2514
Feng, S., Cokus, S. J., Zhang, X., Chen, P.-Y., Bostick, M., Goll, M. G., et al. (2010). Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. U.S.A. 107, 8689–8694. doi: 10.1073/pnas.1002720107
Foret, S., Kucharski, R., Pellegrini, M., Feng, S., Jacobsen, S. E., Robinson, G. E., et al. (2012). DNA methylation dynamics, metabolic fluxes, gene splicing, and alternative phenotypes in honey bees. Proc. Natl. Acad. Sci. U.S.A. 109, 4968–4973. doi: 10.1073/pnas.1202392109
Fu, L., Guerrero, C. R., Zhong, N., Amato, N. J., Liu, Y., Liu, S., et al. (2014). Tet-mediated formation of 5-hydroxymethylcytosine in RNA. J. Am. Chem. Soc. 136, 11582–11585. doi: 10.1021/ja505305z
Galbraith, D. A., Kocher, S. D., Glenn, T., Albert, I., Hunt, G. J., Strassmann, J. E., et al. (2016). Testing the kinship theory of intragenomic conflict in honey bees (Apis mellifera). Proc. Natl. Acad. Sci. U.S.A. 113, 1020–1025. doi: 10.1073/pnas.1516636113
Gelfman, S., Cohen, N., Yearim, A., and Ast, G. (2013). DNA-methylation effect on cotranscriptional splicing is dependent on GC architecture of the exon-intron structure. Genome Res. 23, 789–799. doi: 10.1101/gr.143503.112
Gibson, J. D., Arechavaleta-Velasco, M. E., Tsuruda, J. M., and Hunt, G. J. (2015). Biased allele expression and aggression in hybrid honeybees may be influenced by inappropriate nuclear-cytoplasmic signaling. Front Genet. 6:343. doi: 10.3389/fgene.2015.00343
Glastad, K. M., Chau, L. M., and Goodisman, M. A. D. (2015a). Chapter seven-epigenetics in social insects. Adv. Insect Phys. 48, 227–269. doi: 10.1016/bs.aiip.2014.12.007
Glastad, K. M., Goodisman, M. A., Yi, S. V., and Hunt, B. G. (2015b). Effects of DNA methylation and chromatin state on rates of molecular evolution in Insects. G3 (Bethesda) 6, 357–363. doi: 10.1534/g3.115.023499
Glastad, K. M., Hunt, B. G., and Goodisman, M. A. (2015c). DNA methylation and chromatin organization in insects: insights from the ant Camponotus floridanus. Genome Biol. Evol. 7, 931–942. doi: 10.1093/gbe/evv039
Glastad, K. M., Hunt, B. G., and Goodisman, M. A. D. (2013). Evidence of a conserved functional role for DNA methylation in termites. Insect Mol. Biol. 22, 143–154. doi: 10.1111/imb.12010
Glastad, K. M., Hunt, B. G., Yi, S. V., and Goodisman, M. A. D. (2011). DNA methylation in insects: on the brink of the epigenomic era. Insect Mol. Biol. 20, 553–565. doi: 10.1111/j.1365-2583.2011.01092.x
Goll, M. G., and Bestor, T. H. (2005). Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 74, 481–514. doi: 10.1146/annurev.biochem.74.010904.153721
Goll, M. G., Kirpekar, F., Maggert, K. A., Yoder, J. A., Hsieh, C. L., Zhang, X. Y., et al. (2006). Methylation of tRNA(AsP) by the DNA methyltransferase homolog Dnmt2. Science 311, 395–398. doi: 10.1126/science.1120976
Hartfelder, K., Guidugli-Lazzarini, K. R., Cervoni, M. S., Santos, D. E., and Humann, F. C. (2015). Chapter one-old threads make new tapestry—rewiring of signalling pathways underlies caste phenotypic plasticity in the honey bee, Apis mellifera L. Adv. Insect Phys. 48, 1–36. doi: 10.1016/bs.aiip.2014.12.001
Herb, B. R. (2014). Epigenetics as an answer to Darwin's “special difficulty.” Front. Genet 5:321. doi: 10.3389/fgene.2014.00321
Herb, B. R., Wolschin, F., Hansen, K. D., Aryee, M. J., Langmead, B., Irizarry, R., et al. (2012). Reversible switching between epigenetic states in honeybee behavioral subcastes. Nat. Neurosci. 15, 1371–1373. doi: 10.1038/nn.3218
Heyn, H., Li, N., Ferreira, H. J., Moran, S., Pisano, D. G., Gomez, A., et al. (2012). Distinct DNA methylomes of newborns and centenarians. Proc. Natl. Acad. Sci. U.S.A. 109, 10522–10527. doi: 10.1073/pnas.1120658109
Hölldobler, B., and Wilson, E. O. (1990). The Ants. Harvard University Press. doi: 10.1007/978-3-662-10306-7
Hölldobler, B., and Wilson, E. O. (2009). The Superorganism: the Beauty, Elegance, and Strangeness of Insect Societies. WW Norton & Company.
Hunt, B. G., Brisson, J. A., Yi, S. V., and Goodisman, M. A. D. (2010). Functional conservation of DNA methylation in the pea aphid and the honeybee. Genome Biol. Evol. 2, 719–728. doi: 10.1093/gbe/evq057
Hunt, B. G., Glastad, K. M., Yi, S. V., and Goodisman, M. A. (2013). Patterning and regulatory associations of DNA methylation are mirrored by histone modifications in insects. Genome Biol. Evol. 5, 591–598. doi: 10.1093/gbe/evt030
Isles, A. (2015). Neural and behavioral epigenetics; what it is, and what is hype. Genes, Brain Behav. 14, 64–72. doi: 10.1111/gbb.12184
Jaenisch, R., and Bird, A. (2003). Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet. 33, 245–254. doi: 10.1038/ng1089
Jirtle, R. L., and Skinner, M. K. (2007). Environmental epigenomics and disease susceptibility. Nat. Rev. Genet. 8, 253–262. doi: 10.1038/nrg2045
Jones, P. A. (2012). Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 13, 484–492. doi: 10.1038/nrg3230
Kamakura, M. (2011). Royalactin induces queen differentiation in honeybees. Nature 473, 478–483. doi: 10.1038/nature10093
Kapheim, K. M., Pan, H. L., Li, C., Salzberg, S. L., Puiu, D., Magoc, T., et al. (2015). Genomic signatures of evolutionary transitions from solitary to group living. Science 348, 1139–1143. doi: 10.1126/science.aaa4788
Kocher, S. D., Li, C., Yang, W., Tan, H., Yi, S. V., Yang, X. Y., et al. (2013). The draft genome of a socially polymorphic halictid bee, Lasioglossum albipes. Genome Biol. 14:R142. doi: 10.1186/gb-2013-14-12-r142
Kocher, S. D., Tsuruda, J. M., Gibson, J. D., Emore, C. M., Arechavaleta-Velasco, M. E., Queller, D. C., et al. (2015). A search for parent-of-origin effects on honey bee gene expression. G3 (Bethesda) 5, 1657–1662. doi: 10.1534/g3.115.017814
Kohli, R. M., and Zhang, Y. (2013). TET enzymes, TDG and the dynamics of DNA demethylation. Nature 502, 472–479. doi: 10.1038/nature12750
Kronforst, M. R., Gilley, D. C., Strassmann, J. E., and Queller, D. C. (2008). DNA methylation is widespread across social Hymenoptera. Curr. Biol. 18, R287–R288. doi: 10.1016/j.cub.2008.02.015
Kucharski, R., Maleszka, J., Foret, S., and Maleszka, R. (2008). Nutritional control of reproductive status in honeybees via DNA methylation. Science 319, 1827–1830. doi: 10.1126/science.1153069
Law, J. A., and Jacobsen, S. E. (2010). Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 11, 204–220. doi: 10.1038/nrg2719
Levenson, J. M., Roth, T. L., Lubin, F. D., Miller, C. A., Huang, I.-C., Desai, P., et al. (2006). Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J. Biol. Chem. 281, 15763–15773. doi: 10.1074/jbc.M511767200
Libbrecht, R., Oxley, P. R., Keller, L., and Kronauer, D. J. C. (2016). Robust DNA Methylation in the Clonal Raider Ant Brain. Curr. Biol. 26, 391–395. doi: 10.1016/j.cub.2015.12.040
Libbrecht, R., Oxley, P. R., Kronauer, D., and Keller, L. (2013). Ant genomics sheds light on the molecular regulation of social organization. Genome Biol. 14:212. doi: 10.1186/gb-2013-14-7-212
Li-Byarlay, H., Li, Y., Stroud, H., Feng, S., Newman, T. C., Hou, K. K., et al. (2013). RNA interference knockdown of DNA methyl-transferase 3 affects gene alternative splicing in the honey bee. Proc. Natl. Acad. Sci. U.S.A. 110, 12750. doi: 10.1073/pnas.1310735110
Liu, L., Li, Y., and Tollefsbol, T. O. (2008). Gene-environment interactions and epigenetic basis of human diseases. Curr. Issues Mol. Biol. 10, 25.
Lockett, G. A., Helliwell, P., and Maleszka, R. (2010). Involvement of DNA methylation in memory processing in the honey bee. Neuroreport 21, 812–816. doi: 10.1097/WNR.0b013e32833ce5be
Lockett, G., Kucharski, R., and Maleszka, R. (2012). DNA methylation changes elicited by social stimuli in the brains of worker honey bees. Genes Brain Behav. 11, 235–242. doi: 10.1111/j.1601-183X.2011.00751.x
Lyko, F., Foret, S., Kucharski, R., Wolf, S., Falckenhayn, C., and Maleszka, R. (2010). The honey bee epigenomes: differential methylation of brain DNA in queens and workers. PLoS Biol. 8:e1000506. doi: 10.1371/journal.pbio.1000506
Lyko, F., and Maleszka, R. (2011). Insects as innovative models for functional studies of DNA methylation. Trends Genet. TIG 27, 127–131. doi: 10.1016/j.tig.2011.01.003
Maleszka, R. (2014). The social honey bee in biomedical research: realities and expectations. Drug Discov. Today Dis. Models 12, 7–13. doi: 10.1016/j.ddmod.2014.06.001
Maleszka, R. (2016). Epigenetic code and insect behavioural plasticity. Curr. Opin. Insect Sci. 15, 45–52. doi: 10.1016/j.cois.2016.03.003
Mao, W., Schuler, M. A., and Berenbaum, M. R. (2015). A dietary phytochemical alters caste-associated gene expression in honey bees. Sci. Adv. 1:e1500795. doi: 10.1126/sciadv.1500795
Marbaniang, C. N., and Vogel, J. (2016). Emerging roles of RNA modifications in bacteria. Curr. Opin. Microbiol. 30, 50–57. doi: 10.1016/j.mib.2016.01.001
Maunakea, A. K., Nagarajan, R. P., Bilenky, M., Ballinger, T. J., D'souza, C., Fouse, S. D., et al. (2010). Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466, 253–257. doi: 10.1038/nature09165
Meloni, M. (2015). Epigenetics for the social sciences: justice, embodiment, and inheritance in the postgenomic age. New Genet. Soc. 34, 125–151. doi: 10.1080/14636778.2015.1034850
Menzel, R., and Muller, U. (1996). Learning and memory in honeybees: from behavior to neural substrates. Annu. Rev. Neurosci. 19, 379–404. doi: 10.1146/annurev.ne.19.030196.002115
Miller, C. A., Campbell, S. L., and Sweatt, J. D. (2008). DNA methylation and histone acetylation work in concert to regulate memory formation and synaptic plasticity. Neurobiol. Learn. Mem. 89, 599–603. doi: 10.1016/j.nlm.2007.07.016
Miller, C. A., and Sweatt, J. D. (2007). Covalent modification of DNA regulates memory formation. Neuron 53, 857–869. doi: 10.1016/j.neuron.2007.02.022
Miller, G. (2010). The seductive allure of behavioral epigenetics. Science 329, 24–27. doi: 10.1126/science.329.5987.24
Moczek, A. P. (2010). Phenotypic plasticity and diversity in insects. Philos. Trans. R. Soc. B Biol. Sci. 365, 593–603. doi: 10.1098/rstb.2009.0263
Mohn, F., Weber, M., Schübeler, D., and Roloff, T.-C. (2009). Methylated DNA immunoprecipitation (medip). DNA Methyl. Methods Protoc. 507, 55–64. doi: 10.1007/978-1-59745-522-0_5
Mukherjee, K., Twyman, R. M., and Vilcinskas, A. (2015). Insects as models to study the epigenetic basis of disease. Prog. Biophys. Mol. Biol. 118, 69–78. doi: 10.1016/j.pbiomolbio.2015.02.009
Nanty, L., Carbajosa, G., Heap, G. A., Ratnieks, F., Van Heel, D. A., Down, T. A., et al. (2011). Comparative methylomics reveals gene-body H3K36me3 in Drosophila predicts DNA methylation and CpG landscapes in other invertebrates. Genome Res. 21, 1841–1850. doi: 10.1101/gr.121640.111
Negre, N., Brown, C. D., Ma, L. J., Bristow, C. A., Miller, S. W., Wagner, U., et al. (2011). A cis-regulatory map of the Drosophila genome. Nature 471, 527–531. doi: 10.1038/nature09990
Numata, S., Ye, T., Hyde, T. M., Guitart-Navarro, X., Tao, R., Wininger, M., et al. (2012). DNA methylation signatures in development and aging of the human prefrontal cortex. Am. J. Hum. Genet. 90, 260–272. doi: 10.1016/j.ajhg.2011.12.020
Patalano, S., Hore, T. A., Reik, W., and Sumner, S. (2012). Shifting behaviour: epigenetic reprogramming in eusocial insects. Curr. Opin. Cell Biol. 24, 367–373. doi: 10.1016/j.ceb.2012.02.005
Patalano, S., Vlasova, A., Wyatt, C., Ewels, P., Camara, F., Ferreirab, P. G., et al. (2015). Molecular signatures of plastic phenotypes in two eusocial insect species with simple societies. Proc. Natl. Acad. Sci. U.S.A. 112, 13970–13975. doi: 10.1073/pnas.1515937112
Rasmussen, E. M., and Amdam, G. V. (2015). Cytosine modifications in the honey bee (Apis mellifera) worker genome. Front. Genet. 6:8. doi: 10.3389/fgene.2015.00008
Rehan, S. M., and Toth, A. L. (2015). Climbing the social ladder: the molecular evolution of sociality. Trends Ecol. Evol. 30, 426–433. doi: 10.1016/j.tree.2015.05.004
Remnant, E. J., Ashe, A., Young, P. E., Buchmann, G., Beekman, M., Allsopp, M. H., et al. (2016). Parent-of-origin effects on genome-wide DNA methylation in the Cape honey bee (Apis mellifera capensis) may be confounded by allele-specific methylation. BMC Genomics 17:226. doi: 10.1186/s12864-016-2506-8
Ruden, D. M., Cingolani, P. E., Sen, A., Qu, W., Wang, L., Senut, M.-C., et al. (2015). Epigenetics as an answer to Darwin's “special difficulty,” Part 2: natural selection of metastable epialleles in honeybee castes. Front. Genet. 6:60. doi: 10.3389/fgene.2015.00060
Shi, Y. Y., Yan, W. Y., Huang, Z. Y., Wang, Z. L., Wu, X. B., and Zeng, Z. J. (2013). Genomewide analysis indicates that queen larvae have lower methylation levels in the honey bee (Apis mellifera). Naturwissenschaften 100, 193–197. doi: 10.1007/s00114-012-1004-3
Shukla, S., Kavak, E., Gregory, M., Imashimizu, M., Shutinoski, B., Kashlev, M., et al. (2011). CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 479, 74–79. doi: 10.1038/nature10442
Smith, C. D., Zimin, A., Holt, C., Abouheif, E., Benton, R., Cash, E., et al. (2011). Draft genome of the globally widespread and invasive Argentine ant (Linepithema humile). Proc. Natl. Acad. Sci. U.S.A. 108, 5673–5678. doi: 10.1073/pnas.1008617108
Smith, C. R., Mutti, N. S., Jasper, W. C., Naidu, A., Smith, C. D., and Gadau, J. (2012). Patterns of DNA Methylation in development, division of labor and hybridization in an ant with genetic caste determination. PLoS ONE 7:e42433. doi: 10.1371/journal.pone.0042433
Spannhoff, A., Kim, Y. K., Raynal, N. J. M., Gharibyan, V., Su, M. B., Zhou, Y. Y., et al. (2011). Histone deacetylase inhibitor activity in royal jelly might facilitate caste switching in bees. EMBO Rep. 12, 238–243. doi: 10.1038/embor.2011.9
Standage, D., Berens, A., Glastad, K., Severin, A., Brendel, V., and Toth, A. (2016). Genome, transcriptome, and methylome sequencing of a primitively eusocial wasp reveal a greatly reduced DNA methylation system in a social insect. Mol. Ecol. 25, 1769–1784. doi: 10.1111/mec.13578
Suen, G., Teiling, C., Li, L., Holt, C., Abouheif, E., Bornberg-Bauer, E., et al. (2011). The Genome sequence of the leaf-cutter ant Atta cephalotes reveals insights into its obligate symbiotic lifestyle. PLoS Genet. 7:e1002007. doi: 10.1371/journal.pgen.1002007
Terrapon, N., Li, C., Robertson, H. M., Ji, L., Meng, X., Booth, W., et al. (2014). Molecular traces of alternative social organization in a termite genome. Nat. Commun. 5:3636. doi: 10.1038/ncomms4636
Todd, E. V., Black, M. A., and Gemmell, N. J. (2016). The power and promise of RNA-seq in ecology and evolution. Mol. Ecol. 25, 1224–1241. doi: 10.1111/mec.13526
Tost, J., and Gut, I. G. (2007). DNA methylation analysis by pyrosequencing. Nat. Protoc. 2, 2265–2275. doi: 10.1038/nprot.2007.314
Von Frisch, K. (1967). The Dance Language and Orientation of Bees. Cambridge, MA: Harvard University Press.
Wang, X., Fang, X., Yang, P., Jiang, X., Jiang, F., Zhao, D., et al. (2014). The locust genome provides insight into swarm formation and long-distance flight. Nat. Commun. 5:2957. doi: 10.1038/ncomms3957
Wang, Y., Jorda, M., Jones, P. L., Maleszka, R., Ling, X., Robertson, H. M., et al. (2006). Functional CpG methylation system in a social insect. Science 314, 645–647. doi: 10.1126/science.1135213
Wang, Y., and Li-Byarlay, H. (2015). Physiological and molecular mechanisms of nutrition in Honey Bees. Adv. Insect Phys. 49, 25–58. doi: 10.1016/bs.aiip.2015.06.002
Waterland, R. A., and Jirtle, R. L. (2003). Transposable elements: targets for early nutritional effects on epigenetic gene regulation. Mol. Cell. Biol. 23, 5293–5300. doi: 10.1128/MCB.23.15.5293-5300.2003
Wedd, L., Kucharski, R., and Maleszka, R. (2016). Differentially methylated obligatory epialleles modulate context-dependent LAM gene expression in the honey bee Apis mellifera. Epigenetics 11, 1–10. doi: 10.1080/15592294.2015.1107695
Weiner, S. A., Galbraith, D. A., Adams, D. C., Valenzuela, N., Noll, F. B., Grozinger, C. M., et al. (2013). A survey of DNA methylation across social insect species, life stages, and castes reveals abundant and caste-associated methylation in a primitively social wasp. Naturwissenschaften 100, 795–799. doi: 10.1007/s00114-013-1064-z
Weiner, S. A., and Toth, A. L. (2012). Epigenetics in social insects: a new direction for understanding the evolution of castes. Genet. Res. Int. 2012:609810. doi: 10.1155/2012/609810
Welch, M., and Lister, R. (2014). Epigenomics and the control of fate, form and function in social insects. Curr. Opin. Insect Sci. 1, 31–38. doi: 10.1016/j.cois.2014.04.005
Wilson, E. O. (1979). The evolution of caste systems in social insects. Proc. Am. Philos. Soc. 123, 204–210.
Wojciechowski, M., Rafalski, D., Kucharski, R., Misztal, K., Maleszka, J., Bochtler, M., et al. (2014). Insights into DNA hydroxymethylation in the honeybee from in-depth analyses of TET dioxygenase. Open Biol. 4:140110. doi: 10.1098/rsob.140110
Wolffe, A. P., and Matzke, M. A. (1999). Epigenetics: regulation through repression. Science 286, 481–486. doi: 10.1126/science.286.5439.481
Wurm, Y., Wang, J., Riba-Grognuz, O., Corona, M., Nygaard, S., Hunt, B. G., et al. (2011). The genome of the fire ant Solenopsis invicta. Proc. Natl. Acad. Sci. U.S.A. 108, 5679–5684. doi: 10.1073/pnas.1009690108
Xu, M., Li, X., and Korban, S. S. (2000). AFLP-based detection of DNA methylation. Plant Mol. Biol. Report. 18, 361–368. doi: 10.1007/BF02825064
Yan, H., Bonasio, R., Simola, D. F., Liebig, J., Berger, S. L., and Reinberg, D. (2015). DNA methylation in social insects: how epigenetics can control behavior and longevity. Annu. Rev. Entomol. 60, 435–452. doi: 10.1146/annurev-ento-010814-020803
Yan, H., Simola, D. F., Bonasio, R., Liebig, J., Berger, S. L., and Reinberg, D. (2014). Eusocial insects as emerging models for behavioural epigenetics. Nat. Rev. Genet. 15, 677–688. doi: 10.1038/nrg3787
Yearim, A., Gelfman, S., Shayevitch, R., Melcer, S., Glaich, O., Mallm, J.-P., et al. (2015). HP1 is involved in regulating the global impact of DNA methylation on alternative splicing. Cell Rep. 10, 1122–1134. doi: 10.1016/j.celrep.2015.01.038
Zemach, A., Mcdaniel, I. E., Silva, P., and Zilberman, D. (2010). Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 328, 916–919. doi: 10.1126/science.1186366
Zhang, G. Q., Huang, H., Liu, D., Cheng, Y., Liu, X. L., Zhang, W. X., et al. (2015). N-6-Methyladenine DNA Modification in Drosophila. Cell 161, 893–906. doi: 10.1016/j.cell.2015.04.018
Keywords: epigenetics, epigenomics, behavior, social insects, caste, experiment design
Citation: Li-Byarlay H (2016) The Function of DNA Methylation Marks in Social Insects. Front. Ecol. Evol. 4:57. doi: 10.3389/fevo.2016.00057
Received: 16 March 2016; Accepted: 03 May 2016;
Published: 19 May 2016.
Edited by:
Graham J. Thompson, Western University, CanadaReviewed by:
Michael Goodisman, Georgia Tech, USACopyright © 2016 Li-Byarlay. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongmei Li-Byarlay, aGxpYnlhckBuY3N1LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.