 Caleb D. Phillips
Caleb D. Phillips Robert J. Baker
Robert J. Baker- 1Department of Biological Science, Texas Tech University, Lubbock, TX, USA
- 2Natural Science Research Laboratory, The Museum of Texas Tech University, Lubbock, TX, USA
Regulatory evolution is thought to be fundamental to adaptive evolution. However, the identification of specific regulatory changes responsible for adaptation are sparse. Bats of the family Phyllostomidae, owing to their unparalleled rate of ecological and morphological evolution, represent an ideal system to identify regulatory evolution of adaptive significance. Among ecological niche leaps observed in this family, the most dramatic is the evolution of obligate sanguivory by vampire bats, which due to their highly derived phenotype, the sporadic phylogenetic occurrence of blood-feeding, and the adaptive potential of salivary glands, has enabled the development of hypotheses about adaptive molecular phenotypes. Using comparative transcriptomics of vampire bat, outgroup insectivorous bats, and sanguivorous leeches we identify genes that have been convergently recruited as secretory products of salivary glands of vampire bats and leeches. Comparisons of vampire bat to lineages maintaining the primitive chiropteran condition of insectivory indicated gene recruitment of alternative splice variants, and 5′ exon evolution, as the mechanisms producing secretory expression in vampire bats, but not in the insectivorous bats Macrotus and Myotis. Biochemical functions of hypothesized recruited genes explain adaptive benefits to sanguivory by modulating host hemostasis and neural signaling. It is difficult to identify how complex phenotypic change and rapid ecological transition, such as that observed in vampire bats, evolved over a short evolutionary timescale. Results indicate that regulatory evolution controlling tissue-specific splicing patterns has been important to successful adaptation of this lineage. Future studies that leverage emerging long sequence-read technologies, increased sample sizes, and expression and sequence comparisons across other sanguivore lineages will further elucidate roles of alternative splicing and gene recruitment in the remarkable evolution of sanguivory.
Introduction
Among higher vertebrates obligate blood-feeding has evolved only once, producing three species of New World vampire bats (Baker et al., 2012; subfamily Desmodontinae, family Phyllostomidae). The phylogeny of family Phyllostomidae (Baker et al., 2003; Datzmann et al., 2010; Dumont et al., 2011) establishes that vampire bats diverged from an insectivorous ancestor in a short evolutionary time (~5 my). Transition from obligate insectivory to sanguivory represents an extreme leap in niche space involving modifications to aspects of vampire bat anatomy and physiology including dental, sensory, renal, and secretory anatomy and function (Busch, 1988; Schäfer et al., 1988; Gracheva et al., 2011; Jones et al., 2013; Francishetti et al., 2013; Phillips et al., 2013). Indeed, it is difficult to conceptualize the sequence of adaptations allowing a niche transition from insect foraging to blood-feeding on vertebrate hosts, and the genomic mechanisms that facilitated such a shift in niche space are largely unknown. Recently, a proteomic and transcriptomic investigation was conducted to understand the secretory products in vampire bat saliva (Desmodus rotundus) functioning to support success in this life history strategy (Francishetti et al., 2013). Although that study did not perform comparisons to insectivorous bat lineages, a diversity of vampire bat anticoagulants, vasodilators, anti-inflammatory proteins, neural-disruptors, and antimicrobial agents were identified. These authors postulate a complex series of chemical reactions by which vampire bat saliva modulates prey vascular system by preventing clotting and wound formation, thus enhancing the efficiency of parasitized blood meals.
The model for vampire bat salivary function is an important adaptive component to the successful evolutionary niche leap into sanguivory. An intriguing observation is that among secretory products of the so-called “vampirome,” 17% were unclassified or unknown proteins, leaving the other 83% as known proteins. These known proteins are widely distributed across the tree of life, and have been previously identified as protein products from tissues other than salivary glands. These findings indicate the importance, and perhaps common role, for gene recruitment as a genetic mechanism contributing to the evolution of vampire bats and other examples of niche leaps. In agreement with these observations, it has been hypothesized that gene recruitment has been more common for increasing genomic complexity than has the creation of new genes (Spirov et al., 2012). In the context of venoms specifically, studies of snakes and other sanguivorous lineages have indicated that many proteins providing venom function have been recruited from other biological functions, rather than through de novo gene evolution specific to venom (Fry, 2005). Recruitment to new functions can conceivably arise through a variety of mutational mechanisms, however, the relative contributions of specific mechanisms enabling gene recruitment are lacking.
All animal phyla and many extant gene families originated more than 500 million years ago during or before the Cambrian Explosion, resulting in the distribution of orthologous gene families across most or all animal phyla (Miyata and Suga, 2001). The distribution of orthologs across the tree-of-life has allowed for the evolutionary phenomenon of convergent gene recruitment. Convergent gene recruitment has been identified in specific instances, which are distributed at various taxonomic levels including, within genera (Hovav et al., 2008; Colinet et al., 2013), within classes (Emera et al., 2011), within phyla (Hughes, 2013), within kingdoms (Wistow, 1993; Ruder et al., 2013), and within domains (Elde et al., 2005). Convergent gene recruitment has also been shown to contribute to functional similarities for a broad taxonomic distribution of animal venoms (Fry et al., 2009). The cited examples constitute a considerable portion of the literature on convergent gene recruitment in an adaptive evolutionary context. Yet, the specialization of tissues and biochemical pathways across phylogeny, in addition to complex spatio-temporal expression patterns observed for many proteins (Miyata and Suga, 2001), necessarily involved gene recruitment, and convergence of recruitment, at some point in their evolutionary histories. In addition, previous work has demonstrated considerable diversity and tissue specificity of alternative splice variants for many genes (Yeo et al., 2004; Wang et al., 2008; Pan et al., 2009), indicating that recruitment of alternative splice variants is a fundamental component of tissue specialization. Moreover, through comparisons of homologous organs among vertebrate lineages it has been demonstrated that evolution of alternative splicing occurs at a more rapid rate than does evolution of gene expression levels (Barbosa-Morais et al., 2012). Thus, tissue-specific convergent gene recruitment, and recruitment of alternative splice variants are potentially powerful evolutionary mechanisms that should be expected discoveries during the study of adaptive evolution and diversification.
This study was developed to identify candidate gene recruitments supporting vampire bat sanguivory, and to characterize how they became expressed as salivary gland secretory molecules. The sporadic phylogenetic occurrence of sanguivory across animals, in addition to the adaptive potential of salivary glands (Phillips et al., 1977), provides a unique experimental design with comparisons to elucidate such events. Namely, comparison among sanguivores and their non-sanguivorous relatives can be used to build hypotheses about sanguivore adaptations. In this study we compare sanguivorous leeches, vampire bats, and two closely related insectivorous bats. Results describe an adaptive role for recruitment of alternative secretory splice variants to vampire bat salivary glands, novel instances of convergent gene recruitment, as well as novel sanguivore venom proteins. For these molecules, biochemical hypotheses about the antagonistic action on prey hemostatic and neurologic signaling pathways are provided.
Results
In order to develop suitable comparisons for this study a series salivary gland transcriptomes were collected and characterized. For phylogenetic sampling of leech diversity, partial salivary gland transcriptomes for three sanguivorous leeches, Hirudo verbana (European medicinal leech; family Hirudindae), Aliolimnatis fenestrata (African medicinal leech; family Hirudindae), and Macrobdella decora (North American medicinal leech; family Semiscolecidae), were mined from the literature (Kvist et al., 2013; Genbank accession numbers JZ183761–188441). An annotated vampire bat, D. rotundus, salivary gland transcriptome was retrieved from the supplemental web table from Francishetti et al. (2013). For comparisons to bats maintaining the primitive dietary condition of insectivory, new submandibular salivary gland transcriptomes were generated for Macrotus californicus and Myotis lucifugus. Selection of insectivorous taxa was made to include close phylogenetic relatives to vampire bats and to provide a polarized phylogenetic comparison. Raw transcriptome data for each insectivorous taxon was generated from one lane of 75 bp paired end sequencing performed on an Illumina HiSeq 2000 instrument. Sequencing effort resulted in approximately 30 million pairs of reads for each taxon (Table 1). Transcript discovery was maximized by implementing both Trinity (Grabherr et al., 2011; Li and Dewey, 2011; Marçais and Kingsford, 2011; Haas et al., 2013) and Abyss (Simpson et al., 2009) assembly approaches. Downstream annotation of transcripts consisted of ORF identification using TransDecoder (Haas et al., 2013), predicted protein identifications using blastp (Altschul et al., 1997) and SwissProt protein database (The UniProt Consortium, 2012), signal peptide prediction using Signalp (Petersen et al., 2011), protein domain identifications using Hmmr (Eddy, 2011), and exon discovery using Spidey (Wheelan et al., 2001) and Ensembl M. lucifugus genome assembly 2.0 release 74 (Flicek et al., 2013). Sampling, sequencing, and bioinformatic processing steps are further described in Section Materials and Methods.
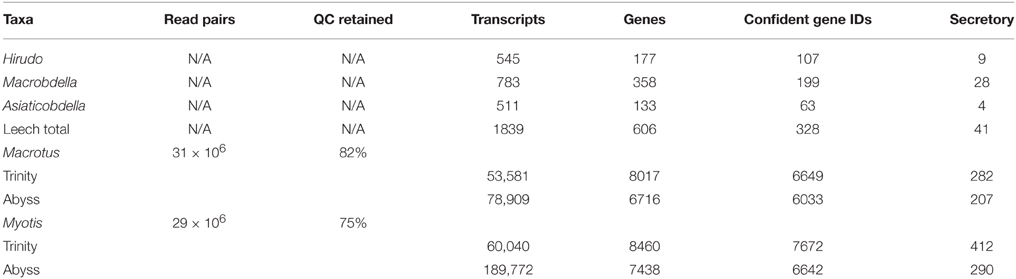
Table 1. Assembly and annotation statistics.
Transcriptome Comparison
Queries of 41 putative leech salivary products against the vampire database recovered 16 putative orthologs, among which 10 were recovered below significance thresholds (Table 2). Six of the ten orthologs were identified as containing secretory signal peptide sequences, and categorization by relevance to sanguivory based on UniProt functional annotations and literature reviews identified three orthologs (Entpd1, ectonucleoside triphosphate diphosphohydrolase 1; Lrp1, low density lipoprotein receptor related protein 1; Nptn, neuroplastin) possess biochemical functions that would be beneficial to sanguivory (Table 3). Biological functions for these genes were described by regulation of hemostasis (Entpd1, Lrp1) or neural signaling (Nptn). Thus, genes identified as co-expressed and beneficial to sanguivory most likely function antagonistically to host clotting by interference with proinflammatory response pathways, or by neural disruption. Two additional putative leech and vampire bat secretory orthologs (Acha, acetylcholine receptor subunit alpha; Cant1, soluble calcium-activated nucleotidase 1) were recovered with unacceptable E-values for the vampire bat; however known biological functions of the proteins encoded by these genes indicated benefits to sanguivory (regulation of hemostasis for Cant1, regulation of neural signaling for Acha). Orthologs excluded from the list of strong candidates likely beneficial to sanguivory are involved in widespread cellular processes such as aerobic respiration, chromatin remodeling, were common salivary products also identified as secretory products in insectivorous lineages, or were proteins for which no secretory signal peptide sequence was identified regardless of potential benefit to sanguivory (Table 3).
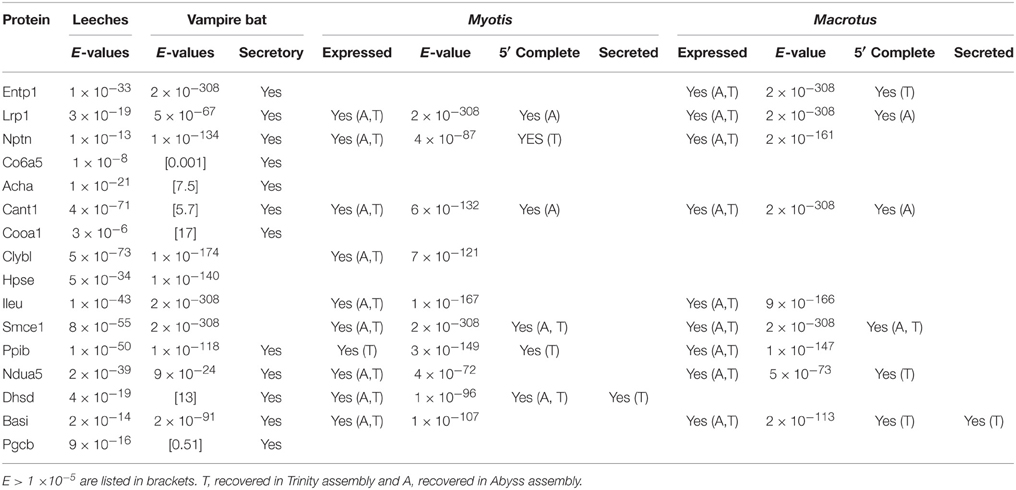
Table 2. Summary information for proteins inferred for both leech and vampire bat salivary gland transcriptomes and frequencies of occurrence in Macrotus and Myotis transcriptomes.
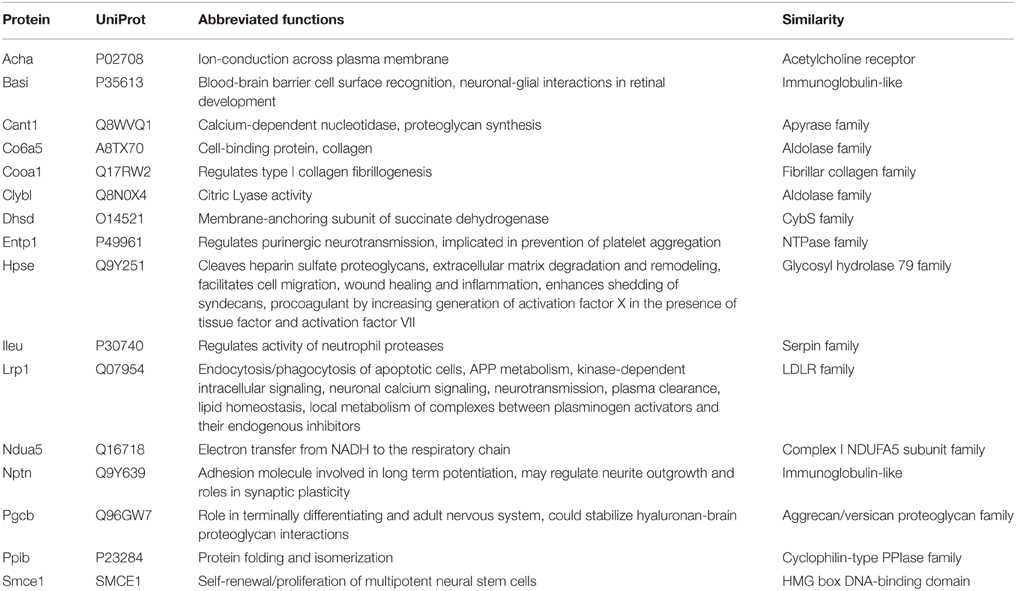
Table 3. Description of shared leech and vampire bat salivary gland products.
Cross-queries to Macrotus and Myotis databases recovered nine and 10 of the 16 putative orthologs expressed in both leeches and vampire bat, respectively. For the three confidently identified genes which would be beneficial to sanguivory, all three were expressed in Macrotus, and two (Lrp1, Nptn) were expressed in Myotis. In contrast to that observed for vampire bat, none of the insectivore orthologs were inferred to contain secretory signal peptide sequences. For the two putatively sanguivory-relevant genes recovered above significance thresholds, Acha was not detected in either insectivore database, and Cant1 was expressed, but inferred non-secretory in Macrotus.
Sequence Comparison
Transcript alignment and genomic mapping was next conducted in order to characterize the transcriptional differences resulting in predicted secretion of vampire bat but not insectivore orthologs. Alignment of bat orthologs generally identified orthology at 3′ ends and non-orthology at 5′ ends (Supplementary Material 1; alignment among bats and leeches was not conducted due to extensive inter-phyla sequence divergence). Exon discovery by mapping transcripts to M. lucifugus genome assembly revealed that vampire bat and Myotis genome inferred Entpd1 transcripts each consisted of nine exons, whereas Macrotus Entpd1 consisted of seven exons. The genomic coordinates of exon 1 were unique for each isoform and only exon 1 incorporated into vampire bat Entpd1 contained the secretory signal peptide sequence identified through previous analyses (Figure 1; Supplementary Material 2). Although the genomic coordinates for exons 1 from vampire bat and Myotis Entpd1 orthologs overlapped, sequence examination revealed a 79 bp microdeletion within vampire bat exon 1 relative to Myotis (Figure 2). The location of the vampire bat microdeletion places the vampire bat start codon adjacent to a downstream region of Entpd1 recognizable as a novel secretory signal peptide sequence and at the same time maintains proper reading-frame.
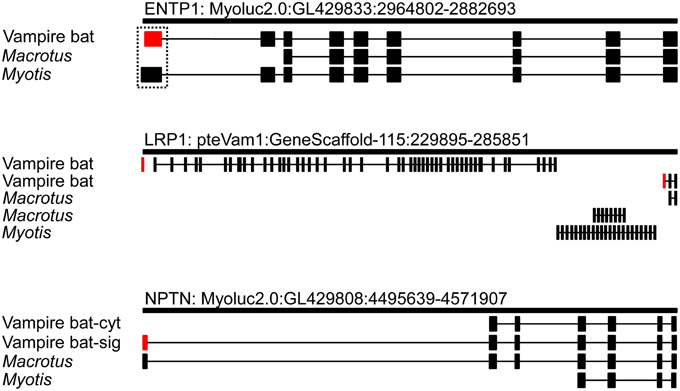
Figure 1. Exon mapping of recruited vampire bat secretory genes to orthologous genomic regions. Vampire bat exons encoding secretory signal peptide sequences are indicated in red. Exon 1 of vampire bat long Lrp1 isoform did not map, and uncertainty on this exon's position is indicated by the absence of intron 1 designation (see text for details).
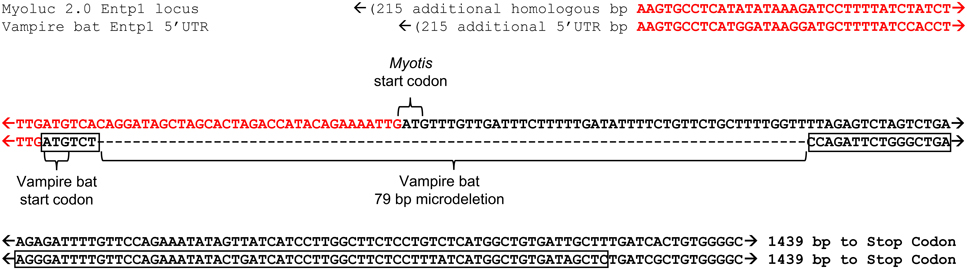
Figure 2. Alignment of Entpd1 5′ region between vampire bat and M. lucifugus (dashed box in Figure 1). Sequence of vampire bat 5′UTR and orthologous M. lucifugus genomic coordinates are displayed in red font and the location of the vampire bat mRNA sequence encoding the secretory signal peptide sequence is boxed. The 79 bp microdeletion observed in vampire bat, which results in the N-terminal location of the novel secretory signal peptide sequence, maintains downstream reading frame.
The vampire bat transcriptome included two Lrp1 alternative transcripts containing secretory signal peptide sequences, and these transcripts were encoded by mutually exclusive exons (Figure 1). In comparison, Macrotus and Myotis transcriptomes included two and three Lrp1 alternative transcripts which incorporated exons not observed in either vampire bat transcript. Lrp1 transcripts were next mapped to the Pteropus genome assembly (Pteropus genome assembly was used for Lrp1 mapping because no M. lucifugus Lrp1 containing scaffold is available), through which all bat transcripts, with the exception of the first 67 bp of the longer vampire bat Lrp1 transcript, mapped within the Lrp1 locus of Pteropus vampyrus. Failure to map the first 67 bp of this transcript was due to a scaffold gap, or absence of this region in the Pteropus genome (i.e., this region successfully mapped within the human LRP1 locus). Alignment of all Lrp1 transcripts was not possible as there was considerable variation in exon structure, with transcripts being composed of two to 48 exons (similar isoform diversity is observed for human and mouse Lrp1; Flicek et al., 2013). Among bat alternative transcripts, only the two isoforms expressed in the vampire bat submandibular gland contained secretory signal peptide sequences. Factor Xa inhibitory domains were also identified in one of the vampire bat alternative transcripts, whereas none were identified in any of the insectivore transcripts (Supplementary Material 3).
For Nptn, similar to that observed for Entpd1, alignment revealed 3′ orthology but 5′ non-orthology among bat orthologs. Exon discovery by genomic mapping disclosed a similar pattern to that observed for both Entpd1 and Lrp1 in which variation in selection of 5′ exons among isoforms was observed; bat Nptn isoforms consisted of four to seven exons, with all variation in exon use occurring at the 5′ end of transcripts (Figure 1). The vampire bat transcriptome included both secretory and a non-secretory isoforms. Vampire bat secretion of Nptn was determined to be the result of production of an Nptn isoform incorporating an exon 1 with a genomic location approximately 50 kbp upstream relative to vampire bat non-secretory isoform. Although assembly and mapping presented the possibility that Macrotus also expresses a secretory isoform similar to vampire bat, the Macrotus transcript was 9′ bp longer on the 5′ end and was inferred to be 5′ incomplete.
In order to characterize the extent to which the inferred transcript diversity and exon use reflects bat specific splicing patterns or ancestral splicing diversity, annotations for each locus were surveyed in human genome annotation GRCh37.74 and mouse genome annotation GRCm38.74 (Flicek et al., 2013; these genomes were selected because of their extensive supporting expression data and distant mammalian relationship to bats). Overall, similar exon structure and alternative splicing diversity was observed in comparisons among bat, human, and mouse (Supplementary Material 4; see Ensembl annotations for additional information). However, lineage-specific expression patterns were also observed which commonly involved variation in 5′ exon incorporation. These patterns indicate the diversity of submandibular gland-expressed bat isoforms as recruitments and modifications from a largely existing repertoire of mammalian alternative transcripts.
Discussion
The bat family Phyllostomidae is the greatest evolutionary diversification of extant forms resulting from a common ancestor in the Eocene (Baker et al., 2012). Vampires, multiple independent origins of nectar feeders, fruit eaters, carnivores, and a broad array of omnivores (classified into 11 subfamilies, 55 genera, 160 species) all evolved under the constraints of natural selection over a 40 million year period from a common ancestor that was primarily an insectivore that may have taken some plant material. What genetic mechanisms and genomic landscapes facilitated making a vampire bat from an insectivorous bat? Our study suggests that recruiting existing genes from other biological functions has been critical for the vampire bat niche leap.
Recruitment of Alternative Splicing
The inclusion of outgroup insectivorous bats provided comparisons for understanding aspects of transcription evolution enabling recruitment of specific genes as vampire bat salivary gland secretory products. For each locus of interest it is notable that expression was documented in vampire bat, leeches, and one or both insectivores; however insectivores were not found to express any secretory isoform for any locus. Although we cannot unequivocally exclude the possibility that insectivorous taxa are capable of expressing the putative sanguivore-adaptive isoforms in their submandibular glands, none were detect through analysis of ~4.5 Gb of sequencing data per taxon. It is also possible that these isoforms were not detected in insectivores due to opportunistic tissue sampling in a period of depressed expression of these isoforms. Future inclusion of experimental replicates could distinguish these possibilities, however, the current data indicate that salivary gland secretory capacity of the identified genes is a derived vampire bat molecular phenotype. Furthermore, while it is straight forward to develop hypotheses for the adaptive value of these gene products in sanguivory, benefits to insectivory are not clearly identifiable. In all cases recruitment of secretory expression in vampire bats is derived from the expression of isoforms incorporating first exons which include secretory signal peptide sequences (Figures 1, 2, Supplementary Material 2). Thus, the identified transcriptional modifications which have been adaptive components of vampire bat evolution all arose via the same general mechanism of recruitment of secretory isoforms.
A survey of the literature reporting on work in model organisms has recently described extensive use of alternative splicing to generate cellular diversity. It has been estimated that 95% of human multiexon genes are alternatively spliced, resulting in the expression of at least 100,000 alternative transcripts in major human tissues (Pan et al., 2009). About 85% of genes have minor isoforms with expression frequencies >15%, and this expression strongly correlates with tissue type (Wang et al., 2008). Thus, these data alone demonstrate an evolutionary history involving extensive recruitment of alternative splice variants in the evolution of tissue function. Alternative splicing is disproportionately mediated by alternative exon use in 5′ and 3′ regions (Yeo et al., 2004), which is in agreement with the observation that all recruitments of secretory vampire bat products identified in this study involved incorporation of alternative 5′ exons not observed in either insectivore salivary gland transcriptome.
Both secretory and non-secretory isoforms of Lrp1 and Nptn have been described in other tissues of other organisms (The UniProt Consortium, 2012). Observed patterns including a lack of detecting secretory isoforms in insectivores, presence of secretory isoforms in vampire bats, and observing conserved exon structure of isoforms among bat, human and mouse, described recruitment of existing alternative splicing mechanisms, rather than de novo origination of alternative exon usage as the mechanism by which vampire bats evolved salivary gland expression of these proteins. In comparison, although Entpd1 exon structure and isoform diversity was generally conserved among bats, human, and mouse, secretory expression of Entpd1 is a vampire bat apomorphy. Entpd1 has not previously been characterized as secretory, and the structure of vampire bat exon 1, which contains the 79 bp microdeletion and secretory signal peptide sequence, is not observed in any other lineage. The recruitment of Enptd1 as a vampire bat secretory molecule represents a rare instance in which a previously non-secretory protein has been recruited to a role as a secretory venom protein (Fry, 2005).
Observations of the current study demonstrate the recurring involvement of recruitment of alternative splice variants in the evolution of vampire bat saliva. Quite remarkably, a survey of other known molecular details regarding vampire bat evolution, also involves recruitment of alternative splicing. It was previously demonstrated (Gracheva et al., 2011) that evolution of thermodetection in vampire bats was mediated by recruitment of a specific TRPV1 alternative splice variant to trigeminal ganglia (nerves embedded in nasal pits imparting vampire bat thermodetection capability). This transcript is also expressed in fruit bats and other mammals, albeit at much lower expression levels, and not concentrated within nasal pits. Thus, thermodetection adaptation in vampire bats evolved through recruitment of existing alternative splice variants, similar to that observed for the secretory proteins identified in the current study. Our current results as well at those reported by Gracheva et al. (2011), provide multiple examples highlighting the importance of regulatory evolution in adaptation (see below for further discussion).
Adaptive Function
The adaptive benefit of convergently recruited Entpd1 was initially indicated by the previous identification of Entpd1 in leech saliva (Rigbi et al., 1996). One of the well-characterized functions of Entpd1 is regulation of vascular thrombosis (Christoforidis et al., 1995; Kaczmarek et al., 1996). Entpd1 is usually expressed on membranes of vascular endothelial cells. Entpd1 functions antagonistically to the proinflammatory response by hydrolyzing the circulating inflammatory mediators ATP and ADP, thereby inhibiting platelet activation. In the event of vascular injury, cellular internalization of Entpd1 and other endothelial surface proteins initiates the proinflammatory response (Robson et al., 1997). Thus, host response to vascular injury by leech or vampire bat is removal of Entpd1 from cell surfaces. However, the presence of salivary Entpd1 at wound site would function antagonistically to host response by inhibiting platelet activation (nucleoside phosphatase domains where detected in both leech and vampire bat Entpd1, suggesting conserved roles with respect to enzymatic activity, Dataset S3 in Supplementary Material). The identification of Entpd1 in plasma microparticles, in which Enpt1 maintains anti-hemostatic properties, confirms that Entpd1 not associated with cell membranes functions in a conserved biochemical capacity (Banz et al., 2008).
Lrp1 is known to function in several metabolic capacities, among which is interaction with thrombospondin, a protein that when released from platelets in response to vascular injury promotes clot formation by binding to fibrin and other clotting molecules (Bale et al., 1985). Lrp1 in conjunction with heparin-sulfate proteoglycans functions antagonistically to the clotting cascade through binding, and subsequent internalization of thrombospondin (Wang et al., 2004). Results identifying Lrp1 presence in both leech and vampire bat salivary transcriptomes suggests an anticoagulation function through interaction with host thrombospondin. Supporting a more complex role for salivary Lrp1 in anticoagulation, protein domain analysis of the two vampire bat Lrp1 isoforms identified multiple coagulation Factor Xa inhibitory sites in one of the isoforms. Factor Xa catabolizes prothrombin to the active pro-coagulant thrombin. Therefore, vampire bat Lrp1 potentially also functions in an anticoagulant capacity by impeding host thrombin formation.
The observed expression, but incomplete status of Macrotus Nptn transcript potentially excludes confident conclusions regarding derived benefits to sanguivory. However, a review of established metabolic function of Nptn does support adaptive benefits of this molecule to sanguivory. Nptn is a neural membrane adhesion molecule capable of regulating long-term potentiation of neurons. Specifically, investigation of hippocampal signaling has demonstrated that increasing levels of Nptn result in a reduction in the level of potentiation (Empson et al., 2006). These findings indicate that Nptn functions in leech and vampire bat saliva by modulating host neural signaling, in which the increase of Nptn at wound site would reduce host nervous system response. Both leech and vampire bat Nptn orthologs retain multiple immunoglobulin domains characteristic of this protein. Reduced nerve sensation at wound site is a characteristic of both leech and vampire bat inflictions (Schutt, 2008), and the presence of salivary Nptn likely contributes to this response.
The biological functions for the tentatively identified expression products Acha and Cant1 are notable, and likely represent highly derived and previously uncharacterized adaptively recruited genes. For example, Acha regulates neurotransmission through acetylcholine binding, and may therefore function as a neurodisruptor in leech and vampire bat saliva, similar to that proposed for Nptn (however vampire bat Acha lacked an expected neurotransmitter-gated ion channel ligand binding domain). Perhaps functioning in a capacity similar to Entpd1, Cant1 was previously identified as a salivary product in sanguivorous arthropods, where it was hypothesized to prevent coagulation by catabolism of ATP, and ADP molecules (Valenzuela et al., 2001; suggesting convergent recruitment among vampire bats, leeches and arthropods). Assuming the vampire bat expression product identified as Cant1 is correct, it is extensively derived as compared to Cant1 identified in Macrotus and Myotis transcriptomes, both of which lacked secretory signal peptide sequences.
Hypotheses on Alternative Splicing and Transcriptome Evolution
Inferences from this study indicate evolution of alternative splicing as the mechanism by which vampire bats evolved salivary expression of proteins that would be adaptive to sanguivory. The diversity of alternative isoforms, and the dynamism of alternative splicing across tissue types reflects the complex cellular regulation of alternative splicing. For example, it is known that alternative splicing is regulated by a multitude of factors including promoter choice, intron structure, sequence of intronic and exonic enhancer and silencer motifs, chromatin structure, tissue-specific expression of protein and nucleic acid spliceosomal components, histone modification, among others (Matlin et al., 2005; Kornblihtt et al., 2013). Although not possible from the current data, results indicate future efforts to identify the underlying genomic mechanisms responsible for lineage and tissue-specific transcriptional diversity will be important. We hypothesize that the importance of this course of study partially relates to genetic load. For example, a substantial genetic load would be expected to accumulate during the rapid and concerted modification of multiple phenotypic traits (such as that observed in vampire bats) if protein functional modification via non-synonymous mutation were the primary source of genetic variation. That is, because many genes are expressed in multiple tissues and organ systems, a new amino acid variant (in a constitutively or broadly expressed exon) can simultaneously be selectively advantageous, neutral, and deleterious depending on site of expression, and the probability of fixation or loss would be influenced by the overall selection coefficient for that variant. However, because of the complex inheritance underlying alternative splicing, mutations that would adaptively modify splicing in a specific tissue could be more likely to exhibit neutral (rather than deleterious) consequences on the function of the same gene when expressed in another tissue which operates under a different combination of splicing interactions. The mutability of alternative splicing indicated by repeated inference of alternative exon use in sanguivore adaptation (this study; Gracheva et al., 2011), and observing the vast abundance of alternative transcripts in RNA-seq data (which greatly exceeds the number of genes), may support this hypothesis.
The above discussion on relative roles of alternative splicing and non-synonymous evolution are not meant to preclude the importance of the latter. In fact, recent work has clearly demonstrated major roles for non-synonymous mutation in adaptation (Parker et al., 2013). However, adaptive roles for Entpd1, Lrp1, and Nptn in sanguivore evolution would not have been identified if analyses were confined to sequence-based selection tests. For example, a post hoc analysis of Entpd1 and Nptn (genes with orthologous alignments for all bats included in this study) using the Mixed Effects Model of Evolution (Murrell et al., 2012) identified two putative codons under selection in Macrotus for Entpd1, and no sites under selection for Nptn (data not shown). In reality, increasing knowledge of isoform diversity in non-model organisms is complementary to the discovery of sequence selection. Currently, exon annotations for most non-model genomes are based on prediction rather than supporting expression data, and the inclusion of previously unidentified exons would be expected to reduce the Type II error rate associated with identifying genes manifesting a recent history of diversifying or convergent sequence selection. It is also notable that previous work has demonstrated that variance in alternative splicing patterns among RNA-seq data sets sampled from multiple organs across broadly divergent vertebrate taxa is largely explained by taxon as opposed to organ, suggesting a trend for ubiquitous expression of apomorphic splicing patterns (Barbosa-Morais et al., 2012). However, this result is not contradictory to the observation that isoform expression is also highly tissue-specific (Yeo et al., 2004; Wang et al., 2008; Pan et al., 2009). Combined, these patterns document cellular-specificity of splicing, but also suggest, at minimum, a frequent evolutionary tolerance to the introduction of new exons expressed in a variety of tissues. It would be expected that in the absence of strong positive selection for a novel exon's expression across tissues, newly introduced motifs would be required to be nearly neutral, or only mildly deleterious, in many tissues in order to persist. In this regard, selection regimes acting on alternative splicing would be similar to those proposed to act on non-synonymous variation.
Conclusions
Comparisons of the evolutionarily divergent sanguivorous leeches and vampire bats allowed the identification of novel and convergently recruited venom proteins. Given that the leech database was developed from a shallow transcriptomic sampling, the findings indicate that additional deep sequencing and comparison will identify more novel and convergently recruited genes, and future comparisons among other sanguivores and their related non-sanguivorous relatives will also contribute to this goal. In addition, the subfamily Desmodontinae includes three genera of vampire bats (which exhibit variation in selecting mammalian or avian hosts), and previous work discovered extensive copy number and sequence evolution for the plasminogen activators among these genera (Tellgren-Roth et al., 2008). Future studies among this assemblage will be valuable for identifying lineage-specific vampire bat adaptations and the timeframes under which they arose. In general, the involvement of gene recruitment and alternative splicing is a viable hypothesis to help explain how the diverse family of phyllostomid bats, from which vampires, nectar-feeders, fruit-eaters, carnivores, and omnivores, all evolved from the same common ancestor, from the same ancestral genome, and under the constraints of natural selection. Future comparative studies among this group will be valuable for elucidating the rate of transcriptome evolution in this rapid adaptive radiation.
Materials and Methods
Vampire bats are a basal radiation in the family Phyllostomidae, with the insectivorous genus Macrotus putatively as the basal most divergence within the family (Baker et al., 2003, 2012; Datzmann et al., 2010; Dumont et al., 2011). Macrotus californicus was identified as a suitable control group due to its close phylogenetic relationship to vampire bats and the primarily insectivorous dietary strategy maintained by Macrotus (a male M. californicus was collected at Picacho Peak Mine, Arizona under permit #SP658736 issued to Robert J. Baker by Arizona Fish and Game). To provide a polarized phylogenetic comparison, the obligate insectivore Myotis lucifugus (family Vespertilionidae) was also included (a male M. lucifugus was collected at Shaver's Creek Environmental Center, Huntingdon County, Pennsylvania under scientific collecting permit #00098 issued to Michael Gannon by the Pennsylvania Game Commission). Because it is thought that sanguivory is the symplesiomorphic condition in leeches, as saliva from non-sanguivorous leech lineages possess anticoagulant properties (Kvist et al., 2011), control comparisons were not made among leeches. Directly following euthanasia submandibular glands were excised and frozen in liquid nitrogen and RNA was subsequently isolated following manufacturer protocols (Trizol Reagent; Life Technologies, Carlsbad, California, USA). TruSeq RNA Sample preparations and one lane of 75 bp paired-end sequencing on a HiSeq 2000 instrument for each sample was conducted at Prognosis Biosciences, Inc. (San Diego, CA). Reads were quality filtered using Trimmomatic with the following constraints: TRAILING:25 SLIDINGWINDOW:5:25 MINLEN:60 (Bolger et al., 2014).
De novo transcriptome assembly was conducted using Trinity under default settings (Grabherr et al., 2011; Li and Dewey, 2011; Marçais and Kingsford, 2011; Haas et al., 2013). Abyss assemblies were also conducted as a means of cross-validation using all even values of k between 20 and 40 (Simpson et al., 2009; Macrotus, k = 38; N50 = 1428; Myotis, k = 32, N50 = 1259). Open-reading frames and predicted peptide translations were recovered using TransDecoder (Haas et al., 2013) and protein identifications were made by blastp queries against SwissProt protein database establishing E < 1 × 10−5 and minimum alignment length of 40 bp as high confidence identifications (Altschul et al., 1997; The UniProt Consortium, 2012). Secretory signal peptide sequences were identified using SignalP (Petersen et al., 2011). Protein domains were identified using Hmmr (Eddy, 2011). Assembly and annotation statistics are available in Table 1. Although expression validation is generally lacking for Myotis lucifugus, ENSEMBL Genome Assembly 2.0 release 74 (Flicek et al., 2013) was queried for putative protein products of relevance (see Section Results), which were also analyzed for the presence of secretory signal peptide sequences.
To develop a database of leech salivary products we leveraged the recently published medicinal leech partial transcriptome prepared from leech salivary gland dissections (Kvist et al., 2013; Genbank accession numbers JZ183761–188441). Data consisted of 1523 mRNA sequences from Hirudo verbana (European medicinal leech), 1604 from Macrobdella decora (North American medicinal leech), and 1555 from Aliolimnatis fenestrata (African medicinal leech). Annotation followed the same bioinformatic workflow described above. Owing to the number of mRNA sequences per leech species, annotations for the three leech species were pooled and treated as a composite partial salivary transcriptome (Table 1).
For the vampire bat, the supplemental web table from Francishetti et al. (2013) was downloaded and converted to a relational database. Secretory proteins inferred for leech saliva meeting significance thresholds were queried against the vampire database, and matches were considered orthologs when significance thresholds were also met for vampire bat expression products. Matches with unacceptable E-values were retained and considered individually based on potential relevance to sanguivory as determined by UniProt functional annotations and literature review. Protein domains were identified as described above. Number of reads mapping to a given transcript are available in supplemental web table from Francishetti et al. (2013).
Expressed products inferred to be secreted in leeches and vampire bats were cross-queried to Macrotus and Myotis databases. Presence or absence in these databases was documented, and for transcripts present in the insectivore databases documentation of the completeness and presence of secretory signal peptide sequences was assessed (Table 2). Alignments (Dataset S1 in Supplementary Material) were made among orthologs from vampire bat, Macrotus and/or Myotis to characterize orthology and exon discovery via mapping to Ensembl Myotis lucifugus genome assembly 74.2 or Pteropus vampyrus genome assembly 74.1 was conducted using Spidey (Wheelan et al., 2001). Comparisons among bat, human, and mouse were conducted in a similar fashion.
Data Deposition
Sequence data generated for this study can be accessed at Sequence Read Archive under accession numbers SRP031492 and SRP032466.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Natural Science Research Laboratory at the Museum of Texas Tech University for curation and tissue loans. We thank Cibele Caio and Carl Phillips for field assistance, and Jim Bull Boyd Butler, David Ray, and Lewis Held for valuable criticisms of this manuscript. The bat salivary gland project was conceived in part by participants at a Texas Tech University Genomics Symposium funded by the Office of the Vice President for Research. We acknowledge Robert Bradley, Adam Freedman, Scott Edwards, Mohamed Noor, Federico Hoffmann, Anton Nekrutenko, Oliver Ryder, Enrique Lessa, and John Novembre for discussions on experimental design. This work was supported by the Texas Biological Database Program.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fevo.2015.00122
References
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Baker, R., Bininda-Emonds, O., Manilla-Meluk, H., Porter, C., and van den Busche, R. A. (2012). “Molecular timescale of diversification of feeding strategy and morphology in New World leaf-nosed bats (Phyllostomidae): a phylogenetic perspective,” in Evolutionary History of Bats: Fossils, Molecules and Morphology, eds G. Gunnell and N. Simmons (New York, NY: Cambridge University Press), 385–409.
Baker, R. J., Hoofer, S. R., Porter, C. A., and van den Bussche, R. A. (2003). Diversification among New World Leaf-Nosed Bats: an evolutionary hypothesis and classification inferred from digenomic congruence of DNA sequence. Oc. Pap. Mus. Tex. Tech. Univ. 230, 1–32.
Bale, M. D., Westrick, L. G., and Mosher, D. F. (1985). Incorporation of thrombospodin into fibrin clots, J. Biol. Chem. 260, 7502–7508.
Banz, Y., Beldi, G., Wu, Y., Atkinson, B., Usheva, A., and Robson, S. C. (2008). CD39 is incorporated into plasma microparticles where it maintains functional properties and impacts endothelial activation. Br. J. Haematol. 142, 627–637. doi: 10.1111/j.1365-2141.2008.07230.x
Barbosa-Morais, N. L., Irimia, M., Pan, Q., Xiong, H. Y., Gueroussov, S., Lee, L. J., et al. (2012). The evolutionary landscape of alternative splicing in vertebrate species. Science 338, 1587–1593. doi: 10.1126/science.1230612
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Busch, C. (1988). Consumption of blood, renal function and utilization of free water by the vampire bat, Desmodus rotundus. Comp. Biochem. Physiol. 90, 141–146. doi: 10.1016/0300-9629(88)91019-5
Christoforidis, S., Papamarcaki, T., Galaris, D., Kellner, R., and Tsolas, O. (1995). Purification and properties of human placental ATP diphosphohydrolase. Eur. J. Biochem. 234, 66–74. doi: 10.1111/j.1432-1033.1995.066_c.x
Colinet, D., Deleury, E., Anselme, C., Cazes, D., Poulain, J., Azema-Dossat, C., et al. (2013). Extensive inter- and intraspecific venom variation in closely related parasites targeting the same host: the case of Leptopilina parasitoids of Drosophila. Insect Biochem. Mol. Biol. 43, 601–611. doi: 10.1016/j.ibmb.2013.03.010
Datzmann, T., von Helversen, O., and Mayer, F. (2010). Evolution of nectarivory in phyllostomid bats (Phyllostomidae Gray, 1825, Chiroptera: Mammalia). BMC Evol. Biol. 4:165. doi: 10.1186/1471-2148-10-165
Dumont, E. R., Dávalos, L. M., Goldberg, A., Santana, S. E., Rex, K., and Voigt, C. C. (2011). Morphological innovation, diversification and invasion of a new adaptive zone. Proc. Biol. Sci. 7, 1797–1805. doi: 10.1098/rspb.2011.2005
Eddy, S. R. (2011). Accelerated profile HMM searches. PLoS Comput. Biol. 7:e1002195. doi: 10.1371/journal.pcbi.1002195
Elde, N. C., Morgan, G., Winey, M., Sperling, L., and Turkewitz, A. P. (2005). Elucidation of clathrin-mediated endocytosis in Tetrahymena reveals an evolutionarily convergent recruitment of dynamin. PLoS Genet. 1:e52. doi: 10.1371/journal.pgen.0010052
Emera, D., Casola, C., Lynch, V. J., Wildman, D. E., Agnew, D., and Wagner, G. P. (2011). Convergent evolution of endometrial prolactin expression in primates, mice, and elephants through the independent recruitment of transposable elements. Mol. Biol. Evol. 29, 239–247. doi: 10.1093/molbev/msr189
Empson, R. M., Buckby, L. E., Kraus, M., Bates, K. J., Crompton, M. R., Gundelfinger, E. D., et al. (2006). The cell adhesion molecule neuroplastin-65 inhibits hippocampal long-term potentiation via a mitogen-activated protein kinase p38-dependent reduction in surface expression of GluR1-containing glutamate receptors. J. Neurochem. 99, 850–860. doi: 10.1111/j.1471-4159.2006.04123.x
Flicek, P., Ahmed, I., Amode, M. R., Barrell, D., Beal, K., Brent, S., et al. (2013). Ensembl 2013. Nucleic Acids Res. 41, D48–D55. doi: 10.1093/nar/gks1236
Francishetti, I. M. B., Assumpção, T. C., Ma, D., Li, Y., Vicente, E. C., Uieda, W., et al. (2013). The “vampirome”: transcriptome and proteome analysis of the principal and accessory submaxillary glands of the vampire bat Desmodus rotundus, a vector of human rabies. J. Proteomics 82, 288–319. doi: 10.1016/j.jprot.2013.01.009
Fry, B. G. (2005). From genome to ”venome”: molecular origin and evolution of the snake venom proteome inferred from phylogenetic analysis of toxin sequences and related body proteins. Genome Res. 15, 403–420. doi: 10.1101/gr.3228405
Fry, B. G., Roelants, K., Champagne, D. E., Scheib, H., Tyndall, J. D. A., King, G. F., et al. (2009). The toxicogenomic multiverse: convergent recruitment of proteins into animal venoms. Annu. Rev. Genomics Hum. Genet. 10, 483–511. doi: 10.1146/annurev.genom.9.081307.164356
Grabherr, M. G., Hass, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Gracheva, E. O., Cordero-Morales, J. F., González-Carcacía, J. A., Ingolia, N. T., Manno, C., Aranguren, C. I., et al. (2011). Ganglion-specific splicing of TRPV1 underlies infrared sensation in vampire bats. Nature 476, 88–91. doi: 10.1038/nature10245
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Hovav, R., Chaudhary, B., Udall, J. A., Flagel, L., and Wendel, J. F. (2008). Parallel domestication, convergent evolution and duplicated gene recruitment in allopolyploid cotton. Genetics 179, 1725–1733. doi: 10.1534/genetics.108.089656
Hughes, A. L. (2013). Evolution of the salivary apyrases of blood-feeding arthropods. Gene 527, 123–130. doi: 10.1016/j.gene.2013.05.087
Jones, G., Teeling, E. C., and Rossiter, S. J. (2013). From the ultrasonic to the infrared: molecular evolution and the sensory biology of bats. Front Physiol. 4:117. doi: 10.3389/fphys.2013.00117
Kaczmarek, E., Koziak, K., Sévigny, J., Siegel, J. B., Anrather, J., Beaudoin, A. T., et al. (1996). Identification and characterization of CD39/vascular ATP diphosphohydrolase. J. Biol. Chem. 271, 33116–33122. doi: 10.1074/jbc.271.51.33116
Kornblihtt, A. R., Schor, I. E., Alló, M., Dujardin, G., Petrillo, E., and Munoz, M. J. (2013). Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat. Rev. Mol. Cell Biol. 14, 153–165. doi: 10.1038/nrm3525
Kvist, S., Min, G. S., and Siddall, M. E. (2013). Diversity and selective pressures of anticoagulants in three medicinal leeches (Hirudinida: Hirudinidae, Macrobdellidae). Ecol. Evol. 3, 918–933. doi: 10.1002/ece3.480
Kvist, S., Sarkar, I. N., and Siddall, M. E. (2011). Genome-wide search for leech antiplatelet proteins in the non-bloodfeeding leech Helobdella robusta (Rhyncobdellida: Glossiphonoodae) reveals evidence of secreted anticoagulants. Invert. Biol. 130, 344–350. doi: 10.1111/j.1744-7410.2011.00238.x
Li, B., and Dewey, C. N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323
Marçais, G., and Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770. doi: 10.1093/bioinformatics/btr011
Matlin, A. J., Clark, F., and Smith, C. W. J. (2005). Understanding alternative splicing: towards a cellular code. Nature 6, 386–398. doi: 10.1038/nrm1645
Miyata, T., and Suga, H. (2001). Divergence pattern of animal gene families and relationship with the Cambrian explosion. Bioessays 23, 1018–1027. doi: 10.1002/bies.1147
Murrell, B., Wertheim, J. O., Moola, S., Weighill, T., Scheffler, K., and Kosakovsky Pond, S. L. (2012). Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 8:e1002764. doi: 10.1371/journal.pgen.1002764
Pan, Q., Shai, O., Lee, L. J., Frey, B. J., and Blencowe, B. J. (2009). Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 40, 1413–1415. doi: 10.1038/ng.259
Parker, J., Tsagkogeorga, G., Cotton, J. A., Lio, Y., Provero, P., Stupka, E., et al. (2013). Genome-wide signatures of convergent evolution in echolocating mammals. Nature 502, 228–231. doi: 10.1038/nature12511
Petersen, T. N., Brunak, S., von Heijne, G., and Nielsen, H. (2011). SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786. doi: 10.1038/nmeth.1701
Phillips, C. D., Butler, B., Fondon, J. W., Mantilla-Meluk, H., and Baker, R. J. (2013). Contrasting evolutionary dynamics of the developmental regulator PAX9, among bats, with evidence for a novel post-translational regulatory mechanism. PLoS ONE. 8:e57649. doi: 10.1371/journal.pone.0057649
Phillips, C. J., Grimes, G. W., and Forman, G. L. (1977). “Oral biology,” in Biology of bats of the New World Phyllostomatidae. Part, I. I. I., eds R. J. Baker, J. K. Jones, and D. C. Carter (Lubbock, TX: Texas Tech University Press), 121–203.
Rigbi, M., Orevi, M., and Eldor, A. (1996). Platelet aggregation and coagulation inhibitors in leech saliva and their roles in leech therapy. Semin. Thromb. Hemost. 22, 273–278. doi: 10.1055/s-2007-999019
Robson, S. C., Kaczmarek, E., Siegel, J. B., Candinas, D., Koziak, K., Millan, M., et al. (1997). Loss of ATP diphosphohydrolase activity with endothelial cell activation. J Exp. Med. 185, 153–163. doi: 10.1084/jem.185.1.153
Ruder, T., Sunagar, K., Undheim, E. A., Ali, S. A., Wai, T. C., Low, D. H., et al. (2013). Molecular phylogeny and evolution of the proteins encoded by coleoid (cuttlefish, octopus, and squid) posterior venom glands. J. Mol. Evol. 76, 192–204. doi: 10.1007/s00239-013-9552-5
Schäfer, K., Braun, H. A., and Kûrten, L. (1988). Analysis of cold and warm receptor activity in vampire bats and mice. Pflug. Arch. 412, 188–194.
Schutt, B. (2008). Dark Banquet: Blood and the Curious Lives of Blood-feeding Creatures. New York, NY: Random House Digital, Inc.
Simpson, J. T., Wong, K., Jackman, S. D., Schein, J. E., Jones, S. J., and Birol, I. (2009). ABySS: a parallel assembler for short read sequence data. Genome Res. 19, 1117–1123. doi: 10.1101/gr.089532.108
Spirov, A. V., Sabirov, M. A., and Holloway, D. M. (2012). In Silico evolution of gene cooption in pattern-forming gene networks. ScientificWorldJournal. 2012:560101. doi: 10.1100/2012/560101
Tellgren-Roth, A., Dittmar, K., Massey, S. E., Kemi, C., Tellgren-Roth, C., Savolainen, P., et al. (2008). Keeping the blood flowing—plasminogen activator genes and feeding behavior in vampire bats. Naturwissenschaften 96, 39–47. doi: 10.1007/s00114-008-0446-0
The UniProt Consortium (2012). Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Res. 40, D71–D75. doi: 10.1093/nar/gkr981
Valenzuela, J. G., Belkaid, Y., Rowton, E., and Ribeiro, J. M. (2001). The salivary apyrase of the blood-sucking sand fly Phlebotomus papatasi belongs to the novel Cimex family of apyrases. J. Exp. Biol. 204, 229–237.
Wang, E. T., Sandberg, R., Luo, S., Khrebtukova, I., Zhang, L., Mayr, C., et al. (2008). Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476. doi: 10.1038/nature07509
Wang, S., Herndon, M. E., Ranganathan, S., Godyna, S., Lawler, J., Argraves, W. S., et al. (2004). Internalization but not binding of thrombospondin-1 to low density lipoprotein receptor-related protein-1 requires heparan sulfate proteoglycans. J. Cell Biochem. 91, 766–776. doi: 10.1002/jcb.10781
Wheelan, S. J., Church, D. M., and Ostell, J. M. (2001). Spidey: a tool for mRNA-to-genomic alignments. Genome Res. 11, 1952–1957.
Wistow, G. (1993). Lens crystallins: gene recruitment and evolution dynamism. Trends Biochem. Sci. 18, 301–306. doi: 10.1016/0968-0004(93)90041-K
Keywords: vampire bat, convergent evolution, alternative splicing, gene recruitment, phyllostomid bats
Citation: Phillips CD and Baker RJ (2015) Secretory gene recruitments in vampire bat salivary adaptation and potential convergences with sanguivorous leeches. Front. Ecol. Evol. 3:122. doi: 10.3389/fevo.2015.00122
Received: 28 April 2015; Accepted: 09 October 2015;
Published: 18 November 2015.
Edited by:
Bernd Schierwater, TiHo Hannover, GermanyReviewed by:
Dmitry Yurievich Sherbakov, Limnological Institute, RussiaMark Siddall, American Museum of Natural History, USA
Copyright © 2015 Phillips and Baker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Caleb D. Phillips, Y2FsZWIucGhpbGxpcHNAdHR1LmVkdQ==