
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Earth Sci., 27 October 2021
Sec. Biogeoscience
Volume 9 - 2021 | https://doi.org/10.3389/feart.2021.722215
 Jonathan Schenk1*
Jonathan Schenk1* Henrique O. Sawakuchi1
Henrique O. Sawakuchi1 Anna K. Sieczko1
Anna K. Sieczko1 Gustav Pajala1
Gustav Pajala1 David Rudberg1
David Rudberg1 Emelie Hagberg1Kjell Fors1
Emelie Hagberg1Kjell Fors1 Hjalmar Laudon2Jan Karlsson3
Hjalmar Laudon2Jan Karlsson3 David Bastviken1
David Bastviken1Methane (CH4) is an important component of the carbon (C) cycling in lakes. CH4 production enables carbon in sediments to be either reintroduced to the food web via CH4 oxidation or emitted as a greenhouse gas making lakes one of the largest natural sources of atmospheric CH4. Large stable carbon isotopic fractionation during CH4 oxidation makes changes in 13C:12C ratio (δ13C) a powerful and widely used tool to determine the extent to which lake CH4 is oxidized, rather than emitted. This relies on correct δ13C values of original CH4 sources, the variability of which has rarely been investigated systematically in lakes. In this study, we measured δ13C in CH4 bubbles in littoral sediments and in CH4 dissolved in the anoxic hypolimnion of six boreal lakes with different characteristics. The results indicate that δ13C of CH4 sources is consistently higher (less 13C depletion) in littoral sediments than in deep waters across boreal and subarctic lakes. Variability in organic matter substrates across depths is a potential explanation. In one of the studied lakes available data from nearby soils showed correspondence between δ13C-CH4 in groundwater and deep lake water, and input from the catchment of CH4 via groundwater exceeded atmospheric CH4 emissions tenfold over a period of 1 month. It indicates that lateral hydrological transport of CH4 can explain the observed δ13C-CH4 patterns and be important for lake CH4 cycling. Our results have important consequences for modelling and process assessments relative to lake CH4 using δ13C, including for CH4 oxidation, which is a key regulator of lake CH4 emissions.
Organic matter (OM) transported from catchments is actively processed and transformed in lakes, leading to burial in sediments, carbon (C) gas release to the atmosphere, or downstream transport via rivers ultimately to the sea (Cole et al., 2007; Tranvik et al., 2018). Methane (CH4) production (methanogenesis) plays an important role in the overall processing of OM in lakes by allowing large amounts of OM buried in the sediments to be remineralized, with methanogenesis found to correspond to 20–56% of whole lake C mineralization (Bastviken, 2009). Remineralized OM can subsequently either be emitted to the atmosphere, directly as CH4 (Bastviken et al., 2004) or as carbon dioxide (CO2) after oxidation of CH4 by methanotrophic bacteria, or it can be reintroduced in lake food webs via the methanotrophic biomass (Bastviken et al., 2003; Jones and Grey, 2011; Grey, 2016). With respect to emissions, lakes represent one of the largest natural sources of atmospheric CH4 (Saunois et al., 2020), yet extensive in-lake CH4 oxidation (MOX) is believed to mitigate as much as 50% and up to more than 90% of the potential CH4 emissions, making MOX a key process for the global CH4 cycling (Bastviken, 2009; Reeburgh, 2014). From the food web perspective, CH4-C has been found important to zooplankton during some seasons (Taipale et al., 2009) and has been detected in fish biomass (Sanseverino et al., 2012).
The measurement of the stable isotope ratio of 13C to 12C in different compartments of the C cycle in lakes has been a powerful tool in the past decades to study the fate of C derived from methanogenesis (e.g., Kankaala et al., 2006; Tsuchiya et al., 2020). The 13C:12C ratio is often expressed relative to a standard (Vienna Peedee belemnite with a 13C:12C ratio of 0.0112372) according to
The quality of δ13C as a tracer of C derived from methanogenesis stems from the fact that biogenic CH4 is highly depleted in δ13C, such that δ13C values in biogenic CH4 (δ13C-CH4) are low (−110‰ to −50‰; Whiticar, 1999) compared to atmospheric CO2 (−7‰; Peterson and Fry, 1987) and primary producers obtaining their C from atmospheric CO2, like terrestrial plants (−27‰ in average for C3 plants; Kohn, 2010). Even in algae assimilating C that has been depleted in 13C isotope by prior metabolic processes, δ13C values stay above −45‰ in lakes (de Kluijver et al., 2014). Figure 1 shows the range of δ13C values observed in several sources of organic matter to the sediments of lakes. All these values are higher than −45‰. Therefore, microbially produced CH4 is one of the most 13C-depleted C-compounds found in nature, facilitating the study of its fates based on measurements of δ13C.
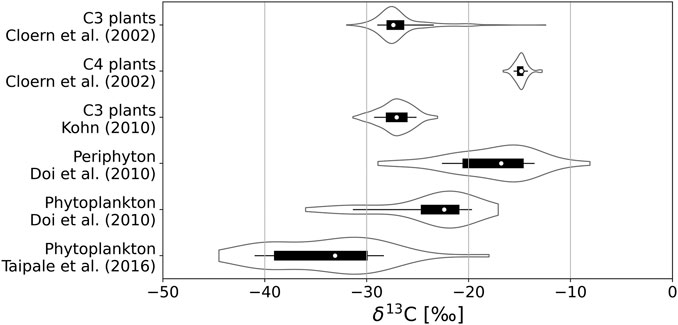
FIGURE 1. Ranges of δ13C values observed in C3 and C4 plants, in phytoplankton and in benthic algae (periphyton) in previous studies. In each distribution, the white dot indicates the median, the thick black bar indicates the interquartile range, the thin black line indicates the range of values between the 10- and 90-percentiles, and the wavy curves indicate the Gaussian kernel density estimate of the distribution of values.
An important methodological requirement for studies using δ13C to investigate C cycling in lakes is to determine a reference δ13C-CH4 value for the source of CH4, sometimes referred to as the endmember δ13C signal representative of the CH4 produced in the sediments. Just as the enzyme systems involved in methanogenesis discriminate strongly against 13C, this is also the case for enzymatic reactions by methanotrophic microorganisms leading to MOX. As a result, the pool of CH4 that remains after being exposed to MOX gradually gets more enriched in 13C (i.e., δ13C-CH4 becomes less negative) the greater proportion is oxidized while CO2 and microbial biomass formed by MOX get more 13C depleted. Accordingly, the difference between endmember δ13C-CH4 and δ13C of partially oxidized CH4 pools can be used to estimate the fraction of the original CH4 being oxidized. In addition, δ13C measured in C pools can be compared to the source δ13C-CH4 to estimate the fraction of C that has a methanogenic origin. Therefore, δ13C analyses are very useful to quantify complex ecosystem processes involving CH4 from measurements of limited numbers of samples. However, the success of this approach critically depends on accurate and reliable knowledge of the relevant endmember δ13C-CH4 values.
Several factors can affect the endmember δ13C-CH4 in sediments of lakes. One is the bacterial community composition, and more specifically the share of different methanogenic pathways that are active (Conrad, 2005). The two main methanogenic pathways are based on CO2 reduction (hydrogenotrophic methanogenesis; HM) and acetate dependent (acetoclastic methanogenesis; AM), and different enzyme systems can fractionate (i.e., discriminate) differently against 13C. A second aspect is the δ13C composition of the substrates used by methanogens (Conrad, 2005). This applies to organic substrates, and to the CO2 or dissolved inorganic C (DIC) used in HM (dissolved CO2 is connected to the whole DIC pool via the carbonic acid chemical equilibrium system). Bacterial communities and substrate composition differ from one lake to another, leading to varying δ13C-CH4 endmember values in different systems (Rinta et al., 2015).
Recent results from a few subarctic lakes also indicate pronounced differences in endmember δ13C-CH4 within lakes, e.g., between littoral and profundal regions (Cadieux et al., 2016; Thompson et al., 2016; Wik et al., 2020). Such variability would have important implications for the applicability of the δ13C approach to trace CH4-related C cycling but has rarely been investigated systematically across different lake types and it is not clear if the observed patterns are widespread and consistent features of lakes or specific to the few subarctic systems studied so far. In this study, we compare δ13C-CH4 values in littoral sediments, where the water was approximately 1 m deep, and in the deep, anoxic hypolimnion in the profundal zone, representing commonly used endmembers for estimation of MOX near the oxyclines of sediments or water columns, of six boreal lakes with different characteristics. We also carry out more detailed studies of one of the lakes where information about CH4 input via groundwater (GW) from an adjacent mire could be generated. We hypothesize that δ13C-CH4 values in deep, anoxic water are more negative than in littoral sediments, as previous observations mentioned above have shown, but that the magnitude of the difference increases in lakes with higher contrasts in the composition of OM reaching littoral and profundal sediments.
Samples were collected in six lakes across Sweden during the ice-free season in 2017, 2018, and 2019 (see Table 1 for their characteristics). STJ, LJR, and NAS are small lakes, located in the same region in the north of Sweden, that cover a range of dissolved organic C (DOC) levels. They are surrounded by coniferous forests (Scots pine and Norwegian spruce) with some presence of birch. Human activities around these lakes are connected to forestry. PAR and VEN are located in the southeastern part of Sweden. PAR is a small humic lake surrounded by a forest where Scots pine and Norwegian spruce dominate, and birch is also present. VEN is a relatively big and eutrophic lake. It is surrounded by some forested areas and agricultural land. SGA is a middle-size, deep, and clear lake located in the southwestern part of Sweden. It is surrounded by forests with a majority of Scots pines and Norwegian spruces and some birches.
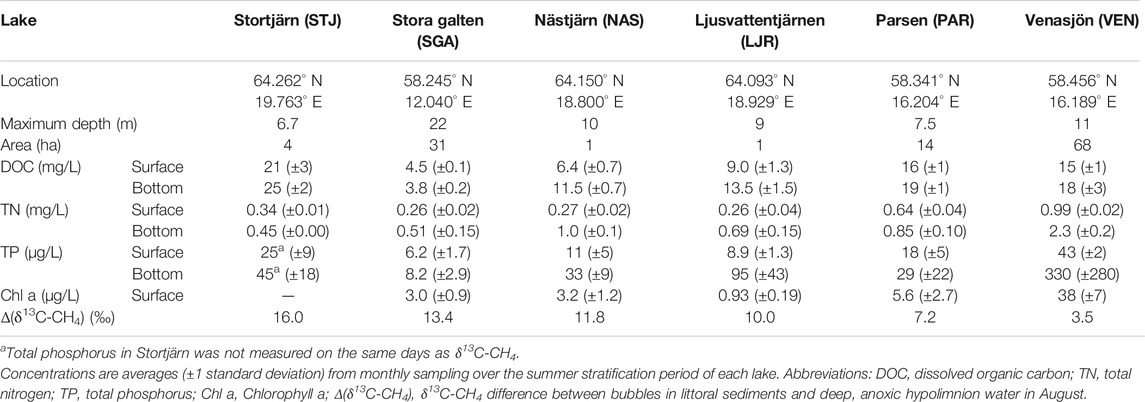
TABLE 1. Characteristics of the lakes investigated in the present study.
All lakes were sampled monthly during the summer stratification period (except STJ, where sampling was conducted several times but only in August) after some CH4 had accumulated in anoxic parts of the water column close to the bottom to ensure a minimum influence of MOX on the deep water CH4 sampled.
The δ13C-CH4 at the bottom of the lakes was measured in anoxic water less than 1 m above the sediments, near the deepest point of each lake. A possibility exists that δ13C values measured in dissolved CH4 in the water column above the sediments differ from δ13C-CH4 values in the sediment bubbles if anaerobic oxidation of CH4 (AOM) occurs at the interface between sediments and water column. However, the net effect of AOM would be to increase δ13C-CH4 values in the water overlaying the sediments compared to δ13C-CH4 values in the sediments themselves (Rinta et al., 2015; Marcek et al., 2021). Therefore, using our initial assumption that δ13C-CH4 values are lower in the deep water of the profundal zone than in littoral sediment bubbles, differences in δ13C-CH4 values between littoral sediment bubbles and dissolved CH4 at the bottom of the water column provide a conservative estimate of the difference in δ13C-CH4 values between profundal and littoral sediment bubbles.
Headspace extraction was used to collect δ13C-CH4 samples from the deep water. Three methods were applied for the extraction. A first method consisted of sampling water at a given depth using a plastic Ruttner-type water sampler equipped with a tubing at the bottom. The water was then transferred to a 1.2 L plastic bottle by inserting the tube of the water sampler down to the bottom of the bottle. To prevent the build-up of air bubbles in the bottle and the exchange of gas between the water and the air during the transfer, the bottle was overflown with 2 L of water. The bottle was then closed with a rubber stopper equipped with two tubes attached to three-way luer-lock valves on the outside. A headspace was created in the bottle by injecting 60 ml of synthetic air containing only molecular nitrogen (N2), O2 and argon, and by simultaneously retrieving 60 ml of water through the tubes. The synthetic air used for the headspace was the same as the gas used as carrier by the instrument on which these samples were analyzed (see below). The bottle was then shaken manually for 3 min to equilibrate the gas in the water with the newly created headspace. Finally, the equilibrated gas in the headspace was retrieved from the bottle using a 60 ml syringe (Becton Dickinson, Franklin Lakes, New Jersey, United States) and transferred to a glass vial capped with a 10 mm butyl rubber stopper. The vial was manually evacuated using another 60 ml syringe right before transferring the sample. The residual CH4 concentration in the vial after evacuation did not affect the measured δ13C-CH4 because of the high CH4 concentration in samples stored in manually evacuated vials (concentrations in the vials ranged between 76 ppm and 137000 ppm, and the residual gas only contained atmospheric CH4 concentration). Also, the presence of O2 in the vial did not cause CH4 to be oxidized because only gas and no water was injected. Upon sample injection in the vial, an overpressure was created in the vial by injecting a larger volume of gas than the volume of the vial. A second extraction method consisted of sampling 60 ml of water at a given depth either through a long tube attached to a weight and connected to a 60 ml syringe at the surface, or by transferring water collected with the Ruttner sampler to a 60 ml syringe. Water was then transferred from the syringe to a 118 ml vial that had been flushed, filled with N2, and amended with 0.2 ml of 85% H3PO4 prior to sampling. The acid was added because the vials were also used to collect samples for DIC, and to prevent biological production and oxidation of CH4 in the vials. When prefilling the vials with N2 an overpressure was created, which was released just before injecting the sample. Thereby, the total pressure of the vial headspace after sample injection could be calculated from the total barometric pressure when sampling, the sample volume, and temperature, and the equilibrated headspace constituted the gas sample analyzed. The third extraction method consisted in collecting 30 ml of water and 30 ml of air in a 60 ml syringe. CH4 was extracted from the water to the headspace by shaking the syringe for at least 1 min and the headspace air was transferred to a manually pre-evacuated vial. When using this method, a sample of atmospheric air was taken separately to correct for the effect of atmospheric CH4 on the concentration and δ13C-CH4 in the sample extracted from the water.
Bubbles from littoral sediments were collected on the same days as deep-water samples. Locations where bubble samples were taken were not always similar among sampling occasions in each lake. The sampling procedure consisted in holding a plastic funnel connected to a 60 ml syringe upside down under water, physically disturbing the sediment under the funnel, and collecting released bubbles in the funnel syringe as they rose towards the surface. The collected gas was injected to a manually pre-evacuated vial (12–30 ml). Enough gas was transferred to the vial to create a minimum of 10 ml overpressure.
δ13C-CH4 samples were analyzed on a cavity ring-down spectrometer (G2201-I Isotopic Analyzer, Picarro, Santa Clara, California, United States). Samples with high concentration of CH4 were diluted with high purity synthetic air (80% N2 and 20% O2) to fall in the optimal range of concentrations for the instrument. To correct for non-linearity and guarantee the accuracy of the measurements in this range of concentrations, calibration curves were derived from the analysis of two standards with δ13C-CH4 values of −66.5 ± 0.2‰ and −23.9 ± 0.2‰ (Isometric Instruments, Victoria, British Columbia, Canada), that were diluted to 20–25 different concentrations ranging logarithmically from 1 to 200 ppm CH4. All values obtained after the analysis were corrected according to the calibration curves.
In all lakes except for STJ, where water chemistry is routinely monitored by the research station managing the site, water samples for DOC, DIC, total nitrogen (TN), total phosphorus (TP), and Chlorophyll a (Chl a) analysis were collected on the same days as the δ13C-CH4 samples. Water was collected near the deepest point of each lake at 1 m depth and 1 m above the sediments using a Ruttner sampler equipped with a tubing at the bottom. For each depth, water was transferred to one 4 L cubitainer for DOC, DIC, TN, and Chl a analysis and two 125 ml HDPE bottles for TP analysis. The samples were kept in cooling boxes in the field. Within the same day as collection, a part of the water from the cubitainers was directly transferred to 50 ml PP tubes (Sarstedt, Nümbrecht, Germany) and stored at 4°C for subsequent TOC and TN analysis. Another part of the water from the cubitainers was filtered using pre-combusted 0.7 µm pore size grade GF/F filters (Whatman, Maidstone, United Kingdom) and the filtrate was stored in 50 ml PP tubes at 4°C for subsequent DOC and DIC analysis. The remaining water was filtered using 1.2 µm nominal pore size grade GF/C (Whatman) filters that were frozen immediately after filtration for storing them until subsequent Chl a analysis.
DOC, DIC, and TN samples were analyzed with a TOC/TN analyzer (Shimadzu, Kyoto, Japan). Samples containing more than 20 mg TOC/L were diluted with deionized water before analysis. Standard samples containing 10 mg TOC/L and 1, 2 or 5 mg TN/L were also analyzed and were used to correct the results from the lake samples. TP samples were analyzed on a continuous flow analyzer (AutoAnalyzer II, Bran + Luebbe) according to the AutoAnalyzer method no. G-175-96 Rev. 16 based on Murphy and Riley (1962). Chl a was extracted from the GF/C filters in methanol and measured by spectrophotometry following the Swedish Standard SS 28170, which is based on Richards and Thompson (1952).
Based on the measured concentrations of DIC, a value expressing the relative contribution of respired C to the DIC pool in the hypolimnion, the hypolimnetic excess DIC, was calculated as:
where DICbottom and DICsurface are the concentrations of DIC at the bottom and surface of the lake, respectively.
At STJ extended sampling was carried out warranting additional description. STJ is located in the Krycklan Catchment Study area in northern Sweden (Laudon et al., 2013). The lake has a maximum and mean depth of 6.7 and 2.7 m, respectively (Denfeld et al., 2018). It is separated in two main basins along a north-south axis, the western basin being about half the size of the eastern basin (Figure 2). More than half of the shoreline of the lake is covered by a Sphagnum mire (Figure 2). The lake catchment has as surface area of 65 ha and is composed at 39.5% of wetland and at 54% of forest (Figure 2; Laudon et al., 2013). The main types of soils in the catchment are till deposits covered locally by thin forest soil and peat. Average yearly precipitation in the catchment is 614 mm (Laudon et al., 2013).
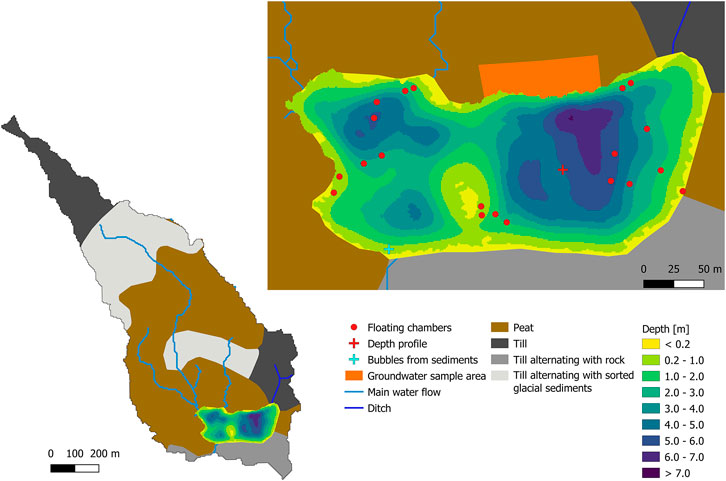
FIGURE 2. Left: Types of soil and theoretical water flow direction in the catchment of Stortjärn. The outlet of the lake is located at the southwestern shore. The water flow direction in the mire on the northwestern side of the lake is calculated based on the DEM of the catchment. A small ditch forms a small inlet in the northeastern corner of the lake. Right: Bathymetric map of the lake with locations where groundwater, water column depth profiles and bubbles in littoral sediments were sampled, and positions of the floating chambers used to measure CH4 fluxes at the surface of the lake. ©Lantmäteriet, i2012/901.
The additional mapping of lake CH4 fluxes of relevance carried out in STJ included water samples taken in the mire to measure δ13C-CH4 in the GW, measurements of daily CH4 emissions at the water-atmosphere interface of the lake using floating chambers, and estimation of daily input of CH4 from the mire using a water balance of the lake.
Water samples for CH4 concentration were collected using a 10 ml plastic syringe (Becton Dickinson). After rinsing the syringe, 5 ml of water were collected at 10 cm depth making sure that no air bubble was introduced in the syringe. The water was transferred to a 22 ml vial capped with a butyl rubber stopper that had been amended with 0.1 ml of 85% H3PO4 for sample preservation and then flushed and filled with N2 prior to sampling. The vials were prepared with a N2 overpressure that was released just prior to injecting the water sample to equilibrate the pressure towards the present barometric pressure and ensure that vials did not leak before used for samples. The concentration of dissolved CH4 in the water was calculated from the sum of 1) the CH4 content in the vial headspace measured with gas chromatography (GC System 7890A with a 1.8 m × 3.175 mm Porapak Q 80/100 column from Supelco, a methanizer and a flame ionization detector, Agilent Technologies, Santa Clara, California, United States) and determined using the common gas law, and 2) remaining CH4 dissolved in the water in the vial determined from headspace concentration using Henry’s law and the temperature adjusted Henry’s solubility constant for CH4 in freshwater.
The rate of CH4 emissions at the lake’s surface was measured at different locations using flux chambers (Bastviken et al., 2004). These flux chambers were made of plastic buckets (6 L, 800 cm2 opening area) equipped with floats and placed at the surface of the lake. The flux of CH4 between the surface water and the atmosphere was determined by measuring the change in CH4 concentration in the chamber over time. Each chamber was attached to a separate float connected to an anchor to allow the chambers to move freely with waves and water level changes. When chambers were deployed, atmospheric air samples and surface water samples were collected near some of the chambers. Atmospheric air samples taken 20 cm above the surface of the water were used to represent initial concentration in the chambers. Surface water samples were collected as described previously. After a deployment period, each chamber headspace was sampled through a polyurethane tube (5 and 3 mm outer and inner diameter, respectively) equipped with a three-way luer-lock valve. These samples were collected prior to redeploying the chambers to start a new measurement cycle. The deployment period of the chambers ranged from 4 to 48 h during the sampling period. Note that CH4 was highly supersaturated in the studied lakes and that equilibration with chambers therefore took much longer than 48 h—in contrast to CO2 which typically equilibrates in flux chambers within a few hours and therefore demands much shorter chamber deployments than CH4. Samples of gas from the chambers and of ambient air were collected using three 60 ml syringes for having a total volume of 180 ml of gas. They were transferred to 22 ml vials with butyl rubber stoppers. Each vial was flushed using 165 ml of the collected gas. The remaining 15 ml of gas were added after removing the gas outlet needle from the vial stopper, creating an overpressure in the vials. All CH4 gas samples were analyzed with gas chromatography as above. The overpressure in the vials containing the samples was checked as a leakage indicator and was released to equilibrate vials with atmospheric pressure just before the analysis.
GW samples for δ13C-CH4 analyses were collected in the mire on the north side of STJ with the use of piezometers (Figure 2). GW samples were collected at different distances from the lake and at 1 m depth in the mire. After discarding the water that was trapped in the tubing, 60 ml of water were collected using a 60 ml syringe. This water was transferred to a 118 ml vial prepared as explained in the description of the second method used to collect δ13C-CH4 water samples.
Air temperature, relative humidity, barometric pressure, and wind speed were measured by a weather station located on a platform in the center of the lake. Data were averaged and logged every 10 min. Daily precipitation data were provided by a rain gauge located 2 km away from the lake. Water temperature was logged every 10 min by sensors positioned every 25 cm from the surface to the bottom of the lake at the platform where weather parameters were measured. The water level in the lake was averaged and logged every 10 min. Daily discharge in the outlet of the lake was measured at a V-notch weir located 30 m downstream of the lake.
CH4 fluxes at the surface of the lake were measured using the method described in Bastviken et al. (2004). The calculation of diffusive flux relies on Fick’s law of diffusion:
where F is the flux of CH4 across the water-air interface (mmol m−2 d−1), k is the piston velocity (m d−1), Cw is the concentration of CH4 in the surface water (µmol L−1) and Cfc is the calculated equilibrium concentration of CH4 in water corresponding to the flux chamber headspace concentration (µmol L−1). Using this equation and measurements of Cw and Cfc after some time, k is solved for by accounting for the gradual change in flux due to the increasing Cfc over time, i.e., using a non-linear regression over the measurement cycle as described in Bastviken et al. (2004). Then, k for chambers receiving only diffusive flux (see below) was used with Cw to estimate instantaneous diffusive flux, setting Cfc to the initial air background CH4 level.
Floating chambers accumulate CH4 emitted by both diffusion and ebullition. Their relative contribution to the measured total flux is estimated based on the method given in Bastviken et al. (2004). The values of k derived for different chambers from the measured flux and water concentrations during the same period were divided by the minimum k value observed for this period. Chambers for which this k ratio was below a certain threshold [we used a threshold of 2 in this study based on independent findings in Bastviken et al. (2004); Schilder et al. (2013)] were assumed to receive CH4 only by diffusion. Therefore, it was assumed that the diffusive part of the flux is rather homogeneous across the lake surface (within the 2-fold variability on k combined with variability on Cw) and that not all chambers collect CH4 bubbles. The contribution of ebullition in the chambers where the k ratio was above the threshold was calculated by subtracting the average flux from the chambers collecting only diffusive flux from the total flux of each chamber.
The water balance is used here to estimate the potential lateral input of CH4 to STJ, which can be approximated by the volume of water flowing to the lake multiplied by the concentration of CH4 in this water. The water balance of the lake expressed for the inflow of water to the lake is:
where Inflow is the total amount of water entering the lake from the catchment, Outflow is the water leaving the lake in the outlet, E is the evaporation at the lake surface, A is the surface area of the lake, ∆h is the water level change in the lake and Plake is the precipitation falling directly in the lake. The surface area of the lake was assumed to be constant over the range of water level changes. Evaporation was calculated according to Sartori (2000). All terms on the right-hand side of the equation could be calculated from parameters measured routinely in or around the lake.
To estimate the lateral CH4 input to the lake, we assume that the water input is primarily in the form of GW flowing through the mire. This assumption is supported by the catchment distribution (Figure 2), the absence of any stream inlet to STJ, except for a small ditch in the northeastern corner of the lake, and the negligible input of CH4 with GW from the forested shoreline, confirmed by previous measurements of GW CH4 (Denfeld et al., 2020).
In four lakes (STJ, SGA, NAS, LJR), the δ13C-CH4 in the anoxic hypolimnion ranged from −80‰ to −76‰ during the measurement period (Figure 3). SGA is deep and oligotrophic, whereas the other three lakes are small and moderately to highly humic. By contrast, in PAR, which is also a humic lake located farther south but with higher TN concentrations and potential influence from more nutrient rich soils, δ13C-CH4 at the bottom ranged between −75‰ and −73‰. In VEN, a relatively large and eutrophic lake, the corresponding values ranged between −71‰ and −69‰. In all lakes, δ13C-CH4 values from littoral sediments ranged between −71‰ and −58‰, except for LJR where lower values of −78‰ to −72‰ were observed between June and August. In the second half of August, when lakes were likely to have been stratified for an extended period and the only time when data was available for all lakes, the difference between δ13C-CH4 measured in the littoral sediment bubbles and in the anoxic hypolimnion water (Δ(δ13C-CH4)LSB-AHW) was 16.0‰ in STJ, 13.4‰ in SGA, 11.8‰ in NAS, 10.0‰ in LJR, 7.2‰ in PAR, and 3.5‰ in VEN (Table 1). δ13C-CH4 was lower in the anoxic hypolimnion than in the littoral sediments in all six lakes investigated (Figure 3).
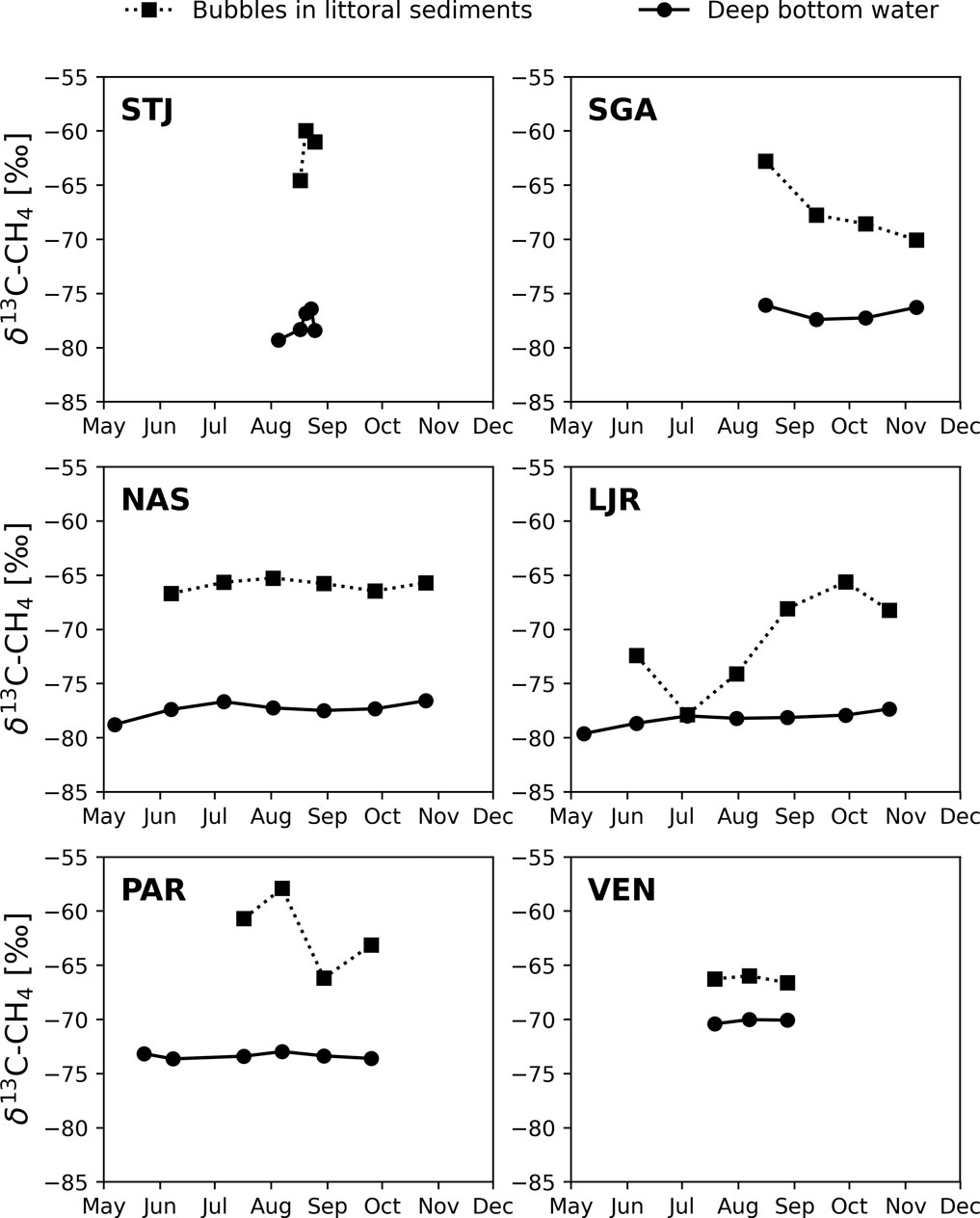
FIGURE 3. δ13C-CH4 in the anoxic water close to the bottom at the deepest point (dots with continuous lines) and in the bubbles in littoral sediments (squares with dotted lines) in six lakes.
Δ(δ13C-CH4)LSB-AHW showed a tendency to decrease with increasing surface water concentration of TP if considering all times where both these variables were available (Figure 4). This tendency is partly a result of clearly lowest Δ(δ13C-CH4)LSB-AHW in the most nutrient rich lake VEN. In addition, some lakes showed decreasing Δ(δ13C-CH4)LSB-AHW with increasing TP when comparing different sampling times within the same lake (Figure 4). More generally, TP may not be an optimal indicator of lake productivity, e.g., as TP can be bound to DOC in humic boreal lakes, but we also observe that the two lakes with the smallest Δ(δ13C-CH4)LSB-AHW in August (VEN and PAR), presumably reflecting the integrated result of the summer productivity, had highest TP, TN, and Chl a levels (Table 1).
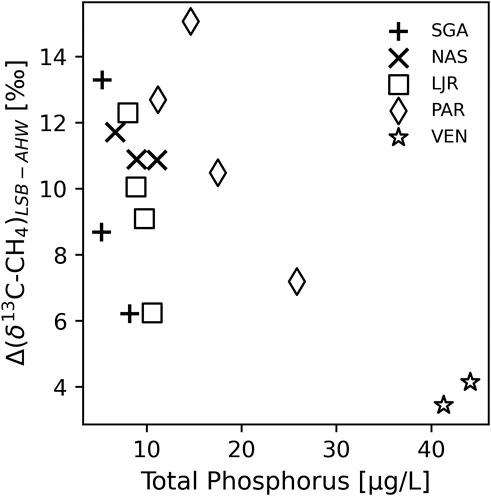
FIGURE 4. Difference in δ13C-CH4 between littoral sediment bubbles (LSB) and anoxic hypolimnion water (AHW) compared to total phosphorus concentration in surface water in the middle of each lake. Each point corresponds to 1 day when both δ13C-CH4 and total phosphorus were sampled.
We also observed that δ13C-CH4 in the anoxic, hypolimnetic water was lower in lakes with higher hypolimnetic excess DIC (defined in Eq. 2; Figure 5A). In VEN, where DIC concentrations were relatively homogeneous over the water column (hypolimnetic excess DIC = 1–2), the δ13C-CH4 values at the bottom were around −70‰ (Figure 5A). PAR was an intermediate case with hypolimnetic excess DIC of 2–4 and bottom δ13C-CH4 values between −73‰ and −74‰ (Figure 5A). The other lakes all had hypolimnetic excess DIC of 3–10 and bottom δ13C-CH4 values between −76‰ and −80‰ (Figure 5A). However, no particular relation between Δ(δ13C-CH4)LSB-AHW and hypolimnetic excess DIC could be observed (Figure 5B).
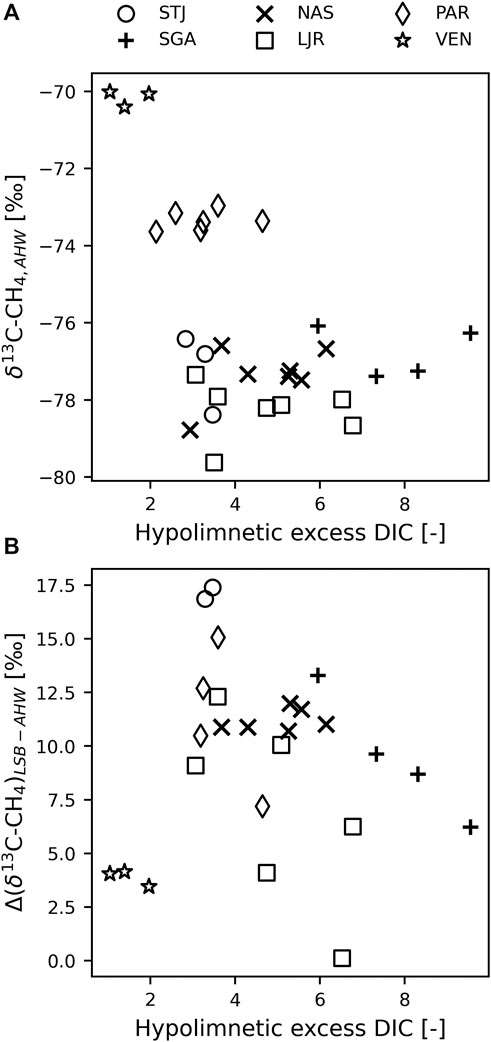
FIGURE 5. (A) δ13C-CH4 in anoxic hypolimnion water (AHW) compared to the hypolimnetic excess dissolved inorganic carbon (DIC), calculated as the difference in DIC concentration between bottom and surface water normalized by the concentration at the surface. (B) δ13C-CH4 difference between AHW and littoral sediment bubbles (LSB) compared to the hypolimnetic excess DIC. In both panels, each point corresponds to 1 day when both δ13C-CH4 and DIC were sampled.
Overall, it was clear and consistent across all lakes that bottom anoxic water δ13C-CH4 was more negative (more depleted in 13C) than littoral CH4 bubbles, with indications of possible links to lake productivity (here estimated via proxies), and a proxy for in lake respiration (hypolimnetic excess DIC).
The δ13C-CH4 values measured in the surface water of STJ ranged between −57.2‰ and −47.4‰ with an average of −53.6‰ (Figure 6). In the bubbles collected in the littoral sediments, δ13C-CH4 ranged between −64.9‰ and −58.9‰ with an average of −62.6‰ (Figure 6). In the GW, δ13C-CH4 ranged between −81.4‰ and −70.1‰ with an average of −76.2‰ (Figure 6). The water samples taken at different depths at the deepest point of the lake show that δ13C-CH4 decreased with depth. The average value was −56.2‰ between 0 and 1.5 m and went down to −76.8‰ at depths below 3 m (Figure 6).
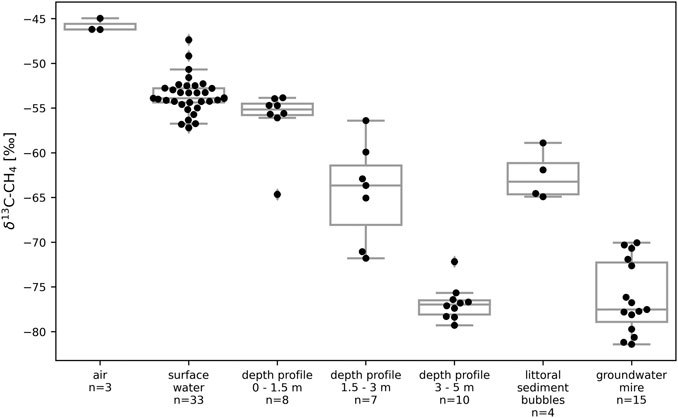
FIGURE 6. Boxplots and individual measurements of δ13C-CH4 values in the air, in lake water (surface and depth profiles) and littoral sediments, and in the mire groundwater in Stortjärn in August. “Surface water” data represents spatial variability at the surface of the lake while “depth profile” data shows short term variability at different depths in the central part of the lake. Figure 2 shows a map with all sampling locations. Boxes extend from the lower quartile to the upper quartile. Whiskers extend to the nearest measurements within bounds corresponding to one and a half time the width of the boxes.
Precipitation measurements in the catchment of the lake and the water budget of the lake show that three major rain events occurred during the sampling period, each lasting between one and 2 days and releasing decreasing amounts of water to the lake over several days after each rain event. The daily input of CH4 from the mire to the lake was calculated by multiplying the daily water inflow from the mire with the average concentration of CH4 that was measured in the GW of the mire (178 µM; very close to the average concentration measured in the same mire over the entire ice-free period in another study; Denfeld et al., 2020). The daily input of CH4 from the mire ranged from 0.14 to 6.8 kg CH4 with an average of 1.6 kg CH4 (Figure 7).
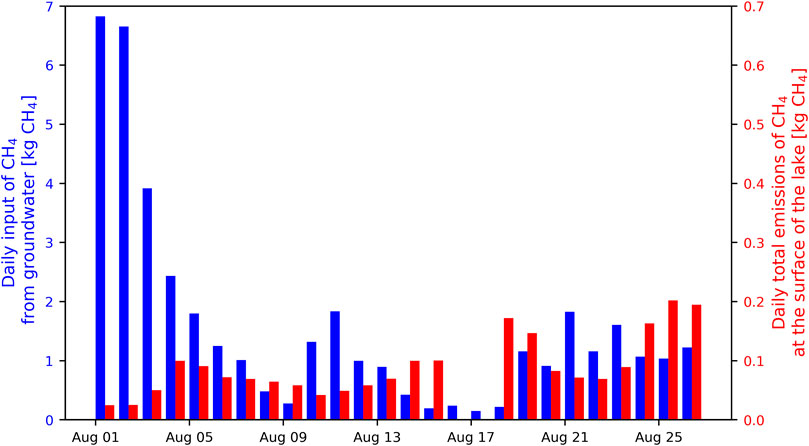
FIGURE 7. Comparison between the estimated daily input of CH4 from the mire to Stortjärn (blue bars) and the total amount of CH4 emitted each day at the surface of the lake (red bars; note the tenfold difference between left and right y-axes scales). Emissions of CH4 at the surface of the lake were not measured on 16–17 August.
Fluxes of CH4 measured at the surface of the lake ranged between 0.01 and 64 mg CH4 m−2d−1, with mean and median values of 2.9 and 1.4 mg CH4 m−2d−1, respectively. Daily whole-lake emissions of CH4, calculated as the average of all chamber measurements for each day multiplied by the surface area of the lake, ranged from 24 g CH4 to 200 g CH4 with an average of 90 g CH4. Flux rates were one to two orders of magnitude smaller than the calculated daily CH4 input from the mire (Figure 7).
The thermal stratification in the lakes resulted in typical depth profiles of CH4 concentrations and δ13C-CH4 values, with low CH4 concentrations and high δ13C-CH4 values in the oxic epilimnion, and high CH4 concentrations and low δ13C-CH4 values in the anoxic hypolimnion. The δ13C-CH4 values we observed in the anoxic hypolimnion of all six lakes are in the same range as observations previously reported in other lakes (Rinta et al., 2015; Thompson et al., 2016). Our results also show that CH4 was more 13C enriched in littoral sediment bubbles of all six lakes than in anoxic water in the hypolimnion (Figure 3). Considering that CH4 in profundal sediment bubbles would most likely be more δ13C depleted than in the overlaying water column (Rinta et al., 2015; Marcek et al., 2021), particularly in cases where significant AOM occurs, it implies that even larger differences in δ13C values than the ones we observed can be expected between CH4 in profundal and littoral sediment bubbles. Lower δ13C-CH4 values in the profundal compared to the littoral zones have also been observed in sediments of three small subarctic lakes in western Greenland (Cadieux et al., 2016; Thompson et al., 2016) and in one small subarctic lake located on the border of a permafrost complex in northern Sweden (Wik et al., 2020). However, as such studies were made on a limited number of lakes with similar properties, it was unclear whether such differences in endmember δ13C-CH4 are common across lakes. Our results indicate that patterns of highly variable endmember δ13C-CH4 values, but with a consistent difference between littoral and profundal zones is a common property of boreal and subarctic lakes. This observation is critical for how to use and sample δ13C-CH4 values to assess CH4 cycling, MOX, and other related lake ecosystem processes or food web interactions.
It is also remarkable that δ13C-CH4 values in the anoxic hypolimnion water were relatively similar among sampling occasions in all lakes while littoral sediment δ13C-CH4 values were more variable. The variability was observed as variability over time but could in fact also represent variability in space because sediment bubbles were not collected at identical locations every time and the temporal variability was asynchronous between lakes, even in similar lakes like NAS and LJR. This means that local conditions in the littoral sediments, for example sources of organic matter, benthic processes, and vegetation cover, can vary in ways that could influence endmember δ13C-CH4 signals substantially.
We discuss below a range of potential explanations for the difference in δ13C-CH4 we observed between littoral sediment bubbles and the anoxic hypolimnion.
Δ(δ13C-CH4)LSB-AHW can be related to differences in δ13C of the substrates used in the anoxic degradation of OM in sediments of lakes. δ13C-CH4 is determined by the initial δ13C of the substrate and by the fractionation imposed by metabolic reactions, such that δ13C of the substrate plays an important role in setting δ13C of CH4 released in the water column (Conrad, 2005). The dominant sources of OM to the sediments of lakes include autochthonous primary production by phytoplankton, benthic algae, or littoral vascular plants on one hand, and allochthonous terrestrial OM on the other hand. Terrestrial OM is derived from the vegetation growing in the catchment of the lake, which is predominantly constituted of C3 plants in boreal regions (Cerling et al., 1997). δ13C for C3 plants are typically between −37‰ and −20‰ and most commonly near −27‰ (Kohn, 2010). In general, values of −27‰ or −28‰ are commonly used for allochthonous C in boreal lakes (Meili et al., 1993; Pace et al., 2004). δ13C values of littoral vascular plants are also predominantly in the range between −30‰ and −20‰ (Cloern et al., 2002; Lammers et al., 2017; Figure 1). On the other hand, δ13C values for different phytoplankton species have been reported in the range −44.5‰ to −18‰ in several boreal lakes, most of them being in the range −39‰ to −30‰ (Taipale et al., 2016), and measurements in several lentic systems across a broad range of latitudes have revealed δ13C values ranging between −36‰ and −17‰ in phytoplankton and between −29‰ and −8‰ in benthic algae, with values usually 2‰–11‰ lower (6‰ in average) in phytoplankton than in benthic algae for a given system (Doi et al., 2010), as illustrated in Figure 1. Therefore, benthic algae seem usually enriched in 13C compared to phytoplankton and allochthonous OM. There are indications that methanogens preferentially use autochthonous freshly produced OM when possible (West et al., 2012).
Hence, in low-nutrient lakes, the littoral where light is available may represent a zone with concentrated production of fresh, autochthonous OM (Karlsson et al., 2009). Nutrients released from sediments may be the main nutrient source in such low-nutrient lakes, favoring benthic algae relative to phytoplankton. If so, littoral δ13C-CH4 in low-nutrient lakes may be influenced by benthic algae, while profundal CH4 production is reflecting δ13C from allochthonous OM (or respired DIC as discussed further below), which given this scenario could lead to a large Δ(δ13C-CH4)LSB-AHW. In more nutrient rich lakes, settling phytoplankton may contribute labile autochthonous OM fueling methanogenesis everywhere in the lake (in both littoral and profundal sediments) leading to lower Δ(δ13C-CH4)LSB-AHW. The wide range of δ13C values in phytoplankton reported in previous studies (Doi et al., 2010; Taipale et al., 2016) and illustrated in Figure 1 does not necessarily contradict this explanation, as measured δ13C-CH4 values reflect average δ13C values in the sediments, and the mean δ13C value in phytoplankton is significantly lower than the mean δ13C value in benthic algae (Wilcoxon signed-rank test on the data from Doi et al. (2010); N = 49, W = 1.0, p < 0.001). However, the wider range of phytoplankton δ13C values can add to the variability among lakes. Although the substrate explanation is a speculation at this stage it is realistic given available scientific evidence and could explain our observations of lower Δ(δ13C-CH4)LSB-AHW in lakes with highest TP, TN, and Chl a values (Table 1).
In addition, DIC is the C substrate for HM, and it is possible that DIC in profundal sediments under isolated hypolimnion is primarily generated by OM respiration which causes depletion in 13C compared to DIC in littoral parts of lakes where continuous exchange with atmospheric CO2 is possible (Miyajima et al., 1997; Karlsson et al., 2007). A few measurements in the lakes studied here confirm this tendency and indicate that δ13C-CO2 at the bottom of the water column is often between 2‰ and 10‰ lower than in surface water (data not shown). Therefore, δ13C values of CH4 produced by HM should decrease at the bottom of lakes with larger differences between bottom and surface DIC concentrations (expressed as hypolimnetic excess DIC in the Results) being a sign of a larger share of accumulated respired C in deep waters. Accordingly, we observed the most positive hypolimnion δ13C-CH4 values in VEN, which also had smallest DIC accumulation in deeper water. Comparing lakes with distinctively different δ13C-CH4 values in the hypolimnion, we also noted that increasing hypolimnetic DIC accumulation was linked with lower δ13C-CH4 values in the hypolimnion (Figure 5A). This indicates that some CH4 was produced by HM in the hypolimnion of the studied lakes.
The subsequent question is if a depletion of δ13C-CH4 from HM using respired C could explain the observed Δ(δ13C-CH4)LSB-AHW. If this was the case, we would expect larger Δ(δ13C-CH4)LSB-AHW when a greater share of respired C were present in the hypolimnion, i.e., we would expect a positive relationship between Δ(δ13C-CH4)LSB-AHW and the hypolimnion excess DIC. However, no such strong pattern could be observed (Figure 5B). Therefore, we conclude that while differences in types of OM substrates (terrestrial/macrophytes vs. phytoplankton vs. periphyton) could provide explanations, CH4 production from respired C does not seem to be a main direct driver for the observed Δ(δ13C-CH4)LSB-AHW.
If the balance between methanogenic pathways differs between littoral and profundal sediments, δ13C-CH4 values could be affected because HM has been suggested to generate more depleted δ13C-CH4 than AM (Whiticar, 1999). The lower δ13C-CH4 values that we report in the deep hypolimnion compared to littoral sediments could then be explained if a greater share of HM is taking place in profundal sediments compared to littoral ones. Our data on δ13C-CH4 and DIC measurements discussed above indicates that HM occurs and consumes respired CO2 in the deep hypolimnion. While we do not have direct estimates of AM and HM, several factors influencing the share of CH4 produced by HM and AM have recently been reviewed by Conrad (2020). They include temperature, pH, and the quality of the available OM. More specifically, previous observations have shown that the HM:AM ratio decreases at lower temperature (Schulz and Conrad, 1996; Nozhevnikova et al., 2007) while low pH values can limit the availability of H2 resulting in a smaller contribution from HM (Goodwin et al., 1988). In addition, Liu et al. (2017) have shown that the HM:AM ratio increased in deeper layers of sediment cores from a mountain lake, which was explained by the decreasing quality of the OM in older sediments. Given present literature information, our data is not clearly compatible with conditions that would favor HM in profundal zones and AM in the littoral, as some variables such a temperature conditions could be expected to yield the opposite result. Hence, although this HM vs. AM hypothesis cannot be excluded as a potential explanation for our results, we find it unlikely that it is of main importance.
A difference in rates of MOX in sediments across depths needs to be considered as a potential explanation for the spatial variability of δ13C-CH4 because MOX leaves 13C enriched CH4 by preferentially consuming lighter 12C-CH4. More MOX is expected to happen in littoral sediments than in deep, anoxic hypolimnia as MOX in littoral sediments can be supported by the transport of suitable electron acceptors into the sediments via plant roots or via diffusion or percolation of O2-rich water. However, if the collected bubbles in the littoral sediments were released from sediment layers below the zone where suitable electron acceptors for MOX or AOM were depleted, δ13C-CH4 values measured in these bubbles would not have been affected by oxidation. Unfortunately, in the absence of measurements of δ13C-CH4 profiles in the pore water of the sediments, it is not possible to confirm if this was the case in the present study. Measurements in a small, eutrophic kettle lake in central Europe, which included collection of sediment bubbles using a similar approach as the one described here, as well as profiles of pore water δ13C-CH4, showed that bubbles released by physically disturbing the sediments originated from sediment layers where CH4 was not or marginally affected by oxidation (Langenegger et al., 2019). If the same is true in the littoral and profundal sediments of the lakes studied here, it would indicate that MOX is not a likely explanation for the higher δ13C-CH4 values observed in littoral sediment bubbles compared to in anoxic, deep water in the profundal zone of the lakes. Direct measurements of CH4 concentrations and δ13C-CH4 values along pore water profiles in the sediments in littoral and profundal zones could provide more conclusive results by allowing a more detailed analysis of the active processes in different sediment layers and should be included in future studies.
GW often contains high concentration of CH4 compared to lake water due to anoxic conditions prevailing in soils (Bugna et al., 1996). Moreover, GW can also be a significant source of CH4 to some lakes (Einarsdottir et al., 2017; Lecher et al., 2017; Dabrowski et al., 2020). A previous study indicates that large amounts of CH4 might be transported to STJ via GW inputs (Denfeld et al., 2020). The GW flowing into STJ passes through a mire and CH4 emitted from wetlands and mires often displays δ13C-CH4 values lower than −60‰ (Quay et al., 1988; Lansdown et al., 1992), especially in Scandinavian wetlands where average values are as low as −75‰ (Ganesan et al., 2018). Our measurements show that δ13C-CH4 in GW from the mire (−82‰ to −70‰) matches δ13C in the dissolved CH4 (−79‰ to −72‰) under the oxycline in the middle of the lake near the deepest point (Figure 6). The deep water δ13C-CH4 in the profundal zone is at the same time different from littoral sediment bubble δ13C-CH4 values that were measured on the opposite side of the lake (−65‰ to −59‰; Figure 6). Together with the fact that the GW is expected to enter the lake in the deeper layer due to its low temperature (higher density) compared to the epilimnion, it points to a possible mire GW origin of CH4 in the deeper water layer of the lake. On the other hand, most of the littoral sediment bubbles CH4 is probably derived from sediment CH4 production and less influenced by the mire CH4. The GW explanation might also apply to other lakes than STJ. Unfortunately, the absence of GW measurements near the five other studied lakes prevents us to establish any link between GW inputs of CH4 depleted in 13C and the lower δ13C-CH4 values observed in the anoxic hypolimnion of these lakes. Alternatively, the low δ13C-CH4 values observed in the anoxic hypolimnion of STJ might also arise for other reasons described previously, like the variability in the composition of the substrate available in different zones of lakes. It is likely that the suggested mechanisms have a combined effect on the difference in δ13C values in endmember CH4 between littoral and profundal zones of lakes.
Furthermore, the amount of CH4 transported by GW to the lake was estimated to be much larger than the amount of CH4 emitted by the lake to the atmosphere (Figure 7). It implies that large quantities of CH4 can be transported from the mire to the lake and most of this CH4 is oxidized in the lake before reaching the surface. It suggests that the importance of GW CH4 contributions to lakes CH4 cycling may have been underestimated. In fact, these observations indicate that GW CH4 inputs could be high enough to sustain most dissolved CH4 in some lakes—meaning that hypolimnetic CH4 and episodic fluxes of dissolved CH4 upon lake mixing would be attributed to GW CH4, while ebullition is derived from intrinsically produced CH4 from the lake sediments. These findings deserve further studies and illustrate that catchment processes and hydrological transport should be considered in assessments of factors controlling CH4 cycling in lakes. Accurate and relevant estimates of the volume of various sources and sinks of water to and from a lake, and of the CH4 concentration in each of these sources and sinks are important requirements for this method to be successful in assessing the contribution of external inputs of CH4 to the lake. Also, the temperature of the groundwater entering a lake is critical in determining in which layer of the lake the groundwater will end up.
The main result of our study is that there seems to be a systematic difference in endmember δ13C-CH4 between littoral sediments and anoxic profundal zones of subarctic and boreal lakes and that it is important to consider which parts of the lake CH4 cycling can be represented by different source δ13C-CH4 values. We also discuss potential explanations to the observed pattern (Figure 8) and argue that several mechanisms are likely to contribute at different degrees to the offset in endmember δ13C-CH4 between littoral and profundal zones of lakes, with no conclusive evidence for one specific mechanism against all others based on our observations.
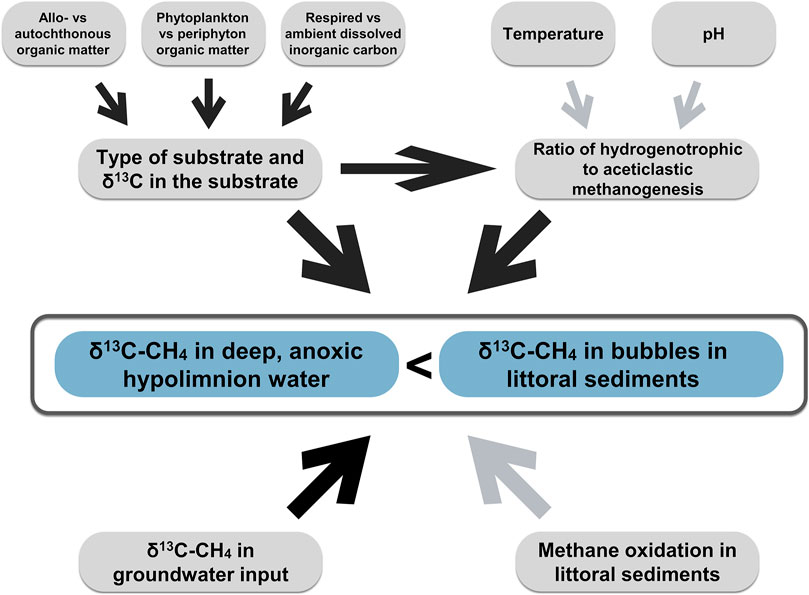
FIGURE 8. Conceptual figure summarizing the different processes discussed in the text as potential explanations for the observed variability in δ13C-CH4 between the anoxic hypolimnion water and the littoral sediments of lakes. Dark arrows indicate relations supported by our measurements. Light arrows indicate relations that are not supported by our observations. See text for details.
It has been shown that stratified lakes in essence have two separated CH4 cycles—one epilimnetic and one hypo-metalimnetic (Bastviken et al., 2008). In the epilimnetic CH4 cycle, CH4 produced in littoral sediments is released to the atmosphere by bubbling or diffuses into the epilimnion where it can be oxidized or emitted. The epilimnetic CH4 cycle contributes most of the CH4 emission during open water periods and the best source δ13C-CH4 value there may be measured in littoral sediment bubbles. In the meta-hypolimnetic CH4 cycle, CH4 primarily leaves the profundal sediment as dissolved CH4, and a concentration gradient develops between the surface of the sediments and the water layer where suitable electron acceptors for MOX are available and where most of the CH4 is oxidized. For estimating MOX in this part of the lake, dissolved CH4 in the anoxic bottom water layer of the lake or CH4 contained in profundal sediments may be the best source δ13C-CH4 value. Distinguishing these two CH4 cycles would represent an important progress and contribute to increased knowledge about the regulation and feedbacks of lake CH4 and C cycling and emissions.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: DiVA (https://doi.org/10.48360/5PPN-C440).
The study was designed by HS, JS, and DB. Field work was performed by JS, HS, AS, GP, DR, EH, and KF. Sample analyses were performed by JS, HS, AS, GP, DR, EH, and KF. Funding for this study was provided through projects led by DB, HL, and JK. The article was drafted by JS with contributions from all authors.
This work was funded by the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (Grant agreement No 725546; METLAKE), the Swedish Research Council (Grant No 2016-04829), FORMAS (Grant No 2018-01794), and the Knut and Alice Wallenberg Foundation (Grant No 2016.0083).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Ingrid Sundgren, Thanh Duc Nguyen, and Kim Lindgren for invaluable assistance, the Swedish Infrastructure for Ecosystem Science (SITES), the Skogaryd Research Catchment (SRC), and inhabitants at Stora Galten, Parsen, and Venasjön for valuable support, interest, and patience with our work.
Bastviken, D., Cole, J. J., Pace, M. L., and Van de Bogert, M. C. (2008). Fates of Methane from Different lake Habitats: Connecting Whole-lake Budgets and CH4emissions. J. Geophys. Res. 113:G02024. doi:10.1029/2007JG000608
Bastviken, D., Cole, J., Pace, M., and Tranvik, L. (2004). Methane Emissions from Lakes: Dependence of lake Characteristics, Two Regional Assessments, and a Global Estimate. Glob. Biogeochem. Cycles 18:GB4009. doi:10.1029/2004GB002238
Bastviken, D., Ejlertsson, J., Sundh, I., and Tranvik, L. (2003). Methane as a Source of Carbon and Energy for Lake Pelagic Food Webs. Ecology 84, 969–981. doi:10.1890/0012-9658(2003)084[0969:maasoc]2.0.co;2
Bastviken, D. (2009). “Methane,” in Encyclopedia of Inland Waters. Editor G. E. Likens (Oxford: Academic Press), 783–805. doi:10.1016/B978-012370626-3.00117-4
Bugna, G. C., Chanton, J. P., Cable, J. E., Burnett, W. C., and Cable, P. H. (1996). The Importance of Groundwater Discharge to the Methane Budgets of Nearshore and continental Shelf Waters of the Northeastern Gulf of Mexico. Geochim. Cosmochim. Acta 60, 4735–4746. doi:10.1016/S0016-7037(96)00290-6
Cadieux, S. B., White, J. R., Sauer, P. E., Peng, Y., Goldman, A. E., and Pratt, L. M. (2016). Large Fractionations of C and H Isotopes Related to Methane Oxidation in Arctic Lakes. Geochim. Cosmochim. Acta 187, 141–155. doi:10.1016/j.gca.2016.05.004
Cerling, T. E., Harris, J. M., MacFadden, B. J., Leakey, M. G., Quade, J., Eisenmann, V., et al. (1997). Global Vegetation Change through the Miocene/Pliocene Boundary. Nature 389, 153–158. doi:10.1038/38229
Cloern, J. E., Canuel, E. A., and Harris, D. (2002). Stable Carbon and Nitrogen Isotope Composition of Aquatic and Terrestrial Plants of the San Francisco Bay Estuarine System. Limnol. Oceanogr. 47, 713–729. doi:10.4319/lo.2002.47.3.0713
Cole, J. J., Prairie, Y. T., Caraco, N. F., McDowell, W. H., Tranvik, L. J., Striegl, R. G., et al. (2007). Plumbing the Global Carbon Cycle: Integrating Inland Waters into the Terrestrial Carbon Budget. Ecosystems 10, 172–185. doi:10.1007/s10021-006-9013-8
Conrad, R. (2020). Importance of Hydrogenotrophic, Aceticlastic and Methylotrophic Methanogenesis for Methane Production in Terrestrial, Aquatic and Other Anoxic Environments: A Mini Review. Pedosphere 30, 25–39. doi:10.1016/S1002-0160(18)60052-9
Conrad, R. (2005). Quantification of Methanogenic Pathways Using Stable Carbon Isotopic Signatures: a Review and a Proposal. Org. Geochem. 36, 739–752. doi:10.1016/j.orggeochem.2004.09.006
Dabrowski, J. S., Charette, M. A., Mann, P. J., Ludwig, S. M., Natali, S. M., Holmes, R. M., et al. (2020). Using Radon to Quantify Groundwater Discharge and Methane Fluxes to a Shallow, Tundra lake on the Yukon-Kuskokwim Delta, Alaska. Biogeochemistry 148, 69–89. doi:10.1007/s10533-020-00647-w
de Kluijver, A., Schoon, P. L., Downing, J. A., Schouten, S., and Middelburg, J. J. (2014). Stable Carbon Isotope Biogeochemistry of Lakes along a Trophic Gradient. Biogeosciences 11, 6265–6276. doi:10.5194/bg-11-6265-2014
Denfeld, B. A., Klaus, M., Laudon, H., Sponseller, R. A., and Karlsson, J. (2018). Carbon Dioxide and Methane Dynamics in a Small Boreal Lake during Winter and Spring Melt Events. J. Geophys. Res. Biogeosci. 123, 2527–2540. doi:10.1029/2018JG004622
Denfeld, B. A., Lupon, A., Sponseller, R. A., Laudon, H., and Karlsson, J. (2020). Heterogeneous CO2 and CH4 Patterns across Space and Time in a Small Boreal lake. Inland Waters 10, 348–359. doi:10.1080/20442041.2020.1787765
Doi, H., Kikuchi, E., Shikano, S., and Takagi, S. (2010). Differences in Nitrogen and Carbon Stable Isotopes between Planktonic and Benthic Microalgae. Limnology 11, 185–192. doi:10.1007/s10201-009-0297-1
Einarsdottir, K., Wallin, M. B., and Sobek, S. (2017). High Terrestrial Carbon Load via Groundwater to a Boreal lake Dominated by Surface Water Inflow. J. Geophys. Res. Biogeosci. 122, 15–29. doi:10.1002/2016JG003495
Ganesan, A. L., Stell, A. C., Gedney, N., Comyn‐Platt, E., Hayman, G., Rigby, M., et al. (2018). Spatially Resolved Isotopic Source Signatures of Wetland Methane Emissions. Geophys. Res. Lett. 45, 3737–3745. doi:10.1002/2018GL077536
Goodwin, S., Conrad, R., and Zeikus, J. G. (1988). Influence of pH on Microbial Hydrogen Metabolism in Diverse Sedimentary Ecosystems. Appl. Environ. Microbiol. 54, 590–593. doi:10.1128/aem.54.2.590-593.1988
Grey, J. (2016). The Incredible Lightness of Being Methane-Fuelled: Stable Isotopes Reveal Alternative Energy Pathways in Aquatic Ecosystems and beyond. Front. Ecol. Evol. 4, 8. doi:10.3389/fevo.2016.00008
Jones, R. I., and Grey, J. (2011). Biogenic Methane in Freshwater Food Webs. Freshw. Biol. 56, 213–229. doi:10.1111/j.1365-2427.2010.02494.x
Kankaala, P., Taipale, S., Grey, J., Sonninen, E., Arvola, L., and Jones, R. I. (2006). Experimental d13C Evidence for a Contribution of Methane to Pelagic Food Webs in Lakes. Limnol. Oceanogr. 51, 2821–2827. doi:10.4319/lo.2006.51.6.2821
Karlsson, J., Byström, P., Ask, J., Ask, P., Persson, L., and Jansson, M. (2009). Light Limitation of Nutrient-Poor lake Ecosystems. Nature 460, 506–509. doi:10.1038/nature08179
Karlsson, J., Jansson, M., and Jonsson, A. (2007). Respiration of Allochthonous Organic Carbon in Unproductive forest Lakes Determined by the Keeling Plot Method. Limnol. Oceanogr. 52, 603–608. doi:10.4319/lo.2007.52.2.0603
Kohn, M. J. (2010). Carbon Isotope Compositions of Terrestrial C3 Plants as Indicators of (Paleo)ecology and (Paleo)climate. Proc. Natl. Acad. Sci. 107, 19691–19695. doi:10.1073/pnas.1004933107
Lammers, J. M., Reichart, G. J., and Middelburg, J. J. (2017). Seasonal Variability in Phytoplankton Stable Carbon Isotope Ratios and Bacterial Carbon Sources in a Shallow Dutch lake. Limnol. Oceanogr. 62, 2773–2787. doi:10.1002/lno.10605
Langenegger, T., Vachon, D., Donis, D., and McGinnis, D. F. (2019). What the Bubble Knows: Lake Methane Dynamics Revealed by Sediment Gas Bubble Composition. Limnol. Oceanogr. 64 (4), 1526–1544. doi:10.1002/lno.11133
Lansdown, J. M., Quay, P. D., and King, S. L. (1992). CH4 Production via CO2 Reduction in a Temperate Bog: A Source of 13C-depIeted CH4. Geochim. Cosmochim. Acta 56, 3493–3503. doi:10.1016/0016-7037(92)90393-W
Laudon, H., Taberman, I., Ågren, A., Futter, M., Ottosson-Löfvenius, M., and Bishop, K. (2013). The Krycklan Catchment Study-A Flagship Infrastructure for Hydrology, Biogeochemistry, and Climate Research in the Boreal Landscape. Water Resour. Res. 49, 7154–7158. doi:10.1002/wrcr.20520
Lecher, A. L., Chuang, P.-C., Singleton, M., and Paytan, A. (2017). Sources of Methane to an Arctic lake in Alaska: An Isotopic Investigation. J. Geophys. Res. Biogeosci. 122, 753–766. doi:10.1002/2016JG003491
Liu, Y., Conrad, R., Yao, T., Gleixner, G., and Claus, P. (2017). Change of Methane Production Pathway with Sediment Depth in a lake on the Tibetan Plateau. Palaeogeogr. Palaeoclimatol. Palaeoecol. 474, 279–286. doi:10.1016/j.palaeo.2016.06.021
McIntosh Marcek, H. A., Lesack, L. F. W., Orcutt, B. N., Wheat, C. G., Dallimore, S. R., Geeves, K., et al. (2021). Continuous Dynamics of Dissolved Methane over 2 Years and its Carbon Isotopes (δ 13 C, Δ 14 C) in a Small Arctic Lake in the Mackenzie Delta. J. Geophys. Res. Biogeosci. 126, e2020JG006038. doi:10.1029/2020jg006038
Meili, M., Fry, B., and Kling, G. W. (1993). Fractionation of Stable Isotopes (13C, 15N) in the Food Web of a Humic lake. SIL Proc. 1922-2010 25, 501–505. doi:10.1080/03680770.1992.11900175
Miyajima, T., Yamada, Y., Wada, E., Nakajima, T., Koitabashi, T., Hanba, Y. T., et al. (1997). Distribution of Greenhouse Gases, Nitrite, and δ13C of Dissolved Inorganic Carbon in Lake Biwa: Implications for Hypolimnetic Metabolism. Biogeochemistry 36, 205–221. doi:10.1023/A:1005702707183
Murphy, J., and Riley, J. P. (1962). A Modified Single Solution Method for the Determination of Phosphate in Natural Waters. Analytica Chim. Acta 27, 31–36. doi:10.1016/S0003-2670(00)88444-5
Nozhevnikova, A. N., Nekrasova, V., Ammann, A., Zehnder, A. J. B., Wehrli, B., and Holliger, C. (2007). Influence of Temperature and High Acetate Concentrations on Methanogenensis in lake Sediment Slurries. FEMS Microbiol. Ecol. 62, 336–344. doi:10.1111/j.1574-6941.2007.00389.x
Pace, M. L., Cole, J. J., Carpenter, S. R., Kitchell, J. F., Hodgson, J. R., Van de Bogert, M. C., et al. (2004). Whole-lake Carbon-13 Additions Reveal Terrestrial Support of Aquatic Food Webs. Nature 427, 240–243. doi:10.1038/nature02227
Peterson, B. J., and Fry, B. (1987). Stable Isotopes in Ecosystem Studies. Annu. Rev. Ecol. Syst. 18, 293–320. doi:10.1146/annurev.es.18.110187.001453
Quay, P. D., King, S. L., Lansdown, J. M., and Wilbur, D. O. (1988). Isotopic Composition of Methane Released from Wetlands: Implications for the Increase in Atmospheric Methane. Glob. Biogeochem. Cycles 2, 385–397. doi:10.1029/GB002i004p00385
Reeburgh, W. S. (2014). “Global Methane Biogeochemistry,” in Treatise on Geochemistry. Editors H. D. Holland, and K. K. Turekian. Second Edition (Oxford: Elsevier), 71–94. doi:10.1016/B978-0-08-095975-7.00403-4
Richards, F. A., and Thompson, T. G. (1952). The Estimation and Characterization of Plankton Populations by Pigment Analyses II. A Spectrophotometric Method for the Estimation of Plankton Pigments. J. Mar. Res. 11, 156–172.
Rinta, P., Bastviken, D., van Hardenbroek, M., Kankaala, P., Leuenberger, M., Schilder, J., et al. (2015). An Inter-regional Assessment of Concentrations and δ13C Values of Methane and Dissolved Inorganic Carbon in Small European Lakes. Aquat. Sci. 77, 667–680. doi:10.1007/s00027-015-0410-y
Sanseverino, A. M., Bastviken, D., Sundh, I., Pickova, J., and Enrich-Prast, A. (2012). Methane Carbon Supports Aquatic Food Webs to the Fish Level. PLOS ONE 7, e42723. doi:10.1371/journal.pone.0042723
Sartori, E. (2000). A Critical Review on Equations Employed for the Calculation of the Evaporation Rate from Free Water Surfaces. Solar Energy 68, 77–89. doi:10.1016/S0038-092X(99)00054-7
Saunois, M., Stavert, A. R., Poulter, B., Bousquet, P., Canadell, J. G., Jackson, R. B., et al. (2020). The Global Methane Budget 2000–2017. Earth Syst. Sci. Data 12, 1561–1623. doi:10.5194/essd-12-1561-2020
Schilder, J., Bastviken, D., van Hardenbroek, M., Kankaala, P., Rinta, P., Stötter, T., et al. (2013). Spatial Heterogeneity and lake Morphology Affect Diffusive Greenhouse Gas Emission Estimates of Lakes. Geophys. Res. Lett. 40, 5752–5756. doi:10.1002/2013GL057669
Schulz, S., and Conrad, R. (1996). Influence of Temperature on Pathways to Methane Production in the Permanently Cold Profundal Sediment of Lake Constance. FEMS Microbiol. Ecol. 20, 1–14. doi:10.1111/j.1574-6941.1996.tb00299.x
Taipale, S. J., Vuorio, K., Brett, M. T., Peltomaa, E., Hiltunen, M., and Kankaala, P. (2016). Lake Zooplankton δ 13 C Values Are Strongly Correlated with the δ 13 C Values of Distinct Phytoplankton Taxa. Ecosphere 7, e01392. doi:10.1002/ecs2.1392
Taipale, S., Kankaala, P., Hämäläinen, H., and Jones, R. I. (2009). Seasonal Shifts in the Diet of lake Zooplankton Revealed by Phospholipid Fatty Acid Analysis. Freshw. Biol. 54, 90–104. doi:10.1111/j.1365-2427.2008.02094.x
Thompson, H. A., White, J. R., Pratt, L. M., and Sauer, P. E. (2016). Spatial Variation in Flux, δ13C and δ2H of Methane in a Small Arctic lake with Fringing Wetland in Western Greenland. Biogeochemistry 131, 17–33. doi:10.1007/s10533-016-0261-1
Tranvik, L. J., Cole, J. J., and Prairie, Y. T. (2018). The Study of Carbon in Inland Waters-From Isolated Ecosystems to Players in the Global Carbon Cycle. Limnol. Oceanogr. 3, 41–48. doi:10.1002/lol2.10068
Tsuchiya, K., Komatsu, K., Shinohara, R., Imai, A., Matsuzaki, S. I. S., Ueno, R., et al. (2020). Variability of Benthic Methane‐derived Carbon along Seasonal, Biological, and Sedimentary Gradients in a Polymictic lake. Limnol. Oceanogr. 65, 3017–3031. doi:10.1002/lno.11571
West, W. E., Coloso, J. J., and Jones, S. E. (2012). Effects of Algal and Terrestrial Carbon on Methane Production Rates and Methanogen Community Structure in a Temperate lake Sediment. Freshw. Biol. 57, 949–955. doi:10.1111/j.1365-2427.2012.02755.x
Whiticar, M. J. (1999). Carbon and Hydrogen Isotope Systematics of Bacterial Formation and Oxidation of Methane. Chem. Geol. 161, 291–314. doi:10.1016/S0009-2541(99)00092-3
Keywords: stable carbon isotope, methane, lake, groundwater, endmember
Citation: Schenk J, Sawakuchi HO, Sieczko AK, Pajala G, Rudberg D, Hagberg E, Fors K, Laudon H, Karlsson J and Bastviken D (2021) Methane in Lakes: Variability in Stable Carbon Isotopic Composition and the Potential Importance of Groundwater Input. Front. Earth Sci. 9:722215. doi: 10.3389/feart.2021.722215
Received: 08 June 2021; Accepted: 12 October 2021;
Published: 27 October 2021.
Edited by:
Volker Brüchert, Stockholm University, SwedenCopyright © 2021 Schenk, Sawakuchi, Sieczko, Pajala, Rudberg, Hagberg, Fors, Laudon, Karlsson and Bastviken. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonathan Schenk, am9uYXRoYW4uc2NoZW5rQGxpdS5zZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.