 Eleanor L. Jones1,2
Eleanor L. Jones1,2 Andrew J. Hodson2,3
Andrew J. Hodson2,3 Steven F. Thornton4*
Steven F. Thornton4* Kelly R. Redeker5Jade Rogers1
Kelly R. Redeker5Jade Rogers1 Peter M. Wynn6
Peter M. Wynn6 Timothy J. Dixon7Simon H. Bottrell7
Timothy J. Dixon7Simon H. Bottrell7 H. Brendan O’Neill8
H. Brendan O’Neill8- 1Department of Geography, The University of Sheffield, Sheffield, United Kingdom
- 2Department of Arctic Geology, The University Centre in Svalbard, Longyearbyen, Norway
- 3Department of Environmental Sciences, Western Norway University of Applied Sciences, Sogndal, Norway
- 4Department of Civil and Structural Engineering, The University of Sheffield, Sheffield, United Kingdom
- 5Department of Biology, University of York, York, United Kingdom
- 6Lancaster Environment Centre, Lancaster University, Lancaster, United Kingdom
- 7School of Earth and Environment, University of Leeds, Leeds, United Kingdom
- 8Geological Survey of Canada, Natural Resources Canada, Ottawa, ON, Canada
Warming of ground is causing microbial decomposition of previously frozen sedimentary organic carbon in Arctic permafrost. However, the heterogeneity of the permafrost landscape and its hydrological processes result in different biogeochemical processes across relatively small scales, with implications for predicting the timing and magnitude of permafrost carbon emissions. The biogeochemical processes of iron- and sulfate-reduction produce carbon dioxide and suppress methanogenesis. Hence, in this study, the biogeochemical processes occurring in the active layer and permafrost of a high Arctic fjord valley in Svalbard are identified from the geochemical and stable isotope analysis of aqueous and particulate fractions in sediment cores collected from ice-wedge polygons with contrasting water content. In the drier polygons, only a small concentration of organic carbon (<5.40 dry weight%) has accumulated. Sediment cores from these drier polygons have aqueous and solid phase chemistries that imply sulfide oxidation coupled to carbonate and silicate dissolution, leading to high concentrations of aqueous iron and sulfate in the pore water profiles. These results are corroborated by δ34S and δ18O values of sulfate in active layer pore waters, which indicate the oxidative weathering of sedimentary pyrite utilising either oxygen or ferric iron as oxidising agents. Conversely, in the sediments of the consistently water-saturated polygons, which contain a high content of organic carbon (up to 45 dry weight%), the formation of pyrite and siderite occurred via the reduction of iron and sulfate. δ34S and δ18O values of sulfate in active layer pore waters from these water-saturated polygons display a strong positive correlation (R2 = 0.98), supporting the importance of sulfate reduction in removing sulfate from the pore water. The significant contrast in the dominant biogeochemical processes between the water-saturated and drier polygons indicates that small-scale hydrological variability between polygons induces large differences in the concentration of organic carbon and in the cycling of iron and sulfur, with ramifications for the decomposition pathway of organic carbon in permafrost environments.
Introduction
Permafrost regions account for 24% of the land area of the Northern Hemisphere and store immense quantities (1330 to 1580 Pg) of organic carbon (Tarnocai et al., 2009; Hugelius et al., 2014). Permafrost ecosystems are currently a net sink of carbon, due to the drawdown of carbon dioxide exceeding emissions of methane and carbon dioxide (Schaefer et al., 2011; Kirschke et al., 2013; Parmentier et al., 2013). As rapid climate change occurs in the high Arctic, rising permafrost temperatures and progressively deeper active layers are exposing previously frozen soil organic carbon to microbial decomposition (Romanovsky et al., 2010; Koven et al., 2011; Schuur et al., 2015). This releases carbon dioxide and methane to the atmosphere and is predicted to cause permafrost ecosystems to become a net carbon source by the mid-2020s (Lee et al., 2012; Elberling et al., 2013; Schuur et al., 2015).
To predict the impact of permafrost thaw on biogeochemical cycling, it is necessary to understand the spatial heterogeneity of biogeochemical processes in Arctic soils. The distribution of stored organic carbon varies across the permafrost landscape, and with depth (Kuhry et al., 2010). Organic carbon accumulation and storage in permafrost is maximised in regions where waterlogged conditions dominate, as under these conditions, net primary production exceeds decomposition (Kolka et al., 2015). Hydrology exerts a powerful influence on the type of gaseous products released from decomposing permafrost organic carbon. For example, drained soil allows previously frozen organic carbon to decompose rapidly under oxic conditions, producing carbon dioxide (Liljedahl et al., 2012; Elberling et al., 2013), whereas water saturation limits the oxygen supply and enhances anaerobic respiration and methane production (Turetsky et al., 2008; Lipson et al., 2012; Olefeldt et al., 2013). However, under anaerobic conditions, alternative electron acceptors such as nitrate, manganese, iron and sulfate may be used preferentially in the microbial respiration of organic carbon, and inhibit the production of methane (Kristjansson and Schönheit, 1983; Dise and Verry, 2001). The use of alternative electron acceptors increases carbon dioxide production relative to methane, and hence the coupling between hydrology and the availability of alternative electron acceptors plays a crucial role in determining the magnitude of permafrost carbon emissions.
Hodson et al. (2016) conducted hydrological monitoring that demonstrated how lowland runoff draining the reactive, fine-grained sediments of Svalbard’s Central Tertiary Basin (i.e., Adventdalen, the site of the present study) acquires reactive iron from pyrite oxidation. Pyrite oxidation exerted a major control upon the composition of this runoff, as evidenced by the presence of high sulfate and cation concentrations (Hodson et al., 2016). This process has also been documented in several nearby catchments (Fardalen, Bolterdalen and Longyeardalen; Yde et al., 2008; Rutter et al., 2011; Hindshaw et al., 2016). Therefore, sediments and water in Adventdalen and the surrounding area have an abundance of alternative electron acceptors, such as ferric iron and sulfate, for the microbial oxidation of organic carbon. The abundance of ferric iron and sulfate in the waters draining through Adventdalen is important in the context of greenhouse gas emissions because Fe(III)- and sulfate-reduction are thermodynamically favourable, and therefore competitive, relative to methanogenesis. The availability of Fe(III) has been reported to decrease methanogenesis in the permafrost landscape of Barrow, Alaska (Lipson et al., 2012; Miller et al., 2015) and the availability of sulfate can decrease methane production in wetlands (e.g., Pester et al., 2012). Hodson et al. (2016) used the decrease in concentrations of sulfate and ∗Fe (i.e., dissolved and colloidal iron that passed through a 0.45 μm filter) at low flow to infer removal by sulfate reduction and precipitation of pyrite (FeS2), iron monosulfide (FeS) and possibly elemental sulfur in ground waters draining through an alluvial sediment fan in Endalen (a tributary to Adventdalen). However, the processes removing ∗Fe and sulfate from these waters were hypothesised to be limited by a lack of organic matter (e.g., Raiswell and Canfield, 2012). A scarcity of organic matter has also been observed to limit the removal of ∗Fe and sulfate from pore water in fjord sediment cores in Svalbard (Wehrmann et al., 2014). From these findings, it is clearly necessary to understand if the accumulation of organic carbon in such environments results in enhanced removal of iron and sulfate from solution.
The objective of this study is to determine how the vertical and lateral heterogeneity of organic carbon accumulation in a high Arctic fjord valley influences the biogeochemical processes in the active layer and shallow permafrost sediments and pore water. This study presents and discusses geochemical analyses of pore water and sediment sampled from cores collected from the active layer and shallow permafrost of ice-wedge polygonal terrain in a high Arctic fjord valley.
Materials and Methods
Field Site Description
Adventdalen (78°19′N, 15°93′E) is a 12 km × 4 km valley, oriented NW-SE, in central Svalbard. The van Mijenfjord and Adventdalen Groups, which contain sedimentary rocks (sandstones, shales, and carbonates), comprise the lithology of the Adventdalen catchment. The sandstones and shales contain iron-bearing mineral phases, such as pyrite, siderite and glauconite (Dallmann et al., 1999; Riber, 2009; Svinth, 2013). There is also a small amount of iron in biotite and chlorite (chamosite; Hodson et al., 2016).
Glacial retreat and postglacial isostatic rebound influence landscape evolution (Milne and Shennan, 2007). Consequently, permafrost aggradation in many high Arctic fjord valleys has involved the freezing of reactive, fine-grained sediment originally deposited on the fjord floor via glacio-marine sedimentation and delta progradation throughout deglaciation (Haldorsen et al., 2010; Gilbert et al., 2018). During the Last Glacial Maximum, Adventdalen was filled with an erosive ice stream; this caused permafrost to thaw (Humlum et al., 2003; Humlum, 2005; Landvik et al., 2005). After retreat of the ice sheet, the glacial isostatic rebound of Svalbard meant that relative sea level fell, and a Gilbert-type delta prograded into Adventfjorden (Lønne and Nemec, 2004). In Adventdalen, the deeper permafrost is epigenetic, as it formed after the progradation of the delta (Gilbert et al., 2018). The shallower syngenetic permafrost in the aeolian terraces of Adventdalen aggraded concurrently with aeolian sedimentation (Gilbert et al., 2018). Although permafrost only aggraded since deglaciation, it is continuous in Svalbard, where it reaches a thickness of 80−100 m near the coast (Brown et al., 1997; Humlum, 2005). The seasonally thawed layer, or “active layer,” can be 0.4−6 m thick in Svalbard (Christiansen, 2005). In Adventdalen, the active layer thickness is around 1 m (Christiansen et al., 2010). This study focuses on the zone 0−2 m depth, which involves sampling the active layer and shallow permafrost.
The climate of Svalbard is polar tundra (Kottek et al., 2006) and in Longyearbyen between 1981 and 2010, the mean annual air temperature (MAAT) was −4.6°C (Førland et al., 2011). Between 1989 and 2011, MAAT increased by 1.25°C per decade (Førland et al., 2011) and in 2018, the MAAT was −1.8°C (eklima.met.no). The mean annual ground temperature (MAGT) ranges from −5.6°C to −3.2°C in the Adventdalen area (Christiansen et al., 2010) and the rising air temperatures have caused a recent increase in permafrost temperatures (Isaksen et al., 2019). The vegetation cover in the valley varies between 1.3 g m–2 and 27.2 g m–2 (moss) and 2.8 g m–2 and 9.6 g m–2 (vascular plants), with the amount of moss dependent on the soil moisture (Sjögersten et al., 2006).
The study sites (Figures 1A–C) were selected with the aim of sampling the heterogeneity of permafrost environments in the fjord valley. Table 1 lists the sample locations with details of the sampling methods. The sites are covered with Late Holocene loess (aeolian) deposits overlying alluvial and deltaic deposits (Cable et al., 2017; Gilbert et al., 2018). The aeolian deposits and alluvial fans dominate the syngenetic permafrost aggradation, whilst the permafrost beneath constitutes the epigenetic permafrost aggradation (following downward freezing; Gilbert et al., 2018; Toft-Hornum et al., In Review). The study sites are situated in areas of low-centred ice-wedge polygons, which are common in Adventdalen (Sørbel and Tolgensbakk, 2002). Ice Wedge North (Figure 1B) is a water-saturated wetland, mainly fed by springs, whereas Ice Wedge South (Figure 1C) is mainly precipitation-fed and only sporadically inundated by local snowmelt and rainfall.
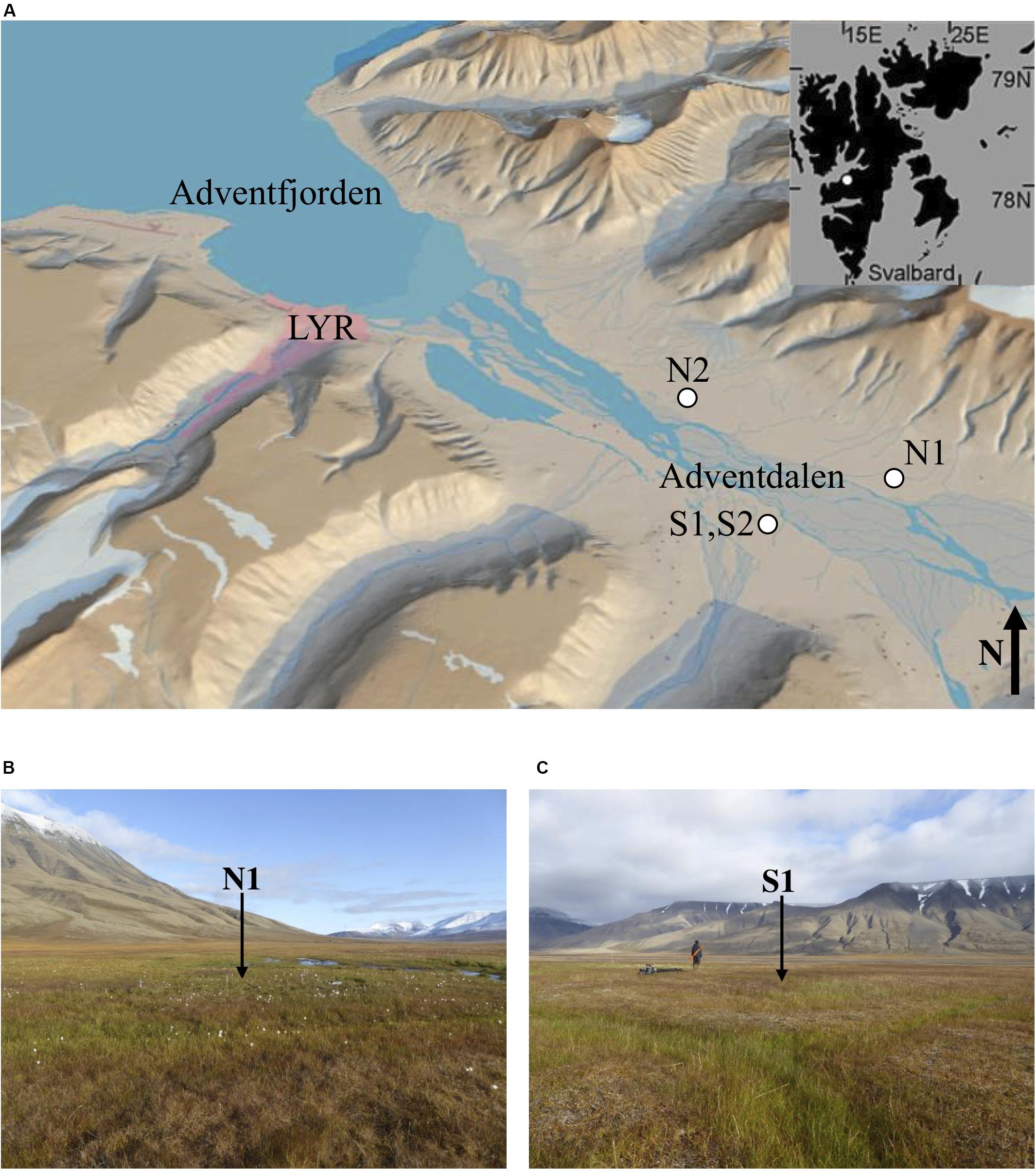
Figure 1. (A) The Svalbard archipelago (inset) and Adventdalen, with a white circle indicating each site where ice-wedge polygons were sampled (courtesey of the Norwegian Polar Institute), (B) overlooking polygon N1, facing east, and (C) overlooking polygon S1, facing north. Photo credit: E. L. Jones.
Table 1. Sampling locations, in UTM zone 33X.
Coring and Core Subdivision
Coring was undertaken before the onset of thaw, when air temperatures were below 0°C. Sediment cores of the frozen active layer and shallow permafrost were extracted in segments (5 cm diameter; between 5 and 50 cm length) to a depth of 2 m using motorised hand drilling equipment (a Stihl BT 130 drilling engine with a cylindrical drill head and rods). The core segments were extruded into sterile Whirl Pak®bags, which were sealed and frozen during transport to the University of Sheffield, where they were stored at −18°C. Cores were subdivided by sawing into 2 cm depth slices while frozen. The freshly cut surfaces were scraped with a scalpel, and the outer 2 cm removed with a hollow brass tube (3 cm diameter), to prevent contamination. The sawblade, scalpel and brass tube were cleaned with 70% isopropanol between slices.
Pore Water Extractions
A pore water extraction method was adapted from Spence et al. (2005). Vials containing samples were transferred to a Coy Vinyl Anaerobic chamber with a N2 atmosphere (0 ppm oxygen). Each vial was weighed to determine the sample mass. Nitrogen-sparged de-ionised water (Milli-Q) was added to fill each vial. The vials were reweighed to determine the mass and volume of water added. A 3 mL volume of water was subsequently removed from the top of the vial to create a headspace. The vials were crimp-capped, inverted and stored for 5 days at 4°C whilst submerged in water (to prevent gas diffusion across the septa). This storage time enabled the de-ionised water to equilibrate with the sediment pore water (e.g., Spence et al., 2005). 7 days after first saturation of the sample, the vials were centrifuged at 7750 rpm for 5 minutes and transferred back to the anaerobic chamber. The equilibrated supernatant was filtered (0.22 μm nylon syringe filter) for chemical analysis and the sediment remaining in the vials was weighed after drying at 105°C for 24 h (Kokelj and Burn, 2003; Ernakovich et al., 2017). The vials were then reweighed to determine the initial moisture content of the samples.
Major ions (Ca2+, Mg2+, Na+, K+, Cl–, , and ) were determined by ion chromatography (Dionex ion chromatograph, DX 90; limit of detection or “LOD” = 0.02 mg L–1 for the lowest, undiluted analysis; precision <5% for the mid-range standards). Trace metals (Fe, Mn) were determined on a 5 mL sample acidified with 50 μL reagent grade HNO3 (Fisher Scientific Trace Metal Grade), using Inductively Coupled Plasma Mass Spectrometry or ICPMS (PerkinElmer Elan DRC II, MA, United States). The precision errors for repeat analyses of mid-range standards were <5%, and the detection limits were 1.0 μg L–1. Analyte concentrations were corrected for blank analyses and corrected for the dilution during the pore water extraction.
Solid Phase Analyses
Carbon and nitrogen elemental abundance were determined by drying sediment samples at 105°C, acidifying in 6 M HCl, rinsing, drying, homogenising, weighing between 25 and 50 mg of each sample into a tin capsule and analysing on an Elementar vario EL cube (Animal and Plant Sciences Department, University of Sheffield). Elemental concentrations were checked with acetanilide standards (C8H9NO; Merck; n = 28, % C = 70.96 ± 0.67, % N = 10.34 ± 0.10), with 2 blanks and 2 acetanilide standards run every 15 samples.
Acid-volatile sulfur (AVS) and chromium-reducible sulfur (CRS) were determined at the University of Leeds. A two-step distillation method was applied to freeze-dried and milled sediment samples, first using 6 M HCl and then boiling 3 M CrCl2 solution (Canfield et al., 1986; Fossing and Barker Jørgensen, 1989). In each extraction, H2S was precipitated as Ag2S, filtered, dried, and sulfide was determined gravimetrically. The stoichiometry of the phase was used to convert the mass to weight percent (FeS for AVS; FeS2 for CRS).
Different operationally defined iron mineral phases were targeted with a four-step sequential extraction procedure applied to 100 mg freeze-dried and milled sediment samples. To extract amorphous and nanoparticulate iron (oxyhydr)oxide phases (Feascorbate), each sample was shaken for 24 h with 10 mL ascorbate solution (50 g L–1 sodium citrate, 50 g L–1 sodium bicarbonate and 10 g L–1 of ascorbic acid; buffered at pH 7.5; Raiswell et al., 2008). To target iron bound in carbonates (Feacetate), the residual sample was shaken for 48 h at 50°C with 10 mL sodium acetate solution (1 M sodium acetate solution buffered with acetic acid to pH 4.5; Poulton and Canfield, 2005). To target crystalline iron (oxyhydr)oxides (Fedithionite), the residual sample was then shaken for 2 h with 10 mL dithionite solution (50 g L–1 sodium dithionate buffered to pH 4.8 with acetic acid and sodium citrate; Poulton and Canfield, 2005). Lastly, for magnetite (Feoxalate), the residual sample was shaken with 10 mL ammonium oxalate solution (0.2 M ammonium oxalate/0.17 mL oxalic acid buffered with ammonium hydroxide to pH 3.2) for 6 h (Poulton and Canfield, 2005). These extractions were performed at room temperature (except for Feacetate at 50°C). After centrifugation (4000 rpm for 4 min at 21°C), the supernatant from each extraction was stored at 4°C until analysis by atomic absorption spectroscopy (AAS). Recent research has shown that mineralogical associations based on sequential chemical extractions need to be treated with caution (Oonk et al., 2017; Slotznick et al., 2020; Hepburn et al., 2020). For instance, the efficacy of the Feacetate stage in extracting iron bound in carbonates ranges from 3 to 85% (Oonk et al., 2017; Hepburn et al., 2020). The solubility of the targeted minerals depends upon their grain size, crystallinity and mineralogical association (Slotznick et al., 2020). Therefore, this study draws only tentative links between the extraction steps and the specific minerals represented by each step.
In situ Pore Water Sampling and Analyses
In late summer 2017, pore waters from the S1 polygon at Ice Wedge South and the N1 polygon at Ice Wedge North were sampled in situ using MacroRhizon soil moisture samplers (Van Walt Ltd.). Water samples for the analysis of δ18O-SO4 and δ34S-SO4 were collected to fill 50 mL centrifuge tubes and were stored at 4°C until processing and analysis at Lancaster Environment Centre, Lancaster University, United Kingdom. Water samples for the analysis of δ18O-H2O were collected to fill Eppendorf tubes and were stored at 4°C until analysis at the University of East Anglia (UEA). Water samples for the analysis of δ18O-SO4 and δ34S-SO4 were loaded onto ion exchange resins (SupeliteTM DAX-8 for removal of dissolved organic matter; Dowex 50 W-X8 for removal of cations; and Dowex AG2 X8 for removal of anions). Anions were eluted from the Dowex® AG2 X8 using aliquots of 1 M ultrapure HCl to a total volume of 1.5 mL. A 0.2 mL volume of 1 M BaCl2 was added to the eluted sample, and the samples were left for 48 h at 4°C to allow BaSO4 to precipitate. Each sample was rinsed three times with Milli-Q de-ionised water using centrifugation and re-suspension to remove any interfering products (e.g., chlorides). The samples were dried at 40°C.
The dry samples were weighed into tin or silver capsules for analysis of δ34S-SO4 and δ18O-SO4, respectively. Isotopic analysis was undertaken by Elemental Analyser (Elementar Pyrocube) linked to a continuous flow isotope ratio mass spectrometer (Isoprime 100 with dual inlet capability for injection of monitoring gases), following methods in Wynn et al. (2015). Combustion of BaSO4 within tin capsules yielded SO2 for determination of δ34S. Analytical conditions demanded the use of vanadium pentoxide as an oxidizing agent and a combustion temperature of 1120°C. Pyrolysis of BaSO4 at 1450°C within silver capsules and in the presence of carbon black, yielded CO for the determination of δ18O. δ34S values were corrected against Vienna Cañon Diablo Troilite (VCDT) using within run analyses of international standards NBS-127 and SO5. δ18O values were corrected to Vienna Standard Mean Ocean Water (VSMOW) using within-run analyses of NBS-127 and SO6. Internal standard MLSG (a subglacial meltwater precipitate of BaSO4) was used to monitor drift and precision within each run as well as external precision between analytical sequences. Within-run standard replication (1SD) was better than ± 0.3‰ for both sulfur and oxygen isotope values.
Water samples for the analysis of δ18O-H2O were analysed at the University of East Anglia (UEA) with a Picarro 1102i analyser, by direct injection of 2.6 μL of water. Samples were measured together with two United States Geological Survey (USGS) standards: USGS 64444 and USGS 67400 and a UEA Norwich Tap Water (NTW) internal laboratory standard. Using the calibration line defined by the USGS standards, the true isotopic composition of the samples was calculated, relative to VSMOW.
Precipitation Correction
Pre-melt snowpack chemistry data from Svalbard were compiled (Hodgkins et al., 1997; Wynn et al., 2006; Tye and Heaton, 2007; Yde et al., 2008) to calculate the mean X/Cl ratios (where X is a major anion or cation). The following ratios in the snowpack were established: Na/Cl = 0.82; K/Cl = 0.02; Mg/Cl = 0.10; Ca/Cl = 0.08; SO4/Cl = 0.11. By assuming that chloride behaves conservatively and originates only from precipitation (pd), these ratios were used to calculate the fraction of each anion or cation in pore water (pw) derived from precipitation (fXpd; Equation 1):
The residual (fXnpd) represents the fraction of each anion or cation in pore water derived from weathering (Equation 2):
The δ34S-SO4 values were corrected for snow inputs, following Equation (3) (from Hindshaw et al., 2016) and using the mean snowpack δ34S-SO4 value of 17.5‰ from the compilation of pre-melt snowpack chemistry from Svalbard.
Where f is the fraction of sulfate derived from snowmelt. The δ18O-SO4 values were corrected in the same way as in Equation (3), using the mean snowpack δ18O-SO4 value of 9.28‰ from the compilation of pre-melt snowpack chemistry from Svalbard.
Bedrock Sulfide δ34S Sampling and Analyses
Bedrock samples (sandstones, siltstones, shales) from each geological formation in the Adventdalen Group and the Carolinefjellet Formation were obtained from cores 13/2013 (33X E522859 N8685197, Dirigenten), BH9/05 (Urdkollbreen, 33X E528365 N8647669) and outcrops in Adventdalen. Rock samples were cut into small blocks using a lapidary trim saw (Lortone, United States) and weathered surfaces removed using a grinding surface (Saphir 330, ATM, Germany). Rocks were washed with de-ionised water in an ultrasonic bath before being dried in an oven at 50°C. Dried rocks were pulverised in a steel pestle and mortar into small chips which were further crushed to a fine powder in an agate disc mill (Tema, United Kingdom). Crushing equipment was washed with water, dried with compressed air and cleaned with ethanol between samples to prevent contamination. Chromium-reducible sulfur (CRS) was extracted from ground rock using the method previously described (Section “Solid Phase Analyses”). The resultant Ag2S precipitates were weighed into tin capsules for δ34S analysis via combustion in an Elementar Pyrocube elemental analyser coupled to an Isoprime continuous flow mass spectrometer at the University of Leeds. Samples were combusted at 1150°C to SO2 in the presence of pure oxygen (N5.0) into a stream of helium (CP grade). The SO2 produced flowed through tungstic oxide packed into the combustion column to ensure quantitative conversion. Excess oxygen was removed by reaction with hot copper wires at 850°C and water was removed in a Sicapent trap. Duplicate aliquots of silver sulfide were prepared because of a small sulfur isotope memory effect incurred during SO2 processing in the pyrocube. δ34S values were corrected to the VCDT scale using international standard IAEA S-3 (−32.06‰) and an inter-lab chalcopyrite standard CP-1 (−4.56‰). Reproducibility of a within-run check standard (BaSO4) was ± 0.24‰ (1 SD).
Data Analyses
Data analyses were performed in Microsoft Excel and R Core Team (2017) and graphics were prepared with Excel or the ggplot2 R package. Prior to testing correlations between variables, Shapiro Wilk’s method was used to test whether data were normally distributed. Many variables were not normally distributed and their inter-relationships were non-linear. Therefore, monotonous relationships between the variables were tested using Spearman correlation, which is appropriate for non-normally distributed data and is particularly robust for dealing with skewed distributions and outliers (du Prel et al., 2010).
Results
Figure 2 shows that the Ice Wedge North cores had a significantly higher gravimetric water content (g water g–1 dry sediment) than the Ice Wedge South cores according to Welch’s t-test (Table 2). The mean gravimetric water content in the N1 core was 2.79 g g–1 (maximum was 9.14 g g–1) and was highest both at the surface and below 90 cm depth, whereas N2 had a mean gravimetric water content of 6.05 g g–1, with no clear patterns with depth. In S1, however, the mean water content was 0.57 g g–1, with peaks near the surface, and at 105, 141 and 169 cm, and the maximum water content was 1.32 g g–1. The mean gravimetric water content of S2a and S2b was similar (0.58 and 0.45 g g–1, respectively). Both these cores had an increased water content near the surface.
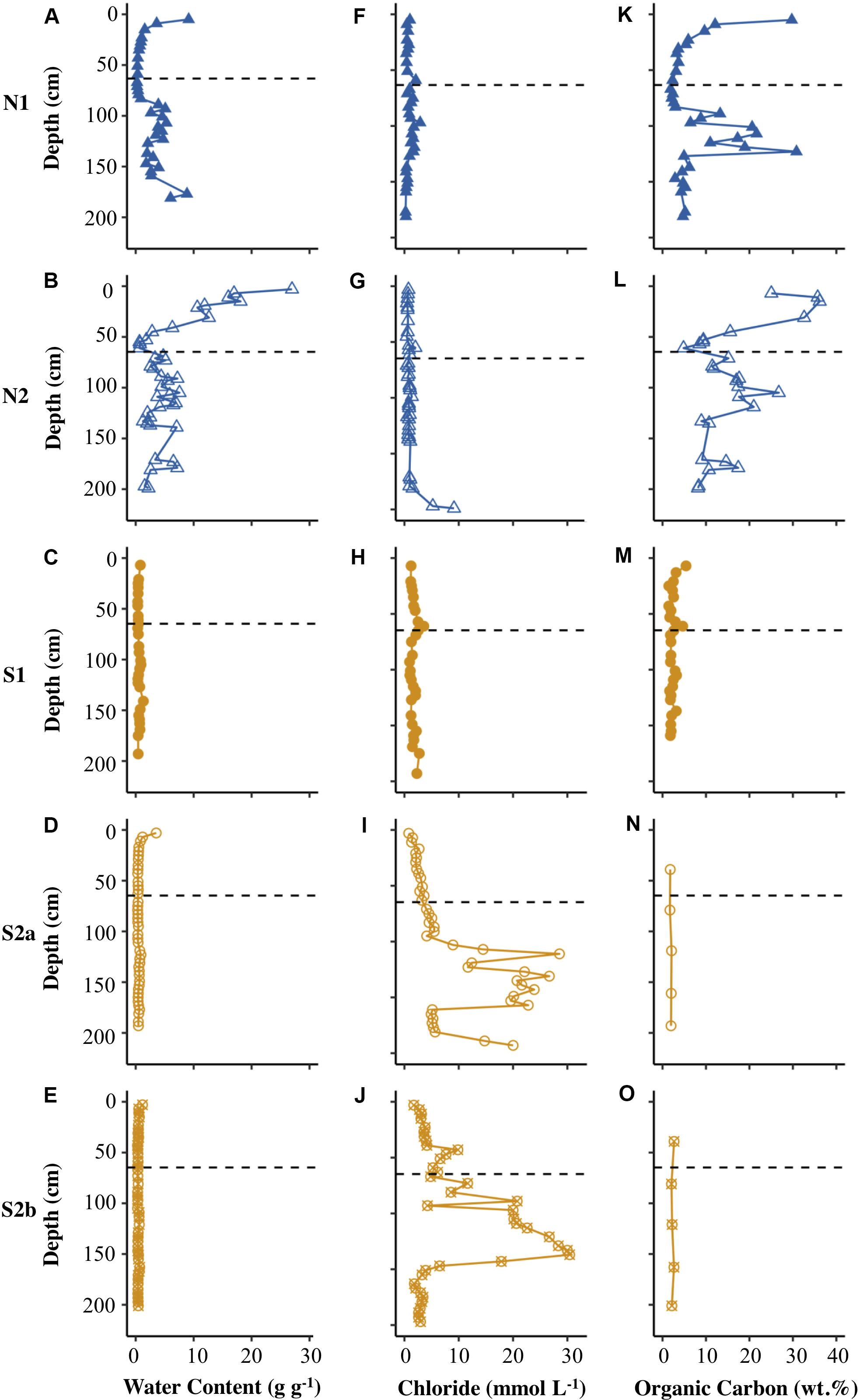
Figure 2. Depth profiles of water content (A–E), chloride concentration (F–J), and concentration of organic carbon (K–O) for N1, N2, S1, S2a, and S2b. The horizontal dashed line on each plot represents the base of the active layer in 2017.
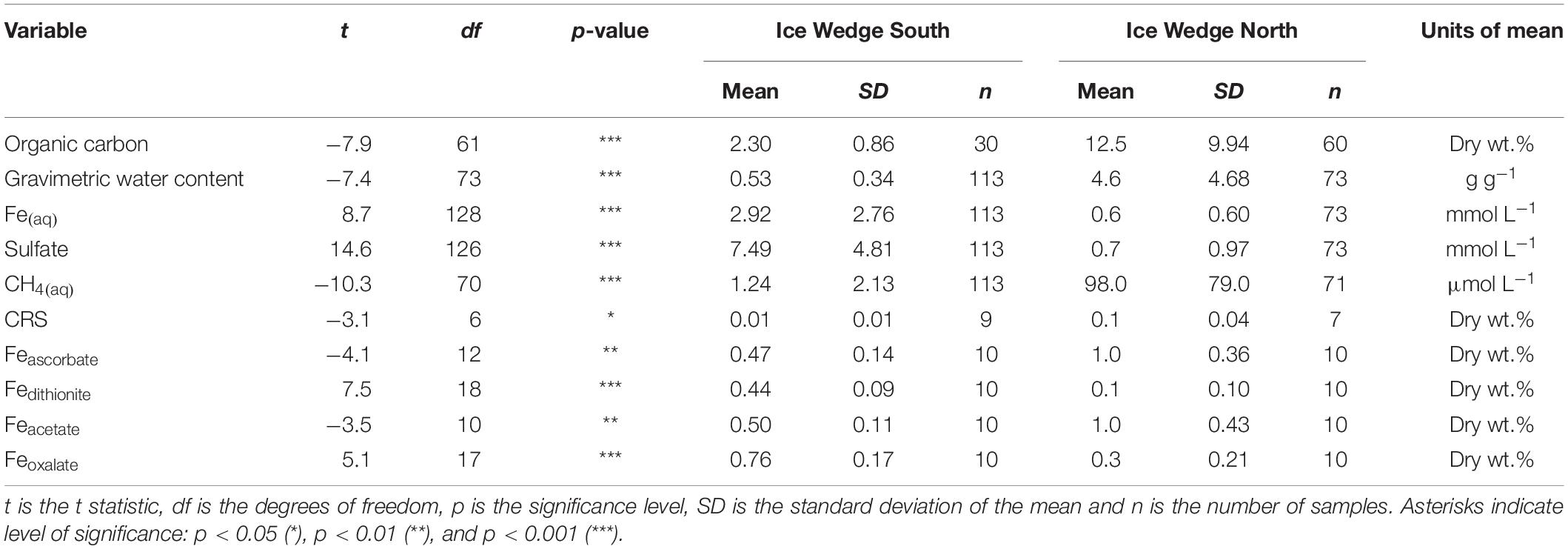
Table 2. Results of Welch’s t-tests comparing organic carbon, gravimetric water content, Fe(aq), sulfate, CH4(aq), CRS, Feascorbate, Fedithionite, Feacetate, and Feoxalate for Ice Wedge South and Ice Wedge North.
The concentration of chloride was higher in the cores from Ice Wedge South, compared to those from Ice Wedge North (Figures 2F–J). The concentration of chloride peaked close to the base of the active layer in cores from both sites (Figures 2F–J). There was also a peak in chloride concentration near the ground surface at N1 (Figure 2F). There was an accumulation of chloride in the underlying permafrost of cores N2, S2a and S2b (Figures 2G–J).
The sedimentary organic carbon content for the two sites also differed significantly according to Welch’s t-test (Table 2). Figure 2 and Table 2 show that Ice Wedge North had a higher organic carbon content than Ice Wedge South. The surface of N1 had a high organic carbon content (29.7 wt.%). In the permafrost, the organic carbon content was high, but variable (low at 30−70 cm depths, peaking at 100−140 cm depth). S1, however, had a more uniform and lower organic carbon content (mean of 2.38 wt.%), but with a peak near the surface (5.40 wt.%) and at 61 cm depth (4.65 wt.%).
The concentrations of Fe(aq) and sulfate in pore waters differed significantly between the two sites, according to Welch’s t-test (Table 2). Figures 3A,B show that the Fe(aq) in N1 and N2 was <3.0 mmol L–1. In N1, peaks in aqueous iron occurred at 31, 67, 93, and 155 cm depth. In N2, the peaks were broader and at depths of 15−73 cm, 109−139 cm, and 197 cm. Figures 3F,G show that while N2 had a sulfate concentration <1.3 mmol L–1, N1 had up to 4.7 mmol L–1 in the peak at the active layer base. N1 had a clear pattern in sulfate, increasing from the surface toward the base of the active layer, and then decreasing into the permafrost (with a small peak ∼150 cm).
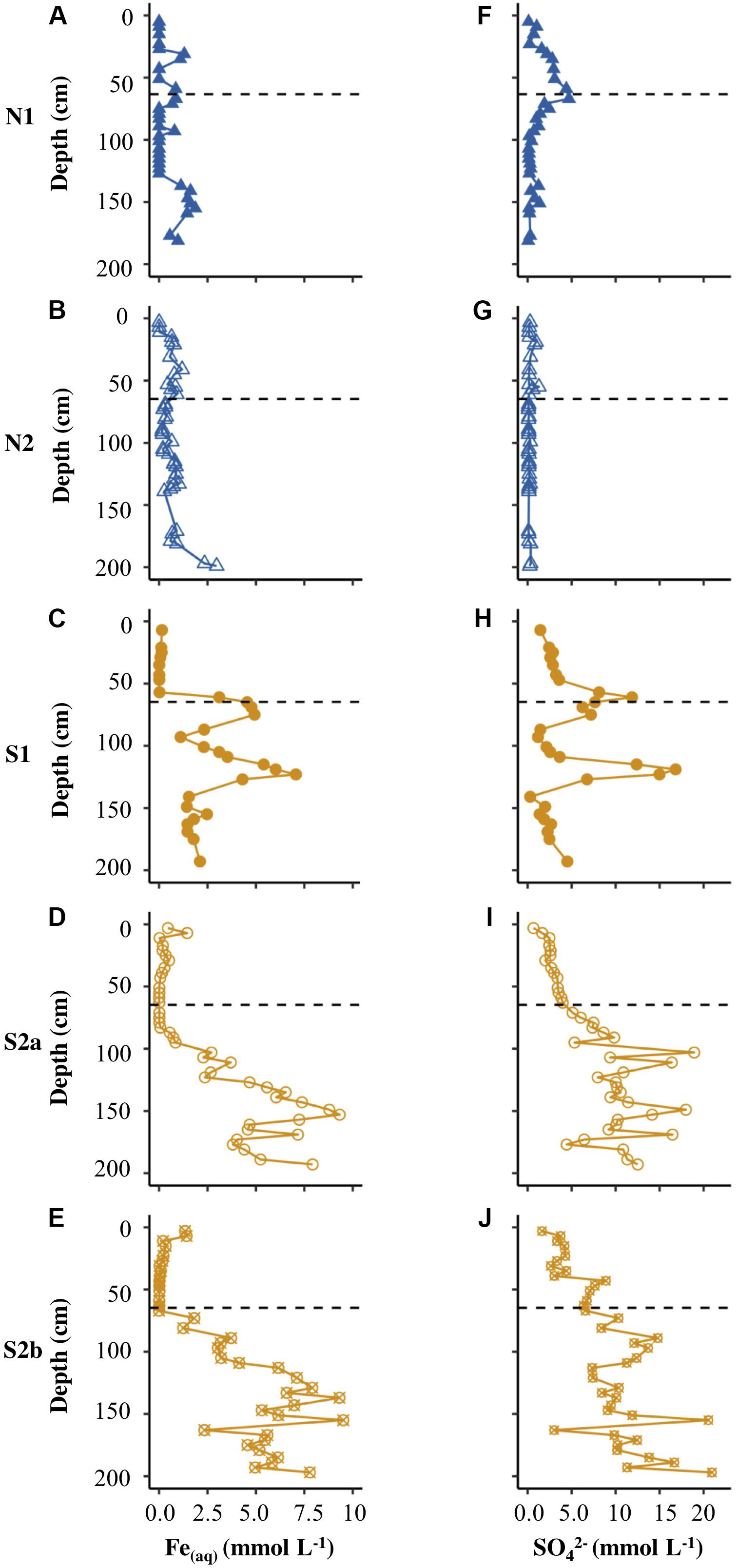
Figure 3. Depth profiles of Fe(aq) (A–E) and (F–J) for N1, N2, S1, S2a, and S2b. The horizontal dashed line on each plot represents the base of the active layer in 2017.
Figures 3C–E show that the concentration of Fe(aq) was low (<1.5 mmol L–1) in the active layer of all the Ice Wedge South cores. Fe(aq) in S1 peaked at 75 cm and 123 cm depth. The Fe(aq) concentration in the permafrost of S2a and S2b had less distinct peaks, but increased with depth, reaching over 7.5 mmol L–1. Figures 3H–J show that sulfate followed similar depth trends to Fe(aq) for all three Ice Wedge South cores, reaching >20 mmol L–1 in core S2b. The sulfate concentration in the active layer was higher than the Fe(aq) concentration.
Figures 4A–F show that N1 had only low concentrations of calcium and magnesium in pore water (<6 mmol L–1), with the highest values of both cations in the uppermost core sample. N2 had similarly low concentrations of calcium and magnesium (Figures 4B,G). S1 displayed distinct peaks in all cations at the base of the active layer and >1 m depth in the permafrost (Figures 4, 5C,H). S2a and S2b show the greatest range in concentrations of calcium and magnesium (reaching ∼15 mmol L–1), with higher concentrations in the permafrost (Figures 4D,E,I,J). The concentration of potassium was generally <2.5 mmol L–1 in N2, S1, S2a and S2b (Figure 5), but reached >20 mmol L–1 in N2. The sodium concentration was <10 mmol L–1 in all the cores (Figures 5F–J).
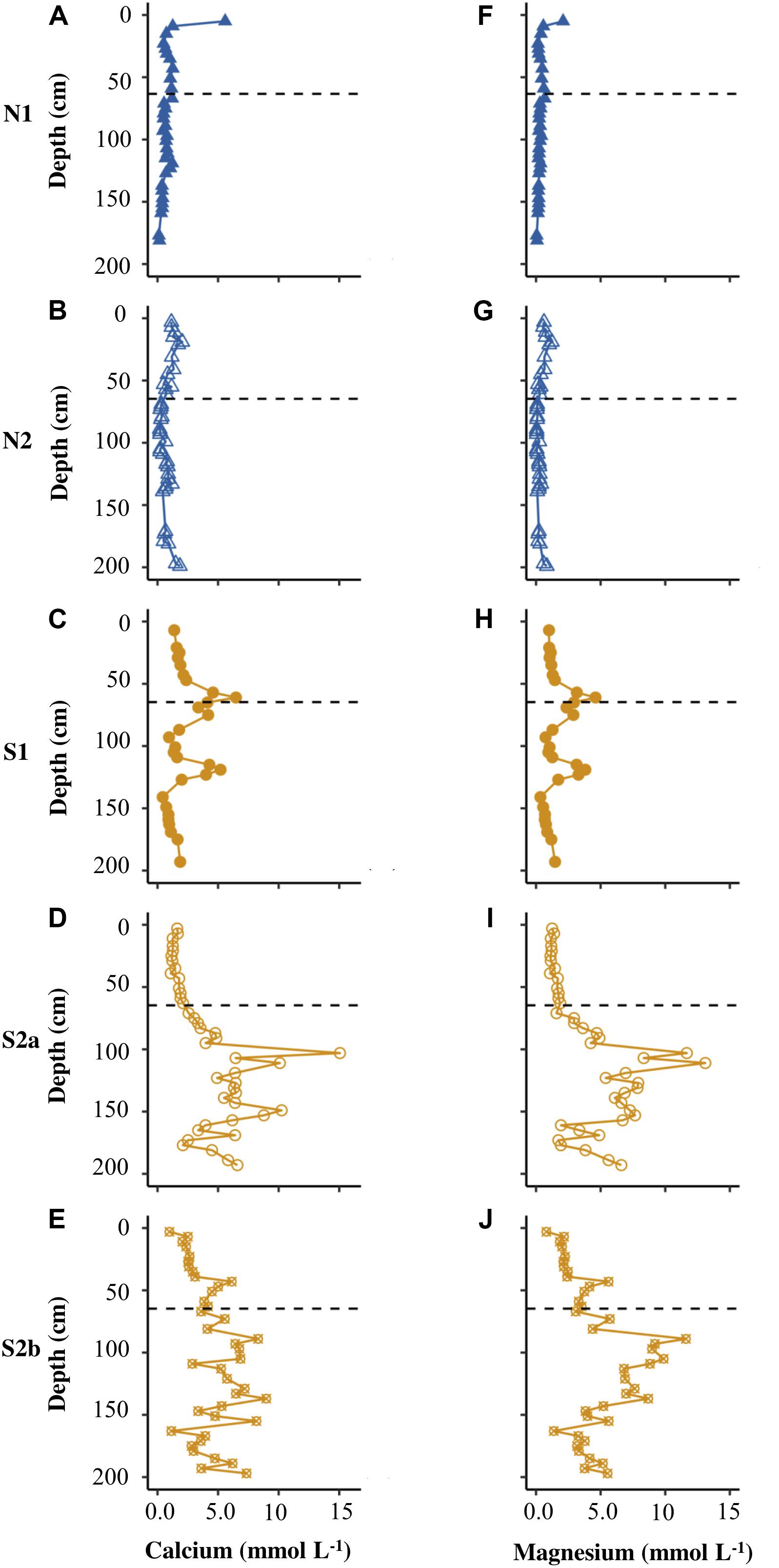
Figure 4. Depth profiles of calcium (A–E) and magnesium (F–J) for N1, N2, S1, S2a, and S2b. The horizontal dashed line on each plot represents the base of the active layer in 2017.
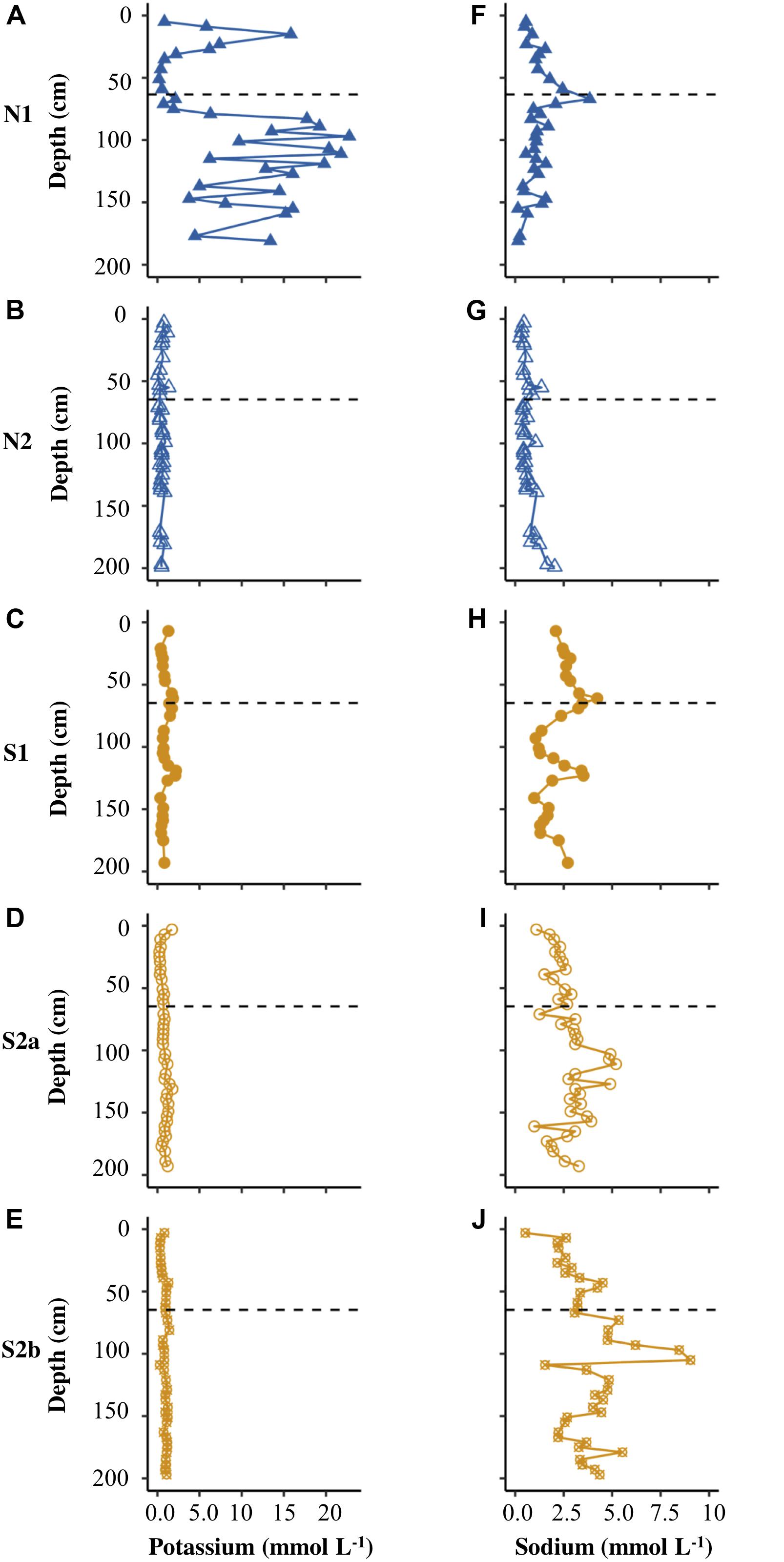
Figure 5. Depth profiles of potassium (A–E) and sodium (F–J) for N1, N2, S1, S2a, and S2b. The horizontal dashed line on each plot represents the base of the active layer in 2017.
The concentration of solid phase iron species was variable in the N1 core, with concentrations: Feascorbate < 1.6 wt.%, Fedithionite < 0.35 wt.%. Feacetate was the dominant extracted iron phase at N1, reaching a maximum of 1.57 wt.% (Table 3 and Supplementary Figures S1A,C,E). Feoxalate at N1 was < 0.71 wt.% (Supplementary Figure S1G). N1 had the highest CRS at 15 cm depth and at 115 cm depth (Supplementary Figure S1K), which tracked the organic carbon content. AVS was detected at 15 cm in N1, and also in all three samples measured between 114 and 156 cm (Supplementary Figure S1I). Compared with the porewater Fe(aq) and sulfate profiles, the concentration of solid phase iron species was constant with depth in S1 (Supplementary Figures S1B,D,F,H,J,L). Feascorbate was <0.8 wt.%, Fedithionite was <0.6 wt.%, Feacetate was <0.8 wt.%, Feoxalate was <1 wt.%, CRS was <0.05 wt.%, and no AVS was detected in this core (Table 3). Overall, N1 had less Fedithionite and Feoxalate than S1. N1 contained an order of magnitude more CRS than S1 (Table 3).
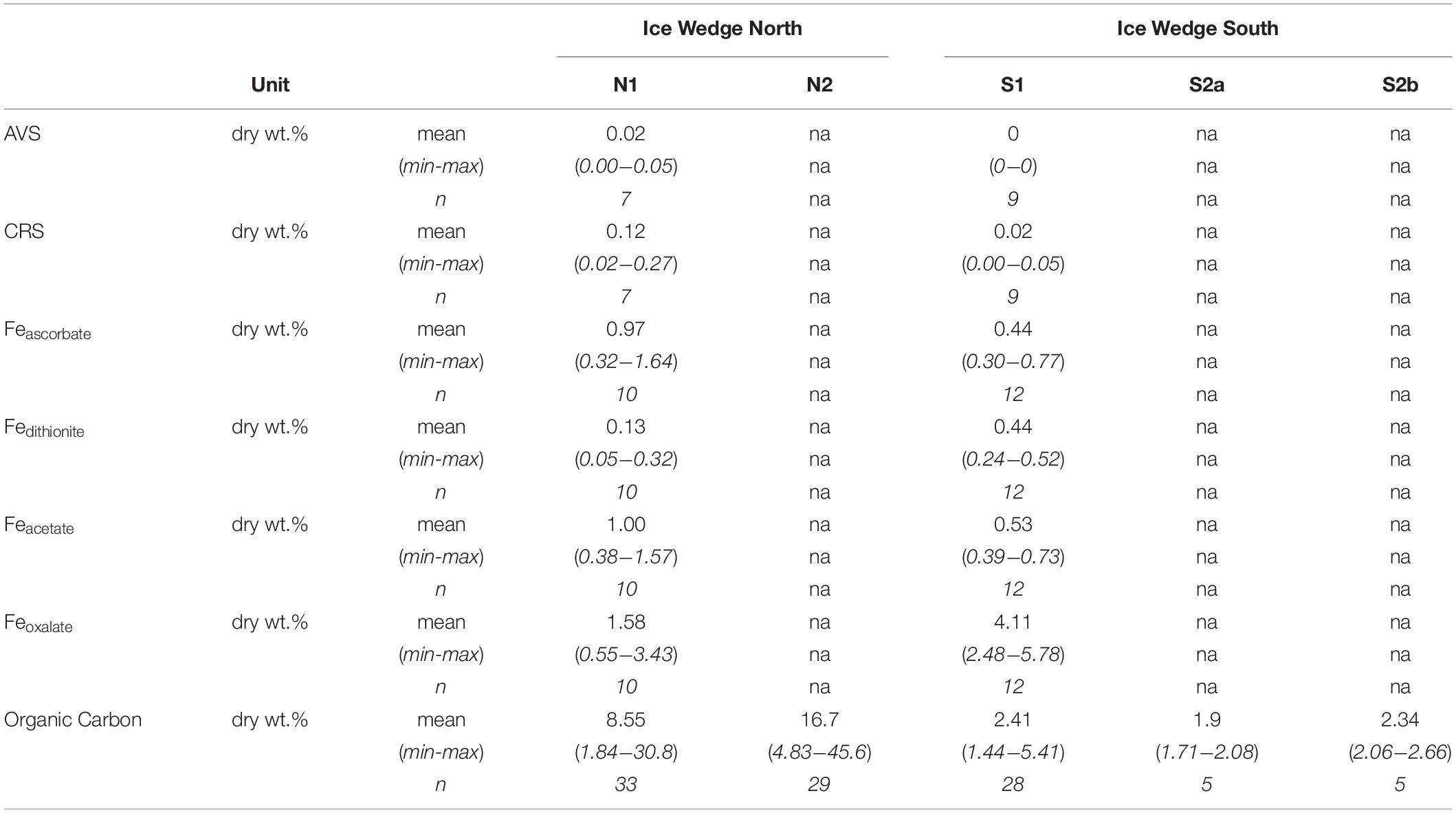
Table 3. Summary statistics calculated for the length of each core for the solid phase data of the cores S1 and N1 from Ice Wedge South and Ice Wedge North, including AVS, CRS, Feascorbate, Fedithionite, Feacetate, Feoxalate, and organic carbon.
Table 4 summarises the δ34S-SO4 results obtained from samples of water within the active layer. Overall, water from Ice Wedge North water was more enriched in 34S than water from Ice Wedge South. The Ice Wedge North δ34S-SO4 was increasingly depleted in 34S with increasing depth. At Ice Wedge South, the water δ34S-SO4 was most enriched in 34S at 30 cm depth, whereas the δ34S-SO4 was more depleted in 34S at depths of 9 cm and 60 cm.
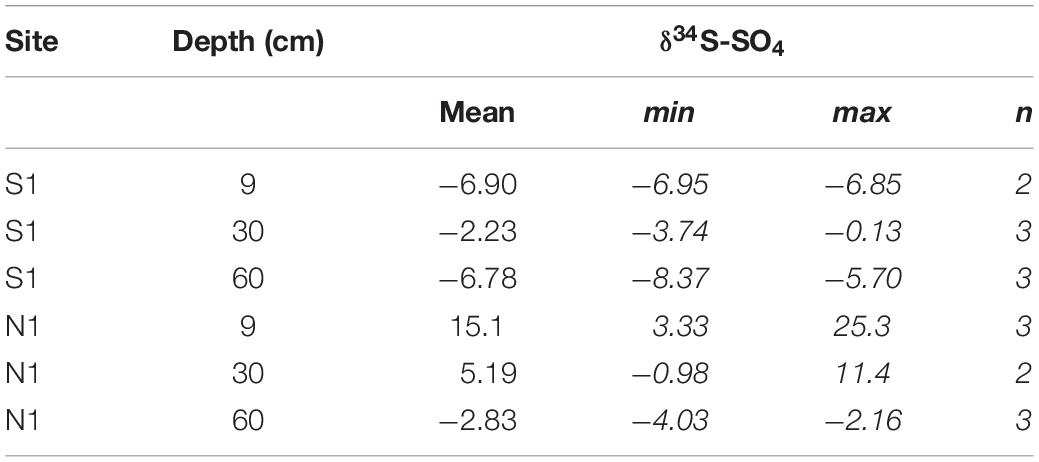
Table 4. δ34S-SO4 in water from the active layer in polygons S1 and N2.
Discussion
Since emergence from the sea during the Holocene epoch, permafrost aggradation and ongoing organic carbon accumulation have most likely caused significant changes in the biogeochemical processes and mineral precipitation reactions within this high Arctic floodplain. The two sites described above help understand these changes because their contrasting hydrological regimes result in marked differences in the accumulation and decomposition of permafrost organic carbon, in spite of their proximity within the same valley. Below, we describe how the quantity of organic carbon regulates the consumption of alternative electron acceptors used for microbial oxidation of organic carbon, thus causing a switch in the water-saturated areas of the floodplain to iron- and sulfate-reduction, with net iron and carbon storage via increased CRS and Feacetate precipitation. In contrast, the drier areas store far less organic carbon and instead remain dominated by the biogeochemical signatures of pyrite oxidation. These processes seem most likely during the earlier stages of floodplain development, when they play a dominant role in the weathering of fresh mineral surfaces, as expressed by the composition of glacial meltwaters in the nearby Endalen, Bolterdalen and Longyeardalen catchments (Yde et al., 2008; Rutter et al., 2011; Hodson et al., 2016, respectively). In the following discussion, we therefore explore first of all the dominant weathering processes that are linked to pyrite oxidation, before examining the other processes associated with sulfur and iron biogeochemistry that better explain the later stages of biogeochemical evolution, once more organic carbon has become available.
Dominant Weathering Reactions
The pore water profiles in Figures 2–5 represent the following attributes of both the active layer and the underlying permafrost: (1) the in situ distribution of extractable or adsorbed solutes, reactive mineral phases, and gases, and (2) the products of additional rock-water-microorganism interactions following thaw. Across both sites, except for N1 where potassium dominated, calcium and magnesium were the dominant cations (Figures 4, 5), indicating that carbonate dissolution prevails over silicate dissolution, despite a low carbonate content in the sediments (< 0.8 dry wt.% at Ice Wedge South) and low volumetric carbonate contents (∼0.3 to 1.8%, but up to 10.7%) in rocks from the Todalen and Endalen endmembers (Svinth, 2013). This contrasts with a study in a nearby unglaciated catchment (Fardalen) that found a relatively high proportion of silicate weathering, suggested to be the result of a combination of relatively rapid leaching of carbonate phases from the active layer and low rates of physical weathering in the sediments, failing to expose fresh carbonates to weathering (Hindshaw et al., 2016). However, carbonate weathering has previously been shown to control the water chemistry of both glacial and non-glacial watersheds, even where the bedrock is predominantly silicate, with only trace amounts of carbonate (Blum et al., 1998; Horton et al., 1999). This strongly suggests that although the low carbonate content coupled with the potential for active layer leaching might limit the importance of carbonate weathering in Adventdalen to some degree, the reactivity of the carbonate phases (compared to silicates) compensated for these factors.
The weathering processes responsible for the acquisition of Ca2+ and Mg2+ in the sediment pore water are most likely to be represented by the following reactions (after Tranter et al., 2002; Yde et al., 2008; Hindshaw et al., 2016):
(1) Carbonate dissolution with carbonic acid (where x is equal to 1 or 0):
(2) Sulfide oxidation coupled to carbonate weathering (where x is equal to 1 or 0):
The covariance between ions is used here to identify the dominant weathering processes in the entire active layer and permafrost. Evidence for dolomite weathering as a source of both Ca2+ and Mg2+ was therefore provided by the strong positive correlation between these ions at Ice Wedge South (p < 0.0001, ρ = 0.95) and a regression slope close to unity (0.89). When corrected for precipitation inputs (rain and snow) of both cations, the slope remained unchanged (Figure 6), but the intercept decreased significantly toward zero (0.24 mmol L–1). Therefore dolomite represents a credible, common source for both ions.
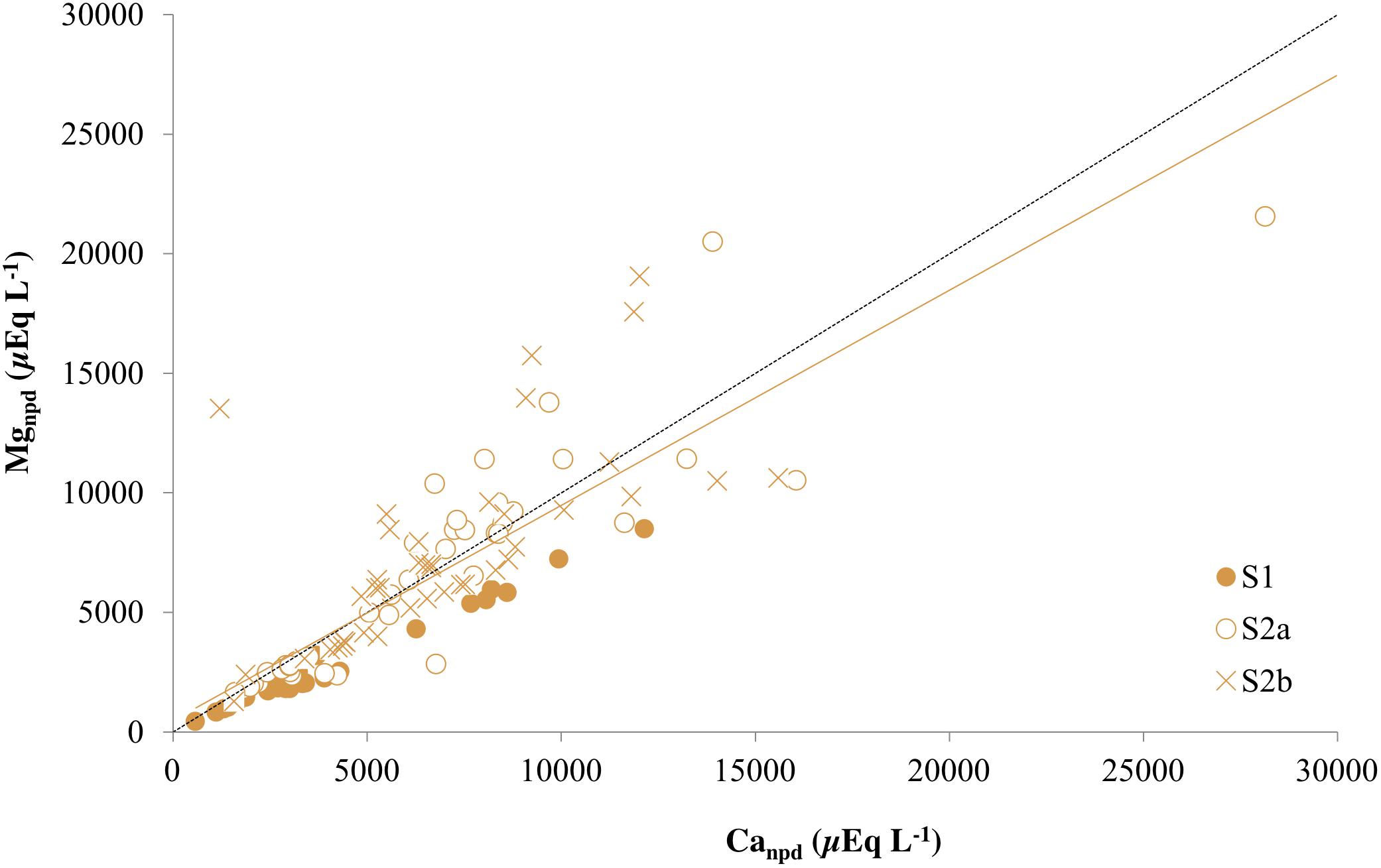
Figure 6. Non-precipitation-derived magnesium (Mgnpd) and non-precipitation-derived calcium (Canpd) in the three cores from Ice Wedge South. The dashed black line corresponds to a 1:1 relationship between Canpd and Mgnpd that represents dolomite dissolution. The solid orange line is a regression for all points from the three cores from Ice Wedge South (R2 = 0.69).
Although (Ca + Mg)npd was strongly correlated with npd (p < 0.0001, ρ = 0.81), the regression slope was 0.73, which suggests that some of the sulfate was associated with other processes. Given the presence of silicates in the catchment, sulfide oxidation coupled to silicate dissolution may play a role in making up the deficit. Since silicates in the catchment are mainly present as Na- and K-feldspars, they may be represented by the formulae: NaAlSi3O8 and KAlSi3O8 in the following reactions (Tranter et al., 2002; Hindshaw et al., 2016):
(1) Silicate dissolution with carbonic acid (where x is equal to 1 or 0):
(2) Sulfide oxidation coupled to silicate dissolution (where x is equal to 1 or 0):
When silicate and carbonate dissolution are both driven by sulfide oxidation, the total base cation (Ca2+ + Mg2+ + Na+ + K+) ratio to tends toward unity when precipitation inputs are insignificant (Fairchild et al., 1994; Tranter et al., 2002; Wadham et al., 2010). However, this characteristic signature can be overprinted or masked by gypsum dissolution, ion exchange reactions or mineral precipitation reactions that remove base cations from solution. Of these, gypsum may be ignored, because it is absent from the bedrock (Svinth, 2013). Prior to correction for precipitation inputs, the total base cation ratio to sulfate at Ice Wedge South was 0.98 (r2 = 0.68). The standard correction for precipitation inputs resulted in a lower regression slope of 0.76 (r2 = 0.70; Figure 7A). Of the base cations, sodium was particularly affected by the above masking effects, with non-precipitation inputs appearing negative, similar to that which may be inferred from pore water data for Adventdalen sediment cores presented by Cable et al. (2017). This is indicative of ion exchange reactions or mineral precipitation (e.g., albite) and hence precludes the use of the non-precipitation ion ratios to estimate the importance of sulfide oxidation coupled to silicate dissolution.
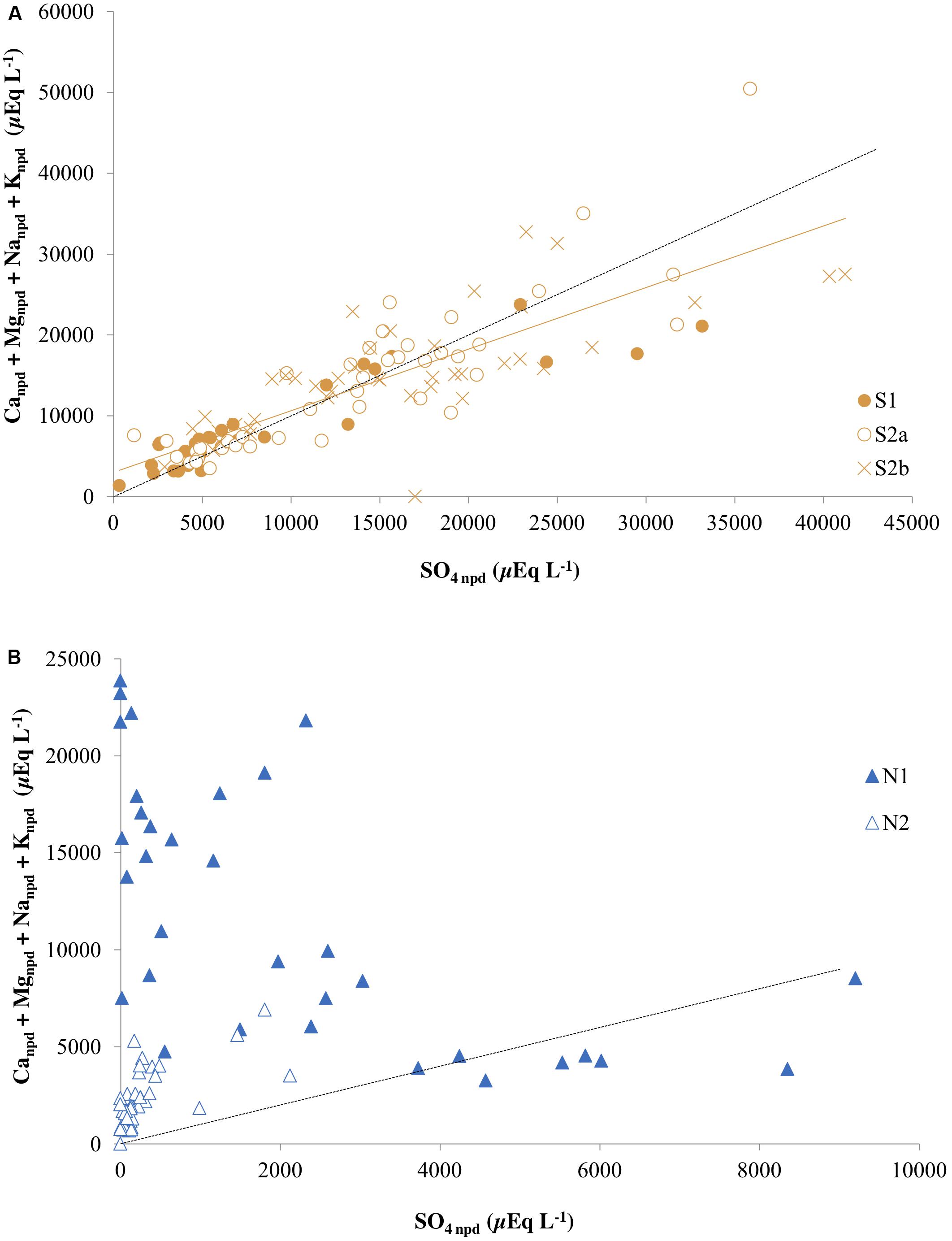
Figure 7. Total non-precipitation-derived base cations and non-precipitation-derived sulfate in (A) the three cores from Ice Wedge South and (B) the two cores from Ice Wedge North. The dashed black line corresponds to a 1:1 relationship between SO4 npd and (Canpd + Mgnpd + Nanpd + Knpd) that represents sulfide oxidation coupled to silicate dissolution. The solid orange line is a regression for all points from the three cores from Ice Wedge South (R2 = 0.70).
Since the concentration of derived from pyrite oxidation was high and developed within a sometimes-anoxic environment, alternative processes of pyrite oxidation to those shown by Equations 5 and 7 require identification. For instance, under acidic conditions, the Fe(OH)3 produced via Equations 5 and 7 dissociates to form Fe3+, the reduction of which could result in a significant contribution of ferrous iron to the total cations in solution (Raiswell and Canfield, 2012; Hodson et al., 2016; Raiswell et al., 2018).
However, the reduction of iron (oxyhydr)oxide need not be achieved in combination with sulfide oxidation (Equation 9).
In contrast to Ice Wedge South, Figure 7B shows that the Ice Wedge North pore waters generally do not plot on the 1:1 line, and so there is no relationship between sulfate and total cations for these. Despite this, some samples from this site have a sulfate to chloride ratio greater than the snowpack sulfate to chloride ratio. This indicates that sulfide oxidation has enhanced the sulfate concentrations. The absence of a 1:1 relationship between total cations and sulfate at this site therefore strongly suggests that the sulfate produced by sulfide oxidation is removed in part by sulfate reduction during respiration of organic matter (Equation 10; Wadham et al., 2004).
The major ion ratios show that sulfide oxidation coupled to carbonate dissolution can contribute substantial quantities of sulfate to the sediment pore water at Ice Wedge South. The weathering mechanisms here are analogous to those reported in glacial catchments of the area, which is intuitive when the source of the sediments is considered (i.e., the aeolian deposition of glaciofluvial sediments dessicated during early winter). The sulfuric acid produced by sulphide oxidation may further act as a weathering agent in this system, akin to in glacial catchments, potentially producing carbon dioxide during weathering of carbonates (e.g., Torres et al., 2017). In contrast, processes removing sulfate from the sediment pore water at Ice Wedge North preclude the use of major ion ratios to determine the significance of sulfide oxidation. In spite of this, sulphide oxidation is still very likely to occur because the provenance of the sediments is the same as that at Ice Wedge South.
Sources and Sinks of Sulfate
Sulfur and oxygen isotopic values of sulfate in waters provide compelling evidence for the identification of the sources and sinks of sulfate (e.g., Wynn et al., 2006, 2015; Turchyn et al., 2013; Hindshaw et al., 2016). Pre-melt snowpacks in Svalbard at Midtre Lovenbreen (Wynn et al., 2006) and near Ny Ålesund (Tye and Heaton, 2007) suggest a δ34S range of +17 to +18‰ and a δ18O range of +8.6 to +9.7‰ for the precipitation-derived sulfate contribution to the active layer pore water (i.e., pd). A study of dissolved organosulfur compounds in a raised peat bog showed that atmospheric sulfur in surface water sulfate is also taken up by plants (plant δ34S was 0.1‰ and 4.2‰) and released when they decay, producing humic organosulfur with δ34S values reflecting the precipitation-derived origin of the sulfate (Bottrell et al., 2010). Other sources of sulfate to the pore water include the oxidative weathering of pyrite (OWP), as in Equations 5, 7, and 8. The mass-weighted mean sulfide δ34S values in the geological formations of the study region vary widely, from −40.6‰ in the Grumantbyen Formation to 1.78‰ in the Aspelintoppen Formation (Table 5). In the geological formations measured closest to the sites, the range in mass weighted mean sulfide δ34S values is narrower, from −13.8‰ in the Carolinefjellet Formation to −2.01‰ in the Firkanten Formation. These nearby δ34S values are therefore used to discriminate the rock-derived sulfate from the snowpack-derived sulfate.
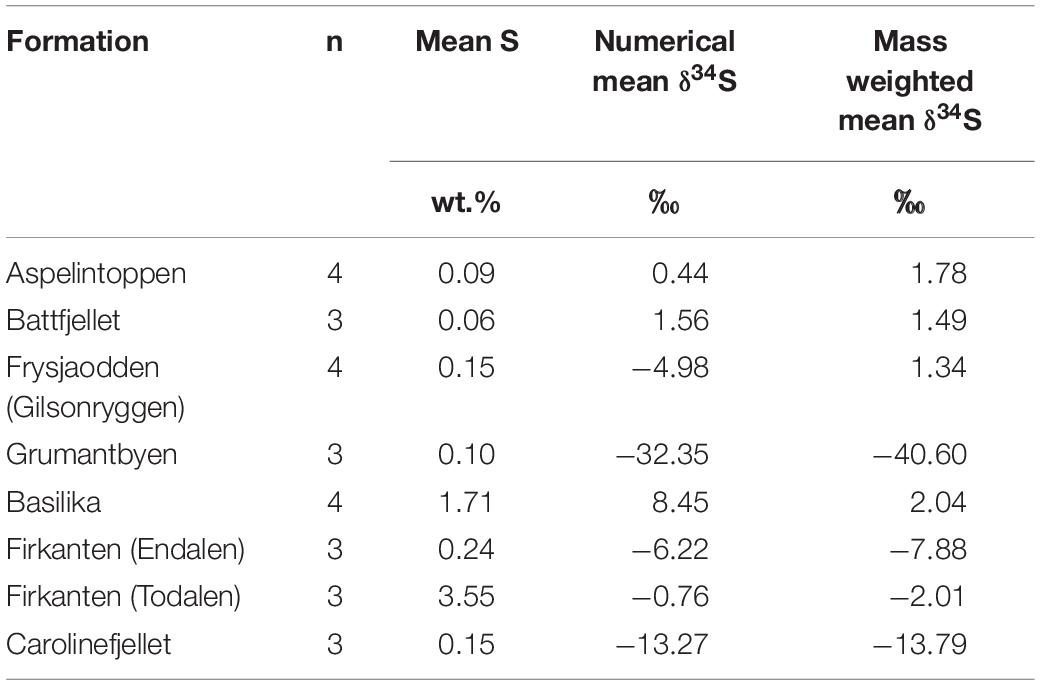
Table 5. The mean quantity of sulfide (wt.%) in bedrock, the numerical mean sulfide δ34S (‰) in bedrock and the mass-weighted mean sulfide δ34S (‰) in bedrock.
The δ18O of sulfate depends on the oxidation pathway; the oxygen atoms in the sulfate can originate either from atmospheric oxygen (+23.5‰) or from the surrounding water (−11‰ to −14‰ at our sites). In sulfate produced by OWP via Fe3+ (Equation 8), the oxygen atoms are derived solely from the surrounding water molecules. Experiments have demonstrated that there is no isotopic discrimination during the incorporation of oxygen atoms from water molecules into sulfate (Lloyd, 1968). In contrast, the incorporation of oxygen atoms from O2 molecules into sulfate molecules during OWP via O2 causes an isotopic fractionation of −8.7‰. Consequently, sulfate produced by OWP via O2 is depleted in 18O by −8.7‰ compared with atmospheric O2, which is strongly enriched in 18O at +23.7‰ (Bottrell and Tranter, 2002). However, during OWP via O2, there can be isotopic exchange between water and oxygen atoms in sulfoxy anions of intermediate valency, obscuring the isotopic signal of atmospheric oxygen (Balci et al., 2007). Hence, even in OWP via O2, three out of four oxygen atoms in the sulfate molecule could show an isotopic signal from water (Bottrell and Tranter, 2002).
To elucidate whether the sulfate in the samples from Adventdalen could derive from OWP only via O2, the approach of Bottrell and Tranter (2002) was applied to the δ18O values of sulfate from the pore waters in the active layer. This conservative approach assumed that only the final oxygen atom incorporated into sulfate will still carry an isotopic signature indicative of its source (water or atmospheric oxygen). Only if there is less than 25% of the oxygen in a sulfate molecule derived from O2 can it be certain that part of the sulfate was produced anoxically, by OWP via Fe3+. Equation 11 uses the measured δ18O-H2O water isotopic compositions (Table 6) to calculate a threshold sulfate δ18O (δ18OTHRESH) for the formation of sulfate with one oxygen atom from O2 and three from water (Bottrell and Tranter, 2002).
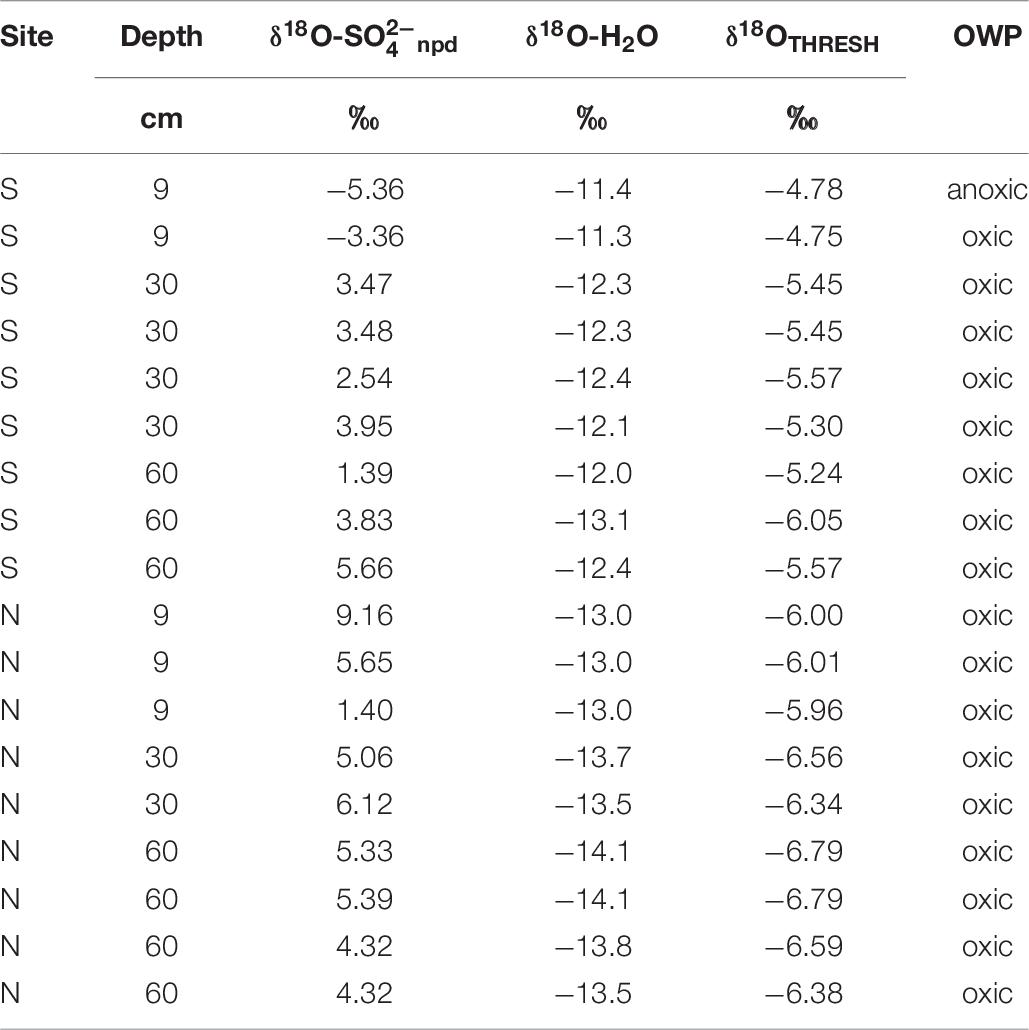
Table 6. Oxygen isotopes of weathering-derived sulfate (δ18O-npd) and water (δ18O-H2O) compared to the threshold for anoxic oxidation of pyrite (δ18OTHRESH).
Comparing δ18O- and δ18OTHRESH- data, only one sample from the Adventdalen active layer pore water falls below the threshold for OWP via Fe3+ (Table 6), indicating that npd in this sample originates from OWP via Fe3+. In the rest of the samples, npd could have originated from OWP via Fe3+, but the isotopic data do not require that and it is probable that npd in the remaining samples instead originated from OWP via O2. This is a surprising result, as these samples are from between 9 and 60 cm below the ground surface and it was anticipated that oxygen penetration would decrease with profile depth. However, it is possible that radial oxygen loss from the roots of wetland plants may have provided an oxygen source to this deeper pore water (e.g., Johnston et al., 2014). In addition, ice-wedge cracking and shallower cracking restricted to the active layer (O’Neill and Christiansen, 2018) provide a route for ingress of oxygenated rain and meltwater.
Figure 8 shows how plotting sulfate δ18O and δ34S in sulfur and oxygen isotopic space can provide a unique solution to elucidating the sulfate sources. The stoichiometric stage of pyrite oxidation results in sulfur isotopic fractionation between pyrite and sulfate (εSO4–pyrite) of −1.3‰ to −0.6‰ (Balci et al., 2007; Pisapia et al., 2007; Brunner et al., 2008). The mass-weighted mean sulfide δ34S of the nearby Firkanten and Carolinefjellet Formations, combined with the sulfur isotopic fractionation during the stoichiometric stage of sulfide oxidation, indicates that any pore water sample with a δ34S-npd value between −15.1‰ and −2.6‰ is likely to have derived all of its sulfur from the stoichiometric oxidation of pyrite. Figure 8 shows that half of the pore water samples from Adventdalen contain npd within this δ34S-npd range, indicating that the stoichiometric oxidation of pyrite is an important process contributing sulfate to active layer pore water in Adventdalen. This corroborates the evidence from the δ18O- results presented earlier.
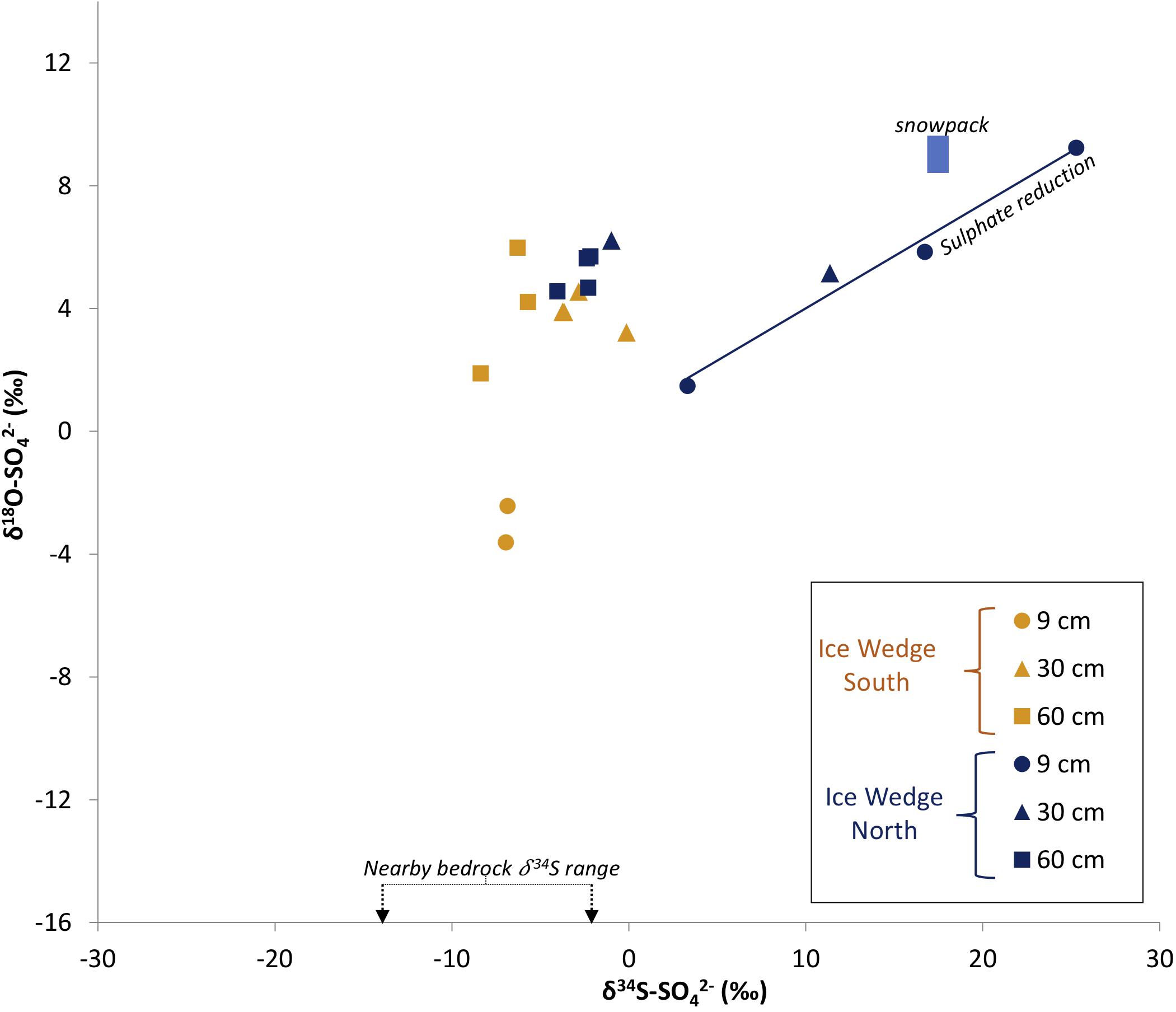
Figure 8. Water samples from the active layer of Ice Wedge South and Ice Wedge North in δ18O-SO4 and δ34S-SO4 isotope space. The blue box represents the isotopic range of pre-melt snowpacks in Svalbard at Midtre Lovenbreen (Wynn et al., 2006) and near Ny Ålesund (Tye and Heaton, 2007). The solid blue line is a regression line illustrating reduction of sulphate by SRB (R2 = 0.98). The nearby bedrock range is of the mass-weighted mean sulfide δ34S in the Firkanten and Carolinefjellet Formations.
Although all samples except for the most 34S-enriched sample have a δ34S range between that of the bedrock and snowpack δ34S values (Figure 8), the origin of sulfate in these samples in 34S and 18O cannot be explained purely as a mixing of sulfate derived from these two sources. There are two lines of evidence for this. Firstly, if mixing between (relatively 34S-enriched) precipitation-derived sulfate and (relatively 34S-depleted) weathering-derived sulfate were solely responsible for the δ34S values of pore water sulfate in the active layer, a negative linear correlation between the concentration and δ34S values of sulfate would result. There is no such negative linear correlation in the pore water samples from Adventdalen (R2 < 0.2). Secondly, a negative correlation between δ34S- and the sulfate-to-chloride molar ratio (/Cl–) would result from mixing between weathering-derived sulfate (high /Cl–, relatively 34S-depleted) and precipitation-derived sulfate (low /Cl–, relatively 34S-enriched). In fact, there is a positive correlation between δ34S- and /Cl– (R2 = 0.49). Neither line of evidence supports mixing between snowmelt and pyrite oxidation as the sole reason for the δ34S- values observed in the pore water samples from Adventdalen. Hence, these statistical tests indicate that sulfate removal from the pore water by sulfate-reducing bacteria (SRB) may be a factor in enriching the remaining sulfate in these samples. SRB preferentially reduce the lighter isotopes of sulfur and oxygen, leading to isotopic enrichment of the residual sulfate. It is common for the product H2S to be incorporated into iron sulfides or organic matter (Brown, 1985, 1986; Blodau et al., 2007). This causes both δ34S and δ18O values in the remaining sulfate to increase, producing a positive correlation between them (Mandernack et al., 2003). Since there is a positive correlation (R2 = 0.98) between δ34S and δ18O in four samples from Ice Wedge North, and δ34S in one sample exceeds the snowpack δ34S, it seems highly likely that SRB are active in the pore water of the active layer at Ice Wedge North.
Iron and Sulfur Mineral Precipitation
The δ34S and δ18O values of pore water sulfate in the active layer are indicative of sulfate reduction at Ice Wedge North. The sulfate concentration was relatively low at Ice Wedge North (< 4.7 mmol l–1; Figures 3F,G), supporting the isotopic evidence for sulfate reduction. To produce a distinctive δ18O-δ34S signature of sulfate reduction, the reduced sulfur must be sequestered in the solid phase (iron sulfide or carbon-bonded sulfur). The reactions of iron with hydrogen sulfide can be expressed with the following simplified equation scheme, where Equation 12 represents hydrogen sulfide reacting with iron oxides to form iron monosulfide (AVS) and sulfur, and Equation 13 represents the formation of the more stable pyrite (CRS) from the metastable iron monosulfide and sulfur (Raiswell and Canfield, 2012):
The AVS formed in Equation 12 consists mainly of the iron sulfide minerals mackinawite, greigite and amorphous FeS. These phases are usually only stable for short periods before their re-oxidation or further reduction to pyrite (Chesworth, 2008). Table 3 and Supplementary Figure S1I show that the AVS concentrations in the Ice Wedge North sediments were low, but detectable, in just over half the samples, reaching a maximum of 0.05 dry weight percent (equivalent to 6.02 μmoles g–1 dry sediment; 115 cm depth). The low concentrations of AVS indicate that AVS is not a significant long-term store of the products of iron and sulfate reduction. The CRS includes pyrite, which is the most thermodynamically stable iron sulfide (Berner, 1967). Concentrations of CRS are higher than AVS, reaching 0.27 dry weight percent (equivalent to 22.4 μmoles g–1 dry sediment; 115 cm depth) at Ice Wedge North (Table 3 and Supplementary Figure S1K).
The Ice Wedge North sediments contain abundant iron in Feascorbate, Fedithionite, Feoxalate, Feacetate and CRS. Ferrous iron in the porewaters is probably derived from the dissimilatory reduction of iron (oxyhydr)oxides (Equation 9), as well as the oxidation of allogenic and authigenic pyrite (Equation 8). In addition to reacting with hydrogen sulfide to form AVS or CRS, the dissolved ferrous iron reacts with bicarbonate ions to form Feacetate (iron bound in carbonates). Siderite (FeCO3) is an iron carbonate, and tends to occur in reducing, CO2-rich, hydromorphic environments, such as peatlands (Chesworth, 2008). Table 3 shows that Feacetate reaches >1.5 dry weight% in the sediments at Ice Wedge North, indicating that it is a more significant sink of ferrous iron than CRS. As the precipitation of Feacetate dominates over the precipitation of CRS, it is possible that an additional sink for the hydrogen sulfide is carbon-bonded sulfur (CBS), which has previously been shown to be an important sink for reduced sulfur in peat soils (Spratt and Morgan, 1990; Blodau et al., 2007). Although CBS was not measured in these cores, a strong positive correlation between organic carbon content and CRS (ρ = 0.9; p < 0.001) and also Feacetate (ρ = 0.67; p < 0.01) indicates that where the organic carbon content is high, sulfate reduction, CRS precipitation and Feacetate precipitation occur. Given the high concentration of sedimentary organic carbon, it seems likely that CBS exists and is forming at this location.
In contrast to Ice Wedge North, the δ34S and δ18O values of sulfate in pore water from the drier active layer of S1 indicate primarily OWP via O2, and some OWP via Fe3+. Evidence for a mostly oxidised active layer at S1 is in the mostly low Fe(aq) concentration in the pore water from the active layer and the low water table (summer 2017). CRS and AVS concentrations at this site are low, corroborating the isotopic evidence that sulfate reduction is negligible at this site. In addition, the concentration of iron bound in carbonate (Feacetate) is lower than at Ice Wedge North. Finally, Figures 3C,H show that aqueous iron and sulfate co-vary at this site, which is indicative of pyrite oxidation (e.g., Hodson et al., 2016).
The quantity of sedimentary organic carbon exerts a strong control on biogeochemical processes and mineral precipitation across both sites. Firstly, the organic carbon content is strongly positively correlated with CRS (ρ = 0.90; p < 0.001), Feacetate (ρ = 0.67; p < 0.01), and Feascorbate (ρ = 0.75; p < 0.001). In addition, the sedimentary organic carbon content is negatively correlated with pore water sulfate (ρ = −0.68; p < 0.001) and aqueous iron (ρ = −0.61; p < 0.001). This further supports the mechanisms discussed above, whereby in organic carbon-poor sediment, the oxidation of pyrite produces aqueous iron and sulfate that are not reduced to form authigenic CRS (primarily pyrite) and Feacetate (primarily siderite). In contrast, in organic carbon-rich sediment, the dissolved iron and sulfate are reduced, forming Feacetate and CRS. Finally, sedimentary organic carbon content appears to influence the formation of Fedithionite (primarily crystalline iron (oxyhydr)oxides) and Feoxalate (primarily magnetite). Organic carbon content was negatively correlated with Fedithionite (ρ = −0.85; p < 0.001) and Feoxalate (ρ = −0.89; p < 0.001). A plausible explanation is a combination of cycling redox conditions and dissolved oxygen levels at Ice Wedge South, which increase the crystallinity of ferrihydrite, coupled with an inhibition of the change from Feascorbate (primarily poorly crystalline ferrihydrite) to Fedithionite (primarily crystalline goethite) by organic compounds at Ice Wedge North (e.g., Schwertmann and Murad, 1988; Thompson et al., 2006; Amstaetter et al., 2012; Herndon et al., 2017). These significant relationships demonstrate that the quantity of sedimentary organic carbon exerts a landscape-scale control on the active layer and permafrost biogeochemistry.
Variations in Pore Water Geochemistry Due to Physical Processes
The distribution of chemical species described above is partly governed by a set of complex physical processes, including hydrological inputs to the active layer (precipitation, advection and ground ice melt; Throckmorton et al., 2016), hydrological outputs from the active layer (evaporation, freezing and advection) and the diffusion of ions from regions of high concentration to regions of low concentration. Each of these physical processes can influence the distribution of chemical constituents in the active layer and their signatures commonly overlap or mask one another. For instance, both evaporation of water and ion freeze-out from the active layer concentrate the pore water chemistry and enrich the δ18O and δD of the remaining water (Throckmorton et al., 2016). However, these physical processes are secondary to the ion ratio interpretations discussed earlier.
The sediments in this study were dominantly fine-grained, with the median grain size in the cores at Ice Wedge South ranging from 34 to 60 μm. In fine-grained sediments, migration of unfrozen water (and solutes) occurs along temperature-induced pressure gradients toward colder ground (Kokelj and Burn, 2003, 2005), and forms segregated ice lenses behind the freezing front. During active layer freezeback, the migration is upward toward the freezing front descending from the surface, and downward toward the permafrost table during upward freezing from the permafrost table (Cheng, 1983). This results in the formation of ice lenses and the concentration of solutes close to the ground surface, in the transient layer and at the top of permafrost, whereas the middle of the active layer becomes desiccated (Mackay, 1983). During thawing of the active layer in summer, unfrozen water and solute migration occurs downward into frozen ground below the advancing thaw front (Cheng, 1983). As the thaw front reaches its maximum depth, moisture and solutes can migrate into the top of permafrost. The downward migration into the top of permafrost in summer is greater than the upward migration out in winter when the temperature and pressure gradients are reversed, because the unfrozen water content and hydraulic conductivity are greatly reduced at lower temperature (Cheng, 1983). Consequently, there is a net annual downward migration that enriches the top of permafrost with segregated ice and solutes following repeated freeze-thaw cycles (Cheng, 1983; Kokelj and Burn, 2003, 2005).
There are multiple lines of evidence that indicate that these mechanisms contribute to redistributing solutes at the study sites. Segregated ice lenses were observed at the base of the active layer in cores from Ice Wedge South, and the top of permafrost was enriched with segregated ice at Ice Wedge North, indicated by the elevated water contents (Figures 2A,B). Assuming that chloride behaves conservatively, without participating in dissolution or precipitation reactions, its concentration profile in each core can be used to establish the net effect of moisture distribution by repeated freeze-thaw in the sediments (Jessen et al., 2014). The peak in the concentration of chloride close to the base of the active layer in cores from both sites coincides with the presence of ice lenses and is likely to have been caused by unfrozen water and solute migration during upward freezing of the active layer (Figures 2F,G,H,J). The peak in chloride concentration, coincident with an increase in water content near the ground surface at N1, may be indicative of upward moisture migration during active layer freezing.
The upper permafrost at Ice Wedge South formed syngenetically as the permafrost table rose in conjunction with sediment deposition (Gilbert et al., 2018), which means that the present-day permafrost is comprised of material formerly in the active layer. The accumulation of chloride in the permafrost is thus probably due to a combination of moisture migration into the top of permafrost at the end of summer (Cheng, 1983) and incorporation into permafrost of solutes from the base of the active layer during permafrost aggradation (Figures 2H–J). Cores S2a and S2b have significantly more concentrated chloride in the permafrost compared with the active layer (Table 7), and the chloride concentration in the active layer is greater than in the other cores. The formation of efflorescent salts on the ground surface at Ice Wedge South (Mora et al., 2015) further highlights the role of solute migration along potential gradients at the site.
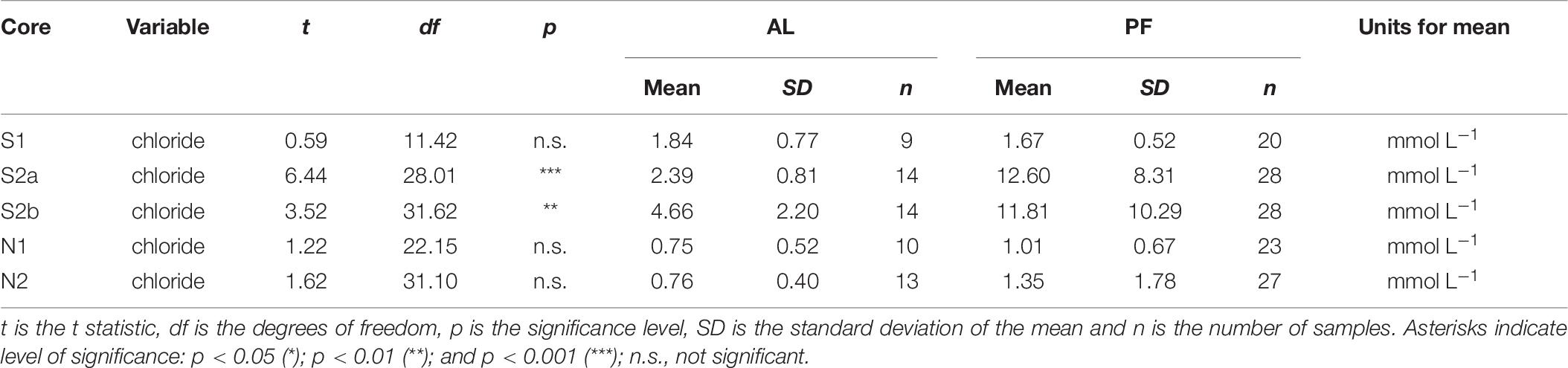
Table 7. Results from Welch’s t-test, which was used to test whether the concentration of chloride was significantly different for permafrost (PF) versus active layer (AL) in each core.
The higher concentration of chloride in the cores from Ice Wedge South, compared to those from Ice Wedge North, could be the result of diffusion of chloride from the underlying deltaic sediments. Nearby cores display an increase in chloride concentration from a mean of ∼1 mmol L–1 in the loess sediments to a mean of ∼68 mmol L–1 in the underlying deltaic sediments (Cable et al., 2017). Diffusion upward from these marine sediments may enhance the pore water chloride concentration at shallower depths in some locations, although the reasons for the location-specific diffusion are not clear. Additionally, it is likely that the topography of the sites has changed over time with the development of depositional landforms and ice-wedge polygons (Gilbert et al., 2018; O’Neill and Christiansen, 2018). Changes in topography are likely to have influenced patterns of water movement through the active layer, while variations in active layer thickness changed the amount of ground subject to advection over time. The data do not enable enhanced chloride concentration due to upward diffusion to be distinguished from enhanced chloride concentration due to low advection rates. However, greater rates of advection most likely occur at Ice Wedge North due to the spring-fed hydrologic regime and high water content.
In summary, unfrozen water and solute migration along potential gradients contribute to water content and solute variations with depth observed in the cores. The complex depositional and periglacial history at the sites makes further interpretation of chloride concentration patterns difficult. Ice Wedge South cores display evidence of chloride diffusion from underlying sediments and/or variations in rates of advection as the aeolian terrace aggraded. Despite the complexity introduced by these physical processes, the use of covariance between ions has enabled the dominant biogeochemical processes in Adventdalen to be elucidated.
Conclusion
This study highlights the importance of landscape evolution and demonstrates that permafrost aggradation and organic carbon accumulation have caused significant changes in the biogeochemical processes and mineral precipitation reactions within this high Arctic floodplain. The contrasting hydrological regimes of the study sites result in marked differences in the accumulation and decomposition of permafrost organic carbon. The drier areas of the floodplain store little organic carbon and are representative of the earlier stages of floodplain development. These areas are dominated by the biogeochemical signatures of pyrite oxidation and the weathering of fresh mineral surfaces, similar to glacial meltwaters in nearby catchments. In contrast, the water-saturated areas of the floodplain represent the later stages of floodplain development, where the accumulation of organic carbon causes a switch to iron- and sulfate-reduction, with net iron and carbon storage via increased CRS (FeS2) and Feacetate (siderite) precipitation. In addition, contrasting chloride concentrations demonstrate the geochemical contrast between sediments of marine and aeolian origin. As air temperatures continue to rise in the high Arctic and as thaw progresses deeper into the permafrost, there are likely to be major changes in the iron, sulfur and carbon cycling in this valley, depending on how permafrost thaw impacts the geomorphology and hydrology of the ice-wedge polygonal terrain.
Data Availability Statement
Jones (2019). Permafrost and active layer biogeochemical data from Adventdalen (2015–2017) (Version 1.0) in the UK Polar Data Centre, Natural Environment Research Council, UK Research & Innovation (https://doi.org/10.5285/4c90d954-3db2-4084-9fe9-e050c839a6fe).
Author Contributions
EJ wrote the manuscript. EJ, AH, and HO’N collected the cores of permafrost and water from the active layer. EJ and JR processed and analysed the cores of permafrost. TD analysed the rock cores. ST and PW facilitated the laboratory analyses of permafrost cores and active layer waters. All authors interpreted data and contributed feedback on this manuscript.
Funding
The authors acknowledge the Joint Programming Initiative (JPI-Climate Topic 2: Russian Arctic and Boreal Systems) Award No. 71126, a NERC studentship NE/L002450/1 to EJ, a Research Council of Norway grant (NRC no. 294764), and NRCan contribution number 20200053. Open Access Support was received from the University of Sheffield.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the two reviewers for their constructive reviews and Sharon Smith for a helpful internal review. Gwilym Jones, Gunnar Mallon, Graham Gilbert, Ebbe Bak, Dotan Rotem, Yishai Weinstein, Sarah St Germain, Simon Norum, and Knut Lindland Tveit are thanked for their field assistance, and Alan Smalley, Rob Ashurst, Joe Hufton, Andy Fairburn, Dave Hughes, Roscoe Blevins, Anthony Turner, Gerd-Irene Signeres, Andy Hobson, and Alina Marca are thanked for their assistance with the laboratory analyses. The content of this manuscript appears online in EJ’s Ph.D. thesis (Jones, 2019).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/feart.2020.00342/full#supplementary-material
References
Amstaetter, K., Borch, T., and Kappler, A. (2012). Influence of humic acid imposed changes of ferrihydrite aggregation on microbial Fe(III) reduction. Geochim. Cosmochim. Acta 85, 326–341. doi: 10.1016/J.GCA.2012.02.003
Balci, N., Shanks, W. C., Mayer, B., and Mandernack, K. W. (2007). Oxygen and sulfur isotope systematics of sulfate produced by bacterial and abiotic oxidation of pyrite. Geochim. Cosmochim. Acta 71, 3796–3811. doi: 10.1016/j.gca.2007.04.017
Berner, R. A. (1967). Thermodynamic stability of sedimentary iron sulfides. Am. J. Sci. 265, 773–785. doi: 10.2475/ajs.265.9.773
Blodau, C., Mayer, B., Peiffer, S., and Moore, T. R. (2007). Support for an anaerobic sulfur cycle in two Canadian peatland soils. J. Geophys. Res. Biogeosci. 112:G02004. doi: 10.1029/2006JG000364
Blum, J. D., Gazis, C. A., Jacobson, A. D., and Chamberlain, C. P. (1998). Carbonate versus silicate weathering in the Raikhot watershed within the High Himalayan crystalline series. Geology 26:411.
Bottrell, S. H., Hatfield, D., Bartlett, R., Spence, M. J., Bartle, K. D., and Mortimer, R. J. G. (2010). Concentrations, sulfur isotopic compositions and origin of organosulfur compounds in pore waters of a highly polluted raised peatland. Organ. Geochem. 41, 55–62. doi: 10.1016/j.orggeochem.2009.07.005
Bottrell, S. H., and Tranter, M. (2002). Sulphide oxidation under partially anoxic conditions at the bed of the Haut Glacier d’Arolla, Switzerland. Hydrol. Process. 16, 2363–2368. doi: 10.1002/hyp.1012
Brown, J., Ferrians, O. J. Jr., Heginbottom, J. A., and Melnikov, E. S. (1997). Circum-Arctic Map of Permafrost and Ground-Ice Conditions. U.S. Geological Survey Map CP-45. Washington, DC: U.S. Department of the Interior.
Brown, K. A. (1985). Sulfur distribution and metabolism in waterlogged peat. Soil Biol. Biogeochem. 17, 39–45. doi: 10.1016/0038-0717(85)90088-4
Brown, K. A. (1986). Formation of organic sulfur in anaerobic peats. Soil Biol. Biogeochem. 18, 131–140. doi: 10.1016/0038-0717(86)90017-9
Brunner, B., Yu, J.-Y., Mielke, R. E., MacAskill, J. A., Madzunkov, S., McGenity, T. J., et al. (2008). Different isotope and chemical patterns of pyrite oxidation related to lag and exponential growth phases of Acidithiobacillus ferrooxidans reveal a microbial growth strategy. Earth Planet. Sci. Lett. 270, 63–72. doi: 10.1016/j.epsl.2008.03.019
Cable, S., Elberling, B., and Kroon, A. (2017). Holocene permafrost history and cryostratigraphy in the High-Arctic Adventdalen Valley, central Svalbard. Boreas 47, 423–442. doi: 10.1111/bor.12286
Canfield, D. E., Raiswell, R., Westrich, J. T., Reaves, C. M., and Berner, R. A. (1986). The use of chromium reduction in the analysis of reduced inorganic sulfur in sediments and shales. Chem. Geol. 54, 149–155. doi: 10.1016/0009-2541(86)90078-1
Cheng, G. (1983). The mechanism of repeated-segregation for the formation of thick layered ground ice. Cold Reg. Sci. Technol. 8, 57–66. doi: 10.1016/0165-232X(83)90017-4
Christiansen, H. H. (2005). Thermal regime of ice-wedge cracking in Adventdalen, Svalbard. Permafrost Periglac. Process. 16, 87–98. doi: 10.1002/ppp.523
Christiansen, H. H., Etzelmüller, B., Isaksen, K., Juliussen, H., Farbrot, H., Humlum, O., et al. (2010). The thermal state of permafrost in the nordic area during the international polar year 2007-2009. Permafrost Periglac. Process. 21, 156–181. doi: 10.1002/ppp.687
Dallmann, W. K., Midbø, P. S., Nøttvedt, A., and Steel, R. J. (1999). Lithostratigraphic Lexicon Of Svalbard: Review And Recommendations For Nomenclature Use: Upper Palaeozoic to Quaternary Bedrock. Tromsø, NO: Norsk Polarinstitutt.
Dise, N. B., and Verry, E. S. (2001). Suppression of peatland methane emission by cumulative sulfate deposition in simulated acid rain. Biogeochemistry 53, 143–160.
du Prel, J.-B., Röhrig, B., Hommel, G., and Blettner, M. (2010). Choosing statistical tests: part 12 of a series on evaluation of scientific publications. Deutsch. Arzteblatt Intern. 107, 343–348. doi: 10.3238/arztebl.2010.0343
Elberling, B., Michelsen, A., Schädel, C., Schuur, E. A. G., Christiansen, H. H., Berg, L., et al. (2013). Long-term CO2 production following permafrost thaw. Nat. Clim. Chang. 3, 890–894. doi: 10.1038/nclimate1955
Ernakovich, J. G., Lynch, L. M., Brewer, P. E., Calderon, F. J., and Wallenstein, M. D. (2017). Redox and temperature-sensitive changes in microbial communities and soil chemistry dictate greenhouse gas loss from thawed permafrost. Biogeochemistry 134, 183–200. doi: 10.1007/s10533-017-0354-5
Fairchild, I. J., Bradby, L., Sharp, M., and Tison, J. –L. (1994). Hydrochemistry of carbonate terrains in alpine glacial settings. Earth Surf. Process. Landf. 9, 33–54. doi: 10.1002/esp.3290190104
Førland, E. J., Benestad, R., Hanssen-Bauer, I., Haugen, J. E., and Skaugen, T. E. (2011). Temperature and precipitation development at Svalbard 1900-2100. Adv. Meteorol. 2011, 1–14. doi: 10.1155/2011/893790
Fossing, H., and Barker Jørgensen, B. (1989). Measurement of bacterial sulfate reduction in sediments: evaluation of a single-step chromium reduction method. Biogeochemistry 8, 205–222. doi: 10.1007/BF00002889
Gilbert, G. L., O’Neill, H. B., Nemec, W., Thiel, C., Christiansen, H. H., and Buylaert, J.-P. (2018). Late Quaternary sedimentation and permafrost development in a Svalbard fjord-valley, Norwegian High Arctic. Sedimentology 65, 2531–2558. doi: 10.1111/sed.12476
Haldorsen, S., Heim, M., Dale, B., Landvik, J. Y., van der Ploeg, M., Leijnse, A., et al. (2010). Sensitivity to long-term climate change of subpermafrost groundwater systems in Svalbard. Q. Res. 73, 393–402. doi: 10.1016/j.yqres.2009.11.002
Hepburn, L. E., Butler, I. B., Boyce, A., and Schröder, C. (2020). The use of operationally-defined sequential Fe extraction methods for mineralogical applications: a cautionary tale from Mössbauer spectroscopy. Chem. Geol. 543:e0119584. doi: 10.1016/j.chemgeo.2020.119584
Herndon, E., AlBashaireh, A., Singer, D., Roy Chowdhury, T., Gu, B., and Graham, D. (2017). Influence of iron redox cycling on organo-mineral associations in Arctic tundra soil. Geochim. Cosmochim. Acta 207, 210–231. doi: 10.1016/j.gca.2017.02.034
Hindshaw, R. S., Heaton, T. H. E., Boyd, E. S., Lindsay, M. R., and Tipper, E. T. (2016). Influence of glaciation on mechanisms of mineral weathering in two high Arctic catchments. Chem. Geol. 420, 37–50. doi: 10.1016/j.chemgeo.2015.11.004
Hodgkins, R., Tranter, M., and Dowdeswell, J. A. (1997). Solute provenance, transport and denudation in a high Arctic glacierized catchment. Hydrol. Process. 11, 1813–1832.
Hodson, A., Nowak, A., and Christiansen, H. (2016). Glacial and periglacial floodplain sediments regulate hydrologic transfer of reactive iron to a high arctic fjord. Hydrol. Process. 30, 1219–1229. doi: 10.1002/hyp.10701
Horton, T. W., Chamberlain, C. P., Fantle, M., and Blum, J. D. (1999). Chemical weathering and lithologic controls of water chemistry in a high-elevation river system: Clark’s Fork of the Yellowstone river, Wyoming and Montana. Water Resour. Res. 35, 1643–1655. doi: 10.1029/1998WR900103
Hugelius, G., Strauss, J., Zubrzycki, S., Harden, J. W., Schuur, E. A. G., Ping, C.-L., et al. (2014). Estimated stocks of circumpolar permafrost carbon with quantified uncertainty ranges and identified data gaps. Biogeosciences 11, 6573–6593. doi: 10.5194/bg-11-6573-2014
Humlum, O. (2005). Holocene permafrost aggradation in Svalbard. Geol. Soc. Lond. Spec. Public. 242, 119–129. doi: 10.1144/GSL.SP.2005.242.01.11
Humlum, O., Instanes, A., and Sollid, J. L. (2003). Permafrost in Svalbard: a review of research history, climatic background and engineering challenges. Polar Res. 22, 191–215. doi: 10.1111/j.1751-8369.2003.tb00107.x
Isaksen, K., Christiansen, H. H., and Westermann, S. (2019). Climate in Svalbard 2100-a knowledge base for climate adaptation, Report Chapter 7.1. Norwegian Cent. Clim. Serv. Rep. Num. 2019, 113–118.
Jessen, S., Holmslykke, H. D., Rasmussen, K., Richardt, N., and Holm, P. E. (2014). Hydrology and pore water chemistry in a permafrost weland, Ilulissat, Greenland. Water Resourc. Res. 50, 1–15. doi: 10.1002/2013WR014376
Johnston, S. G., Burton, E. D., Aaso, T., and Tuckerman, G. (2014). Sulfur, iron and carbon cycling following hydrological restoration of acidic freshwater wetlands. Chem. Geol. 371, 9–26. doi: 10.1016/j.chemgeo.2014.02.001
Jones, E. L. (2019). The Biogeochemistry of the Active Layer and Shallow Permafrost in a High Arctic Fjord Valley, Svalbard. Ph. D. thesis, University of Sheffield, Sheffield.
Kirschke, S., Bousquet, P., Ciais, P., Saunois, M., Canadell, J. G., Dlugokencky, E. J., et al. (2013). Three decades of global methane sources and sinks. Nat. Geosci. 6, 813–823. doi: 10.1038/ngeo1955
Kokelj, S. V., and Burn, C. R. (2003). Ground ice and soluble cations in near-surface permafrost, Inuvik, Northwest Territories, Canada. Permafrost Periglac. Process. 14, 275–289. doi: 10.1002/ppp.458
Kokelj, S. V., and Burn, C. R. (2005). Geochemistry of the active layer and near-surface permafrost, Mackenzie delta region, Northwest Territories, Canada. Can. J. Earth Sci. 42, 37–48. doi: 10.1139/e04-089
Kolka, R., Bridgham, S., and Ping, C.-L. (2015). “Soils of peatlands: histosols and gelisols,” in Wetlands Soils: Genesis, Hydrology, Landscapes And Classification, eds M. J. Vepraskas and C. L. Craft (Boca Raton, FL: Lewis Publishing), 277–310. doi: 10.1201/b18996-13
Kottek, M., Grieser, J., Beck, C., Rudolf, B., and Rubel, F. (2006). World map of the Köppen-geiger climate classification updated. Meteorol. Zeitschrift 15, 259–263. doi: 10.1127/0941-2948/2006/0130
Koven, C. D., Ringeval, B., Friedlingstein, P., Ciais, P., Cadule, P., Khvorostyanov, D., et al. (2011). Permafrost carbon-climate feedbacks accelerate global warming. Proc. Natl. Acad. Sci. U.S.A. 108, 14769–14774. doi: 10.1073/pnas.1103910108
Kristjansson, J. K., and Schönheit, P. (1983). Why do sulfate-reducing bacteria outcompete methanogenic bacteria for substrates? Oecologia 60, 264–266. doi: 10.1007/BF00379530
Kuhry, P., Dorrepaal, E., Hugelius, G., Schuur, E. A. G., and Tarnocai, C. (2010). Potential remobilization of belowground permafrost carbon under future global warming. Permafrost Periglac. Process. 21, 208–214. doi: 10.1002/ppp.684
Landvik, J. Y., Ingólfsson, Ó, Mienert, J., Lehman, S. J., Solheim, A., Elverhøi, A., et al. (2005). Rethinking Late Weichselian ice-sheet dynamics in coastal NW Svalbard. Boreas 34, 7–24. doi: 10.1080/03009480510012809
Lee, H., Schuur, E. A. G., Inglett, K. S., Lavoie, M., and Chanton, J. P. (2012). The rate of permafrost carbon release under aerobic and anaerobic conditions and its potential effects on climate. Glob. Chang. Biol. 18, 515–527. doi: 10.1111/j.1365-2486.2011.02519.x
Liljedahl, A., Hinzman, L. D., and Schulla, J. (2012). “Ice-wedge polygon type controls low-gradient watershed-scale hydrology,” in Proceedings of the Tenth International Conference on Permafrost, Zurich.
Lipson, D. A., Zona, D., Raab, T. K., Bozzolo, F., Mauritz, M., and Oechel, W. C. (2012). Water-table height and microtopography control biogeochemical cycling in an Arctic coastal tundra ecosystem. Biogeosciences 9, 577–591. doi: 10.5194/bg-9-577-2012
Lloyd, R. M. (1968). Oxygen isotope behaviour in the Sulfate-Water System. J. Geophys. Res. 73, 6099–6110. doi: 10.1029/jb073i018p06099
Lønne, I., and Nemec, W. (2004). High-arctic fan delta recording deglaciation and environment disequilibrium. Sedimentology 51, 553–589. doi: 10.1111/j.1365-3091.2004.00636.x
Mackay, J. R. (1983). Downward water movement into frozen ground, western arctic coast, Canada. Can. J. Earth Sci. 20, 120–134. doi: 10.1139/e83-012
Mandernack, K. W., Krouse, H. R., and Skei, J. M. (2003). A stable sulfur and oxygen isotopic investigation of sulfur cycling in an anoxic marine basin, Framvaren Fjord, Norway. Chem. Geol. 195, 181–200. doi: 10.1016/S0009-2541(02)00394-7
Miller, K. E., Lai, C. T., Friedman, E. S., Angenent, L. T., and Lipson, D. A. (2015). Methane suppression by iron and humic acids in soils of the Arctic Coastal Plain. Soil Biol. Biochem. 83, 176–183. doi: 10.1016/j.soilbio.2015.01.022
Milne, G., and Shennan, I. (2007). “Sea Level Studies | Isostasy,” in Encyclopedia of Quaternary Science, eds S. A. Elias and C. J. Mock (Amsterdam: Elsevier), 3043–3051. doi: 10.1016/b0-44-452747-8/00142-3
Mora, C., Vieira, G., Pina, P., Lousada, M., and Christiansen, H. H. (2015). Land cover classification using high-resolution aerial photography in Adventdalen, Svalbard. Swedish Soc. Anthropol. Geogr. 97, 473–488. doi: 10.1111/geoa.12088
Olefeldt, D., Turetsky, M. R., Crill, P. M., and Mcguire, A. D. (2013). Environmental and physical controls on northern terrestrial methane emissions across permafrost zones. Glob. Chang. Biol. 19, 589–603. doi: 10.1111/gcb.12071
O’Neill, H. B., and Christiansen, H. H. (2018). Detection of ice wedge cracking in permafrost using miniature accelerometers. J. Geophys. Res. Earth Surf. 123, 642–657. doi: 10.1002/2017JF004343
Oonk, P. B. H., Tsikos, H., Mason, P. R. D., Henkel, S., Staubwasser, M., Fryer, L., et al. (2017). Fraction-specific controls on the trace element distribution in iron formations: implications for trace metal isotope proxies. Chem. Geol. 474, 17–32. doi: 10.1016/j.chemgeo.2017.10.018
Parmentier, F.-J. W., Christensen, T. R., Sørensen, L. L., Rysgaard, S., McGuire, A. D., Miller, P. A., et al. (2013). The impact of lower sea-ice extent on Arctic greenhouse-gas exchange. Nat. Clim. Chang. 3, 195–202. doi: 10.1038/nclimate1784
Pester, M., Knorr, K.-H., Friedrich, M. W., Wagner, M., and Loy, A. (2012). Sulfate-reducing microorganisms in wetlands – fameless actors in carbon cycling and climate change. Front. Microbiol. 3:72. doi: 10.3389/fmicb.2012.00072
Pisapia, C., Chaussidon, M., Mustin, C., and Humbert, B. (2007). O and S isotopic composition of dissolved and attached oxidation products of pyrite by Acidithiobacillus ferrooxidans: comparison with abiotic oxidations. Geochim. Cosmochim. Acta 71, 2474–2490. doi: 10.1016/J.GCA.2007.02.021
Poulton, S. W., and Canfield, D. E. (2005). Development of a sequential extraction procedure for iron: implications for iron partitioning in continentally derived particulates. Chem. Geol. 214, 209–221. doi: 10.1016/J.CHEMGEO.2004.09.003
R Core Team (2017). A Language And Environment For Statistical Computing. Vienna: R Foundation for Statistical Computing.
Raiswell, R., Benning, L. G., Tranter, M., and Tulaczyk, S. (2008). Bioavailable iron in the Southern Ocean: the significance of the iceberg conveyor belt. Geochem. Transact. 9, 1–9. doi: 10.1186/1467-4866-9-7
Raiswell, R., and Canfield, D. E. (2012). The iron biogeochemical cycle past and present. Geochem. Perspect. 1:220. doi: 10.7185/geochempersp.1.1
Raiswell, R., Hawkings, J., Elsenousy, A., Death, R., Tranter, M., and Wadham, J. (2018). Iron in glacial systems: speciation, reactivity, freezing behaviour, and alteration during transport. Front. Earth Sci. 6:222. doi: 10.3389/feart.2018.00222
Riber, L. (2009). Paleogene Depositional Conditions And Climatic Changes Of The Frysjaodden Formation In Central Spitsbergen (Sedimentology And Mineralogy). MSc thesis, University of Oslo, Oslo.
Romanovsky, V. E., Drozdov, D. S., Oberman, N. G., Malkova, G. V., Kholodov, A. L., Marchenko, S. S., et al. (2010). Thermal state of permafrost in Russia. Permafrost Periglac. Process. 21, 136–155. doi: 10.1002/ppp.689
Rutter, N., Hodson, A., Irvine-Fynn, T., and Solås, M. K. (2011). Hydrology and hydrochemistry of a deglaciating high-Arctic catchment, Svalbard. J. Hydrol. 410, 39–50. doi: 10.1016/J.JHYDROL.2011.09.001
Schaefer, K., Zhang, T., Bruhwiler, L., and Barrett, A. P. (2011). Amount and timing of permafrost carbon release in response to climate warming. Tellus Ser. B Chem. Phys. Meteorol. 63, 165–180. doi: 10.1111/j.1600-0889.2011.00527.x
Schuur, E. A. G., McGuire, A. D., Schädel, C., Grosse, G., Harden, J. W., Hayes, D. J., et al. (2015). Climate change and the permafrost carbon feedback. Nature 520, 171–179. doi: 10.1038/nature14338
Schwertmann, U., and Murad, E. (1988). The nature of an iron oxide—organic iron association in a peaty environment. Clay Miner. 23, 291–299. doi: 10.1180/claymin.1988.023.3.06
Sjögersten, S., Van Der Wal, R., and Woodin, S. J. (2006). Small-scale hydrological variation determines landscape CO2 fluxes in the high Arctic. Biogeochemistry 80, 205–216. doi: 10.1007/s10533-006-9018-6
Slotznick, S. P., Sperling, E. A., Tosca, N. J., Miller, A. J., Clayton, K. E., van Helmond, N. A. G. M., et al. (2020). Unraveling the mineralogical complexity of sediment iron speciation using sequential extractions. Geochem. Geophys. Geosyst. 21:8666. doi: 10.1029/2019GC008666
Sørbel, L., and Tolgensbakk, J. (2002). Ice-wedge polygons and solifluction in the Adventdalen area, Spitsbergen, Svalbard. Norsk Geografisk Tidsskrift Norwegian J. Geogr. 56, 62–66. doi: 10.1080/002919502760056369
Spence, M. J., Thornton, S. F., Bottrell, S. H., and Spence, K. H. (2005). Determination of interstitial water chemistry and porosity in consolidated aquifer materials by diffusion equilibrium-exchange. Environ. Sci. Technol. 39, 1158–1166. doi: 10.1021/es049401v
Spratt, H. G., and Morgan, M. D. (1990). Sulfur cycling in a cedar-dominated, freshwater wetland. Limnol. Oceanogr. 35, 1586–1593. doi: 10.4319/lo.1990.35.7.1586
Svinth, A. A. G. (2013). A Sedimentological and Petrographical Investigation of the Todalen Member and the Boundary Beds of the Endalen Member.: Within the Firkanten Formation (Paleocene) in the Central Basin of Spitsbergen, Svalbard. Masters thesis, Norwegian University, Trondheim, NO.
Tarnocai, C., Canadell, J. G., Schuur, E. A. G., Kuhry, P., Mazhitova, G., and Zimov, S. (2009). Soil organic carbon pools in the northern circumpolar permafrost region. Glob. Biogeochem. Cycles 23:3327.
Thompson, A., Chadwick, O. A., Rancourt, D. G., and Chorover, J. (2006). Iron-oxide crystallinity increases during soil redox oscillations. Geochim. Cosmochim. Acta 70, 1710–1727. doi: 10.1016/J.GCA.2005.12.005
Throckmorton, H. M., Newman, B. D., Heikoop, J. M., Perkins, G. B., Feng, X., Graham, D. E., et al. (2016). Active layer hydrology in an arctic tundra ecosystem: quantifying water sources and cycling using water stable isotopes. Hydrol. Process. 30, 4972–4986. doi: 10.1002/hyp.10883
Torres, M. A., Moosdorf, N., Hartmann, J., Adkins, J. F., and West, J. (2017). Glacial weathering, sulfide oxidation, and global carbon cycle feedbacks. Proc. Natl. Acad. Sci. U.S.A. 33, 8716–8721. doi: 10.1073/pnas.1702953114
Tranter, M., Sharp, M. J., Lamb, H. R., Brown, G. H., Hubbard, B. P., and Willis, I. C. (2002). Geochemical weathering at the bed of Haut Glacier d’Arolla, Switzerland—a new model. Hydrol. Process. 16, 959–993. doi: 10.1002/hyp.309
Turchyn, A. V., Tipper, E. T., Galy, A., Lo, J. K., and Bickle, M. J. (2013). Isotope evidence for secondary sulfide precipitation along the Marsyandi River, Nepal, Himalayas. Earth Planet. Sci. Lett. 374, 36–46. doi: 10.1016/j.epsl.2013.04.033
Turetsky, M. R., Treat, C. C., Waldrop, M. P., Waddington, J. M., Harden, J. W., and McGuire, A. D. (2008). Short-term response of methane fluxes and methanogen activity to water table and soil warming manipulations in an Alaskan peatland. J. Geophys. Res. 113:G00A10. doi: 10.1029/2007JG000496
Tye, A. M., and Heaton, T. H. E. (2007). Chemical and isotopic characteristics of weathering and nitrogen release in non-glacial drainage waters on Arctic tundra. Geochim. Cosmochim. Acta 71, 4188–4205. doi: 10.1016/j.gca.2007.06.040
Wadham, J. L., Bottrell, S., Tranter, M., and Raiswell, R. (2004). Stable isotope evidence for microbial sulphate reduction at the bed of a polythermal high Arctic glacier. Earth Planet. Sci. Lett. 219, 341–355. doi: 10.1016/S0012-821X(03)00683-6
Wadham, J. L., Tranter, M., Skidmore, M., Hodson, A. J., Priscu, J., Lyons, W. B., et al. (2010). Biogeochemical weathering under ice: size matters. Glob. Biogeochem. Cycles 24:3688. doi: 10.1029/2009GB003688
Wehrmann, L. M., Formolo, M. J., Owens, J. D., Raiswell, R., Ferdelman, T. G., Riedinger, N., et al. (2014). Iron and manganese speciation and cycling in glacially influenced high-latitude fjord sediments (West Spitsbergen, Svalbard): evidence for a benthic recycling-transport mechanism. Geochim. Cosmochim. Acta 141, 628–655. doi: 10.1016/j.gca.2014.06.007
Wynn, P. M., Hodson, A., and Heaton, T. (2006). Chemical and isotopic switching within the subglacial environment of a High Arctic glacier. Biogeochemistry 78, 173–193. doi: 10.1007/s10533-005-3832-0
Wynn, P. M., Morrell, D. J., Tuffen, H., Barker, P., Tweed, F. S., and Burns, R. (2015). Seasonal release of anoxic geothermal meltwater from the Katla volcanic system at Sólheimajökull, Iceland. Chem. Geol. 396, 228–238. doi: 10.1016/J.CHEMGEO.2014.12.026
Keywords: permafrost, biogeochemistry, iron-sulfur, carbon, Svalbard
Citation: Jones EL, Hodson AJ, Thornton SF, Redeker KR, Rogers J, Wynn PM, Dixon TJ, Bottrell SH and O’Neill HB (2020) Biogeochemical Processes in the Active Layer and Permafrost of a High Arctic Fjord Valley. Front. Earth Sci. 8:342. doi: 10.3389/feart.2020.00342
Received: 06 April 2020; Accepted: 21 July 2020;
Published: 02 September 2020.
Edited by:
Melissa Jean Murphy, University College London, United KingdomReviewed by:
Christian Schröder, University of Stirling, United KingdomZiming Yang, Oakland University, United States
Copyright © 2020 Jones, Hodson, Thornton, Redeker, Rogers, Wynn, Dixon, Bottrell and O’Neill. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Steven F. Thornton, cy5mLnRob3JudG9uQHNoZWZmaWVsZC5hYy51aw==