 Muammar Mansor
Muammar Mansor Christopher Winkler2
Christopher Winkler2 Jie Xu
Jie Xu- 1NanoGeoBio Laboratory, Department of Geological Sciences, The University of Texas at El Paso, El Paso, TX, United States
- 2Virginia Tech National Center for Earth and Environmental Nanotechnology Infrastructure, Blacksburg, VA, United States
- 3Energy and Environment Directorate, Pacific Northwest National Laboratory, Richland, WA, United States
The precipitation of nickel with sulfide is an important process governing the bioavailability of Ni in natural waters, and this process has the potential to effectively remove aqueous Ni contaminants in near-surface environments. In this study, we use experimental approaches to investigate the diversity of Ni-hosting phases precipitated in sulfidic environments across a range of aqueous Ni-to-Fe ratios ([Ni]aq/[Fe]aq) and in the presence or absence of the sulfate-reducing bacteria (SRB), Desulfovibrio vulgaris. In the absence of Fe(II), the initial precipitates in abiotic experiments are found to consist primarily of polyphasic Ni-sulfides (average sizes <20 nm) with millerite (trigonal NiS) cores and amorphous shells. The precipitates’ crystallinity is enhanced noticeably over a period of ∼6 days, forming larger-sized hexagonal α-NiS, and observations of defects such as twinning and stacking faults implicate a formation pathway via reassembly of fine nanoparticulate precursors. By comparison, in the presence of SRB and in the absence of Fe, more crystalline phases such as polydymite (Ni3S4) and vaesite (NiS2) are also precipitated in addition to the monosulfide phases. The observed difference suggests that the presence of SRB enables the transformation of polyphasic precursors to more crystalline structures through the combined effects of bacterial metabolites and localized precipitation within a low pH micro-environment around the cell walls. The addition of Fe(II) (i.e., [Ni]aq/[Fe]aq = 5:1) leads to formation of less crystalline Ni-sulfides in both biotic and abiotic systems, indicating crystal structure distortion caused by substitution of Ni with Fe. With decreasing [Ni]aq/[Fe]aq, Ni-sulfides become rarer, mixed Ni-Fe phases start to appear, and finally Ni-rich mackinawite (FeS) becomes the primary Ni-hosting phase at the lowest ratio tested ([Ni]aq/[Fe]aq = 1:5). We propose that whether aqueous Ni forms discrete Ni-S phases or is incorporated into dominantly Fe-S phases is primarily determined by the precipitation kinetics, and our experiments at [Ni]aq/[Fe]aq = 1:1 suggest that Ni-sulfide precipitation kinetics is comparable or higher than Fe-sulfides at this condition. Overall, our study allows for prediction on the phases and biogeochemical factors controlling Ni removal and availability in sulfidic environments.
Introduction
Nickel (Ni) is a bio-essential trace metal that is required for several geochemically relevant metabolisms. These include the cellular defense mechanism against reactive oxygen species, the production of ammonia from urea, the interconversion of di-hydrogen and protons, and the microbially mediated production of greenhouse gasses such as methane and carbon monoxide (Ragsdale, 2009). Nickel concentrations are low in the modern ocean with a range from 2 to 12 nM, and aqueous Ni shows a nutrient-like distribution with water depth due to depletion in the photic zone as a result of biological uptake (Sclater et al., 1976; Bruland et al., 2013). However, elevated concentrations up to 2 mM can be found in streams draining nickel-bearing minerals such as pyrite (FeS2) and millerite (trigonal NiS), as well as streams receiving industrial and mining wastes (reviewed in Rinklebe and Shaheen, 2017). High concentrations of Ni can adversely affect human health and the ecosystem; therefore, the World Health Organization recommended an upper limit of 0.001 mM (1 μM) for Ni concentrations in drinking water (WHO, 2007). Consequently, the removal mechanism of aqueous Ni from natural waters is of great interest both in terms of improving understanding of natural biogeochemical processes and in applying that information to improve water treatment quality.
In oxygen-rich environment, aqueous Ni in the water column is initially scavenged by Fe-Mn-oxides and organic matter and subsequently transported to the sediments (Sclater et al., 1976; Gall et al., 2013). Development of anoxic conditions below the sediment-water interface leads to the reduction of Fe-Mn-oxides and the oxidation of organic matter, prompting the release of Ni back into solution (Tribovillard et al., 2006; Weber et al., 2009; Hindersmann and Mansfeldt, 2014). In euxinic (i.e., anoxic and sulfidic) zones generated by the metabolism of sulfate-reducing microbes, the released Ni can be re-sequestered either by co-precipitation with Fe-sulfides such as mackinawite (FeS) and pyrite (FeS2) (Huerta-Diaz and Morse, 1992; Abraitis et al., 2004; Algeo and Maynard, 2004; Noël et al., 2015; Houben et al., 2017; Ikogou et al., 2017) or by precipitation as discrete Ni-sulfides (Ferris et al., 1987). Between these two mechanisms, co-precipitation with Fe-sulfides is typically the more important removal mechanism due to the high abundance of Fe (∼5 wt% Fe vs. 0.005 wt% Ni in Earth’s upper crust; Rudnick and Gao, 2003) and the slower water exchange kinetics of Ni compared to Fe, leading to preferential Ni incorporation into the faster-precipitating Fe-sulfides (Morse and Luther, 1999).
Discrete Ni-sulfides are rarely observed in nature (see Huang et al., 2010 for a brief review of their occurrences). Nonetheless, Ni-sulfides readily form in experiments employing high concentrations of Ni comparable to those found in polluted streams (Lewis and Swartbooi, 2006; Karbanee et al., 2008; Cao et al., 2009; Sampaio et al., 2010; Reis et al., 2013; Kiran et al., 2015). Additionally, geochemical observations indicate that Ni removal is decoupled from Fe precipitation in sulfidic and weakly acidic (pH 4–6) pore waters, suggesting the formation of discrete Ni-sulfides under these conditions (Huerta-Diaz et al., 1998; Frohne et al., 2011). Therefore, depending on the environment, Ni-sulfides may be an important host for Ni. The question of whether Ni is hosted in discrete Ni-sulfides or within Fe-sulfides, and the relative importance of each, matters because each phase has different solubilities and reactivities, which subsequently governs the fate and bio-availability of Ni in nature (Thoenen, 1999).
Fortunately, the understanding on the low temperature formation of Ni-sulfides has developed considerably over the past 20 years. Most of the efforts have been focused on abiotic precipitation with Ni as the sole metal species, yielding Ni-sulfides such as hydrated NiS (Ni1.1⋅1.5 H2O), millerite, α-NiS, polydymite (Ni3S4), heazlewoodite (Ni3S2), and vaesite (NiS2) (Wang et al., 1997; Jeong and Manthiram, 2001; Huang et al., 2009; Wilkin and Rogers, 2010; Table 1). The formed Ni-sulfides are almost exclusively nanocrystalline. While these past experiments provide a foundation for studies on Ni-sulfides, they are not representative of natural systems where precipitation occurs in complex solutions containing other metals and microbial life. Specifically, the effects of both aqueous Fe(II) and sulfate-reducing bacteria (SRB) (both of which should be present in euxinic zones) on Ni-sulfide precipitation are relatively understudied. The presence of Fe(II) during copper precipitation for example has been shown to modify the mineralogy and increase the crystallinity of copper-sulfides (Mansor et al., 2019). The presence of SRB also modifies the formation mechanism, size, shape, phase, and aggregation state of various metal sulfides (Gramp et al., 2006; Moreau et al., 2007; Xu et al., 2016; Picard et al., 2018). Specific to Ni, Sitte et al. (2013) found that crystalline α-NiS formation was favored in cultures of SRB compared to abiotic controls in which only amorphous NiS was precipitated. The difference in the crystallinity between biogenic and abiogenic precipitates can have important implications to the availability of Ni in euxinic environments.
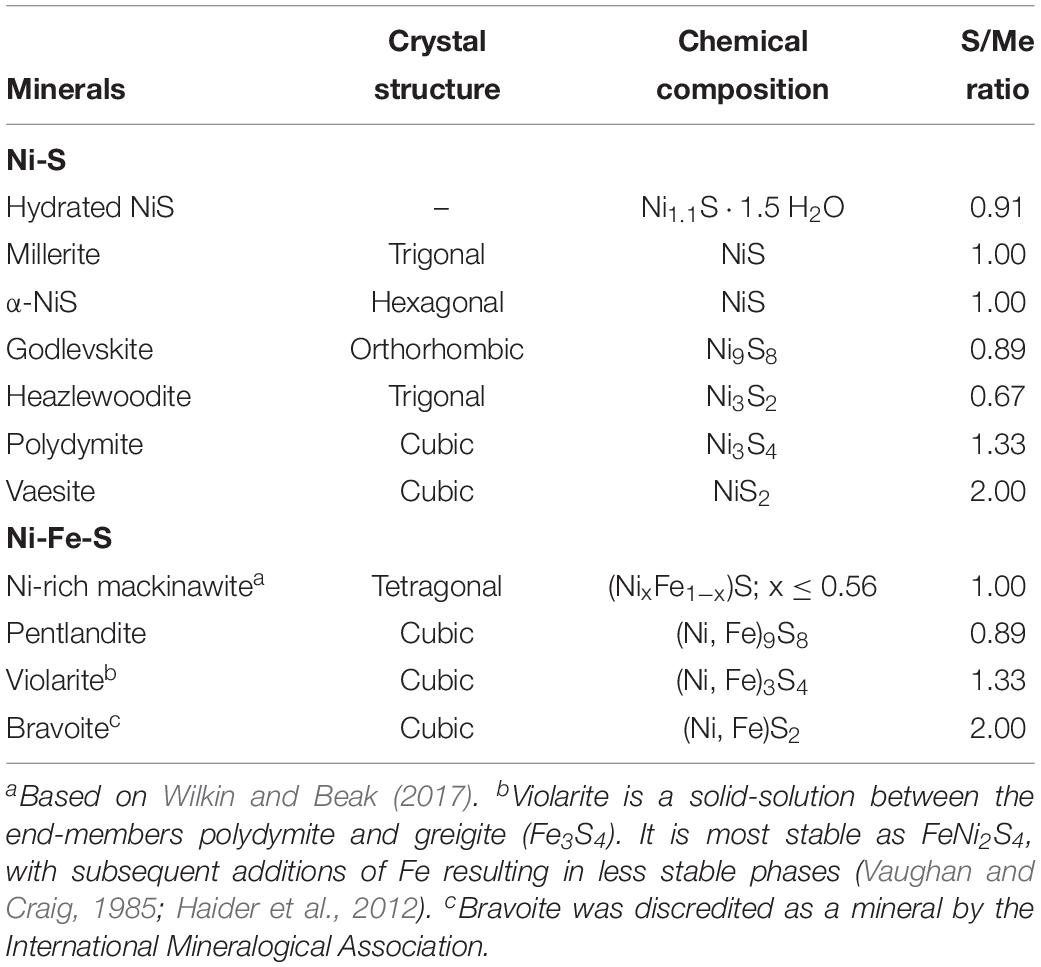
Table 1. Crystal chemistry for Ni-sulfide and Ni-Fe-sulfide minerals.
The current study is motivated by the need to identify and characterize the Ni-hosting phases that can form in natural euxinic environments. Correspondingly, we performed a systematic set of experiments that mimic the biogeochemical diversity of natural environments, where Ni is precipitated with excess sulfide in both Fe-poor and Fe-rich systems [from “No Fe” to aqueous Ni/Fe ratios ([Ni]aq/[Fe]aq) of 1:5 to 5:1] and in the presence versus absence of SRB at near-neutral pH. We conclude that across the [Ni]aq/[Fe]aq ranges typically observed in natural environments, the primary Ni-hosting phase(s) in Fe-poor systems will be Ni-sulfides (such as polyphasic precursors, α-NiS and vaesite) while in Fe-rich systems, the primary Ni-hosting phase will be mackinawite. The presence of SRB cells promotes the growth and transformation of fine Ni-sulfide nanoparticles into more crystalline phases, thus likely reducing the probability of Ni re-mobilization into solution.
Materials and Methods
All experimental procedures were performed under anoxic conditions and using acid-cleaned containers unless otherwise specified. Anoxic solutions were prepared by boiling for 20 min and degassing with N2 gas for another 20 min, followed by storage under pure N2 or 97% N2 – 3% H2 headspace. The syntheses were performed in a modified metal toxicity medium (MTM) that contains (per liter): 3.8 ml 80% lactic acid, 2.23 g Na2SO4, 0.06 g CaCl2.2H2O, 1.0 g NH4Cl, 1.0 g MgSO4.7H2O, 0.05 g yeast extract, 0.5 g tryptone and 2.83 g HEPES. Initial pH was adjusted to 7.2 by the addition of NaOH. The initial concentrations of Ni and Fe were adjusted by amending base MTM with metal stock solutions of either 20 mM NiCl2.6H2O or 10 mM FeSO4.7H2O (pH adjusted to 2 with HCl to minimize oxidation). The total metal concentration was kept constant at 500 μM, while the initial aqueous Ni/Fe ratios ([Ni]aq/[Fe]aq) were varied from “No Fe,” 5:1, 1:1, and 1:5.
Biotic Precipitation
Biotic syntheses were conducted in the presence of Desulfovibrio vulgaris strain DSM 644 (also known as ATCC 29579 or strain Hildenborough), which gradually produce sulfide as the end-product of sulfate reduction. The culture was initially maintained over a period of months at 4°C in modified Postgate medium #63 (media composition detailed in Xu et al., 2016). Prior to each round of synthesis, the culture was inoculated at 1% v/v into MTM and allowed to grow at 30°C while shaking. After 72 h, the cells were inoculated at 1% v/v into 100 ml of Ni- and/or Fe-amended MTM for the actual synthesis experiments. After 6–25 days of incubation, the culture and associated mineral precipitates were aliquoted into two separate portions for X-ray diffraction (XRD; ∼80 ml) and transmission electron microscopy (TEM; ∼5 ml) analyses, respectively. Both aliquots were centrifuged at 15,000 g for 5 min followed by removal of the supernatant via pipetting. Mineral pellets from the XRD aliquot were rinsed once with 10 ml H2O, resuspended in 0.8 ml H2O and then allowed to dry on a glass slide as a thin film. Mineral pellets for TEM were treated for 1 h at 45°C in a 10 ml solution containing 0.1% Triton-X and 100 μg/ml each of lysozyme and proteinase K to lyse the bacterial cells. Soluble organics were then separated from the mineral fraction by centrifugation at 15,000 g for 15 min. The mineral pellets were rinsed twice more with 10 ml H2O to remove traces of organics before being set aside for TEM analyses.
Abiotic Precipitation
For abiotic synthesis, 50 ml of 15 mM Na2S.9H2O solution was titrated at a rate of 2 ml/h into 50 ml of Ni- and/or Fe-amended MTM. The initial concentration of chemical components in the MTM were doubled so that after Na2S addition, the final concentration was similar as in the biotic synthesis. A Teflon-coated magnetic stir bar was used to mix the solution during titration. After 5–36 days of incubation, nanoparticle aggregates were separated from solution by filtration through a 0.2 μm pore size mixed cellulose ester membrane. The solution remained clear during this step. The precipitates were then resuspended in 1 ml H2O and aliquoted for XRD (∼0.8 ml dried onto glass slide) and TEM (∼0.2 ml stored in sealed tubes) analysis.
Characterization
The samples were characterized using a combination of XRD and TEM-based methods. The XRD patterns were collected using a Rigaku Miniflex II from 10 to 60° 2θ angle with a step size of 0.05° and a scan speed of 0.5°/min, totaling a collection time of about 1 h and 45 min per sample. Samples for TEM were diluted in H2O in the anaerobic chamber and shipped in rubber-capped and aluminum-sealed glass serum vials to the NanoEarth National Center at Virginia Tech. On site, the samples were sonicated for up to 1 h to disperse aggregates prior to mounting onto gold-coated TEM grids (product #01824G, Ted Pella, Inc.). Microscopy images and selected area electron diffraction patterns (SADP) were obtained using a JEOL-2100 TEM operating at 200 kV, and elemental ratios [e.g., S/(MeFe+Ni) and Ni/Fe] were obtained with the accessory energy-dispersive X-ray spectroscopy (EDS) system coupled to a silicon drift detector. Spot EDS analysis used beam sizes of 20–500 nm in diameter, while mapping analysis was conducted in scanning-TEM mode with a resolution of ∼10 nm. The inherent error of ≤7% for the standardless EDS analysis method (Newbury and Ritchie, 2014), compounded by the possibility of sulfur adsorption onto the nanoparticles’ surface (Rickard et al., 2006; Huang et al., 2010; Picard et al., 2016) may contribute to the deviation of the measured elemental ratios from the real ones, especially for nanoparticles with substantially high surface areas. Thus, the EDS data were considered semi-quantitative. There were no observable differences in the nanoparticles’ characteristics between the samples that were dried under anoxic conditions versus in air (<10 min drying time). There were also no obvious enhanced dissolution (e.g., less yield over time, etch pits, noticeably different morphology) for the samples that were stored in H2O for up to 2 months prior to mounting versus samples that were mounted to TEM grids immediately following preparation. Some Fe-rich samples that were stored in vacuum for >1 week after mounting onto TEM grids did show morphological changes that are likely caused by oxidation to Fe-oxides. All the samples in this study were therefore analyzed within 24 h of mounting onto TEM grids (while kept within a dry vacuum chamber during this period). Particle size distributions were obtained by measuring the diameter of >50 individual nanoparticles manually in ImageJ1.
Results
Within a day of the start of incubation, black or dark brownish precipitates are readily formed under all tested conditions. Starting from an initial pH of 7.2, the final pH values of the biotic experiments decrease slightly to 7.15 ± 0.10. In contrast, the final solution pH values in all of the abiotic experiments are about one unit higher, with a range of 8.03–8.24 (Table 2).
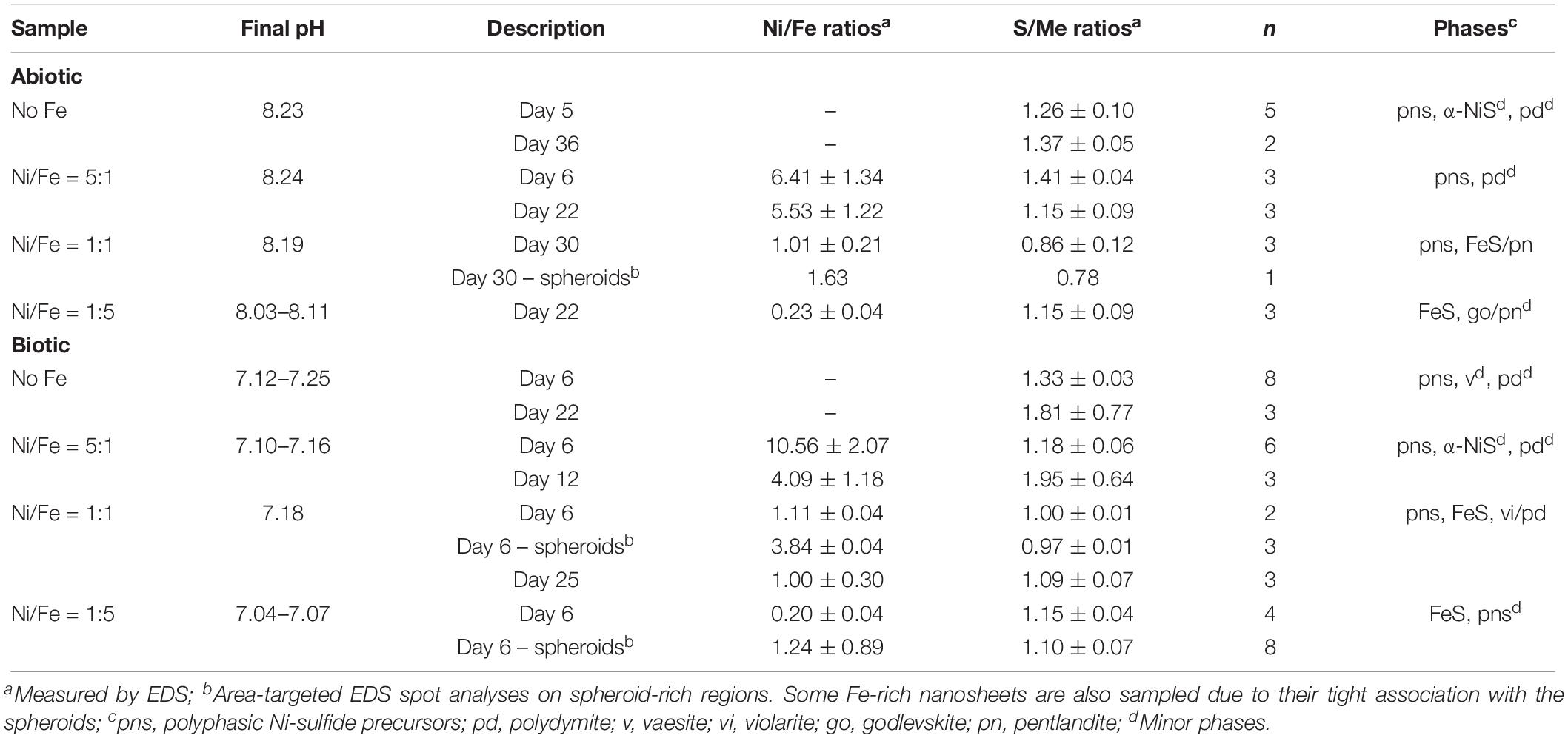
Table 2. Solution pH, solid-phase molar elemental ratios and identity of mineral phases (determined by XRD and SADP) in the samples.
Characterization of Nanoparticles Precipitated in Fe-Free and Fe-Poor Systems
Analyses of XRD patterns of precipitates formed in the Fe-free and Fe-poor abiotic and biotic systems (“No Fe” and [Ni]aq/[Fe]aq = 5:1) indicate that the precipitates are largely amorphous (Figure 1). Microscopy observations of early-stage (<1 week old) samples reveal a predominance of Ni-sulfide spheroids (Figure 2), with particle sizes typically <10 nm and extending up to ∼20 nm (Figure 3). The SADP derived from these nanoparticles reveal two major diffuse rings with d-spacing ranges of 2.44–2.83 Å and 1.64–1.99 Å (Figure 2c). The d-spacings are consistent with those of millerite, although we cannot rule out the presence of other phases such as α-NiS, godlevskite and polydymite (Supplementary Table S1). The average sulfur-to-metal ratios for these spheroids are slightly above 1.00 (Table 2), which is likely due to excess sulfur adsorption to nanoparticles’ surfaces (Rickard et al., 2006; Huang et al., 2010; Picard et al., 2016). High-resolution TEM images reveal the polyphasic nature (as defined by French et al., 2012) of the early-stage Ni-sulfide spheroids, in that they contain crystalline areas ∼1–3 nm in size within an amorphous matrix (Figure 2b). From here on, we will refer to these spheroidal nanoparticles as “polyphasic Ni-sulfide precursors.” With increasing incubation time, the polyphasic precursors display enhanced crystallinity and a distribution toward larger sizes (Figures 2d,e, 3). The biotic precipitates in particular are generally more crystalline and larger than abiogenic precipitates. Distinct diffraction spots become noticeable in the SADP of the aged or biogenic crystalline nanoparticles (Figure 2f). The measured d-spacings from the spots are 2.91, 2.82, 2.54, 2.40–2.43, 2.21, 1.90, and 1.70 Å, which can be best explained by the reflections from millerite’s (101), (300), (021), (220), (211), (131), and (401) planes, respectively.
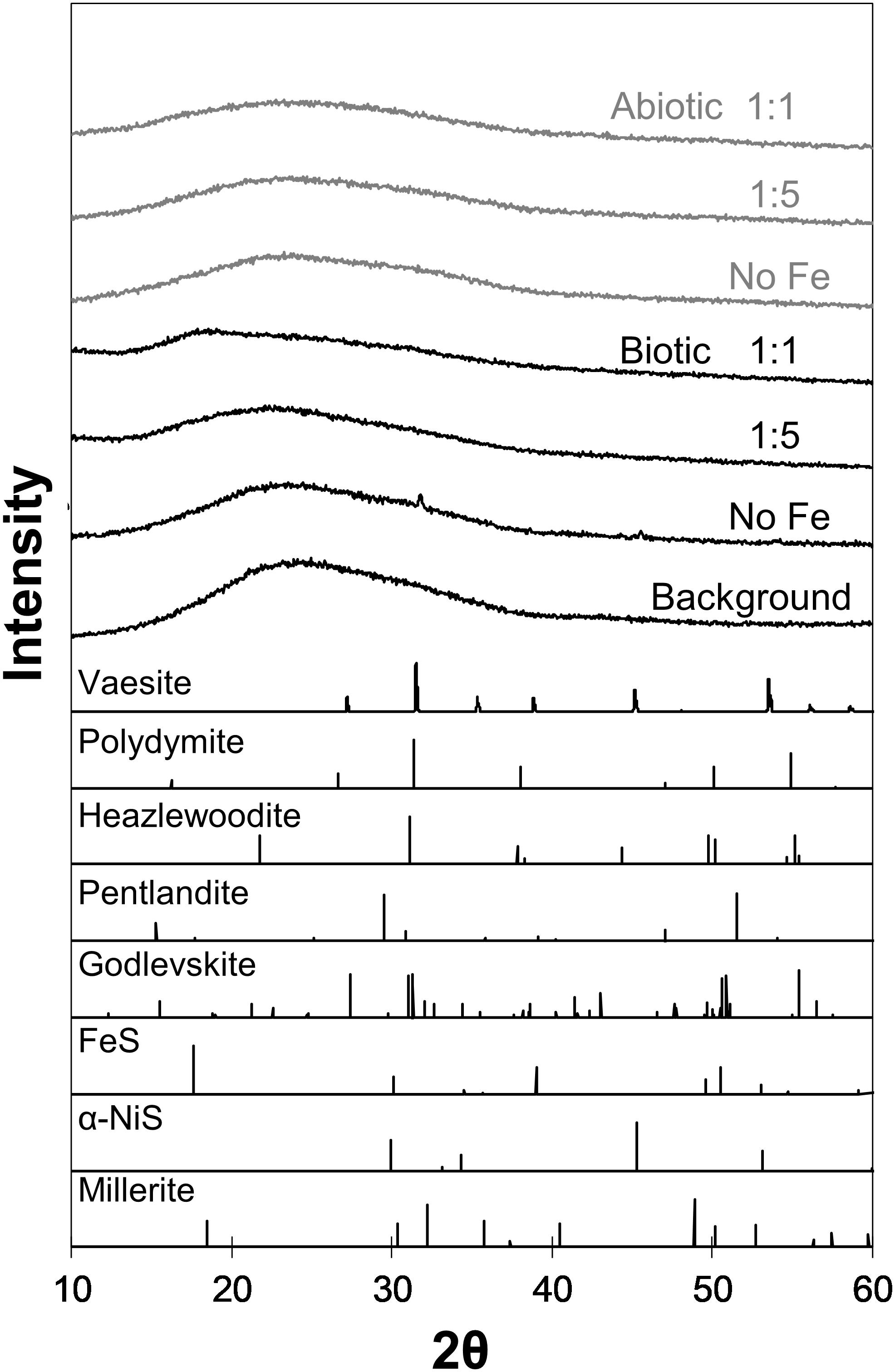
Figure 1. XRD patterns of biogenic (black) and abiogenic (gray) precipitates shown at the upper portion of the figure, with corresponding standard patterns of select Ni-Fe-sulfides (from American Mineralogist database accessed on February 2019; Downs and Hall-Wallace, 2003) shown at the bottom portion. Background signal contributed by the glass slide is also included in the figure. Most samples are indistinguishable from the background except “Biotic No Fe” and “Biotic 1:1 Ni:Fe” samples.

Figure 2. Left to right: low resolution images, high resolution images and SADPs derived from the spheroidal nanoparticles. Top row (a–c): spheroidal nanoparticles observed in abiotic “No Fe” systems during the early periods of incubation (≤1 week old). These nanoparticles are polyphasic, in that they display crystalline nano-domains ∼1–3 nm in size within an amorphous matrix. The corresponding SADP displays only diffuse rings. Bottom row (d–f): spheroidal nanoparticles typically observed in the biotic “No Fe” systems. These nanoparticles are larger in size and are more crystalline. Similar nanoparticles are also observed in the abiotic “No Fe” systems after aging for >1 week. The corresponding SADP starts to resolve into distinct spots with a set of d-spacing values that are most consistent with those of millerite. Annotated SADPs are available in Supplementary Material.
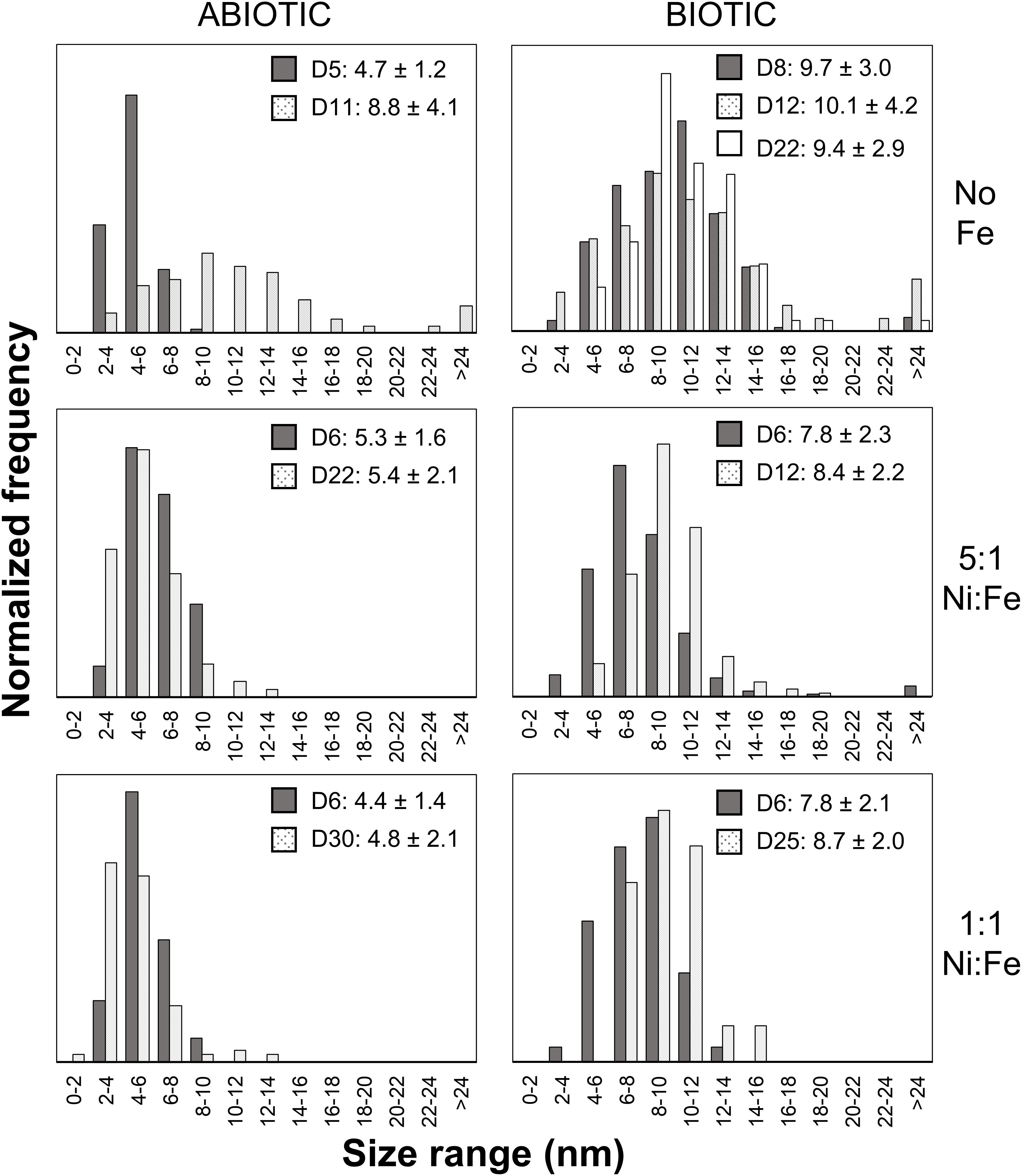
Figure 3. Particle size distributions of Ni-rich nanoparticles. Over 50 particles were analyzed per sample. The legends denote the coloring for samples from different sampling day (D#) along with the average size and standard deviation in nanometers.
Other Ni-sulfide phases such as vaesite, α-NiS and polydymite/violarite are also positively identified in the samples. Vaesite occurs as ∼20-nm spheroidal nanoparticles or 100- to 300-nm euhedral nanocrystals only in the biotic system and in the absence of Fe (Figures 4a,b). The presence of vaesite is confirmed via both XRD (small peaks at 31.5°, 41.5°, and 54° 2θ in Figure 1) and SADP (insets of Figures 4a,b) analyses. The α-NiS phase occurs as ∼20- to 80-nm hexagonal nanoplates and is detected in aged (>1 week old) abiogenic precipitates in the absence of Fe and in biogenic precipitates at [Ni]aq/[Fe]aq = 5:1 (Figure 4c). Finally, polydymite (Ni3S4)/violarite (FeNi2S4) occurs as ∼10–20 nm irregularly shaped nanoparticles in all of the Ni-rich samples (Figure 4d). This phase is identified based on high-resolution TEM images that reveal lattice fringes with 3.32–3.38 Å d-spacing corresponding to the (220) planes of polydymite/violarite (Supplementary Table S1).

Figure 4. TEM images of identified Ni-sulfide phases besides millerite. Insets show the corresponding SADPs. (a) Aggregates containing vaesite and polyphasic Ni-sulfide precursors. (b) Euhedral vaesite. (c) Larger-sized α-NiS nanoplate (denoted by the arrow) clustering with smaller polyphasic Ni-sulfide precursors and polydymite. (d) Irregularly shaped polydymite. Annotated SADPs are available in Supplementary Material.
Characterization of Nanoparticles Precipitated in Fe-Rich Systems
Analysis of TEM and EDS data of biotic and abiotic samples precipitated at [Ni]aq/[Fe]aq = 1:1 indicate the presence of Ni-rich spheroids (Ni/Fe ratio up to 3.84) that are tightly associated with extensive Fe-Ni-sulfide nanosheets (Ni/Fe = 1.01–1.11) (Figures 5a,b, 6). Compared to those formed in the Fe-poor systems, the Ni-rich spheroidal nanoparticles here have lower crystallinity and are finer in size (Figure 3). The mineralogy of the spheroids and nanosheets are complex. Morphological similarities suggest that the Ni-rich spheroids are polyphasic Ni-sulfide precursors whereas the Fe-Ni-sulfide nanosheets are Ni-containing mackinawite (see Mansor et al., 2019 for images of Ni-free mackinawite formed under comparable conditions). Based on the XRD patterns, these precipitates are largely amorphous except that a tentative shoulder occurs at 17° 2θ (∼5 Å), attributable to the (001) planes of mackinawite, in the biogenic samples (Figure 1). In the abiogenic precipitates, SADP collected from spheroid-rich regions indicate a diffuse ring with a d-spacing range of 2.40–2.82 Å, and additional diffraction spots with d-spacings of 2.51, 2.43, and 1.70 Å (Figure 5a, inset); these d-spacings are most consistent with those of millerite. Comparatively in the biogenic precipitates, SADP collected from spheroid-rich regions display diffraction rings with measured d-spacings of 3.38, 2.85, 2.42, 1.84, and 1.71 Å (Figure 5b, inset) that match the reflections from major planes of violarite/polydymite. Additional d-spacings obtained from imaging of lattice fringes are: (a) 5.4–5.7 Å from the nanosheets, most likely corresponding to reflections from the (001) plane of disordered mackinawite (Wolthers et al., 2003) while not ruling out overlapping reflections from pentlandite (111) and/or violarite (111) planes, and (b) ∼1.5 Å from the Ni-rich spheroids, corresponding to the (012) planes of millerite. Overall, the precipitates are likely mixtures of the aforementioned phases with varying degrees of crystallinity at various combination ratios.

Figure 5. (a,b) TEM images of abiogenic (left) and biogenic (right) precipitates from [Ni]aq/[Fe]aq = 1:1 systems. Ni-rich spheroids (examples denoted by arrows) are tightly associated with Fe-rich nanosheets. (c,d) TEM images of abiogenic (left) and biogenic (right) precipitates from [Ni]aq/[Fe]aq = 1:5 systems. The samples are dominated by Fe-rich nanosheets. Small Ni-rich spheroids (examples denoted by arrows) are also present in the biogenic precipitates but not in the abiogenic precipitates. Insets correspond to representative SADP collected from the samples. Annotated SADPs are available in Supplementary Material.
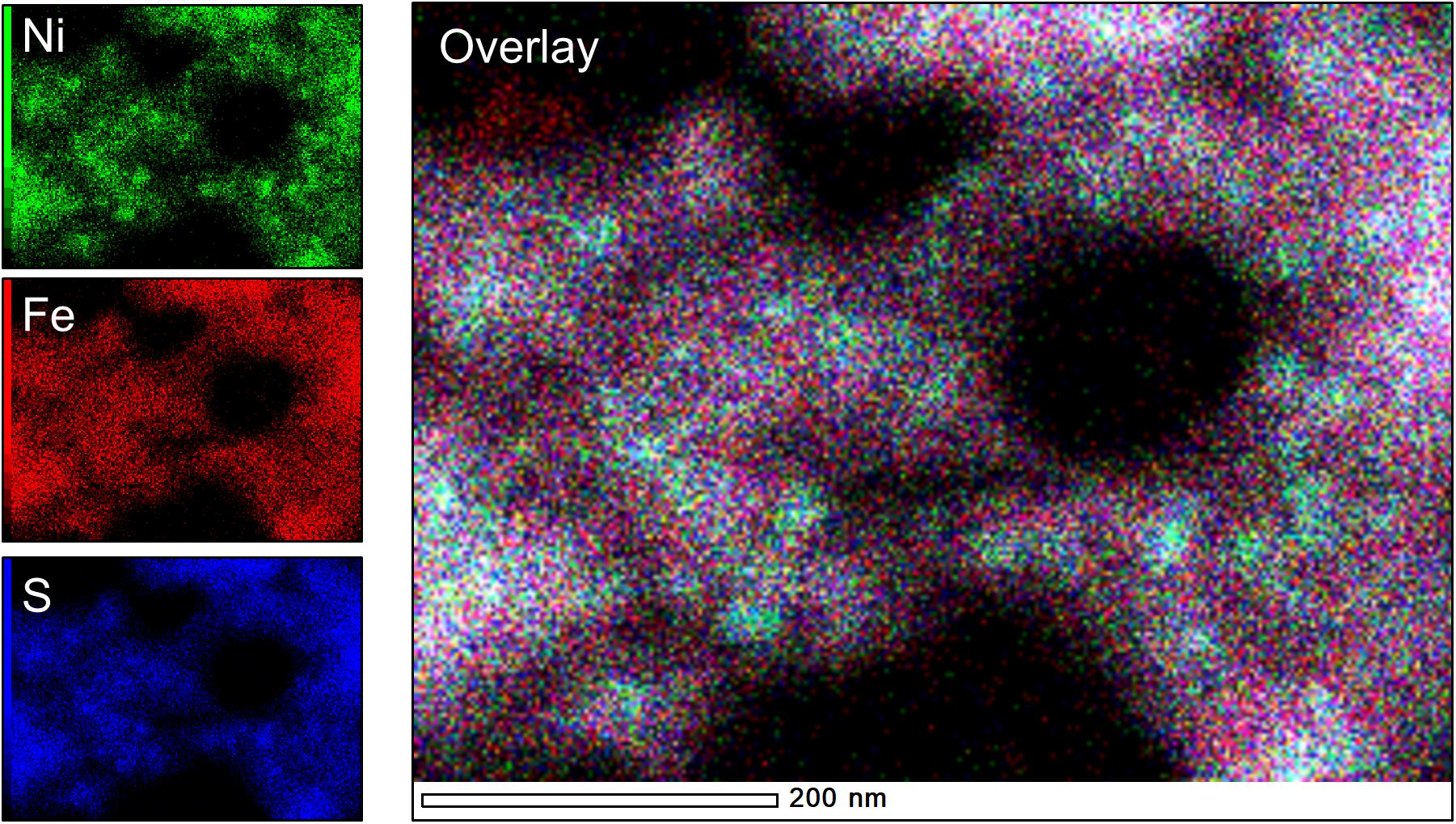
Figure 6. Elemental maps of “Biotic 1:1 Ni:Fe” sample, highlighting the presence of Ni-rich spheroids (teal-colored hotspots in the overlay image) within a more diffuse Fe-rich matrix.
As the initial relative [Fe]aq is increased to a [Ni]aq/[Fe]aq ratio of 1:5, the precipitates become dominated by Fe-rich nanosheets (with a Ni/Fe ratio of ∼0.20) (Figures 5c,d). Analyses of these samples’ XRD patterns confirm the presence of mackinawite (Figure 7). The SADP analyses are also consistent with the reflections of mackinawite especially for the biogenic precipitates (Figures 5c,d, insets). For the abiogenic precipitates, additional rings with corresponding d-spacings of 3.57 and 2.89 Å indicate the presence of pentlandite or godlevskite along with mackinawite. Interestingly, while no discrete Ni-sulfides are observed in the abiogenic precipitates, the biogenic precipitates still contain a significant number of Ni-rich spheroids (Ni/Fe ratio = 1.24 ± 0.89; n = 8). These spheroidal nanoparticles have a mean size of 11.9 ± 3.2 nm (n = 8). Therefore, the presence of D. vulgaris plays a role in favoring the precipitation of discrete Ni-sulfides even in Fe-rich systems.
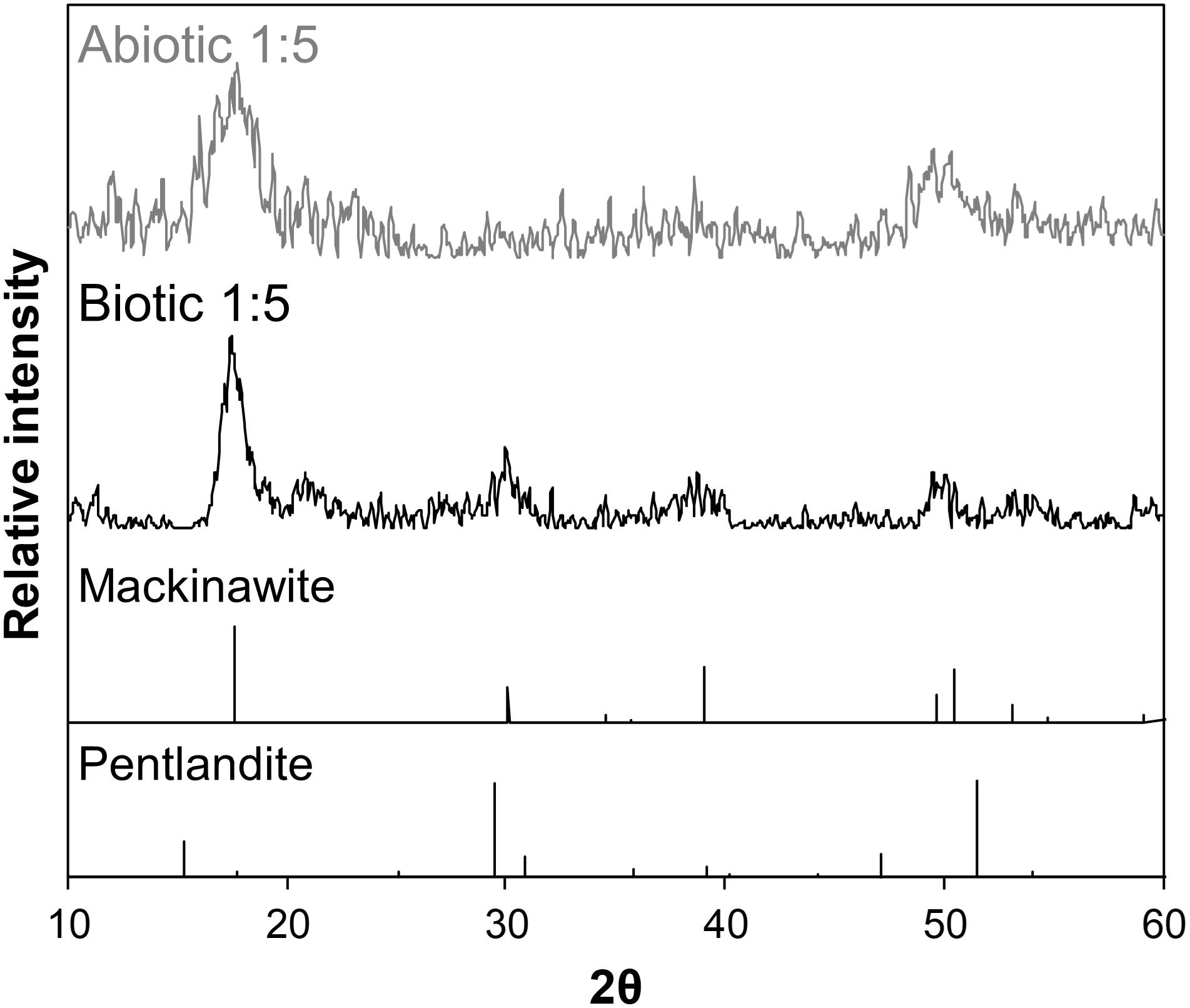
Figure 7. Background-subtracted XRD patterns of biogenic (black) and abiogenic (gray) precipitates at [Ni]aq/[Fe]aq = 1:5 shown at the upper portion of the figure, with corresponding standard patterns of mackinawite and pentlandite (from American Mineralogist database; Downs and Hall-Wallace, 2003) shown at the bottom portion. The sample patterns are most consistent with mackinawite.
Discussion
Formation Mechanism of Ni-Sulfides
Through the various techniques employed in our study, we have identified several phases of Ni-sulfides including polyphasic Ni-sulfide precursors, millerite, α-NiS, polydymite and vaesite. Each of these phases has different solubility and formation energy, with vaesite being predicted as the thermodynamically most stable phase at room temperature (Wilkin and Rogers, 2010). Since all of the Ni-sulfides are precipitated as nanoparticles, their solubilities are also affected by their sizes and crystallinity (Gilbert and Banfield, 2005; Hochella et al., 2008; Caraballo et al., 2015). Understanding the factors that control the formation of a specific Ni-sulfide phase, as well as their size and crystallinity, can help us better predict the behavior and fate of Ni in euxinic environments. Below, we discuss the formation pathway(s) of Ni-sulfides in the absence and presence of SRB, and how these pathways may be modified in the presence of Fe(II).
Abiotic Formation of Ni-Sulfides
In our study, the first Ni-sulfide phase to precipitate out of solution is XRD-invisible polyphasic Ni-sulfide precursors. Previous abiotic studies indicate that the first product between aqueous Ni and sulfide at 25°C is also XRD-invisible Ni-sulfides (Wang et al., 1997; Jeong and Manthiram, 2001; Wilkin and Rogers, 2010). This phase is later determined through a combined TEM and pair distribution function analysis to be nanoparticulate Ni-sulfides with a core-shell structure (Huang et al., 2009, 2010). The 1–3 nm core is composed of crystalline millerite while the 3–5 nm thick shell is composed of slightly Ni-rich material with structural H2O in an amorphous configuration, leading to the chemical composition of Ni1.1S⋅1.5H2O. Overall, the structure and size of this hydrated phase is similar to the polyphasic Ni-sulfide precursors identified in our study.
Over time in the abiotic systems, we observe the development of early-stage polyphasic Ni-sulfide precursors to larger and more crystalline nanoparticles up to α-NiS within a few days to weeks. Wilkin and Rogers (2010) observed the formation of α-NiS within similar timescales at room temperature, and its formation rate is accelerated to within a day at 60°C. The presence of stacking faults and twinning within the nanoparticles in our study (Figure 8) implicate crystal growth mechanism through particle attachment of smaller nanoparticles or soluble nanoclusters, with potential subsequent atomic rearrangements to higher stability structures (De Yoreo et al., 2015). This is consistent with solubility consideration; since most metal sulfides have low solubilities, the degree of supersaturation during precipitation is extremely high (Rickard and Luther, 2006). Initial mineral formation is therefore dominated by nucleation events, and additional growth can only proceed through attachment and re-assembly of the initially formed nanoparticles. Hexagonal α-NiS is generally considered as a metastable phase but once formed, it is stable for over 6 months in aqueous solution at room temperature (Wang et al., 2006; Wilkin and Rogers, 2010).
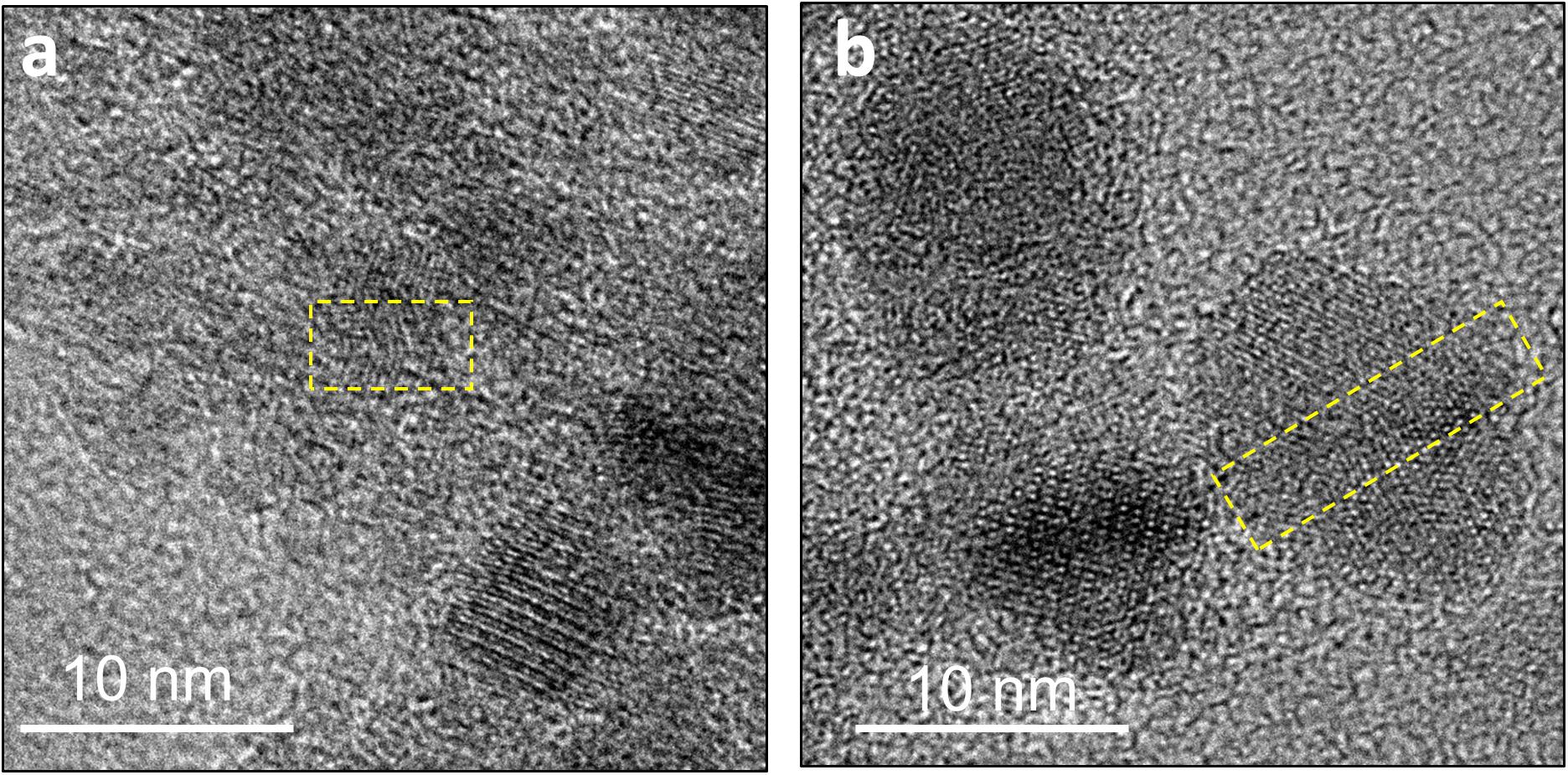
Figure 8. Defects in the crystal structure of Ni-sulfides due to attachment of two or more nanoparticles. (a) A stacking fault in a ∼5 nm particle. (b) Twinning in a ∼12 nm particle.
Careful examination of the abiogenic precipitates in our study also indicates the presence of trace amounts of polydymite, but not vaesite in the Fe-poor systems. Wilkin and Rogers (2010) have shown that in the presence of elemental sulfur, α-NiS is rapidly transformed to vaesite through polydymite as an intermediate (Figure 9). The added elemental sulfur acts as an oxidant to facilitate the partial oxidation of Ni (from Ni2+ in α-NiS to Ni3+ in polydymite) and to produce polysulfides (e.g., S22– in vaesite) by reacting with H2S. Thus, the pathway of vaesite formation is comparable to one of the pathways to forming pyrite in the Fe-S system, in which mackinawite is transformed to pyrite via greigite intermediate (Wang and Morse, 1996; Benning et al., 2000; Hunger and Benning, 2007):
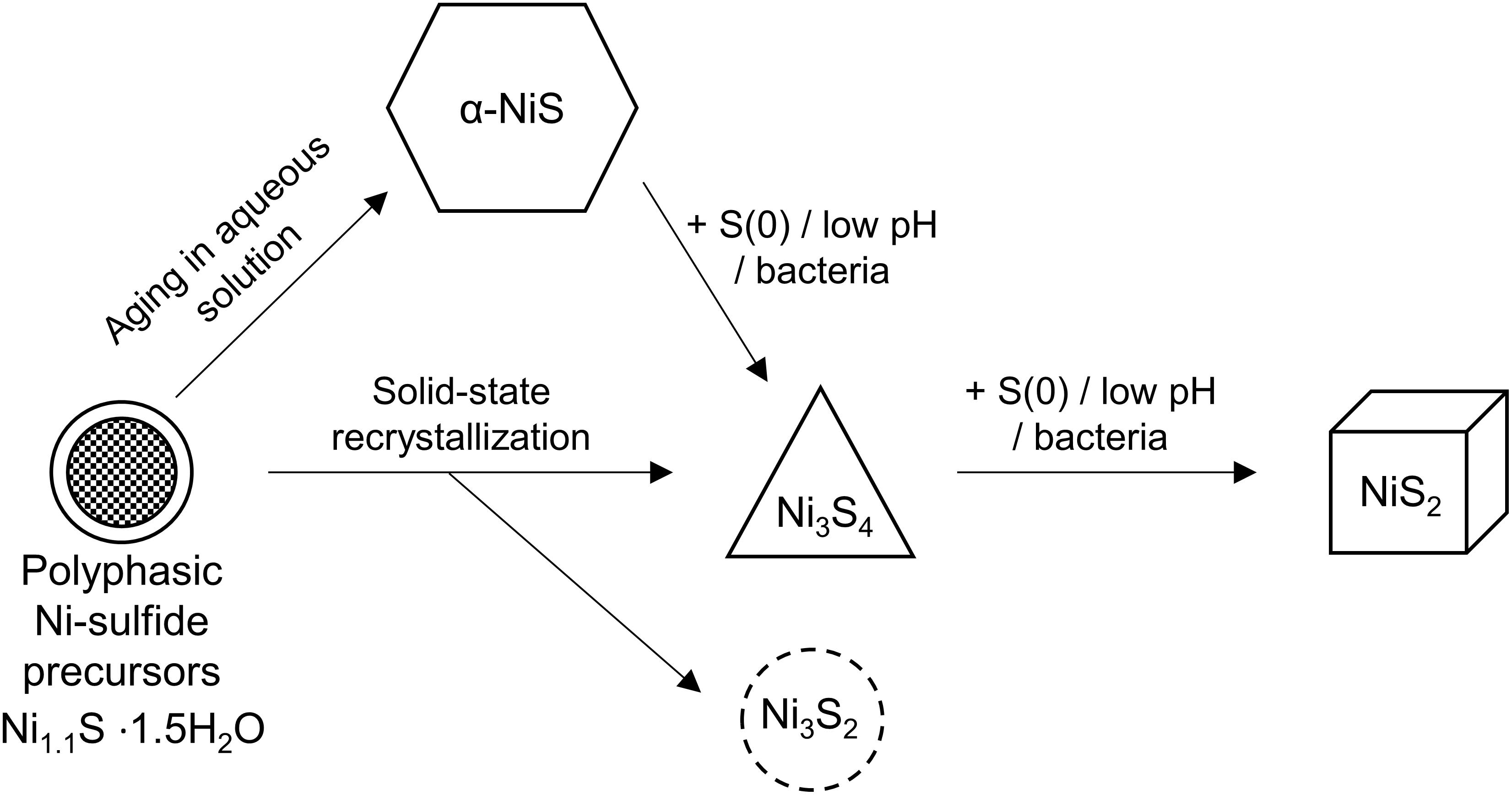
Figure 9. Diagram depicting the crystallization sequence of Ni-sulfides. The initial precipitate is polyphasic Ni-sulfide precursors. Metastable α-NiS can form naturally within a few hours to days. In the presence of S(0) or bacteria or acidic pH, α-NiS recrystallizes to form polydymite (Ni3S4) and vaesite (NiS2). Polydymite and heazlewoodite (Ni3S2) can also form through solid-state recrystallization of hydrated NiS under an anoxic atmosphere.
The formation of polydymite and vaesite are favored at acidic pH (Wang et al., 1997; Huang et al., 2009). By analogy to the Fe-S system, this preference is attributed to (i) protons acting as oxidants for the transformation of MeS to Me3S4 (Bourdoiseau et al., 2011) and (ii) higher rate of dissolution-reprecipitation reactions to form MeS2 from either MeS or Me3S4 (Schoonen and Barnes, 1991; Wang and Morse, 1996). The lack of vaesite in the abiotic systems in our study is consistent with predictions for the near-neutral pH experimental conditions and suggests that negligible amounts of polysulfides are formed (these species may form in the presence of trace oxygen). In contrast, the trace amount of polydymite found in all the Fe-poor abiotic samples is rather surprising. Previous understanding suggests that this phase is primarily an artifact of sample preparation and is formed during the brief period of oxidation either during sample mounting or during transport to the TEM rather than being an actual experimental product. This is similar to the multiple reports of greigite detection in experimental Fe-S systems, which has been attributed to the fast oxidation of mackinawite to greigite in air (Boursiquot et al., 2001) and even under vacuum (reviewed by Rickard and Luther, 2007). Indeed, solid-state oxidation of hydrated NiS to polydymite has been shown to occur even under an anoxic atmosphere with purportedly <2 ppm O2 (Huang et al., 2009).
Formation of More Crystalline Biogenic Ni-Sulfides
Precipitation in the presence of D. vulgaris leads to more crystalline structures of Ni-sulfides as indicated by two lines of observations. First, higher stability phases such as vaesite (in “no Fe” systems) and polydymite/violarite (at [Ni]aq/[Fe]aq = 1:1) are more prominent in the biotic compared to abiotic systems. Second, biogenic Ni-sulfide nanoparticles are larger in size and more crystalline than their abiogenic counterparts precipitated at comparable [Ni]aq/[Fe]aq and incubation time. These observations imply a faster growth and transformation rate of initially formed Ni-sulfides to more crystalline structures when precipitation occurs in the presence of SRB.
We first consider if the slightly lower pH in the biotic experiments (final pH ∼7) compared to the abiotic experiments (final pH ∼8) may increase the transformation rate of Ni-sulfides. The pH difference can be understood by considering the protons and buffering capacity generated during microbial sulfate reduction reaction (Pankhania et al., 1986; Voordouw, 2002; Heidelberg et al., 2004):
compared to the protons consumed during the dissociation reaction of sodium sulfide in the abiotic systems:
Although the lower pH in the biotic systems may contribute to the more crystalline structures of biogenic Ni-sulfides by (i) favoring the formation of polydymite and vaesite (see section “Abiotic Formation of Ni-Sulfides”) and (ii) favoring growth to larger-sized Ni-sulfides by slowing down the nucleation rate (metal solubility tends to increase with decreasing pH; Lewis, 2010), we argue that the observed pH difference is insufficient to cause these effects. First, previous abiotic experiments have shown that Ni-sulfides precipitated at room temperature from pH 3–9 share similar initial mineralogy (Huang et al., 2009). Second, the abiotic formation of polydymite and vaesite requires a pH range that is much more acidic (pH ≤5; Wang et al., 1997; Huang et al., 2009) than that measured for the biotic systems. Third, the most recent model of Ni-sulfide solubility in the presence of excess sulfide indicates that the solubility does not change appreciably across pH 7–8 (Figure 10). Considering all of these, we argue that the formation of more crystalline biogenic Ni-sulfides are not mainly caused by the slight variation in the bulk solution pH. Other effects of SRB to Ni-sulfide precipitation must be considered.
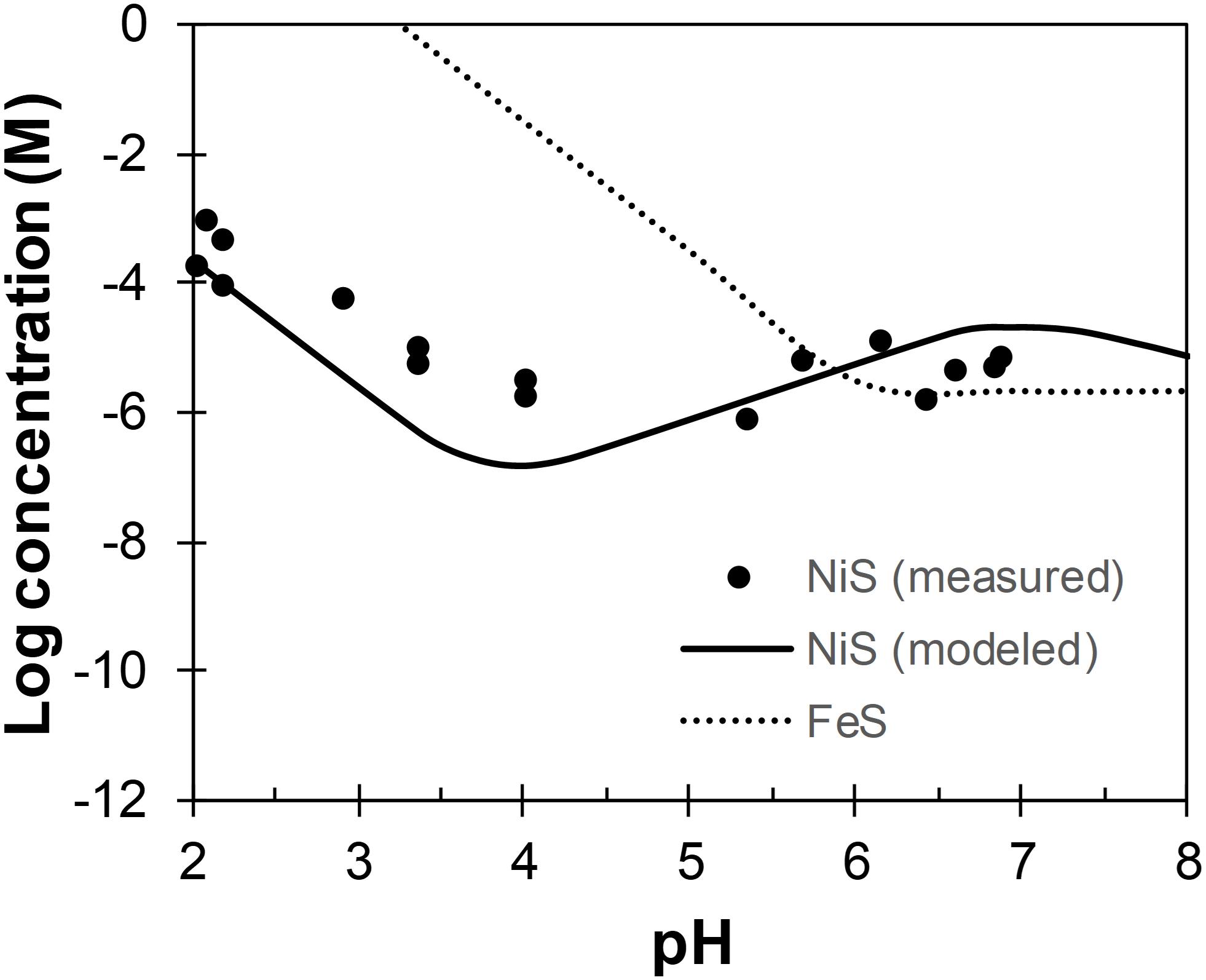
Figure 10. Concentrations of Ni and Fe across pH in equilibrium with α-NiS (Wilkin and Rogers, 2010) and mackinawite (Rickard, 2006), respectively. Total sulfide concentration is assumed to be 1 mM.
Besides altering the inorganic chemistry of the solution, the presence and metabolism of SRB are also known to provide additional precipitation sites for metal sulfides. Binding of Ni to cell walls and extracellular polymeric substances (EPS) secreted by bacteria are well-known (Beveridge and Murray, 1976; Beveridge and Koval, 1981; Ferris et al., 1989; Wuertz et al., 2000; Guibaud et al., 2005; Quintelas et al., 2009) and Ni-sulfides have been observed to precipitate on bacterial cell walls under both experimental (Fortin et al., 1994; Sitte et al., 2013) and natural (Ferris et al., 1987) conditions. During the initial nucleation event, it is important to recognize that the (sub)micro-environments around the cell walls are chemically distinct from the bulk solution due to limitations in diffusion at small scales (Purcell, 1977). A prominent feature of this micro-environment is a high concentration of sulfide, which creates local supersaturation zones that favor metal sulfide precipitation (Southam, 2000). Additionally, the protons and organics (e.g., acetate) produced as by-products of sulfate reduction (Eq. 3) will be concentrated in this micro-environment. In other bacterial cultures and natural biofilms, the micro-environment pH has been shown to reach levels as low as 3.5 compared to the near-neutral bulk solution (Mera et al., 1992; De Los Ríos et al., 2003; Hunter and Beveridge, 2005; Hidalgo et al., 2009). A combination of micro-environmental low pH and the presence of soluble organics that can bind Ni are likely contributing to the distinct characteristics of biogenic Ni-sulfides.
As previously mentioned, the formation of vaesite and polydymite are favored at acidic pH (pH ≤5), which plausibly exist in the micro-environments around SRB cells. The fact that neither of these phases are the most abundant phase in the biotic systems indicates some chemical/physical limitations to their formation, largely in line with the micro-environment formation hypothesis. Once formed, these phases may detach from the cell walls but are nevertheless stable at the near-neutral pH of the bulk solution (Wilkin and Rogers, 2010). Furthermore, precipitation in an acidic micro-environment may also explain the larger sizes of biogenic Ni-sulfides compared to their abiogenic counterparts. An analogy can be made using the example of mackinawite and pyrite, both of which tend to be larger in size when precipitated at acidic pH (Schoonen and Barnes, 1991; Wang and Morse, 1996). This phenomena is attributed to the increase in metal solubility with decreasing pH, leading to a decrease in the degree of supersaturation that results in slower nucleation rates and faster growth rates compared to the scenarios at higher pH (Lewis, 2010). The solubility of Ni, however, changes in complex fashion across pH and only increases with decreasing pH below pH 4 in the presence of sulfide (Figure 10). Therefore, the formation of more crystalline Ni-sulfides likely requires a micro-environment pH ≈4, which remains to be confirmed experimentally for SRB.
In addition to pH modification, the organics secreted by SRB can also affect the dynamics of Ni-sulfide precipitation. For example, bacterial metabolites have been implicated in mediating attachments between ZnS nanoparticles, allowing biogenic ZnS to achieve larger sizes compared to abiogenic ZnS (Xu et al., 2016). Such a mechanism may also operate in our study, but we note that there are no indications of more particle attachment events (e.g., defects such as stacking faults and twinning) in the biogenic Ni-sulfides compared to abiogenic Ni-sulfides. Secreted proteins may also decrease the availability of free Ni(II) ion by complexation (Fortin et al., 1994), resulting in lower degree of supersaturation and contributing to larger Ni-sulfides by altering the relative kinetics of Ni-sulfide growth over nucleation. Overall, we propose that the combination of precipitation within acidic micro-environments and the interaction of Ni/Ni-sulfides with secreted organics act in tandem to generate more crystalline Ni-sulfides in the biotic systems.
Metal Sulfide Formation During Co-existence of Fe and Ni: Effects of Cation Sizes and Reaction Kinetics
In the presence of appreciable quantities of Fe(II)aq, a diversity of Ni-hosting phases is formed including Ni-sulfides, mackinawite and (minor) mixed Ni-Fe phases such as pentlandite and violarite. Pentlandite is generally considered a high temperature phase of Fe-Ni-sulfides that weathers to form violarite at low temperatures (e.g., Grguric, 2002; Tenailleau et al., 2006; Xia et al., 2009), but its stability field could be enhanced at the nano-scale due to changes in the relative stability of minerals as a function of particle size (Navrotsky et al., 2008). The preferential formation of all of the aforementioned phases and their respective properties are strongly influenced by the solution’s initial [Ni]aq/[Fe]aq ratios and can be understood by considering the differences in the bond chemistry, reaction kinetics and potential post-precipitation recrystallization of Ni- and Fe-sulfides.
When the initial concentration of Ni exceeds Fe ([Ni]aq/ [Fe]aq = 5:1), our TEM analyses indicate that Fe is associated with Ni-sulfides rather than forming separate Fe-sulfide phases. This is consistent with the observation of the presence of Fe impurities in natural millerite and polydymite (Belkin and Luo, 2008). It is apparent through our experiments that the addition of Fe(II) leads to the formation of smaller, less crystalline Ni-sulfide nanoparticles (Figure 3). We hypothesize that this can be explained if Fe is incorporated into the Ni-sulfide crystal lattice. Due to the ∼9% larger ionic radius of Fe(II) than Ni(II) (Shannon, 1976), the incorporation of Fe into Ni-sulfides can lead to structural instability that prevents or delays the formation of more crystalline Ni-sulfides. Confirmation of this factor depends on a more comprehensive approach using density functional theory to account for complex bonding energetics of transition metals in sulfide minerals (Kwon et al., 2015).
At equimolar concentrations of Ni and Fe ([Ni]aq/ [Fe]aq = 1:1), two classes of minerals are appreciably present: Ni-sulfide spheroids (predominantly polyphasic Ni-sulfide precursors of millerite and minor polydmite/violarite) and Fe-rich nanosheets (predominantly mackinawite and likely some pentlandite based on the electron diffraction patterns). The preferential partitioning of Ni and Fe into two separate phases is consistent with the notion that millerite and mackinawite are not likely to be miscible based on the differences in their S-Me bond distances and coordination numbers (Wilkin and Beak, 2017). In the absence of significant post-precipitation mixing, metal partitioning is therefore more likely to be governed by the relative early-stage precipitation kinetics of Ni- and Fe-sulfides. The water exchange kinetics of Ni is about two orders of magnitude slower than Fe, prompting suggestions that Ni should preferably be incorporated into Fe-sulfides rather than forming discrete Ni-sulfides (Morse and Luther, 1999). However, the abundance of Ni-sulfides in our experiments indicates that Ni precipitates faster than (or at least comparable to) Fe at equimolar concentration. This implies that the simple kinetic approach taken previously can lead to erroneous predictions. Rather, a more sophisticated kinetic model that incorporates other factors such as the degree of supersaturation, surface areas and activation energies need to be developed for metal sulfides.
Finally, at the lowest ratio tested ([Ni]aq/[Fe]aq = 1:5), mackinawite is the dominant phase in these systems. In the abiotic systems, Ni is fully associated with mackinawite either by adsorption or by incorporation to form a solid-solution (Fe1–xNixS where maximum x = 0.56; Wilkin and Beak, 2017). This observation implies that mackinawite forms more rapidly than Ni-sulfides in the abiotic systems at this concentration ratio. Interestingly though, discrete Ni-sulfides are still present in the biotic systems at this concentration ratio. This implies that the precipitation kinetics of Ni is enhanced in the presence of D. vulgaris, which is consistent with the micro-environment precipitation theory. As previously discussed, Ni-sulfide solubility decreases with lower pH between pH 4 and 7 while Fe-sulfide solubility increases over the same pH range (Figure 10). This will cause an increase of Ni precipitation kinetics relative to Fe, thus allowing the formation of some discrete Ni-sulfides that will persist once formed.
Environmental Implications
This study explores the form and attributes of Ni-hosting phases in euxinic settings that can precipitate across a range of aqueous Ni-to-Fe ratios and in the presence and absence of SRB. The variables tested in this study are chosen so as to capture the biogeochemical diversity of potential Ni precipitation sites in natural settings. For example, SRB is known to be a primary producer of sulfide in low temperature environments and are expected to be intimately associated with metal sulfide precipitation (Picard et al., 2016). Experiments, however, have shown that even when SRB are present, a significant fraction of the metals may still be precipitated in the bulk solution away from the cell surfaces (Picard et al., 2018; Stanley and Southam, 2018). Therefore, it is relevant to consider and expect that both abiotic and biotic precipitation will occur in tandem in nature. In terms of metal concentrations, we utilize high concentrations that are more similar to highly polluted streams rather than pristine environments. The chosen initial aqueous Ni-to-Fe ratios are comparable to those observed in nature, ranging from Ni-rich and Fe-poor streams receiving inputs from industrial wastewaters, mining wastes and Ni-rich bedrocks (e.g., Periasamy and Namasivayam, 1995; Heikkinen et al., 2002; Moreton et al., 2009; Azizullah et al., 2011) to relatively Fe-rich and Ni-poor water bodies as observed primarily in pristine environments (oceans, rivers, groundwaters) and also in some polluted streams (Thoenen, 1999; Lee et al., 2002; Rahman et al., 2015). The results of our study therefore allow predictive capabilities on the phases controlling Ni availability in euxinic environments across a range of aqueous Ni/Fe ratios (Figure 11).
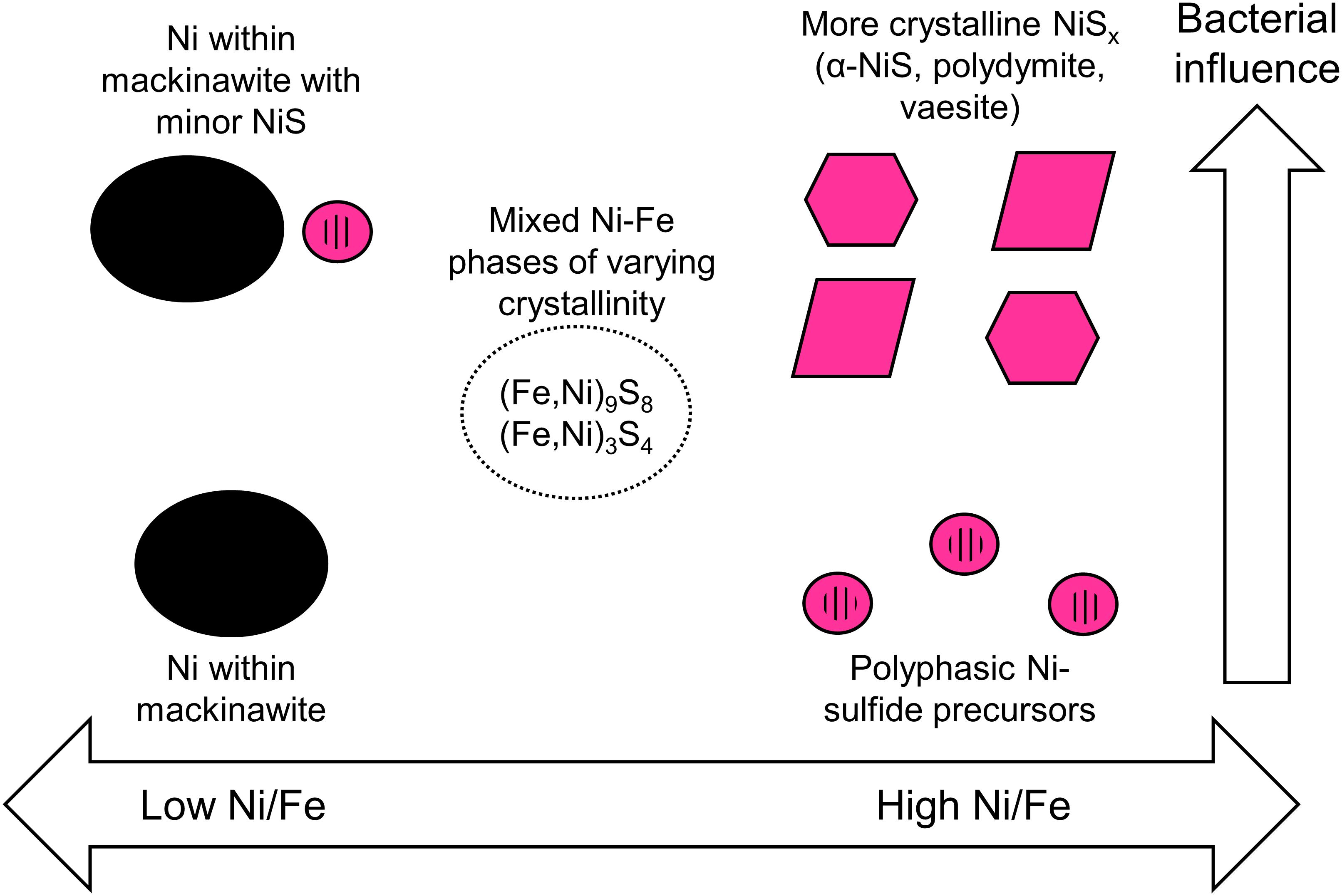
Figure 11. Schematic summary of the predicted Ni-hosting phases in euxinic environments based on the initial [Ni]aq/[Fe]aq ratios in solution and the degree of bacterial influence.
Based on our study, we will expect nanoparticulate Ni-sulfides to be important Ni-hosting phases in Fe-poor euxinic environments. These Ni-sulfides will age and develop over time to more crystalline phases particularly in the presence of SRB. Trace amounts of Fe, however, will likely delay the development to more crystalline phases, making it more likely for Ni to be re-released into solution especially under conditions of rapidly alternating redox potential.
As Fe concentration is increased relative to Ni, one needs to consider the relative fraction of Ni sequestered in Ni-sulfides versus those in association with Fe-sulfides. Our experimental results at [Ni]aq/[Fe]aq = 1:1 indicate that Ni-sulfides will be the primary Ni-hosting phase under this condition, with other Fe-sulfide phases being of secondary importance. When Fe concentration exceeds Ni, one should expect Fe-sulfides such as mackinawite to be the primary Ni-hosting phase. Even though discrete Ni-sulfides are still precipitated in the biotic systems at [Ni]aq/[Fe]aq = 1:5, mass balance dictates that this phase contain at most 7% of the initial Ni fraction (assuming all metals precipitated, a maximum Ni/Fe ratio of 3.84 for the Ni-sulfides and minimum Ni/Fe ratios of 0.16 for the nanosheets). It is important to note that Ni incorporation increases the growth kinetics and thermodynamic stability of mackinawite, with potential implications to the fate of Ni and other various trace metals in natural environments (Kwon et al., 2015; Ikogou et al., 2017).
Data Availability
All datasets generated for this study are included in the manuscript and/or the Supplementary Files.
Author Contributions
MM and JX designed the study. MM performed all the experiments and wrote the first draft of the manuscript. MM and CW conducted the analyses. All authors contributed to manuscript revision and read and approved the submitted version.
Funding
This study was funded by the grant DOE-BES DE-FG02-06ER15786 bestowed by the United States Department of Energy to MH, Mitsu Murayama, and JX, and also by the start-up package provided to JX by The University of Texas at El Paso.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
For permitting us to use their shared facilities, we are grateful to the Virginia Tech National Center for Earth and Environmental Nanotechnology Infrastructure (NanoEarth), a member of the National Nanotechnology Coordinated Infrastructure (NNCI) network, supported by the NSF (NNCI 1542100). NanoEarth is housed at Virginia Tech’s Institute for Critical Technology and Applied Sciences (ICTAS). We thank Debora Berti and Elizabeth Cantando for their help in characterizing the samples.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/feart.2019.00151/full#supplementary-material
Footnotes
References
Abraitis, P. K., Pattrick, R. A. D., and Vaughan, D. J. (2004). Variations in the compositional, textural and electrical properties of natural pyrite: a review. Int. J. Miner. Process. 74, 41–59. doi: 10.1016/j.minpro.2003.09.002
Algeo, T. J., and Maynard, J. B. (2004). Trace-element behavior and redox facies in core shales of Upper Pennsylvanian Kansas-type cyclothems. Chem. Geol. 206, 289–318. doi: 10.1016/j.chemgeo.2003.12.009
Azizullah, A., Khattak, M. N. K., Richter, P., and Häder, D. P. (2011). Water pollution in Pakistan and its impact on public health - a review. Environ. Int. 37, 479–497. doi: 10.1016/j.envint.2010.10.007
Belkin, H. E., and Luo, K. (2008). Late-stage sulfides and sulfarsenides in Lower Cambrian black shale (stone coal) from the Huangjiawan mine, Guizhou Province, People’s Republic of China. Mineral. Petrol. 92, 321–340. doi: 10.1007/s00710-007-0201-9
Benning, L. G., Wilkin, R. T., and Barnes, H. L. (2000). Reaction pathways in the Fe-S system below 1000C. Chem. Geol. 167, 25–51. doi: 10.1016/S0009-2541(99)00198-9
Beveridge, T. J., and Koval, S. F. (1981). Binding of metals to cell envelopes of Escherichia coli K-12. Appl. Environ. Microbiol. 42, 325–335.
Beveridge, T. J., and Murray, R. G. E. (1976). Uptake and retention of metals by cell walls of Bacillus subtilis. J. Bacteriol. 127, 1502–1518.
Bourdoiseau, J. A., Jeannin, M., Rémazeilles, C., Sabot, R., and Refait, P. (2011). The transformation of mackinawite into greigite studied by Raman spectroscopy. J. Raman Spectrosc. 42, 496–504. doi: 10.1002/jrs.2729
Boursiquot, S., Mullet, M., Abdelmoula, M., Génin, J.-M., and Ehrhardt, J.-J. (2001). The dry oxidation of tetragonal FeS1-x mackinawite. Phys. Chem. Miner. 28, 600–611.
Bruland, K. W., Middag, R., and Lohan, M. C. (2013). “Controls of Trace Metals in Seawater,” in Treatise on Geochemistry, 2nd Edn eds H. D. Holland and K. K. Turekian (Amsterdam: Elsevier Ltd.).
Cao, J., Zhang, G., Mao, Z., Fang, Z., and Yang, C. (2009). Precipitation of valuable metals from bioleaching solution by biogenic sulfides. Miner. Eng. 22, 289–295. doi: 10.1016/j.mineng.2008.08.006
Caraballo, M. A., Michel, F. M., and Hochella, M. F. (2015). The rapid expansion of environmental mineralogy in unconventional ways: beyond the accepted definition of a mineral, the latest technology, and using nature as our guide. Am. Mineral. 100, 14–25. doi: 10.2138/am-2015-4749
De Los Ríos, A., Wierzchos, J., Sancho, L. G., and Ascaso, C. (2003). Acid microenvironments in microbial biofilms of antarctic endolithic microecosystems. Environ. Microbiol. 5, 231–237. doi: 10.1046/j.1462-2920.2003.00417.x
De Yoreo, J. J., Gilbert, P. U., Sommerdijk, N. A., Penn, R. L., Whitelam, S., Joester, D., et al. (2015). Crystallization by particle attachment in synthetic, biogenic, and geologic environments. Science 349:aaa6760. doi: 10.1126/science.aaa6760
Downs, R. T., and Hall-Wallace, M. (2003). The American mineralogist crystal structure database. Am. Mineral. 88, 247–250.
Ferris, F., Schultze, S., Witten, T., Fyfe, W., Beveridge, T., and Schultz, S. (1989). Metal interactions with microbial biofilms in acidic and neutral pH environments. Appl. Environ. Microbiol. 55, 1249–1257.
Ferris, F. G., Fyfe, W. S., and Beveridge, T. J. (1987). Bacteria as nucleation sites for authigenic minerals in a metal-contaminated lake sediment. Chem. Geol. 63, 225–232.
Fortin, D., Southam, G., and Beveridge, T. J. (1994). Nickel sulfide, iron-nickel sulfide and iron sulfide precipitation by a newly isolated Desulfotomaculum species and its relation to nickel resistance. FEMS Microbiol. Ecol. 14, 121–132. doi: 10.1111/j.1574-6941.1994.tb00099.x
French, R. A., Caraballo, M., Kim, B., Rimstidt, J. D., Murayama, M., and Hochella, M. F. (2012). The enigmatic iron oxyhydroxysulfate nanomineral schwertmannite: morphology, structure, and composition. Am. Mineral. 97, 1469–1482. doi: 10.2138/am.2012.4032
Frohne, T., Rinklebe, J., Diaz-Bone, R. A., and Du Laing, G. (2011). Controlled variation of redox conditions in a floodplain soil: impact on metal mobilization and biomethylation of arsenic and antimony. Geoderma 160, 414–424. doi: 10.1016/j.geoderma.2010.10.012
Gall, L., Williams, H. M., Siebert, C., Halliday, A. N., Herrington, R. J., and Hein, J. R. (2013). Nickel isotopic compositions of ferromanganese crusts and the constancy of deep ocean inputs and continental weathering effects over the Cenozoic. Earth Planet Sci. Lett. 375, 148–155. doi: 10.1016/j.epsl.2013.05.019
Gilbert, B., and Banfield, J. F. (2005). Molecular-scale processes involving nanoparticulate minerals in biogeochemical systems. Rev. Min. Geochem. 59, 109–155.
Gramp, J. P., Sasaki, K., Bigham, J. M., Karnachuk, O. V., and Tuovinen, O. H. (2006). Formation of covellite (CuS) under biological sulfate-reducing conditions. Geomicrobiol. J. 23, 613–619. doi: 10.1080/01490450600964383
Grguric, B. A. (2002). Hypogene violarite of exsolution origin from Mount Keith, Western Australia: field evidence for a stable pentlandite–violarite tie line. Mineral. Mag. 66, 313–326. doi: 10.1180/0026461026620032
Guibaud, G., Comte, S., Bordas, F., Dupuy, S., and Baudu, M. (2005). Comparison of the complexation potential of extracellular polymeric substances (EPS), extracted from activated sludges and produced by pure bacteria strains, for cadmium, lead and nickel. Chemosphere 59, 629–638. doi: 10.1016/j.chemosphere.2004.10.028
Haider, S., Grau-Crespo, R., Devey, A. J., and De Leeuw, N. H. (2012). Cation distribution and mixing thermodynamics in Fe/Ni thiospinels. Geochim. Cosmochim. Acta 88, 275–282. doi: 10.1016/j.gca.2012.04.007
Heidelberg, J. F., Seshadri, R., Haveman, S. A., Hemme, C. L., Paulsen, I. T., Kolonay, J. F., et al. (2004). The genome sequence of the anaerobic, sulfate-reducing bacterium Desulfovibrio vulgaris Hildenborough. Nat. Biotechnol. 22, 554–559. doi: 10.1038/nbt959
Heikkinen, P. M., Korkka-Niemi, K., Lahti, M., and Salonen, V. P. (2002). Groundwater and surface water contamination in the area of the Hitura nickel mine, Western Finland. Environ. Geol. 42, 313–329. doi: 10.1007/s00254-002-0525-z
Hidalgo, G., Burns, A., Herz, E., Hay, A. G., Houston, P. L., Wiesner, U., et al. (2009). Functional tomographic fluorescence imaging of pH microenvironments in microbial biofilms by use of silica nanoparticle sensors. Appl. Environ. Microbiol. 75, 7426–7435. doi: 10.1128/AEM.01220-09
Hindersmann, I., and Mansfeldt, T. (2014). Trace element solubility in a multimetal-contaminated soil as affected by redox conditions. Water Air Soil Pollut. 225:2158. doi: 10.1007/s11270-014-2158-8
Hochella, M. F., Lower, S. K., Maurice, P. A., Penn, R. L., Sahai, N., Sparks, D. L., et al. (2008). Nanominerals, mineral nanoparticles, and earth systems. Science 319, 1631–1635. doi: 10.1126/science.1141134
Houben, G. J., Sitnikova, M. A., and Post, V. E. A. (2017). Terrestrial sedimentary pyrites as a potential source of trace metal release to groundwater – A case study from the Emsland. Germany. Appl. Geochem. 76, 99–111. doi: 10.1016/j.apgeochem.2016.11.019
Huang, S., Harris, K. D. M., Lopez-Capel, E., Manning, D. A. C., and Rickard, D. (2009). “Amorphous nickel sulfide” is hydrated nanocrystalline NiS with a core-shell structure. Inorg. Chem. 48, 11486–11488. doi: 10.1021/ic901512z
Huang, S., Lopez-Capel, E., Manning, D. A. C., and Rickard, D. (2010). The composition of nanoparticulate nickel sulfide. Chem. Geol. 277, 207–213. doi: 10.1016/j.chemgeo.2010.08.001
Huerta-Diaz, M. A., and Morse, J. W. (1992). Pyritization of trace metals in anoxic sediments. Geochim. Cosmochim. Acta 56, 2681–2702.
Huerta-Diaz, M. A., Tessier, A., and Carignan, R. (1998). Geochemistry of trace metals associated with reduced sulfur in freshwater sediments. Appl. Geochem. 13, 213–233. doi: 10.1016/S0883-2927(97)00060-7
Hunger, S., and Benning, L. G. (2007). Greigite: a true intermediate on the polysulfide pathway to pyrite. Geochem. Trans. 8, 1–20. doi: 10.1186/1467-4866-8-1
Hunter, R. C., and Beveridge, T. J. (2005). Application of a pH-sensitive gluoroprobe (C-SNARF-4) for pH microenvironment analysis in Pseudomonas aeruginosa biofilms. Appl. Environ. Microbiol. 71, 2501–2510. doi: 10.1128/AEM.71.5.2501
Ikogou, M., Ona-Nguema, G., Juillot, F., Le Pape, P., Menguy, N., Richeux, N., et al. (2017). Long-term sequestration of nickel in mackinawite formed by Desulfovibrio capillatus upon Fe(III)-citrate reduction in the presence of thiosulfate. Appl. Geochem. 80, 143–154. doi: 10.1016/j.apgeochem.2017.02.019
Jeong, Y. U., and Manthiram, A. (2001). Synthesis of nickel sulfides in aqueous solutions using sodium dithionite. Inorg. Chem. 40, 73–77. doi: 10.1021/ic000819d
Karbanee, N., Van Hille, R. P., and Lewis, A. E. (2008). Controlled nickel sulfide precipitation using gaseous hydrogen sulfide. Ind. Eng. Chem. Res. 47, 1596–1602. doi: 10.1021/ie0711224
Kiran, M. G., Pakshirajan, K., and Das, G. (2015). Heavy metal removal using sulfate-reducing biomass obtained from a lab-scale upflow anaerobic-packed bed reactor. Environ. Eng. 142, 1–8. doi: 10.1061/(ASCE)EE.1943-7870.0001005
Kwon, K. D., Refson, K., and Sposito, G. (2015). Transition metal incorporation into mackinawite (tetragonal FeS). Am. Mineral. 100, 1509–1517. doi: 10.2138/am-2015-5211ccbyncnd
Lee, G., Bigham, J. M., and Faure, G. (2002). Removal of trace metals by coprecipitation with Fe, Al and Mn from natural waters contaminated with acid mine drainage in the Ducktown Mining District, Tennessee. Appl. Geochem. 17, 569–581. doi: 10.1016/S0883-2927(01)00125-1
Lewis, A., and Swartbooi, A. (2006). Factors affecting metal removal in mixed sulfide precipitation. Chem. Eng. Technol. 29, 277–280. doi: 10.1002/ceat.200500365
Lewis, A. E. (2010). Review of metal sulphide precipitation. Hydrometallurgy 104, 222–234. doi: 10.1016/j.hydromet.2010.06.010
Mansor, M., Berti, D., Hochella, M. F., Murayama, M., and Xu, J. (2019). Phase, morphology, elemental composition and formation mechanisms of biogenic and abiogenic Fe-Cu-sulfide nanoparticles: a comparative study on their occurrences under anoxic conditions. Am. Mineral. 104, 703–717.
Mera, M. U., Kemper, M., Doyle, R., and Beveridge, T. J. (1992). The membrane-induced proton motive force influences the metal binding ability of Bacillus subtilis cell walls. Appl. Environ. Microbiol. 58, 3837–3844.
Moreau, J. W., Weber, P. K., Martin, M. C., Gilbert, B., Hutcheon, I. D., and Banfield, J. F. (2007). Extracellular proteins limit the dispersal of biogenic nanoparticles. Science 316, 13–16. doi: 10.1126/science.1141064
Moreton, B. M., Fernandez, J. M., and Dolbecq, M. B. D. (2009). Development of a field preconcentration/elution unit for routine determination of dissolved metal concentrations by ICP-OES in marine waters: application for monitoring of the New Caledonia Lagoon. Geostand. Geoanal. Res. 33, 205–218. doi: 10.1111/j.1751-908X.2009.00899.x
Morse, J. W., and Luther, G. W. (1999). Chemical influence on trace metal-sulfide interactions in anoxic sediments. Geochim. Cosmochim. Acta 63:3378.
Navrotsky, A., Mazeina, L., and Majzlan, J. (2008). Size-driven structural and thermodynamic complexity in iron oxides. Science 319, 1635–1638. doi: 10.1126/science.1148614
Newbury, D. E., and Ritchie, N. W. M. (2014). Performing elemental microanalysis with high accuracy and high precision by scanning electron microscopy/silicon drift detector energy-dispersive X-ray spectrometry (SEM/SDD-EDS). J. Mater. Sci. 50, 493–518. doi: 10.1007/s10853-014-8685-2
Noël, V., Morin, G., Juillot, F., Marchand, C., Brest, J., Bargar, J. R., et al. (2015). Ni cycling in mangrove sediments from New Caledonia. Geochim. Cosmochim. Acta 169, 82–98. doi: 10.1016/j.gca.2015.07.024
Pankhania, I. P., Gow, L. A., and Hamilton, W. A. (1986). The effect of hydrogen on the growth of Desulfovibrio vulgaris (Hildenborough) on lactate. J. Gen. Microbiol. 132, 3349–3356. doi: 10.1099/00221287-132-12-3349
Periasamy, K., and Namasivayam, C. (1995). Removal of nickel(II) from aqueous solution and nickel plating industry wastewater using an agricultural waste: Peanut hulls. Water Manag. 15, 63–68. doi: 10.1016/0956-053X(94)00071-S
Picard, A., Gartman, A., Clarke, D. R., and Girguis, P. R. (2018). Sulfate-reducing bacteria influence the nucleation and growth of mackinawite and greigite. Geochim. Cosmochim. Acta 220, 367–384. doi: 10.1016/j.gca.2017.10.006
Picard, A., Gartman, A., and Girguis, P. R. (2016). What do we really know about the role of microorganisms in iron sulfide mineral formation? Front. Earth Sci. 4:68. doi: 10.3389/feart.2016.00068
Quintelas, C., Rocha, Z., Silva, B., Fonseca, B., Figueiredo, H., and Tavares, T. (2009). Biosorptive performance of an Escherichia coli biofilm supported on zeolite NaY for the removal of Cr(VI), Cd(II), Fe(III) and Ni(II). Chem. Eng. J. 152, 110–115. doi: 10.1016/j.cej.2009.03.039
Ragsdale, S. W. (2009). Nickel-based enzyme systems. J. Biol. Chem. 284, 18571–18575. doi: 10.1074/jbc.R900020200
Rahman, M. M., Dong, Z., and Naidu, R. (2015). Concentrations of arsenic and other elements in groundwater of Bangladesh and West Bengal, India: potential cancer risk. Chemosphere 139, 54–64. doi: 10.1016/j.chemosphere.2015.05.051
Reis, F. D., Silva, A. M., Cunha, E. C., and Leão, V. A. (2013). Application of sodium- and biogenic sulfide to the precipitation of nickel in a continuous reactor. Sep. Purif. Technol. 120, 346–353. doi: 10.1016/j.seppur.2013.09.023
Rickard, D. (2006). The solubility of FeS. Geochim. Cosmochim. Acta 70, 5779–5789. doi: 10.1016/j.gca.2006.02.029
Rickard, D., Griffith, A., Oldroyd, A., Butler, I. B., Lopez-Capel, E., Manning, D. A. C., et al. (2006). The composition of nanoparticulate mackinawite, tetragonal iron(II) monosulfide. Chem. Geol. 235, 286–298. doi: 10.1016/j.chemgeo.2006.07.004
Rickard, D., and Luther, G. W. (2006). Metal sulfide complexes and clusters. Rev. Mineral. Geochem. 61, 421–504. doi: 10.2138/rmg.2006.61.8
Rickard, D., and Luther, G. W. (2007). Chemistry of iron sulfides. Chem. Rev. 107, 514–562. doi: 10.1021/cr0503658
Rinklebe, J., and Shaheen, S. M. (2017). Redox chemistry of nickel in soils and sediments: a review. Chemosphere 179, 265–278. doi: 10.1016/j.chemosphere.2017.02.153
Rudnick, R. L., and Gao, S. (2003). Composition of the continental crust. Treat. Geochem. 3, 1–64. doi: 10.1016/B0-08-043751-6/03016-4
Sampaio, R. M. M., Timmers, R. A., Kocks, N., André, V., Duarte, M. T., Van Hullebusch, E. D., et al. (2010). Zn-Ni sulfide selective precipitation: the role of supersaturation. Sep. Purif. Technol. 74, 108–118. doi: 10.1016/j.seppur.2010.05.013
Schoonen, M. A., and Barnes, H. L. (1991). Reactions forming pyrite and marcasite from solution: II. Via FeS precursors below 100°C. Geochim. Cosmochim. Acta 55, 1505–1514. doi: 10.1016/0016-7037(91)90123-M
Sclater, F. R., Boyle, E. A., and Edmond, J. M. (1976). On the marine geochemistry of nickel. Earth Planet. Sci. Lett. 31, 119–128.
Shannon, R. D. (1976). Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. 32, 751–767.
Sitte, J., Pollok, K., Langenhorst, F., and Küsel, K. (2013). Nanocrystalline nickel and cobalt sulfides formed by a heavy metal-tolerant, sulfate-reducing enrichment culture. Geomicrobiol. J. 30, 36–47. doi: 10.1080/01490451.2011.653082
Southam, G. (2000). “Bacterial surface-mediated mineral formation,” in Environmental Microbe-Metal Interactions, ed. D. R. Lovley (Washington, DC: American Society of Microbiology), 257–276.
Stanley, W., and Southam, G. (2018). The effect of gram-positive (Desulfosporosinus orientis) and gram-negative (Desulfovibrio desulfuricans) sulfate-reducing bacteria on iron sulfide mineral precipitation. Can. J. Microbiol. 64, 629–637. doi: 10.1139/cjm-2017-0545
Tenailleau, C., Pring, A., Etschmann, B., Brugger, J., Grguric, B., and Putnis, A. (2006). Transformation of pentlandite to violarite under mild hydrothermal conditions. Am. Mineral. 91, 706–709. doi: 10.2138/am.2006.2131
Thoenen, T. (1999). Pitfalls in the use of solubility limits for radioactive waste disposal: the case of nickel sulfidic groundwaters. Nucl. Technol. 126, 75–87. doi: 10.13182/NT99-A2959
Tribovillard, N., Algeo, T. J., Lyons, T., and Riboulleau, A. (2006). Trace metals as paleoredox and paleoproductivity proxies: an update. Chem. Geol. 232, 12–32. doi: 10.1016/j.chemgeo.2006.02.012
Vaughan, D. J., and Craig, J. R. (1985). The crystal chemistry of iron-nickel thiospinels. Am. Mineral. 70, 1036–1043. doi: 10.1016/j.apsusc.2007.07.027
Voordouw, G. (2002). Carbon monoxide cycling by Desulfovibrio vulgaris hildenborough. J. Bacteriol. 184, 5903–5911. doi: 10.1128/JB.184.21.5903
Wang, H., Pring, A., Ngothai, Y., and O’Neill, B. (2006). The kinetics of the α→β transition in synthetic nickel monosulfide. Am. Mineral. 91, 171–181. doi: 10.2138/am.2006.1962
Wang, L., Schultz, M., and Matijeviæ, E. (1997). Preparation and properties of uniform amorphous and crystalline colloidal nickel sulfide. Colloid Polym. Sci. 275, 593–598. doi: 10.1007/s003960050123
Wang, Q., and Morse, J. W. (1996). Pyrite formation under conditions approximating those in anoxic sediments I. pathway and morphology. Mar. Chem. 52, 99–121. doi: 10.1016/0304-4203(95)00082-8
Weber, F. A., Voegelin, A., and Kretzschmar, R. (2009). Multi-metal contaminant dynamics in temporarily flooded soil under sulfate limitation. Geochim. Cosmochim. Acta 73, 5513–5527. doi: 10.1016/j.gca.2009.06.011
WHO (2007). Nickel in Drinking-Water: Background Document for Development of WHO Guidelines for Drinking-Water Quality. Geneva: WHO.
Wilkin, R. T., and Beak, D. G. (2017). Uptake of nickel by synthetic mackinawite. Chem. Geol. 462, 15–29. doi: 10.1016/j.chemgeo.2017.04.023
Wilkin, R. T., and Rogers, D. A. (2010). Nickel sulfide formation at low temperature: initial precipitates, solubility and transformation products. Environ. Chem. 7, 514–523. doi: 10.2166/wrd.2014.068
Wolthers, M., Van der Gaast, S. J., and Rickard, D. (2003). The structure of disordered mackinawite. Am. Mineral. 88, 2007–2015. doi: 10.1021/acs.est.5b04281
Wuertz, S., Muller, E., Spaeth, R., Pfleiderer, P., and Flemming, H.-C. (2000). Detection of heavy metals in bacterial biofilms and microbial flocs with the fluorescent complexing agent Newport Green. J. Ind. Microbiol. Biotechnol. 24, 116–123. doi: 10.1038/sj.jim.2900784
Xia, F., Brugger, J., Chen, G., Ngothai, Y., O’Neill, B., Putnis, A., et al. (2009). Mechanism and kinetics of pseudomorphic mineral replacement reactions: a case study of the replacement of pentlandite by violarite. Geochim. Cosmochim. Acta 73, 1945–1969. doi: 10.1016/j.gca.2009.01.007
Xu, J., Murayama, M., Roco, C. M., Veeramani, H., Michel, F. M., Rimstidt, J. D., et al. (2016). Highly-defective nanocrystals of ZnS formed via dissimilatory bacterial sulfate reduction: a comparative study with their abiogenic analogues. Geochim. Cosmochim. Acta 180, 1–14. doi: 10.1016/j.gca.2016.03.036
Keywords: nickel sulfides, millerite, polydymite, vaesite, sulfate-reducing bacteria, polyphasic, sulfidic environments, mackinawite
Citation: Mansor M, Winkler C, Hochella MF Jr and Xu J (2019) Nanoparticulate Nickel-Hosting Phases in Sulfidic Environments: Effects of Ferrous Iron and Bacterial Presence on Mineral Formation Mechanism and Solid-Phase Nickel Distribution. Front. Earth Sci. 7:151. doi: 10.3389/feart.2019.00151
Received: 08 March 2019; Accepted: 28 May 2019;
Published: 26 June 2019.
Edited by:
Alejandro Fernandez-Martinez, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Mariette Wolthers, Utrecht University, NetherlandsLars Ehm, Stony Brook University, United States
Copyright © 2019 Mansor, Winkler, Hochella and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Muammar Mansor, bWJtYW5zb3JAdXRlcC5lZHU=; bXVhbW1hcjEwQGdtYWlsLmNvbQ==; Jie Xu, anh1MkB1dGVwLmVkdQ==